
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChallenges and future prospects of graphene-based hybrids for solar fuel generation: moving towards next generation photocatalysts
Kommula
Bramhaiah
 * and
Santanu
Bhattacharyya
*
* and
Santanu
Bhattacharyya
*
Indian Institute of Science Education and Research (IISER), Govt. ITI Building (Transit campus), Engg. School Road, Berhampur 760010, Odisha, India. E-mail: bramhaiah@iiserbpr.ac.in; santanub@iiserbpr.ac.in
First published on 17th November 2021
Abstract
Considering the current global energy crisis and the most alarming environmental pollution issues, moving towards renewable energy resources instead of fossil fuels should be the ultimate goal of modern civilization. Alternative solar fuel generation by applying heterogenous photocatalysis is one of the major approaches in renewable energy research. After the discovery of the amazing material graphene and its derivatives, it has become one of the most promising materials for efficient photocatalysis in terms of solar water splitting and CO2 photo-reduction. However, a lot of challenges are still there in terms of efficiency, product selectivity, fabrication techniques, etc. Herein, we have provided deeper insights into the fundamental aspects of graphene/graphene oxide and their derivatives in terms of structural, electronic, and thermodynamic considerations for the fabrication of new generation photocatalysts. Finally, we have discussed the future directions and provided strategic insights to overcome the current challenges for sustainable production of solar fuels.
 Kommula Bramhaiah | Dr Kommula Bramhaiah received his MSc from the Department of Chemistry in 2011 at Jawaharlal Nehru Technological University Anantapur, Andhra Pradesh, India. He then joined Dr Neena S. John's group as a PhD student. He received his PhD in 2018 from the Centre for Nano and Soft Matter Sciences, Bangalore, India. Then, he moved to the Indian Institute of Science Education and Research (IISER), Berhampur, India, for his postdoctoral research under the supervision of Dr Santanu Bhattacharyya. His research interests are mainly focused on strategic materials design for energy applications including water splitting, energy storage, and catalysis. |
 Santanu Bhattacharyya | Dr Santanu Bhattacharyya received his PhD (Science) from the Indian Association for the Cultivation of Science, Kolkata, in 2013. He was subsequently a postdoctoral scientist in IMDEA Nanociencia, Madrid, and later in 2015, he joined the Chair for Photonics and Optoelectronics, LMU, Munich, as an Alexander von Humboldt fellow. After finishing his postdoctoral stay in Germany, he joined as an Assistant Professor in the Department of Chemical Sciences, IISER Berhampur, in August 2018. His group mainly focuses on several light-induced processes in fluorescent nanomaterials and the optimization of environmentally sustainable nanomaterials for solar fuel generation. |
1. Introduction
The increasing global energy demand and the increase in environmental pollution are the two major concerns for modern civilization.1–4 These two problems are interconnected, as the excessive use of fossil fuels largely increases greenhouse gases, especially CO2. Considering this, various research groups have been working extensively to address the energy crisis and environmental issues simultaneously, followed by the development of alternatives for depleting fossil fuel reserves.1–3,5 Hence, there is an urgent need for eco-friendly, sustainable, and technologically advanced methods to harvest clean energy and conversion of CO2 emissions into transportable fuels and other value-added products.1–3,5 Molecular hydrogen (H2) is recognized as a potentially significant alternative form of storable and clean energy for the future because of its pollution-free, storage, and cost-effective advantages. Currently, H2 can be produced industrially by three main pathways.6 These are steam methane reforming, coal gasification, and water electrolysis methods. In these approaches, almost 95% of the total H2 gas is generated by steam methane reforming and coal gasification, while only 4% of the H2 gas is produced by water electrolysis. However, these conventional approaches for H2 generation are limited either by the emission of CO2, a greenhouse gas, as a by-product or by consumption of more electricity. Therefore, it is essential to develop carbon-free and low-cost approaches for vigorous and efficient H2 generation to support the emerging hydrogen economy. Among these three methods, water electrolysis still offers hope as it can sustainably generate H2 gas from the most abundant and renewable feedstock, i.e. – water.3,5–7 However, it is also energy-consuming, and, definitely, we need a greener approach. Hydrogen production through photocatalytic solar water splitting can be one of the best alternatives to address future energy issues.3,6–8 On the other hand, solar light-induced transformation of CO2 into renewable chemicals or fuels, such as CO, CH4, CH3OH, C2H5OH, and HCOOH, is another promising strategy to solve the issues of an energy crisis, global warming, environmental pollution, and climate change together. Therefore, the efficient conversion of solar energy to chemical energy or solar fuels employing the photocatalytic process has been considered as one of the most efficient and long-term solutions to solve the global energy crisis and environmental problems.9–11As of now, various metal oxide-based semiconducting nanostructures (including ZnO, TiO2, WO3, and CdS) have been employed as photocatalysts for both solar water splitting and CO2 photoreduction.9,12 These inorganic semiconductors display relatively high stability, are low cost, and absorb light with photon energy equal to or higher than their bandgap energy. However, the practical use of these materials for overall photocatalysis is very limited due to the fast recombination of photo-induced charge carriers and lack of visible light absorption.13,14 To overcome these problems, various strategies have been developed including bandgap engineering, morphology control, loading of co-catalysts, and loading of metals and non-metals.5,15 More specifically, the hunt for new semiconductors as efficient photocatalysts is focused on the following points: (a) increasing the valence band energy to decrease the bandgap; (b) moving the conduction band to modulate the required reduction potential; (c) improving the quantum efficiency of exciton formation and suppressing the recombination of photo-induced charge carriers; and (d) developing nanoscale diverse morphologies to provide the outermost surface area with multiple photocatalytically active sites.3,5,14–16 In this regard, new semiconductors with carbon-based nanomaterials and their hybrids can be promising alternatives to traditional metal-based semiconductors. Carbon is one of the most abundant and versatile elements on earth and it is the basis of life on the planet. It is well known to exist in three forms, such as amorphous carbon, graphite, and diamond; as we know, their properties mainly depend on how the carbon atoms are arranged.3,5,15–17 The C–C sp3 hybridization makes a diamond the natural hardest material. Graphite is a solid lubricant due to the loose interlamellar coupling between the sheets. In the past three decades, various new carbon nanomaterials have been discovered including fullerenes, carbon nanotubes, carbon quantum dots, and graphene.3,5,16,18 These carbon materials have found potential applications in the field of solar fuel generation. In this context, graphene has numerous advantages compared with other related materials such as carbon nanotubes (CNTs) and carbon nitride (g-C3N4), including high electron mobility at room temperature, high thermal conductivity, and superior mechanical properties with high Young's moduli.3,5,16–19 Also, the bandgap of graphene can be tuned to produce semiconducting behavior, which is as an essential requirement for it to act as a catalyst. Furthermore, its shape, size, and chemical structure can also be tuned to expand its applications further.3,5,17 Over the last few years, considerable attention has been devoted to graphene-based photocatalysts, especially for hydrogen production via water-splitting reaction and CO2 reduction to value-added products. However, the fundamental understandings related to the performance, durability, and stability of these materials are still major challenges for practical applications. Various groups have been working towards improving the light-harvesting as well as the photocatalytic performance of graphene-based semiconducting materials by employing various strategies like designing appropriate morphologies, doping with suitable heteroatoms, loading of noble metal nanostructures, and forming semiconducting hetero-structure composites.17,18,20 Kamat and his group fabricated GO–TiO2 nanocrystalline composites employing a simple sonication approach where TiO2 NPs and GO are in ethanol solvent and showed the feasibility of using graphene as an electron-transfer medium in graphene/TiO2 composite photocatalysts.21,22 Further, Zhang and his group reported graphene–P25 TiO2 composites prepared using a hydrothermal approach and they exhibited a significantly enhanced photocatalytic activity for the degradation of dyes in aqueous medium.23 Nag and co-workers synthesized a graphene-based BiVO4 composite and demonstrated the photo-electrochemical water-splitting reaction under visible light illumination.24,25 These pioneering studies have excited researchers to do more extensive research on the fabrication, modification, and demonstration of these materials for various applications including solar water splitting for hydrogen generation and CO2 reduction to value-added products.
In this review, we have provided the fundamental insight into graphene-based nanomaterials and highlighted several scopes from a fundamental point of view for their development as efficient photocatalysis. In addition to these, this review includes several strategies for their fabrication and their tunability. This review not only covers the recent advancements in graphene-based materials for artificial photosynthesis (in terms of H2 fuel generation and CO2 photoreduction), but we have also highlighted the current challenges and possible solutions to overcome these problems.
2. Basic features
Graphene has exceptional electronic properties, which arise from its single atom thick, two-dimensional honeycomb-like lattice structure. It has a two-atom unit cell, resulting in an electronic structure with a point where the bands touch the so-called Dirac point.26,27 Further notably, the energy dispersion displays a linear relation close to the Dirac point, where electrons resemble massless Dirac fermions but with an effective speed of light of c/300 (where c is the speed of light) (Fig. 1). Hence, the charge carriers in graphene are realistic particles rather than normal charge carriers present in semiconductors and metals.15,18,26–28 P. R. Wallace first reported the band structure of graphene and the low energy band due to the π electrons. Bonding π states form the valence band, while the conduction band is formed by antibonding π* orbitals.29 These bonding and antibonding states are orthogonal and touch at six points known as Dirac points. Thus, graphene is considered to be a zero bandgap semiconductor. Hence, the charge carriers can travel without scattering. Various exceptional properties are derived from its unique electronic features, which include high charge carrier mobility, ambipolar field effects, anomalous quantum Hall effects, ballistic transport, chirality, the Klein paradox, and weak anti-localization.26–28,30 Graphene exhibits a very high charge mobility due to its electronic structure, and over a long period, the mobility of graphene is mostly limited to 2000–15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1 due to microscopic ripples, scattering, and defects.15,18,26,27,30 Nevertheless, recent studies reported that graphene suspended on a clean surface exhibited a carrier mobility of up to 200000 cm2 V−1 s−1, which is primarily because of the decrease in the scattering of graphene with the help of the substrate, resulting in high carrier mobility and ballistic transport.15,26,30 Further, similar to its electronic properties, the optical properties are also strongly related to its two-dimensional, single atom thick, and honeycomb-like lattice structure. The optical transparency of graphene is defined by its fine structure constant; α = e2ℏc, where e is the electron charge, ℏ is the Dirac constant, and c is the speed of light. Single-layer graphene absorbs 2.3% of white light with a mere reflectance of 0.01%, and the absorbance varies linearly with the number of layers of graphene from 1 to 5.3.15,26,27,31 Along with these, the material exhibits some interesting properties including a large surface area (2630 m2 g−1), very high thermal conductivity (>3000 W mK−1), and a high Young's modulus (1 TPa) compared with other materials (Fig. 1b).26,32
000 cm2 V−1 s−1 due to microscopic ripples, scattering, and defects.15,18,26,27,30 Nevertheless, recent studies reported that graphene suspended on a clean surface exhibited a carrier mobility of up to 200000 cm2 V−1 s−1, which is primarily because of the decrease in the scattering of graphene with the help of the substrate, resulting in high carrier mobility and ballistic transport.15,26,30 Further, similar to its electronic properties, the optical properties are also strongly related to its two-dimensional, single atom thick, and honeycomb-like lattice structure. The optical transparency of graphene is defined by its fine structure constant; α = e2ℏc, where e is the electron charge, ℏ is the Dirac constant, and c is the speed of light. Single-layer graphene absorbs 2.3% of white light with a mere reflectance of 0.01%, and the absorbance varies linearly with the number of layers of graphene from 1 to 5.3.15,26,27,31 Along with these, the material exhibits some interesting properties including a large surface area (2630 m2 g−1), very high thermal conductivity (>3000 W mK−1), and a high Young's modulus (1 TPa) compared with other materials (Fig. 1b).26,32
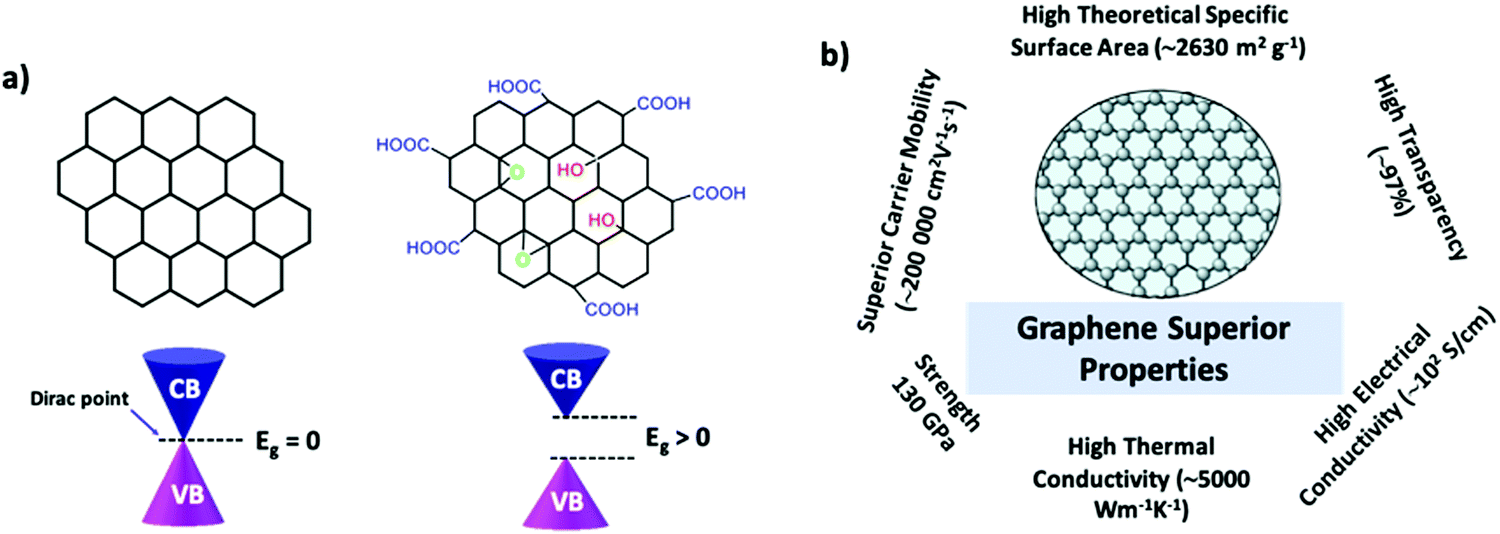 | ||
| Fig. 1 (a) Structures of graphene-based materials such as pristine graphene and graphene oxide along with band structures and (b) their superior properties. | ||
In the context of photocatalysis, the electrical and optical properties are two of the major constituents for the development of advanced photocatalytic materials. As discussed earlier, perfect π conjugated single sheet graphene lacks electronic bandgaps and is not photoluminescent. However, in the case of GO, photoluminescence emissions can be observed and are mainly due to the bandgap transitions corresponding to the conjugated π domains and defects in the graphene structures.5,14,16–18 As we know, the lack of a bandgap in graphene is advantageous in nanoelectronics, and on the other hand, the research community is more interested in graphene optical properties. Making the π-networks in graphene from infinity to finite opens up the electronic bandgaps, which is useful in photocatalysis for the generation of solar fuels (see Fig. 1a). These properties can be tuned by introducing heteroatoms/oxygen functionalities. The oxygen functionalities remove the equivalence of two carbon sublattices in graphene by forming C–O covalent bonds, which can damage original orbitals such as bonding π-orbitals and π*-antibonding orbitals.15,33 Introducing more oxygen functionalities over graphene leads to the enlargement of the bandgap, which includes changes in the valence band maximum (VBM) from the π-orbitals of the graphene to the 2p orbital of the O2, and the π* orbitals remain as the conduction band minimum.33–35 The bandgap depends on the extent of oxidation of the material. Therefore, by tuning the oxidation level, the bandgap of GO can be controlled. The formation of sp2 clusters in the sp3 matrix is likely to produce quantum confinement effects, which results in the opening of the bandgap at the Fermi level. Theoretical studies also showed that the carbon material containing a mixture of sp2 and sp3 hybridized carbons exhibits photoluminescence. The radiative recombination of electron–hole pairs of sp2 embedded in the sp3 matrix can give rise to fluorescence.17,34,36 GO is a P-doped material because oxygen atoms are more electronegative than carbon atoms, and by replacing the oxygen, functionalities bind at the edges of GO with a few nitrogen-containing groups that transform GO into an n-type semiconductor.15,17,37,38 Fully oxidized GO can act as an electrical insulator and partially oxidized GO acts as a semiconductor. The chemical reduction process can eliminate more functional groups from graphene oxide and form reduced graphene oxide (rGO) with the least functional groups. Although rGO has lower electrical conductivity than graphene, it has so many advantages like low production costs, large-scale production, and easy processing.34,36,37 Moreover, it can form stable aqueous dispersions through electrostatic stabilization, and shows higher hydrophobicity, greater surface area, a higher adsorption ability for reactants, and more defect sites as active sites. Hence, it is considered to be a versatile material for photocatalysis applications.
3. Intriguing functionalities for photocatalysis
Typical carbon-based 2D materials exhibit some unique mechanical, chemical, and physical properties, which can increase the efficiency of overall photocatalysis, especially upon combining with efficient photosensitizers. This can be briefly explained as follows.14,18,39 (a) The 2D structure of π-conjugated carbon-based materials (graphene, graphene oxide, reduced graphene oxide, and graphene/carbon quantum dots) provides an efficient template/platform for the controlled decoration of semiconducting nanomaterials.14,33 (b) Carbon materials exhibit a high surface area, adsorption capacity, and excellent stability. (c) Extended aromatic domains can facilitate free carrier transport, which eventually enhances the photocatalysis processes. (d) Extended π-conjugated domains of these 2D-materials can enhance the adsorption process of organic pollutants utilizing strong π–π interaction, and thus the photocatalytic degradation of the pollutant molecules. (e) Heterojunction hybrids made of π-conjugated 2D carbon-based materials can tune the band gaps of attached semiconductors by creating additional states. (f) They further help in avoiding agglomeration of semiconductors as well as enhancing the effective surface area. Furthermore, they can also hinder the leaching of the semiconducting counterpart on their surface during photocatalysis. (g) Additionally, 2D carbon-based materials as well as their composites exhibit enhanced cycling stability, robustness, easy recycling, extended light absorption, and enhanced adsorption properties, and hinder the electron–hole recombination processes during photocatalysis.14,37,39 All are schematically represented in Fig. 2.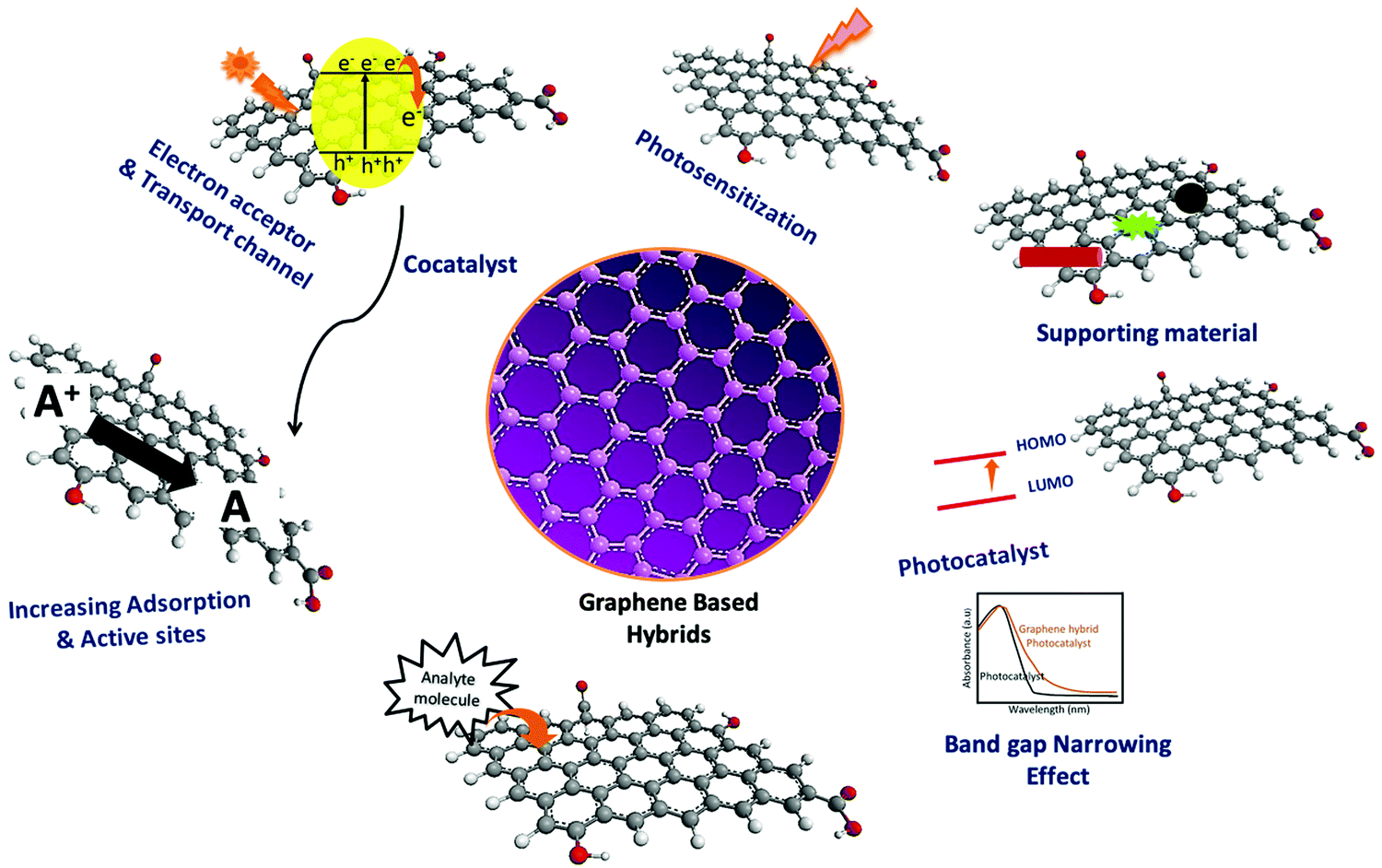 | ||
| Fig. 2 Various roles of graphene in hybrid systems. | ||
4. Strategies to build up composite materials with inorganic semiconductors
In continuation to the previous discussion, to achieve an efficient photocatalyst we have to merge two basic processes together: (a) harvesting a large extent of solar light absorbed using an efficient photosensitizer and (b) photoinduced exciton generation followed by efficient charge separation as well as free carrier accumulation on the active catalytic sites accessible for photocatalysis.3,4,17,18 Inorganic semiconductors are well known to act as efficient photocatalysts.40 In general, bare photocatalysts made of inorganic QDs suffer from several drawbacks, e.g. their large bandgap which inhibits visible light absorption, competitive recombination processes which reduce the number of free carriers accessible for photocatalysis, aggregation-induced competitive relaxation processes, and heavy metal-based toxicity. Considering this, making hybrids by combining inorganic semiconductors with π-conjugated 2D carbon-based nanomaterials can overcome almost all the difficulties. Several wet chemical approaches are presented in the literature for the preparation of graphene-based nanocomposites (Fig. 3).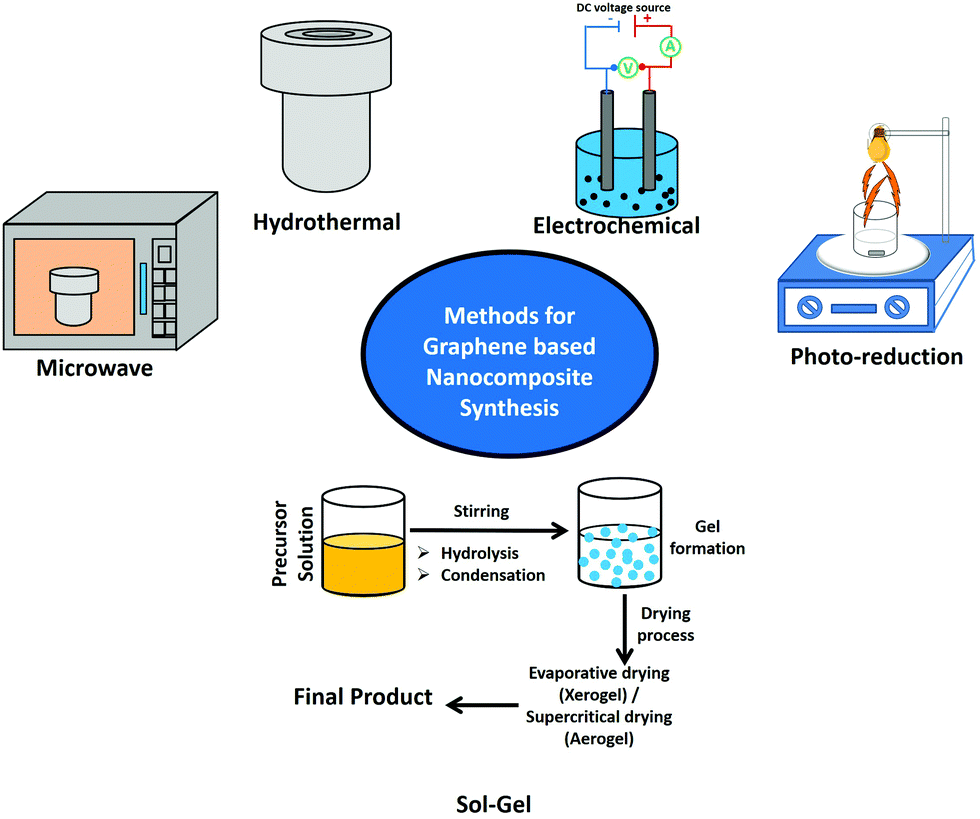 | ||
| Fig. 3 Schematic representation of various wet chemical approaches for the synthesis of graphene-based nanocomposite materials. | ||
Wet chemical methods such as hydrothermal, microwave, sol–gel, electrochemical, and photoreduction methods along with a few other approaches have been successfully employed for the integration of a variety of semiconductor nanostructures over graphene sheets. In this connection, the hydrothermal approach is a widely used synthesis technique for the preparation of various nanomaterials. The chemical reactions could be performed in solvent containing sealed vessels, known as autoclaves, pressure vessels, or high-pressure bombs. In autoclaves, the temperature can be increased above the boiling point of water, and the pressure of vapor saturation can be attained.41,42 It is basically the crystallization process directly from the solution containing reactants and solvent, which usually involves two steps: formation of nuclei and subsequent growth.42,43 By controlling synthesis parameters such as pH, temperature, solvent, reactant concentration, and additives, the final products could be fabricated with desired particle sizes and morphologies. Furthermore, this method is facile, very low-cost, and environmentally friendly, and can be used to synthesize aqueous dispersible reduced graphene oxide and rGO based composites in excessive amounts.43,44 Another approach is microwave irradiation, which is a rapid and facile method to provide energy for chemical reactions. The microwave approach is particularly advantageous for large-scale synthesis without using any complicated preparation conditions.45–47 The quick transfer of energy and fast decomposition of precursors provided by microwave sources would result in highly effective local reaction temperatures, resulting in significant reaction kinetics.45–47 One more approach is the sol–gel process, which involves hydrolysis of a metal oxide followed by cascade condensation and polycondensation reactions. Among the available wet-chemical approaches, the sol–gel process is the low-cost and facile one.48,49 Mainly, two chemical ways are presently used to form reliable products. They are the metal–organic route, which involves taking metal alkoxides in organic solvents, and the inorganic approach, which involves taking metal salts like chlorides, nitrates, and sulfides in aqueous solutions. The route using the alkoxide precursor appears to be the most versatile technique. Various inorganic and organic precursors can be used to make hybrid nanostructures.48,49 Electrochemical techniques are known as useful tools for modifying the intrinsic optoelectronic features of materials by altering the external power source to change the Fermi levels of so-called electrode materials. An electrochemical approach involves the direct deposition of inorganic nanomaterials over graphene-coated substrates.50,51 This method does not require any additional post-synthesis treatments like annealing and transferring. Primarily, electrochemical approaches offer the advantages of simplicity, time efficiency, and environmental friendliness. Thus, they will not result in the contamination of a reducing agent. The nanostructures of ZnO, Cu2O, and CdSe have been successfully deposited on the surface of rGO or CVD-grown graphene.50,51 Photochemical reduction is another fundamental approach for the synthesis of graphene-based semiconductor nanocomposites. Williams et al. prepared an rGO–TiO2 nanocomposite by a photoreduction approach.15,21,52 A mixture of GO and colloidal TiO2 was taken in ethanol solution and illuminated under UV light irradiation. During the reaction process, a color change was observed from light brown to dark brown followed by black. The reduction of GO was further confirmed using absorption spectra. Without TiO2 NPs, no significant change was observed for the GO solution, which confirmed the role of TiO2 in the reduction of GO to rGO.21 The GO dispersed in ethanol solution undergoes reduction by accepting the electron from the photoexcited TiO2 NPs. The TiO2 has highly negative conduction band energy upon UV light irradiation, and transfer of conduction band electrons takes place efficiently to the GO, resulting in reduction of GO. Before the reduction, the carboxylic groups on the GO surface can interact with the hydroxyl groups on the surface of TiO2 by a charge transfer mechanism. This hybrid structure is retained even after the GO reduction. The direct interaction between the TiO2 and rGO sheets hinders the collapse of exfoliated graphene sheets.21 A few approaches have been reported, which include the solution mixing approach, ultrasonic and sonochemical method, and liquid/liquid interface self-assembly approach. Recently, a liquid–liquid interface has been utilized for the self-assembly of graphene and its composites, and this is an inexpensive and facile route to obtain atomically thin films. Bramhaiah et al. have also employed a liquid–liquid interface approach for the fabrication of various nanocomposites including rGO–noble metal (Ag, Au, Pd, and Os),53,54 rGO–semiconductor (Fe2O3, Ag2S, ZnO, CuO and SnO2),55–57 and rGO–Ni(OH)2 nanostructures57 for various applications such as surface-enhanced Raman spectroscopy (SERS), heterogeneous catalysis, reusable SERS, supercapacitors, electrochemical sensors, and electrochemical water splitting reactions.
