
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning compatibility and water uptake by protein charge modification in melt-polymerizable protein-based thermosets†
Emil
Andersen‡
abc,
Wui Yarn
Chan‡
 a,
Sarah
Av-Ron
a,
Hursh V.
Sureka
a and
Bradley D.
Olsen
*a
a,
Sarah
Av-Ron
a,
Hursh V.
Sureka
a and
Bradley D.
Olsen
*a
aDepartment of Chemical Engineering, MIT, 25 Ames Street, Cambridge, MA 02142, USA. E-mail: bdolsen@mit.edu
bLEGO System A/S, 16 Kløvermarken, Billund 7190, Denmark
cFaculty of Technical Sciences, Aarhus University, 40 Åbogade, Aarhus 8000, Denmark
First published on 24th January 2022
Abstract
Suppressing the influence of humidity in protein-based materials is central to their use in a variety of applications. It is believed that protein charge plays a key role in water uptake. Therefore, in this work, whey protein was neutralized, supercharged, and superneutralized to examine the effects of protein modification on moisture absorption in protein copolymers. The charge-modified proteins were formulated into thermoset elastomers through a three-step process: methacrylation, complexation with various surfactants, and co-polymerization with n-butyl acrylate. Compatibility of the protein and hydrophobic acrylate monomer can be tuned through changes in surfactant type, ratio between surfactant and protein, and protein charge modification. Using benzalkonium chloride as the surfactant compatibilizer, elastomers with the various modified proteins were prepared using a melt polymerization approach. Acetylation and esterification of whey protein, which neutralize charged functional groups, resulted in the reduction of the proteins’ water uptake relative to unmodified whey. Once incoporated into elastomers, all copolymers regardless of protein modifications have similar moisture contents. However, elastomers with superneutralized proteins demonstrated a lowered mechanical dependence on humidity, presented as a smaller change in elongation at break and tensile strength compared to a copolymer based on non-charge modified whey.
Introduction
The plastic waste crisis has prompted interest in material manufacturing from sustainable feedstocks.1,2 Bio-derived waste streams are an economical solution for substituting fossil-based sources with more sustainable ones at scale; for example, more than 90 million tons (MT) of primary crop residues are yearly generated in the US, with the largest contributor being zein protein from corn. Global yearly whey protein production has also already surpassed 240 MT,3,4 making proteins interesting candidates for material formulations. Pure protein materials are typically brittle and demonstrate poor mechanical properties. Therefore, application of proteins in materials requires modification for increasing their flexibility, commonly through plasticizers. Typical plasticizers used include small molecules such as water5 ethylene glycol, sorbitol,6 and glycerol.7 Plasticizers increase the free volume of the protein, creating a tougher and more malleable material.The mechanical properties of protein-based materials can also be improved by controlling the microphase structure. Natural materials such as silk,8 feathers,9 and teeth10 achieve excellent mechanical properties from hierarchical structures and interfacial hydrogen bonding. Silks have alternate amorphous and crystalline regions along the fibers, making the soft–hard domain interfacial energy critical in achieving high mechanical strength.11 In commercial engineering plastics such as polyurethanes, microphase separation of hard and soft domains has been leveraged to provide excellent and tunable mechanical properties.12,13 Proteins contain many functional groups that enable further functionalization to modulate hierarchal structure, and as a result, control of mechanical properties. Therefore, the self-assembling properties of proteins and their hydrogen bonding nature makes them possible substitutes as the hard component in an engineering plastic.
Because proteins are composed of amino acids that are often charged or polar, proteins are strongly hydrophilic.14 This makes protein-based materials sensitive to water as they are heavily plasticized by water uptake.15,16 As a result, there is a non-zero optimal humidity level for mechanical performance of protein-based materials, limiting applications to within a narrow humidity range.17 Water also acts as a plasticizer in commodity plastics, for example polyesters18 and polyamides,19 where the plasticization is attributed to increased flexibility from weakening the interchain hydrogen bonding of the polymers. The significant sensitivity to humidity in protein-based materials is further compounded by the use of hygroscopic small molecule plasticizers.20
A strategy to limit water uptake in protein-based materials is to modify the protein.21,22 One method is to lower the charge of the protein by modifying functional groups of the polar amino acids that are known to coordinate water.23 Succinylation,24 acetylation,25 and esterification26 are common reactions that modify the protein charged functional groups (Scheme 1). A second strategy is incorporating hydrophobic components in the form of copolymers and plasticizers into the materials.27–29 This second strategy was demonstrated in a previous work on melt polymerizable protein-based thermosets, where surfactant compatibilized mixtures of polymerizable protein macromonomer were copolymerized with a hydrophobic acrylate comonomer, resulting in thermosets containing hydrophobic polymer segments.31 While these materials exhibited lowered water uptake, humidity sensitivity was not eliminated and still posed as a major challenge to material performance.31 However, this synthetic platform offers opportunities for further tuning material properties as various protein modifications and surfactant types can be accommodated.
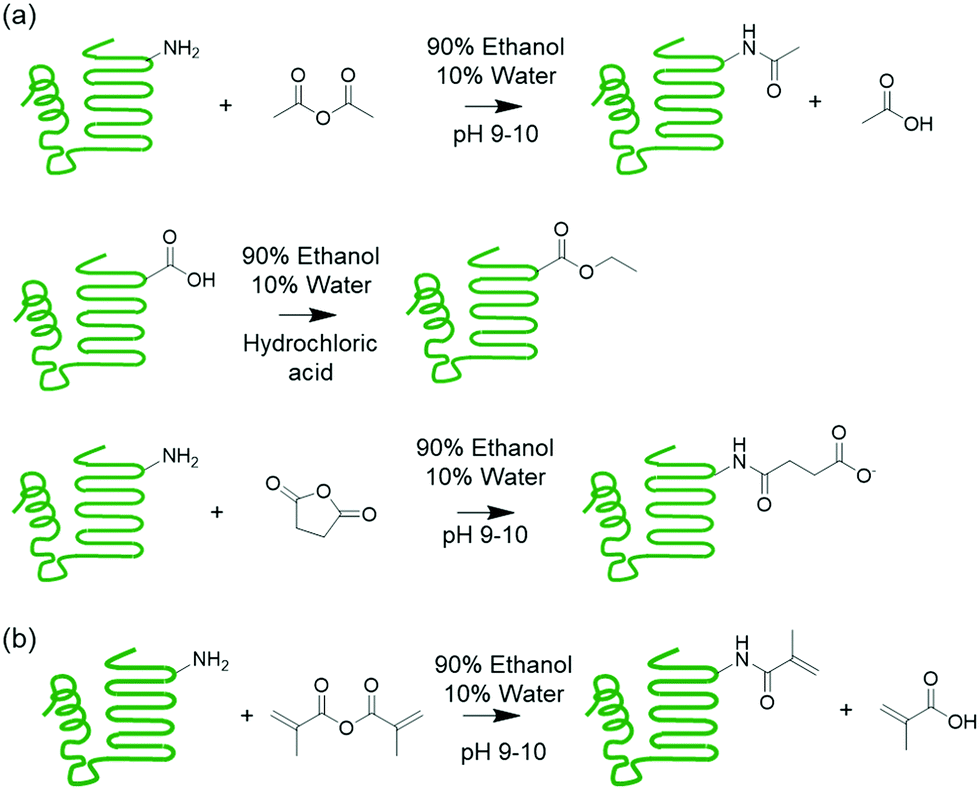 | ||
| Scheme 1 (a) Supercharging of proteins by acetylation, esterification, and succinylation. (b) Methacrylation of protein to install polymerizable groups. | ||
This work explores chemically modifying the charged groups of a protein in the preparation of protein copolymer thermosets using the aforementioned synthetic platform, with a particular focus on understanding surfactant compatibility and the influence of water on mechanical properties. Melt polymerizable thermosets were prepared using whey protein isolate (WPI) and n-butyl acrylate as the model protein and comonomer, and surfactants were used to compatibilize the otherwise immiscible protein–monomer mixtures following the previous procedure. To produce both cationic and anionic proteins with a wide range of net charges, the whey protein was succinylated, acetylated, or esterified. These modifications installed negative or neutral groups on amines, and neutral groups on carboxylic acids. A superneutralized protein was also synthesized through successive acetylation and sucinylation reactions, which reduced the number of ionizable primary amines and carboxylic acids. The effectiveness of anionic, cationic, and nonionic surfactants as compatibilizers was then evaluated. Preparation of crosslinked networks from the ternary mixtures was demonstrated, and the water sensitivity of the resulting materials was explored through exposure to different levels of relative humidity followed by mechanical testing.
Materials and methods
n-Butyl acrylate (n-BA) and acetic anhydride were obtained from Sigma-Aldrich. tert-Butyl peroxyacetate and succinic anhydride were purchased from Acros Organics and Alfa Aesar, respectively. The surfactants QS-44 and Triton XN-45S were gifts from Dow Chemical. Finally, whey protein isolate (WPI), benzalkonium chloride (BAC), and pyridinium chloride were purchased from Bipro USA, MP Biomedicals, and Spectrum Chemical Mfg Corp, respectively.Protein modification
Charge-modified whey proteins were prepared using acetylatation, esterification, or succinylation reactions by heterogeneous suspension reactions. Protein acetylation was performed by suspending 10 g protein in 100 mL 90/10 v/v ethanol/water solution. Then 300 μL sodium hydroxide solution in water (30 wt%) and 1.125 mL acetic anhydride were added to the protein solution while stirring. Protein succinylation was performed following the same procedure as protein acetylation except for the addition of 1.125 g succinic anhydride instead of acetic anhydride to the protein suspension. Protein esterification was performed following a procedure adapted from Sitohy et al.32 10 g whey protein was suspended in 100 mL ethanol, 1 mL water, and 16.25 mL concentrated hydrochloric acid (∼37%). The three modification reactions were performed overnight, and the modified proteins were recovered from the suspension by vacuum filtration. The reaction products were washed with 3 × 30 mL ethanol. Superneutral protein was first acetylated, then lyophilized, and finally esterified.After charge modification, some of the modified proteins were methacrylated using methacrylic anhydride with a procedure modified from previous work.30 A stirred 10 wt% protein suspension was prepared in 90/10 v/v ethanol/water and adjusted to pH 9–10 using sodium hydroxide. Then methacrylic anhydride was added with an anhydride-to-protein mole ratio of 5, calculated based on the molecular weight of β-lactoglobulin, and stirred overnight. Finally, the product was washed with 3 × 30 mL ethanol per 10 g starting material to remove the methacrylic acid and salt before lyophilization for 48 h and was stored in an enclosed bottle at −20 °C when not in use.
Estimating the protein solubility in water
Water solubility of the modified protein was determined by gravimetry. A 1 mg mL−1 protein solution was prepared by dissolving 20.0 ± 3.0 mg protein in ultrapure water in a scintillation vial, followed by titration with sodium hydroxide or hydrogen chloride to pH 7.0 ± 0.1. This solution was stirred overnight, and the non-dissolved protein was transferred to a separate scintillation vial. Both vials were lyophilized for 48 h, then the mass of the scintillation vial and protein were noted. The protein was then washed out of both scintillation vials with deionized water, and the vials were lyophilized for 24 h and weighed for the reference mass of the vial.Estimating the degree of modification
Determination of the degree of acetylation, esterification, succinylation, and methacrylation was done using liquid chromatography-mass spectrometry (LC-MS) on proteins dissolved in water at 0.5 mg mL−1 and pH = 7, using a method described in previous work.30 The mass spectral deconvolution was performed with molecular weight limits of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–25000 Da, and 20 maximum charges. Charge modifications were obtained from non-methacrylated proteins (WheyAc, WheyEt, WheySA). As methacrylation was performed after charge modification, the level of charge modification was assumed to be unchanged from the precursor. Averages and variances were calculated from all species with relative abundance >20% relative to the most abundant species. Standard errors reflect batch-to-batch variation across three replicates.
000–25000 Da, and 20 maximum charges. Charge modifications were obtained from non-methacrylated proteins (WheyAc, WheyEt, WheySA). As methacrylation was performed after charge modification, the level of charge modification was assumed to be unchanged from the precursor. Averages and variances were calculated from all species with relative abundance >20% relative to the most abundant species. Standard errors reflect batch-to-batch variation across three replicates.
Estimating protein surface charge
Surface charge of the modified proteins was evaluated by zeta-potential. Data was acquired on protein solutions in ultrapure water at soluble protein concentrations of 0.5 mg mL−1. Solutions were titrated with 0.1 M HCl and NaOH solutions to pH = 7. After stirring overnight, solutions were filtered, transferred to a polycarbonate capillary cell, ultrasonicated for 15 seconds, and immediately inserted into the Zetasizer Nano ZS90, with the laser attenuation adjusted for 100% transmission.Protein–surfactant complex preparation and copolymerization
Protein–surfactant complexes were prepared, and copolymerization of the protein and monomer was performed, using a method modified from previous work.17 Proteins and surfactants were co-dissolved in water and lyophilized to form protein–surfactant complexes. No intermediate purification steps were performed to separate the soluble and insoluble protein fractions. The dried protein–surfactant complexes were mixed 1:1 by mass with n-butyl acrylate at 110 °C, then melt polymerized in a hot press at 121 °C with t-butyl peroxyacetate at monomer:initiator mole ratios of 80:1. After removing the copolymers from the mold, they were vacuum dried for 24 h at 60 °C to remove any residual monomer. The pure protein, protein:surfactant complex, and copolymer were equilibrated at 4, 20, 35, 50, 70, or 90% relative humidity (RH) at 23 °C for at least 72 h prior characterization. 4% RH was achieved in a desiccator with Drierite while 20–90% RH was controlled in a Memmert HPP 110 climate control chamber. In all analyses for which humidity is not an independent variable, samples were equilibrated at 50% RH.
Moisture absorption was determined gravimetrically, after equilibrating the materials at the specified relative humidity levels. A 24-factorial design33 (with acetylation, esterification, methacrylation and surfactant complexation as factors) was used to investigate the influence of the chemical modifications and surfactant complexation on the water uptake of the pure proteins and protein–surfactant complexes.
Gel fraction and swelling ratio for copolymers were quantified by weighing vacuum dried samples followed by submersion in dimethyl sulfoxide (DMSO) overnight. The swollen samples were weighed and reported as ratio of swollen mass over dry mass. The samples were then vacuum dried for 24 h at 60 °C and weighed to provide gel fraction, reported as the dry mass before over dry mass after equilibration in DMSO.
Miscibility evaluation
Miscibility of different modified whey proteins with n-butyl acrylate after complexation with various surfactants was determined by mixing the protein–surfactant complex with n-butyl acrylate at 110 °C. Samples that formed a continuous opaque or clear phase were labeled miscible while phase separated clear n-butyl acrylate solution and opaque protein–surfactant complex were labeled immiscible.Mechanical evaluation
Tensile tests were performed according to previous work at a strain rate of 100% min−1.17Chemical characterization
Attenuated total reflectance Fourier-transform infrared spectroscopy (ATR-FTIR) spectra were collected from 700–4000 cm−1 on a Bruker Alpha II FTIR spectrometer with a Diamond Crystal ATR, in Opus v.7.8. The ratio between protein and n-butyl acrylate in the copolymer was evaluated by the peak area ratio of the carbonyl band centered at 1731 cm−1 and the amide I band at 1641 cm−1. The α-helix and β-sheet contents of the protein in the copolymer were evaluated by deconvolution, decomposing the amide I band into two peaks centering at ∼1630 and ∼1652 cm−1. Deconvolution was performed by fitting two Gaussian functions to the background subtracted 1600–1700 cm−1 region, with the peak positions, heights, and widths as fitting parameters (Fig. S5, ESI†). The ∼1630 cm−1 peak was assigned to β-sheet, while the ∼1652 cm−1 peak was assigned to α-helix and unordered structures, and the relative areas are presented.Morphological characterization
Low-vacuum Scanning Electron Microscopy, Energy-Dispersive X-ray Spectroscopy (SEM-EDX) was performed at a working distance of 8.5 mm and an accelerating voltage of 10 kV with a variable pressure secondary electron detector (VPSE G3). The sample was imaged and scanned by EDX at 1000× and 3000× magnification in an 800 × 600 pixel resolution with 100 μs pixel dwell time and 16 total scans. Atomic force microscopy (AFM) images were captured using a MikroMasch HQ:NSC16/AL BS tip, and phase images were postprocessed using first order flattening corrections.Small-angle and wide-angle X-ray scattering (SAXS and WAXS) data were acquired in transmission mode using a Rigaku 002 microfocus X-ray source with Cu Kα radiation (0.154 nm) with a sample-to-detector distance of 109.1 mm for WAXS and 950 mm for SAXS. Data was acquired using a DECTRIS Pilatus 300 K hybrid panel array with an exposure time of 5 min for SAXS and 2 min for WAXS. Two-dimensional diffraction images were background corrected, azimuthally averaged, and plotted as one-dimensional scattering profiles.
Results and discussion
The effects of charged functional groups and net charge on absorption of water by the materials were investigated using whey protein isolate as a model protein, as it contains the full range of hydrophilic charged, polar uncharged, amphiphatic, and hydrophobic amino acid residues. To obtain proteins with highly net negative, less net negative, and highly net positive charges, a set of reactions were performed to modify the ionizable amino and carboxyl side chain groups. The amine-anhydride succinylation (WheySA) and acetylation (WheyAc) reactions installed negatively charged succinate carboxylic acids and uncharged amide groups, respectively, onto the amines, resulting in an increase of net negative protein charges (Scheme 1). Esterification (WheyEt) installed uncharged ethyl esters onto carboxylic groups on aspartic and glutamic acid residues, reducing the net negative charge of the whey protein. Ethanol was used as both reactant and solvent to produce highly esterified proteins. A 90/10 v/v ethanol/water mixture was used as solvent for all protein modifications, which allows all reaction products to be recovered by simply filtering the protein suspension. Superneutralized whey protein was prepared by first acetylating, and then esterifying the protein, neutralizing both amines and carboxylic groups. The supercharged and superneutralized proteins were methacrylated to install polymerizable functional groups onto proteins to allow copolymerization with an acrylate comonomer. The extents of protein reactions were estimated by quantifying the number of modifications on β-lactoglobulin, the primary protein species in whey, via LC-MS. Each succinylation, acetylation, esterification, and methacrylation reaction results in molar mass increases of 100, 73, 28, and 68 Da respectively, enabling extents of reaction to be quantified via mass spectral deconvolution (Table 1). According to the amino acid sequence of bovine β-lactoglobulin, a total of 16 amino groups from lysine and the N-terminus, 3 guanidino groups from arginine, and 27 carboxylic groups from aspartic acid, glutamic acid and the C-terminus are expected.34 All charge reactions result in large numbers of modified groups, with esterification having the largest number, consistent with the higher abundance of carboxylic acid groups. As the anhydride reactions do not go to full completion for succinylated and acetylated proteins, the remaining few amine groups allow for further reactions with methacrylic anhydride to install the polymerizable groups. High variances in extent of modification were observed due to the abundance of reactive functional groups and non-specificity of the reactions (Table 1 and Fig. S1, S2, ESI†). This was also previously observed for succinylated proteins evaluated using mass spectral deconvolution,35 and for esterification reactions, where the heterogeneity manifested in widening of protein bands in isoelectric focusing gels.36 Variances are expected to decrease with increasing molar excess of reactants, as proteins converge on the maximum degree of modification.35| Whey5MA | WheyAc5MA | WheyEt5MA | WheySA5MA | WheyAcEt5MA | |
|---|---|---|---|---|---|
| a WheyAcEt, WheyEt5MA, and WheyAcEt5MA did not run in the LC-MS due to low solubilities. | |||||
| Modification (Ac, Et, SA) | None | 16.0 ± 0.30 | 21.0 ± 0.17 | 12.1 ± 0.19 | NDa |
| Variance | None | 19.6 ± 6.36 | 142.4 ± 6.00 | 25.7 ± 0.57 | NDa |
| Methacrylation | 2.25 ± 0.10 | 1.63 ± 0.09 | NDa | 3.21 ± 0.04 | NDa |
| Variance | 0.10 ± 0.01 | 0.42 ± 0.07 | NDa | 1.22 ± 0.19 | NDa |
Solubility profiles and zeta-potentials of modified proteins reflect changes in charged functional groups from chemical modification (Fig. 1A and B). The non-denatured whey protein isolate has a high water solubility and is comprised mainly of β-lactoglobulin, a globular protein with an isoelectric point of 5.2.37 Whey protein was therefore negatively charged at pH 7, as were other modified whey proteins except for esterified whey (WheyEt) (Fig. 1B). WheyEt maintained similar high solubility at pH 7, even though some reports have shown decreases in solubility ethyl esterified whey protein, which was attributed to the more hydrophobic ethyl esters.37 However, esterification generally improves solubility over the acidic pH range due to the increased isoelectric points, and net effects on solubility is likely driven by the balance between introduction of hydrophobic groups and charge effects. On the other hand, succinylation, acetylation, and methacrylation resulted in the formation of insoluble precipitates, even though succinylation and acetylation were also previously reported to increase protein water solubility.38 Solubility may be affected by multiple factors, including net charge, charge density, and the partial unfolding of the proteins in ethanolic solutions.39 Therefore, the interplay between charge and structure effects, which are highly dependent on the protein environment and processing history, may have contributed to differences in observed solubilities. Proteins that underwent modifications on both carboxylic acids and amines (WheyAcEt, WheyEt5MA, and WheyAcEt5MA) had the lowest solubility, and the reduced solubility prohibited characterization by MS deconvolution due to low signal-to-noise ratio (Fig. S3, ESI†).
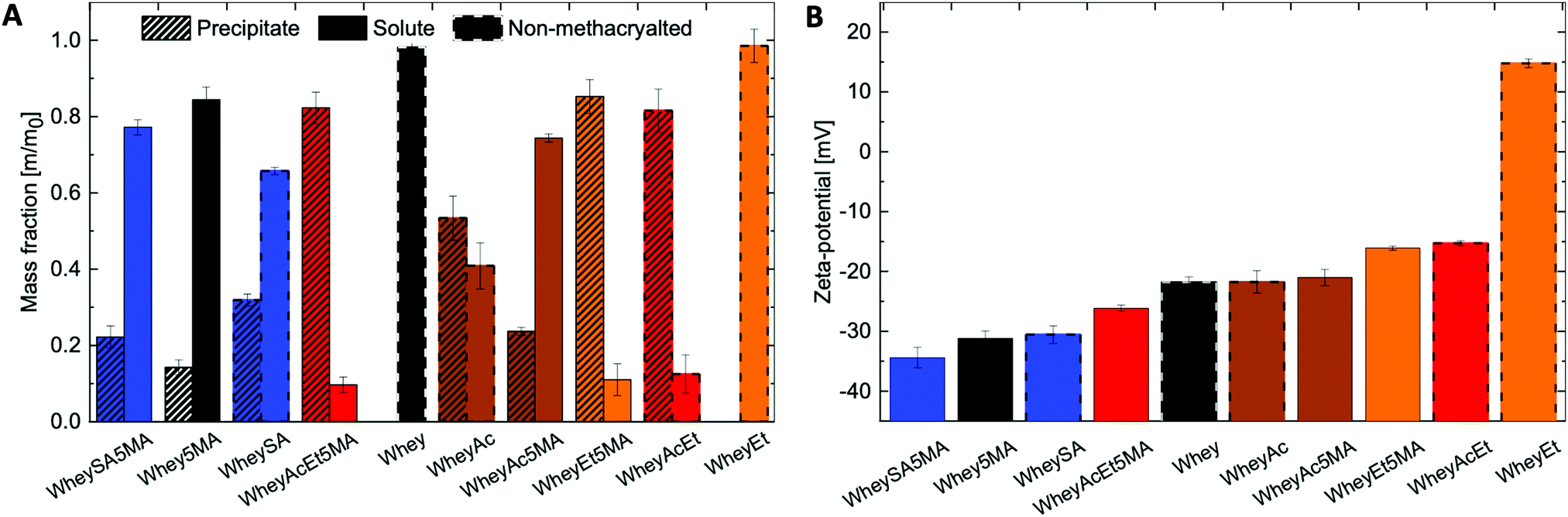 | ||
| Fig. 1 (A) Fractions of soluble and insoluble modified proteins in water, at a concentration of 1 mg mL−1 and pH of 7, (B) zeta-potential characterization of soluble protein fractions at pH 7 in water, presenting changing protein surface charge. | ||
In general, succinylated proteins had large net negative charges, acetylated proteins had less net negative charges, and esterified proteins had net positive charges at pH 7, as measured by electrophoresis zeta-potential (Fig. 1B). However, not all modified proteins follow the expected trend in net charge based on the type of modification (note the large net negative charge of WheyAcEt5MA). This may be due to the characterization of water-soluble fractions only during zeta-potential measurements, which can artificially increase the charge magnitude if the water-soluble fractions are not representative of the entire protein population. In addition, the protein surface charge may deviate from the net protein charge, as a result of amphiphilic behavior burying charged groups.21,40
Thermoset elastomers comprised of proteins as the reinforcing component and poly(n-butyl acrylate) as the rubbery component were prepared with methacryalted supercharged and superneutralized proteins following a method described in previous work,17 where the surfactant performs the role of a plasticizer and a compatibilizer. Complexation by co-lyophilizing the protein with a model surfactant, benzalkonium chloride (BAC), enabled the proteins regardless of charge state to be softened at elevated temperatures in the absence of solvent. As no intermediate purification step was performed to separate water insoluble proteins from soluble fractions, the protein–surfactant complexes contained both. They were then mixed with the comonomer to form miscible three component mixtures with final 1:1:2 protein:surfactant:n-butyl acrylate mass ratios. Mixtures were then melt copolymerized via free radical polymerization to form the elastomers, where methacrylated groups on proteins enable them to be incorporated into the network. The average number of functionalized methacrylate groups ranged from 1.6 to 3.2 across the various modified proteins (Table 1), enabling networks with similar crosslink densities to be prepared, as determined from swelling experiments (Table 2). Unlike copolymers that swell in good solvents, blends prepared by polymerizing n-butyl acrylate in the presence of non-methacrylated proteins and surfactant were previously shown to dissolve in DMSO, indicating that the methacrylated proteins are functioning as macrocrosslinkers.17 In addition, since poly(n-butyl acrylate) is a liquid at room temperature, the copolymers’ tensile strength and mechanical integrity are derived from the protein domains. Blends were also previously observed to have significantly lower elongation at break than copolymers, suggesting that the protein and polyacrylate domains have poor adhesion, and that the covalent bonds linking the two play an important role in stress transfer.17 Tensile properties of copolymers after equilibration at 50% RH exhibit dependence on protein modification, as shown in Fig. 2. A tradeoff between tensile strength and elongation at break was observed for materials prepared with the negatively charged proteins (Whey5MA, WheySA5MA, WheyAc5MA, WheyAcEt5MA), where copolymers with lower tensile strength exhibited higher elongation at break. On the other hand, the WheyEt5MA-based copolymer has distinctly smaller elongation at break and lower tensile strength, because of protein–surfactant complex incompatibility with n-butyl acrylate. The immiscible mixture of cationically modified protein, cationic surfactant, and n-butyl acrylate was observed to undergo macrophase separation. Because of the incompatibility, WheyEt5MA is excluded in mechanical comparisons moving forward.
| Copolymer | Swelling [ms/md] | Gel [m0/md] |
|---|---|---|
| Whey5MA | 1.64 ± 0.02 | 0.73 ± 0.003 |
| WheyAc5MA | 2.65 ± 0.14 | 0.64 ± 0.004 |
| WheyEt5MA | 2.17 ± 0.06 | 0.70 ± 0.040 |
| WheySA5MA | 2.85 ± 0.12 | 0.59 ± 0.003 |
| WheyAcEt5MA | 1.87 ± 0.01 | 0.70 ± 0.001 |
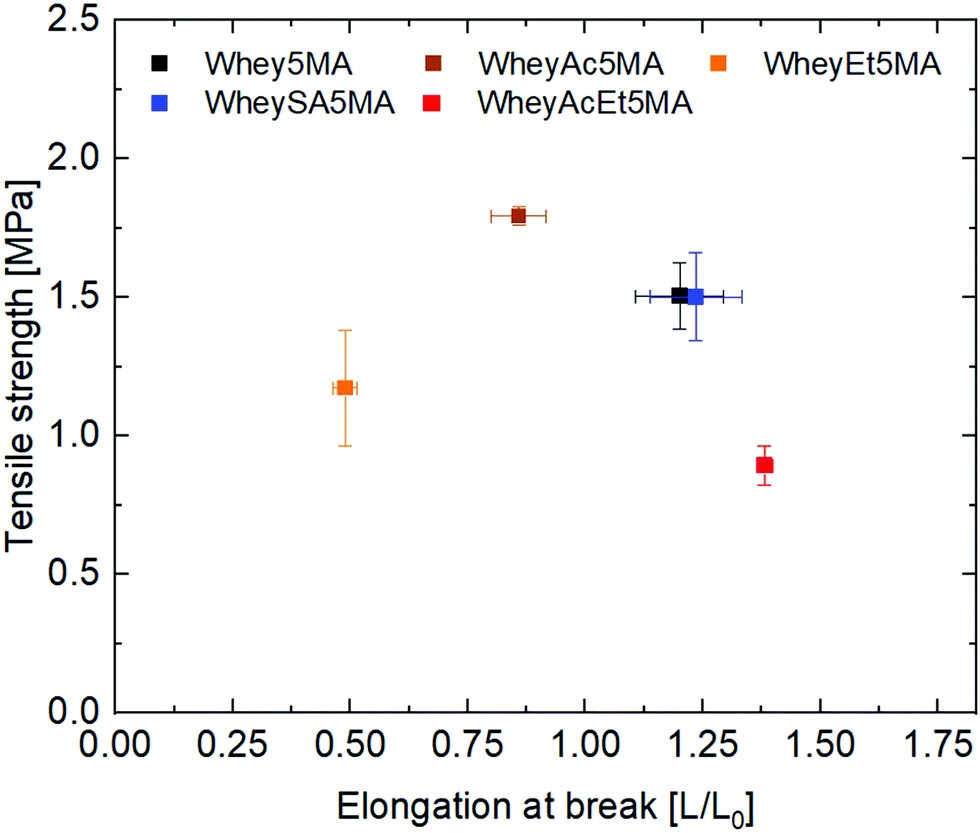 | ||
| Fig. 2 Ultimate tensile strength (Y) and elongation at break (X) of copolymers with methacrylated protein:benzalkonium chloride:n-butyl acrylate mass ratios of 1:1:2. All materials equilibrated at 50% RH. | ||
The influence of electrostatic interactions on material formulation and properties was investigated using a panel of ionic surfactants, demonstrating a broad set of protein–surfactant combinations suitable for material preparation (Table 3). Melt-polymerizable and dispersible mixtures of protein, surfactant, and n-butyl acrylate could be prepared not only with benzalkonium chloride as shown in previous work,17 but also a range of cationic, anionic, and non-ionic surfactants (Table 3 and Table S1, ESI†). However, polymeric materials prepared with non-ionic surfactants were too brittle to handle and test mechanically, suggesting weak interaction between charged protein, uncharged surfactant, and hydrophobic n-butyl acrylate. Miscibility studies shown in Table 3 further demonstrate that the presence of charged groups in proteins improved miscibility of the protein–ionic surfactant complex with n-butyl acrylate. Theoretical net protein charges at pH 7 were estimated based on the extent of protein modification (Table S2, ESI†). Immiscible mixtures were observed more frequently when the protein had net positive charge, e.g. for WheyAcEt and WheyEt5MA with calculated net positive charges of 6.2 and 14.2, respectively. In general, modification of both amines and carboxylic acids lead to reduced propensities to form miscible protein–surfactant–monomer mixtures. However, there are no clear correlations between the type of surfactant (cationic or anionic) and protein charge (positive or negative at pH 7), suggesting net charge and compatible charges were not the dominating factors for determining miscibility in the ternary system. In addition, although methacrylated and non-methacrylated species have relatively small differences in number of modified amine groups and thus have similar theoretical charges, their dispersibilities varied, e.g. WheyEt and WheyAcEt vs. their methacrylated counterparts. This indicates that the presence of charged groups alone was not the defining factor in obtaining a dissolvable protein–surfactant complex.41 In general, the miscibility panel (Table 3) confirms that the protein–surfactant complexation strategy allows protein-based resins to be processed, polymerized, and cured in the absence of solvents. This strategy is generalizable to various charged proteins and surfactants, regardless of surfactant and protein charge type.
:surfactant complex and n-butyl acrylate
| Protein:surfactant mass ratio |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Whey | WheyAc | WheyEt | WheySA | WheyAcEt | |||||||||||
| a Cation surfactant. b Anionic surfactant. | |||||||||||||||
| Surfactant | 1:1 |
1:2 |
1:3 |
1:1 |
1:2 |
1:3 |
1:1 |
1:2 |
1:3 |
1:1 |
1:2 |
1:3 |
1:1 |
1:2 |
1:3 |
| Benzalkonium chloridea | Y | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | Y | Y | Y | Y |
| Cetylpyridinium chloridea | N | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | Y | N | N | Y |
| QS44b | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | N | N | N |
| XN45Sb | Y | Y | Y | N | Y | Y | Y | Y | Y | N | Y | Y | Y | N | Y |
| Protein:surfactant mass ratio |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Whey5MA | WheyAc5MA | WheyEt5MA | WheySA5MA | WheyAcEt5MA | |||||||||||
| Surfactant | 1:1 |
1:2 |
1:3 |
1:1 |
1:2 |
1:3 |
1:1 |
1:2 |
1:3 |
1:1 |
1:2 |
1:3 |
1:1 |
1:2 |
1:3 |
| Benzalkonium chloridea | Y | Y | Y | Y | Y | Y | N | N | Y | Y | Y | Y | Y | Y | Y |
| Cetylpyridinium chloridea | N | Y | Y | N | Y | Y | N | N | N | N | N | Y | N | Y | Y |
| QS44b | Y | Y | Y | N | Y | Y | Y | Y | Y | Y | Y | Y | N | Y | Y |
| XN45Sb | Y | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | N | Y | Y | Y |
Cationic benzalkonium chloride enabled miscible mixtures of protein–surfactant complexes and n-butyl acrylate at a 1:1 ratio to be prepared for most methacrylated and charge modified proteins, except for WheyEt5MA (Table 3). The compatibility allowed free-radical polymerization to form stable crosslinked copolymers of protein and n-butyl acrylate. Based on the initial reactant ratios, the copolymer is expected to be made up of 25 wt% protein, 25 wt% benzalkonium chloride, and 50 wt% poly(n-butyl acrylate). However, during the molding and polymerization process at high temperature and pressure, some n-butyl acrylate was observed to discharge from the polymerization mixture. The extent of monomer loss varies in materials prepared from different modified proteins and is particularly prominent in formulations containing WheyEt5MA. The variability observed was inferred to be a manifestation of compatibility differences between the protein–surfactant complexes and n-butyl acrylate, as the discharge may be caused by syneresis, which was also observed in polymer–diluent systems where the diluent has low compatibility with the growing network.42 In addition, the molding conditions may have shifted phase boundaries such that some of the initially miscible mixtures became unstable. As discharge of the volatile monomer leads to a lower n-butyl acrylate to protein ratio than the initial feed ratio, the acrylate content relative to protein was used as a proxy to measure compatibility. Protein to poly(n-butyl acrylate) relative ratios in the final copolymers were determined by ATR-FTIR, by the area of amide I band (1647 cm−1) relative to the ester carbonyl peak (1737 cm−1) (Fig. 3A and B). Comparisons of relative n-butyl acrylate content suggest that among all crosslinked samples, superneutralized whey was the most compatible in the three component mixture, while acetylated, esterified, and succinylated whey were less compatible than the protein without charge modification. The most neutralized, methacrylated, and charge-modified protein, WheyAcEt5MA, contains the most poly(n-butyl acrylate) in the final copolymer compared to the other methacrylated proteins (Fig. 3A and B). The compatibility of protein–surfactant complex and n-butyl acrylate was therefore inferred to be highest when the protein is least charged. While the addition of large numbers of negatively charged groups in succinylated protein WheySA5MA would be expected to increase favorable interactions with the cationic surfactant, the protein–surfactant–acrylate mixture was revealed to be less miscible than the non-charge modified Whey5MA (Fig. 3A and B). The effect of charge modification on compatibility was also studied in blend materials, where non-methacrylated proteins were used in polymerization mixtures (Fig. S4, ESI†). The incompatibility observed for blends containing positively charged WheyEt was even more pronounced. These studies suggest that when a positively charged surfactant was used as compatibilizer, large protein charge magnitudes lead to higher degrees of incompatibility, with net positive protein charges having larger detrimental impacts on miscibility than negative charges. However, charge effects on compatibility may also be convoluted by changes in protein secondary structure from charge modification.
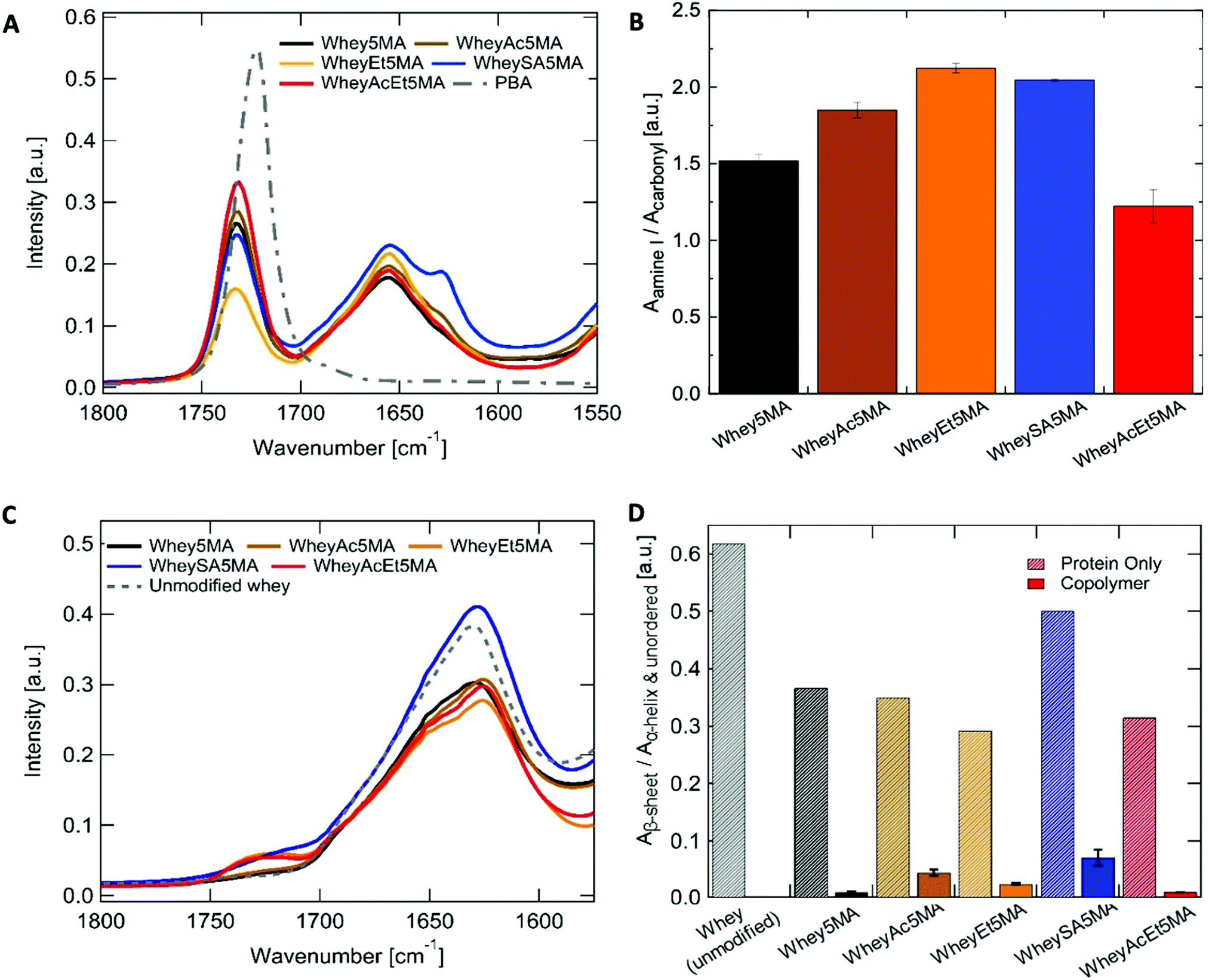 | ||
| Fig. 3 (A) ATR-FTIR spectra of the copolymer (WPI:BAC:n-BA, 25:25:50 expected based on feed) and poly(n-BA) homopolymer. (B) Ratios between protein and poly(n-BA), measured as area ratios of the protein amide I band (centered at 1647 cm−1) to the poly(n-butyl acrylate) carbonyl peak (centered at 1737 cm−1), n = 3. (C) ATIR-FTIR spectra of protein powder before and after charge modifications and methacrylation. (D) Amide I band in the protein powder and copolymer spectra deconvoluted to obtain ratio of the low frequency (∼1630 cm−1, β-sheet) to the high frequency component (∼1652 cm−1, α-helix and unordered structures). N = 1 for protein powders, N = 5 for copolymers. | ||
Whey protein underwent structural changes and denaturation during modification, thermal processing and free-radical copolymerization. After charge modification and methacrylation, the proteins retain most of their highly β-sheet enriched character, as indicated by the peak maxima at ∼1630 cm−1 (Fig. 3C and Table S3, ESI†). Deconvolution of the amide I peak enables evaluation of secondary structure changes (Fig. S5, ESI†) through comparisons of peak area ratios between the low frequency band (∼1630 cm−1), assigned to β-sheet, to the high frequency band (∼1652 cm−1), assigned to α-helix and unordered structures. β-Sheet content decreased for all modified proteins when compared to whey protein. In addition, peak shifts were also observed for the low frequency β-sheet band from 1628 cm−1 for unmodified whey protein to 1623–1625 cm−1 for the charge modified proteins (Table S3, ESI†), which may be due to stronger intermolecular bonds and protein aggregation.43 On the other hand, more pronounced disruption of the protein's predominant β-sheet structure was observed in copolymerized methacrylated proteins, where features corresponding to unordered or α-helix structures (∼1652 cm−1) increased, shifting the amide I peak maxima to higher frequencies after melt polymerization (Fig. 3A). The ∼1630/1652 cm−1 peak area ratios were significantly smaller in all copolymers when compared to the modified protein powder (Fig. 3D). This may be attributed to protein denaturation caused by surfactant complexation, mixing and heating. In addition, structural changes may also be caused by incorporation of protein into the polyacrylate network, as protein denaturation during reactions has been reported in crosslinking reactions of protein by methacrylation.44 Variations in the ∼1630/1652 cm−1 peak area ratios indicate that protein structures differ as a function of charge modification, as the ratio was notably larger for WheySA5MA protein powder and copolymer compared to other modified proteins. As secondary structure differences do not exhibit the same trend as compatibility (Fig. 3B and D), protein structural effects likely do not dominate differences in compatibility.
The presence of microphase separated hard and soft domains in the crosslinked materials was confirmed via AFM (Fig. 4). Phase images show distinct light (poly(n-BA)) and dark (protein) phases as well as differences in domain size and shape across modifications. Whey5MA and WheyAcEt5MA appear the most homogeneous, while WheyAc5MA and WheySA5MA are clearly microphase separated with sharper interfaces. These observations are consistent with the FTIR results, where WheyAcEt5MA was inferred to form the most homogeneous protein–surfactant–monomer mixtures, suggesting that proteins that are more compatible with the monomer mixture are also more miscible in the polymerized polyacrylate phase. In SEM-EDX, embedded sodium chloride (Fig. S6 and S7, ESI†) and surface aluminum impurities introduced during blending and thermal processing were also revealed. Embedded sodium chloride was also observed by AFM and is likely a product of counterions released upon complexation between the surfactant and protein. Despite the observation of clear phase separation by AFM, SAXS (Fig. S8, ESI†) shows a lack of well-defined domain periodicities in the tens of nanometers range. A WAXS peak at 2.3 nm−1 was observed, which was also previously reported for surfactant-containing complexes and copolymers.31 The observed 14 nm−1 peak can be attributed to alkyl tail–tail distance, typically observed in bilayers observed both in pure benzalkonium chloride and poly(n-butyl acrylate). All structural analyses imply successful compatibilization between the protein–surfactant complex and poly(n-butyl acrylate).
 | ||
| Fig. 4 Atomic force microscope phase angle images of (A) Whey5MA, (B) WheyAc5MA, (C) WheyEt5MA, (D) wheySA5MA, (E) WheyAcEt5MA presenting microphase separation between protein and copolymer. * Sub-micron dark domains in WheyAcEt5MA are NaCl crystals. | ||
Influence of protein modification on water uptake and mechanical properties
Effects of chemical modification and surfactant blending on protein moisture absorption were investigated with a particular focus on acetylation and esterification, where the reactions reduced the number of protein charged groups. Fig. S9 (ESI†) shows the moisture absorption of all modified and unmodified proteins and their surfactant complexed counterparts at various relative humidity levels. A 24 factorial design experiment was analyzied using the Yates algorithm,33 with acetylation, esterification, methacrylation, and surfactant complexation as factors, and moisture absorption as the response. Table 4 shows the primary influence of each factor on water uptake, normalized by the average water uptake of pure whey. Fig. S10–S12 (ESI†) show the primary, secondary, and tertiary factor interaction estimates at each relative humidity. Acetylation and esterification have large effects (Table 4), and are able to significantly lower the water uptake of pure protein. However, the two-way interaction results suggest that these modifications have smaller effects when the protein is complexed with surfactant (Fig. S11, ESI†). Methacrylation and surfactant complexation, on the other hand, have relatively limited effects on moisture absorption. Water uptake of charge modified proteins and surfactant complexed proteins were in some cases significantly different (Fig. S9, ESI†), for example, water uptake of WheySA protein was lower than that of non-charge modified whey before surfactant complexation, but much higher when in a protein–surfactant complex. The variation in effectiveness of charge modifications may arise from the plasticizing effect of the surfactants, effectively exposing available hydrophilic groups in the proteins for the water to inhabit near the binding site of the surfactant.5,45| Factor | Water uptake, relative to pure whey [%] |
|---|---|
| Acetylation (Ac) | −6.4 ± 0.3% |
| Esterification (Et) | −5.4 ± 0.3% |
| Methacrylation (5MA) | 1.0 ± 0.2% |
| Surfactant complex. (surf) | 0.8 ± 0.6% |
When the protein–surfactant complex was molded with the hydrophobic monomer, a significant drop in water uptake was observed, e.g. Whey5MA with a moisture absorption reduction from 21.6 wt% to 8.2 wt% at 90% RH (Fig. 5). The significant decrease is mostly due to the copolymer comprising 50% hydrophobic copolymer. A 50% reduction in water uptake would be expected assuming the poly(n-butyl acrylate) took up insignificant amounts of water, but a reduction of more than 50% was observed. The enhanced hydrophobicity from incorporating n-butyl acrylate may be due to the promotion of hydrophobic interactions from crosslinking between the protein and poly(n-butyl acrylate) and the burial of hydrophilic protein functional groups.46 Compared to the protein and protein–surfactant complexes, the water uptake for the various copolymers are more similar to one another (Fig. 5 and Fig. S9, ESI†), regardless of the type of protein charge modification, suggesting that the effects from copolymerizing with the hydrophobic polyacrylate are dominant. This may stem from the diminished effects of protein modification once the proteins are complexed with surfactants, as alluded to in the 24 factorial water uptake study.
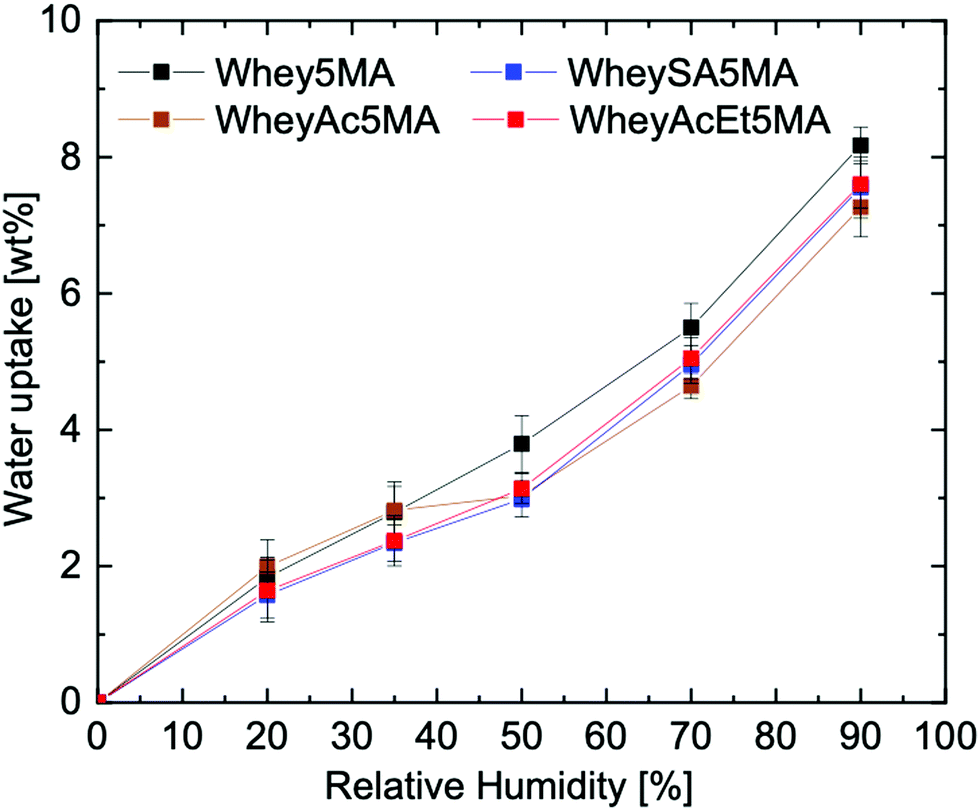 | ||
| Fig. 5 Water uptake of copolymer, protein:surfactant:n-butyl acrylate 25:25:50. | ||
While protein modifications resulted in minor changes in humidity uptake, they altered the humidity sensitivity of the copolymer mechanical properties substantially. Generally, the ultimate tensile strength decrease while elongation at break increase with increasing humidity levels (Fig. 6A), except below 20% RH, where many of the copolymers exhibited extraordinarily brittle behavior. Non-charge modified Whey5MA was significantly plasticized between 4–50% RH, as shown in larger changes in tensile properties with humidity, while at higher relative humidity, it presented stable mechanical performance. Reduced influence of humidity was observed for superneutralized WheyAcEt5MA, as the elongation at break only changed significantly between 4–20% RH. Between 35–90% RH, this material does not follow the general trend of increasing elongation at break with humidity, but instead have mechanical properties that remain relatively constant. On the other hand, the supercharged WheySA5MA was significantly plasticized in the entire humidity range 4–90% RH, presenting an increased humidity sensitivity from increased protein charge density. Copolymer mechanical property dependence on humidity was also quantified by calculating variance of elongation at break and tensile strength from 20–90% RH for each modified protein (Fig. 6B), with the superneutralized WheyAcEt5MA having the lowest variance due to its reduced humidity sensitivity. Large variances are a result of the significant plasticizing effect of water on the protein in the copolymers; however, trends in mechanical property dependence on humidity may not be fully attributable to single factors, as mechanical properties can be influenced by a range of chemical and structural differences including secondary structure, water content, and comonomer content. Acetylated WheyAc5MA had the largest variance, even though it is less negatively charged compared to WheySA5MA, suggesting that the charge itself may not be the dominating factor influencing humidity sensitivity, considering that the protein contents and secondary structures in these copolymers were comparable (Fig. 3B and D). However, the large reduction in mechanical property sensitivity observed for WheyAcEt5MA copolymer even though its water uptake is similar to the others suggest that its protein-rich domains are less susceptible to water plasticization. This is potentially due to greater phase mixing with the hydrophobic poly(n-butyl acrylate), which is supported by the featureless AFM images (Fig. 4) and the much lower stiffness of the WheyAcEt5MA copolymer. Since the polyacrylate is a low Tg polymer, greater phase mixing would reduce the stiffness of the protein domains, but at the same time reduce the moisture induced mechanical property variability due to its hydrophobicity.
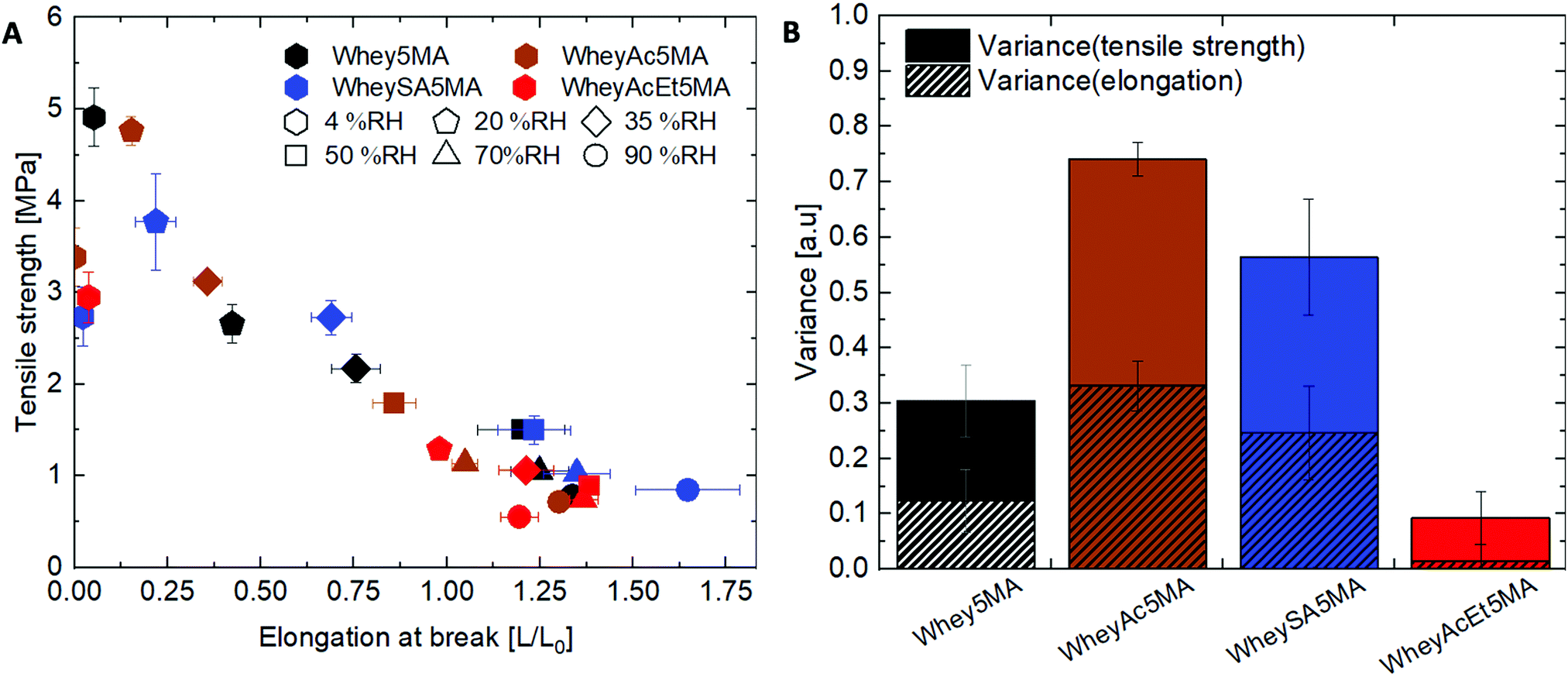 | ||
| Fig. 6 (A) Elongation at break and ultimate tensile strength of copolymers equilibrated at relative humidity levels ranging from 4–90% RH. Copolymers are color coded based on the type of protein modification, and the equilibration conditions are noted with different marker shapes. Copolymers with small mechanical property variations with humidity are clustered closer together. (B) Data clustering from 20–90% RH determined as the variance. | ||
Conclusion
Suppressing the influence of humidity on protein materials is an important step in maturing protein-based materials towards application. This work presents a method of tuning the compatibility and reducing the influence of humidity in protein copolymers by changing surface functional groups and net charge. The net charge was controlled by acetylation, esterification, succinylation and a combination of acetylation and esterification of whey protein isolate. Subsequently, whey protein was methacrylated to allow for copolymerization with the hydrophobic poly(n-butyl acrylate). Incorporation of the charge modified proteins into a copolymer system is possible due to the flexible nature of the employed surfactant compatibilization strategy, where ionic surfactants were shown to be effective compatibilizers and plasticizers that allowed proteins to be blended and copolymerized with vinyl comonomers solvent-free. Variabilities in protein–surfactant–monomer miscibility was observed; however, net protein charge and surfactant charge type were not the dominating factors affecting miscibility. Importantly, superneutralization of the protein by acetylation and esterification to neutralize both positively and negatively charged residues prior methacrylation resulted in similar n-butyl acrylate compatibility as pure protein.Water solubility of proteins was significantly reduced through superneutralization. Both acetylation and esterification reactions were shown to reduce the water uptake of pure proteins, although their effects were diminished after surfactant compatibilization. When incorporated into a copolymer with a hydrophobic comonomer, all materials have reduced water uptake, but differences between the various protein modifications were small. In spite of the small water uptake difference, superneutralization significantly reduced the influence of humidity on mechanical performance. The range of stable mechanical performance increased from a humidity range of 50–70% RH for non-charge modified Whey5MA to a range of 20–90% RH for superneutralized WheyAcEt5MA, which may be a result of improved phase mixing and reduced protein charge density. This provides a clear pathway to reducing the humidity-dependence of biomass-based polymer materials, overcoming a key barrier to their widespread application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by MRSEC Program of the National Science Foundation under Award DMR-1419807 and by the MIT Abdul Latif Jameel Water and Food Systems Lab. The authors acknowledge the MIT Department of Chemistry Instrumentation Facility for providing the infrastructure for ATR-FTIR measurements, the Koch Institute for Integrative Cancer Research and Celestine Hong for zeta-potential measurements, MIT Center for Materials Science and Engineering for SAXS and WAXS, Hursh Sureka for AFM measurements, and LEGO System A/S, Martin Brorholt Sørensen for SEM-EDX measurements.References
- S. K. Bardhan, S. Gupta, M. Gorman and M. A. Haider, Renewable Sustainable Energy Rev., 2015, 51, 506–520 CrossRef CAS.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- Global Whey Protein Market - Growth, Trends And Forecast (2015–2020), https://www.prnewswire.com/news-releases/global-whey-protein-market---growth-trends-and-forecast-2015-2020-300155575.html, (accessed 14/12/2019, 2019).
- U.S. Department of Energy, U.S. Billion-Ton Update: Biomass Supply for a Bioenergy and Bioproducts Industry, R. D. Perlack and B. J. Stokes, ORNL/TM-2011/224, Oak Ridge National Laboratory, Oak Ridge, TN, 2011, 227p.
- M. Pouplin, A. Redl and N. Gontard, J. Agric. Food Chem., 1999, 47, 538–543 CrossRef CAS PubMed.
- T. H. McHugh and J. M. Krochta, J. Agric. Food Chem., 1994, 42, 841–845 CrossRef CAS.
- M. Abdalrazeq, C. V. L. Giosafatto, M. Esposito, M. Fenderico, P. Di Pierro and R. Porta, Coatings, 2019, 9, 322 CrossRef CAS.
- W. Qiu, A. Patil, F. Hu and X. Y. Liu, Small, 2019, 15, 1903948 CrossRef CAS PubMed.
- T. N. Sullivan, B. Wang, H. D. Espinosa and M. A. Meyers, Mater. Today, 2017, 20, 377–391 CrossRef.
- S. H. Hiew and A. Miserez, ACS Biomater. Sci. Eng., 2017, 3, 680–693 CrossRef CAS PubMed.
- S. Sampath, T. Isdebski, J. E. Jenkins, J. V. Ayon, R. W. Henning, J. P. Orgel, O. Antipoa and J. L. Yarger, Soft Matter, 2012, 8, 6713–6722 RSC.
- J. Blackwell and M. Ross, J. Polym. Sci., Polym. Lett. Ed., 1979, 17, 447–451 CrossRef CAS.
- Z. S. Petrović and J. Ferguson, Prog. Polym. Sci., 1991, 16, 695–836 CrossRef.
- L. Zhang and J. Hermans, Proteins: Struct., Funct., Bioinf., 1996, 24, 433–438 CrossRef CAS.
- A. Gennadios, H. Park and C. L. Weller, Trans. ASAE, 1993, 36, 1867–1872 CAS.
- B. Cuq, N. Gontard, C. Aymard and S. Guilbert, Polym. Gels Networks, 1997, 5, 1–15 CrossRef CAS.
- W. Chan, E. King and B. Olsen, ACS Sustainable Chem. Eng., 2019, 7(10), 9103–9111 CrossRef CAS.
- Y. Chen, Z. Lin and S. Yang, J. Therm. Anal. Calorim., 1998, 52, 565–568 CrossRef CAS.
- V. Miri, O. Persyn, J. M. Lefebvre and R. Seguela, Eur. Polym. J., 2009, 45, 757–762 CrossRef CAS.
- T. H. McHugh and J. M. Krochta, J. Agric. Food Chem., 1994, 42, 841–845 CrossRef CAS.
- J. Kester and T. Richardson, J. Dairy Sci., 1984, 67, 2757–2774 CrossRef CAS.
- R. E. Feeney, R. B. Yamasaki and K. F. Geoghegan, Adv. Chem. Ser., 1982, 198, 3–55 CrossRef CAS.
- D. H. Chou and C. V. Morr, J. Am. Oil Chem. Soc., 1979, 56, A53–A62 CrossRef.
- M. Watanabe and S. Arai, Adv. Chem. Ser., 1982, 198, 199–221 CrossRef CAS.
- K. L. Franzen and J. E. Kinsella, J. Agric. Food Chem., 1976, 24, 788–795 CrossRef CAS.
- H. Fraenkel-Conrat and H. S. Olcott, J. Biol. Chem., 1945, 161, 259–268 CrossRef CAS.
- T. Mekonnen, P. Mussone, H. Khalil and D. Bressler, J. Mater. Chem. A, 2013, 1, 13379–13398 RSC.
- C. Andreuccetti, R. A. Carvalho and C. R. F. Grosso, Food Res. Int., 2010, 43, 1710–1718 CrossRef CAS.
- P. Fairley, F. J. Monahan, J. B. German and J. M. Krochta, J. Agric. Food Chem., 1996, 44, 438–443 CrossRef CAS.
- W. Y. Chan, T. Bochenski, J. E. Schmidt and B. D. Olsen, ACS Sustainable Chem. Eng., 2017, 5, 8568–8578 CrossRef CAS.
- W. Chan, E. King and B. Olsen, ACS Sustainable Chem. Eng., 2019, 7, 9103–9111 CrossRef CAS.
- M. Sitohy, J.-M. Chobert and T. Haertle, J. Food Biochem., 2000, 24, 381–398 CrossRef CAS.
- G. E. Box, W. G. Hunter and J. S. Hunter, Statistics for Experimenters, John Wiley and Sons, New York, NY, vol. 2, 1978 Search PubMed.
- https://www.uniprot.org/uniprot/P02754, accessed 18 July 2021.
- A. C. Obermeyer, C. E. Mills, X. H. Dong, R. J. Flores and B. D. Olsen, Soft Matter, 2016, 12, 3570–3581 RSC.
- M. Sitohy, J. M. Chobert and T. Haertle, J. Food Biochem., 2000, 24, 381–398 CrossRef CAS.
- M. Sitohy, J. M. Chobert and T. Haertlé, Food/Nahrung, 2001, 45, 87–93 CrossRef CAS PubMed.
- J. J. Kester and T. Richardson, J. Dairy Sci., 1984, 67, 2757–2774 CrossRef CAS.
- A. Nikolaidis and T. Moschakis, Food Hydrocolloids, 2018, 84, 389–395 CrossRef CAS.
- C. Nick Pace, S. Trevino, E. Prabhakaran and J. Martin Scholtz, Philos. Trans. R. Soc. London, Ser. B, 2004, 359, 1225–1235 CrossRef PubMed.
- D. Otzen, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 562–591 CrossRef CAS PubMed.
- K. Dušek, J. Polym. Sci., Part C: Polym. Symp., 1967, 16, 1289–1299 CrossRef.
- M. Jackson and H. H. Mantsch, Crit. Rev. Biochem. Mol., 1995, 30, 95–120 CrossRef CAS PubMed.
- K. E. Drzewiecki, A. S. Parmar, I. D. Gaudet, J. R. Branch, D. H. Pike, V. Nanda and D. I. Shreiber, Langmuir, 2014, 30, 11204–11211 CrossRef CAS PubMed.
- J.-F. Su, Z. Huang, Y.-H. Zhao, X.-Y. Yuan, X.-Y. Wang and M. Li, Ind. Crops Prod., 2010, 31, 266–276 CrossRef CAS.
- T. Li, C. Wang, T. Li, L. Ma, D. Sun, J. Hou and Z. Jiang, Molecules, 2018, 23, 2383 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00485a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |