
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTesting of drugs using human feto-maternal interface organ-on-chips provide insights into pharmacokinetics and efficacy†
Lauren S. Richardson‡
 a,
Ananth K. Kammala‡a,
Maged M. Costantineb,
Stephen J. Fortunatoc,
Enkhtuya Radnaaa,
Sungjin Kimd,
Robert N. Taylore,
Arum Han*d and
Ramkumar Menon*a
a,
Ananth K. Kammala‡a,
Maged M. Costantineb,
Stephen J. Fortunatoc,
Enkhtuya Radnaaa,
Sungjin Kimd,
Robert N. Taylore,
Arum Han*d and
Ramkumar Menon*a
aDivision of Basic and Translational Research, Department of Obstetrics and Gynecology, Division of Basic Science and Translational Medicine, Department of Obstetrics & Gynecology, The University of Texas Medical Branch at Galveston, 301 University Blvd., Galveston, TX 77555-1062, Texas, USA. E-mail: ra2menon@utmb.edu; Tel: +1 409 772 7596
bDivision of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, The Ohio State University Wexner Medical Center, Columbus, OH, USA
cObstetrics and Gynecology, Maternal-Fetal Medicine, Ochsner Medical Center, New Orleans, LA, USA
dDepartment of Electrical and Computer Engineering, Department of Biomedical Engineering, Texas A&M University, College Station, Texas, USA. E-mail: arum.han@ece.tamu.edu
eDepartment of Obstetrics and Gynecology, Jacobs School of Medicine & Biomedical Sciences, University at Buffalo, Buffalo, New York, USA
First published on 2nd November 2022
Abstract
Objectives: To improve preclinical drug testing during pregnancy, we developed multiple microfluidic organ-on-chip (OOC) devices that represent the structure, functions, and responses of the two feto-maternal interfaces (FMis) in humans (fetal membrane [FMi-OOC] and placenta [PLA-OOC]). This study utilized feto-maternal interface OOCs to test the kinetics and efficacy of drugs during pregnancy. Study design: The FMi-OOC contained amnion epithelial, mesenchymal, chorion trophoblast, and decidual cells. The PLA-OOC contained cytotrophoblasts (BeWo), syncytiotrophoblasts (BeWo + forskolin), and human umbilical vein endothelial cell lines. Therapeutic concentrations of either pravastatin or rosuvastatin (200 ng mL−1), a model drug for these experiments, were applied to either decidua (in FMi-OOC) and syncytiotrophoblasts (in PLA-OOC) chambers under normal and oxidative stress conditions (induced by cigarette smoke extract [CSE 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25]) to evaluate maternal drug exposure during normal pregnancy or oxidative stress (OS) associated pathologies, respectively. We determined statin pharmacokinetics and metabolism (LC-MS/MS), drug-induced cytotoxicity (LDH assay), and efficacy to reduce OS-induced inflammation (multiplex cytokine assay). Results: Both OOCs mimicked two distinct human feto-maternal interfaces. The drugs tested permeated the maternal–fetal cell layers of the FMi-OOC and PLA-OOC within 4 hours and generated cell and time-specific statin metabolites from various cell types without causing any cytotoxicity. OS-induced pro-inflammatory cytokines were effectively reduced by statins by increasing anti-inflammatory cytokine response across the FMi-OOC and PLA-OOC. Conclusion: Two distinct feto-maternal interface OOCs were developed, tested, and validated for their utility to conduct preclinical trials during pregnancy. We demonstrated that the placenta and fetal membranes-decidual interface both are able to transport and metabolize drugs and that the safety and efficacy of a drug can be determined using the anatomical structures recreated on OOCs.
:25]) to evaluate maternal drug exposure during normal pregnancy or oxidative stress (OS) associated pathologies, respectively. We determined statin pharmacokinetics and metabolism (LC-MS/MS), drug-induced cytotoxicity (LDH assay), and efficacy to reduce OS-induced inflammation (multiplex cytokine assay). Results: Both OOCs mimicked two distinct human feto-maternal interfaces. The drugs tested permeated the maternal–fetal cell layers of the FMi-OOC and PLA-OOC within 4 hours and generated cell and time-specific statin metabolites from various cell types without causing any cytotoxicity. OS-induced pro-inflammatory cytokines were effectively reduced by statins by increasing anti-inflammatory cytokine response across the FMi-OOC and PLA-OOC. Conclusion: Two distinct feto-maternal interface OOCs were developed, tested, and validated for their utility to conduct preclinical trials during pregnancy. We demonstrated that the placenta and fetal membranes-decidual interface both are able to transport and metabolize drugs and that the safety and efficacy of a drug can be determined using the anatomical structures recreated on OOCs.
Introduction
Placental-mediated adverse pregnancy outcomes including preterm birth (PTB) and pre-eclampsia (PE) represent a myriad of conditions unique to pregnancy and thought to be mediated by placental dysfunction.1 Worldwide, more than 10.5% of all pregnancies deliver prematurely.2,3 In addition to these maternal and neonatal mortalities and morbidities,2,4 PE and PTB are associated with significant societal and health care financial burden.5 Despite the above, there are limited interventions available to prevent PTB but none to treat PE.6,7 Although there are ongoing clinical trials, no therapeutically effective interventions are currently available for PE. This is partially due to 1) the inability of drugs to cross feto-maternal interface barriers to treat both the mother and her fetus, and 2) proper models to test drug transport, metabolic changes, teratogenicity, and cytotoxicity at the feto-maternal interface.8,9 Consequently, many pregnant women remain as therapeutic orphans due to lack of clinical trials and FDA approved drugs to be used during pregnancy and are often being prescribed “off-label” drugs. PTB and a large subset of PE are associated with oxidative stress (OS) and inflammation as major mechanisms driving preterm labor pathways and contributing to placental vascular pathology, respectively.10 Multiple drugs in preclinical trials have shown that they can reduce inflammation, delay PTB, or reduce the severity of PE.11 However, before these drugs can advance to clinical trials, their efficacy, kinetics, and mechanism of action at the feto-maternal interfaces (FMi) are needed.A recent NIH/NICHD workshop identified several areas that ought to be addressed to improve clinical trial outcomes of pregnancy.6 Key factors include the inability of some drugs to cross the FMi, preventing treatment both the mother and her fetus, or accurate ways of testing drug transport, metabolic changes, teratogenicity, and cytotoxicity. Also not factored often is the fact that the FMi tissues have two distinct interfaces: 1) between the placenta and decidua basalis, and 2) between the fetal membranes and decidua parietalis. Structurally and functionally, these are very different microenvironments, and the passage of drugs and or their metabolites are likely to occur through both interfaces. The second FMi (fetal membrane-decidua parietalis) is often ignored likely due to the avascular nature of fetal membranes. The placenta is considered to be the unique organ during pregnancy as they express several transporter proteins like ATP-binding cassette proteins and solute carrier proteins. Our recent findings have shown the functionally active transporter proteins including phospho-glycoprotein-1, breast cancer receptor protein-1, and organic anion transporter proteins in fetal membranes.12,13
Current study has shown the protein and gene expression of several active influx and efflux transporters proteins and active cytochrome 3A4 enzyme capable of metabolizing drug molecules, in human fetal membrane cell lines (data in press). Therefore, simultaneous testing of both FMis will be necessary to fully understand the effect of a drug's potential impact during pregnancy. Unfortunately, current drug testing models have several limitations: 1) FMi's in various animal models of pregnancy do not structurally mimic those of humans; 2) non-human primates mimic human pregnancy the best, but many animal subjects are needed and are hence cost-prohibitive; 3) ex vivo placental perfusion studies are restricted to the placental–decidual interface, whereas a drug's passage through the fetal membrane–decidual interface is not tested; and 4) perfusion studies are also not reliable as delivered term placentas are often senescent and partly necrotic and do not replicate normal pregnancy physiology.14 Therefore, new methods of pharmaceutical testing of drugs used during pregnancy are needed and in vitro models should test both interfaces and should replicate both placental and fetal membranes histologically and physiologically.
To overcome current limitations, we have designed organ-on-chips (OOCs) that closely mimic the structure and function of the two human FMis.15 We report the development of two OOC systems: fetal membrane–decidua organ-on-chip (FMi-OOC) and placenta organ-on-chip (PLA-OOC) that can be used for pharmaceutical testing during pregnancy. These individual microfluidic devices with multiple human cell types from both fetal membranes and placenta mimick in utero conditions. Using FMi-OOC, we have successfully modeled ascending infection,16 tested kinetics of propagation of infectious and inflammatory mediators,17 cellular changes, and cytotoxicity.18,19 The FMi-OOC model is validated physiologically in animal models confirming their utility in conducting experiments to mimic in utero conditions.19 Several variations of placental OOCs exist and have been reported for drug trials and studying pregnancy physiology.20–22 We have modified current placental models to include a third cell compartment for umbilical cord cells that are also involved in drug transport.
To test the functioning of OOCs, we created normal (healthy) and disease models of both fetal membranes and placenta. To create a disease model, OS and inflammation were induced in the cells as these two pathophysiologies underlie most cases of PTB (i.e., due to underlying infection, systemic inflammation, genetic risk factors, or environmental exposures) and/or PE.10,23–25 To test the drug efficacy in reversing inflammation, we used statins (pravastatin and rosuvastatin), which are in clinical trials to treat PE.26 We previously reported the beneficial effects of hydrophilic (pravastatin)27,28 and lipophilic forms (simvastatin)29 of these drugs in multiple models of infectious and noninfectious PTB as well as PE.29–31 Rosuvastatin shares similarities to simvastatin but has lesser side effects and is more effective in suppressing inflammatory conditions.32 Using our OOC models that represent the structure, functions, and responses of human FMis, we have conducted preclinical testing of statins. These models indicate that both placenta and fetal membranes transport statins with no cytotoxicity and demonstrated efficacy (reduction of OS-induced inflammation). Additionally, we report differences in the metabolic profiles of drugs identified in various cell types within each tissue. This study is not implying to promote statins as drugs to be used during pregnancy based on these experiments alone, but to highlight the importance of studying these drugs in physiologically relevant models.
Materials and methods
Cell line establishment for OOC experiments
Microfluidic design, fabrication, and setup
μm in height, and the width (maternal: decidua – 3000 μm; fetal: chorion – 2000 μm, AMC – 2000 μm, and AEC – 600 μm) of each chamber was designed to mimic the thickness of each maternal and fetal layer as seen in utero.36,37 Thus, this design allows the four different cell types to be cultured in four separate microenvironments (e.g., different culture media). The chambers are interconnected through an array of 24 microchannels (width: 35μm, length: 600μm for CTCs to DECs and AMCs to CTCs, 300μm for AECs to AMCs; all height: 5μm) that performed several functions, such as preventing the movement of cells between compartments during the initial cell loading process, allowing independent localized biochemical treatments to each compartment, enabling independent elution of supernatant from each cell compartment, and allowing biochemicals to diffuse among the chambers to enable cell–cell communication.18,24,36 The number of microchannels, along with the height and width, remained the same throughout all experiments as to not affect the diffusion rate of drugs and products.To model the trophoblast–endothelial interface, a plastic in vitro device was designed and fabricated to mimic the tissue in utero. The microfluidic PLA-OOC is composed of three poly(dimethylsiloxane) (PDMS)17,19,24,36 cell culture chambers mimicking the placenta interface: STB (yellow), CTB (pink), and HUVEC (blue) chambers as shown in Fig. 1. The two trophoblast chambers are connected by an array of 24 microchannels that are 5 μm in height, 30 μm in width, and 300 μm in length allowing for tight cell–cell interfaces between each of these layers. The fetal endothelial cell chambers are connected to the CTB chamber by an array of 24 microchannels (5 μm in height, 30 μm in width, and 600 μm in length) that are coated with Type I collagen to mimic the stroma of the placenta. Each cell chamber within the PLA-OOC is 250 μm in height and is loaded by inlets and outlets. The PLA-OOC was fabricated in PDMS using a two-step photolithography master mold fabrication, followed by soft lithography of the replica mold to create the final PDMS device. These devices were cured at 85 °C for 45–60 min (1:10 mixture, Sylgard 184; DowDuPont, Midland, MI, USA). The master mold was then coated with (tridecafluoro-1,1,2,2-tetrahydro octyl) trichlorosilane (United Chemical Technologies, Bristol, PA, USA) to facilitate PDMS release from the master mold after replication.21,38 With this design, we can fabricate eight devices in one wafer and run about 30–40 chips per experiment. This PLA-OOC model is a modified version of other reported placenta OOC models21,22 by adding distinct STB and CTB layers along with mimicking the stroma of the placenta.
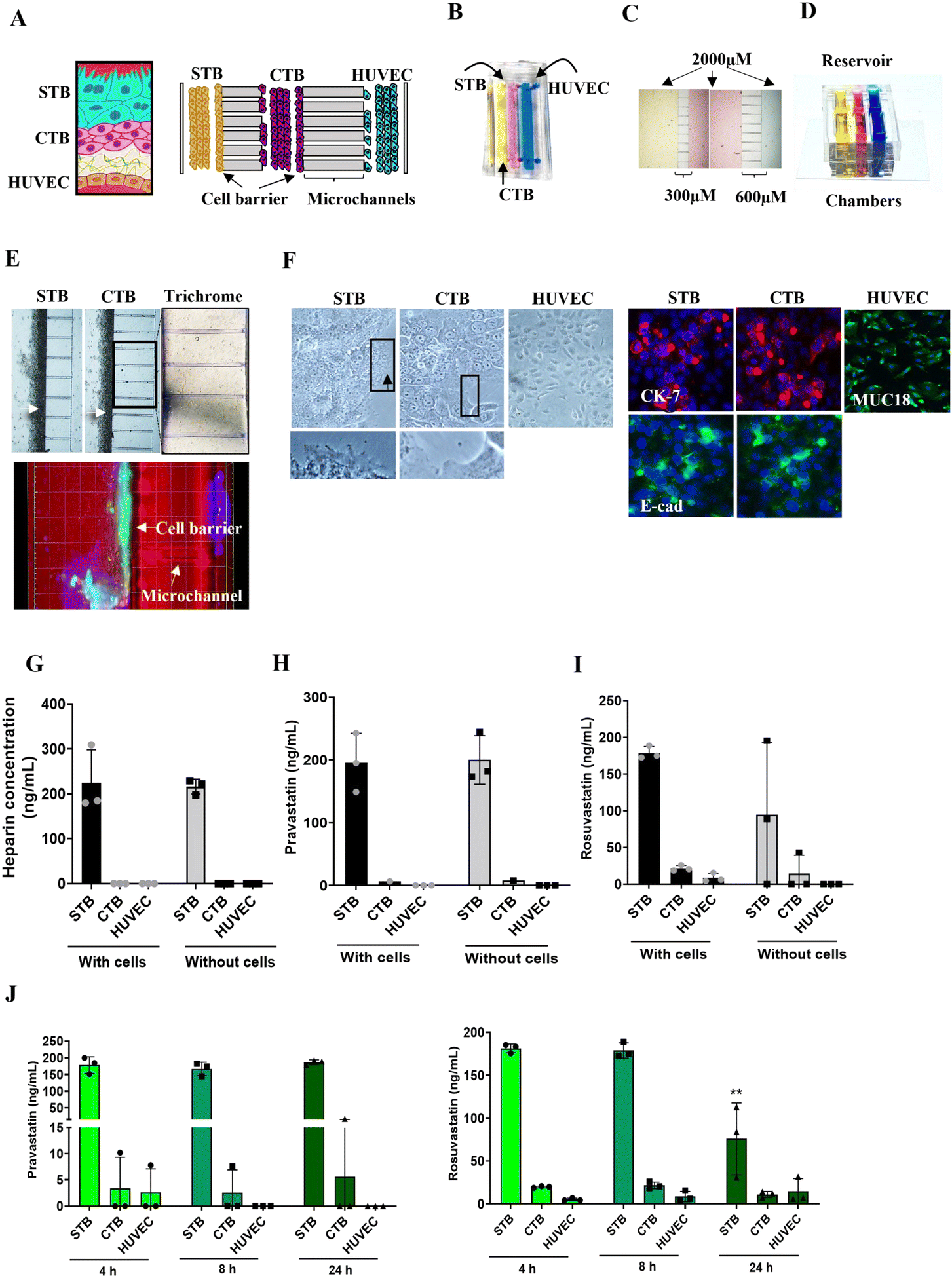 | ||
| Fig. 1 Statin pharmacokinetics across the placenta organ-on-Chip (PLA-OOC). (A) Schematic of the placenta trophoblast–endothelial interface and PLA-OOC. The PLA-OOC contains three rectangular cell culture chambers separated by arrays of microchannels. The cells are seeded as follows, from the left to right: syncytialized BeWo cells forming the STB layer (yellow), the center chamber contains BeWo cells recreating the CTB layer (pink), and the right chamber contains HUVECs forming the endothelial layer (blue). To recreate the trophoblast cell barriers, STB and CTB cells were grown in 3D over the microchannels (black arrows). All cell layers were also grown in 2D cultures adherent to the glass slide. (B) Image of the microfabricated PLA-OOC filled with color dye for easy visualization of each cell culture chamber. (C) Bird's eye view of the cell culture chambers showing the chamber width (2000 μm) and individual microchannel length mimicking the cell interface of the trophoblast layer (300 μm) and the placenta stroma (600 μm). (D) Design of an on-chip media reservoir layer that was aligned on top of the cell loading inlets and outlets (seen in image B) of the main cell culture layer, allowing media perfusion throughout the chamber for 24 h. Effluents from each culture chamber were collected through the outlet reservoirs. (E) Brightfield image showing STB and CTB cell barrier (white arrow) formation covering the microchannels. The cell barriers were confirmed to contain tight junction marker E-cadherin (green) expression confirming their functionality. Additionally, microchannels between the CTB and HUVEC chamber are filled with type I collagen to recreate the placenta stroma. Collagen was stained with Masson trichome for visualization (blue color, right image). Black and white scale bars are 100 μm. (F) A variety of in utero characteristics were measured to determine if cells grown within the PLA-OOC retained their in vivo characteristics. These measurements included cell morphology, microvilli expression (black arrow), cytoskeletal markers (cytokeratin-7 [CK-7]; red); tight junction marker (E-cadherin; green); and endothelial cell marker (MUC18; green). Scale bars are 10um. (G) In PLA-OOC with or without cells, heparin did not cross the device after 8 h. Values are expressed as mean intensities ± SD (n = 3). (H) Pravastatin and (I) rosuvastatin propagated across the PLA-OOC device within 8 h only in the presence of cells (black vs. grey bars), suggesting they are contributing to statin transport. Values are expressed as mean intensities ± SD (n = 3). (J) Targeted mass spectrometry showed that pravastatin and rosuvastatin can cross the PLA-OOC within 4 h. Rosuvastatin levels in the STB chamber decreased over time (p = 0.006), while pravastatin levels did not change over 24 h. Values are expressed as mean intensities ± SD (n = 3). | ||
These devices also contain an on-chip reservoir block, where each reservoir is aligned on top of the inlets and outlets of each chamber. The designed platform was fabricated in PDMS (1:10 mixture, Sylgard 184; DowDuPont, Midland, MI, USA) using a two-step photolithography master mold fabrication process, followed by a soft lithography process of replica-molding the final PDMS device from the master mold.38 To improve the bonding of the PDMS layer onto the glass substrate the PDMS layers were treated with oxygen plasma (Harrick Plasma, Ithaca, NY, USA) for 90s, followed by bonding the layer onto a glass substrate. This process was repeated to bond the PDMS reservoir layer on top of the device. The assembled device was stored dry (for up to 1 month).
:25 in media) or collagen type 1, to model the placenta stroma (Gibco, collagen 1 rat tail; 1:25 in DMEM/F12). The Matrigel filled the microchannels connecting the AEC compartment to the AMC compartment and filled the microchannels between the AMC compartment and the CTC compartment, mimicking the amnion and chorion basement membranes in utero, respectively within the FMi-OOC. Type 1 collagen was loaded into the cytotrophoblast chamber of the PLA-OOC and applying suction pressure from the HUVEC chamber recreated the placenta stroma. The device was then incubated overnight at 37 °C in a 5% CO2 environment. After this process, the cell chambers were rinsed with PBS to remove extra Matrigel or collagen, and then the cells were introduced.000 DEC cells for chamber one, 200000 CTCs + 5% primary collagen33 + 25% Matrigel for chamber two, 62500 AMCs + 20% primary collagen + 25% Matrigel for chamber three, and 120000 AECs for chamber four), similar to the concentration ratios seen in utero. STB and CTB cells were trypsinized and loaded into the PLA-OOC device first (60000 each) and the device was flipped on its side for 1 h inside a standard cell incubator to allow for cell barrier formation. The number of cells chosen is expected to mimic late second-trimester trophoblasts layers which have even amounts of STBs and CTBs. After 1 h, the devices were flipped to the original orientation, and an additional 60000 STB and CTB cells were added to each chamber, and 120000 HUVECs were loaded. Cell number and the size of the cell chambers, as well as distinct Oxygen environments experienced at different trimesters, could be addressed in the future to model different trimesters of the placenta. Following cell seeding, both the FMi-OOC and PLA-OOC devices were placed in 6-wells and were incubated at 37°C with 5% CO2 overnight before statin treatment. 200 ng mL−1 of pravastatin or rosuvastatin were added to the DEC chamber (to mimic maternal drug ingestion) of the FMi-OOC or the STB chamber of the PLA-OOC for up to 24 h.Imaging and cell marker analysis
:400 in 1× PBS for 2 h in the dark. The PLA-OOCs were washed with 1× PBS, and then also treated with NucBlue® Fixed ReadyProbes Reagent (R37606; Thermo Fisher Scientific, Waltham, MA) to stain the nucleus. OOCs were fixed in 4% paraformaldehyde for 15 min and stained using the Masson Trichrome method to identify collagen within the microchannels. Three microscopic fields for each condition were captured at 10×.Endpoint assays
Mass spectrometry analysis
000 rpm for 10 min. The supernatant was passed through a 0.2 μm membrane filter, and 20 μL of the filtrate was injected for the LC-MS/MS analysis.000 resolution (200 m/z) with a scan range of 50–750 m/z. Full MS followed by ddMS2 scans were obtained at 35000 (MS1) and 17500 resolutions (MS2) with a 1.5 m/z isolation window and a stepped NCE (20, 40, 60). Samples were maintained at 4 °C before injection. The injection volume was 10 μL. Chromatographic separation was achieved on a Hypersil Gold 5 μm, 50 mm × 2.1 mm C18 column (Thermo Scientific) maintained at 30 °C using a solvent gradient method. Solvent A was 0.1% formic acid in water. Solvent B was 0.1% formic acid in acetonitrile. The gradient method used was 0–1 min (20% B to 60% B), 1–2 min (60% B to 95% B), 2–4 min (95% B), 4–4.1 min (95% B to 20% B), 4.1–5 min (20% B). The flow rate was 0.5 mL min−1. Sample acquisition was performed by Xcalibur (Thermo Scientific). Data analysis was performed with Compound Discoverer 3.1 (Thermo Scientific).Statistical analyses
All data were analyzed using Prism 7 software (GraphPad Software, La Jolla, CA, USA). The Shapiro–Wilk test was conducted to check for the normality of the data. Student's t-test was used to compare results with two means. Ordinary one-way analysis of variance followed by Tukey's multiple comparison test was used to compare normally distributed data with at least three means. The Kruskal–Wallis test with Dunn's multiple comparison test was used for data that were not normally distributed. Asterisks denote p values as follows: *p < 0.05; **p < 0.01; ***p < 0.001, ***p < 0.0001.Results
Maternal–fetal drug transport during pregnancy requires passage through the placenta and or fetal membranes, which are two distinct feto-maternal interfaces (FMi). The placenta is considered the major transport route for drugs as it contains multiple transport proteins and is directly connected to the feto-maternal circulation.39,40 The fetal membranes surround and protect the fetus throughout gestation and form the FMi covering the inner intrauterine surface that is in contact with vascular decidua.41,42 Traditionally, the fetal membranes have been ignored in clinical drug or transporter protein studies. However, recently it has been shown that the fetal membrane also expresses functional transporter proteins to similar levels as the placenta (data in press). Thus, both FMi's were tested in our study.Establishing a novel tri-culture placenta on-chip model
In the human placenta, the maternal decidua is connected to the fetal syncytiotrophoblasts [STB] and cytotrophoblast [CTB] by anchoring villi extending from the villous tree.43 This interface act as a barrier and functions as a selective passage route for nutrients and drugs to reach the fetal endothelial cells (HUVECs) and eventually into the fetal circulation.44 To recreate the placenta (trophoblast–endothelial interface) in vitro (Fig. 1A) we developed a tri-culture PLA-OOC device, that contains three cell culture chambers connected by an array of microchannels (Fig. 1A–F). From the left to the right, the left chamber contains syncytialized BeWo cells (cultured with forskolin) forming the STB layer (yellow), the center chamber contains BeWo cells recreating the CTB layer (pink), and the right chamber contains HUVECs forming the endothelial layer (blue) (Fig. 1A and B). The rectangular chambers are connected by arrays of 24 microchannels (5 μm in height, 30 μm in width, and 300 or 600 μm in length) to mimic tight cell interfaces in placental stroma (Fig. 1C). A cell media reservoir was placed on top of the device to allow gravity-dependent drug diffusion for long-term culture (Fig. 1D) to model the human trophoblast–endothelial interface of the placenta.Each PLA-OOC was placed inside a single well of a conventional 6-well plate, and the CTB-HUVEC microchannels were filled with type I collagen (stained blue using Masson trichome stain), mimicking the placenta stroma seen in utero (Fig. 1E). Microchannels are permeable to biochemical products but do not allow free movement of cells, except for supporting active cell migration, as we have previously described.18 To recreate the trophoblast barrier, STB and CTB cells were loaded into the PLA-OOC and rotated on their side for 1 h to allow for the 3D formation of a cell barrier covering the microchannels (Fig. 1E – white arrows).
The PLA-OOC was first validated by characterizing whether in utero cellular characteristics were maintained during culture. STB cultured on-chip maintained their epithelial morphology, contained multiple nuclei and microvilli (black arrow; crop), and had decreased cytokeratin-7 (CK-7) expression, confirming syncytialization had occurred (Fig. 1F). The expression of tight junctional marker E-cadherin (green) in the cell barrier (E) and the general cell population (F) suggests the cells' barrier function was retained across the microchannels (Fig. 1E and F). CTB cultured on-chip maintained their epithelioid morphology, CK-7 expression, and expressed E-cadherin at their junctions (Fig. 1F). HUVEC cultured within the PLA-OOC contained an endothelial morphology and expressed cell adhesion molecule MUC18 (Fig. 1F). These results validate that the cellular and collagen components of the PLA-OOC model retain their key characteristics as seen in the placenta in vivo.
Diffusion of statins across the placenta on-chip
In the tri-chamber PLA-OOC, we studied fluid diffusion characteristics between cells to determine the intercellular propagation of drugs, metabolites, and biomarkers. Experimental time points at 4–24 h were set, based on our initial analysis of drug propagation kinetics (Fig. S1†). The ability to maintain fluidic separation between the three chambers of the PLA-OOC was tested with and without cells loaded into the devices. Heparin was loaded into the STB chamber, and a plate reader was used to monitor the rate of perfusion between the three chambers over 8 h. Heparin did not diffuse across PLA-OOCs without cells, suggesting it takes longer than 8 hours to observe the diffusion of high molecular weight molecules. Minimal diffusion was detected across the device and the trophoblast cell barrier prevented the heparin from reaching the HUVEC layer (Fig. 1G) as seen in utero and as expected due to heparin's high molecular weight.45 Next, statins were tested in this system to determine the rate of diffusion. Both pravastatin and rosuvastatin were transported across the PLA-OOC within 8 h compared to devices that did not contain any cells where no transport occurred (control OOCs) (Fig. 1H and I). To determine statin pharmacokinetics over time, a therapeutic concentration (200 ng mL−1) of either of the statins was added to the STB chamber and media were collected from each chamber after 4, 8, or 24 h and analyzed by targeted mass spectrometry. Pravastatin levels in the STB chamber remained rather steady over the 24 h, reached HUVEC by 4 h, and then decreased after 8 h (Fig. 1J). Conversely, rosuvastatin significantly decreased over time in the STB chamber (p = 0.006), reached the HUVEC chamber in 4 h, where levels remained constant over the 24 h (Fig. 1J). Additionally, neither statin treatment affected cell viability throughout these experiments (Fig. S2A†), suggesting this dose is safe at this concentration. These data validated the use of the PLA-OOC to study drug kinetics. Additionally, these data suggested that over time, cells within this device could metabolize the parent compounds, leading to a decrease in statin concentration. To determine this, the same samples were analyzed for documented pravastatin or rosuvastatin metabolites.Metabolism of statins within the placenta on-chip
Though both are a part of the statin family, pravastatin and rosuvastatin have very different metabolic characteristics. This is partially due to their unique compositions but also due to different first or second-pass metabolism rates, and the cellular availability of modifying CYP enzymes.46–48 Pravastatin is primarily metabolized (3′α-iso pravastatin, 6′-epipravastatin, 3′α, 5′ β-dihydro-pravastatin, desacyl-dehydro pravastatin, 3′-hydroxy-pravastatin) by glucuronidation and minimally involve CYP enzymes.49–51 Rosuvastatin is predominantly metabolized (N-desmethyl rosuvastatin and rosuvastatin-5S-lactone) by CYP 3A4 and 2D6 enzymes.52 To note, pravastatin metabolites are inactive and often lead to increased excretion, whereas rosuvastatin metabolites are active and carry out its mechanism of action.53 To determine each statin drugs' metabolism within different cellular layers of the placenta, media from the PLA-OOC chamber after 4, 8, or 24 h from above experiments were analyzed by untargeted mass spectrometry. All cell layers metabolized pravastatin in a time-depended manner to form the isomer within 24 h (Fig. 2A; Table 1). After 8 h, STB cells also formed desacyl-dehydro pravastatin (Fig. 2A; Table 1). Both trophoblast layers metabolized rosuvastatin into N-desmethyl rosuvastatin and rosuvastatin-5S-lactone after 8 and 24 h (Fig. 2B; Table 1); possibly explaining the decrease in the parent compound over time. HUVEC also metabolized rosuvastatin in a time-dependent manner into rosuvastatin-5S-lactone, but not into N-desmethyl rosuvastatin, possibly due to the lack of CYP enzymes required for this byproduct (Fig. 2B). These results highlight the utility of the PLA-OOC as determination of time and cell-specific statin metabolites is difficult with other in vitro and in vivo model systems (Table 2).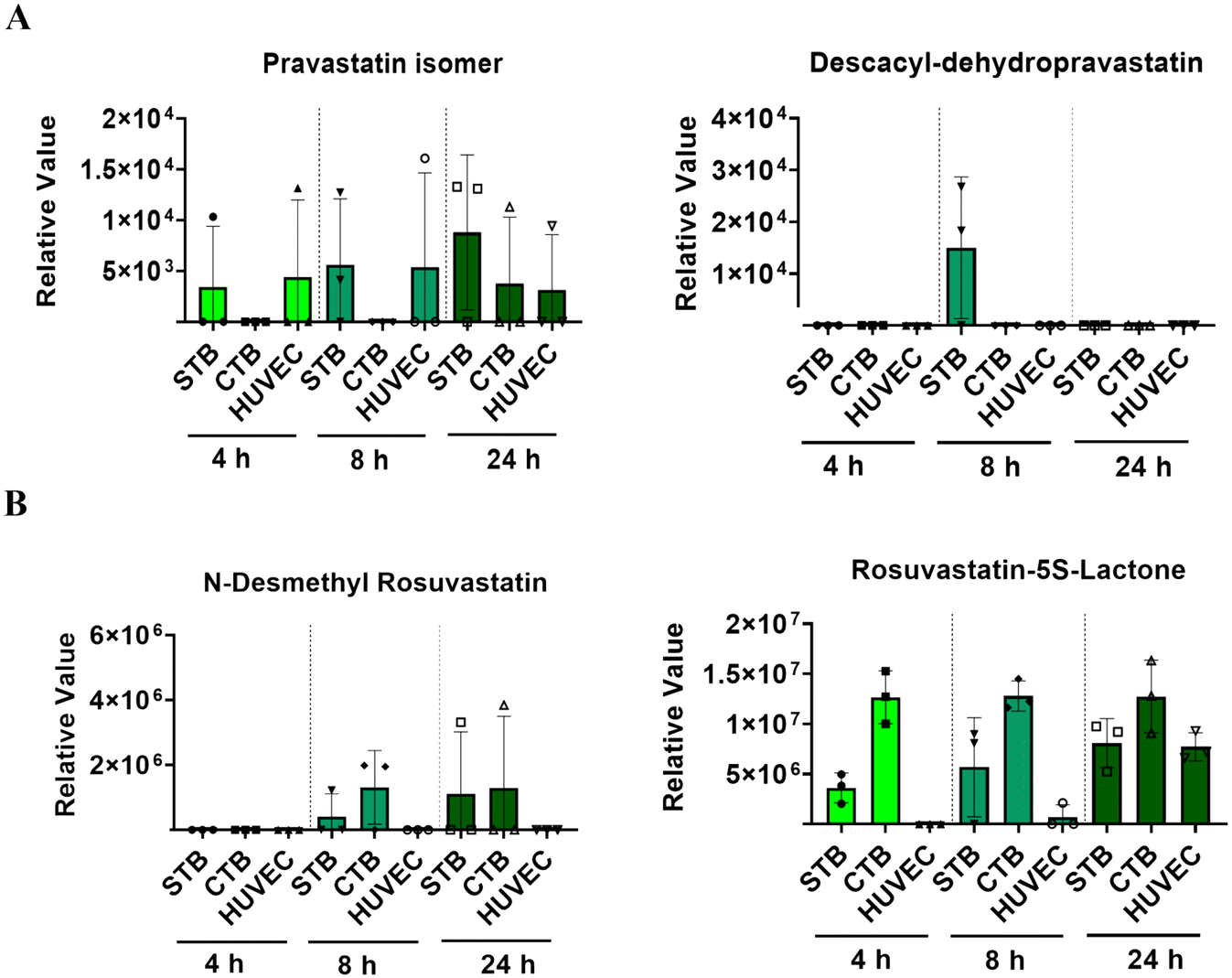 | ||
| Fig. 2 Statin metabolism across the PLA-OOC. (A) Untargeted mass spec analysis identified known pravastatin metabolites within media from the PLA-OOC chambers. Two metabolites, a pravastatin isomer and desacyl-dehydro pravastatin, were both identified to have time and cell-dependent expression. Values are expressed as mean intensities ± SD (n = 3). (B) Untargeted mass spec analysis identified known rosuvastatin metabolites from the PLA-OOC chambers. N-Desmethyl rosuvastatin and rosuvastatin-5S-lactone were both identified at different time points and in different cell chambers. Values are expressed as mean intensities ± SD (n = 3). | ||
| Formula | Name | Molecular weight | Transformations | Composition change |
|---|---|---|---|---|
| C23H36O7 | Parent-pravastatin | 424.5 | ||
| C23H36O7 | Pravastatin isomer | 424.2461 | ||
| C18H28O5 | Desacyl-dehydropravastatin | 324.19367 | Dehydration, reduction | –(C5H8O2) |
| C22H28FN3O6S | Parent-rosuvastatin | 481.1683 | ||
| C21H22FN3O6S | N-Desmethyl-rosuvastatin | 463.1213 | Desaturation, desaturation | –(CH6) |
| C21H25FN3O5S | Rosuvastatin-5S-lactone | 450.1499 | Dehydration, reduction | –(CH3O) |
| Cell culture | Trans wells | Placenta perfusion | Organ-on-chip | |
|---|---|---|---|---|
| Anatomy | One cell type from a specific organ | Multiple cell types from a specific organ | Whole placenta | Multiple cell types and organs can be combined on-chip |
| Cellularity | Single cell | Multi cell | Multi cell | Multi cell/multi organ |
| Viability | >Week | 48–72 hours | <4 hours | >Week |
| Ability to test PK | No | Yes | Yes | Yes |
| Ability to test efficacy | Yes | Yes | Yes | Yes |
| Limitations | Does not mimic a full system | Prone to tearing (tissue) or gaps in confluency (cells) | • Prone to leaks and tissue damage | Requires special set up |
| • Can not mimic disease states | ||||
| • Requires special set up |
Determining statins' efficacy in reducing OS induced inflammatory changes
After determining statin diffusion and metabolism within the PLA-OOC device, we determined their efficacy in reducing oxidative stress induced inflammation. To mimic the oxidative stress-associated inflammatory state often reported in PE, the STB chamber of the PLA-OOC was treated with cigarette smoke extract (CSE; 1:25), a validated laboratory reagent to induce oxidative stress.54 After 48 h of CSE treatment, either pravastatin or rosuvastatin was added for 24 h. Media were collected from each cell chamber and analyzed for pro-and anti-inflammatory cytokines to determine the inflammatory state. Compared to controls, CSE treatment increased IL-6 within the PLA-OOC, validating our disease model (STB: IL-6 – p = 0.034) (Fig. 3A). In the CTB and HUVEC chambers, pravastatin increased pro-inflammatory cytokines IL-6 (CTB: IL-6 – p = 0.004) (HUVEC: IL-6 – p = 0.041) (Fig. 3B), as well as anti-inflammatory cytokines IL-4 and IL-10 (CTB: IL-4 – p = 0.049) (HUVEC: IL-4 – p = 0.010) compared to CSE (Fig. 3C). Conversely, rosuvastatin did not change pro-inflammatory cytokines (Fig. 3B) but increased the production of anti-inflammatory cytokine IL-4, but not IL-10, in the CTB (IL-4 – p = 0.035) and HUVEC chambers (IL-4 – p = 0.048) (Fig. 3C).
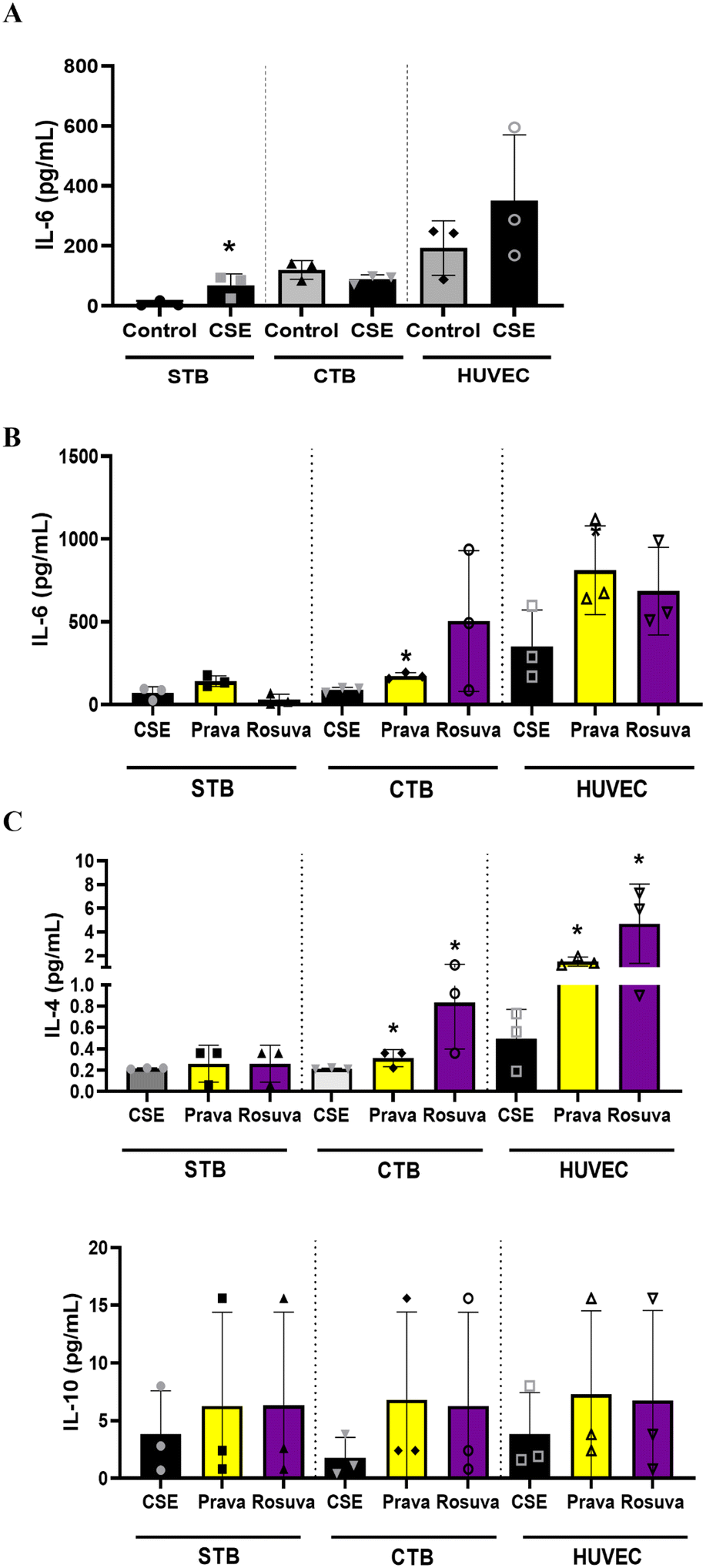 | ||
| Fig. 3 Determining the efficacy of statins within the PLA-OOC. The inflammatory status of the PLA-OOC was determined by measuring pro- and anti-inflammatory cytokines from media with a Luminex multiplex cytokine panel. (A) 48 h cigarette smoke extract (CSE) treatment in the STB chamber induced a pro-inflammatory environment in the STB, CTB, and HUVEC chambers as noted by increased IL-6 and IL-8 levels (STB: IL-6 – p = 0.034; IL-8 – p = 0.040). Values are expressed as mean intensities ± SD (n = 3). (B) Compared to CSE treatment, pravastatin significantly increases pro-inflammatory cytokines IL-6 and IL-8 in the CTB and HUVEC chambers (CTB: IL-6 – p = 0.004) (HUVEC: IL-6 – p = 0.041; IL-8 – p = 0.039). Rosuvastatin did not change pro-inflammatory cytokine levels across the PLA-OOC. Values are expressed as mean intensities ± SD (n = 3). (C) Both statins significantly increase anti-inflammatory cytokines IL-4 and IL-10 in the CTB and HUVEC chambers compared to CSE treatment (pravastatin: CTB: IL-4 – p = 0.049; HUVEC: IL-4 – p = 0.010) (rosuvastatin: CTB: IL-4 – p = 0.035; HUVEC: IL-4 – p = 0.048). Values are expressed as mean intensities ± SD (n = 3). | ||
As statins' mechanism of action is shown to increase anti-inflammatory cytokines,55 here we document that both statins can increase anti-inflammatory cytokines within the PLA-OOC device. Pravastatin produced a balanced response whereas rosuvastatin caused a pronounced anti-inflammatory response. For the first time, we report effectively testing pravastatin and rosuvastatin's propagation, metabolism, and efficacy utilizing the PLA-OOC (Table 3).
| Pravastatin | Rosuvastatin | |||
|---|---|---|---|---|
| Placenta OOC | Fetal membranes OOC | Placenta OOC | Fetal membranes OOC | |
| Pharmacokinetics | ∼4 h (1.3%) | ∼4 h (2%) | ∼4 h (2.6%) | ∼4 h (1.1%) |
| Metabolism | Pravastatin isomer | Descacyl-dehydropravastatin | Rosuvastatin-5S-lactone & N-desmethyl rosuvastatin | Rosuvastatin-5S-lactone & N-desmethyl rosuvastatin |
| Efficacy | 4.36 ± 1.34 fold | 7.19 ± 4.21 fold | 4.83 ± 1.63 fold | 4.51 ± 1.13 fold |
Diffusion of statins across the fetal membrane on-chip
Although these findings with PLA-OOC are novel, a report without considering the second FMi that can potentially passage the drug is incomplete. Therefore, we repeated these experiments and investigated statin transport and function in fetal membranes using FMi-OOC.To test this further, we used the experimental model for FMi-OOC and tested statin's role in the fetal membrane cell. Using FMi-OOC pravastatin and rosuvastatin propagation, metabolism, and efficacy were tested. Experiments conducted on PLA-OOC were repeated using FMi-OOC. The previously validated FMi-OOC is composed of four concentric chambers with cells and collagen, designed to mimic the thickness and cell density of the FMi in vivo36 (Fig. 4). Fig. 4 shows images of the developed FMi-OOC device, with four different color dyes loaded into each chamber (Fig. 4A and B). Starting at the center, the first chamber contains maternal decidual cells (red), the next chamber contains fetal CTCs (yellow), the third chamber contains AMCs (green), and the outermost chamber contains AECs (purple). Each layer is connected by arrays of microchannels coated with type IV collagen to recreate the basement membrane of the amniochorion (Fig. 4C). Cellular characteristics (viability, morphology, production of nascent collagen, cellular transitions-EMT, and migration) (Fig. 4D) in the OOC have been previously documented to be similar to those seen in utero,42,56,57 validating the physiological relevance of the FMi-OOC.
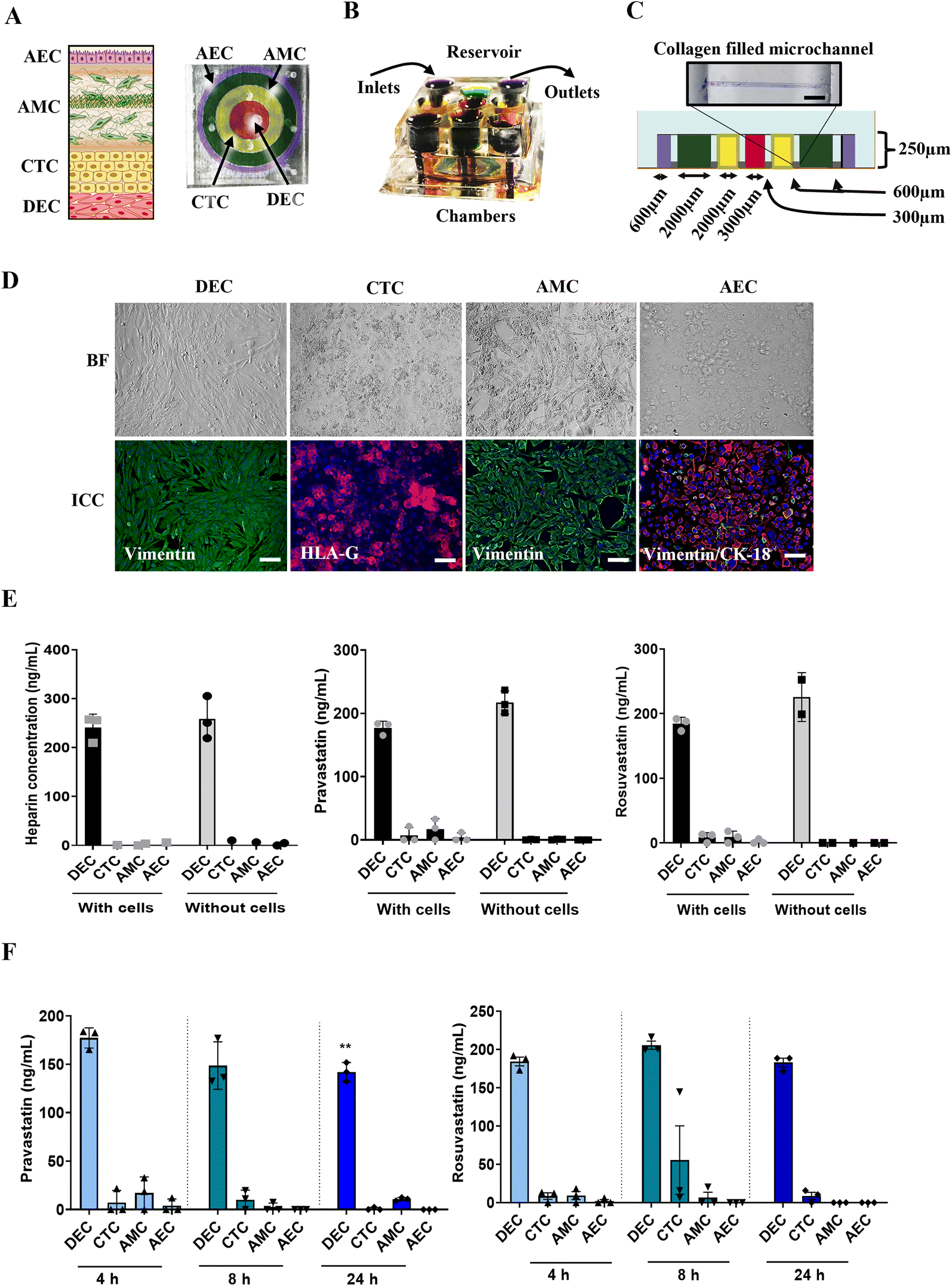 | ||
| Fig. 4 Statin pharmacokinetics across the fetal membrane OOC. (A) Schematic of the feto-maternal interface (FMi-OOC) device designed to mimic the fetal membrane amniochorion-decidua interface. The FMi-OOC contains four concentric circular cell culture chambers separated by arrays of microchannels. The cells are seeded as follows, from the center to the outside: decidua cells (red), CTCs (yellow), AMCs (green), and AECs (purple), respectively. (B) The FMi-OOC device with an integrated media reservoir filled with color dye in each of the corresponding cell culture layers. (C) Schematic showing the width and height of cell chambers and microchannels within the FMi-OOC. Microchannels between the CTC-AMC and AMC-AEC chambers are filled with type 4 collagen to recreate the chorion and amnion basement membrane. Collagen was stained with Masson trichome for visualization (blue color). Scale bars are 100 μm. (D) In utero cell specific markers were measured to determine if cells grown within the FMi-OOC retained their in vivo characteristics. These measurements included cell morphology (brightfield [BF]), cytoskeletal markers (vimentin green; cytokeratin-18 [CK-18] red), and human leukocyte antigen G (HLA-G) in the chorion. Scale bars are 50um. (E) In FMi-OOC devices with or without cells, heparin did not cross the decidua-amniochorion interface after 8 h. Values are expressed as mean intensities ± SEM (n = 3). Pravastatin and rosuvastatin propagated across the FMi-OOC device within 8 h only in the presence of cells (black vs. grey bars), suggesting they are contributing to statin transport. Values are expressed as mean intensities ± SD (n = 3). (F) Targeted mass spectrometry showed that pravastatin can cross the FMi-OOC within 4 h. Pravastatin also significantly decreases in the DEC chamber over 24 h. Values are expressed as mean intensities ± SD (n = 3). Targeted mass spectrometry showed that rosuvastatin can cross the FMi-OOC within 4 h. Values are expressed as mean intensities ± SD (n = 3). | ||
To test the fluidic separation between the four chambers of the FMi-OOC, heparin was loaded into the DEC chamber and monitored for 8 h. Minimal diffusion was detected across the device with and without cells (Fig. 4E) as expected. Next, statins were tested in the FMi-OOC to determine the pharmacokinetics across the fetal membranes. Pravastatin and rosuvastatin in the FMi-OOC were transported across the device within 8 h compared to devices that did not contain cells (Fig. 4E). To determine statin propagation over time, a therapeutic concentration (200 ng mL−1) of either statin was added to the DEC chamber. Media samples were collected from each chamber after 4, 8, or 24 h and analyzed by targeted mass spectrometry. Pravastatin levels in the DEC chamber decreased over the 24 h (p = 0.006), reached the AEC chamber by 4 h, which was then decreased after 8 h (Fig. 4F). Conversely, rosuvastatin levels did not change substantially over time in the DEC chamber, reached the AEC chamber in 4 h, and then decreased by 8 h. Additionally, neither statin affected cell viability throughout the experiments (Fig. S2B†).
These data support the hypothesis that the fetal membranes, like the placenta, are important for drug transport during pregnancy. Additionally, the FMi-OOC can be used to study drug kinetics.
Metabolism of statins by fetal membrane cells on-chip
Like the PLA-OOC findings reported above, data from FMi-OOC suggest that over time cells within the fetal membranes metabolize the parent statin compounds. 200 ng mL−1 of each statin was added to the DEC chamber and media were analyzed for known pravastatin or rosuvastatin metabolites after 4, 8, or 24 h. All cell layers metabolized pravastatin to form desacyl-dehydro pravastatin within 4 h and levels decreased over 24 h (Fig. 5A; Table 1). Additionally, all cells metabolized rosuvastatin into rosuvastatin-5S-lactone after 4 h, while only DECs and AMCs produced N-desmethyl rosuvastatin after 8 h (Fig. 5B; Table 1). These data show differential pravastatin and rosuvastatin metabolite expression on-chip.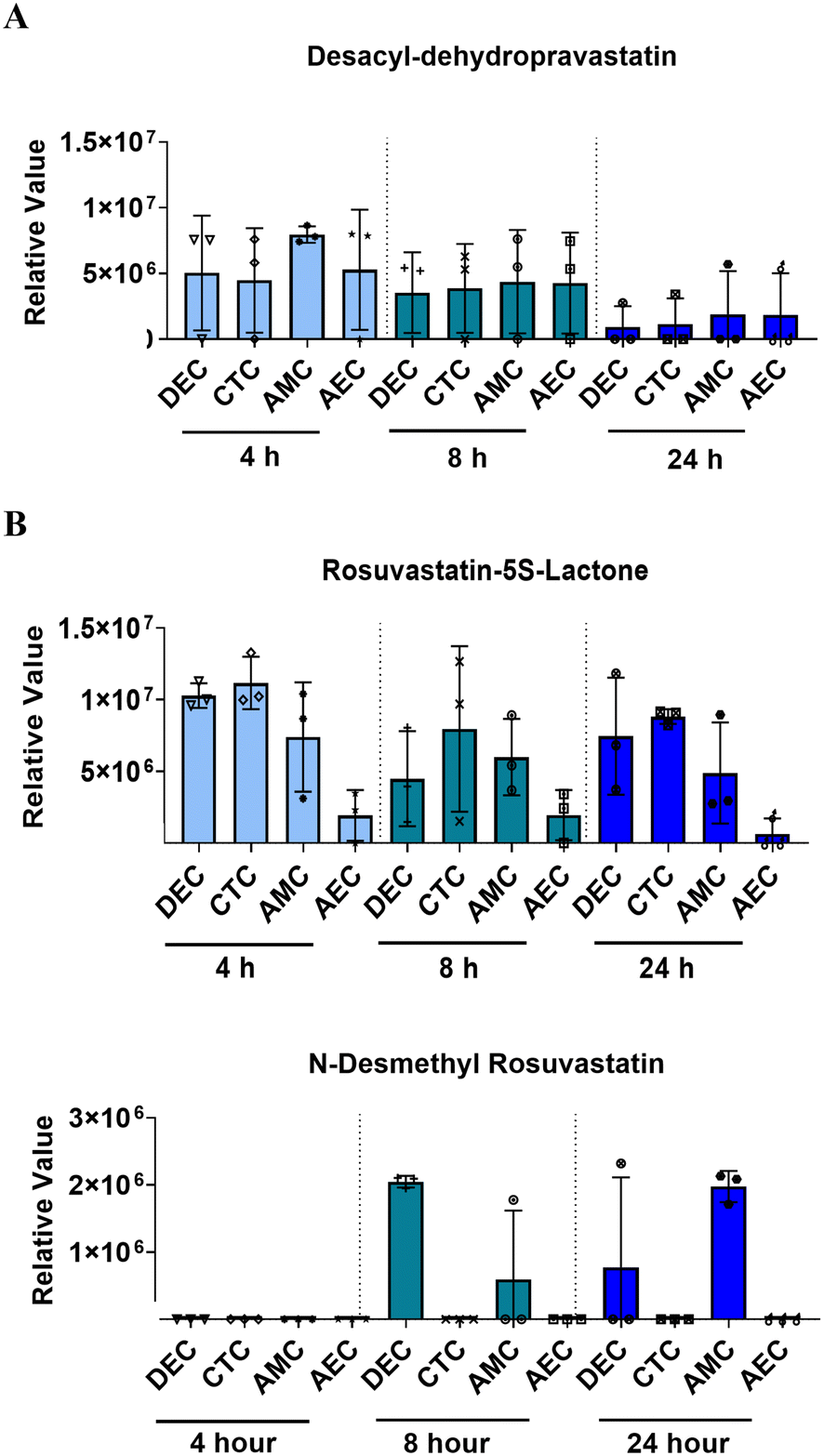 | ||
| Fig. 5 Statin metabolism across the FMi-OOC. (A) Untargeted mass spec analysis identified known pravastatin metabolites within media from the FMi-OOC chambers. Only one metabolite, desacyl-dehydro pravastatin, was identified all cells at all time points. Values are expressed as mean intensities ± SEM (n = 3). (B) Untargeted mass spec analysis identified known rosuvastatin metabolites from the FMi-OOC chambers media. N-Desmethyl rosuvastatin and rosuvastatin-5S-lactone were both identified at different time points and in different cell chambers. Values are expressed as mean intensities ± SD (n = 3). | ||
Determining statin efficacy within the fetal membrane on-chip
Once we determined that statins cross fetal membrane cells and undergo cellular metabolism, we evaluated their efficacy to mitigate OS observed in adverse states of pregnancy.58 The DEC chamber of the FMi-OOC was treated with CSE (1:25) for 48 h and then pravastatin or rosuvastatin was added for an additional 24 h. Media were collected from each cell chamber after statin treatment and analyzed for pro-and anti-inflammatory cytokines. Compared to the controls, CSE increased the production of pro-inflammatory cytokines in CTC (IL-6: p = 0.03), AMC, and AEC (Fig. 6A). After 24 h, pravastatin significantly reduced IL-6 compared to CSE alone (Fig. 6B) and increased anti-inflammatory IL-4 compared to CSE treatment (CTC: p = 0.03; AMC: p = 0.001) (Fig. 6C). When compared to CSE treatment, rosuvastatin increased IL-4 and IL-10 in CTC (IL-4: p = 0.029), AMC (IL-4: p = 0.005), and AEC (IL-10: p < 0.0001) (Fig. 6C).
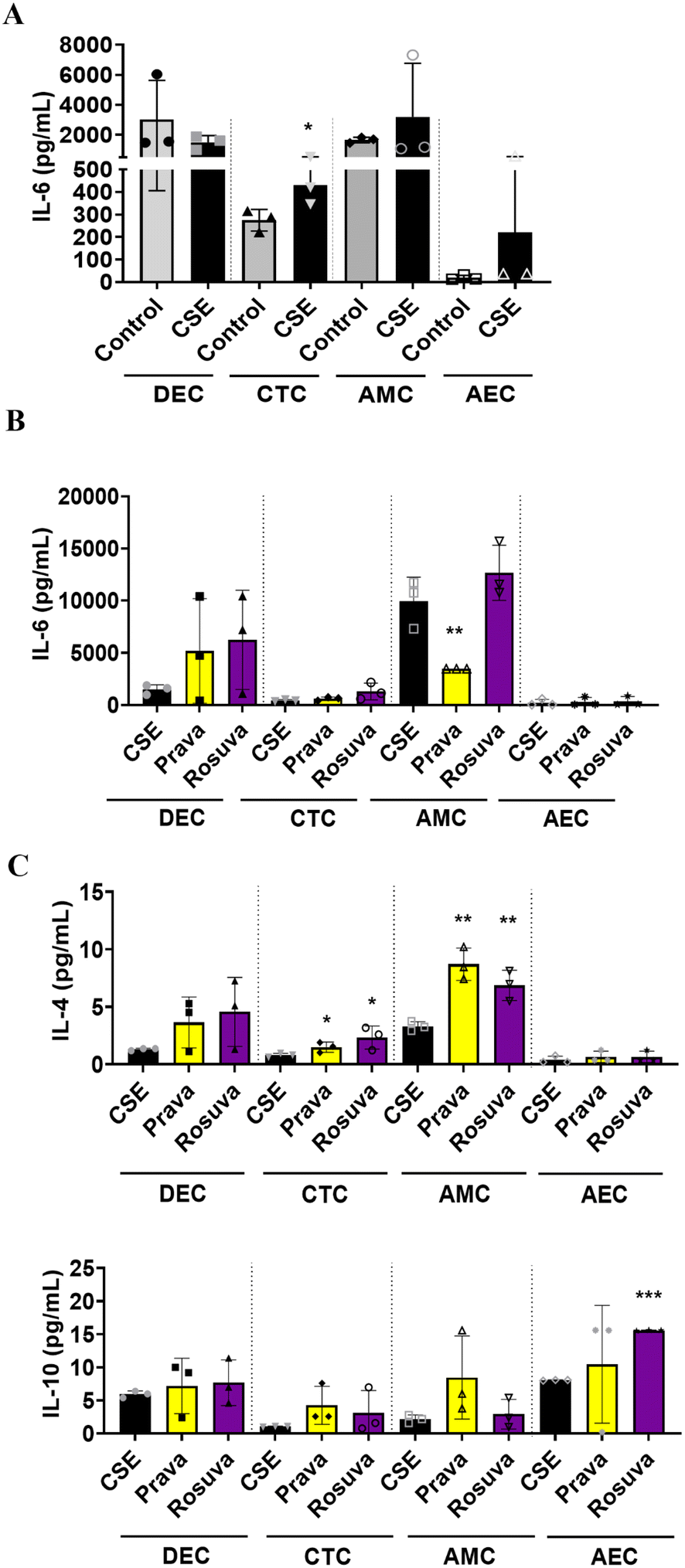 | ||
| Fig. 6 Determining the efficacy of statins within the FMi-OOC. The inflammatory status of the FMi-OOC was determined by measuring pro- and anti-inflammatory cytokines from media with a Luminex multiplex cytokine panel. (A) CSE treatment in the DEC chamber induced an increase in pro-inflammatory cytokines IL-6 in DEC, CTC (IL-6: p = 0.029), AMC and AEC chambers. Values are expressed as mean intensities ± SD (n = 3). (B) Both statins significantly increase anti-inflammatory cytokines IL-4 and IL-10 in the CTC, AMC, and AEC chambers compared to CSE treatment (pravastatin: CTC [IL-4: p = 0.032]; AMC [IL-4: p = 0.001]) (rosuvastatin: CTC [IL-4: p = 0.029], AMC [IL-4: p = 0.005], and AEC [IL-10: p < 0.0001]). Values are expressed as mean intensities ± SD (n = 3). | ||
In this study, we have documented for the first time that pravastatin and rosuvastatin are transported and metabolized across the fetal membranes, utilizing the novel FMi-OOC (Table 3).
Discussion
Assessment of drug and their metabolite transport in human pregnancy is difficult and current models projects numerous challenges.59 Multiplicity of cell types and complex FMis and physiological changes during pregnancy alter drug pharmacokinetics across gestation.47,60 Improvements are needed to provide safer treatments during pregnancy. Many traditional in vitro models have ignored human fetal membrane–decidual interface as a potential entryway for drugs.61 The recent discovery of multiple drug transporter proteins and drug-metabolizing enzymes in fetal membranes prompted us to speculate that the fetal membrane–decidual interface along with the placenta may also transport drugs.12,62 To overcome current limitations and to test the potential of a 2nd FMi' to transport drugs, we used two distinct models of the OOCs and tested statin drug transport kinetics, metabolism, and efficacy. Statins were used as a model drug for testing our devices based on our in vitro and in vivo experiences with this drug. This study is not implying to promote statins as drugs to be used during pregnancy based on these experiments alone.The five major achievements of this study were: (1) to develop and validate OOC devices to study drug pharmacokinetics at two FMis; (2) to demonstrate that neither of the statins tested caused cytotoxicity in any cell type represented in the OOC device; (3) to show that statins are transported across both the FMi's producing distinct pharmacokinetics patterns; (4) to identify statin metabolites across the different layers of fetal membranes and placenta, suggesting that cells from both tissues produce functionally active drug-metabolizing enzymes; and (5) to determine in our oxidative stress paradigm that statins minimize inflammation by a combination of reduced pro-inflammatory mediators and increased anti-inflammatory cytokines in a cell type-dependent fashion. Overall, our findings suggest that OOC can be utilized in preclinical studies for the assessment of drugs during pregnancy.
Here we document the pharmacokinetics of pravastatin and rosuvastatin across PLA-OOC and FMi-OOC and that they each have distinct characteristics. Interestingly, due to these drugs hydrophilic properties and use of transporter proteins, pravastatin and rosuvastatin propagated across the FMi-OOC quicker than infectious stimuli,17 exosomes, and environmental toxicants.63 Pravastatin, as expected for a soluble, hydrophilic compound, was transported across both interfaces and detectable even in the HUVEC chamber at 4 h. However, the inactive metabolites of pravastatin showed differential profiles in each OOCs. Pravastatin isomer accumulated in cells of the PLA-OOC but not in FM-OOC cells. Conversely, diacyl-dehydro pravastatin was not present in placental cells (except at 8 h in STB), however, appeared in all the cell types of the FMi-OOC at all time points. Rosuvastatin, which is dependent on transporter proteins for its propagation,64 was transported across both OOCs. Rosuvastatin's active metabolites (N-desmethyl rosuvastatin and 5S-lactone) were also seen in placental cells but not in the HUVEC layer, suggesting that the drug and its metabolites do not cross the placental barrier to reach HUVEC (fetal side). Decidual cells also retained rosuvastatin and produced both metabolites. Similarly, 5S-lactone was seen in all fetal membrane cell supernatants whereas N-desmethyl rosuvastatin was limited to the AMC layer, suggesting differential kinetics and metabolic capability by cells of the fetal membranes. Though documenting metabolism on-chip is an important step for understanding drug pharmacokinetics, it is prohibitive in quantitating parent drug that reaches each chamber. Real-time mass spec analysis of supernatants in each chamber could alleviate this limitation in the future.
To determine the factors associated with the differential kinetic effects, we screened drug transporter proteins and metabolizing enzymes. This confirmed the existence of functional machinery capable of drug transport and to rule out artifacts generated in OOC experiments. Pravastatin and rosuvastatin uptake were facilitated by the organic anion transporting polypeptide transporters and breast cancer resistance protein respectively.49,52,65 breast cancer resistance protein is highly expressed in the human placenta and facilitates the transport of a wide variety of drugs.40,66 Since rosuvastatin was efficiently transported by BCRP, the concentration of the rosuvastatin in the STB chamber was decreased. The transport of rosuvastatin to fetal membrane layers indicates that decidua also possesses proteins that facilitate transport of rosuvastatin to the membranes. Although breast cancer resistance protein expression is limited in decidua, it is highly expressed in CTC and AECs emphasizing the potential role of chorion trophoblast in propagating the drug further towards the fetus.
In addition, we have confirmed that fetal membrane cells express cytochrome 3A4 enzyme and convert pravastatin and rosuvastatin into inactive and active metabolites.51,52,67 The placenta along with fetal organs are the only organ reported to metabolize drugs during pregnancy.48 However, for the first time, we showed that fetal membrane cells also metabolize drugs to form either active or inactive metabolites. Our studies strongly suggest that drug modification during pregnancy is not only the effect of the placenta, but also by fetal membrane cells. Drugs to be used during pregnancy need to be screened for their metabolite formation across FMis. We conclude that OOCs can be used to determine the metabolic profiles of drugs at FMi barriers. Though, slight expression level changes might be observed due to experimental set up (i.e., dynamic vs. static flow), metabolism of drugs should be dependent on cell availability of CYP enzyme expression. Another interesting conclusion can also be derived from studies on transporter proteins in fetal membranes. Accumulation of drugs or other toxic metabolites and molecules can be refluxed by fetal membrane cells using BCRP and other efflux proteins. This scenario could be the opposite of what we have shown in this study, specifically, when the drugs or other toxic molecules accumulate in the amniotic fluid via systemic circulation. Since amnion epithelial layers will be in direct contact with these substances in the amniotic fluid, fetal membranes may detoxify them by functioning as an efflux center to reduce teratogenicity.
We have shown that OOCs can be used to estimate the pharmacokinetics of statin drugs, as well as their efficacy. In models of pregnancy where CSE induces oxidative stress,19,24,36,57 CSE increased inflammatory cytokines from fetal membrane cells and STB in PLA-OOC. Co-treatment of CSE with either of the statins did not change inflammatory IL-6 significantly, but both pravastatin and rosuvastatin substantially increased anti-inflammatory cytokines IL-4 and IL-10 in a cell type-specific manner. This effect of rosuvastatin was reported previously using fetal membrane explant models and in animal models.9,27,68 Although we have reported cellular levels changes by CSE on cells tested using FMi-OOC, we have not conducted any studies to determine the restoration of cellular level derangements by statins in our models. Determination of these changes is a part of our ongoing studies.
Statins are competitive inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme-A reductase (HMG-CoA reductase), clinically used to lower total and low-density lipoprotein cholesterol.69 We tested the effects of a hydrophilic statin (pravastatin)30 and a lipophilic one (simvastatin)29 with a potential to treat PTB as well as in PE.29–31 To this point, pravastatin has advanced to clinical trials (Identifier: NCT03944512) thought, rosuvastatin is the more potent compound with high antioxidant and anti-inflammatory properties. Rosuvastatin has also showed better effectiveness in reducing inflammation compared to simvastatin in our prior in vitro studies.70 Pravastatin also reduced inflammation, crossed the placenta and minimized PE-like symptoms in animal models.6,28,71 Here expand on this work by utilizing in vitro tool to determine the pharmacokinetics, metabolism, and efficacy of statins in each cell layer of the placenta and fetal membranes. These data show pravastatin and rosuvastatin metabolites measurable in elutes from different cellular compartments. Using our devices, we were able to show not only the drug transportation at maternal fetal interface but also their toxicity profile at different cell layers and pharmacological activity at cellular levels.
For the first time in this study, we have screened the expression of transporter proteins and CYP enzymes of different fetal membrane cells (under standard culture conditions). These are critical components that allow for drug transport across the FMis in vitro and in vivo. The discovery of drug transporter proteins and metabolic enzymes in the fetal membranes opens up new avenues of research in regard to drug propagation and metabolism. This is critical as traditionally only the placenta is studied in these aspects. Additionally, this study documents the utility of OOC devices as key tools for preclinical trial research that should be expanded upon in the future.
Utilizing two different OOC devices representing two distinct FMis, we determined the drug propagation, metabolic profile, and therapeutic efficacy across the FMi barriers. Although it has several advantages compared to other transwell or OOC systems, the major limitation of current devices is the low number of replicates and the lack of a dynamic flow. Our devices are currently being modified to include dynamic flow to properly assess the pharmacokinetics of drugs and metabolite production. Additionally, these devices can be adapted to model different trimesters of pregnancy by containing trimester-specific cell types (i.e., extra villous trophoblasts), cell ratios, chamber widths and providing appropriate oxygen environment. These components could all effect the pharmacokinetics and metabolism of drugs on-chip.
In summary, we demonstrate the following: (1) fetal membranes are functionally important in drug transport during pregnancy and their activity mimics placental drug transport; (2) fetal membranes express transporter proteins and functional CYP enzymes, like those seen in the placenta; and (3) OOC devices can be used to test drug transport effectiveness across FMis. These data corroborate and extend those obtained using in silico or in vivo approaches.
Glossary
| ATCC | American Type Culture Collection |
| AEC | Amnion epithelial cell |
| AMC | Amnion mesenchymal cell |
| CTC | Chorion trophoblast cell |
| CSE | Cigarette smoke extract |
| CYP | Cytochrome |
| CK | Cytokeratin |
| CTB | Cytotrophoblast |
| DEC | Decidua cell |
| FBS | Fetal bovine serum |
| FMi-OOC | Fetal membrane organ-on-chip |
| FMi | Feto-maternal interface |
| HUVEC | Human umbilical vein endothelial cells |
| IL | Interleukin |
| LDH | Lactate dehydrogenase |
| OOC | Organ-on-chip |
| OS | Oxidative stress |
| PBS | Phosphate buffered saline |
| PLA-OOC | Placenta organ-on-chip |
| PDMS | Poly(dimethylsiloxane) |
| PE | Pre-eclampsia |
| PTB | Preterm birth |
| RNA | Ribonucleic acid |
| STB | Syncytiotrophoblast |
Funding
This study was supported by R01HD100729-01S1 (NIH/NICHD) and UG3TR003283 (NCATS/NICHD) to Dr. Ramkumar Menon and Dr. Arum Han. Dr. Richardson is supported by a research career development award (K12HD052023: Building Interdisciplinary Research Careers in Women's Health Program-BIRCWH; Berenson, PI) from the National Institutes of Health/Office of the Director (OD)/National Institute of Allergy and Infectious Diseases (NIAID), and Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.Data and materials availability
All data and materials used in the analysis must be available in some form to any researcher for purposes of reproducing or extending the analysis.Author contributions
Project ideas were conceived, and funding was provided by RM and AH. AK, LR, ER, and SK conducted the experiments. MMC (reproductive pharmacology), SJF (fetal membrane functions and perinatal medicine) and RNT (placental and decidual functions and biology) provided expert opinions and helped with the manuscript draft. LR, AK, AH, and RM helped with data analysis and interpretation and prepared the manuscript.Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.References
- S. E. Parker and M. M. Werler, Epidemiology of ischemic placental disease: a focus on preterm gestations, Semin. Perinatol., 2014, 38(3), 133–138, DOI:10.1053/j.semperi.2014.03.004; J. M. Roberts, Pathophysiology of ischemic placental disease, Semin. Perinatol., 2014, 38(3), 139–145, DOI:10.1053/j.semperi.2014.03.005; J. M. Roberts and C. A. Hubel, The two stage model of preeclampsia: variations on the theme, Placenta, 2009, 30 Suppl A, S32–S37, DOI:10.1016/j.placenta.2008.11.009 , From NLM.
- S. Beck, D. Wojdyla, L. Say, A. P. Betran, M. Merialdi, J. H. Requejo, C. Rubens, R. Menon and P. F. Van Look, The worldwide incidence of preterm birth: a systematic review of maternal mortality and morbidity, Bull. W. H. O., 2010, 88(1), 31–38, DOI:10.2471/BLT.08.062554.
- C. V. Ananth, Ischemic placental disease: a unifying concept for preeclampsia, intrauterine growth restriction, and placental abruption, Semin. Perinatol., 2014, 38(3), 131–132, DOI:10.1053/j.semperi.2014.03.001.
- E. R. McCabe, G. E. Carrino, R. B. Russell and J. L. Howse, Fighting for the next generation: US Prematurity in 2030, Pediatrics, 2014, 134(6), 1193–1199, DOI:10.1542/peds.2014-2541.
- Institute of Medicine Committee on Understanding Premature Birth and Assuring Healthy, O. The National Academies Collection: Reports funded by National Institutes of Health, in Preterm Birth: Causes, Consequences, and Prevention, ed. R. E. Behrman and A. S. Butler, National Academies Press (US), National Academy of Sciences, 2007 Search PubMed.
- J. V. Ilekis, E. Tsilou, S. Fisher, V. M. Abrahams, M. J. Soares, J. C. Cross, S. Zamudio, N. P. Illsley, L. Myatt and C. Colvis, et al., Placental origins of adverse pregnancy outcomes: potential molecular targets: an Executive Workshop Summary of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, Am. J. Obstet. Gynecol., 2016, 215(1 Suppl), S1–S46, DOI:10.1016/j.ajog.2016.03.001.
- ACOG Practice Bulletin No. 202: Gestational Hypertension and Preeclampsia, Obstet. Gynecol., 201913311 DOI:10.1097/AOG.0000000000003018.
- R. Thadhani and C. G. Solomon, Preeclampsia--a glimpse into the future?, N. Engl. J. Med., 2008, 359(8), 858–860, DOI:10.1056/NEJMe0804637.
- M. Ma'ayeh, K. M. Rood, D. Kniss and M. M. Costantine, Novel Interventions for the Prevention of Preeclampsia, Curr. Hypertens. Rep., 2020, 22(2), 17, DOI:10.1007/s11906-020-1026-8.
- M. B. Tenório, R. C. Ferreira, F. A. Moura, N. B. Bueno, A. C. M. de Oliveira and M. O. F. Goulart, Cross-Talk between Oxidative Stress and Inflammation in Preeclampsia, Oxid. Med. Cell. Longevity, 2019, 2019, 8238727, DOI:10.1155/2019/8238727; L. Myatt and R. P. Webster, Vascular biology of preeclampsia, J. Thromb. Haemostasis, 2009, 7(3), 375–384, DOI:10.1111/j.1538-7836.2008.03259.x.
- M. A. Elovitz, Anti-inflammatory interventions in pregnancy: now and the future, Semin. Fetal Neonatal Med., 2006, 11(5), 327–332, DOI:10.1016/j.siny.2006.03.005; A. Guarini, M. P. Pereira, A. van Baar and A. Sansavini, Special Issue: Preterm Birth: Research, Intervention and Developmental Outcomes, Int. J. Environ. Res. Public Health, 2021, 18(6) DOI:10.3390/ijerph18063169; G. C. Di Renzo, L. Cabero Roura, F. Facchinetti, H. Helmer, C. Hubinont, B. Jacobsson, J. S. Jørgensen, R. F. Lamont, A. Mikhailov and N. Papantoniou, et al., Preterm Labor and Birth Management: Recommendations from the European Association of Perinatal Medicine, J. Matern.-Fetal Neonat. Med., 2017, 30(17), 2011–2030, DOI:10.1080/14767058.2017.1323860.
- E. Ganguly, A. K. Kammala, M. Benson, L. S. Richardson, A. Han and R. Menon, Organic Anion Transporting Polypeptide 2B1 in Human Fetal Membranes: A Novel Gatekeeper for Drug Transport During Pregnancy?, Front. Pharmacol., 2021, 12(3538), 771818, DOI:10.3389/fphar.2021.771818 , Original Research; A. Kammala, M. Benson, E. Ganguly, L. Richardson and R. Menon, Functional role and regulation of permeability-glycoprotein (P-gp) in the fetal membrane during drug transportation, Am. J. Reprod. Immunol., 2021, e13515, DOI:10.1111/aji.13515.
- A. Kammala, M. Benson, E. Ganguly, E. Radnaa, T. Kechichian, L. Richardson and R. Menon, Fetal Membranes Contribute to Drug Transport across the Feto-Maternal Interface Utilizing the Breast Cancer Resistance Protein (BCRP), Life, 2022, 12(2), 166, DOI:10.3390/life12020166.
- Z. Sultana, K. Maiti, J. Aitken, J. Morris, L. Dedman and R. Smith, Oxidative stress, placental ageing-related pathologies and adverse pregnancy outcomes, Am. J. Reprod. Immunol., 2017, 77(5) DOI:10.1111/aji.12653.
- L. Richardson, S. Kim, R. Menon and A. Han, Organ-On-Chip Technology: The Future of Feto-Maternal Interface Research?, Front. Physiol., 2020, 11, 715, DOI:10.3389/fphys.2020.00715.
- O. A. G. Tantengco, L. S. Richardson, P. M. B. Medina, A. Han and R. Menon, Organ-on-chip of the cervical epithelial layer: A platform to study normal and pathological cellular remodeling of the cervix, FASEB J., 2021, 35(4), e21463, DOI:10.1096/fj.202002590RRR.
- L. S. Richardson, S. Kim, A. Han and R. Menon, Modeling ascending infection with a feto-maternal interface organ-on-chip, Lab Chip, 2020, 20(23), 4486–4501, 10.1039/d0lc00875c.
- L. Richardson, S. Jeong, S. Kim, A. Han and R. Menon, Amnion membrane organ-on-chip: an innovative approach to study cellular interactions, FASEB J., 2019, fj201900020RR, DOI:10.1096/fj.201900020RR , From NLM.
- E. Radnaa, L. S. Richardson, S. Sheller-Miller, T. Baljinnyam, M. de Castro Silva, A. Kumar Kammala, R. Urrabaz-Garza, T. Kechichian, S. Kim, A. Han and R. Menon, Extracellular vesicle mediated feto-maternal HMGB1 signaling induces preterm birth, Lab Chip, 2021, 21(10), 1956–1973, 10.1039/D0LC01323D.
- Y. Pu, J. Gingrich and A. Veiga-Lopez, A 3-dimensional microfluidic platform for modeling human extravillous trophoblast invasion and toxicological screening, Lab Chip, 2021, 21(3), 546–557, 10.1039/d0lc01013h; M. Horii, O. Touma, T. Bui and M. M. Parast, Modeling human trophoblast, the placental epithelium at the maternal fetal interface, Reproduction, 2020, 160(1), R1–R11, DOI:10.1530/REP-19-0428; N. Arumugasaamy, K. D. Rock, C. Y. Kuo, T. L. Bale and J. P. Fisher, Microphysiological systems of the placental barrier, Adv. Drug Delivery Rev., 2020, 161-162, 161–175, DOI:10.1016/j.addr.2020.08.010; R. L. Pemathilaka, D. E. Reynolds and N. N. Hashemi, Drug transport across the human placenta: review of placenta-on-a-chip and previous approaches, Interface Focus, 2019, 9(5), 20190031, DOI:10.1098/rsfs.2019.0031.
- B. Mosavati, A. V. Oleinikov and E. Du, Development of an Organ-on-a-Chip-Device for Study of Placental Pathologies, Int. J. Mol. Sci., 2020, 21(22), 8755, DOI:10.3390/ijms21228755; F. Yin, Y. Zhu, M. Zhang, H. Yu, W. Chen and J. Qin, A 3D human placenta-on-a-chip model to probe nanoparticle exposure at the placental barrier, Toxicol. In Vitro, 2019, 54, 105–113, DOI:10.1016/j.tiv.2018.08.014.
- J. S. Lee, R. Romero, Y. M. Han, H. C. Kim, C. J. Kim, J. S. Hong and D. Huh, Placenta-on-a-chip: a novel platform to study the biology of the human placenta, J. Matern.-Fetal Neonat. Med., 2016, 29(7), 1046–1054, DOI:10.3109/14767058.2015.1038518.
- L. Myatt and A. Maloyan, Obesity and Placental Function, Semin. Reprod. Med., 2016, 34(1), 42–49, DOI:10.1055/s-0035-1570027; R. Menon, F. Behnia, J. Polettini and L. S. Richardson, Novel pathways of inflammation in human fetal membranes associated with preterm birth and preterm pre-labor rupture of the membranes, Semin. Immunopathol., 2020, 42(4), 431–450, DOI:10.1007/s00281-020-00808-x; A. M. Borzychowski, I. L. Sargent and C. W. Redman, Inflammation and pre-eclampsia, Semin. Fetal Neonatal Med., 2006, 11(5), 309–316, DOI:10.1016/j.siny.2006.04.001; C. W. Redman and I. L. Sargent, Immunology of pre-eclampsia, Am. J. Reprod. Immunol., 2010, 63(6), 534–543, DOI:10.1111/j.1600-0897.2010.00831.x; J. Polettini, L. S. Richardson and R. Menon, Oxidative stress induces senescence and sterile inflammation in murine amniotic cavity, Placenta, 2018, 63, 26–31, DOI:10.1016/j.placenta.2018.01.009; Y. C. Lien, Z. Zhang, G. Barila, A. Green-Brown, M. A. Elovitz and R. A. Simmons, Intrauterine Inflammation Alters the Transcriptome and Metabolome in Placenta, Front. Physiol., 2020, 11, 592689, DOI:10.3389/fphys.2020.592689.
- L. S. Richardson, E. Radnaa, R. Urrabaz-Garza, N. Lavu and R. Menon, Stretch, scratch, and stress: Suppressors and supporters of senescence in human fetal membranes, Placenta, 2020, 99, 27–34, DOI:10.1016/j.placenta.2020.07.013.
- J. Jin, L. Richardson, S. Sheller-Miller, N. Zhong and R. Menon, Oxidative stress induces p38MAPK-dependent senescence in the feto-maternal interface cells, Placenta, 2018, 67, 15–23, DOI:10.1016/j.placenta.2018.05.008.
- A. Vahedian-Azimi, L. Karimi, Ž. Reiner, S. Makvandi and A. Sahebkar, Effects of statins on preeclampsia: A systematic review, Pregnancy Hypertens., 2021, 23, 123–130, DOI:10.1016/j.preghy.2020.11.014 , From NLM.
- S. K. Basraon, R. Menon, M. Makhlouf, M. Longo, G. D. Hankins, G. R. Saade and M. M. Costantine, Can statins reduce the inflammatory response associated with preterm birth in an animal model?, Am. J. Obstet. Gynecol., 2012, 207(3), 224.e221–224.e227, DOI:10.1016/j.ajog.2012.06.020.
- A. R. Carver, E. Tamayo, J. R. Perez-Polo, G. R. Saade, G. D. Hankins and M. M. Costantine, The effect of maternal pravastatin therapy on adverse sensorimotor outcomes of the offspring in a murine model of preeclampsia, Int. J. Dev. Neurosci., 2014, 33, 33–40, DOI:10.1016/j.ijdevneu.2013.11.004.
- S. K. Basraon, M. M. Costantine, G. Saade and R. Menon, The Effect of Simvastatin on Infection-Induced Inflammatory Response of Human Fetal Membranes, Am. J. Reprod. Immunol., 2015, 74(1), 54–61, DOI:10.1111/aji.12372.
- S. K. Basraon, R. Menon, M. Makhlouf, M. Longo, G. D. Hankins, G. R. Saade and M. M. Costantine, Can statins reduce the inflammatory response associated with preterm birth in an animal model?, Am. J. Obstet. Gynecol., 2012, 207(3), 224.e221–224.e227, DOI:10.1016/j.ajog.2012.06.020.
- M. T. Ayad, B. D. Taylor and R. Menon, Regulation of p38 mitogen-activated kinase-mediated fetal membrane senescence by statins, Am. J. Reprod. Immunol., 2018, e12999, DOI:10.1111/aji.12999.
- B. C. Fellstrom, A. G. Jardine, R. E. Schmieder, H. Holdaas, K. Bannister, J. Beutler, D. W. Chae, A. Chevaile, S. M. Cobbe, C. Gronhagen-Riska, J. J. De Lima, R. Lins, G. Mayer, A. W. McMahon, H. H. Parving, G. Remuzzi, O. Samuelsson, S. Sonkodi, D. Sci, G. Süleymanlar, D. Tsakiris, V. Tesar, V. Todorov, A. Wiecek, R. P. Wüthrich, M. Gottlow, E. Johnsson and F. Zannad, Rosuvastatin and cardiovascular events in patients undergoing hemodialysis, N. Engl. J. Med., 2009, 360(14), 1395–1407, DOI:10.1056/NEJMoa0810177; T. Rezen, M. Hafner, S. Kortagere, S. Ekins, V. Hodnik and D. Rozman, Rosuvastatin and Atorvastatin Are Ligands of the Human Constitutive Androstane Receptor/Retinoid X Receptor alpha Complex, Drug Metab. Dispos., 2017, 45(8), 974–976, DOI:10.1124/dmd.117.075523.
- R. Menon, E. Radnaa, F. Behnia and R. Urrabaz-Garza, Isolation and characterization human chorion membrane trophoblast and mesenchymal cells, Placenta, 2020, 101, 139–146, DOI:10.1016/j.placenta.2020.09.017.
- S. Sheller, J. Papaconstantinou, R. Urrabaz-Garza, L. Richardson, G. Saade, C. Salomon and R. Menon, Amnion-Epithelial-Cell-Derived Exosomes Demonstrate Physiologic State of Cell under Oxidative Stress, PLoS One, 2016, 11(6), e0157614, DOI:10.1371/journal.pone.0157614 , From NLM.
- E. Radnaa, R. Urrabaz-Garza, N. D. Elrod, M. Castro Silva, R. Pyles, A. Han and R. Menon, Generation and characterization of human Fetal membrane and Decidual cell lines for reproductive biology experiments, Biol. Reprod., 2022, 106(3), 568–582, DOI:10.1093/biolre/ioab231.
- L. Richardson, J. Gnecco, T. Ding, K. Osteen, L. M. Rogers, D. M. Aronoff and R. Menon, Fetal Membrane Organ-On-Chip: An Innovative Approach to Study Cellular Interactions, Reprod. Sci., 2020, 27(8), 1562–1569, DOI:10.1007/s43032-020-00184-9.
- J. S. Gnecco, T. Ding, C. Smith, J. Lu, K. L. Bruner-Tran and K. G. Osteen, Hemodynamic forces enhance decidualization via endothelial-derived prostaglandin E2 and prostacyclin in a microfluidic model of the human endometrium, Hum. Reprod., 2019, 34(4), 702–714, DOI:10.1093/humrep/dez003.
- E. S. Park, D. Jang, J. Lee, Y. J. Kim, J. Na, H. Ji, J. W. Choi and G. T. Kim, Maskless optical microscope lithography system, Rev. Sci. Instrum., 2009, 80(12), 126101, DOI:10.1063/1.3266965.
- M. Afrouzian, R. Al-Lahham, S. Patrikeeva, M. Xu, V. Fokina, W. G. Fischer, S. Z. Abdel-Rahman, M. Costantine, M. S. Ahmed and T. Nanovskaya, Role of the efflux transporters BCRP and MRP1 in human placental bio-disposition of pravastatin, Biochem. Pharmacol., 2018, 156, 467–478, DOI:10.1016/j.bcp.2018.09.012; H. Wang, L. Zhou, A. Gupta, R. R. Vethanayagam, Y. Zhang, J. D. Unadkat and Q. Mao, Regulation of BCRP/ABCG2 expression by progesterone and 17beta-estradiol in human placental BeWo cells, Am. J. Physiol., 2006, 290(5), E798–E807, DOI:10.1152/ajpendo.00397.2005; E. M. Leslie, R. G. Deeley and S. P. Cole, Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense, Toxicol. Appl. Pharmacol., 2005, 204(3), 216–237, DOI:10.1016/j.taap.2004.10.012.
- L. W. Han, C. Gao and Q. Mao, An update on expression and function of P-gp/ABCB1 and BCRP/ABCG2 in the placenta and fetus, Expert Opin. Drug Metab. Toxicol., 2018, 14(8), 817–829, DOI:10.1080/17425255.2018.1499726; Q. Mao, BCRP/ABCG2 in the placenta: expression, function and regulation, Pharm. Res., 2008, 25(6), 1244–1255, DOI:10.1007/s11095-008-9537-z.
- R. Menon and J. J. Moore, Fetal Membranes, Not a Mere Appendage of the Placenta, but a Critical Part of the Fetal-Maternal Interface Controlling Parturition, Obstet. Gynecol. Clin. North Am., 2020, 47(1), 147–162, DOI:10.1016/j.ogc.2019.10.004.
- R. Menon, L. S. Richardson and M. Lappas, Fetal membrane architecture, aging and inflammation in pregnancy and parturition, Placenta, 2019, 79, 40–45, DOI:10.1016/j.placenta.2018.11.003.
- B. Huppertz, The anatomy of the normal placenta, J. Clin. Pathol., 2008, 61(12), 1296–1302, DOI:10.1136/jcp.2008.055277; M. Panigel, The human placenta. Anatomy and morphology, Clin. Obstet. Gynecol., 1986, 13(3), 421–445 CrossRef.
- J. Stefulj, U. Panzenboeck, T. Becker, B. Hirschmugl, C. Schweinzer, I. Lang, G. Marsche, A. Sadjak, U. Lang and G. Desoye, et al., Human endothelial cells of the placental barrier efficiently deliver cholesterol to the fetal circulation via ABCA1 and ABCG1, Circ. Res., 2009, 104(5), 600–608, DOI:10.1161/CIRCRESAHA.108.185066.
- W. Ageno, S. Crotti and A. G. Turpie, The safety of antithrombotic therapy during pregnancy, Expert Opin. Drug Saf., 2004, 3(2), 113–118, DOI:10.1517/eods.3.2.113.27343; A. Omri, J. F. Delaloye, H. Andersen and F. Bachmann, Low molecular weight heparin Novo (LHN-1) does not cross the placenta during the second trimester of pregnancy, Thromb. Haemostasis, 1989, 61(1), 55–56 CrossRef.
- J. F. Robinson, E. G. Hamilton, J. Lam, H. Chen and T. J. Woodruff, Differences in cytochrome p450 enzyme expression and activity in fetal and adult tissues, Placenta, 2020, 100, 35–44, DOI:10.1016/j.placenta.2020.07.009.
- D. L. Shuster, T. K. Bammler, R. P. Beyer, J. W. Macdonald, J. M. Tsai, F. M. Farin, M. F. Hebert, K. E. Thummel and Q. Mao, Gestational age-dependent changes in gene expression of metabolic enzymes and transporters in pregnant mice, Drug Metab. Dispos., 2013, 41(2), 332–342, DOI:10.1124/dmd.112.049718.
- P. Myllynen, E. Immonen, M. Kummu and K. Vähäkangas, Developmental expression of drug metabolizing enzymes and transporter proteins in human placenta and fetal tissues, Expert Opin. Drug Metab. Toxicol., 2009, 5(12), 1483–1499, DOI:10.1517/17425250903304049; M. R. Syme, J. W. Paxton and J. A. Keelan, Drug transfer and metabolism by the human placenta, Clin. Pharmacokinet., 2004, 43(8), 487–514, DOI:10.2165/00003088-200443080-00001.
- J. B. Wagner, M. Ruggiero, J. S. Leeder and B. Hagenbuch, Functional Consequences of Pravastatin Isomerization on OATP1B1-Mediated Transport, Drug Metab. Dispos., 2020, 48(11), 1192–1198, DOI:10.1124/dmd.120.000122.
- J. Zarek, M. K. DeGorter, A. Lubetsky, R. B. Kim, C. A. Laskin, H. Berger and G. Koren, The transfer of pravastatin in the dually perfused human placenta, Placenta, 2013, 34(8), 719–721, DOI:10.1016/j.placenta.2013.05.002.
- T. Hatanaka, Clinical pharmacokinetics of pravastatin: mechanisms of pharmacokinetic events, Clin. Pharmacokinet., 2000, 39(6), 397–412, DOI:10.2165/00003088-200039060-00002.
- P. D. Martin, M. J. Warwick, A. L. Dane, S. J. Hill, P. B. Giles, P. J. Phillips and E. Lenz, Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers, Clin. Ther., 2003, 25(11), 2822–2835, DOI:10.1016/s0149-2918(03)80336-3.
- A. El-Zailik, L. K. Cheung, Y. Wang, V. Sherman and D. S. Chow, Simultaneous LC-MS/MS analysis of simvastatin, atorvastatin, rosuvastatin and their active metabolites for plasma samples of obese patients underwent gastric bypass surgery, J. Pharm. Biomed. Anal., 2019, 164, 258–267, DOI:10.1016/j.jpba.2018.10.045; H. Son, H. Roh, D. Lee, H. Chang, J. Kim, C. Yun and K. Park, Pharmacokinetics of rosuvastatin/olmesartan fixed-dose combination: a single-dose, randomized, open-label, 2-period crossover study in healthy Korean subjects, Clin. Ther., 2013, 35(7), 915–922, DOI:10.1016/j.clinthera.2013.05.016.
- Y. Hamdi, H. Madfai, R. Belhareth, M. Mokni, O. Masmoudi-Kouki and M. Amri, Prenatal exposure to cigarette smoke enhances oxidative stress in astrocytes of neonatal rat, Toxicol. Mech. Methods, 2016, 26(4), 231–237, DOI:10.3109/15376516.2016.1156205; A. Shinjo, W. Ventura, K. Koide, K. Hori, J. Yotsumoto, R. Matsuoka, K. Ichizuka and A. Sekizawa, Maternal smoking and placental expression of a panel of genes related to angiogenesis and oxidative stress in early pregnancy, Fetal Diagn. Ther., 2014, 35(4), 289–295, DOI:10.1159/000357704; A. Agarwal, A. Aponte-Mellado, B. J. Premkumar, A. Shaman and S. Gupta, The effects of oxidative stress on female reproduction: a review, Reprod. Biol. Endocrinol., 2012, 10, 49, DOI:10.1186/1477-7827-10-49.
- C. H. Fan, Y. Hao, Y. H. Liu, X. L. Li, Z. H. Huang, Y. Luo and R. L. Li, Anti-inflammatory effects of rosuvastatin treatment on coronary artery ectasia patients of different age groups, BMC Cardiovasc. Disord., 2020, 20(1), 330, DOI:10.1186/s12872-020-01604-z; D. Kata, I. Földesi, L. Z. Feher, L. Hackler, L. G. Puskas and K. Gulya, Rosuvastatin enhances anti-inflammatory and inhibits pro-inflammatory functions in cultured microglial cells, Neuroscience, 2016, 314, 47–63, DOI:10.1016/j.neuroscience.2015.11.053; S. Dursun, S. Cuhadar, M. Köseoğlu, A. Atay and S. Günden Aktaş, The anti-inflammatory and antioxidant effects of pravastatin and nebivolol in rat aorta, Anadolu Kardiyol. Derg., 2014 DOI:10.5152/akd.2014.4708; S. Steffens and F. Mach, Anti-inflammatory properties of statins, Semin. Vasc. Med., 2004, 4(4), 417–422, DOI:10.1055/s-2004-869599.
- S. Sen, R. Rao and G. Chaudhuri, Endothelial cell function in utero-placental circulation physiology and pathophysiology, Curr. Vasc. Pharmacol., 2013, 11(5), 730–736, DOI:10.2174/1570161111311050010.
- L. S. Richardson, R. N. Taylor and R. Menon, Reversible EMT and MET mediate amnion remodeling during pregnancy and labor, Sci. Signaling, 2020, 13(618), eaay1486, DOI:10.1126/scisignal.aay1486.
- O. A. G. Tantengco, M. de Castro Silva, H. Shahin, G. F. C. Bento, G. C. Cursino, S. Cayenne and M. G. da Silva, The role of nuclear factor erythroid 2-related factor 2 (NRF2) in normal and pathological pregnancy: A systematic review, Am. J. Reprod. Immunol., 2021, 86(6), e13496, DOI:10.1111/aji.13496; R. Menon, Initiation of human parturition: signaling from senescent fetal tissues via extracellular vesicle mediated paracrine mechanism, Obstet. Gynecol. Sci., 2019, 62(4), 199–211, DOI:10.5468/ogs.2019.62.4.199; R. Menon, Oxidative stress damage as a detrimental factor in preterm birth pathology, Front. Immunol., 2014, 5, 567, DOI:10.3389/fimmu.2014.00567.
- M. Feghali, R. Venkataramanan and S. Caritis, Pharmacokinetics of drugs in pregnancy, Semin. Perinatol., 2015, 39(7), 512–519, DOI:10.1053/j.semperi.2015.08.003.
- M. M. Costantine, Physiologic and pharmacokinetic changes in pregnancy, Front. Pharmacol., 2014, 5, 65, DOI:10.3389/fphar.2014.00065.
- R. Menon, Human fetal membranes at term: Dead tissue or signalers of parturition?, Placenta, 2016, 44, 1–5, DOI:10.1016/j.placenta.2016.05.013.
- I. L. Aye, J. W. Paxton, D. A. Evseenko and J. A. Keelan, Expression, localisation and activity of ATP binding cassette (ABC) family of drug transporters in human amnion membranes, Placenta, 2007, 28(8–9), 868–877, DOI:10.1016/j.placenta.2007.03.001.
- S. Kim, L. Richardson, E. Radnaa, Z. Chen, I. Rusyn, R. Menon and A. Han, Molecular mechanisms of environmental toxin cadmium at the feto-maternal interface investigated using an organ-on-chip (FMi-OOC) model, J. Hazard. Mater., 2021, 422, 126759, DOI:10.1016/j.jhazmat.2021.126759 , From NLM.
- W. J. Hua, W. X. Hua and H. J. Fang, The role of OATP1B1 and BCRP in pharmacokinetics and DDI of novel statins, Cardiovasc. Ther., 2012, 30(5), e234–e241, DOI:10.1111/j.1755-5922.2011.00290.x.
- X. Zhang, D. I. Vernikovskaya, X. Wang, T. N. Nanovskaya, M. Costantine, G. D. Hankins and M. S. Ahmed, Quantitative determination of pravastatin and its metabolite 3α-hydroxy pravastatin in plasma and urine of pregnant patients by LC-MS/MS, Biomed. Chromatogr., 2016, 30(4), 548–554, DOI:10.1002/bmc.3581; C. E. Halstenson, J. Triscari, A. DeVault, B. Shapiro, W. Keane and H. Pan, Single-dose pharmacokinetics of pravastatin and metabolites in patients with renal impairment, J. Clin. Pharmacol., 1992, 32(2), 124–132, DOI:10.1002/j.1552-4604.1992.tb03816.x.
- F. Deng, S. K. Tuomi, M. Neuvonen, P. Hirvensalo, S. Kulju, C. Wenzel, S. Oswald, A. M. Filppula and M. Niemi, Comparative Hepatic and Intestinal Efflux Transport of Statins, Drug Metab. Dispos., 2021, 49(9), 750–759, DOI:10.1124/dmd.121.000430.
- T. Komai, K. Kawai, T. Tokui, Y. Tokui, C. Kuroiwa, E. Shigehara and M. Tanaka, Disposition and metabolism of pravastatin sodium in rats, dogs and monkeys, Eur. J. Drug Metab. Pharmacokinet., 1992, 17(2), 103–113, DOI:10.1007/BF03188778.
- A. F. Saad, Z. M. Diken, T. B. Kechichian, S. M. Clark, G. L. Olson, G. R. Saade and M. M. Costantine, Pravastatin Effects on Placental Prosurvival Molecular Pathways in a Mouse Model of Preeclampsia, Reprod. Sci., 2016, 23(11), 1593–1599, DOI:10.1177/1933719116648218; A. F. Saad, T. Kechichian, H. Yin, E. Sbrana, M. Longo, M. Wen, E. Tamayo, G. D. Hankins, G. R. Saade and M. M. Costantine, Effects of pravastatin on angiogenic and placental hypoxic imbalance in a mouse model of preeclampsia, Reprod. Sci., 2014, 21(1), 138–145, DOI:10.1177/1933719113492207.
- E. Yeatman, Simvastatin, Aust. Nurses J., 1991, 21(1), 29–30 CAS.
- T. K. Allen, M. N. Nazzal, L. Feng, I. A. Buhimschi and A. P. Murtha, Progestins Inhibit Tumor Necrosis Factor α-Induced Matrix Metalloproteinase 9 Activity via the Glucocorticoid Receptor in Primary Amnion Epithelial Cells, Reprod. Sci., 2015, 26(9), 1193–1202, DOI:10.1177/1933719118811646 , From NLM; G. Luo, V. M. Abrahams, S. Tadesse, E. F. Funai, E. J. Hodgson, J. Gao and E. R. Norwitz, Progesterone inhibits basal and TNF-alpha-induced apoptosis in fetal membranes: a novel mechanism to explain progesterone-mediated prevention of preterm birth, Reprod. Sci., 2010, 17(6), 532–539, DOI:10.1177/1933719110363618 , From NLM.
- M. M. Costantine, Pravastatin to ameliorate early-onset pre-eclampsia: promising but not there yet, BJOG, 2020, 127(4), 489, DOI:10.1111/1471-0528.16067; M. M. Costantine and K. Cleary, Eunice Kennedy Shriver National Institute of Child, H.; Human Development Obstetric--Fetal Pharmacology Research Units, N. Pravastatin for the prevention of preeclampsia in high-risk pregnant women, Obstet. Gynecol., 2013, 121(2 Pt 1), 349–353, DOI:10.1097/aog.0b013e31827d8ad5; K. A. Fox, M. Longo, E. Tamayo, T. Kechichian, E. Bytautiene, G. D. Hankins, G. R. Saade and M. M. Costantine, Effects of pravastatin on mediators of vascular function in a mouse model of soluble Fms-like tyrosine kinase-1-induced preeclampsia, Am. J. Obstet. Gynecol., 2011, 205(4), 366.e361–366.e365, DOI:10.1016/j.ajog.2011.06.083; C. C. Marrs and M. M. Costantine, Should We Add Pravastatin to Aspirin for Preeclampsia Prevention in High-risk Women?, Clin. Obstet. Gynecol., 2017, 60(1), 161–168, DOI:10.1097/GRF.0000000000000248.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2lc00691j |
| ‡ Authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2022 |