5. Main scopes for enhancing the photocatalytic activity
In general, photocatalytic activity depends on two major aspects: (a) artificial light-harvesting and (b) exciton generation followed by efficient charge separation for photocatalysis.58–61 In the case of graphene-based composite systems, we have room to improve both the counterparts, graphene-based materials and semiconductors as light-harvesting materials and catalysts, respectively. Mostly inorganic semiconductors are active constituents with tunable bandgaps for solar light harvesting, and active sites on the surface facilitate photochemical redox reactions.13,62 Therefore, several strategies have been developed for enhancing photocatalytic activity in semiconductor nanostructures, which include sensitization with various sensitizers such as dyes, polymers, and semiconductors, and coupling with semiconductors. Other approaches include surface complex assisted approaches, bandgap modification by creating oxygen vacancies and oxygen sub-stoichiometry, doping with different elements like metals and non-metals, spatial structuring, and morphology optimization for the improvement of active sites (Fig. 4).63–66 Surface sensitization is an effective method to improve photocatalytic activity. The active compound that can strongly absorb visible light leads to the excitation of electrons followed by an injection into the conduction band of a semiconductor. According to the literature, this electron injection is favorable due to the more negative potential of exciting photosensitizers than the CB of a semiconductor.67–69 Another important approach for the improvement of photocatalysis is the coupling of semiconductor nanostructures, which can change the excitation path and block the electron–hole recombination. Coupling with narrow bandgap semiconductors can improve visible light absorption as a result of the improvement in photocatalytic activity.70–72 The CB and VB of the coupled semiconductor are more negative and less positive, respectively, than the active photocatalyst. The main idea behind this approach is to promote the separation of photo-induced electron–hole pairs by changing the carrier transport pathways. Oxygen vacancies arising from doping or surface redox treatment can efficiently capture photo-induced electrons, which leads to the prevention of electron–hole pair recombination, resulting in an enhancement of quantum efficiency.73,74 The most prominent strategy for enhancing photocatalytic activity under light illumination is the doping approach. Doping with non-metals like nitrogen, fluorine, chlorine, sulfur, boron, and carbon in a semiconductor can form covalent bonds that can lead to enhancement in light absorption.75–77 Furthermore, doping with various transition metal ions including cobalt (Co2+), iron (Fe3+), and copper (Cu2+) ions has different effects on photocatalytic performance.78,79 Besides doping, the crystallinity, phase composition, native defects, and morphology of a photocatalyst are the major factors affecting photocatalytic activity. The crystallinity and native defects can be tuned by annealing, and the morphology and phase composition can be determined by tuning the growth/synthesis conditions.80 The morphology manipulation can affect the charge carrier separation as well as the availability of active sites due to the differences in the terminating facets. In TiO2 nanostructures, the (101) facets are claimed to be more reactive than the (001) and low energy (101) facets.80,81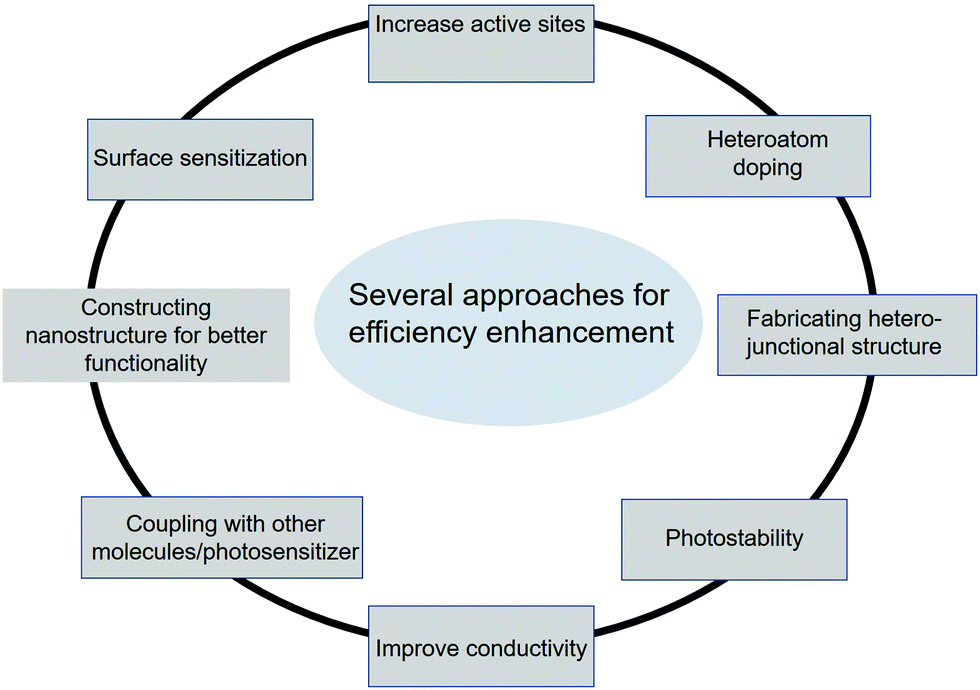 | ||
| Fig. 4 Schematic representation of various methods for the photocatalytic activity enhancement of carbon-based materials. | ||
Depending on the structure–property correlation, graphene-based materials can act as electron acceptors, electron mediators, and sole-photocatalysts for water splitting.17,18 It is well-known that single-layer graphene is a semimetal/zero bandgap semiconductor because of the overlap of the valence band and conduction band at the Dirac points, which results in superior electrical conductivity.60 Nevertheless, the conductivity of the graphene can be tuned by doping (opening of the bandgap). In this context, the chemical doping of graphene has been a promising approach to enhance the performance of graphene-based catalysts.18,82 The chemical doping of graphene is mainly of two types, which are surface transfer doping and substitutional doping. Surface transfer doping occurs primarily due to the additional functional groups over the graphene sheet and the natural tendency of these groups like donating and withdrawing electrons from the graphene, which leads to n or p-type conductivity, respectively.18,60,82,83 Substitutional doping involves replacing the carbon atoms in the graphene lattice with heteroatoms with different numbers of valence electrons like atoms having more valence electrons than the carbon, leading to n-type conductivity, and atoms with fewer valence electrons than the carbon, giving rise to p-type conductivity. Generally, doping in graphene introduces additional states in the density of states due to the formation of additional free charge carriers. Ideal undoped graphene is chemically inert because its unpaired electrons are strongly bound and passivated. The number of active carriers generated in a photocatalytic redox reaction mainly depends on the number of catalytically active sites present over the surface of the photocatalyst. Therefore, the larger the active surface area of the photocatalyst, the more the number of active sites on the surface, which leads to a higher rate of photocatalysis. This can be achieved by either creating more active sites through substitutional doping of graphene or loading co-catalysts as active sites over the graphene. Generally, heteroatom doping like boron, nitrogen, phosphorus, and sulfur doping in graphene can effectively create active sites for various catalytic applications.84,85
Another critical issue of graphene and its hybrids is catalytic stability/reproducibility, which is largely determined by the stability of the photocatalyst. In many cases, photocatalysts can suffer due to the loss of activity by self-oxidation/reduction caused by accumulated photoinduced holes/electrons.86 Akhavan et al. reported the use of reduced graphene oxide (rGO) for the enhancement of the photocatalytic activity of TiO2. They observed a gradual decrease in graphene content along with an increase in UV light irradiation time in the case of GO physically attached with TiO2 (without the Ti–C bond), whereas the chemical bond formed between TiO2 and graphene (Ti–C bond) exhibited better stability.86,87 In general, the strong interfacial interaction between graphene and other semiconducting materials could be the critical factor in stabilizing the graphene during photocatalysis. However, in some cases, the photo-induced charge carriers may degrade the graphene sheets. In contrast, the deposition of co-catalysts over graphene-based systems can protect them from self-decomposition by effectively removing holes/electrons from the graphene, resulting in enhanced photostability. Furthermore, the formation of reactive species such as hydroxyl radicals by the reaction between photoexcited holes and water/hydroxyl groups on the surface of the photocatalyst can also degrade the graphene. However, in some other systems such as CdSe–RGO/TiO2, no obvious degradation of graphene has been observed even with hydroxyl radicals.88 The addition of other species to consume the reactive hydroxyl radicals during photocatalysis is also another feasible approach for the improvement of graphene-based composites.89 Furthermore, the addition of hole scavengers (methanol, EDTA, TEOA, etc.), to photocatalytic systems can eventually increase the charge separation and the stability of the photocatalyst. Moreover, decreasing the exposure area of the isolated graphene in graphene-based composites by rationally controlling the weight ratio of graphene or improving the synthesis approaches can decrease the interaction between graphene and reactive species.86
6. Thermodynamics behind photocatalytic solar fuel generation
The best way to convert solar energy to solar fuels is to mimic the natural photosynthesis of green plants artificially by using heterogeneous photocatalysts. It includes mainly two energetically uphill reactions: (a) photocatalytic solar water splitting into molecular hydrogen and oxygen and (b) CO2 photoreduction to chemical fuels.2–4,15,17,90Fig. 5 illustrates systematically the water-splitting and CO2 photoreduction reactions. The overall water splitting reaction (H2O → H2 + O2) is an energetically uphill reaction requiring a standard free energy change (ΔG°) of 237.2 kJ mol−1 or potential of 1.23 eV per electron. However, the bandgap of 1.23 eV is practically not suitable for photocatalytic water splitting. In the literature, it is well demonstrated that the minimum bandgap required for photocatalytic water splitting is ∼2 eV.3,4,10,62,90 Generally, water splitting under sunlight with the help of a semiconductor involves three major steps: (i) a semiconductor absorbs a light photon and (ii) generates charge carriers (electron and holes), which can be bound by coulombic forces; (iii) these photoinduced charge carriers such as electrons and holes can migrate to the surface of the semiconductor or recombine again in the bulk material; (iv) on the surface of the semiconductor, the holes can oxidize water into O2 and electrons can reduce the protons to H2 molecules.3,4,91 Based on the following equations, two electrons are needed to produce an H2 molecule and four holes for the O2 molecules. In overall water splitting, H2 and O2 are generated simultaneously in a 2:1 stoichiometric ratio.3,4,91| Absorption of light: semiconductor + hν (>Eg) → eCB− + hVB+ |
| Water oxidation (OER): 2H2O + 4hVB+ → O2 + 4H+ | (1) |
| Proton reduction (HER): 2H + 2eCB− → H2 | (2) |
| Overall water splitting reaction: 2H2O(l) → 2H2(g) + O2(g) ΔG° = 237.2 kJ mol−1 | (3) |
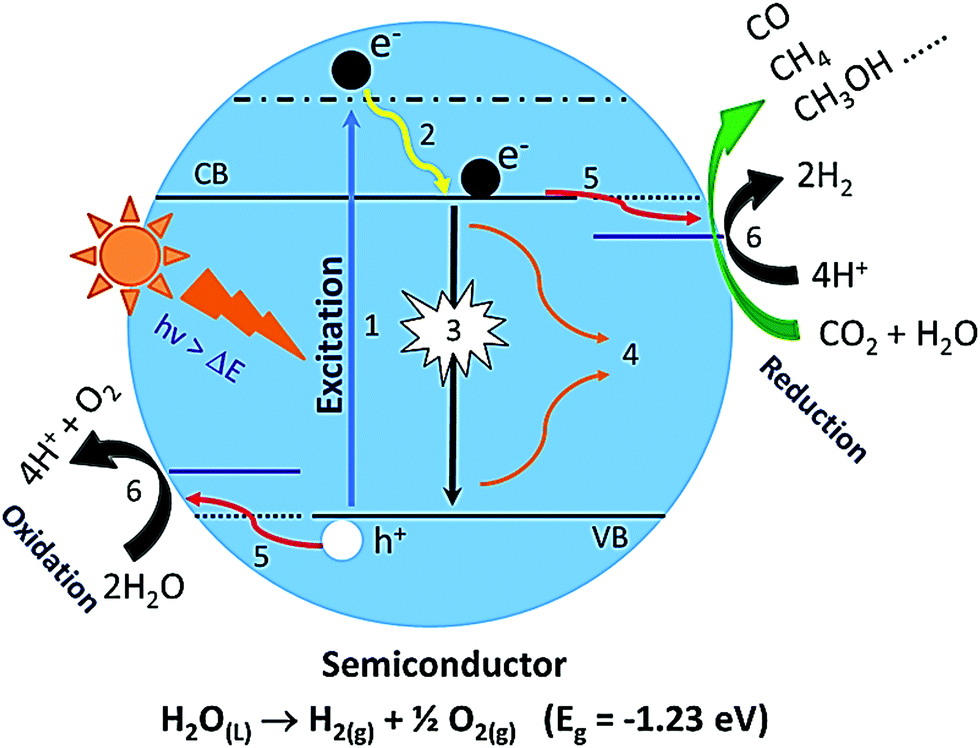 | ||
| Fig. 5 Schematic illustration of various photophysical processes on a semiconducting photocatalyst upon light irradiation: (1) light absorption, (2) thermal relaxation, (3) radiative recombination, (4) non-radiative recombination, (5) diffusion of free carriers towards the surface of the photocatalyst and (6) photochemical redox reactions. | ||
The photon absorption by a semiconductor upon light irradiation initiates a time-dependent non-equilibrium photophysical and photochemical process. The schematic illustration of various plausible photophysical processes happening in the semiconductor upon the light illumination is shown in Fig. 5. As we discussed earlier, large bandgap semiconductor materials are efficient for water splitting. On the other hand, the utilization of full-range solar light (especially visible light) is limited. Hence, one should have to keep in mind these two aspects to develop highly efficient visible light active photocatalysts.3,4,18,62 Under light illumination, photo-induced excitons such as excited electrons are formed in the conduction band (CB) and their counterparts such as holes are formed in the valence band (VB). After the generation of excitons, the electrons in the CB can recombine with the holes in the VB through radiative and non-radiative recombination processes and these are competitive processes. As a result, the availability of free carriers decreases. Overall, the free carriers that survived can reach the interface through diffusion, or an electric field followed by redox reactions.3,4 The times required for these processes to happen are as follows: the generation of excitons in the femtosecond time scale, and the radiative and non-radiative recombination processes happen in the time scale of pico-seconds to nanoseconds, while the recombination through the triplet state may reach a time-scale of a few hundred nanoseconds to milliseconds.3,4,92 The diffusion and charge separation can also happen at a similar time scale. Nevertheless, the photochemical redox reactions over the surface of the photocatalyst are in the microsecond time scale. Consequently, it is always crucial to regulate the overall photophysical and following photochemical processes to enhance the efficiency of the overall solar water splitting reaction. However, the major challenge regarding photocatalytic water splitting is charge (electron–hole pair) recombination. The electron–hole pair recombines and dissipates an unproductive heat. Therefore, it reduces the effectiveness of the H2 production process. Specifically, in pure water, it is difficult to achieve water-splitting reaction for H2 production due to the fast recombination of charge carriers. Hence, photocatalytic water splitting is typically studied in the presence of sacrificial agents like methanol, ethanol, and glycerol, and electrolytes like Na2S and KI. The electrolytes do not undergo any oxidation or reduction by the CB electrons and VB holes. The electrolytes act as transport media for ions and transfer electrons to the adjacent semiconductor, thus improving the efficiency of photocatalysis. The sacrificial reagents or electron donors react with the VB holes as a result of the enhancement of the charge separation. Therefore, the H2 generation from pure water has its limitations. The photoreduction of CO2 over the photocatalyst is a multistep process involving the adsorption and activation of CO2, and dissociation of the C–O bond. However, CO2 is an extremely stable molecule and its C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond holds a higher dissociation energy of 750 kJ mol−1 than any other chemical bonds such as C–H (430 kJ mol−1) and C–C (336 kJ mol−1), which indicates that high energy input is needed for the transformation of CO2 to value-added products. In addition to this, the carbon in CO2 exhibits the highest oxidation state and the reduction of CO2 leads to a large variety of products ranging from CO, CH4, and higher hydrocarbons in the gas phase to oxygenates in the liquid phase such as alcohols, aldehydes, and carboxylic acids.97 The plausible reaction pathways for the photoreduction of CO2 and conceivable formation of main products and their corresponding reduction potentials vs. NHE are given in Table 1. At present, one of the utmost problems of CO2 photoreduction is the low conversion efficiency. Here, some of the key factors which limit the efficiency are listed: (1) mismatch between the absorption ability of the photocatalyst and the solar spectrum, (2) poor photo-induced charge carrier separation, (3) the low solubility of CO2 molecules in water: ∼33 μmoles of CO2 in 1 mL of water at 100 kPa and room temperature, (4) backreaction during the reduction of CO2 and (5) competition reaction of water reduction to hydrogen. The foremost reasons for the limited efficiency of photocatalytic reduction of CO2 with H2O are as follows: (1) the highly unreceptive one-electron transfer to form CO2− that requires a very negative reduction potential: −1.9 V vs. NHE (Table 1); (2) the strong oxidation power of the photoexcited holes and OH− radicals that induce the backward reaction of intermediates and products formed by the CO2 photoreduction; (3) the fast recombination rates of photo-induced electron–hole pairs (e−–h+) in the semiconductor photocatalyst; and (4) the limitations in the harvesting of visible light due to the wide-bandgap of the semiconductor photocatalyst.
O bond holds a higher dissociation energy of 750 kJ mol−1 than any other chemical bonds such as C–H (430 kJ mol−1) and C–C (336 kJ mol−1), which indicates that high energy input is needed for the transformation of CO2 to value-added products. In addition to this, the carbon in CO2 exhibits the highest oxidation state and the reduction of CO2 leads to a large variety of products ranging from CO, CH4, and higher hydrocarbons in the gas phase to oxygenates in the liquid phase such as alcohols, aldehydes, and carboxylic acids.97 The plausible reaction pathways for the photoreduction of CO2 and conceivable formation of main products and their corresponding reduction potentials vs. NHE are given in Table 1. At present, one of the utmost problems of CO2 photoreduction is the low conversion efficiency. Here, some of the key factors which limit the efficiency are listed: (1) mismatch between the absorption ability of the photocatalyst and the solar spectrum, (2) poor photo-induced charge carrier separation, (3) the low solubility of CO2 molecules in water: ∼33 μmoles of CO2 in 1 mL of water at 100 kPa and room temperature, (4) backreaction during the reduction of CO2 and (5) competition reaction of water reduction to hydrogen. The foremost reasons for the limited efficiency of photocatalytic reduction of CO2 with H2O are as follows: (1) the highly unreceptive one-electron transfer to form CO2− that requires a very negative reduction potential: −1.9 V vs. NHE (Table 1); (2) the strong oxidation power of the photoexcited holes and OH− radicals that induce the backward reaction of intermediates and products formed by the CO2 photoreduction; (3) the fast recombination rates of photo-induced electron–hole pairs (e−–h+) in the semiconductor photocatalyst; and (4) the limitations in the harvesting of visible light due to the wide-bandgap of the semiconductor photocatalyst.
| Photocatalyst + hν → e− + h+ | ||
| e− + h+ → heat (recombination) | ||
| Photooxidation reactions | ||
| Water oxidation/decomposition | H2O + 2h+ → 1/2O2 + 2H+ | +0.82 E0 redox (V vs. NHE) |
| Hydrogen peroxide formation | O2 + 2H+ + 2e− → H2O2 | |
| Photoreduction reactions | ||
|---|---|---|
| Product | Reaction | E 0 redox (V vs. NHE) |
| Hydrogen formation | 2H+ + e− → H2 | −0.41 |
| CO2 radical formation | CO2 + e− → CO2˙− | −1.90 |
| Formic acid formation | CO2 + 2H+ + 2e− → HCO2H | −0.61 |
| Carbon monoxide formation | CO2 + 2H+ + 2e− → CO + H2O | −0.53 |
| Formaldehyde formation | CO2 + 4H+ + 4e− → HCHO + H2O | −0.48 |
| Methanol formation | CO2 + 6H+ + 6e− → CH3OH + H2O | −0.38 |
| Methane formation | CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.24 |
| Ethanol formation | 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O | −0.16 |
As discussed previously, one of the most significant limitations of photocatalytic water decomposition is utilizing pure water (hard to perform). This is usually related to the fact that the simultaneous reduction and oxidation of water is a complex multi-step reaction process involving four electrons. Using sacrificial agents as electron donors or acceptors can remarkably accelerate the H2 production rate (Fig. 6). In the case of electron donors, such as methanol, the holes are scavenged and get oxidized to CO2, through sequential steps. As a result, the photoinduced charge carrier recombination is greatly reduced and the CB electrons exhibit higher lifetimes and are better utilized for the reduction. Moreover, the production of O2 is minimized as a result of the oxidation of sacrificial reagents by the OH radicals; the back reaction (recombination of O2 and H2 gases) to produce water is suppressed, increasing the H2 yield; and the gas separation stage can be avoided. Nevertheless, it can be noted that the H2 production rate can subsequently be reduced by the competitive reduction reactions with the products formed upon oxidation of sacrificial agents. Electron donor sacrificial reagents can be divided into two categories: inorganic agents such as EDTA and Na2S/Na2SO3, and organic agents like alcohols, organic acids, and hydrocarbons used as hole scavengers (electron donors) for photocatalytic H2 generation by water splitting.98
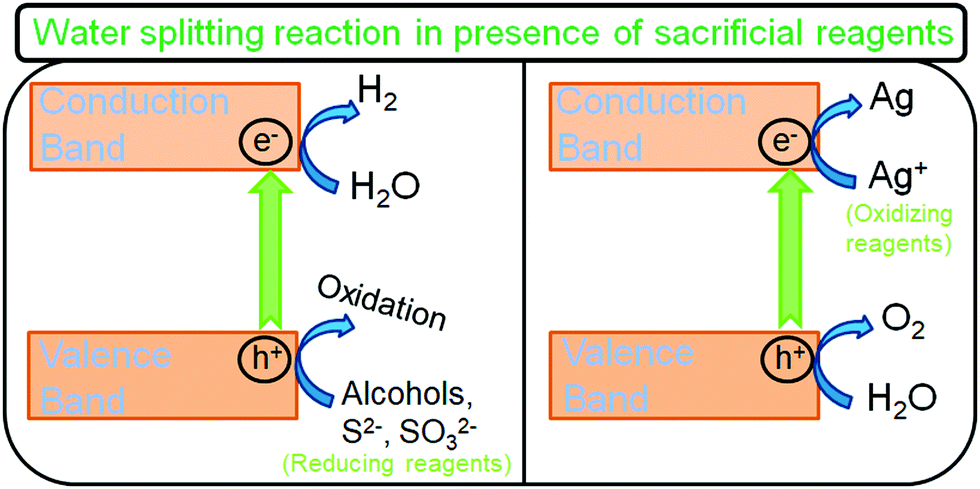 | ||
| Fig. 6 Schematic illustration of the water splitting reaction in the presence of sacrificial reagents. | ||
On the other hand, electron scavenging reagents like persulfate, silver (Ag+), and iron (Fe3+) ions act exactly in the opposite manner to hole scavengers and holes are utilized for oxidation (Fig. 6).
7. Development of graphene-based composites for photocatalytic solar-fuel generation
Considering its superior electron mobility, surface area, viable assembly, and tunable electronic band structure, pure GO can act as a photocatalyst for solar-driven water splitting. Yeh et al. demonstrated the photocatalytic water splitting activity of GO with a bandgap of 2.4–4.3 eV for hydrogen generation under a mercury lamp.38 Under UV or visible light, this GO catalyst steadily catalyzes H2 production from 20 vol% aqueous methanol and pure water, even without any metal co-catalyst. Incident UV light caused a π–π* transition and generated electron–hole pairs on GO sheets. The photogenerated electrons reduce the oxygen functional groups to form defect carbon sites, which act as active sites for water reduction to H2. All GO sheets have a valence band and a conduction band suitable for O2 and H2 evolution from complete water splitting. However, the experimental results show that the higher degree of oxidation in GO catalyzed O2 evolution, whereas less oxygenated GO did not catalyze O2 evolution. As we know, GO displays p-type conductivity, which hinders the hole transfer for an oxidative half-reaction for O2 generation. To overcome this obstacle, Yeh et al. introduced amino and amide groups on the surface of GO and demonstrated that the ammonia-modified GO displays n-type conductivity and can catalyze H2 and O2 evolution separately.99On the other hand, while making a composite of GO with TiO2, GO layers are particularly useful in separating the charges on the TiO2 surface because of its 2D structure as well as conjugated backbone. Additionally, the unpaired π electrons in the GO can form a bond (Ti–O–C) with Ti atoms of TiO2. This eventually extends the light absorption of TiO2 into the visible region. The transient photovoltage measurements showed that the photovoltaic response of TiO2–graphene is positive and the mean lifetime of electron–hole pairs is prolonged from ∼10−7 to ∼10−5 ns in comparison to similarly-sized bare TiO2.100 From these reports, one can conclude that the photoinduced electrons could transfer from TiO2 to the graphene sheets for suppressing the recombination. Hence, graphene can act as an electron acceptor/transporter and can separate the photoinduced charges in the TiO2 semiconductor as well as also transport the photogenerated electron—hole pairs to the active sites, resulting in an improvement of the H2 production. Patra and his group have extensively studied the excited state dynamics of graphene-based hybrids, especially explaining the electron-accepting properties of graphene/rGO while combining with organic/inorganic semiconducting materials for light-energy conversion devices.20,101–103 Furthermore, these functions of graphene could be affected by the graphene content in the composite, interfacial interaction, and formation of intimate heterojunctions between the graphene and semiconductor materials. Moreover, the electron transfer and acceptance role of graphene in the composite is significantly affected by the interfacial interaction between the graphene and semiconductor nanoparticles and it can be critically controlled by typical synthetic techniques. In this regard, Fan et al. synthesized graphene–TiO2 (P25) nanocomposites by several techniques including UV-assisted photocatalytic reduction, hydrazine reduction, and hydrothermal reduction approaches, and further applied them as photocatalysts for the evolution of H2 from alcoholic solution under UV-visible light irradiation. A comparison of the photocatalytic performances of the various nanocomposites is shown in Fig. 7a. The incorporation of rGO into the TiO2 (P25) significantly enhanced the photocatalytic H2 evolution, and the graphene–TiO2 (P25) nanocomposite prepared by the hydrothermal reduction approach exhibited the best performance when the optimum mass ratio of TiO2 (P25) to rGO in the nanocomposite was 1:0.5. The contact between the TiO2 and graphene is different for the nanocomposites prepared by the different approaches including the photoreduction, hydrazine reduction, and hydrothermal approaches. In the case of the hydrothermal approach, the TiO2 NPs are highly dispersed on the graphene sheets and less aggregated, leading to strong interaction, while in the case of photo- and hydrazine reduction, the TiO2 NPs were aggregated. This aggregation makes a difference in the catalytic performances of the nanocomposites. The stronger interaction between the TiO2 (P25) and rGO accelerates the charge transfer of photogenerated electrons on TiO2 (P25) to rGO, resulting in the suppression of the charge recombination and thus improvement of photocatalytic performance.104 Li et al. reported the synthesis of a reduced TiO2–GO heterostructure using a chemical reduction agent-free one-step laser ablation in a liquid approach. The heterostructure exhibited an extended optical response range from the UV to visible region, which is due to the formation of Ti–C and Ti–O bonds, leading to the creation of shallow traps into the band of TiO2 and narrowing of the bandgap of TiO2 due to the introduction of plenty of intermediate energy levels within the bandgap of TiO2. The evolution of the hydrogen mechanism over the reduced TiO2–GO heterostructure is illustrated in Fig. 7b. The introduction of graphene and Ti3+ as shallow trap states highly enhances the charge transfer efficiency of electrons and results in efficient charge separation (Fig. 7b). The photoexcited electrons migrate to the conduction band, and some of them flow to the graphene sheets as well and holes in the valence band (VB). The generated holes would react with the methanol molecules (CH3OH + h+ → CO2 + H2O) and the electrons would reduce the H+ (H+ + e− → H2), producing H2.105
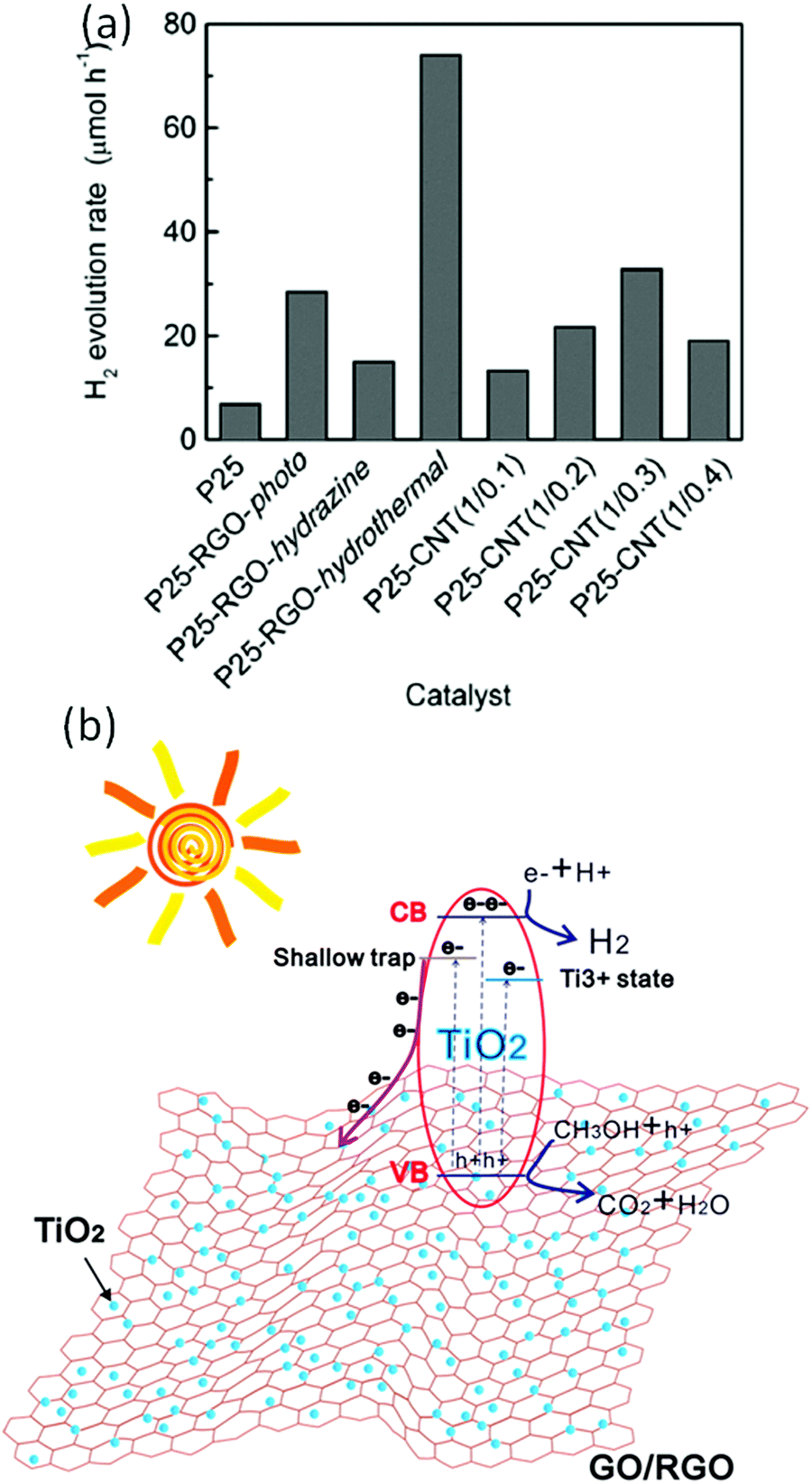 | ||
| Fig. 7 (a) Comparison of the photocatalytic performances of various nanocomposites synthesized using different methods.104 (b) Schematic illustration of H2 production over the TiO2–rGO nanocomposite.105 Copyright 2011, American Chemical Society. | ||
As a step forward, the integration of semiconductors with graphene and plasmonic nanomaterials provides a synergic contribution to enhanced photocatalytic activity. By decorating the surfaces of semiconductors with noble metal nanostructures such as Ag, Au, and Cu, an enhanced visible-light photocatalytic activity of the semiconductor is achieved due to the surface plasmon resonance (SPR) of noble metal particles, which can directly transfer the plasmon-induced electrons to the semiconductor through the metal–semiconductor junction.106–108 The plasmonic metal NPs form a Schottky barrier at the metal–semiconductor interface, and it can serve as an effective electron trap, causing a high density of states at the Fermi level of the interface and minimizing the charge carrier recombinations.106 Hence, designing such multifunctional materials provides various pathways for the transport of electrons arising from noble metal–semiconductor, semiconductor–graphene, and graphene–noble metal interfaces.
Khalid et al. fabricated a Ag–TiO2/graphene nanocomposite via a microwave hydrothermal approach and demonstrated its application for photocatalytic water splitting for the generation of H2 under visible light irradiation.46 Compared with bare TiO2, Ag–TiO2, and graphene–TiO2, the Ag–TiO2/graphene nanocomposite exhibited an enhanced photocatalytic performance. Ag and graphene played vital roles in the enhanced photocatalysis (Fig. 8a). After the Ag loading on the nanocomposite, the H2 production rate becomes higher, and as the concentration of Ag increases up to 0.09%, the H2 generation increases (Fig. 8a and b). Upon further increasing the Ag concentration, the H2 production rate decreases because the Ag could act as a recombination center for electron–hole pairs. The schematic of the photocatalytic reaction mechanism of the Ag–TiO2/graphene composites under visible light is shown in Fig. 8. The Fermi level of the Ag is lower than the CB band of TiO2. During the SPR excitation, the photo-excited electrons of Ag nanoparticles in the composite can overcome the Schottky barrier and tunnel through the TiO2/graphene interface into the CB of TiO2. At the same time, some of the energetic electrons in the CB of TiO2 reduce the H2O to H2, and the remaining electrons can transfer to the graphene (Fig. 8a). The Fermi level of graphene is lower than the CB of TiO2 and slightly higher than the potential of H+/H2. Therefore, the CB of TiO2 and graphene act as active sites for the reduction of H2O to H2. The overall photocatalytic efficiency of the Ag–TiO2/graphene composites for the water splitting is attributed to the following reasons: (1) suppressed charge recombinations due to the quick electron transfer; (2) enhanced visible light absorption due to the plasmonic band of Ag nanoparticles; and (3) the strong covalent bond between the TiO2 and graphene (TiO–C bond), which virtually facilitates the easy and quick transfer of energetic electrons from the CB of TiO2 to graphene (Fig. 8a).45,46 Recently, Tudu et al. studied the catalytic influence of the type of carbon support and the form of composite material (thin film form and powder form) on photocatalytic water splitting for the production of H2 under solar light. For this study, they synthesized two varieties of composites including Au–Pd/C/TiO2 (carbon prepared using local plant materials) and Au–Pd/rGO/TiO2 (1:1 Au–Pd ratio) nanocomposites employing an eco-friendly hydrothermal approach. For comparison studies, thin films were prepared from the dispersions of powder samples through a simple drop-casting approach. The thin-film form of the nanocomposite exhibited 30 times higher H2 production than the powder form. In general, the thin film on a nonconductive simple glass substrate improves the connectivity between the catalyst particles and facilitates easy electron transfer locally. The improved performance of the thin film form is due to suppressed light scattering and thus enhanced light absorption, which leads to a high number of charge carriers, while in the case of the powder form, the light scattering is predominant. In the present case, the light scattered within the film is subsequently absorbed and leads to an enhanced SPR effect, and this contributes to the generation of more charge carriers. On the other hand, aggregation occurs in the case of powder suspension under light irradiation, which leads to predominant light scattering, resulting in poor light absorption and generation of a small number of charge carriers, which consequently leads to the generation of a small amount of H2. Furthermore, with a higher concentration of particulate matter, reduction in photocatalytic activity is observed, mainly due to the unequal exposure of the particles to the light irradiation. Therefore, for a better photocatalytic activity for the generation of H2 from water-splitting, one might choose a thin-film form.109
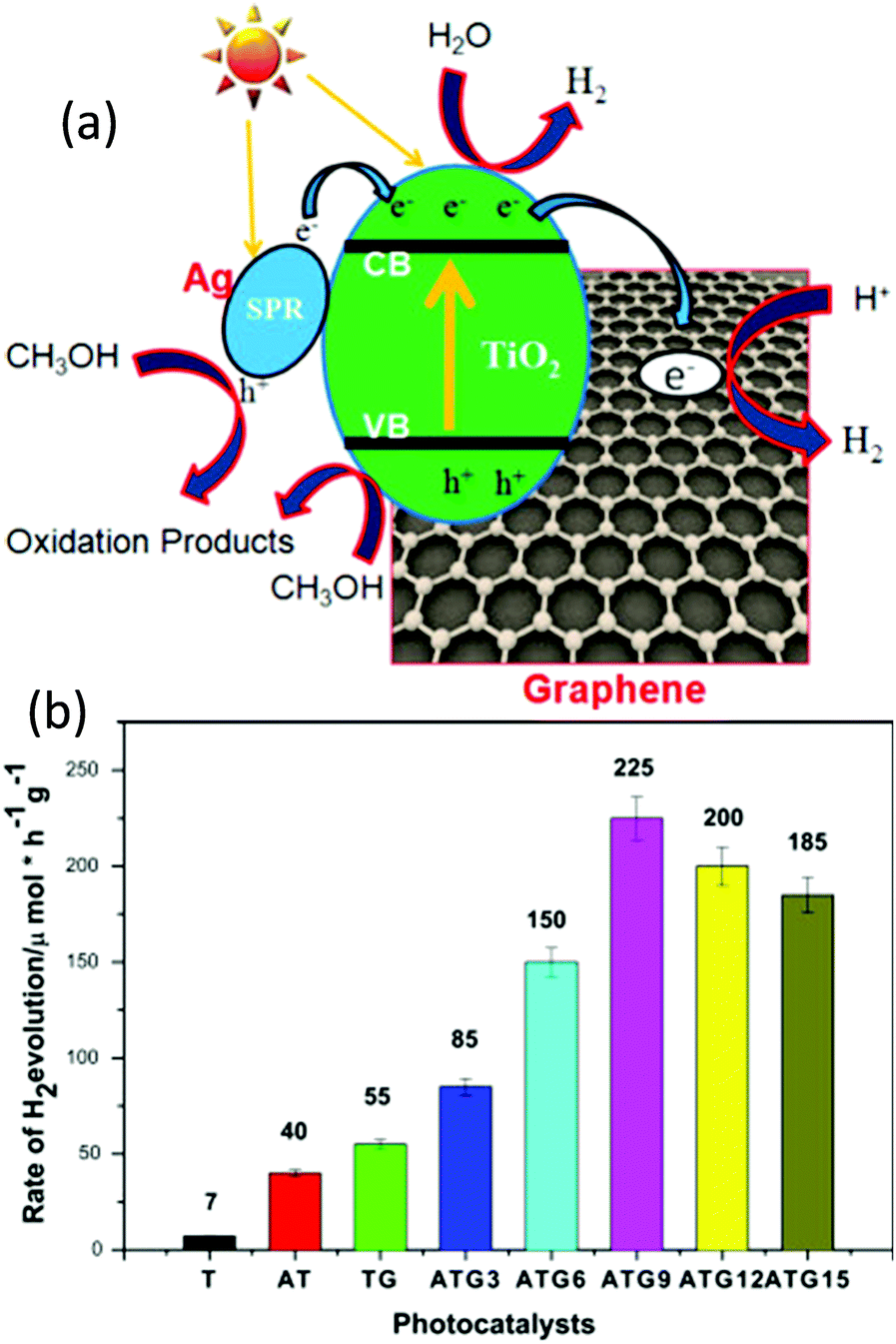 | ||
| Fig. 8 (a) Schematic illustration of H2 generation over the Ag–TiO2/graphene composite under visible light and (b) histogram of H2 evolution of various Ag–TiO2/graphene composites under visible light (λ ≥ 420).46 Copyright 2016, Elsevier. | ||
Although several advances have been made on TiO2 based composites, a few fundamental drawbacks are still there regarding their large bandgaps, defect state induced recombination, etc. As an efficient alternative to TiO2, perovskite, and spinel-type structures have drawn much attention from the scientific community as photocatalysts due to their excellent optical response, exceptional stability, and flexibility towards substitution which induce new properties such as magnetic, electrical, and optical behavior. Typically, perovskites are binary metal oxides with the general formula ABO3.110,111 The crystal structure of the perovskite is a cubic structure with a Pmm or monoclinic or triclinic space group and distorted orthorhombic, rhombohedral, or tetragonal symmetry. The radii of the A site cations are generally more prominent than those of the B site cations. Eight BO6 octahedra with shared corners form a 3D framework of a cubic cell, and the center of this framework is occupied by the A-site cations. These perovskite materials exhibit excellent photocatalytic activity under visible light irradiation. In this regard, various perovskite-like materials such as YInO3, SrTiO3, LaFeO3, NaTaO3, LaCrO3, and KNbO3111–113 have been reported. Although all the above-mentioned perovskite materials show outstanding photocatalytic activity, the photo-corrosion phenomena are the major challenges, which lower the overall photocatalytic performance of these materials. Ding et al. reported the synthesis of a graphene–YInO3 composite by a facile solvothermal approach, in which the reduction of graphene oxides and deposition of YInO3 nanoparticles over graphene sheets are achieved simultaneously, and demonstrated it as a photocatalyst for H2 evolution under visible light irradiation. The addition of graphene can act as a co-catalyst and narrow the bandgap of YInO3 to the visible region by forming a chemical bond. It can also help in prolonging the separation and lifetime of photogenerated electron–hole pairs. Under visible light irradiation, an H2 evolution rate of ∼400 μmol h−1 g−1 is attained for a graphene content of 0.5 wt%. This value is 127 and 3.7 times higher than those of bare YInO3 and the Pt–YInO3 composite, respectively. The graphene–YInO3 composite is highly stable and produces H2 continuously up to 40 h without decay in the activity.114 Bai et al. fabricated size controlled ZnGa2O4 nanospheres by adjusting the trisodium citrate concentration and assembled these ZnGa2O4 nanospheres over 2D N-doped graphene layers through the facile hydrothermal approach and studied the effect of the size of the particles on the photocatalytic activity. As the diameter of the spheres increased, the H2 production rate increased and reached a maximum value of 38.94 μmol h−1 g−1 for 230 nm particles. Upon further increasing the size of the ZnGa2O4 nanospheres, the H2 rate decreased, which could be ascribed to the crystallinity, morphology, and surface area of the nanospheres. Moreover, by introducing N-doped rGO into the system of ZnGa2O4 nanospheres, the photocatalytic activity improved and the production rate was almost ∼4.2 times that of bare ZnGa2O4.115
Chemical doping with heteroatoms is one of the most feasible approaches to achieve diverse properties of graphene. Heteroatom doping of graphene can simultaneously introduce a bandgap and endow graphene with catalytically active sites, which leads to intrinsic sole photocatalytic activity on its own. Initially, N-doped graphene was used as a supporting matrix of semiconductor-based photocatalysts for enhanced activity. The N-doped graphene helps in effective photo-induced charge separation, migration, and collection, which can be greatly improved by the formation of effective interfacial interactions and the formation of a p–n heterojunction between the semiconductor and N-doped graphene. The presence of nitrogen atoms modifies the electronic band structure of graphene, which becomes a semiconductor, rather than a conductor like all-carbon graphene.116 Theoretical studies also confirm that doping with nitrogen alters the electronic band structure of the graphene sheet depending on the percentage of nitrogen substitution. The bandgap can continuously increase from 1.0 to 0.7 eV for 2 to 10% nitrogen doping. According to these calculations, it is predicted that the n-doped graphene can act as a sole photocatalyst.85 In this connection, Lavorato et al. reported the use of N-doped graphene as a visible light photocatalyst for H2 generation. In this study, the N-doped graphene was synthesized employing the pyrolysis of chitosan at higher temperatures. Chitosan is a nitrogen-containing biopolymer and can be used as a single source of carbon and nitrogen precursors. It was found that upon increasing the pyrolysis temperature, the nitrogen content in N-doped graphene decreased, with the highest weight percentage of nitrogen (16.2%) at 200 °C, and the lowest weight percentage of nitrogen (5.4%) at 900 °C. N-doped graphene synthesised at higher temperatures exhibited a higher photocatalytic activity with low N-loading. The authors observed that the photocatalytic activity largely depends on the crystallinity of the material and pyrolysis temperature.117
Transition metal chalcogenide (sulfide or selenide)-based photocatalysts are widely investigated due to their unique physical and chemical properties. In comparison to metal oxide nanostructures, metal sulfide semiconductors have a narrow bandgap, and thus hold more promise as photocatalysts for H2 generation by water splitting.118 While designing 2D material-based composite photocatalysts, manipulating the dimensionalities of individual counterparts is an effective way to achieve enhanced or superior photocatalysts. The dimensionalities of all components and interfacial contacts between them influence the efficiency of the photo-generated electron transfer process and separation and thus the performance of photocatalytic activity. 2D/2D heterostructure composites exhibit a better photocatalytic performance due to better coupling at hetero-interfaces, which facilitates effective charge transfer and separation, leading to enhanced photocatalytic activity compared with 0D/2D and 1D/2D heterostructures. Two-dimensional metal sulfides including MoS2 and WS2 combined with rGO sheets have been reported for the water-splitting reaction. A MoS2–rGO composite was synthesized via a solvothermal approach by taking (NH4)2MoS4, hydrazine, and mildly oxidized graphene in dimethylformamide solvent. Interestingly, the MoS2–rGO hybrid showed superior catalytic activity over other MoS2 catalysts. This remarkable performance is due to the availability of numerous catalytically active edge sites in the MoS2 and electronic coupling between the rGO and MoS2.119 To increase the photocatalytic efficiency, Xiang et al. fabricated a new composite material composed of TiO2 nanocrystals over the surface of a layered MoS2–rGO hybrid, and high photocatalytic HER performance was observed. The high photocatalytic HER performance is attributed to the synergic effect and effectively suppresses the charge recombination and increases the charge transfer as well as creates more active adsorption sites.120 Recently Xia et al. compared the photocatalytic performances of various carbon material composites for solar-driven water splitting for H2 evolution. Carbon materials with different dimensionalities including 0D carbon quantum dots (CQDs), 1D carbon nanotubes (CNTs), and 2D rGO are combined with ZnIn2S4 nanosheets under the same experimental conditions, and a systematic study was conducted. The morphology of the ZnIn2S4 nanosheets is determined by the type of carbon material. In case of low dimensional carbon materials (CQDs, CNT etc.) flower like 3D morphologies are obtained, while 2D r-GO maintain the sheet like structure of ZnIn2S4. The authors observed better performance for rGO based ZnIn2S4 nanosheets, and the order of the different composites is as follows: rGO > CQDs > CNTs. The better performance due to the large exposed surface of the rGO provides more nucleation sites, more active sites, a higher visible light absorption ability, and large space for charge carrier transfer.121
Typically, Z-scheme photocatalytic systems or two-step photoexcitation systems are composed of two semiconductors, one producing H2 and the other one producing O2 with the aid of an electron shuttle, and are considered to function very similarly to natural photosynthesis for efficient water splitting and also have greater potential to work under sunlight. As we know, the O2 and H2 photocatalysts can function independently to split water into H2 and O2. When these two photocatalysts are combined, the electrons generated in the O2 photocatalyst can be transferred to holes in the excited H2 photocatalyst, allowing the oxidation and reduction of water over the O2 and H2 photocatalysts, respectively. The first report was by Sayama et al. in 1997 on water-splitting for the generation of stoichiometric H2 and O2 through the Z-scheme approach.58 They employed the RuO2 loaded WO3 photocatalyst suspended in an aqueous solution consisting of Fe3+/Fe2+ redox systems for water splitting (Fig. 9). Under visible light, O2 gas is evolved and Fe3+ ions get reduced to Fe2+ ions over the WO3 powder catalyst. On the other hand, these Fe2+ ions are oxidized to Fe3+ ions with UV light irradiation and the evolution of H2 gas takes place, resulting in continuous decomposition of water into H2 and O2 gases (Fig. 9). These two reactions occur effectively, continuously, and simultaneously. Recent reports suggest that the electron transfer between the two photocatalysts will be the rate-determining step. Commonly, ionic redox couples such as IO3−/I− and Fe3+/Fe2+ are employed as electron mediators in Z-scheme photocatalytic systems to promote electron transfer between the two semiconductors. However, it is very difficult to maintain the ionic redox couples under stable conditions for a long time, leading to a reduction in photocatalytic activity. Hence, the development of Z-scheme systems without ionic redox couples is highly desirable.
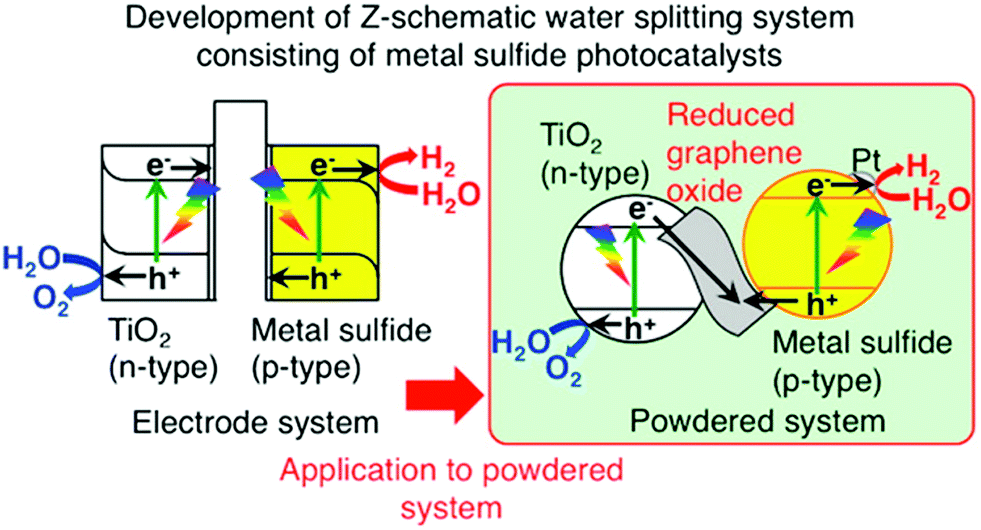 | ||
| Fig. 9 Schematic illustration of water splitting for O2 and H2 generation through Z-scheme photocatalytic systems.122 Copyright 2015, American Chemical Society. | ||
To address the above disadvantage, lately, a solid-state electron mediator has been introduced in Z-scheme photocatalytic systems and is more favorable for the recovery of photocatalysts and reclamation of clean water. Recently, Iwashina et al. reported a Z-scheme system for water splitting using rGO as a conductive solid-state material, promoting electron transfer between photocatalysts. They used a metal sulfide, which is a p-type material, as an H2 evolution photocatalyst, and an rGO–TiO2 composite, which is an n-type material, as an O2 evolution photocatalyst. The schematic of the Z-scheme photocatalytic system is shown in Fig. 9. In the system, the photogenerated electrons in the TiO2 will be transferred to the metal sulfide through the rGO layers for water splitting, and it is very much feasible to use the system in solar light. Moreover, without the rGO, the photocatalytic activity was low. The evolution of H2 and O2 was not in a stoichiometric ratio, which indicates that the rGO was necessary for the smooth transfer of electrons from the O2 photocatalyst to the H2 photocatalyst.122 Iwashi et al. constructed a Z-scheme system consisting of photo-reduced rGO as a solid-state electron mediator, Ru/SrTiO3:Rh and BiVO4, and it showed a better H2 evolution rate. In this system, the rGO acts as a stable electron mediator for 24 h, transferring the electron from the CB of BiVO4 to the impurity levels of Ru/SrTiO3:Rh. The electrons in Ru/SrTiO3:Rh can reduce water to H2 with the Rh cocatalyst. On the other hand, the hole present in BiVO4 simultaneously oxidizes the water to O2, achieving complete water splitting.123 A detailed list of the photocatalytic performances of various graphene based hybrids for hydrogen production through water splitting reaction is provided in Table 2.
| Nanocomposite | Method of synthesis | Light source | Sacrificial reagents | H2 production (μmol h−1) | Ref. |
|---|---|---|---|---|---|
| Graphene–TiO2 nanocomposite | Sol–gel (calcination) | 500 W Xe lamp | Na2S & Na2SO3 | 8.6 | 154 |
| Reduced TiO2–GO heterostructures | Laser ablation in liquid | 300 W Xe lamp | Methanol | 16 | 105 |
| Mesoporous anatase TiO2 nanospheres/graphene sheets | Self-assembly | 300 W Xe lamp | Methanol | 16 | 155 |
| rGO–TiO2 (P25) nanocomposite | Hydrothermal | 300 W Xe lamp | Methanol | 74 | 104 |
| Photoreduction | ∼30 | ||||
| Hydrazine | ∼15 | ||||
| Ag–TiO2/graphene composite | Microwave-hydrothermal | 300 W Xe light (λ > 420 nm) | Methanol | 225 | 46 |
| Graphene/Au–TiO2 composite | Microwave-hydrothermal | 420 nm-LEDs (3 W) | Methanol | 296 | 45 |
| Au–Pd/carbon/TiO2 nanocomposite | Hydrothermal | 300 W Xe light | Methanol | 6.42 | 109 |
| Au–Pd/rGO/TiO2 nanocomposite | 21.50 | ||||
| Spherical YInO3–graphene nanocomposite | Solvothermal | 300 W Xe light (λ > 420 nm) | Na2S & Na2SO3 | 400.4 | 114 |
| m-rGO–Au–NaTaO3 NP composite | Exo-template & solid-state | 150 W Hg lamp | Methanol | 13.78 | 161 |
| N-doped rGO–ZnGa2O4 nanospheres | Hydrothermal | 300 W Xe lamp (λ ≥ 250 nm) | Na2SO3 | 147.61 | 115 |
| ZnGa2O4 nanospheres | 38.94 | ||||
| rGO–MoS2/TiO2 | Hydrothermal | 300 W Xe arc light | Ethanol | 165.3 | 120 |
| rGO–TiO2 | 29.7 | ||||
| ZnS/rGO–MoS2 | Hydrothermal | 300 W Xe light | Na2S & Na2SO3 | 2258 | 167 |
| ZnS–MoS2 | 1185 | ||||
| Nanopetal CdIn2S4–graphene nanocomposite | Hydrothermal | 300 W Xe light (λ > 420 nm) | Na2S & Na2SO3 | 4495 | 168 |
| ZnIn2S4–graphene nanocomposite | Microwave-assisted hydrothermally | 350 W Xe light (λ > 420 nm) | Triethanolamine | 2640.8 | 121 |
| ZnIn2S4–CQD nanocomposite | 1767.7 | ||||
| ZnIn2S4–CNT nanocomposite | 1601.5 | ||||
| CuGaS2–rGO–TiO2 | Solid-state reaction & photoreduction | 350 W Xe light (λ > 420 nm) | K2SO4 | 56.2 | 122 |
| PrGO/BiVO4 and Ru/SrTiO3:Rh |
Solid-state reaction & photoreduction | 300 W Xe light (λ > 420 nm) | Methanol | 11 | 123 |
It is well known that GO exhibits an insulating behavior with a wide bandgap, and the electronic structure depends on the stoichiometric carbon to oxygen atomic ratio. The basal plane of GO was surrounded with several oxygen functional groups like epoxy and hydroxyl groups, and the edges of the sheets were decorated with carboxylic groups.33,37,123 These oxygenated functional groups provide a 2D network of sp2 and sp3 bonded atoms in the GO, which leads to the presence of a finite band gap depending on the isolated sp2 domains. By controlling the ratio of the sp2 and sp3 fractions by reduction chemistry, one can tune the bandgap of GO and transform the insulating GO to a semiconductor and graphene-like semi-metal. For example, Hsu et al. reported the photocatalytic reduction of CO2 with H2O vapor to methanol using GO as a low-cost and effective photocatalyst under simulated solar light irradiation. They synthesized a series of GO samples employing different chemical oxidants and found that GO-3 (Fig. 10a) obtained using the Hummers’ oxidation approach with excess KMnO4 and H3PO4 displayed a higher photocatalytic activity compared with those obtained by the other approaches. The main use of H3PO4 is to distribute the ester groups over the GO basal plane, which could further prevent the further oxidation of GO during the photocatalytic reduction of CO2. Their study concluded that by modulating the oxygenated functional groups, a methanol conversion rate of up to 0.172 μmol gcat−1 h−1 on GO can be achieved, which is six-fold higher than that of P25 TiO2. Besides, to confirm the methanol formation from the CO2 reduction, instead of the photo-dissociation of GO, isotope tracer analysis with 13CO2 was performed. The results showed that CH3OH was formed by the photocatalytic reduction of CO2 instead of any photo-dissociation of GO.17 Overall, the photocatalytic CO2 reduction mechanism is a consecutive combination of H2O oxidation and CO2 reduction (Fig. 10b).
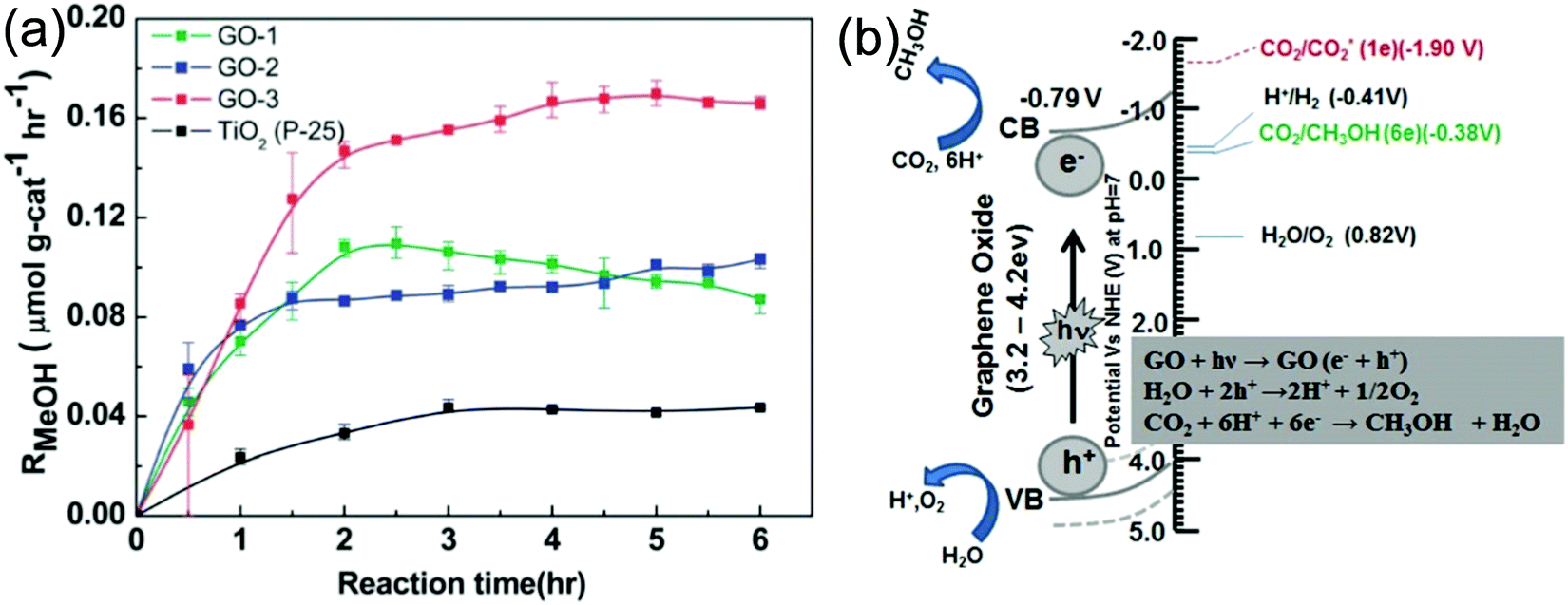 | ||
| Fig. 10 (a) Photocatalytic methanol formation on various GO samples and TiO2 using simulated solar light. (b) Schematic illustration of the photocatalytic CO2 reduction mechanism over the GO.17 Copyright 2013, Royal Society of Chemistry. | ||
The photocatalytic mechanism of the GO was demonstrated for a better understanding of the CO2 photo-reduction process. The isolated sp2 clusters with oxygen functional groups including C–OH and C–O–C in the GO lead to the localization of electron–hole pairs on its basal plane, which further migrate to the surface of the GO and serve as oxidizing and reducing sites, respectively, to react with the adsorbed reactants. The reduction potential of e− in the GO conduction band was at −0.79 V vs. NHE, which is lower than the potential of CO2/CH3OH (−0.38 V vs. NHE), and acts as donors. On the other hand, the oxidation potential of h+ in the GO valence band was around 4 V vs. NHE, which is higher than the potential of H2O/O2, H+ (−0.82 V vs. NHE), and acts as an acceptor. The whole mechanism is shown Fig. 10. The conduction band position (−0.79 V vs. NHE) of GO supports photocatalytic CO2 reduction, which is more feasible for the multi-electron reduction process than the one-electron reduction process.3,17,18,33 Recently, Kuang et al. reported the photocatalytic CO2 reduction ability of light irradiated GO. In their study, the simulated sunlight and UV irradiated GO displayed an enhanced photocatalytic CO2 reduction compared with pristine GO; in particular, the sunlight irradiated GO exhibited a nearly threefold higher efficiency than pristine GO. The irradiation mainly plays the following roles: irradiating GO (1) eliminates the CO released during photolysis from the photocatalytic reaction and (2) increases the defect density and π-conjugated network, which results in improvement in the yield of photo-electrons and prolongs the lifetime of photo-generated charge carriers.60 Addition of hole capturing agents like metals over GO can improve its photocatalytic efficiency. Shown et al. synthesized a series of Cu NP decorated GO materials with different weight ratios of Cu employing a rapid one-pot microwave method and demonstrated their photocatalytic CO2 reduction under visible light irradiation. The Cu/GO composite with 10 wt% Cu displayed the maximum solar fuel formation rate of 6.84 μmol gcat−1 h−1, and the achieved CO2 photocatalytic reduction rate was 60 and 240 times higher than those of pristine GO and commercial P25 TiO2, respectively, under visible light irradiation. The incorporation of Cu effectively tunes the work function of GO due to the spontaneous transfer of photogenerated electrons captured by Cu and then effective transfer of electrons from the Cu d orbital to C–O π* orbital, which results in effective separation of photoinduced charge carriers, consequently leading to the enhanced photocatalytic CO2 reduction activity of the GO–Cu NP hybrid compared with pristine GO.90 Noble metals under light irradiation generate hot plasmonic electrons that can induce photochemical reactions in the adsorbed molecules on the metal surface. The intrinsic ultrafast hot electron relaxation in the metal (<160 fs) can compete with the charge transfer from the metal NPs. Therefore, to maximize the charge separation of hot electrons from the NP surfaces, it must be coupled with an efficient electron collector and high electron mobility materials. In this regard, the coupling of noble metals with graphene or its derivatives can be an agile strategy for efficient electron extraction and mobility. Recently, Kumar et al. reported rGO–Au NPs as excellent photocatalysts for solar fuel applications such as photoreduction of CO2 to HCOOH. The rGO–Au NPs displayed an improved conversion efficiency compared with the Pt–Au NPs due to the enhanced utilization of plasmonic hot electrons of Au NPs under light irradiation. Moreover, the rGO can be an alternative to expensive and scarce Pt. rGO–Au NPs showed a 6 fold higher activity than pristine graphene. The enhanced activity is due to the efficient transfer of hot electrons to the rGO sheets which act as reaction sites. Initially, the main products detected consisted of CH3OH and HCOOH. As the reaction progressed, the HCOOH concentration increased, which was presumed to be due to the consumption of CH3OH as a hole scavenger when the reaction continued. Moreover, the effect of the rGO concentration and the photothermal effect were studied. As the concentration of rGO increased, the productivity increased and then decreased at a point where the rGO concentration inhibited the passage of light to Au NPs and also due to the agglomeration. On the other hand, increasing the temperature during light irradiation speeded up the photoconversion of CO2 due to the accelerated plasmonic electron injection into the rGO sheets.124
The catalytic properties of GO mainly originate from its oxygenated functional groups and unpaired electrons in GO scaffolds and also its high surface area. Recently, Kim et al. reported that GO is a metastable material whose structure and chemical functionalities evolve at room temperature. Under quasi-equilibrium conditions, GO shows a structure deprived of epoxy groups; however, it is rich in hydroxyl groups, with a final reduced C/O ratio of 0.38. The presence of C–H species in the GO favors the reduction of epoxide groups and the formation and release of water molecules.125 Furthermore, an increase in temperature results in a gradual decrease in oxygen functionalities of GO. Consequently, the easy GO aging and reduction during the catalytic reactions at mild to high temperatures raised the issues of recyclability and degrading catalytic activity of GO. To overcome these problems and further to increase the yield by anchoring visible light-absorbing materials onto GO nanosheets, several methodologies have been developed for immobilization of various nanomaterials on GO nanosheets. Kumar et al. synthesized a novel GO immobilized ruthenium trinuclear triazine complex and demonstrated its photocatalytic activity for CO2 reduction to methanol under visible light illumination. Owing to its wide band gap, GO alone could not undergo excitation in visible light. The ruthenium complex, due to its virtuous visible light absorbance and its ability to be excited from the S0 to S1 state, facilitates the transport of photo-excited electrons from the ruthenium complex to the conduction band of GO, which is subsequently used for the reduction of adsorbed CO2 to methanol over the GO.126 Furthermore, the same group fabricated a cobalt phthalocyanine and ruthenium complex bearing 2-thiophenylbenzimidazole ligand immobilized GO for the photocatalytic reduction of CO2 to methanol, and the formation rates were 78.8 μmol g−1 h−1 and 85.4 μmol g−1 after 24 h, respectively.127,128
Hexamolybdenum (Mo6) cluster-based compounds specifically Cs2Mo6Bri8Bra6 and (TBA)2Mo6Bri8Bra6 (TBA = tetrabutylammonium) were immobilized onto GO nanosheets by taking advantage of the high lability of the apical bromide ions with the oxygen-functionalities of GO. During immobilization, the apical bromide ions were replaced by the oxygen functionalities of GO. These composites, including GO–Cs2Mo6Bri8Brax and GO–(TBA)2Mo6Bri8Brax materials, were used for methanol formation from the photocatalytic reduction of CO2 with formation rates of 68.5 and 53.5 μmol g−1 h−1, respectively (Fig. 11a), after 24 h irradiation using 20 W LED spotlight as a visible-light source. Also, both these composites could be easily recovered and provided similar yields of methanol under identical conditions (Fig. 11b), and also the studies suggested that these composites were highly stable and the reactions were truly heterogeneous. The probable photocatalytic reduction of CO2 over these composites on their basis of bandgap is displayed in Fig. 11c. The Mo6 clusters over the GO, due to their low bandgap values, in these composites exhibited good visible light absorbance. Under visible light, the Mo6 clusters transformed into excited state Mo6* by HOMO to LUMO transition followed by effective electron transfer from the excited state Mo6* to the conduction band of GO. Further, these electrons were utilized for the reduction of adsorbed CO2 to methanol. As the position of the GO conduction band is more negative than the reduction potential of CO2/CH3OH (−0.38 V vs. NHE, pH 7), the positively charged Mo6+ clusters get the electrons from the valence band of GO and transform to their original (Mo6) state and the water-splitting reaction takes place at the valence band of GO and provides necessary electrons and protons for the reduction of CO2. Finally, the better charge mobility and high surface area of GO enhance the photocatalytic performance.129
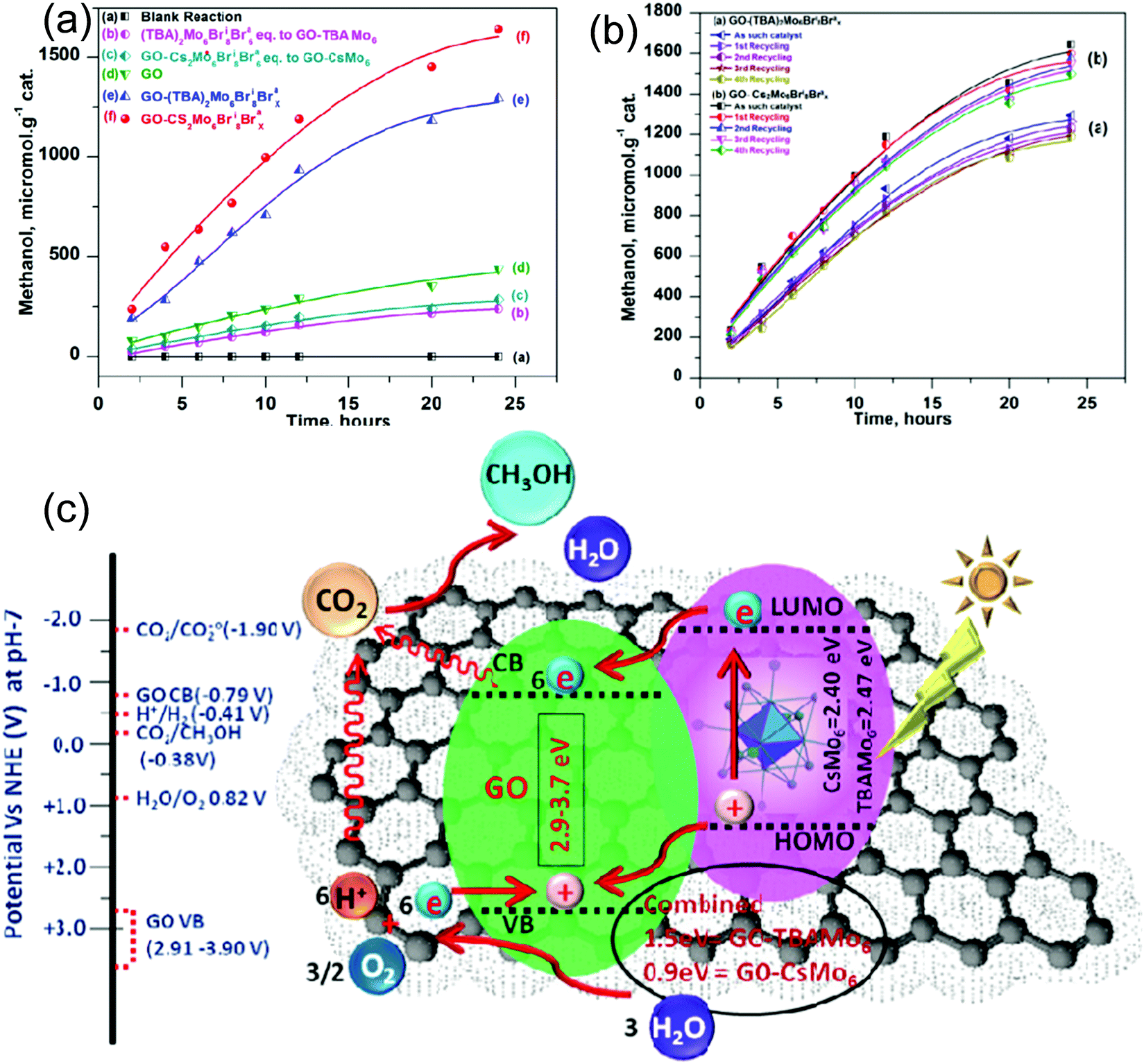 | ||
| Fig. 11 (a) Methanol formation rates, (b) recycling experiments and (c) plausible mechanism of CO2 photoreduction over a GO–hexamolybdenum composite.129 Copyright 2015, Elsevier. | ||
Semiconductors are photocatalysts of the utmost significance due to their photostability, nontoxicity, chemical stability, and low cost. Despite these advantages, the photocatalytic activity of these materials is inadequate due to their wide bandgap energy and higher recombination rates.4,13,62 Hence, these semiconductors can be coupled with graphene for obtaining graphene-based semiconductor composites, which have been verified to display improved photocatalytic performance for the reduction of CO2 to useful products compared to the corresponding bare semiconductors. Among the semiconductors, TiO2 is regarded as the most feasible in terms of its inexpensiveness, non-toxicity, abundance, and high redox potentials. To overcome the competetive recombinations and lack of visible absorption due to wide band gap, specific cations and anions can be incorporated into TiO2. However, these create secondary impurities and oxygen vacancies, which could ultimately reduce the efficiency of the TiO2 photocatalytic performance. Hence it is a necessity to develop an effective way to produce visible light-responsive and stable TiO2 without any oxygen vacancies and secondary impurities. Tan et al. developed a dopant-free approach to preparing a novel-oxygen-rich TiO2 (O2–TiO2) photocatalyst with significantly enhanced photocatalytic performance. By the introduction of oxygen excess defects into the lattice of TiO2, the surface disorderliness caused the upshift of the VB edge, which resulted in a reduction of bandgap energy from 3.2 eV to 2.95 eV. Hence, the O2–TiO2 photocatalyst could absorb visible light and electron–hole pairs were generated on its surface. Even though the O2–TiO2 photocatalyst exhibited an enhanced photocatalytic performance for the reduction of CO2 to CH4, it progressively degraded over time. To overcome this problem, GO could be incorporated into O2–TiO2via a wet chemical impregnation technique. The GO–O2–TiO2 nanocomposite with an optimized loading of GO (5 wt% GO) showed an enhanced photocatalytic reduction activity with a yield of 3.450 μmol gcat−1 after 8 h visible light illumination, which is 14 fold higher than that of commercial P25 TiO2. The enhanced activity is due to the oxygen-rich vacancies and GO as a conductor. The GO served as an electron sink and effective charge transport bridge due to its high conductivity and extended conjugated network. The Schottky barrier formed at the interface of both the components could help in the separation of photo-induced charge carriers and decreased the charge recombination rates, which in turn improved the photostability of the nanocomposite significantly.130 Razzaq et al. fabricated a novel nanocomposite comprised of TiO2 nanotube arrays with a surface coating of rGO platelets that contained within them embedded TiO2 nanoparticles (rGO–TNTNP) for CO2 photoreduction with H2O vapor under simulated solar light illumination. The composite was synthesized employing a two-step process: initially, simultaneous electrophoretic deposition and reduction of GO over Ti foil, followed by electrochemical anodization of the rGO–Ti foil. The rGO–TNTNP composite exhibited a CH4 evolution rate of 5.67 ppm cm−2 h−1, which is 4.4 fold higher than that of bare TiO2 nanotube arrays (1.28 ppm cm−2 h−1).131
Adsorption of CO2 over the nanocomposite plays a crucial role in photocatalytic CO2 reduction performance. As we know, graphene is a promising adsorbent for CO2 capture and storage due to its vast effective surface area and planar structure associated with the 2D lattice structure formed by sp2 carbon atoms. The reports showed that the uptake of CO2 over carbon materials could be significantly improved by selective functionalization with nitrogen dopants. Kemp et al. studied CO2 adsorption over pristine graphene and N-doped graphene materials. The N dopants act as basic sites for the anchoring of CO2. Furthermore, they can significantly alter the electronic properties of graphene and change the local surface reactivity, leading to improved adsorption and catalytic performances.132 Therefore, the functionalization of graphene with the N-dopants can be an effective approach to achieve enhanced CO2 adsorption and CO2 reactivity over graphene-based materials. Recently, Lin et al. synthesized TiO2/N-doped graphene (TiO2/N-rGO) nanocomposites with varying concentrations and bonding configurations of nitrogen employing a one-step urea-assisted hydrothermal approach and studied the TiO2/N-rGO composite as a bifunctional adsorbent and photocatalyst for the gas phase photoreduction of CO2 with H2O vapor. The effect of the N-dopant (including doping quantity and conformation) on the catalytic performance was examined, and TiO2/N-rGO-300 (with a 300:1 mass ratio of urea/GO in the precursor solution) exhibited a higher CO production yield of 356.5 μmol g−1, which is significantly 4.4 and 2.2 fold higher CO production than those of pure TiO2 and TiO2/rGO, respectively (Fig. 12a and b). Remarkably, the presence of N-dopants contributes to the enhancement of the photocatalytic activity and stability of the TiO2/N-rGO composites. The spin density and charge distribution of carbon atoms can be influenced by the neighboring N-dopants, resulting in the formation of activation sites over the graphene nanosheets. Various bonding configurations of N-dopants such as pyridinic-N and pyrrolic-N and quaternary-N can produce diverse active sites. The pyridinic-N and pyrrolic-N have Lewis basicity, and work as active sites for CO2 capture and activation (Fig. 12c). The quaternary-N enhances the electron mobility for the effective transfer of photogenerated electrons from the CB of a semiconductor.133
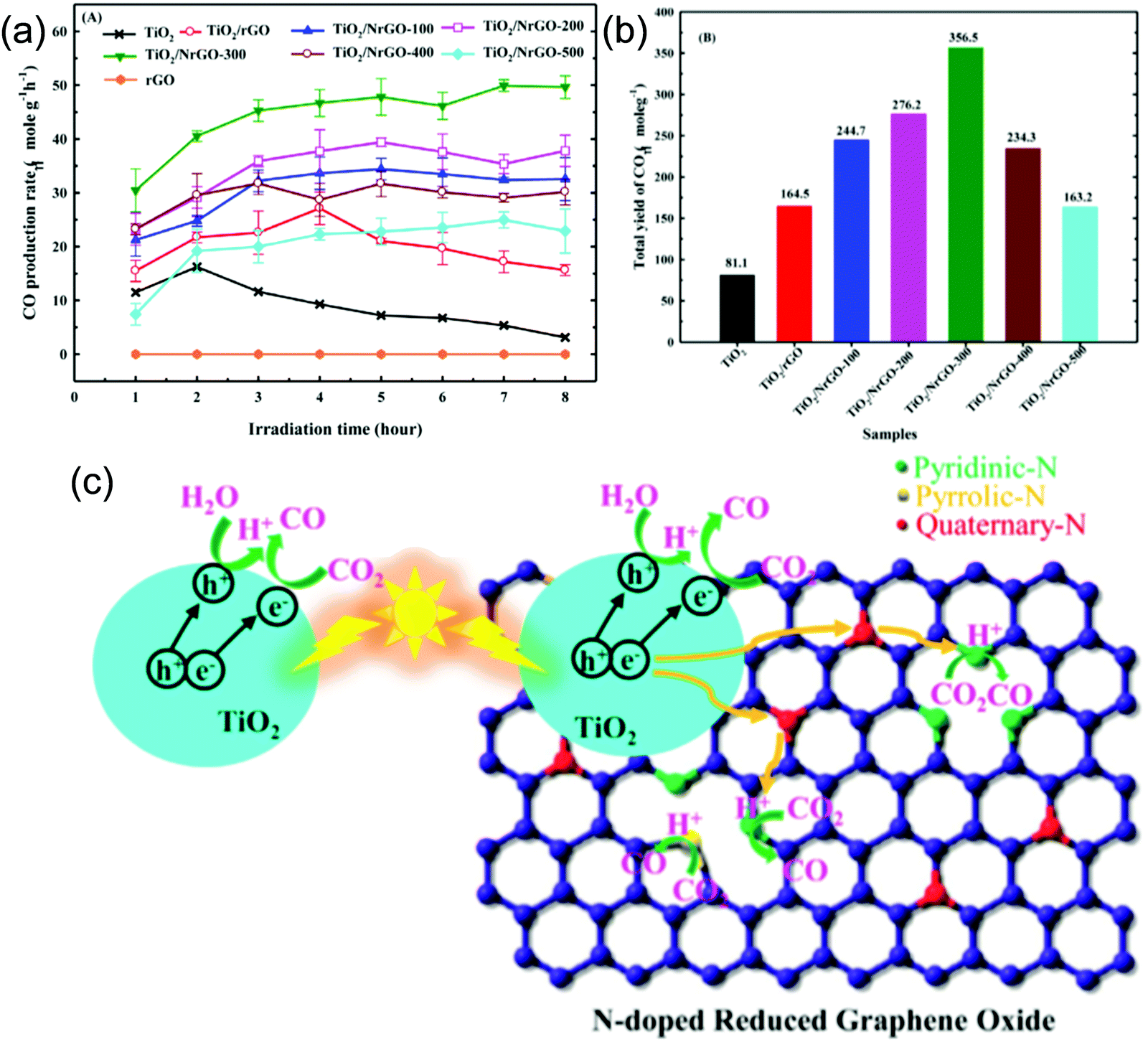 | ||
| Fig. 12 (a) Photoreduction rate of CO vs. time over an N-doped rGO–TiO2 composite, (b) total yield of CO under visible light irradiation and (c) possible photoreduction mechanism of CO with H2O.133 Copyright 2017, Elsevier. | ||
An et al. reported the fabrication of an rGO–Cu2O composite employing a facile one-step microwave-assisted chemical method and demonstrated its application as a photocatalyst for the reduction of CO2. The rGO coating over the Cu2O significantly boosts the photocatalytic activity. The good electron conductivity and large surface area of the rGO inhibit electron–hole recombination, resulting in a large number of active sites in the rGO–Cu2O composite. The rGO–Cu2O composite exhibits better photocatalytic activity than bare Cu2O (Fig. 13a and b). Furthermore, the photocorrosion problem for the Cu2O was prohibited by the loading of rGO. In detail, the bare Cu2O had a high leaching Cu concentration of 2670 ppm after 3 h light irradiation, and the Cu concentration was measured by inductively coupled plasma optical emission spectroscopy (Fig. 13a). The leaching of Cu ions was significantly reduced to 96 ppm by the loading of rGO layers. The presence of rGO in the composite can avoid the direct contact of Cu2O with water, which can prevent the attack of active species on Cu2O (Fig. 13c and d). This study suggests that the loading of rGO layers over the photocatalyst not only increases the performance but also enhances the photostability.134 Recently, Gusain and coworkers prepared rGO–CuO and rGO–Cu2O nanocomposites employing covalent grafting of CuO and Cu2O nanorods over the rGO skeleton and explored their potential application for the reduction of CO2 to methanol under visible light irradiation. The effects of the covalent grafting of CuO nanorods on the rGO skeleton and the oxidation state of copper in the developed nanocomposite on the photocatalytic activity was examined. Pristine CuO nanorods exhibited a very low photocatalytic activity due to the fast recombination of photo-induced charge carriers and yielded 175 μmol g−1 methanol although the rGO–Cu2O and rGO–CuO nanocomposites revealed significantly higher photocatalytic activities, and the yields were five (862 μmol g−1) and seven (1228 μmol g−1) times as much methanol, respectively. The superior photocatalytic activity of the rGO–CuO nanocomposite was attributed to the slow recombination of photo-induced charge carriers and efficient charge transfer through the rGO skeleton. Furthermore, rGO–CuO (Cu2+) exhibited superior photocatalytic activity over rGO–Cu2O (Cu1+) under identical conditions.135
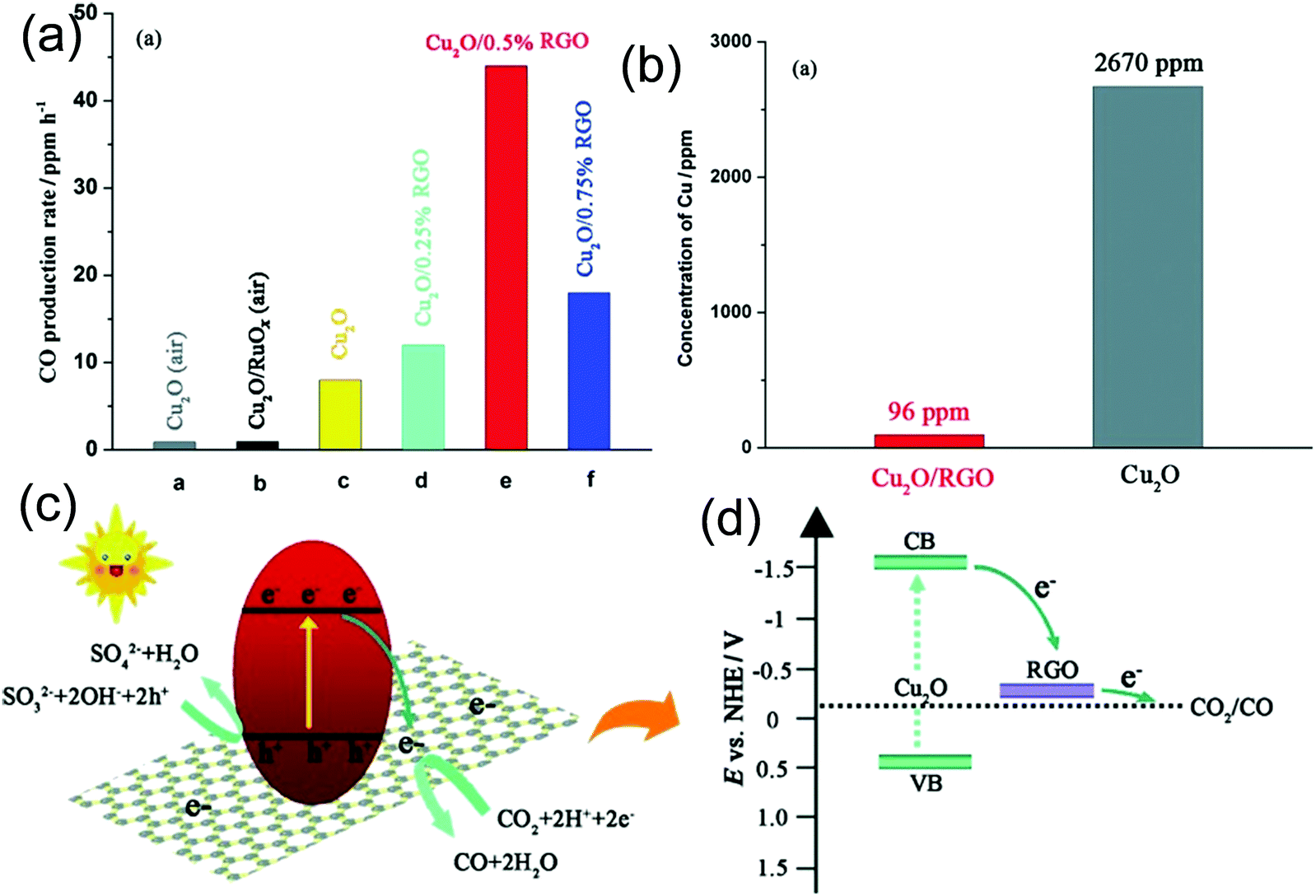 | ||
| Fig. 13 (a) CO yields over various photocatalysts, (b) plot of leaching of Cu caused by photoreduction, and (c) and (d) schematic illustrations of the CO2 reduction mechanism and charge transfer in the rGO–Cu2O composite.134 Copyright 2014, Wiley. | ||
Graphene not only facilitates electron transport but also elevates the position of the semiconductor's conduction band. For example, WO3 is a visible light active semiconductor due to its narrow bandgap (∼2.7 eV). However, the low position of its conduction band (>−1.0 eV vs. NHE, pH = 7) has limited its application for the reduction of CO2 to useful products. Wang et al. prepared a graphene–WO3 nanobelt composite employing a facile in situ hydrothermal approach and demonstrated its application in photocatalytic reduction of CO2 to methanol. By using this system, 0.89 μmol g−1 methanol was obtained after 8 h visible light illumination. From the XPS VB spectra, it was observed that WO3 has a deeper VB maximum of 0.37 eV than graphene–WO3, signifying a stronger oxidation power of photoexcited VB holes and also an elevated CB minimum of the graphene–WO3, which is higher than the redox potential of CH4/CO2 at −0.24 V. Therefore, the photoreduction of CO2 by the photoexcited electrons from graphene–WO3 is thermodynamically feasible.136 In another study, Yu et al. fabricated an rGO–CdS nanorod composite employing a microwave-assisted hydrothermal approach for the photocatalytic reduction of CO2 to CH4.137 The combination of rGO with the CdS nanorods enhanced the photocatalytic reduction activity of CO2 to CH4 up to 10 fold compared with bare CdS NRs. Therefore, the rGO was regarded as an effective cocatalyst for the photocatalytic reduction of CO2 to CH4. The enhancement is attributed to the following reasons: the rGO not only acted as an electron acceptor and transporter and efficiently separated the generated photo-induced charge carriers but also enhanced the adsorption and activation of CO2 molecules over the rGO–CdS NR composite.
Recently, Ti- and Nb-based metal oxides have been pursued extensively as they exhibit nontoxicity, chemical inertness, and high stability under light irradiation. Remarkably, lamellar niobium containing solid acids like KNb3O8 and HNb3O8 show fascinating performance because their conduction band top and valence band bottom are located at necessary potential levels. Mostly, exfoliated single crystal nanosheets display much higher catalytic activities than their bulk counterparts because of their enormously high 2D anisotropy. This can offer an enhanced specific surface area and improved Brønsted and Lewis acidity as well as a reduced transfer path for photogenerated charge carriers. Liu et al. reported the fabrication of a layered HNb3O8–graphene nanohybrid employing the exfoliation–restacking approach. Plentiful interfaces and pores were generated by the alternative stacking of HNb3O8 nanosheets with limited size and graphene nanosheets with buckling and folding features. The photocatalytic reduction of CO2 to renewable energy by the optimized layered HNb3O8–graphene nanohybrid showed 8 fold improvement in CO production compared to commercial TiO2 (P25), 6.1 fold compared to pristine layered HNb3O8 nanosheets, and 8.6 fold compared to bulk HNb3O8 powders (Fig. 14). With increasing the loading of graphene nanosheets the effective absorption of HNb3O8 decreases. Furthermore, the excess graphene nanosheets can block the active sites and favor the recombination centers. The enhanced photocatalytic activity can be attributed to the following reasons: (i) the ultra-thin nature of HNb3O8 nanosheets permits the charge carriers to transport quickly onto the surface active sites to participate in the photoreduction reaction; (ii) the improved specific surface area of the hybrids can offer more active adsorption sites and readily accessible channels for reactants to the active sites; (iii) the intercalated graphene nanosheets in between the HNb3O8 nanosheets provide abundant active sites serving as energetically favorable sites for the fast migration of photogenerated electrons from the HNb3O8 nanosheets to graphene sheets; (iv) introduction of graphene narrows the bandgap of the HNb3O8 nanosheets, resulting in a noticeable redshift of the absorption band in the hybrid system compared with bare HNb3O8 nanosheets; (v) the porous feature and broad pore width permit the fast diffusion of reactant gases in the hybrid system; and (vi) the protonic acidity of exfoliated HNb3O8 nanosheets permits them to be easily hydrated and also results in enhanced strong Brønsted acidity, which favours the separation of electron–hole pairs at the interlayer sites (Fig. 14).138
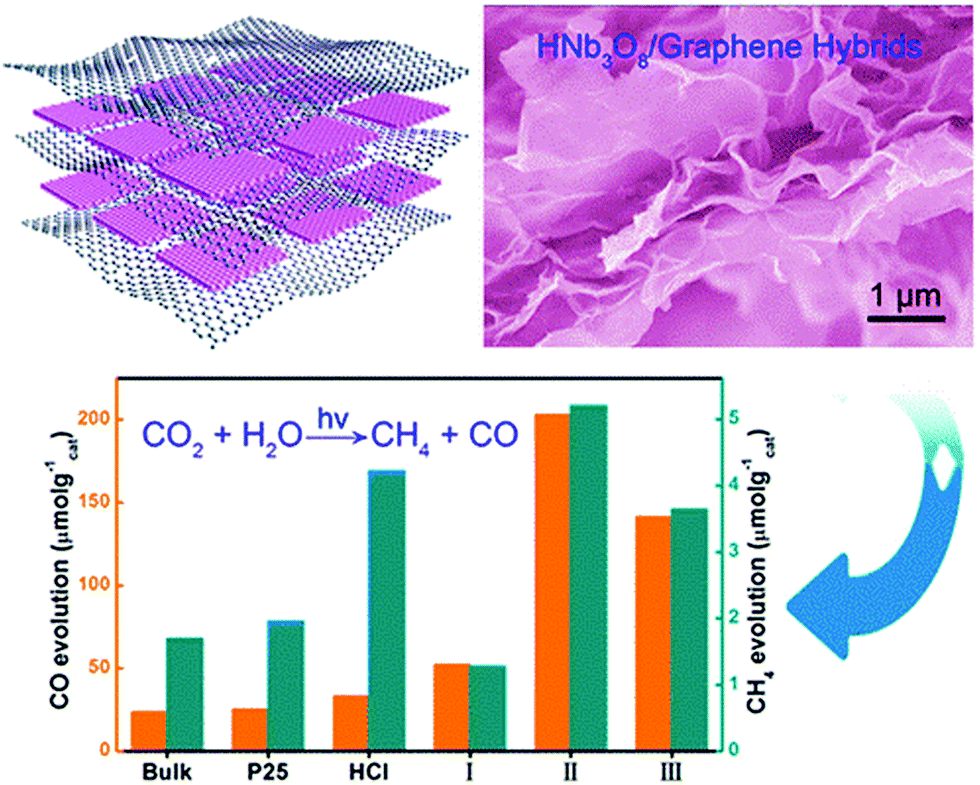 | ||
| Fig. 14 Schematic illustration of the HNb3O8–graphene nanohybrid along with FESEM images and CO & CH4 yields under light irradiation.138 Copyright 2016, American Chemical Society. | ||
The introduction of abundant pores in the layered nanostructures leads to the generation of highly concentrated oxygen vacancies, enhancing the bonding strength of OH* intermediates formed during the water oxidation. Furthermore, creating metallic vacancies in addition to oxygen vacancies may enhance photocatalytic performances. In this connection, recently, Wang et al. reported the exfoliation of a Ti containing layered double hydroxide followed by coupling with GO by an electrostatic assembly approach and demonstrated it as a photocatalyst for the visible-light-driven reduction of CO2. The prepared catalyst with an optimized GO content displayed enhanced photocatalytic performances with CH4 and CO yields of 3.8 and 4.6 μmol h−1 gcat−1, respectively. The generation of abundant Ti3+-V0 sites by exfoliation of TiMgAl layered hydroxides could expand the light absorption range of the composites into the visible region. Owing to the favorable Fermi level equilibrium, the photoexcited electrons from the conduction band of the TiMgAl migrate towards the carbon defects in GO, resulting in the effective separation followed by prolonged lifetimes. Moreover, the obtained electron-rich carbon defects enable the adsorption of CO2 molecules followed by activation of the negative and metastable superoxide radicals (CO2−˙). Simultaneously, the photoinduced holes in the VB of TiMgAl could react with H2O and produce O2, H˙, and OH˙, which further reacts with the CO2−˙, and eventually produced CH4.44
Decorating semiconductor particles with noble metal nanostructures such as Pd, Pt, Au, and Ag has been demonstrated to enhance the photocatalytic conversion of CO2 with H2O into useful solar products. In general, the noble metal nanostructures play the following vital roles in the improved efficiency: (1) they hinder electron–hole recombination by trapping photogenerated conduction band electrons and prolong their lifetime; (2) they enhance photoreduction efficiency owing to the Fermi level equilibrium; (3) they provide thermal catalytic sites for the adsorbed species and intermediates; (4) they exhibit surface plasmon resonance, which enables them to show intense absorption in the visible region of the solar spectrum and (5) they modify the surface properties of the photocatalyst.139,140 Recently, Zhu et al. fabricated a ternary hybrid composed of rGO–CdS decorated with Ag nanoparticles by a combination of the solvothermal process and subsequent photochemical reduction approaches. The composite exhibited enhanced photocatalytic activity for the reduction of CO2 to CO, and the optimum activity was achieved over 1 wt% Ag–3 wt% rGO–CdS. The enhanced performance is due to the multifunctional effects of rGO and Ag: the rGO and Ag can act as electron acceptors, thus (i) enhancing the separation of photogenerated charge carriers and (ii) improving the adsorption and activation sites for CO2, which results in an enhancement in the performance of the photocatalytic photoreduction of CO2 to CO. The authors also concluded that the excessive loading of rGO onto CdS brings about a decrease in photocatalytic activity; this is due to the shielding effect of the rGO.141 Hou et al. developed a novel three-dimensional bimetallic–graphene–semiconductor (Au–Cu/graphene/Cu2O) composite system for the solar-driven conversion of CO2 to methanol, in which the Cu2O nanoarrays on Cu mesh substrates were encapsulated within ultrathin rGO sheets followed by decoration with an optimized combination of Au–Cu nanoalloys. Under visible light irradiation for 6 h, the yield over the graphene–Cu2O arrays is 46.3 ppm cm−2 h−1, which is 3 fold higher than that of the bare Cu2O array (25.4 ppm cm−2 h−1). In particular, the Au–Cu/graphene–Cu2O composite arrays exhibit a higher methanol yield of 120 ppm cm−2 h−1, which is 7.8 times higher than that of bare Cu2O. Without the protection of graphene sheets, the composite shows a lower methanol production, which indicates the pivotal role of graphene in the 3D architecture. The graphene/Cu2O composite decorated with Au, Cu, and Au + Cu prepared by step by step deposition of metal over the supportive matrix exhibits a much lower photocatalytic performance. The significantly enhanced photocatalytic activity of the Au–Cu/graphene–Cu2O composite arrays is ascribed to the following: (1) the introduction of graphene as an electron acceptor and conductive support with a lower energy level than the conduction band of Cu2O leads to the migration of photogenerated electrons of the Cu2O conduction band to the graphene layer and (2) the establishment of a Schottky junction in the Au–Cu/graphene–Cu2O composite arrays results in further migration of electrons from graphene to the Au–Cu nanoalloy. Additionally, the electromagnetic field induced by the surface plasmon resonance effect of the Au–Cu nanoalloys promotes the migration of electrons through the following way: Cu2O nanoarrays → graphene → Au–Cu nanoalloy. Accordingly, the electron–hole pairs are readily separated and the lifetimes of photogenerated charge carriers have been efficiently prolonged in the Au–Cu/graphene–Cu2O composite arrays, which become readily available for the multi-electron CO2 reduction process.142
Tan et al. synthesized a series of ternary composites – noble metal (Pd, Pt, Au, and Ag) decorated rGO–TiO2 composites – employing a facile and environmentally benign two-step approach including solvothermal and polyol methods, without using any toxic and harsh reducing agents like hydrazine and sodium borohydride. In the polyol process, ethylene glycol was used as both solvent and a reducing agent for the additive metal ions to form metal islands over the TiO2 and rGO. The three-component composites displayed an enhanced photocatalytic activity towards reduction of CO2 to CH4 under visible light irradiation. A comparison of all the noble metal decorated rGO–TiO2 composites showed that the Pt–rGO–TiO2 composite exhibited a higher photocatalytic activity of 1.696 μmol g−1 in comparison to the other noble metal decorated rGO–TiO2 systems, i.e. Pd–rGO–TiO2 (1.193 μmol g−1), Ag–rGO–TiO2 (0.998 μmol g−1), Au–rGO–TiO2 (0.756 μmol g−1) and rGO–TiO2 (0.651 μmol g−1). The Pt–rGO–TiO2 composite displayed 2.6 and 13.2 fold higher activity than bare TiO2 (0.126 μmol g−1) and commercially available P25 (0.129 μmol g−1), respectively, after 6 h of visible light irradiation (Fig. 15a). The experimental results showed that the Pt–rGO–TiO2 composite displayed a higher photocatalytic activity than the other noble metal–rGO–TiO2 systems. The activity is mainly because of the photonic efficiency of the noble metal decorated rGO–TiO2 composite and is strongly dependent on the electron affinity and work function of noble metal to form a favorable contact with the TiO2. Pt has a higher work function (−5.62 eV) than the other noble metals Au (−5.1 eV), Ag (−4.7 eV), and Pd (−5.2 eV); therefore the photogenerated electrons can transfer most efficiently from TiO2 to the loaded Pt and also the reversible process is extremely difficult, and hence, it can acts as an efficient electron sink (Fig. 15b). Hence, the work function of the metals played a dominant role in the photocatalytic activity, rather than the surface plasmon effect. Moreover, the adsorption, transportation, and activation of different reactants on the metal surface are quite diverse, and the effect of each noble metal on the photocatalytic reaction may vary with different reactions. The plausible photocatalytic mechanism for the reduction of CO2 is displayed in Fig. 15c. Upon illumination with light, electron–hole pairs are formed. Since Pt nanostructures trap the electrons very effectively, the photogenerated electron would migrate to Pt nanostructures, where the photoreduction of CO2 takes place. In the case of Pt deposited over the rGO layers, the photogenerated electrons migrate from the TiO2 to rGO layers due to the favorable equilibrium Fermi levels followed by shuttles across the 2D sheets and are finally trapped by the Pt active sites. Finally, the improved photocatalytic activity on the Pt–rGO–TiO2 composite could be attributed to the synergetic effect of Pt, rGO, and TiO2, and also rGO provided a large surface area and greatly increased the conductivity of the electrons.Also, rGO served as an excellent supporting material for achieving a high aggregation free dispersion of Pt and increases catalytic efficeincy.143
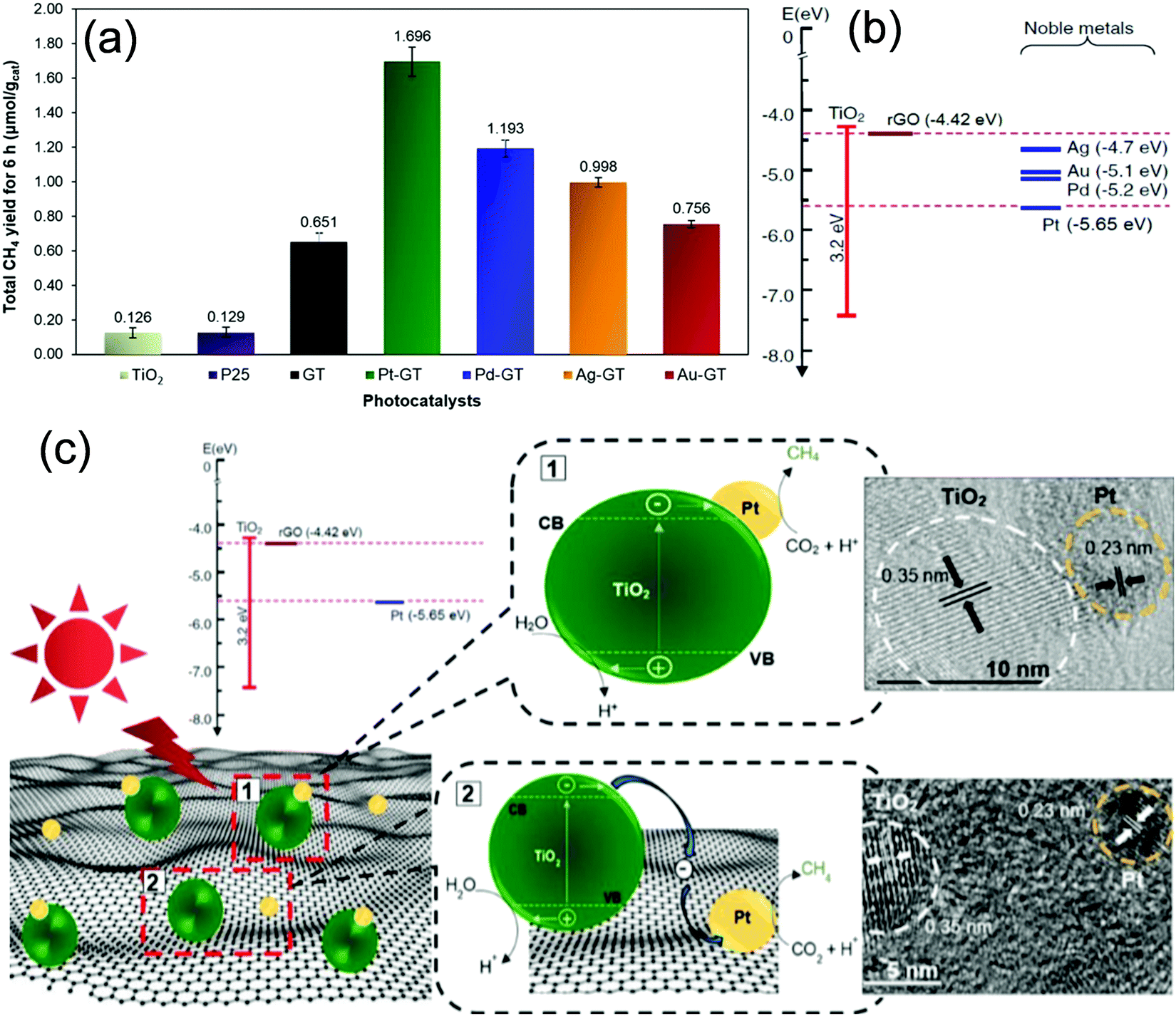 | ||
| Fig. 15 (a) The total yields of CH4 over various noble metal decorated TiO2/rGO composites; (b) energy band position and (c) plausible photocatalytic mechanism for the reduction of CO2.143 Copyright 2015, Elsevier. | ||
Perovskite nanocrystals have emerged as one of the most efficient light-harvesting materials and the major obstacles to these materials are their sensitivity towards polar environments and the strong recombination possibility of photogenerated charge carriers. Hence, creating heterojunctions with highly conducting materials like graphene and graphene derivatives can reduce the possibility of carrier recombination, increase stability, and enhance the efficiency of photocatalytic activity of these materials. By tuning the size and composition of perovskite nanomaterials, the yield and selectivity of the products from CO2 reduction can be greatly enhanced. Xu et al. reported the use of CsPbBr3/GO as a novel photocatalyst to convert CO2 into useful products in non-aqueous media. A facile room temperature anti-solvent precipitation approach was leveraged to synthesize CsPbBr3 and CsPbBr3/GO composite materials (Fig. 16a and b) and utilized these composite materials for the photochemical conversion of CO2. Compared to the individual CsPbBr3 QDs, the rate of electron consumption improved from 23.7 to 29.8 μmol g−1 h−1 after the introduction of GO with a selectivity of 99.3% under AM 1.5G simulated illumination (Fig. 16c and d). To clarify the role of GO in the enhanced photoreduction of CO2, photoluminescence (PL) and electrochemical studies were conducted. The PL intensity of the CsPbBr3/GO composite decreased to a lower level than that of pristine CsPbBr3 because the introduction of GO provided additional energy-transfer pathways in addition to the intrinsic radiative channel for excited-state electron transfer (Fig. 16e). The rapid transfer of photogenerated electrons in the CsPbBr3 to the GO is because the Fermi level of GO is more positive than the conduction band edge of CsPbBr3 and also due to the conductive nature of GO. The photocurrent response of the CsPbBr3/GO composite was 54 μA cm−2, corresponding to a 28.57% enhancement compared to CsPbBr3 (42 μA cm−2) (Fig. 16f).144
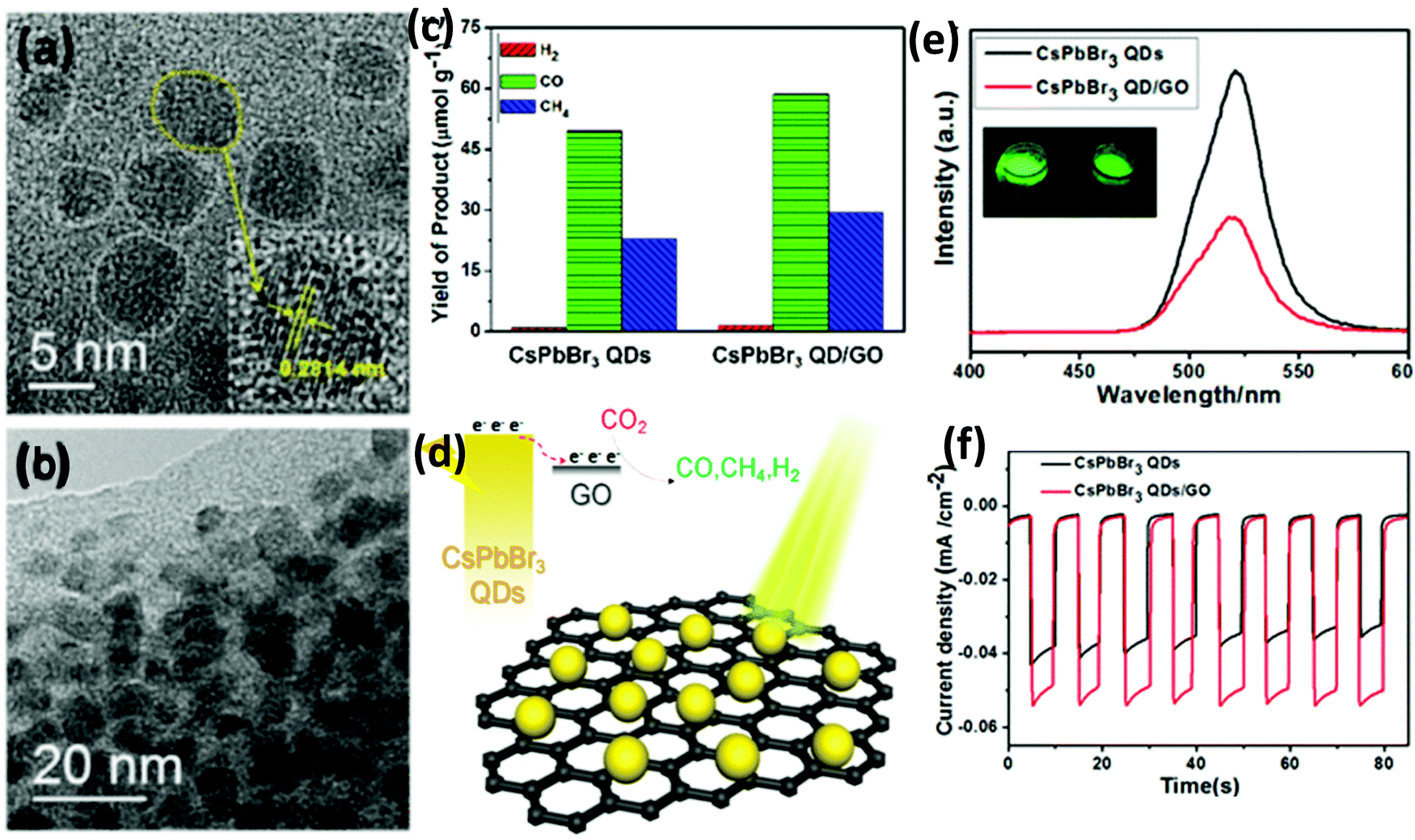 | ||
| Fig. 16 (a) and (b) HRTEM images of the CsPbBr3 QDs, (c) yields of the CO2 reduction products after 12 h of photochemical reaction, (d) schematic illustration of CO2 photoreduction over the CsPbBr3 QD/GO photocatalyst, (e) steady-state PL spectra with an excitation wavelength of 369.6 nm, and (f) amperometric I–t curves plotted at −0.4 V Ag/AgCl under chopped AM 1.5G illumination.144 Copyright 2017, American Chemical Society. | ||
The photocatalytic CO2 reduction efficiency is a key parameter affecting ultimate practical applications. Further, to enhance the CO2 photocatalytic reduction efficiency, the construction of heterojunction structures has been proved to be an effective method, benefiting from the broadened light absorption range, the accelerated charge separation, and the suppressed charge carrier recombinations induced by heterostructures. Jiang et al. skillfully designed a novel hierarchical heterogeneous catalyst with CsPbBr3 nanocrystals anchored onto 1D branched ZnO nanowires/3D mesoporous rGO structures and demonstrated it as a photocatalyst for the reduction of CO2. The 1D ZnO nanowires were selected due to their good electron mobility, appropriate energy bandgap, low cost, and easy synthesis. By in situ growth of ZnO nanowires on the surface of mesoporous rGO, the specific surface area and effective electron transport increases, which is beneficial for the homogenous distribution of CsPbBr3 nanocrystals. Due to the synergistic interactions among 0D CsPbBr3 nanocrystals, 1D ZnO nanowires, and mesoporous rGO, enhanced light-harvesting efficiency, facilitating higher electron transfer rate, improved CO2 capture, and selective CO2 reduction. As a consequence, this hierarchical heterogeneous catalyst exhibited a remarkable photocatalytic activity and CH4 selectivity of 52.02 mmol g−1 h−1 and 96.7%, respectively, under visible-light irradiation.145 Ali et al. fabricated a CuSe2–G–TiO2 heterojunction composite employing an ultrasonication technique for the photocatalytic reduction of CO2. The estimated bandgap energies of the as-synthesized nanocomposites were in the following order: CuSe2–G–TiO2 < CuSe2–G < G–TiO2. Hence, doping with CuSe2 and GO decreased the bandgap of TiO2, indicating that the CuSe2–G–TiO2 nanocomposite exhibited high UV and visible light photocatalytic activity. The methanol yields for the as-synthesized nanocomposites such as Cu2Se–G–TiO2 (12 h), Cu2Se–G–TiO2 (24 h), Cu2Se–G–TiO2 (36 h), and Cu2Se–G–TiO2 (28 h) at various time intervals under visible light were 0.86, 1.15, 1.96, and 2.60 μmol g−1 h−1, and also the methanol yields for Cu2Se–G–TiO2 (12 h), Cu2Se–G–TiO2 (24 h), Cu2Se–G–TiO2 (36 h) and Cu2Se–G–TiO2 (48 h) under UV light were 1.36, 2.01, 3.12 and 3.92 μmol g−1 h−1, respectively, without any sacrificial agent. Furthermore, the authors studied the effect of the Na2S/Na2SO3 scavenger on the photocatalytic activity of the Cu2Se–G–TiO2 nanocomposite, and the methanol yield was two fold higher than that without using the sacrificial agent. Also, the composite displayed good stability and the CH3OH yield rate was approximately in the range of 6.4 to 4.4 μmol g−1 h−1. Hence, it can be used for a continuous photocatalytic reduction system of CO2.146
The decrease in photocatalytic activity might be due to the loss of material during the recycling process. Hence, recycling the photocatalyst without losing the catalyst weight and its efficiency is most desirable. In this connection, Kumar et al. prepared highly efficient, recyclable, and magnetically separable core–shell nanostructured CuZnO@Fe3O4 wrapped with rGO sheets (rGO@CuZnO@Fe3O4) for visible light-mediated CO2 photoreduction. Owing to the synergic effect between the rGO and CuZnO@Fe3O4 in the nanocomposite, significantly enhanced photocatalytic activity compared with the CuZnO@Fe3O4 and GO@CuZnO@Fe3O4 microspheres was observed. The methanol yields for the as-prepared composites including CuZnO@Fe3O4, GO@CuZnO@Fe3O4, and rGO@CuZnO@Fe3O4 were around 858, 1749, and 2656 μmol gcat−1, respectively. The catalyst can easily be recovered from the reaction mixture using an external magnet, and the recovered catalyst exhibits consistent activity for cycles and provides methanol in very similar yields. The probable mechanism for the enhanced photocatalytic activity of the rGO@CuZnO@Fe3O4 nanocomposite is shown in Fig. 17. Fe3O4 displays strong absorption in the visible region, but due to its narrow bandgap (1.16 eV), it is unable to achieve water splitting. ZnO has a wide bandgap (3.2 eV) with apt positions of the conduction band (−0.66 V vs. NHE at pH 7) and valence band (+2.54 V vs. NHE at pH 7) to reduce CO2 to CH4 and oxidize water to O2, respectively. However, only UV light can generate charge carriers. Therefore, neither Fe3O4 nor ZnO alone could reduce CO2. But, when ZnO was coated over the Fe3O4, it could reduce CO2 to CH4 and oxidize water to O2. After absorption of visible light, the photogenerated electrons and holes from the Fe3O4 moved to the conduction and valence bands of the ZnO. But due to the charge carrier recombination, the methanol yield was very poor. To improve the catalytic activity and slow down the electron–hole recombination, Cu particles were added as a cocatalyst on the surface of ZnO@Fe3O4 (Fig. 17a and b). Furthermore, upon adding rGO, the bandgap decreased and the photoefficiency increased followed by electron–hole pairs moving apart, which led to the effective photoreduction of adsorbed CO2 to methanol (Fig. 17c).147
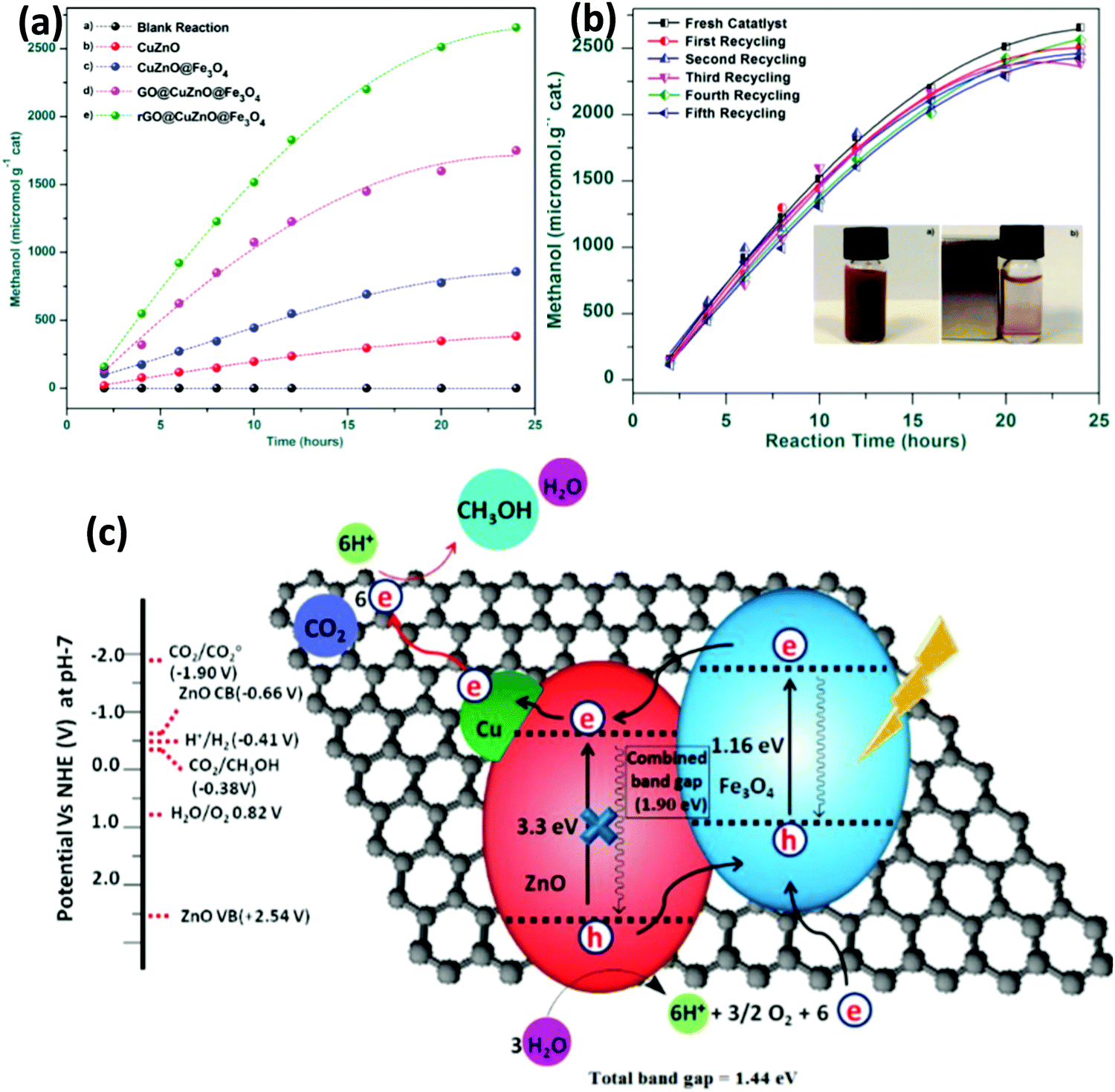 | ||
| Fig. 17 (a) Yield of methanol, (b) recycling experiment for the rGO@CuZnO@Fe3O4 microsphere photocatalyst and (c) plausible CO2 photoreduction over the rGO@CuZnO@Fe3O4 microsphere photocatalyst.147 Copyright 2017, Elsevier. | ||
The construction of Z-scheme heterojunctions is an effective way to isolate photogenerated electron–holes and enhance the activity of semiconductor photocatalysts (Fig. 18a). In this connection, Jiang et al. fabricated an α-Fe2O3/amine rGO/CsPbBr3 hybrid perovskite-based solid-state Z-scheme photocatalyst through a multistep fabrication process for efficient CO2 reduction and water oxidation. CsPbBr3 and α-Fe2O3 are selected as Photosystem I and Photosystem II, respectively, considering their significant visible light absorption, and outstanding reduction and oxidation potentials. Initially, α-Fe2O3 single-crystal nanorods were grown on an FTO substrate using a hydrothermal method followed by a surface coating of amine-functionalized rGO sheets and subsequent deposition of CsPbBr3 nanocrystals using a solvent evaporation deposition approach. CsPbBr3 nanocrystals have a bandgap of ∼2.3 eV with ECB negative enough towards CO2 reduction. On the other hand, α-Fe2O3 shows a bandgap of ∼2.2, and it is documented as the best photocatalyst for the oxidation of water to O2, even though its ECB is too positive to reduce CO2. As shown in Fig. 18b and c, the matched energy band structure enables direct Z-scheme charge transfer from CsPbBr3 nanocrystals to α-Fe2O3 nanorods, leading to boosted charge separation and prolonged lifetimes of charge carriers. The photoelectrons in the CB of α-Fe2O3 can recombine with the VB holes of CsPbBr3, leaving holes in the VB of α-Fe2O3 and electrons in the CB band of CsPbBr3. Furthermore, by introducing amine–rGO with high conductivity and electron affinity, the charge transfer efficiency accelerated and regulated the interfacial interactions in the composite. The α-Fe2O3/amine–rGO/CsPbBr3 composite showed an utmost photocatalytic activity for the reduction of CO2 to CH4 of 181.68 mmol g−1 after 15 h of continuous illumination, which is a much higher value than those of α-Fe2O3/CsPbBr3 (66.64 mmol g−1) and CsPbBr3 nanocrystals (30.55 mmol g−1) (Fig. 18d and e). CH4 was produced as the main product along with small amounts of CO and H2, and the electron selectivity for CH4 was around 93.4%. In addition, the Z-scheme system displayed good stability during the long-time reaction (Fig. 18f).148
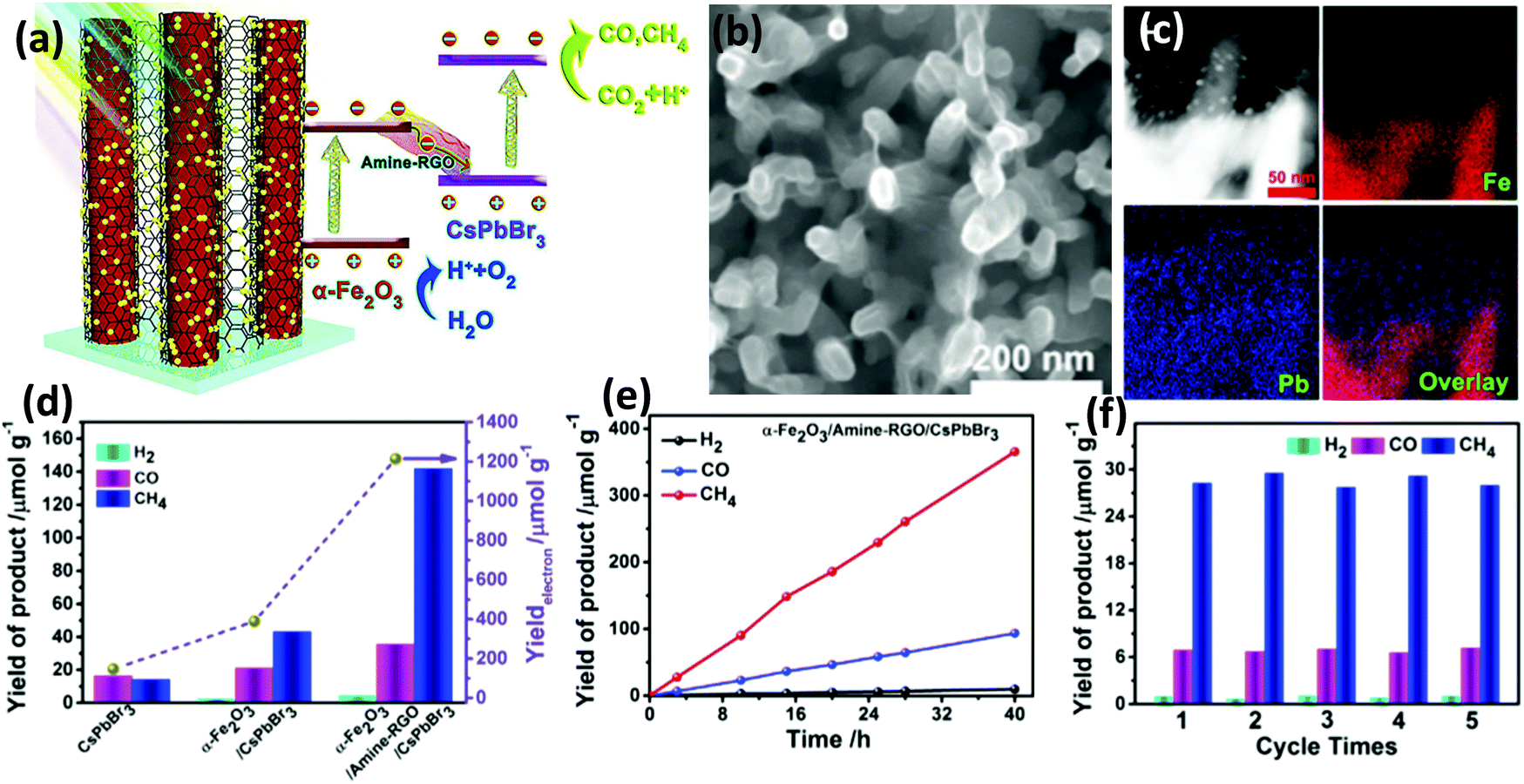 | ||
| Fig. 18 (a) Schematic illustration of charge transfer and CO2 photoreduction over the Fe2O3–amino-rGO–CsPbBr3 composite, (b) FESEM image, (c) EDS elemental mapping, (d) comparison of CO2 photoreduction over various photocatalysts, (e) long-term utilization as a photocatalyst and (f) recycling stability tests of Fe2O3/amine-rGO/CsPbBr3.148 Copyright 2020, Elsevier. | ||
Meng et al. constructed a novel three-component Z-scheme system employing a facile solvothermal approach, in which UiO-66-NH2 and oxygen defective ZnO (O-ZnO) were used as photocatalysts and rGO was used as an electron mediator. This Z-scheme photocatalyst was demonstrated as an excellent photocatalyst for the conversion of CO2 to CH3OH and HCOOH under visible light illumination and exhibited yields of 34.85 and 6.40 μmol g−1 h−1, respectively, which are much higher than those of O-ZnO/UiO-66-NH2 (CH3OH, 19.67 and HCOOH, 4.94 μmol g−1 h−1). The enhancement in the photocatalytic performance was triggered by the formation of a direct Z-scheme heterojunction between the O-ZnO and UiO-66-NH2, thus promoting and separating photoinduced charge carriers and improving the photocatalytic activity, and this was also due to the rGO acting as an effective electronic medium, which can not only enhance the contact area and tightness of the semiconductors but also create a new electron transfer bridge, which is beneficial for the transmission and separation of photoinduced charge carriers. Besides, the oxygen vacancies present in the ZnO were advantageous for the enhancement of the photocatalytic performance for the following reasons: (i) the occurrence of oxygen vacancies favors the adsorption of CO2 onto the surface of the photocatalyst; and (ii) oxygen vacancies can act as trapping centers for photo-induced electrons, thus hindering the recombination of photogenerated electron–hole pairs in ZnO and enhancing the photocatalytic performance.149 All the Z-scheme photocatalysts reported so far were powder systems. For practical applications, the integration of nanosystems of two individual components closely connected by charge transport units such as rGO is desirable. In this connection, Li et al. rationally designed a Z-scheme photocatalyst array built on a stainless steel substrate for the photoconversion of gaseous CO2 to CH4. The photocatalyst consists of Fe2V4O13 nanoribbons and CdS nanoparticles as visible light active materials and a low cost highly conductive rGO interlayer as an electron mediator and also protects the CdS from photocorrosion. It can be easily seen from Fig. 19a–f that a lot of small CdS nanoparticles are uniformly deposited over the rGO sheets over the Fe2V4O13 nanoribbons of 10–20 nm lengths. The photocatalyst displayed good activity for CO2 reduction with a yield of 1.57 μmol h−1 g−1 under visible light irradiation, which is three times higher than those of bare Fe2V4O13 nanoribbons (0.55 μmol h−1 g−1) and Fe2V4O13 nanoribbons/rGO (0.57 μmol h−1 g−1) (Fig. 19g). This enhanced activity is due to the formation of an intimate interface between the Fe2V4O13 nanoribbons, rGO sheets, and CdS nanoparticles. Conspicuously, the production of O2 with a non-stoichiometric molar ratio to CH4 (<2:1) is also detected, which indicates the simultaneous occurrence of water oxidation and photoabsorption of oxygen molecules on the photocatalyst surface. Finally, the obviously enhanced activity can be attributed to the Z-scheme charge separation mechanism as shown in Fig. 19h. The photogenerated electrons of the CdS NPs can readily recombine with the existing holes of the Fe2V4O13 nanoribbons through the rGO mediator, which was further confirmed by the photoluminescence decay time profiles. As a result, the photogenerated holes in the VB of CdS and electrons in the CB of Fe2V4O13 could be available for the O2 evolution and CO2 reduction to CH4, respectively.150 A detailed list of the photocatalytic performances of various graphene based hybrids for CO2 photoreduction to value added products is shown in Table 3.
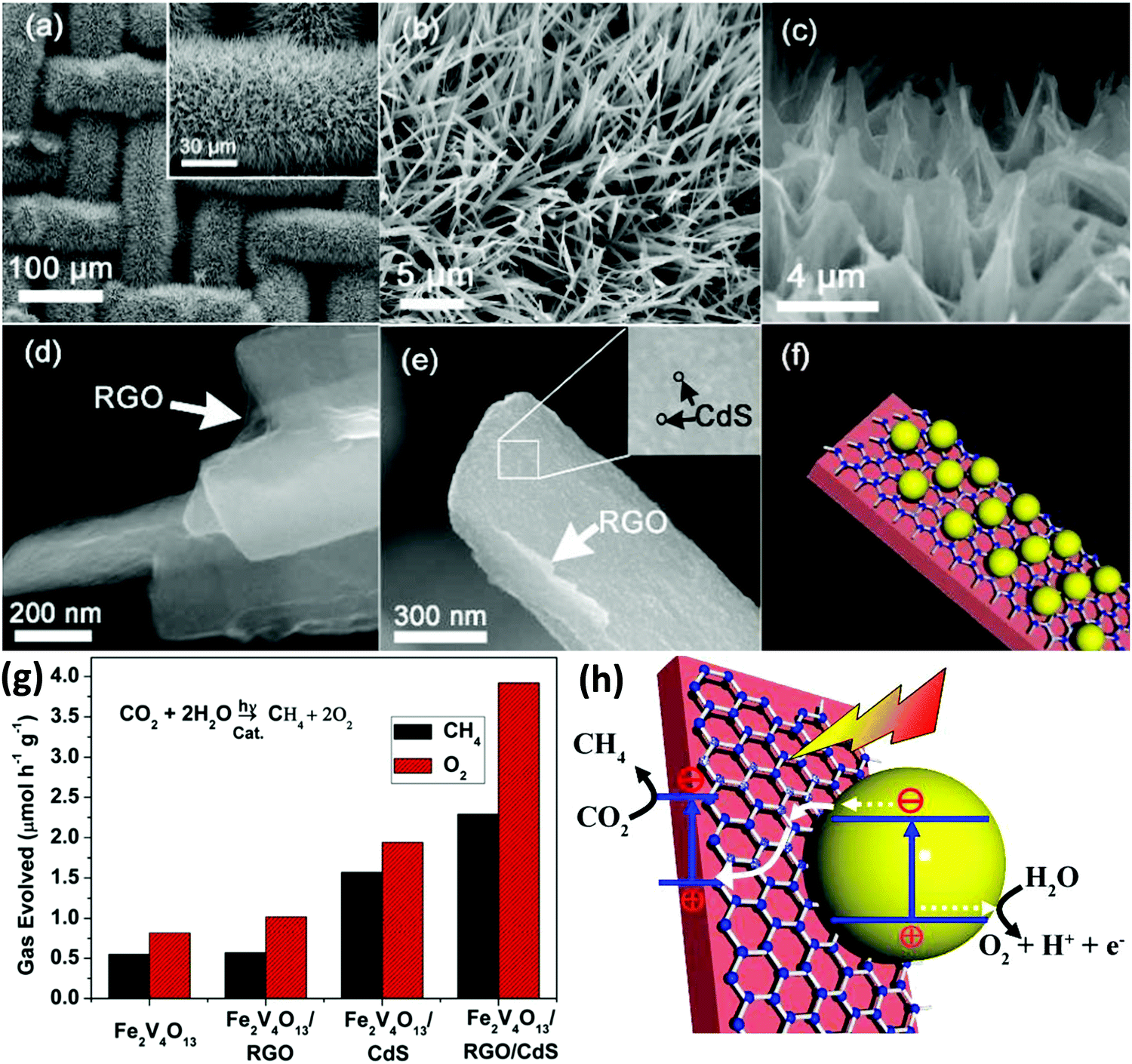 | ||
| Fig. 19 FESEM images of (a and b) Fe2V4O13, (c and d) rGO–Fe2V4O13 and (e) Fe2V4O13–rGO–CdS. (f) Schematic representation of Fe2V4O13–rGO–CdS. (g) Photocatalytic activities of various photocatalysts. (h) Schematic illustration of photocatalytic reduction of CO2 over the Fe2V4O13–rGO–CdS catalyst.150 Copyright 2014, Royal Society of Chemistry. | ||
| Photocatalyst | Light source | Main products | Photocatalytic performance | Ref. |
|---|---|---|---|---|
| GO | 300 W halogen lamp | CH3OH | ∼0.172 mmol gcat−1 h−1 | 17 |
| GO | 500 W xenon lamp | CO | ∼1.23 μmol gcat−1 h−1 | 60 |
| Cu NP/GO hybrids | 300 W halogen lamp | CH3OH, CH3CHO | ∼6.84 μmol gcat−1 h−1 | 90 |
| rGO–Au NPs | Xe lamp (power density: 5.68 W cm−2) | HCOOH | ∼3.12% QY | 124 |
| GO–phenanthroline ligands | 20 W white cold LED flood light | CH3OH | ∼82.86 μmol gcat−1 h−1 | 126 |
| GO | ∼45.86 μmol gcat−1 h−1 | |||
| GO-tethered–CoII phthalocyanine complex | 20 W white cold LED flood light | CH3OH | ∼78.78 μmol gcat−1 h−1 | 127 |
| GO-heteroleptic ruthenium(II) complex | 20 W white cold LED flood light | CH3OH | ∼85.41 μmol gcat−1 h−1 | 128 |
| GO–Cs2Mo6Bri8Brax | 20 W white cold LED flood light | CH3OH | ∼68.5 μmol gcat−1 h−1 | 129 |
| GO–(TBA)2Mo6Bri8Brax | ∼53.91 μmol gcat−1 h−1 | |||
| GO-doped oxygen-rich TiO2 | 500 W xenon lamp | CH4 | ∼0.43 μmol gcat−1 h−1 | 130 |
| GO–TiO2 nanotube arrays | 100 W xenon solar simulator | CH4 | ∼5.67 ppm cm−2 h−1 | 131 |
| N-rGO/TiO2 | 400 W xenon lamp | ∼5.1 μmol gcat−1 h−1 | 133 | |
| Cu2O/rGO | 150 W xenon lamp | CO | ∼50 ppm g−1 h−1 | 134 |
| rGO–CuO/Cu2O | 20 W white cold LED flood light | CH3OH | ∼53.41 μmol gcat−1 h−1 | 135 |
| Graphene–WO3 nanobelts | 300 W Xe lamp | CH4 | ∼0.89 μmol | 136 |
| GO–CdS nanorods | 300 W Xe arc lamp | CH4 | ∼2.51 μmol h−1 g−1 | 137 |
| HNb3O8/graphene hybrids | 300 W xenon arc lamp | CO | ∼50 μmol gcat−1 h−1 | 138 |
| CH4 | ∼1.3 μmol gcat−1 h−1 | |||
| Ag–RGO–CdS | 300 W Xe lamp | CO | ∼1.61 μmol h−1 | 141 |
| Au–Cu/graphene/Cu2O | 400 W Xe lamp | CH4 | ∼18.80 ppm cm−2 h−1 | 142 |
| Pt/graphene/TiO2 | 300 W Xe lamp | CH4 | ∼0.282 μmol gcat−1 h−1 | 143 |
| Pd/graphene/TiO2 | ∼0.198 μmol gcat−1 h−1 | |||
| Ag/graphene/TiO2 | ∼0.166 μmol gcat−1 h−1 | |||
| Au/graphene/TiO2 | ∼0.126 μmol gcat−1 h−1 | |||
| CsPbBr3/GO | 100 W Xe lamp | CH4 | ∼23.7 μmol gcat−1 h−1 | 144 |
| CsPbBr3–ZnO nanowire/mrGO | 150 W Xe lamp | CH4 | ∼52.02 μmol gcat−1 h−1 | 145 |
| rGO@CuZnO@Fe3O4 microspheres | 20 W white cold LED light | CH4 | ∼110.66 μmol gcat−1 h−1 | 147 |
| α-Fe2O3/amine–RGO/CsPbBr3 hybrids | 150 W Xe lamp | CH4 | ∼5.44 μmol gcat−1 h−1 | 148 |
| Fe2V4O13/RGO/CdS | 300 W Xe lamp | CH4 | ∼2.23 μmol gcat−1 h−1 | 150 |
8. Challenges for the commercialization of graphene and its hybrids
Extensive reports have surged over the past few years on graphene and its composites for various applications, and the mass production of these materials has been realized by various industries, which have started to commercialize some of these products already in the market. However, the market for graphene-based materials is still worth exploring and enlarging. So far, only a handful of products which contain graphene have reached the market such as the tennis racket by Head, the battery strap by Vorbeck, and the phone touch screen by Samsung.151,152 The main problem with high-quality or pristine graphene (which is a great conductor) is that it does not have a bandgap; as a result, it can’t be switched off. Hence, the use of graphene in the fabrication of future nano-electronic and optoelectronic devices has been restricted. Thus, the opening of the bandgap in graphene is very essential and it needs to be engineered, which can reduce its electron mobility so that it can replace the silicon in electronic systems shortly. Nevertheless, for the bulk production of graphene and its hybrid materials, wet chemical approaches are suitable methods. However, the produced graphene and its hybrid materials show some discrepancies in the following areas: in the size of graphene flakes, the number of layers in commercially available graphene materials along with substantial alterations in defect states and impurity content. All these wide dissimilarities in the quality of graphene materials restrict them to be commercialized. Here, we have summarized some of the challenges for the commercialization of graphene-based materials according to specific requirements. The basic challenges in the commercialization of these products are mainly because of the following concerns. The basic problem people face is uncontrolled and costly synthesis approaches of graphene since its emergence, which is the key factor for the large-scale production of graphene and its hybrids for industrial use. Specifically, the synthesis of GO using the Hummers’ method (dominated by academia and industry) doesn’t have fine control over the functional groups and size of the GO system; in addition, classification of GO materials as per their defects, functional groups, and size is a challenging task.151–153 Furthermore, efficient separation techniques are highly desirable, because of the utilization of harsh oxidative chemical reagents, dispersant media, and purifying solvents, which makes the production costs of GO much higher compared with other raw materials. Consequently, this tedious process hinders GO and its hybrid materials in further applications in many fields. Furthermore, the properties of GO and its hybrid materials change with synthesis approaches, which currently limits the transfer of academic advancements into real industrial progress. Moreover, the reliability and process repeatability of a particular fabrication technique along with a stable production need to be improved.Another crucial factor is the lack of standardization for the GO and its hybrids in the market, mainly due to expensive characterization techniques such as scanning and transmission electron microscopy, atomic force microscopy, elemental analysis, and X-ray photoelectron spectrometry. All these characterization techniques are time- and cost-consuming methods for industrial users. Therefore, low-cost, simple, fast, and reliable methods on accessible equipment are necessary for small and start-up industries. Additionally, the uncontrolled chemical reduction and functionalization of GO on a large scale are still under debate. Safety issues such as the chronic toxicity and genotoxicity of graphene and its hybrids are not clear. Therefore, further investigations are absolutely necessary.151–153
9. Summary and future perspectives
Photocatalysis can be a significant approach for the generation of solar fuels to overcome energy crises and environmental problems. It includes hydrogen evolution by direct water splitting and CO2 photoreduction to value-added products.3,4,104 However, the selection of a suitable photocatalyst and adoption of an appropriate approach are of primary importance for achieving efficient product formation for practical demands. Several reports are already there based on the fundamental understanding of the process, and several challenges/promises for practical applicabilities. However, the complete photocatalytic water splitting into H2 and O2 in a stoichiometric ratio of 2:1 under light illumination typically displays low efficiency due to the high energy barrier.16,17,62,135 Though the efficiency of H2 production can be improved by the addition of sacrificial agents/specific hole scavengers to complete the redox cycle,137,154 it is not desirable to use additional sacrificial agents. Therefore, the best solution will be to achieve complete water splitting, which is much closer to the greener, cleaner, and renewable energy concept. However, the back oxidation of H2 into water or oxygen reduction reaction to water is one of the major challenges, which can be prevented by surface modification or poisoning. Furthermore, the efficiency of water splitting relies on the innovative design of photocatalysts such as hetero-junction-based materials. In particular, carbon-based hetero-junctional photocatalysts or graphene-based Z-scheme systems are believed to be feasible future effective photocatalysts.148,155 The architecture of hetero-junctions directly influences the activity and could lead to stimulating improvements.
Another energy-related photocatalytic application is the photoreduction of CO2 to value-added products or the conversion of CO2 and H2O into useful products. Mainly there are two major limitations for solar light-driven photocatalytic CO2 conversions: (a) the efficiency is extremely low considering practical applicability and (b) specific product selectivity is also very difficult, especially for higher alkyl products.4,61 As discussed before, the photocatalytic reduction efficiency can be improved by improving light absorption, charge separation, and charge accumulation, and modifying the catalytic sites on the surfaces of heterogeneous catalysts. However, product selectivity is not so easy. During the photoreduction of CO2 to CO, CH4, CH3OH, HCOOH, and other long-chain alkanes, H2O can act as the reductant. In addition, H2 can act as a common feedstock for solar fuel production through photocatalysis. Therefore, it is quite obvious that we can obtain multiple products simultaneously, and an additional separation process is required to achieve a pure product, which increases the overall cost of solar fuel production. The adsorption and activation of CO2 over the surface of the photocatalyst can play vital roles in improving the selectivity towards the desired products. Generally, increasing the catalytic surface area, improving surface defects by defect engineering, and introducing noble metal co-catalysts have been widely used for improving the adsorption and activation of CO2. In this regard, more experimental and computational models may help in obtaining a detailed structural correlation with the electronic features and ongoing photophysical properties, which include in situ time-resolved spectroscopy such as time-resolved fluorescence/absorption spectroscopy (up to the femtosecond time scale), electron paramagnetic resonance spectroscopy, and density functional theory calculations. The rational understanding of the mechanism may give information to design more efficient and highly selective photocatalysts. Furthermore, the products generated from the reduction of CO2 over photocatalysts should be identified by an isotopic labeling experiment with 13CO2. Therefore, we should minimize the hydrocarbon conversion under the process needed for CO2 reduction. Besides, hydrocarbons may be intact over the surface of the photocatalyst during the reduction of CO2. For effective photocatalytic performance, the following requirements are recommended for the photocatalyst: (a) stability, (b) low-cost and large-scale fabrication, (c) availability, (d) efficiency, (e) suitable bandgap, (f) corrosion resistance, etc. In addition, the adsorption of CO2 molecules onto the photocatalyst is a significant factor and it is considered as an initial step for the photocatalytic CO2 reduction process. Furthermore, the feasibility of the electron transfer from the photocatalyst to the CO2 molecules depends on the interaction between the photocatalyst and CO2 molecules. The enhancement in the physical or chemical adsorption of CO2 onto the photocatalyst surface can improve the activation of CO2.156,157 Hence, if more CO2 molecules are adsorbed onto the surface of the photocatalyst, more electrons would potentially reach and reduce the CO2 molecules. Therefore, by increasing the surface area of the photocatalyst we can improve the active sites, resulting in a better CO2 adsorption capacity, which leads to enhanced photocatalytic performance. In this connection, metal–organic frameworks (MOFs), a class of porous crystalline materials, are formed by a network of metal ions linked to the organic molecules through a metal–ligand coordination bond, and have emerged as alternative photocatalysts.158 The three diverse components of the MOF including metal nodes, organic linkers, and pores will help to modify the active sites, bandgap, and feasible pathways to obtain different products. Furthermore, the high surface area and porous architecture further improve the CO2 adsorption on the surface. Moreover, the porous structure facilitates the diffusion channels for the reactants and products.157,159
However, the main challenges regarding heavy metal-based photocatalysts are toxicity, environmental unsustainability, and high cost, which make their practical use questionable. Consequently, it is essential to search for alternative photocatalytic materials that can overcome all the challenges and can subsequently be used for hydrogen production by overall water splitting and CO2 reduction to value-added products. In this context, carbon-based luminescent nanomaterials and their hybrid systems, especially carbon nitride, luminescent carbon dots/graphene quantum dots, soluble graphene oxide, and their derivatives like graphdiyne and carbon nanotubes, etc., are the possible alternatives. These could be very efficient, more robust, environmentally friendly, and low-cost alternatives. Besides, they exhibit fascinating properties including high specific surface area, visible light absorption, and excellent charge carrier mobility. Generally, in semiconductor–graphene hybrids, the extended conjugation of carbon in graphene sheets facilitates better charge transportation on their surface, which leads to efficient charge separation.160,161 However, the lack of a bandgap in graphene sheets restricts their use as sole photocatalysts. The introduction of defects into the lattice of graphene sheets by doping with heteroatoms or oxidation can transform conductive graphene into a semiconductor. Furthermore, the bandgap can be tuned by systematic approaches. However, obtaining photocatalysts with a higher dopant concentration and robustness is still challenging. However, these sp2 hybridized carbon materials including graphene, graphene oxide, and carbon nanotubes do not solely act as effective photocatalysts. Mostly these types of materials can be used as photocatalysts with the help of co-catalysts or charge-transporting materials.
In these connections, 0-dimensional carbon dots (CDs)/graphene QDs are of particular interest as new generation alternative photocatalysts for solar fuel generation. The approaches for the fabrication of this new class of materials are mostly divided into two categories, i.e. top-down and bottom-up methods. The top-down approach is mainly a typical exfoliation technique, followed by cutting larger carbon structures into ultra-small dot like structures with high crystallinity along with a few surface defects by employing various physical approaches such as laser ablation, electrochemical etching, plasma treatment, and arc-discharge methods.3,162–164 On the other hand, the bottom-up synthesis approach refers to the building of dot like structures with carbon cores along with various surface states, defects, and functional groups from small molecule precursors. Based on the availability of a diversity of precursors and synthesis approaches, CDs/GQDs are endowed with widely variable optoelectronic properties and performance parameters.165,166 Besides, they show several advantages including high water solubility, non-toxicity, facile preparation schemes, environmental sustainability, tunable optical properties, and enhanced photostability. Furthermore, CDs/GQDs consist of a heterogeneous distribution of aromatic domains (sp2 hybridized carbons) embedded in amorphous regions (sp3 hybridized carbons) along with various heteroatom functionalities, which facilitate the generation of various discrete energy levels within the CDs, resulting in unique photophysical, chemical, and electronic properties.162,163,165,166 Based on these intriguing optoelectronic properties, CDs have become potentially significant materials for photocatalytic applications. Nevertheless, the practical applicabilities of these luminescent CDs as solely photocatalysts (without co-catalyst) are still under debate due to the lack of studies for the fundamental structure–property correlation. Hence, further research is needed in this direction. In our opinion, the intrinsic structural features at the atomic scale level such as ratiometric proportions of aromatic and amorphous domains, various bonding motifs of heteroatoms, distribution of defect states, and atomic-scale vacancies can critically control the overall photophysics of CDs/GQDs. However, the photocatalytic efficiency of these materials is very limited compared with traditional transition metal-based photocatalysts mainly because of the lack of visible light absorption and competitive recombination processes. Therefore, further research is highly desirable particularly in this direction to improve the photocatalytic efficiency. Usually, visible light absorption can be improved through the following ways: (a) creating mid bandgap states, (b) tuning defects as well as molecular and atomic vacancies and (c) altering surface functionalities.3,162,165,166 In addition, the photocatalytic efficiency can also be improved by enhancing the charge separation followed by charge carrier survival while competing with the other recombination processes and finally charge carrier diffusion to surface or active sites.3,167,168 However, getting a clear picture of the fundamental photophysical and photochemical processes in CDs/GQDs is extremely difficult because of the lack of the structure–property correlation. A complex interplay between the various states of CDs/GQDs such as aromatic/amorphous domains along with surface states and atomic vacancies controls the overall excitonic features and charge separation kinetics.163 In addition, tuning surface states such as –CO and –COOH groups can induce up-ward band bending, resulting in effective charge separation followed by enhanced activity. In addition to this, the defined morphologies can also enhance the photocatalytic activity of CDs by improving the active sites and effective surface area. Therefore, upon combining CDs/GQDs with other carbon-based materials can further increase photocatalytic performance.162,163 Moreover, making intrinsic n–p dyad-like structures inside CDs/GQDs by incorporating various hetero-atoms can increase visible light absorption and effective charge separation, followed by efficient photocatalysis. Therefore, further research is needed in this direction to attain superior photocatalysts. A detailed theoretical understanding using density functional theory (DFT) calculations is very much essential for structural optimization and for unraveling the atomic level surface chemistry of these materials. Therefore, a lot of optimization is needed to utilize these materials solely as photocatalysts. In recent years, use of computational approaches has become an alternative strategy for prior screening of photocatalysts for particular reactions, with parameters including high efficiency, high selectivity, and cost guiding the choice of elements. In particular, DFT-based first-principles calculations are widely utilized to explore the properties of materials and further understand the role of the different components in the photocatalytic mechanism or overall reaction cycle.169 The theoretical simulations provide the framework for understanding the catalysis of bulk materials to their atomic level. Therefore, a fundamental understanding of crystal structures, electronic structures, bandgap energies, carrier mobilities, and reaction energy pathways could help in the fabrication of novel and efficient photocatalytic materials in the future.
Author contributions
Kommula Bramhaiah: conceptualization, writing – original draft. Santanu Bhattacharyya: conceptualization, writing – review & editing, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
K. B. acknowledges IISER Berhampur for providing an institute postdoctoral fellowship. The authors acknowledge DST-SERB (grant numbers/2019/000026) for financial support and S. B. acknowledges IISER Berhampur for an initiation grant (IG/21082018/B0035).References
- X. Chen, C. Li, M. Grätzel, R. Kostecki and S. S. Mao, Chem. Soc. Rev., 2012, 41, 7909–7937 RSC.
- S. S. Mao, S. Shen and L. Guo, Prog. Nat. Sci.: Mater. Int., 2012, 22, 522–534 CrossRef.
- S. Kundu, K. Bramhaiah and S. Bhattacharyya, Nanoscale Adv., 2020, 2, 5130–5151 RSC.
- J. K. Stolarczyk, S. Bhattacharyya, L. Polavarapu and J. Feldmann, ACS Catal., 2018, 8, 3602–3635 CrossRef CAS.
- M.-Q. Yang, N. Zhang, M. Pagliaro and Y.-J. Xu, Chem. Soc. Rev., 2014, 43, 8240–8254 RSC.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- C. L. Muhich, B. D. Ehrhart, I. Al-Shankiti, B. J. Ward, C. B. Musgrave and A. W. Weimer, Wiley Interdiscip. Rev.: Energy Environ., 2016, 5, 261–287 CAS.
- J.-H. Tang and Y. Sun, Mater. Adv., 2020, 1, 2155–2162 RSC.
- J. Z. Y. Tan and M. M. Maroto-Valer, J. Mater. Chem. A, 2019, 7, 9368–9385 RSC.
- J. Di, B. Lin, B. Tang, S. Guo, J. Zhou and Z. Liu, Small Struct., 2021, 2(10), 2100046, DOI:10.1002/sstr.202100046.
- X. Jiao, K. Zheng, L. Liang, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2020, 49, 6592–6604 RSC.
- P. Liao and E. A. Carter, Chem. Soc. Rev., 2013, 42, 2401–2422 RSC.
- M. Marszewski, S. Cao, J. Yu and M. Jaroniec, Mater. Horiz., 2015, 2, 261–278 RSC.
- R. K. Upadhyay, N. Soin and S. S. Roy, RSC Adv., 2013, 4, 3823–3851 RSC.
- S. Badhulika, T. Terse-Thakoor, C. M. Chaves Villarreal and A. Mulchandani, Front. Chem., 2015 DOI:10.3389/fchem.2015.00038.
- J. Low, J. Yu and W. Ho, J. Phys. Chem. Lett., 2015, 6, 4244–4251 CrossRef CAS.
- H.-C. Hsu, I. Shown, H.-Y. Wei, Y.-C. Chang, H.-Y. Du, Y.-G. Lin, C.-A. Tseng, C.-H. Wang, L.-C. Chen, Y.-C. Lin and K.-H. Chen, Nanoscale, 2013, 5, 262–268 RSC.
- X. Li, J. Yu, S. Wageh, A. A. Al-Ghamdi and J. Xie, Small, 2016, 12, 6640–6696 CrossRef CAS PubMed.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- S. Kundu and A. Patra, Chem. Rev., 2017, 117, 712–757 CrossRef CAS PubMed.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS PubMed.
- I. V. Lightcap, T. H. Kosel and P. V. Kamat, Nano Lett., 2010, 10, 577–583 CrossRef CAS PubMed.
- Y. Zhang, Z.-R. Tang, X. Fu and Y.-J. Xu, ACS Nano, 2010, 4, 7303–7314 CrossRef CAS PubMed.
- Y. H. Ng, A. Iwase, A. Kudo and R. Amal, J. Phys. Chem. Lett., 2010, 1, 2607–2612 CrossRef CAS.
- Y. H. Ng, A. Iwase, N. J. Bell, A. Kudo and R. Amal, Catal. Today, 2011, 164, 353–357 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nanoscience and Technology, Macmillan Publishers Ltd, UK, 2009, pp. 11–19 Search PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- K. P. Loh, Q. Bao, P. K. Ang and J. Yang, J. Mater. Chem., 2010, 20, 2277–2289 RSC.
- P. R. Wallace, Phys. Rev., 1947, 71, 622–634 CrossRef CAS.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- L. A. Falkovsky, J. Phys.: Conf. Ser., 2008, 129, 012004 CrossRef.
- A. A. Balandin, Nat. Mater., 2011, 10, 569–581 CrossRef CAS PubMed.
- X.-K. Kong, C.-L. Chen and Q.-W. Chen, Chem. Soc. Rev., 2014, 43, 2841–2857 RSC.
- Y. Jin, Y. Zheng, S. G. Podkolzin and W. Lee, J. Mater. Chem. C, 2020, 8, 4885–4894 RSC.
- M. A. Velasco-Soto, S. A. Pérez-García, J. Alvarez-Quintana, Y. Cao, L. Nyborg and L. Licea-Jiménez, Carbon, 2015, 93, 967–973 CrossRef CAS.
- K. A. Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda, C. Mattevi, S. Miller and M. Chhowalla, Nano Lett., 2009, 9, 1058–1063 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- T.-F. Yeh, J.-M. Syu, C. Cheng, T.-H. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255–2262 CrossRef CAS.
- C. Han, N. Zhang and Y.-J. Xu, Nano Today, 2016, 11, 351–372 CrossRef CAS.
- B. Wang, G. M. Biesold, M. Zhang and Z. Lin, Chem. Soc. Rev., 2021, 50, 6914–6949 RSC.
- J. Strachan, A. F. Masters and T. Maschmeyer, J. Mater. Chem. A, 2021, 9, 9451–9461 RSC.
- M. Shandilya, R. Rai and J. Singh, Adv. Appl. Ceram., 2016, 115, 354–376 CrossRef CAS.
- J. Shen, B. Yan, M. Shi, H. Ma, N. Li and M. Ye, J. Mater. Chem., 2011, 21, 3415–3421 RSC.
- K. Wang, C. Miao, Y. Liu, L. Cai, W. Jones, J. Fan, D. Li and J. Feng, Appl. Catal., B, 2020, 270, 118878 CrossRef CAS.
- Y. Wang, J. Yu, W. Xiao and Q. Li, J. Mater. Chem. A, 2014, 2, 3847–3855 RSC.
- N. R. Khalid, E. Ahmed, M. Ahmad, N. A. Niaz, M. Ramzan, M. Shakil, T. Iqbal and A. Majid, Ceram. Int., 2016, 42, 18257–18263 CrossRef CAS.
- D. H. Youn, J.-W. Jang, J. Y. Kim, J. S. Jang, S. H. Choi and J. S. Lee, Sci. Rep., 2014, 4, 5492 CrossRef CAS PubMed.
- M. Rostami, R. M. Zamani, K. M. Aghajanzadeh and H. Danafar, J. Pharm. Invest., 2018, 48, 657–664 CrossRef CAS.
- A. Giampiccolo, D. M. Tobaldi, S. G. Leonardi, B. J. Murdoch, M. P. Seabra, M. P. Ansell, G. Neri and R. J. Ball, Appl. Catal., B, 2019, 243, 183–194 CrossRef CAS.
- V. M. Varsha and G. Nageswaran, J. Electrochem. Soc., 2020, 167, 155527 CrossRef.
- F. Trivinho-Strixino, J. S. Santos and M. Souza Sikora, in Nanostructures, ed. A. L. Da Róz, M. Ferreira, F. de Lima Leite and O. N. Oliveira, William Andrew Publishing, 2017, pp. 53–103 Search PubMed.
- J. D. Mangadlao, P. Cao, D. Choi and R. C. Advincula, ACS Appl. Mater. Interfaces, 2017, 9, 24887–24898 CrossRef CAS PubMed.
- K. Bramhaiah, I. Pandey, V. N. Singh, C. Kavitha and N. S. John, J. Nanopart. Res., 2018, 20, 56 CrossRef.
- K. Bramhaiah and N. S. John, RSC Adv., 2013, 3, 7765–7773 RSC.
- K. Bramhaiah, V. N. Singh and N. S. John, Phys. Chem. Chem. Phys., 2016, 18, 1478–1486 RSC.
- K. Bramhaiah, V. N. Singh, C. Kavitha and N. S. John, J. Nanosci. Nanotechnol., 2017, 17, 2711–2719 CrossRef CAS PubMed.
- K. Bramhaiah, C. Alex, V. N. Singh and N. S. John, ChemistrySelect, 2019, 4, 2519–2528 CrossRef CAS.
- K. Sayama, R. Yoshida, H. Kusama, K. Okabe, Y. Abe and H. Arakawa, Chem. Phys. Lett., 1997, 277, 387–391 CrossRef CAS.
- X. Wu, J. Lang, Z. Sun, F. Jin and Y. H. Hu, Appl. Catal., B, 2021, 295, 120312 CrossRef CAS.
- Y. Kuang, J. Shang and T. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 3580–3591 CrossRef CAS PubMed.
- S. Protti, A. Albini and N. Serpone, Phys. Chem. Chem. Phys., 2014, 16, 19790–19827 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- X. Chen, Y. Dai and X. Wang, J. Alloys Compd., 2015, 649, 910–932 CrossRef CAS.
- H. Yan, X. Wang, M. Yao and X. Yao, Prog. Nat. Sci.: Mater. Int., 2013, 23, 402–407 CrossRef.
- H. Li, Y. Zhou, W. Tu, J. Ye and Z. Zou, Adv. Funct. Mater., 2015, 25, 998–1013 CrossRef CAS.
- Y. Guo, H. Li, W. Ma, W. Shi, Y. Zhu and W. Choi, Carbon Energy, 2020, 2, 308–349 CrossRef CAS.
- H. Park, Y. Park, W. Kim and W. Choi, J. Photochem. Photobiol., C, 2013, 15, 1–20 CrossRef CAS.
- A. Sułek, B. Pucelik, J. Kuncewicz, G. Dubin and J. M. Dąbrowski, Catal. Today, 2019, 335, 538–549 CrossRef.
- Y. Ben-Shahar, F. Scotognella, N. Waiskopf, I. Kriegel, S. D. Conte, G. Cerullo and U. Banin, Small, 2015, 11, 462–471 CrossRef CAS PubMed.
- R. Daghrir, P. Drogui and D. Robert, Ind. Eng. Chem. Res., 2013, 52, 3581–3599 CrossRef CAS.
- C. Yu, L. Wei, J. Chen, Y. Xie, W. Zhou and Q. Fan, Ind. Eng. Chem. Res., 2014, 53, 5759–5766 CrossRef CAS.
- A. Habibi-Yangjeh, S. Feizpoor, D. Seifzadeh and S. Ghosh, Sep. Purif. Technol., 2020, 238, 116404 CrossRef CAS.
- T.-T. Chen, I.-C. Chang, M.-H. Yang, H.-T. Chiu and C.-Y. Lee, Appl. Catal., B, 2013, 142–143, 442–449 CrossRef CAS.
- A. Kumar and V. Krishnan, Adv. Funct. Mater., 2021, 31, 2009807 CrossRef CAS.
- G. Liu, L. Wang, H. G. Yang, H.-M. Cheng and G. Q. (Max) Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- J. Liu, Y. Song, H. Xu, X. Zhu, J. Lian, Y. Xu, Y. Zhao, L. Huang, H. Ji and H. Li, J. Colloid Interface Sci., 2017, 494, 38–46 CrossRef CAS PubMed.
- R. Marschall and L. Wang, Catal. Today, 2014, 225, 111–135 CrossRef CAS.
- K. Wilke and H. D. Breuer, J. Photochem. Photobiol., A, 1999, 121, 49–53 CrossRef CAS.
- A. Hameed, M. A. Gondal and Z. H. Yamani, Catal. Commun., 2004, 5, 715–719 CrossRef CAS.
- A. B. Djurišić, Y. H. Leung and A. M. C. Ng, Mater. Horiz., 2014, 1, 400–410 RSC.
- J. Yang, J. Wang, X. Li, J. Lang, F. Liu, L. Yang, H. Zhai, M. Gao and X. Zhao, J. Alloys Compd., 2012, 528, 28–33 CrossRef CAS.
- M. Singh, S. Kaushal, P. Singh and J. Sharma, J. Photochem. Photobiol., A, 2018, 364, 130–139 CrossRef CAS.
- J. Gliniak, J.-H. Lin, Y.-T. Chen, C.-R. Li, E. Jokar, C.-H. Chang, C.-S. Peng, J.-N. Lin, W.-H. Lien, H.-M. Tsai and T.-K. Wu, ChemSusChem, 2017, 10, 3260–3267 CrossRef CAS PubMed.
- Y. Li, X. Li, H. Zhang, J. Fan and Q. Xiang, J. Mater. Sci. Technol., 2020, 56, 69–88 CrossRef.
- P. Rani and V. K. Jindal, RSC Adv., 2012, 3, 802–812 RSC.
- N. Zhang, M.-Q. Yang, S. Liu, Y. Sun and Y.-J. Xu, Chem. Rev., 2015, 115, 10307–10377 CrossRef CAS PubMed.
- O. Akhavan and E. Ghaderi, J. Phys. Chem. C, 2009, 113, 20214–20220 CrossRef CAS.
- T. Ghosh, K.-Y. Cho, K. Ullah, V. Nikam, C.-Y. Park, Z.-D. Meng and W.-C. Oh, J. Ind. Eng. Chem., 2013, 19, 797–805 CrossRef CAS.
- X. Ma, Q. Xiang, Y. Liao, T. Wen and H. Zhang, Appl. Surf. Sci., 2018, 457, 846–855 CrossRef CAS.
- I. Shown, H.-C. Hsu, Y.-C. Chang, C.-H. Lin, P. K. Roy, A. Ganguly, C.-H. Wang, J.-K. Chang, C.-I. Wu, L.-C. Chen and K.-H. Chen, Nano Lett., 2014, 14, 6097–6103 CrossRef CAS PubMed.
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- M. Z. Rahman, C. W. Kwong, K. Davey and S. Z. Qiao, Energy Environ. Sci., 2016, 9, 709–728 RSC.
- H. Idriss, Catal. Sci. Technol., 2020, 10, 304–310 RSC.
- T. Hisatomi, K. Takanabe and K. Domen, Catal. Lett., 2015, 145, 95–108 CrossRef CAS.
- S. Yan, L. Wan, Z. Li and Z. Zou, Chem. Commun., 2011, 47, 5632–5634 RSC.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- J. Schneider and D. W. Bahnemann, J. Phys. Chem. Lett., 2013, 4, 3479–3483 CrossRef CAS.
- T.-F. Yeh, S.-J. Chen, C.-S. Yeh and H. Teng, J. Phys. Chem. C, 2013, 117, 6516–6524 CrossRef CAS.
- P. Wang, Y. Zhai, D. Wang and S. Dong, Nanoscale, 2011, 3, 1640–1645 RSC.
- S. Das, A. Dutta, R. Bera and A. Patra, Phys. Chem. Chem. Phys., 2019, 21, 15568–15575 RSC.
- R. Bera, B. Jana, B. Mondal and A. Patra, ACS Sustainable Chem. Eng., 2017, 5, 3002–3010 CrossRef CAS.
- R. Bera, S. Kundu and A. Patra, ACS Appl. Mater. Interfaces, 2015, 7, 13251–13259 CrossRef CAS PubMed.
- W. Fan, Q. Lai, Q. Zhang and Y. Wang, J. Phys. Chem. C, 2011, 115, 10694–10701 CrossRef CAS.
- L. Li, L. Yu, Z. Lin and G. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 8536–8545 CrossRef CAS PubMed.
- X. Zhou, G. Liu, J. Yu and W. Fan, J. Mater. Chem., 2012, 22, 21337–21354 RSC.
- R. S. Haider, S. Wang, Y. Gao, A. S. Malik, N. Ta, H. Li, B. Zeng, M. Dupuis, F. Fan and C. Li, Nano Energy, 2021, 87, 106189 CrossRef CAS.
- Q. Huang, T. D. Canady, R. Gupta, N. Li, S. Singamaneni and B. T. Cunningham, ACS Photonics, 2020, 7, 1994–2001 CrossRef CAS.
- B. Tudu, N. Nalajala, K. P. Reddy, P. Saikia and C. S. Gopinath, ACS Appl. Mater. Interfaces, 2019, 11, 32869–32878 CrossRef CAS PubMed.
- W. Wang, M. O. Tadé and Z. Shao, Chem. Soc. Rev., 2015, 44, 5371–5408 RSC.
- K. S. Schanze, P. V. Kamat, P. Yang and J. Bisquert, ACS Energy Lett., 2020, 5, 2602–2604 CrossRef CAS.
- A. Kumar, A. Kumar and V. Krishnan, ACS Catal., 2020, 10, 10253–10315 CrossRef CAS.
- X. Li, H. Zhao, J. Liang, Y. Luo, G. Chen, X. Shi, S. Lu, S. Gao, J. Hu, Q. Liu and X. Sun, J. Mater. Chem. A, 2021, 9, 6650–6670 RSC.
- J. Ding, W. Yan, W. Xie, S. Sun, J. Bao and C. Gao, Nanoscale, 2014, 6, 2299–2306 RSC.
- X. P. Bai, X. Zhao and W. L. Fan, RSC Adv., 2017, 7, 53145–53156 RSC.
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed.
- C. Lavorato, A. Primo, R. Molinari and H. Garcia, Chem. – Eur. J., 2014, 20, 187–194 CrossRef CAS PubMed.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed.
- Y. Xia, Q. Li, K. Lv, D. Tang and M. Li, Appl. Catal., B, 2017, 206, 344–352 CrossRef CAS.
- K. Iwashina, A. Iwase, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2015, 137, 604–607 CrossRef CAS PubMed.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS PubMed.
- D. Kumar, A. Lee, T. Lee, M. Lim and D.-K. Lim, Nano Lett., 2016, 16, 1760–1767 CrossRef CAS PubMed.
- S. Kim, S. Zhou, Y. Hu, M. Acik, Y. J. Chabal, C. Berger, W. de Heer, A. Bongiorno and E. Riedo, Nat. Mater., 2012, 11, 544–549 CrossRef CAS PubMed.
- P. Kumar, B. Sain and S. L. Jain, J. Mater. Chem. A, 2014, 2, 11246–11253 RSC.
- P. Kumar, A. Kumar, B. Sreedhar, B. Sain, S. S. Ray and S. L. Jain, Chem. – Eur. J., 2014, 20, 6154–6161 CrossRef CAS PubMed.
- P. Kumar, A. Bansiwal, N. Labhsetwar and S. L. Jain, Green Chem., 2015, 17, 1605–1609 RSC.
- P. Kumar, H. P. Mungse, S. Cordier, R. Boukherroub, O. P. Khatri and S. L. Jain, Carbon, 2015, 94, 91–100 CrossRef CAS.
- L.-L. Tan, W.-J. Ong, S.-P. Chai and A. R. Mohamed, Chem. Eng. J., 2017, 308, 248–255 CrossRef CAS.
- A. Razzaq, C. A. Grimes and S.-I. In, Carbon, 2016, 98, 537–544 CrossRef CAS.
- K. C. Kemp, V. Chandra, M. Saleh and K. S. Kim, Nanotechnology, 2013, 24, 235703 CrossRef PubMed.
- L.-Y. Lin, Y. Nie, S. Kavadiya, T. Soundappan and P. Biswas, Chem. Eng. J., 2017, 316, 449–460 CrossRef CAS.
- X. An, K. Li and J. Tang, ChemSusChem, 2014, 7, 1086–1093 CrossRef CAS PubMed.
- R. Gusain, P. Kumar, O. P. Sharma, S. L. Jain and O. P. Khatri, Appl. Catal., B, 2016, 181, 352–362 CrossRef CAS.
- P.-Q. Wang, Y. Bai, P.-Y. Luo and J.-Y. Liu, Catal. Commun., 2013, 38, 82–85 CrossRef CAS.
- J. Yu, J. Jin, B. Cheng and M. Jaroniec, J. Mater. Chem. A, 2014, 2, 3407–3416 RSC.
- H. Liu, H. Zhang, P. Shen, F. Chen and S. Zhang, Langmuir, 2016, 32, 254–264 CrossRef CAS PubMed.
- Y. Pan and M. Wen, Int. J. Hydrogen Energy, 2018, 43, 22055–22063 CrossRef CAS.
- J. Prakash, S. Sun, H. C. Swart and R. K. Gupta, Appl. Mater. Today, 2018, 11, 82–135 CrossRef.
- Z. Zhu, Y. Han, C. Chen, Z. Ding, J. Long and Y. Hou, ChemCatChem, 2018, 10, 1627–1634 CrossRef CAS.
- J. Hou, H. Cheng, O. Takeda and H. Zhu, Angew. Chem., Int. Ed., 2015, 54, 8480–8484 CrossRef CAS PubMed.
- L.-L. Tan, W.-J. Ong, S.-P. Chai and A. R. Mohamed, Appl. Catal., B, 2015, 166–167, 251–259 CrossRef CAS.
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- Y. Jiang, J.-F. Liao, Y.-F. Xu, H.-Y. Chen, X.-D. Wang and D.-B. Kuang, J. Mater. Chem. A, 2019, 7, 13762–13769 RSC.
- A. Ali, D. C. T. Nguyen, K.-Y. Cho and W.-C. Oh, Fullerenes, Nanotubes, Carbon Nanostruct., 2018, 26, 827–836 CrossRef CAS.
- P. Kumar, C. Joshi, A. Barras, B. Sieber, A. Addad, L. Boussekey, S. Szunerits, R. Boukherroub and S. L. Jain, Appl. Catal., B, 2017, 205, 654–665 CrossRef CAS.
- Y. Jiang, J.-F. Liao, H.-Y. Chen, H.-H. Zhang, J.-Y. Li, X.-D. Wang and D.-B. Kuang, Chem, 2020, 6, 766–780 CAS.
- J. Meng, Q. Chen, J. Lu and H. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 550–562 CrossRef CAS PubMed.
- P. Li, Y. Zhou, H. Li, Q. Xu, X. Meng, X. Wang, M. Xiao and Z. Zou, Chem. Commun., 2014, 51, 800–803 RSC.
- L. Lin, H. Peng and Z. Liu, Nat. Mater., 2019, 18, 520–524 CrossRef CAS PubMed.
- K. A. Madurani, S. Suprapto, N. I. Machrita, S. L. Bahar, W. Illiya and F. Kurniawan, ECS J. Solid State Sci. Technol., 2020, 9, 093013 CrossRef CAS.
- Y. Ma, Y. Zheng and Y. Zhu, Sci. China Mater., 2020, 63, 1861–1869 CrossRef.
- X.-Y. Zhang, H.-P. Li, X.-L. Cui and Y. Lin, J. Mater. Chem., 2010, 20, 2801–2806 RSC.
- N. Li, G. Liu, C. Zhen, F. Li, L. Zhang and H.-M. Cheng, Adv. Funct. Mater., 2011, 21, 1717–1722 CrossRef CAS.
- Y. Ban, M. Zhao and W. Yang, Front. Chem. Sci. Eng., 2020, 14, 188–215 CrossRef CAS.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- C. Liu, T. Luo, A. M. Sheveleva, X. Han, X. Kang, S. Sapchenko, F. Tuna, E. J. L. McInnes, B. Han, S. Yang and M. Schröder, Mater. Adv., 2021, 2, 5144–5149 RSC.
- L. B. Hamdy, C. Goel, J. A. Rudd, A. R. Barron and E. Andreoli, Mater. Adv., 2021, 2, 5843–5880 RSC.
- S. Kumar, N. L. Reddy, H. S. Kushwaha, A. Kumar, M. V. Shankar, K. Bhattacharyya, A. Halder and V. Krishnan, ChemSusChem, 2017, 10, 3588–3603 CrossRef CAS PubMed.
- T. Meyer, J. B. Priebe, R. O. da Silva, T. Peppel, H. Junge, M. Beller, A. Brückner and S. Wohlrab, Chem. Mater., 2014, 26, 4705–4711 CrossRef CAS.
- S. Bhattacharyya, S. Kar, B. Kommula and N. S. John, Chem. – Asian J., 2021, 16, 1138–1149 CrossRef PubMed.
- K. Bramhaiah, R. Bhuyan, S. Mandal, S. Kar, R. Prabhu, N. S. John, M. Gramlich, A. S. Urban and S. Bhattacharyya, J. Phys. Chem. C, 2021, 125, 4299–4309 CrossRef CAS.
- R. Bhuyan, K. Bramhaiah and S. Bhattacharyya, J. Colloid Interface Sci., 2022, 605, 364–372 CrossRef CAS PubMed.
- B. Yang, R. Jelinek and Z. Kang, Mater. Chem. Front., 2020, 4, 1287–1288 RSC.
- A. Kalkal, S. Kadian, R. Pradhan, G. Manik and G. Packirisamy, Mater. Adv., 2021, 2, 5513–5541 RSC.
- B. Zhu, B. Lin, Y. Zhou, P. Sun, Q. Yao, Y. Chen and B. Gao, J. Mater. Chem. A, 2014, 2, 3819–3827 RSC.
- M. A. Mahadadalkar, S. B. Kale, R. S. Kalubarme, A. P. Bhirud, J. D. Ambekar, S. W. Gosavi, M. V. Kulkarni, C.-J. Park and B. B. Kale, RSC Adv., 2016, 6, 34724–34736 RSC.
- Y. Zhao, S. Zhang, R. Shi, G. I. N. Waterhouse, J. Tang and T. Zhang, Mater. Today, 2020, 34, 78–91 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |