
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchical α-MnO2 nanowires as an efficient anode material for rechargeable lithium-ion batteries†
Ediga
Umeshbabu
 *ab,
M.
Satyanarayana
c,
Guruprakash
Karkera
b,
Ashok
Pullamsetty
d and
P.
Justin
*e
*ab,
M.
Satyanarayana
c,
Guruprakash
Karkera
b,
Ashok
Pullamsetty
d and
P.
Justin
*e
aDepartment of Chemistry, Indian Institute of Technology Madras, Chennai - 600036, India. E-mail: umeshediga@gmail.com
bHelmholtz Institute Ulm (HIU) for Electrochemical Energy Storage, Helmholtzstraße 11, D-89081 Ulm, Germany
cDepartment of Chemistry, Faculty of Exact Sciences, Bar-Ilan University, Ramat-Gan 5290002, Israel
dDepartment of Physics, Indian Institute of Technology Madras, Chennai - 600036, India
eDepartment of Chemistry, Rajiv Gandhi University of Knowledge Technologies, RK Valley, Kadapa 516330, Andhra Pradesh, India. E-mail: ponjustin@rgukt.in
First published on 15th December 2021
Abstract
In this work, a bulk quantity of α-MnO2 hierarchical nanowires was synthesized by a facile redox reaction between potassium permanganate (KMnO4) and glycine under ambient conditions. The physicochemical characterization results revealed that the MnO2 sample exhibited highly uniform (1D) nanowire morphology and unique surface texture features such as a high specific surface area of 181 m2 g−1 with a beneficial mesoporous structure. Taking the advantage of hierarchical 1D nanowires morphology and improved microstructural properties, a cell with MnO2 nanowires as an anode showed relatively good electrochemical performance with enhanced lithiation degree and improved cycling stability. The initial discharge capacity of as high as 1373 mA h g−1 was achieved at 100 mA g−1 and rendered a reversible capacity of 735 mA h g−1 even after 100 cycles with an average Coulombic efficiency of 97%. The improved electrochemical performance of α-MnO2 is attributed to its hierarchical nanowire morphology and enriched textural surface properties, which provide a good Li-ion diffusion path, large electrode–electrolyte contact area and effective accommodation of the strain generated from volume expansion during repeated lithiation/delithiation processes.
1. Introduction
Among various kinds of electrochemical energy storage devices, lithium-ion batteries (LIBs) have been extensively used in many electronic applications throughout the past decades as primary sources of power. In recent years, they have been explored as promising candidates for electric vehicle applications due to their advantages of high gravimetric/volumetric energy, high power density, prolonged cycle-life, and low self-discharge properties.1–4 Traditionally, graphite is used as an anode material in commercial LIBs due to its layered structure that facilitates the Li+ ion intercalation effectively. The limited theoretical specific capacity and poor rate capability of graphite are inadequate to power high energy demands for electric vehicle applications.5 Therefore, it is imperative to develop highly efficient and advanced anode materials in building next-generation Li batteries with high specific capacity and high energy densities, so as to meet the ever-growing performance demands.3,6–9Transition metal oxides (MOx; M = Ni, Co, Fe, Nb, Mo, and so forth)8–15 have been researched intensively as promising alternative anode materials for LIBs because they are suggested to have extremely high reversible capacities of about three times higher than that of graphite (2–3 times that of graphite) owing to their unique conversion type reactions (MOx + 2xLi+ + 2xe− ↔ M + xLi2O) and ability to tune the working voltage as well as the energy density by selecting the metal type.16 Moreover, these oxides are low-cost and environmentally benign. Among metal oxides, MnO2 is regarded as one of the promising anode material for LIBs due to its inherent advantages such as natural abundance, low-cost, environmental benignity and large theoretical capacity of 1230 mA h g−1.17–19 In addition, MnO2 can crystallize in different kinds of polymorphs, i.e., α-, β-, γ-, λ- δ- and ε-types, which are connected by the basic unit, [MnO6] octahedron, by sharing edges or corners. As a result, they possess tunnels or interlayers with gaps of different magnitudes, which provide effective diffusion pathways for Li+ ions.19–23 Among phases, α-MnO2 shows adequate Li+ charge storage ability since it has a hollandite-type crystal structure with relatively large 1D [2 × 2] tunnels formed via interlinked octahedral [MnO6] units, which enable easier and faster ionic transport and electrode reaction kinetics within the MnO2 framework.24,25 In addition, the hollow-interior structures can minimize the volume expansion during the charging/discharging process. However, several intrinsic problems existing in MnO2 include low electronic conductivity and pulverization of small particles during the Li+ ion insertion/extraction process, which results in poor rate performance and rapid capacity fading during long cycling.18,26
In order to relieve the aforementioned inherent difficulties and thereby improve battery performance with good cycling stability, nanotechnology (fabricating MnO2 in nanostructures or hybrid composites with a conductive matrix such as graphene, polymers, etc.) has become one of the efficient approach.18,26–32 The physicochemical and electrochemical properties of MnO2 are significantly influenced by its crystalline structure, morphology and synthetic methods as well as its compositional characteristics. For example, Li et al. synthesized hollow-urchin-like MnO2 by a low-temperature mild reduction route, which showed a discharge capacity of 481 mA h g−1 after 40 cycles.27 Chen et al. reported MnO2 nanorods using a hydrothermal method that provided a discharge capacity of 930 mA h g−1 after 100 cycles.28 A coaxial MnO2/carbon nanotube array was reported by Reddy et al. fabricated using a chemical vapor deposition (CVD) technique and the hybrid electrode material delivered a very high initial capacity of 2000 mA h g−1 at 50 mA g−1; however, the discharge capacity faded very fast with only 500 mA h g−1, about 25% of the initial capacity at 15th cycle.31 Nonetheless, such methods have multitudinous shortfalls, such as complex time-intensive steps and high equipment costs. Indeed, some of the methods, mainly the utilization of CVD gets lower yield. Therefore, developing MnO2 nanomaterials through simple, environmentally-friendly and cost-effective methods are imperative.
In this work, we employed a simple, inexpensive and straightforward approach to synthesize hierarchical α-MnO2 nanowires through a reaction involving KMnO4 and glycine. This approach involves simple mixing of the precursors in the solution medium and subsequent annealing. Moreover, glycine is a non-toxic, biodegradable and relatively cheap amino acid whose amino group can effectively undergo ionic interaction with Mn7+ ions and reduce KMnO4 successfully to produce MnO2 at room temperature.33 The combined physicochemical and electrochemical measurements were performed on the MnO2 sample to scrutinize the structural and charge/discharge performance. The prepared material exhibits α-phase and hierarchical 1D-nanowire morphology with high specific surface area and incumbent porous structure, which is promising in providing inflated conductive networks with intimate electrode–electrolyte contact and short diffusion lengths for Li+ ions during charge/discharge. Benefiting from such a nanoarchitecture, the α-MnO2 electrode showed a high reversible capacity, excellent cycling stability of over 100 cycles and high rate capability.
2. Experimental section
2.1 Synthesis of α-MnO2 nanowires
All chemical reagents used in this study were of analytical grade and were used without any further purification. α-MnO2 nanowires were prepared by a simple and straightforward in situ redox reaction followed by a heat-treatment. In a typical reaction, 5 mmol of solid KMnO4 crystals were dissolved in 100 mL of deionized (DI) water under vigorous magnetic stirring until a transparent purple color solution appeared. To this, 5 mmol of glycine (amino acid and commonly used as a reducing agent) was separately dissolved in 25 mL DI water was added slowly under vigorous magnetic stirring. The total solution mixture was stirred for another 3 h. The purple-colored KMnO4 solution gradually changed to dark brown residue due to the in situ redox reaction between permanganate and glycine. The obtained precipitate was centrifuged, washed extensively with copious amounts of DI water followed by washing with absolute ethanol to eliminate the unreacted reactants if any. The collected residue was air-dried overnight, and then calcined in a muffle furnace at 400 °C for 3 h at a ramp rate of 5 °C min−1 in the air to engender well-crystalline α-MnO2 nanowires.2.2 Materials characterization
Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) were performed using a TA make TGA Q500V20.10 Build 36 instrument. For TGA/DTA measurement, the sample was heated from room temperature to 750 °C at a ramp rate of 20 °C min−1 in air. XRD was performed on a Bruker AXS D8 advanced diffractometer, using Cu Kα radiation (λ = 1.5418 Å). The phase and lattice constants were identified by Rietveld refinement analysis using the General Structure Analysis System (GSAS) software. A Horiba HR 800UV Raman spectrometer was used with a laser excitation of 632.8 nm to obtain the Raman spectrum. The surface area and pore-size distribution of the sample were obtained using BET (Brunauer–Emmett–Teller) and BJH (Barret–Joyner–Halenda) methods from the data collected on a Micromeritics ASAP 2020 physisorption tool. The microstructure and morphology of the sample were characterized using field emission scanning electron microscopy (FESEM; FEI, Quanta 400). TEM and high-resolution TEM images were obtained using a JEOL JEM 3010 instrument. Prior to TEM measurements, the sample powder was suspended in ethanol by sonication for 5 min and then dispersed on a carbon-coated Cu grid. X-ray photoelectron spectroscopy (XPS) measurements were performed on a PHOIBOS 150 XPS (SPECS-Surface concept) spectrometer with an exciting source of Al Kα = 1486.6 eV. The spectral data were calibrated to 284.6 eV (C 1s).2.3 Electrochemical measurements
The electrochemical properties were measured using galvanostatic charge/discharge (GCD), cyclic voltammetry (CV) and AC impedance spectroscopy in assembled CR2016-type coin cells. The working electrodes were fabricated by mixing 75 wt% α-MnO2 nanowires, 15 wt% acetylene black, and 10 wt% PVDF (polyvinylidene fluoride) binder in a ball-milled with the required amount of N-methyl-2-pyrrolidone (NMP) solvent to form a homogeneous slurry. Subsequently, the slurry was painted over a Cu foil current collector using a doctor blade. Then, the electrodes were dried in a vacuum oven at 80 °C and then punched into disks to meliorate the contact among copper foil, active materials, and conductive carbon. The active material loading on each electrodes was 2.0 mg cm−2. Preliminary cell tests were performed using a CR2025 type coin cell with Li metal as the anode and a Celgard 2500 polypropylene separator. The electrolyte solution was 1 M LiPF6 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio of EC and DMC. The coin cells were assembled in an Ar-filled glove box (O2 and H2O < 0.1 ppm). The cell's discharge–charge cycling was between 0.05 and 3.0 V (vs. Li+/Li) and was conducted on a LAND battery testing system. Cyclic voltammetry (0.005–3.0 V, 1 mV s−1) and impedance spectroscopy (from 0.1 Hz to 1 MHz) were performed on a Versa-STAT (Princeton Applied Research) electrochemical work station. All electrochemical studies were conducted at room temperature.
:1 volume ratio of EC and DMC. The coin cells were assembled in an Ar-filled glove box (O2 and H2O < 0.1 ppm). The cell's discharge–charge cycling was between 0.05 and 3.0 V (vs. Li+/Li) and was conducted on a LAND battery testing system. Cyclic voltammetry (0.005–3.0 V, 1 mV s−1) and impedance spectroscopy (from 0.1 Hz to 1 MHz) were performed on a Versa-STAT (Princeton Applied Research) electrochemical work station. All electrochemical studies were conducted at room temperature.
3. Results and discussion
3.1 Morphology and structural characterization
The schematic illustration for the growth mechanism of α-MnO2 1D nanowires under a solution-processed redox-reaction is shown in Scheme 1. The morphology and crystalline evolution of α-MnO2 nanowires typically involve (i) nucleation of nascent MnOx nuclei through the reduction between permanganate MnO4− and glycine, (ii) progression to nanosized particles, (iii) Ostwald ripening and self-assembling and (iv) expeditious self-transformation to hierarchical nanowires under high-temperature calcination.34,35 The morphology of the uncalcined precursor showed an irregular agglomerated surface structure (Fig. S1A, ESI†). On heat treatment, the agglomerated surface structure transformed into well-defined 1D nanowires of lengths of tens of micrometers and a diameter of 15–25 nm. This unique nanowire morphology can provide better electrochemical accessibility to Li+ ions, leading to better performance towards charge storage characteristics. During the growth process of nanowires, several intrinsic and extrinsic forces and other parameters influence the shape, orientations, growth of crystallites and the final morphology of oxide.21,35–37 | ||
| Scheme 1 A Schematic illustration for the formation process of α-MnO2 nanowires under a solution-processed redox-reaction between permanganate and glycine. | ||
To monitor the thermal stability and phase evaluation of the as-prepared MnOx precursor, we performed thermogravimetric (TGA) and differential thermal analysis (DTA) and the respective patterns are shown in Fig. 1. The TGA profile shows a total weight loss of 16.16% up to a temperature of 470 °C. A steep decrease in weight loss up to 200 °C corresponded to the removal of surface-adsorbed as well as uncoordinated interlayer water and then gradual weight loss from 200 to 470 °C corresponding to the loss of water molecules from the tunnel cavity.38–40 A small weight (0.71%) increment was observed in the sample at 506 °C due to the compensation of released oxygen produced by the oxidation of subvalent Mn cations to Mn(IV) leading to the phase transformation from the layered structure to tetragonal the α-MnO2 phase.40–42 Moreover, the weight loss observed at about 665 °C, corresponds to the phase transformation of MnO2 to Mn2O3.39,41 The XRD pattern of the uncalcined precursor in Fig. S1B (ESI†) exhibits broad diffraction peaks at about 12°, 25°, 37.2° and 66.2°, which can be indexed to amorphous MnO2 phase with poorly crystalline nature.43
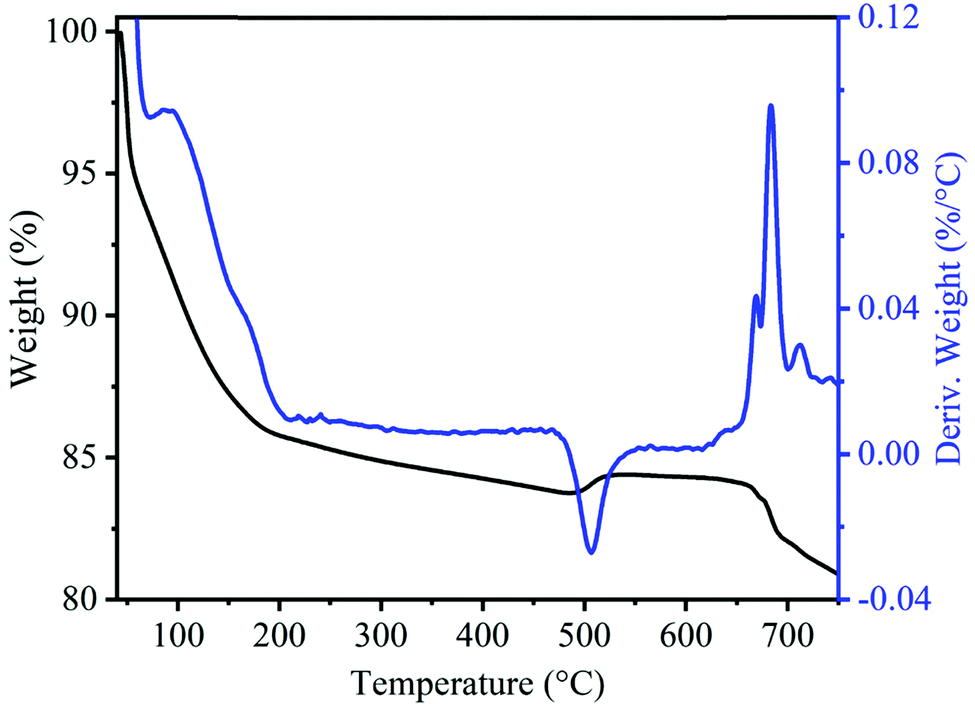 | ||
| Fig. 1 TGA and DTG curves of uncalcined precursors of MnO2 sample. | ||
The crystal structure of α-MnO2, as shown in Fig. 2A and B encompass double chains of the edge-shared MO6 octahedra, which are interconnected at corners to form (2 × 2) and (1 × 1) tunnels in the tetragonal unit cell. The XRD pattern and Rietveld refinement affirmed the phase purity and lattice structure of the prepared MnO2. Rietveld refinement of XRD patterns was executed by the General Structure Analysis System (GSAS) and corresponding results are depicted in Fig. 2C. All reflections in the XRD pattern can be indexed to the tetragonal α-MnO2 with a space group I4/m (JCPDS no# 42-1348). Importantly, we did not find any impurity peaks in the XRD pattern, signifying that the as-prepared α-MnO2 sample is a pure phase.21 The better χ2 (∼2.1) reveals that the Rietveld refinement exercise is a good fit with the experimental data. Rp and Rwp values are 3.9% and 5.4%, respectively. The refined lattice constants are a = b = 9.842 Å and c = 2.858 Å, which are consistent with the literature values of a = b = 9.871 Å and c = 2.858 Å.44 Further, the average crystallite size of α-MnO2 was calculated to be 15.3 nm using Scherrer's equation.21 Raman spectrum of α-MnO2, as shown in Fig. 3, unveils well-resolved Raman bands located at 302, 385, 571 and 633 cm−1 that are characteristic of the α-MnO2 phase.45,46 It is worth noting that all materials with the α-MnO2 crystal phase reported two diagnostic peaks at around 571 and 633 cm−1. The former peak is the M–O stretching vibration in the basal plane of the MnO6 sheet, while the latter corresponds to the M–O symmetric stretching vibration of the MnO6 groups.26,46
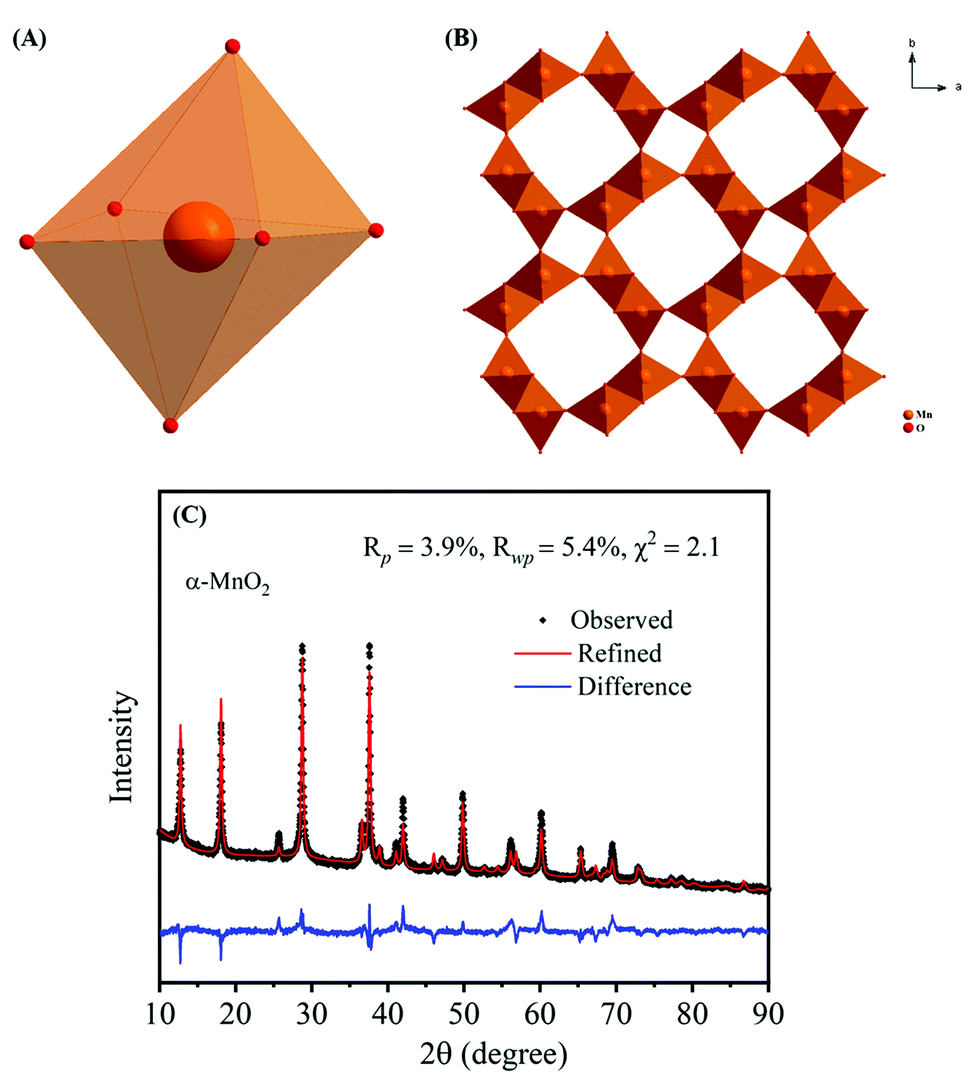 | ||
| Fig. 2 Crystallographic structures of α-MnO2: (A) MnO6 octahedron structural unit and (B) α-MnO2 (2 × 2 tunnels); (C) X-ray Rietveld refinement results of the α-MnO2 sample. | ||
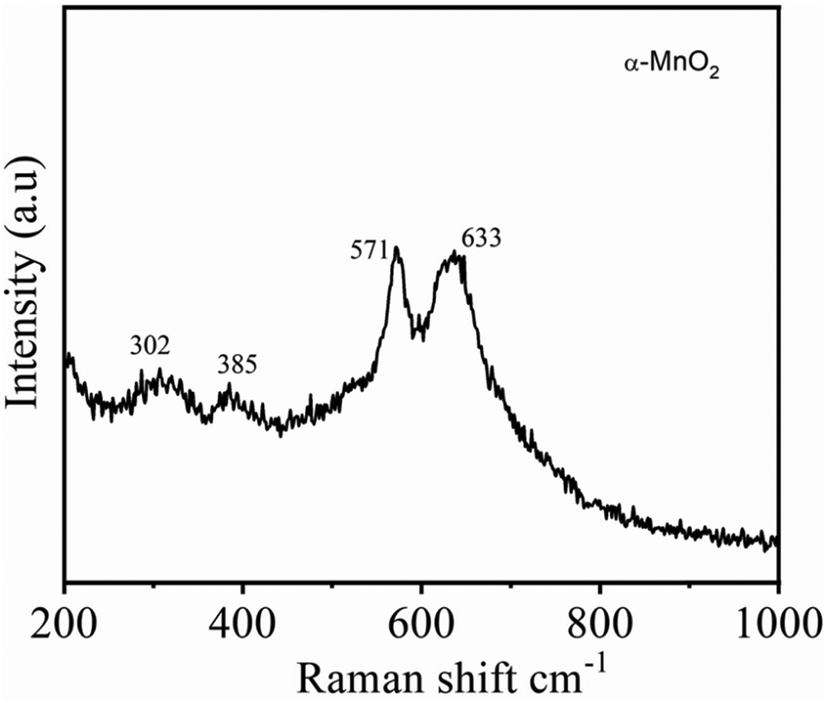 | ||
| Fig. 3 Raman spectra of the α-MnO2 nanowires. | ||
To inspect the morphology and microstructure of the prepared α-MnO2, scanning electron microscopy and transmission electron microscopy were performed and the corresponding results are shown in Fig. 4 and 5, respectively. From the low-magnification SEM images (Fig. 4A and B), the MnO2 sample shows hierarchical ultralong 1D nanowires morphology. The high-magnification SEM in Fig. 4C and D unveils the nanowire's lengths as tens of micrometers and their widths in the range of 15–25 nm. The representative TEM images (Fig. 5A and B) of the MnO2 sample revealed the distribution of nanowires evenly and the average diameter of nanowires as 20 nm. The high-resolution TEM pictures (Fig. 5C–E) display clear lattice fringes with an interplanar spacing between neighboring lattice fringes as 0.685, 0.501, 0.315 and 0.245 nm, all of which are in good agreement with XRD results of (110), (200), (310) and (211) planes of α-MnO2 (JCPDS# 42-1348). The selected area electron diffraction (SAED) pattern (Fig. 5F), reveals typical concentric circles, suggesting the polycrystalline nature of α-MnO2 nanowires.
 | ||
| Fig. 4 (A–D) Field emission scanning electron microscopy (FESEM) images of the α-MnO2 sample at different magnifications. | ||
 | ||
| Fig. 5 Different magnified (A and B) TEM images and (C–E) high-resolution TEM images of the α-MnO2 sample; (F) SAED pattern of the corresponding sample. | ||
To measure the surface textural characteristics of α-MnO2 nanowires, we collected nitrogen adsorption and desorption isotherms (Fig. 6) on the material. As seen in Fig. 6A, the isotherms have type-IV characteristics (based on IUPAC classification) with a capillary condensation step viewing at high relative pressures (P/P0) between 0.4 and 0.9 that distinctly signifies mesoporous nature of the MnO2 sample. The pore distribution profile (Fig. 6B) further revealed a unimodal pore-size distribution with a pore maximum of 2.96 nm. The estimated specific surface area and pore volume of α-MnO2 nanowires are 181 m2 g−1 and 0.257 cm3 g−1, respectively. We note that the specific surface area of our α-MnO2 nanowires is far away from others reported in literature prepared by a different method such as hollow-urchins (132 m2 g−1),27 nanopetals (68 m2 g−1),17 nitrogen-enriched porous carbon–MnOx hybrid (148 m2 g−1)4 and MnO2–polythiophene (136 m2 g−1).32 It has commonly been reported that the electrode materials comprising high specific surface area and large pore-volume as well as the indispensable mesoporous structure could reinforce the electrode–electrolyte contact and afford a facile e− transport as well as a short path for Li+ diffusion during charge/discharge process.47,48 Therefore, we expected, α-MnO2 featuring hierarchical 1D nanowires morphology and favorable textural properties would provide the possibility of efficient transport of electrolyte ions in Li-ion batteries.
 | ||
| Fig. 6 (A) N2 adsorption–desorption isotherms and (B) BJH pore-size distribution profile of the α-MnO2 sample. | ||
The elemental composition and their valence state of Mn ions in α-MnO2 nanowires were investigated using the X-ray photoelectron spectroscopy technique. Fig. 7A shows the XPS survey scan, demonstrating the presence of Mn and O as well as C from the reference. It also confirms the absence of any apparent impurities in the prepared sample. The high-resolution Mn 2p spectrum (Fig. 7B) shows a spin–orbit doublet with binding energy peaks at 642.3 eV (Mn 2p1/2) for and 654.15 eV for (Mn 2p3/2), suggesting that the oxidation state of Mn is 4+.49 In the high-resolution O 1s spectrum (Fig. 7C), the peaks located at binding energies of 529.75 eV and 531.50 eV, which correspond to those for anhydrous (Mn–O–Mn) and hydrated (Mn–O–H) manganese oxides, respectively.50,51
 | ||
| Fig. 7 (A) XPS survey spectrum of α-MnO2; core-level XPS of (B) Mn 2p and (C) O 1s. | ||
3.2 Electrochemical performance
To investigate the electrochemical properties of as-prepared α-MnO2 nanowires as anodes for LIBs in the half-cell configuration, cyclic voltammograms and galvanostatic discharge–charge cycling tests were performed. Fig. 8 shows typical CV profiles of tα-MnO2 nanowires obtained at a 0.1 mV s−1 scan rate within a voltage range of 0.005–3.0 V (vs. Li/Li+). Generally, the reaction between transition metal oxides (e.g., MnO2) and Li is typically a conversion-type mechanism.52 The CV profiles of α-MnO2 nanowires are consistent with those reported for relevant systems,53–56 demonstrating a similar Li storage mechanism is taking place in our system, which is as follows:| MnO2 + 4Li+ + 4e− ↔ Mn + 2Li2O | (1) |
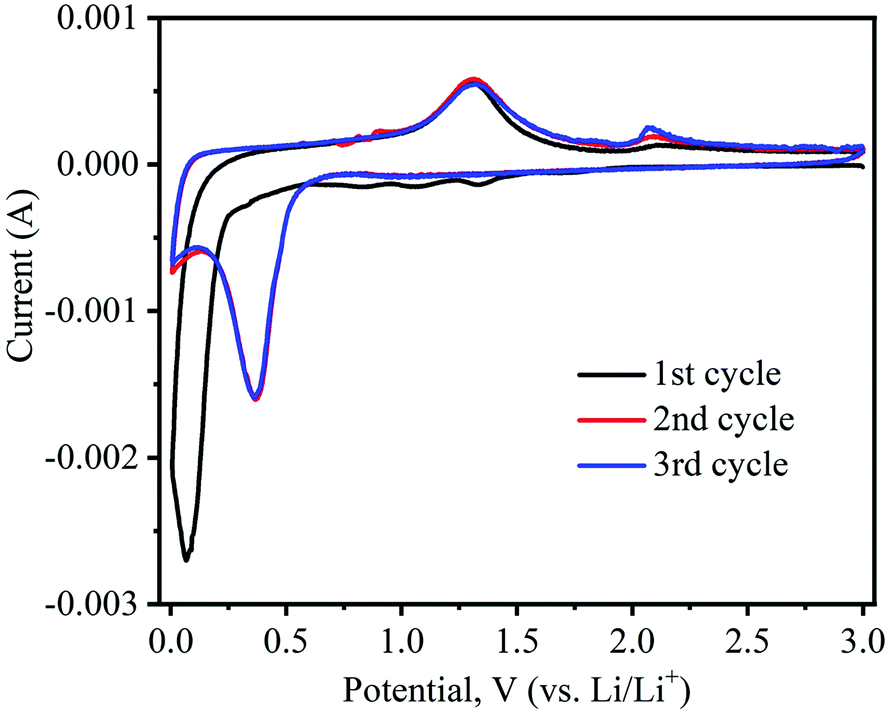 | ||
| Fig. 8 Cyclic voltammetry (CV) curves of α-MnO2 nanowires electrode recorded at a sweep rate of 0.1 mV s−1 and 0.05–3.0 V (vs. Li/Li+) potential range. | ||
Noticeably, the CV shape of the first cathodic curve essentially contrasts with the posterior ones. Upon the first scan in the cathodic (lithiation) process, the prominent irreversible peaks are situated at 1.34 and 1.06 V, caused by electrolyte decomposition leading to the solid electrolyte interphase (SEI) formation on electrode surfaces.52 A small peak appears at 0.84 V, which corresponds to the reduction of Mn4+ to Mn2+.55,56 The large intensity peak at approximately 0.07 V during the first cycle can be imputed to the reduction of Mn2+ to Mn0.54,57 In the following delithiation (anodic) process, two distinctive peaks are noticed at 1.31 and 2.08 V, indicating that reversible oxidation of metallic Mn (i.e., Mn0 to Mn2+ and Mn2+ to Mn4+, respectively).28,54 Interestingly, in the subsequent second and third cycles of the cathodic process, peak position shifts from 0.07 V to 0.36 V, which can be attributed to the structural reconstruction resulting from the formation of Li2O and manganese metal through the conversion reaction, as shown in eqn (1).52 In the second and third anodic cycles, the peaks’ positions were almost identical to those in the first one (1.31 and 2.08 V).
The galvanostatic charge/discharge experiment within a voltage range of 0.05–3.0 V (vs. Li/Li+) was employed to further investigate the electrochemical lithiation/delithiation process of the as-prepared α-MnO2 nanowire electrode. The active material loading was 2.0 mg cm−2. The specific capacity of the cells was estimated from the loading of the active material. Fig. 9A shows typical charge/discharge characteristics (i.e., voltage vs. capacity) of α-MnO2 nanowire electrode at a specific current of 100 mA g−1. During the charge and discharge process, noticeable plateaus at 1.0 and 0.4 V are observed, consistent with literature reports,55,58 suggesting similar lithium intercalation/absorption behaviour occurring in α-MnO2 nanowires. The cell's 1st discharge and charge capacities are 1373 and 852 mA h g−1, respectively, leading to a relatively low Coulombic efficiency of 62%. The discharge capacities for the subsequent cycles (2nd to 10th) are 897, 878, 851, 846, 837, 833, 837, 834 and 836 mA h g−1, respectively. The abnormal high capacity during the 1st discharge is caused by electrolyte decomposition and the inevitable formation of the SEI passivation layer, which is common in most anode materials.54,56
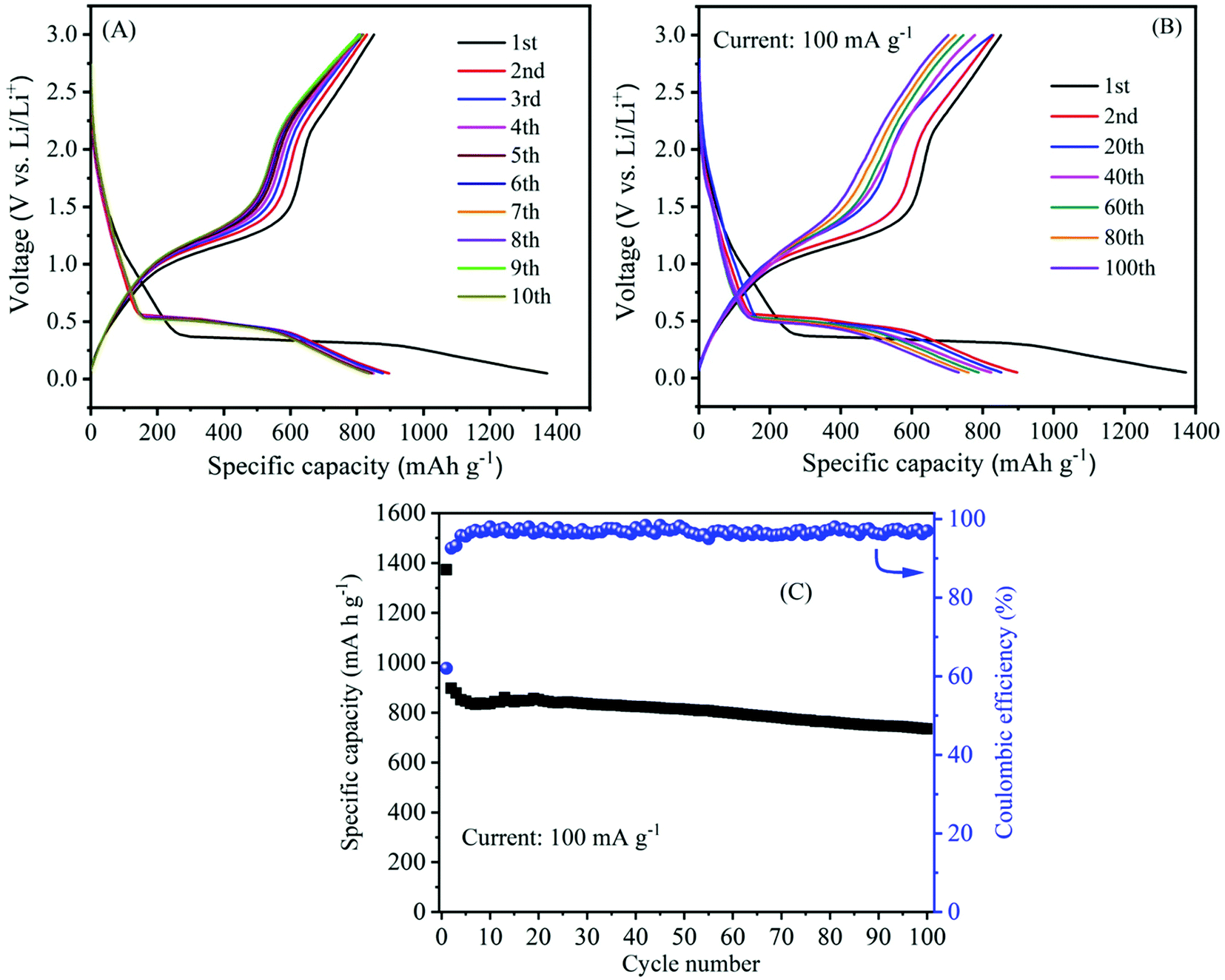 | ||
| Fig. 9 Electrochemical properties of α-MnO2 nanowires electrode: (A) Galvanostatic charge–discharge curves at a current density of 100 mA g−1 in the initial 10 cycles; (B) Typical charge–discharge curves at different cycle numbers and (C) the cycling performance (variation of the capacity with cycle number) at a current density of 100 mA g−1. | ||
The long-term cycling stability is also an important parameter and was measured to determine the strength of the electrode. The galvanostatic charge–discharge technique was employed to evaluate the cycling stability of the α-MnO2 electrode at a current density of 100 mA g−1 in a potential window of 0.05 and 3.0 V. Fig. 9B illustrates the 1st to 100th charge/discharge profiles of α-MnO2 at an applied current of 100 mA g−1. The profiles show a stable performance upon increasing the cycle number up to 100 cycles. As seen from Fig. 9C, the capacity of the cells decreased gradually with cycle number. Remarkably, a high reversible capacity of around 735 mA h g−1 was retained after 100 cycles, demonstrating an excellent cycle performance of the nanowire electrode. Meanwhile, the average Coulombic efficiency of the cell was maintained at about 97%. Moreover, the performance in terms of specific capacity and stability as well as rate capability delivered by α-MnO2 hierarchical nanowires was more significant when compared to that reported on several MnO2 nanostructures and hybrid composites (Table 1). The excellent performance with high capacity and good cycling stability indicates considerable progress compared to other previous reports.
| Synthesis method | Crystal structure | Morphology | S BET (m2 g−1) | 1st discharge–charge capacity (mA h g−1) | Capacity retention (mA h g−1) | Rate performance (mA h g−1) | Ref. |
|---|---|---|---|---|---|---|---|
| Redox reaction | α-MnO2 | Nanoneedles | 181 | 1373/852 | 735@100 cycles | 495 at 1000 mA g−1 | This work |
| Low-temperature mild reduction | α-MnO2 | Hollow Urchins | 132 | 746/650 | 481@40 cycles | — | 27 |
| Solution method | MnO2/carbon nanohorns | Nanoflakes | — | 1190/795 | 565@60 cycles | 385 at 1000 mA g−1 | 30 |
| In situ polymerization | Birnessite-type MnO2/polythiophene | Nanoneedles | 136 | 700 | 500@100 cycles | — | 32 |
| Hydrothermal | Birnessite-type MnO2/CNTs | Nanoflakes | — | 800 | 600@50 cycles | 310 at 4000 mA g−1 | 52 |
| Hydrothermal | γ-MnO2 | Nanocubes | — | 1992/1042 | 602@20 cycles | — | 64 |
| Molten salt method | λ-MnO2 α-MnO2 | Spherical Nanorods | 4.7 38 | 1400/600 1820/910 | 372@50 cycles 845@50 cycles | — | 65 |
| Hydrothermal | α-MnO2 | Cactus-like nanostructure | 75.4 | 196 | 149@25 cycles | — | 66 |
| Ultrasound irradiation | γ-MnO2/CNTs | Mesoporous particles | 206 | 1278/741 | 934@100 cycles | 380 at 1000 mA g−1 | 67 |
| Hydrothermal | Birnessite-MnO2 | Nanosheets | — | 1278/854 | 541@100 cycles | 178 at 1000 mA g−1 | 68 |
| Ultra-filtration | α-MnO2/graphene | Nanotubes | — | 1250/686 | 495@40 cycles | 208 at 1600 mAg−1 | 69 |
| In situ polymerization | MnO2/Conjugated polymer/graphene | Nanorods | — | 1835/1050 | 948@15 cycles | 698 at 400 mA g−1 | 70 |
| Hydrothermal | α-MnO2/graphene | Nanorods | 149 | 971/483 | 595@60 cycles | — | 71 |
| Reflux method | α-MnO2/graphene | Nanoparticles | — | 1589/746 | 752@65 cycles | 304 at 800 mA g−1 | 72 |
| Solvothermal | α-MnO2/MWCNTs | Nanowires | — | 875 | 770@50 cycles | — | 73 |
| Hydrothermal | Birnessite-type MnO2@carbon microbead | Flower-like texture | — | 1480/698 | 525@100 cycles | 230 at 1500 mA g−1 | 74 |
Besides their excellent reversible capacity and cycling stability, the prepared MnO2 nanowire electrode also presents an impressive rate performance. Fig. 10 shows the rate capability curves (variation of the capacity as a function of applied current density) of MnO2 nanowires. The figure clearly shows that the cell's discharge and charge capacities are gradually reduced as the applied current density increases, which is typically posed by the low diffusion rate of Li+ ions into anodes under high current rates.59,60 α-MnO2 nanowires provide discharge capacities of 1369, 727, 652, 586 and 495 mA h g−1 at 100, 250, 500, 750 and 1000 mA g−1, respectively. Even at a high applied current density of 1000 mA g−1, the electrode maintained a discharge capacity of as high as 495 mA h g−1. Moreover, the reversible capacity could be recovered to 890 mA h g−1, when the current density reverses back to 100 mA g−1, demonstrating good stability and rate capacity of α-MnO2 nanowires.
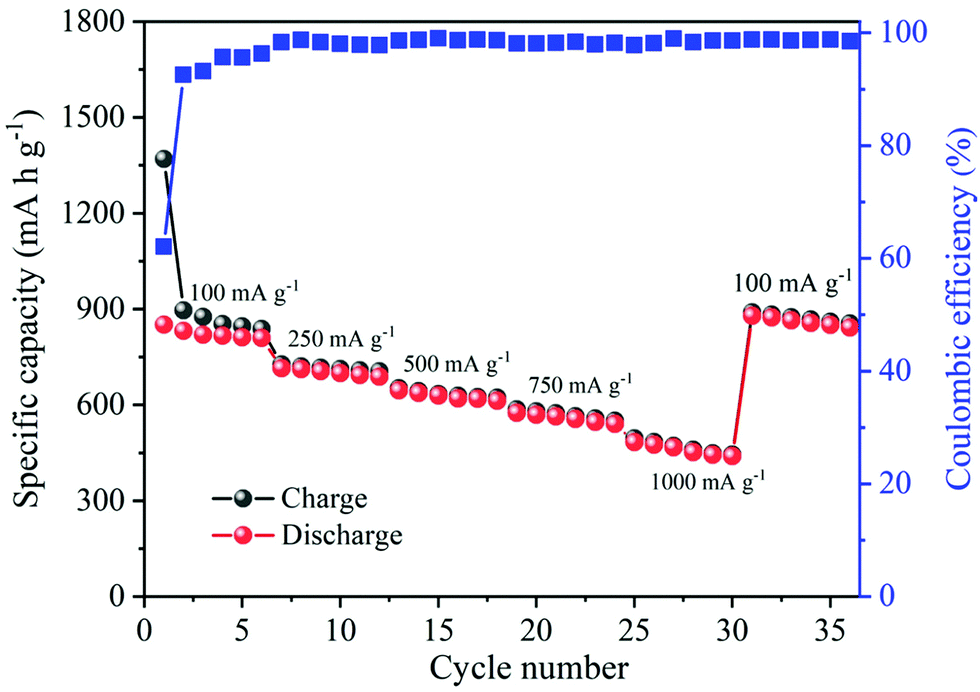 | ||
| Fig. 10 The rate performance of α-MnO2 nanowires at the rates between 100 and 1000 mA g−1. | ||
Electrochemical impedance spectroscopy (EIS) is a powerful analytical technique widely used to analyze the electrode kinetics of α-MnO2 material. The impedance measurements were recorded before and after cycling of the MnO2 electrode over the frequency range of 0.01 Hz–1M Hz and 10 mV amplitude. Fig. 11 compares the Nyquist plots (real, Z′ vs. imaginary, Z′′) of α-MnO2 obtained before and after 100th charge/discharge cycles. A simple equivalent circuit model (inset of Fig. 11) was built to analyze the EIS data. The Nyquist plots obtained before and after cycling are composed of a high-frequency semicircle followed by a low-frequency inclined line. The real (Z′) axis intercept (at x-axis) in the high-frequency region is ascribed to Ohmic electrolyte resistance (Rs), which essentially comes from the electrolyte, electrodes, etc.11,61 The middle-frequency region semicircle corresponds to the interface contact and the charge transfer resistance, Rct.62,63 A spike in the low-frequency region imputes to mass-transfer resistance. The Rct value after 100 cycles is about 178 Ω, which changes only by 19.4% when compared to before cycled one, suggesting better contact between the active material and electrolyte throughout cycling, leading to better electrochemical performance of α-MnO2 nanowires. The inclined line in the lower frequency (Warburg region) reflects the diffusion of Li+ ions into the MnO2 active material.15,63
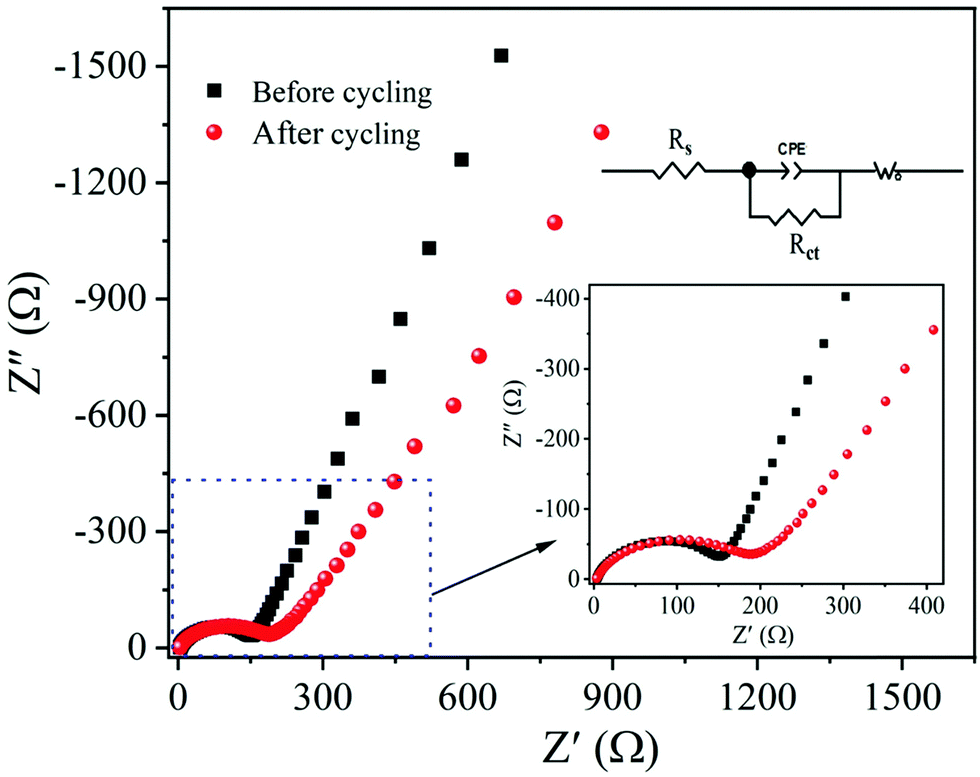 | ||
| Fig. 11 EIS Nyquist plots of the α-MnO2 nanowires electrode obtained before and after 100th charge–discharge cycles: The insets show the equivalent circuit and enlarged view of the high-frequency region. | ||
The excellent battery performance in terms of high reversible capacity, good cycling stability and rate capability of the as-prepared α-MnO2 can be explained as follows. First of all, hierarchical 1D nanowires with a large specific surface area and mesoporous structure can provide open channels for electrolyte penetration, thereby ensuring a good electrode/electrolyte contact area. Secondly, the nanowire architecture can relieve the stress caused by the volume change during the lithiation/delithiation and thus improve the reaction kinetics. Thirdly, the nanoscale diameter of nanowires shortens the diffusion paths for Li+ and e−, thus favouring fast charge transport during cycling. All these merits contributed to the fabulous electrochemical performance of α-MnO2 nanowires, making them a promising candidate for advanced anode materials for high-performance Li-ion batteries.
4. Conclusion
We have reported a simple, inexpensive and eco-friendly approach to the synthesis of large-scale production of α-MnO2 nanowires by a quick redox-reaction between permanganate and glycine, that was investigated as an anode material for Li-ion batteries. The unique structure of hierarchical nanowires with mesoporous structure provides high specific surface area (∼181 m2 g−1) and beneficial pore-volume (∼0.21 cm3 g−1). These factors are highly beneficial in energy storage applications, which allowed facile Li+ ion transport and reinforced the electrode–electrolyte contact area during charge/discharge cycling. As a result, a high discharge capacity of 1373 mA h g−1, good rate capability, and excellent cycling performance are achieved. The electrode retained a high reversible capacity of 735 mA h g−1 after 100 cycles. We believe that the simple, straightforward, low-cost, and quick-redox tunable synthesis method presented here may pave the way for successfully extending the synthesis to other oxide nanostructures. These oxide nanostructures can be effectively employed for interdisciplinary energy conversion and storage applications. It can also be widely used to investigate other potential applications such as sensors, catalysis, and microelectronic devices.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully thank Xiamen University, China for providing the electrochemical instrument facility for battery applications of the materials. Also, we would like to thank IIT Madras SAIF for providing instruments for structural analysis of the samples.References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- S. Goriparti, E. Miele, F. D. Angelis, E. D. Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- J. Wang, C. Zhang and F. Kang, ACS Appl. Mater. Interfaces, 2015, 7, 9185–9194 CrossRef CAS PubMed.
- S. Konar, U. Häusserman and G. Svensson, Chem. Mater., 2015, 27, 2566–2575 CrossRef CAS.
- H. Li, W. Zhang, K. Sun, J. Guo, K. Yuan, J. Fu, T. Zhang, X. Zhang, H. Long, Z. Zhang, Y. Lai and H. Sun, Adv. Energy Mater., 2021, 11, 2100867 CrossRef CAS.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- V. Aravindan, Y.-S. Lee and S. Madhavi, Adv. Energy Mater., 2015, 5, 1402225 CrossRef.
- X. Li, D. Li, Z. Wei, X. Shang and D. He, Electrochim. Acta, 2014, 121, 415–420 CrossRef CAS.
- Y. Luo, K. Wang, S. Luo, F. Zhao, H. Wu, K. Jiang, Q. Li, S. Fan and J. Wang, ACS Appl. Nano Mater., 2018, 1, 2997–3005 CrossRef CAS.
- M. V. Reddy, T. Yu, C.-H. Sow, Z. X. Shen, C. T. Lim, G. V. Subba Rao and B. V. R. Chowdari, Adv. Funct. Mater., 2007, 17, 2792–2799 CrossRef CAS.
- A. Bhaskar, M. Deepa and T. Narasinga Rao, Nanoscale, 2014, 6, 10762–10771 RSC.
- D. Li, J. Shi, H. Liu, C. Liu, G. Dong, H. Zhang, Y. Yang, G. Lu and H. Wang, Sustainable Energy Fuels, 2019, 3, 1055–1065 RSC.
- M. S. Salman, A. R. Park, M. J. Cha, Y. Choi, S. K. Jang, L. Tan, P. J. Yoo and W. S. Choe, ACS Appl. Nano Mater., 2018, 1, 698–710 CrossRef CAS.
- A. Bhaskar, M. Deepa and T. Narasinga Rao, ACS Appl. Mater. Interfaces, 2013, 5, 2555–2566 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J.-M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- P. K. Nayak, T. R. Penki and N. Munichandraiah, J. Electroanal. Chem., 2013, 703, 126–134 CrossRef CAS.
- Y. Li, Q. Zhang, J. Zhu, X.-L. Wei and P. K. Shen, J. Mater. Chem. A, 2014, 2, 3163–3168 RSC.
- Y. Deng, L. Wan, Y. Xie, X. Qin and G. Chen, RSC Adv., 2014, 4, 23914–23935 RSC.
- J. Huang, L. M. Housel, C. D. Quilty, A. B. Brady, P. F. Smith, A. Abraham, M. R. Dunkin, D. M. Lutz, B. Zhang, E. S. Takeuchi, A. C. Marschilok and K. J. Takeuchi, J. Electrochem. Soc., 2018, 165, A2849–A2858 CrossRef CAS.
- E. Umeshbabu, P. Justin and G. Ranga Rao, ACS Appl. Energy Mater., 2018, 1, 3654–3664 CrossRef CAS.
- Y. S. Yun, J. M. Kim, H. H. Park, J. Lee, Y. S. Huh and H.-J. Jin, J. Power Sources, 2013, 244, 747–751 CrossRef CAS.
- C. Guo, H. Liu, J. Li, Z. Hou, J. Liang, J. Zhou, Y. Zhu and Y. Qian, Electrochim. Acta, 2019, 304, 370–377 CrossRef CAS.
- C. S. Johnson, M. F. Mansuetto, M. M. Thackeray, Y. Shao-Horn and S. A. Hackney, J. Electrochem. Soc., 1997, 144, 2279–2282 CrossRef CAS.
- Y. Tang, S. Zheng, Y. Xu, X. Xiao, H. Xue and H. Pang, Energy Storage Mater., 2018, 12, 284–309 CrossRef.
- S. Liu, X. Liu, J. Zhao, Z. Tong, J. Wang, X. Ma, C. Chi, D. Su, X. Liu and Y. Li, RSC Adv., 2016, 6, 85222–85229 RSC.
- B. Li, G. Rong, Y. Xie, L. Huang and C. Feng, Inorg. Chem., 2006, 45, 6404–6410 CrossRef CAS PubMed.
- J. Chen, Y. Wang, X. He, S. Xu, M. Fang, X. Zhao and Y. Shang, Electrochim. Acta, 2014, 142, 152–156 CrossRef CAS.
- Y. Li, D. Ye, B. Shi, W. Liu, R. Guo, H. Pei and J. Xie, Phys. Chem. Chem. Phys., 2017, 19, 7498–7505 RSC.
- H. Lai, J. Li, Z. Chen and Z. Huang, ACS Appl. Mater. Interfaces, 2012, 4, 2325–2328 CrossRef CAS PubMed.
- A. L. Mohana Reddy, M. M. Shaijumon, S. R. Gowda and P. M. Ajayan, Nano Lett., 2009, 9, 1002–1006 CrossRef PubMed.
- W. Xiao, J. S. Chen, Q. Lu and X. W. Lou, J. Phys. Chem. C, 2010, 114, 12048–12051 CrossRef CAS.
- S. Bose, T. Kuila, A. K. Mishra, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22, 9696 RSC.
- S. Bag and C. R. Raj, J. Mater. Chem. A, 2016, 4, 8384–8394 RSC.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS PubMed.
- A. K. Pearce, T. R. Wilks, M. C. Arno and R. K. O’Reilly, Nat. Rev. Chem., 2020, 21, 21–45 CrossRef PubMed.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed.
- M. H. Rossouw, D. C. Liles, M. M. Thackeray, W. I. F. David and S. Hull, Mater. Res. Bull., 1992, 27, 221–230 CrossRef CAS.
- L. Li, Y. Pan, L. Chen and G. Li, J. Solid State Chem., 2007, 180, 2896–2904 CrossRef CAS.
- I. I. Misnon, R. A. Aziz, N. K. M. Zain, B. Vidhyadharan, S. G. Krishnan and R. Jose, Mater. Res. Bull., 2014, 57, 221–230 CrossRef CAS.
- H. T. Zhu, J. Luo, H. X. Yang, J. K. Liang, G. H. Rao, J. B. Li and Z. M. Du, J. Phys. Chem. C, 2008, 112, 17089–17094 CrossRef CAS.
- E. Eren, H. Gumus and A. Sarihan, Desalination, 2011, 279, 75–85 CrossRef CAS.
- H. Huang, G. Sun, J. Hu and T. Jiao, J. Chem., 2015, 8, 629362 Search PubMed.
- W. Li, X. Cui, R. Zeng, G. Du, Z. Sun, R. Zheng, S. P. Ringer and S. X. Dou, Sci. Rep., 2015, 5, 8987 CrossRef CAS PubMed.
- A. M. Hashem, H. Abuzeid, M. Kaus, S. Indris, H. Ehrenberg, A. Mauger and C. M. Julien, Electrochim. Acta, 2018, 262, 74–81 CrossRef CAS.
- S. Liang, F. Teng, G. Bulgan, R. Zong and Y. Zhu, J. Phys. Chem. C, 2008, 112, 5307–5315 CrossRef CAS.
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1, 16023 CrossRef CAS.
- Y. Ren, Z. Ma, R. E. Morris, Z. Liu, F. Jiao, S. Dai and P. G. Bruce, Nat. Commun., 2013, 4, 2015 CrossRef PubMed.
- X. Guo, J. Han, L. Zhang, P. Liu, A. Hirata, L. Chen, T. Fujita and M. Chen, Nanoscale, 2015, 7, 15111–15116 RSC.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2004, 16, 3184–3190 CrossRef CAS.
- J. Deng, L. Chen, Y. Sun, M. Ma and L. Fu, Carbon, 2015, 92, 177–184 CrossRef CAS.
- H. Xia, M. Lai and L. Lu, J. Mater. Chem., 2010, 20, 6896–6902 RSC.
- H. Kim, N. Venugopal, J. Yoon and W.-S. Yoon, J. Alloys Compd., 2019, 778, 37–46 CrossRef CAS.
- S. Z. Huang, Y. Cai, J. Jin, J. Liu, Y. Li, H.-E. Wang, L.-H. Chen, T. Hasan and B. L. Su, J. Mater. Chem. A, 2016, 4, 4264–4272 RSC.
- Z. Li, J. Wang, Z. Wang, Y. Tang, C.-S. Lee and S. Yang, RSC Adv., 2014, 4, 54416–54421 RSC.
- J. Yoon, W. Choi, T. Kim, H. Kim, Y. S. Choi, J. M. Kim and W.-S. Yoon, J. Energy Chem., 2021, 53, 276–284 CrossRef.
- R. Cai, S. Guo, Q. Meng, S. Yang, H. L. Xin, X. Hu, M. Li, Y. Sun, P. Gao, S. Zhang, H. Dong, S. Lei, K. Kim, H. Zeng, L. Sun, F. Xu and Y. Zhu, Nano Energy, 2019, 63, 103840 CrossRef CAS.
- J. Chen, Y. Wang, X. He, S. Xu, M. Fang, X. Zhao and Y. Shang, Electrochim. Acta, 2014, 142, 152–156 CrossRef CAS.
- N. Zhao, S. Wu, C. He, Z. Wang, C. Shi, E. Liu and J. Li, Carbon, 2013, 57, 130–138 CrossRef CAS.
- J. H. Park, W. Y. Choi, S. Lee, T.-S. Kim and J. W. Lee, Electrochim. Acta, 2020, 348, 136310 CrossRef CAS.
- M. Satyanarayana, A. K. Jibin, E. Umeshbabu1, J. James and U. V. Varadaraju, Ionics DOI:10.1007/s11581-021-04297-2.
- Y. Hu, K. Wu, F. Zhang, H. Zhou and L. Qi, ACS Appl. Nano Mater., 2019, 2, 429–439 CrossRef CAS.
- Y. Yang, G. Ma, J. Huang, J. Nan, S. Zhen, Y. Wang and A. Li, J. Solid State Chem., 2020, 286, 121297 CrossRef CAS.
- J. Zhao, Z. Tao, J. Liang and J. Chen, Cryst. Growth Des., 2008, 8, 2799–2805 CrossRef CAS.
- P. Nithyadharseni, M. V. Reddy, H. Fanny, S. Adams and B. V. R. Chowdari, RSC Adv., 2015, 5, 60552–60561 RSC.
- R. Rajagopal and K.-S. Ryu, J. Energy Storage, 2020, 32, 101880 CrossRef.
- Y. S. Yun, J. M. Kim, H. H. Park, J. Lee, Y. S. Huh and H.-J. Jin, J. Power Sources, 2013, 244, 747–751 CrossRef CAS.
- Y. Wang, Z. Han, S. Yu, R. Song, H. Song, K. Ostrikov and H. Yang, Carbon, 2013, 64, 230–236 CrossRef CAS.
- A. Yu, H. W. Park, A. Davies, D. C. Higgins, Z. Chen and X. Xiao, J. Phys. Chem. Lett., 2011, 2, 1855–1860 CrossRef CAS.
- C. Xian Guo, M. Wang, T. Chen, X. W. Lou and C. Ming Li, Adv. Energy Mater., 2011, 1, 736–741 CrossRef.
- H. Liu, Z. Hu, Y. Su, H. Ruan, R. Hu and L. Zhang, Appl. Surf. Sci., 2017, 392, 777–784 CrossRef CAS.
- K. Wen, G. Chen, F. Jiang, X. Zhou and J. Yang, Int. J. Electrochem. Sci., 2015, 10, 3859–3866 CAS.
- S. J. Ee, H. Pang, U. Mani, Q. Yan, S. L. Ting and P. Chen, ChemPhysChem, 2014, 15, 2445–2449 CrossRef CAS PubMed.
- H. Wang, J. Liu, X. Wang, C. Wu, Q. Zhao, Y. Fu, X. Yang and H. Shu, RSC Adv., 2014, 4, 22241–22245 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00755f |
| This journal is © The Royal Society of Chemistry 2022 |