
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ceiling culture chip reveals dynamic lipid droplet transport during adipocyte dedifferentiation via actin remodeling†
Jiwon
Kim‡
a,
Kun-Young
Park‡
b,
Sungwoo
Choi
b,
Ung Hyun
Ko
a,
Dae-Sik
Lim
cd,
Jae Myoung
Suh
*b and
Jennifer H.
Shin
 *a
*a
aDepartment of Mechanical Engineering, Korea Advanced Institute of Science and Technology, Daejeon, Republic of Korea. E-mail: j_shin@kaist.ac.kr
bGraduate School of Medical Science and Engineering, Korea Advanced Institute of Science and Engineering, Daejeon, Republic of Korea. E-mail: jmsuh@kaist.ac.kr
cNational Creative Research Center for Cell Plasticity, Korea Advanced Institute of Science and Technology, Daejeon, Republic of Korea
dDepartment of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon, Republic of Korea
First published on 6th September 2022
Abstract
Adipocyte dedifferentiation has recently gained attention as a process underpinning adipocyte plasticity; however, a lack of suitable experimental platforms has hampered studies into the underlying mechanisms. Here, we developed a microscope-mountable ceiling culture chip that provides a stable yet tunable culture environment for long-term live-imaging of dedifferentiating adipocytes. A detailed spatiotemporal analysis of mature adipocyte dedifferentiation utilizing the culture platform and Cre-recombinase tracers revealed the involvement of dynamic actin remodeling for lipid droplet (LD) secretion during adipocyte dedifferentiation. Additionally, Hippo, Hedgehog, and PPARγ signaling pathways were identified as potent regulators of adipocyte dedifferentiation. Contrary to the belief that adult adipocytes are relatively static, we show that adipocytes are very dynamic, relying on actin-driven mechanical forces to execute LD extrusion and intercellular LD transfer processes.
Introduction
As an endocrine and energy-storing organ, adipose tissues play crucial roles in the maintenance of metabolic homeostasis.1,2 In addition, adipose tissues can undergo adaptive remodeling in response to a wide range of developmental, physiological, and pathological cues. Notably, the cellular plasticity of mature adipocytes is widely recognized to play a critical role in this process.3Adipocytes originate from mesenchymal stem cells and undergo adipogenesis to produce fully differentiated adipocytes. Adipogenesis is divided mainly into two stages: (1) commitment of mesenchymal stem cell (MSC) to the adipocyte lineage and (2) terminal differentiation of the preadipocytes to mature adipocytes.4 Here, the term terminal implies that the process cannot be reversed once differentiation is completed and cells acquire a specific function and morphology. However, recent studies revealed that the reversion, or dedifferentiation, of a mature adipocyte into a cell with greater developmental potential, such as a preadipocyte or MSC, can occur both in vitro and in vivo.1 During pregnancy, for example, adipocytes in the maternal mammary gland have been shown to dedifferentiate into fibroblast-like cells. Furthermore, after weaning, the lactating mammary glands undergo involution, during which dedifferentiated adipocytes re-differentiate into mature adipocytes and displace regressing mammary gland tissue.5
The process of mature adipocyte dedifferentiation is important from both a scientific and translational perspective. Dedifferentiated adipocytes, by definition, acquire broader developmental potential, implying that they can be differentiated into other cells besides adipocytes.6 For example, dedifferentiated adipocyte-derived progeny cells, or DFATs, have been shown to differentiate into osteoblasts,7 chondrocytes,8 smooth muscle cells,9 and endothelial cells.10 DFATs are emerging as an attractive material for tissue engineering due to their multilineage potential and accessibility of adipose tissue. Besides, adipocytes are more accessible than MSCs from bone marrow, making them considerably easier to harvest with fewer adverse effects.11
The phenomena of LD secretion by adipocytes has recently been documented as a central event of the dedifferentiation process whereby adipocytes transform into a delipidated state through a mechanism distinct from lipolysis.12,13 Recently, electron micrographs of fat tissue biopsies from obese individuals revealed the presence of in vivo dedifferentiated adipocytes that had also undergone LD secretion to acquire the delipidated phenotype.14 Despite its physiological importance, however, the molecular and cellular mechanisms of adipocyte dedifferentiation and LD secretion are poorly understood.
The physical resemblance between DFATs and primary fibroblasts, which are inherent to the stromal vascular fraction (SVF) of adipose tissues, is a major hindrance to studying adipocyte dedifferentiation. Therefore, we utilized Adipoq-Cre; mT/mG mice to overcome the issue of fibroblast contamination. Because only adipocytes express EGFP in Adipoq-Cre; mT/mG mice, we were able to unambiguously track the cells that were dedifferentiated from adipocytes, eliminating any concerns regarding fibroblast contamination.
Another challenge of studying primary adipocytes is their low specific gravity, which causes them to float in cell culture media. Various culture techniques have emerged to address this problem since the introduction of Sugihara's ceiling culture method in 1986.15 In 2017, Julie Anne Côté et al. introduced a ceiling culture model that can be inserted into a six-well plate.16 This model consists of plastic bushing and detachable coverslips, allowing more flexible designs of experiments. More recently, Matthew J. Harms et al. adopted the commercialized trans-well to develop a new platform for adipocyte culture, termed MAAC, which significantly improved adipocyte viability in long-term culture.17 Akiyo Yokomizo et al. took a unique approach based on coaxial microfibers where adipocyte-embedded collagen gel was fabricated as a core fiber sheathed by a layer of permeable alginate gel.18 While extant methods provide suitable conditions for in vitro culture of primary adipocytes, real-time live imaging remains challenging due to the opacity in some of these constructs as well as the geometrical constraints due to microscope focal length. Alternately, methods for the in vitro induction of mature adipocytes from adherent cells are being intensively researched. Using the Greek letter ‘σ’-shaped microwells, Kim et al. were able to capture buoyant adipocyte spheroids made from the human adipose-derived stem cells.19 Pope et al. utilized fiber networks to hold differentiated mature adipocytes while allowing them to grow into the size of the ones found in obese adults.20 However, the gene expression profiles of differentiated preadipocytes and primary adipocytes are inextricably distinct,17 and cellular modalities in these limited contexts were only suitable for observing the phenomenon of differentiation, not the reverse “dedifferentiation” process.
Here, we introduce a soft lithography-based, multi-layered ceiling culture platform that allows culturing of non-adherent, floating cell types. To address prior drawbacks, we designed a system where all parts of the chip are transparent, and the height of the ceiling of the cell culture chamber is only 200 μm, which is compatible with the short working distance of high magnification objective lenses. Furthermore, our innovative two-layered structure effectively traps floating adipocytes, preventing cell loss while tracking cellular responses in the designated ROIs. The volume of the chip's channel is also as small as 10 μl, enabling more cost-effective experiments. Using this platform, we explored different ECM conditions and observed that a fibronectin-coated surface promoted spontaneous dedifferentiation of adipocytes, whereas the Matrigel inhibited dedifferentiation. Furthermore, utilizing our high-resolution live imaging platform, we discovered two distinct processes of LD secretion in dedifferentiating adipocytes. The first involved remodeling of the actin cytoskeleton to rapidly expel the large central LD out of the adipocyte. The second was the exchange of LD content between neighboring adipocytes in contact with each other.
Materials and methods
Animals
Adipoq-Cre,21Adipoq-CreERT2,22Ppargflox/flox,23Rosa26-mT/mG,24Lats1flox/flox,25Lats2flox/flox,26 and Rosa26-tdTomato27 mice were bred to generate mice with indicated allele combinations. For all experiments, 6–8-week-old male mice were used. For induction of Cre-mediated recombination in Adipoq-CreERT2 mice, 75 mg kg−1 tamoxifen (Cayman Chemical) dissolved in corn oil (Sigma) was administered via intraperitoneal injection at the indicated time points. Mice were housed in a specific pathogen-free facility within the KAIST Laboratory Animal Resource Center, maintained under a 12 h light–dark cycle, and given free access to food and water. All protocols for mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the Korea Advanced Institute of Science and Technology.Isolation of primary adipocytes
Subcutaneous (scWAT) and visceral (vWAT) fat pads were dissected and processed for mature adipocyte isolation. Briefly, mouse white adipose tissue (WAT) fat pads were dissected and minced finely with a razor blade. The minced tissue was digested in a 37 °C shaking water bath at 120 rpm with Krebs–Ringer–Henseleit (KRH) buffer (30 mM HEPES, pH 7.4, 1 mM CaCl2, 120 mM NaCl, 4 mM KH2PO4, 1 mM MgSO4, 10 mM NaCO3, 200 nM adenosine) containing 1.5% bovine serum albumin (BSA), 0.9 mg ml−1 glucose and 1 mg ml−1 of collagenase type 1 (Worthington) for 30 min. Collagenase digestion was followed by filtration through a 100 μm nylon mesh strainer (Falcon), and adipocytes were allowed to float for approximately 3 minutes. The floating adipocytes were transferred to a 1.5 ml Eppendorf tube using a wide-bore tip and washed 3 times with a culture medium (DMEM/F12, 10% FBS, 1% Penn/Strep) at room temperature while free-floating. For each round of washing, cells were allowed to float for 3 minutes, and the infranatant was removed with a syringe and needle. After removing the last wash, isolated adipocytes were diluted with culture medium and embedded into Matrigel or transferred to the ceiling culture chip for further experiments. For induction of Cre-mediated recombination in adipocytes isolated from Adipoq-CreERT2 mice, 1 μM 4-hydroxytamoxifen (4-OHT) was treated for the indicated time.Labelling of lipid droplets for live cell imaging
Freshly isolated primary adipocytes were incubated in a culture medium including BODIPY (0.2 μg ml−1) overnight to label LDs. Incubated cells were centrifuged at 100 RCF in a benchtop clinical centrifuge and then washed with fresh media several times before being transferred to the ceiling culture chip.Fabrication of ceiling culture chip and cell loading
The standard soft lithography technique was used to fabricate the ceiling culture chip (Fig. 1c). First, the two-layered SU-8 master mold was fabricated on a silicon wafer using photolithography. Both the first and second layers are 100 μm high, making the central cell culture chamber as high as 200 μm. Next, polydimethylsiloxane (Sylgard 184, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was poured with caution to avoid air bubbles and cured in the oven at 80 °C for 2 h. Next, the holes for an inlet and an outlet were punched on the cured PDMS replicas, followed by bonding of the punched PDMS block to a coverslip or a glass-bottom dish using oxygen plasma treatment. The ceiling culture chips were then placed in an 80 °C oven for firm bonding and then rinsed with 70% ethanol and tertiary distilled water. Finally, the ceiling culture chips were sterilized under UV light with a wavelength of 365 nm for 15 min.
:1) was poured with caution to avoid air bubbles and cured in the oven at 80 °C for 2 h. Next, the holes for an inlet and an outlet were punched on the cured PDMS replicas, followed by bonding of the punched PDMS block to a coverslip or a glass-bottom dish using oxygen plasma treatment. The ceiling culture chips were then placed in an 80 °C oven for firm bonding and then rinsed with 70% ethanol and tertiary distilled water. Finally, the ceiling culture chips were sterilized under UV light with a wavelength of 365 nm for 15 min.
 | ||
| Fig. 1 Schematic presentation of the experiment. a Mature adipocytes were isolated from scWAT and vWAT of mice and then transferred to the ceiling culture chip. b Detailed design of the ceiling culture chip. Not drawn to scale in the z-direction. c Fabrication of ceiling culture chip. d Image was taken immediately after loading the mature adipocyte into the channel. Cells trapped in the cell culture chamber cannot escape through the outlet. | ||
For the fibronectin-coated condition, fibronectin was diluted in ice-cold PBS to the concentration of 100 μg ml−1 and loaded into the channel. The channel filled with fibronectin solution was sealed and flipped over to be placed in an incubator overnight. The channel was then rinsed 1–2 times with PBS before 2000 mature adipocytes were loaded through the inlet in the cell culture chamber. For the Matrigel embedding condition, adipocyte suspension was first diluted in the same volume of growth factor reduced Matrigel (Corning) to be loaded in the sterilized ceiling culture chips. The chips were kept in the incubator for 40 min until the Matrigel solidified. After the cells were loaded, 3 ml of complete media was added to each glass-bottom dish.
Live cell imaging
The ceiling culture chips were mounted on an epifluorescence microscope (Zeiss, Axio Observer series) that was equipped with a custom-made microscope-mountable incubator system to control CO2 levels and temperature. Images were obtained at intervals of 15 or 20 min with ZEN blue software.Image processing
The images obtained with ZEN blue software were processed with ImageJ software. Briefly, the outline of cells was marked with the freehand selection tool, and the area, perimeter, position, roundness, circularity, etc., were calculated.Shape index
In this paper, we introduced the shape index to quantify the level of dedifferentiation instead of conventional parameters such as circularity or roundness. The shape index is defined by the formula shown below.We defined the shape index because we empirically found that circularity and roundness did not yield adequate results in our hands. For example, the circularity was consistent with the subjective scoring of cell morphology irregularity, but it did not reproduce the same result on the same cell morphology depending on cell size or image resolution. On the other hand, roundness showed robust results, but it was not consistent with subjective scoring (ESI† Fig. S1). Therefore, we defined a shape index that was more robust than circularity and more intuitive than roundness.

Immunostaining
Cells in the ceiling culture chips were fixed with 4% paraformaldehyde for 15 min at room temperature. Fixed cells were gently washed with PBS 2 times, permeabilized with 0.2% Triton X-100 for 15 min, and blocked with 3% (w/w) bovine serum albumin. Samples were then incubated with phalloidin (Abcam) at 4 °C for 20 min and washed with PBS several times. Lastly, the chips were filled with a mounting medium (Dako) to prevent photobleaching.Confocal microscopy
The confocal microscope (ZEISS, LSM780) and z-stack module of ZEN blue software were used to reconstruct the 3-D structure of cells undergoing dedifferentiation. The pinhole size was adjusted to make the diameter of the airy disc ∼1 μm, and the step size was set to 0.5 μm.Statistical analysis
Origin software was used for statistical analysis. The two-tailed t-test with Welch correction was performed to determine significant differences between pairs of datasets, and one-way analysis of variation (ANOVA) with Fisher LSD post hoc analysis was used when there were more than two datasets. The significance cutoff was 0.05 (*: p < 0.05, **: p < 0.01). All the I-shaped bars drawn on the data represent the mean value ± standard deviation range.Results
Ceiling culture chip facilitates long-term observation of adipocytes under varying ECM conditions
Because adipocytes float and do not attach to the bottom of the cultureware, imaging them is challenging unless they are embedded in a hydrogel. However, many natural/synthetic hydrogels, including Matrigel, are likely to influence the cellular states of adipocytes. To address this concern, we developed a ceiling culture chip system. Due to the two-layer construction that confines buoyant cells within the cell culture chamber, the isolated primary adipocytes in the ceiling culture chip can be readily imaged. Furthermore, the unique chamber design allows imaging of the cells either attached to the ECM-coated ceiling or embedded within hydrogels, enabling us to explore various ECM conditions in both 2D and 3D (Fig. 1a).When viewed from the top, our ceiling culture chip is symmetrical with a cell culture chamber in the middle having an inlet and outlet holes on each end of the chamber, connected by the wings. The ceiling culture chip was fabricated using a two-layered master mold, thereby featuring a step between the wing and the cell culture chamber. The height of the cell culture chamber is 100 μm higher than its two wings (Fig. 1b and c). The step feature on the chamber ceiling keeps adipocytes from escaping through the outlet once loaded in the chamber (Fig. 1d).
Also, since the microscale chip was completely filled with culture media, there was no flow within the chamber. Thus, mature adipocytes with low binding affinity to the substrate remain stationary throughout long-term culture even under the continuous motion of the microscope stage or during the medium change. Furthermore, because the ceiling is only 200 μm high from the bottom of the chamber, the platform could fit within the short working distances of the high magnification imaging system.
Fibronectin promotes adipocyte dedifferentiation while Matrigel inhibits it
The ECM microenvironment significantly impacts adipocyte differentiation and plasticity.28 For example, previous studies have reported that about 60% of SVF from the breast of 25–45-year-old women completed adipogenesis on Matrigel-coated substrate, whereas only 40% and 20% of SVF differentiated on fibronectin-coated or non-coated plastic surfaces.29 Therefore, based on the effect of ECM type on adipogenesis, we hypothesized that ECM type might also influence the reverse process, adipocyte dedifferentiation.To verify this, we first generated a transgenic mouse model by crossing mice with a Cre transgene under control of the adiponectin promoter (Adipoq-Cre) with mice carrying the membrane-targeted tdTomato/membrane-targeted EGFP (Rosa26-mT/mG or mT/mG) Cre reporter allele, which expresses EGFP in adipocytes and tdTomato in others, allowing permanent and subsequent tracing of adipocytes (Fig. 2a). Using this mouse model (hereafter, referred to as Adipoq-Cre; mT/mG), we isolated mature adipocytes from subcutaneous adipose tissue (scWAT) and visceral adipose tissue (vWAT) and cultured them in three different ECM conditions: untreated PDMS (P) as a control, Matrigel-coated (M), and fibronectin-coated (F). The adipocytes in the chamber were imaged once every two days. Since adipocytes undergo a morphological change from a perfectly spherical shape (Fig. 1d) into irregular shapes as dedifferentiation progresses, we employed the shape index as a metric to assess the degree of dedifferentiation. The shape index shown below ranges from 0 to 1, where an index value of 1 indicates a perfect circle. As the shape becomes irregular, the shape index approaches 0 (Fig. 2b).
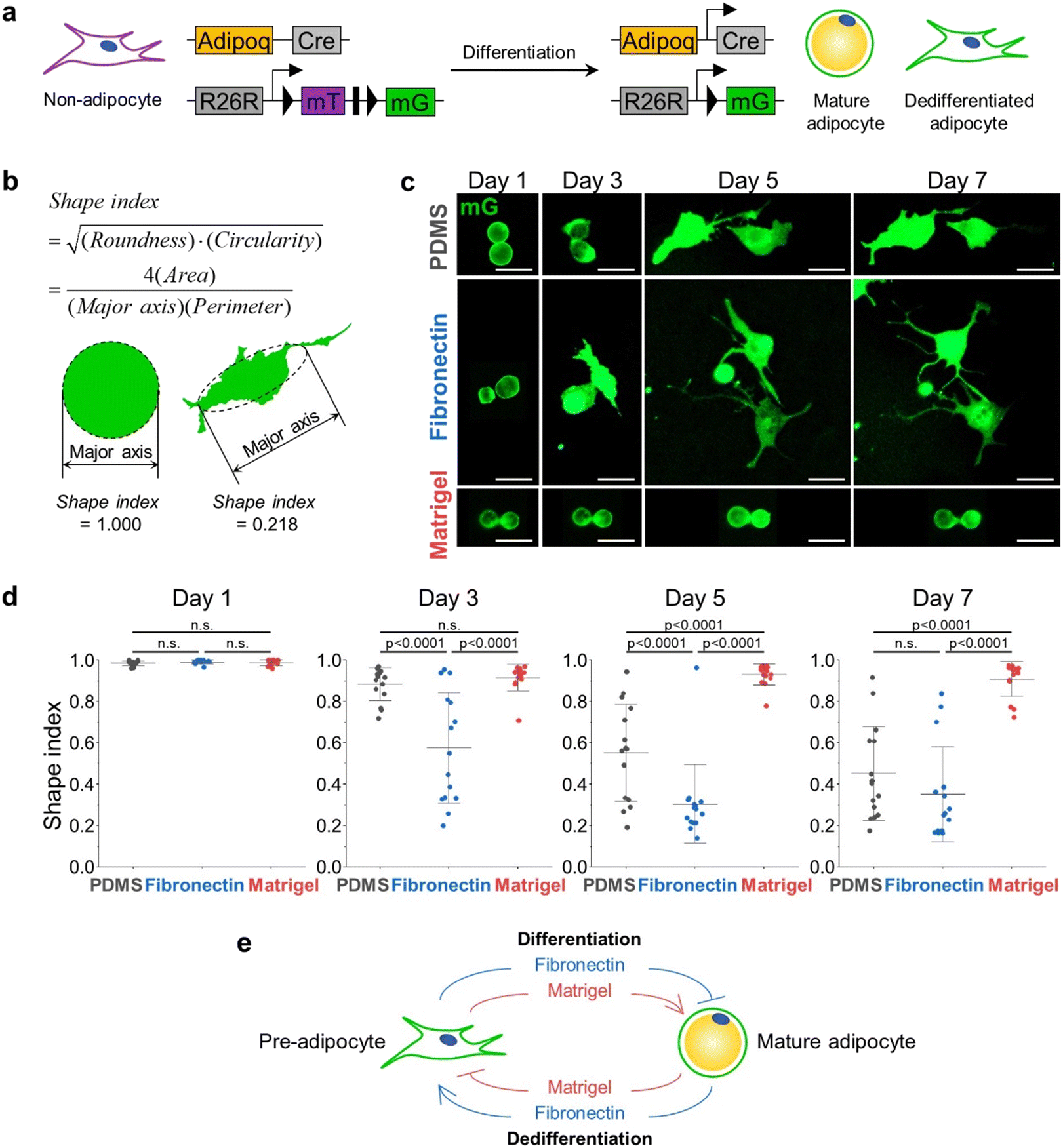 | ||
| Fig. 2 The change in shape index is ECM-dependent. The fibronectin condition promoted the dedifferentiation while the Matrigel condition inhibited it. a Schematic diagram for specific labeling and tracing of adipocytes using Adipoq-Cre; mT/mG mice. b Example of shape index used to quantify the degree to which adipocytes are dedifferentiated. Mature adipocytes have shape indices of 1. c Representative images of primary adipocytes cells undergoing dedifferentiation at each condition on day 1, 3, 5, and 7 (PDMS: adipocytes were embedded in untreated PDMS chamber, fibronectin: adipocytes were seeded in PDMS chip coated with 100 μg ml−1 fibronectin, Matrigel: adipocytes were embedded in 50% Matrigel medium). d Shape index distributions of scWAT adipocytes over time. I-shaped bars represent mean ± s.d. n = 15 each. p-Value from one-way ANOVA with Fisher's LSD post hoc test. e Schematic representation of promoting/inhibiting effect of ECM type on adipocyte differentiation and dedifferentiation. | ||
In the case of mature adipocytes isolated from scWAT, the shape indices of all cells under conditions P, F, and M are approximately 1 on day 1, which means that the cells are mostly circular in shape. On day 3, all the groups have dedifferentiated when compared to the ones on day 1, evidenced by the changes in the shape indices. Though there was no significant difference between the shape indices of cells under the conditions P and M, the cells under condition F showed significantly lower values of shape indices. On day 5, the cells seeded on the untreated PDMS chip exhibited a similar distribution of shape indices to that of cells under condition F on day 3. For condition F, the shape indices of the cells on day 5 became considerably smaller than those on day 3, and their distribution was also significantly narrowed. On the other hand, the cells for condition M did not dedifferentiate further compared to day 3. None of the three conditions made cells further dedifferentiate between days 5 and 7. Collectively, mature adipocytes under condition F dedifferentiated the fastest. However, condition P also induced the cells to dedifferentiate to a similar degree, only taking longer than condition F. Adipocytes embedded in the Matrigel were resistant to dedifferentiation, retaining LDs with high shape indices (Fig. 2c and d).
Adipocytes isolated from vWAT exhibited similar changes in shape indices with those isolated from the scWAT (ESI† Fig. S2). These results suggest that ECM conditions that restrict or promote adipogenesis have an opposite effect on adipocyte dedifferentiation. Notably, the findings establish a set of culture conditions that are either permissive or non-permissive to adipocyte dedifferentiation, as defined by ECM conditions. This result can also be explained by the fact that collagen VI and laminin are the common major components of in vivo adipose tissue and Matrigel,30 suggesting that the physiologically relevant 3D soft microenvironment constructed by Matrigel may contribute to the maintenance of adipocyte integrity, hindering the dedifferentiation process. As summarized in Fig. 2e, we found that the ECM condition that inhibits adipocyte differentiation, specifically fibronectin, promoted adipocyte dedifferentiation.
Suppression of Hippo or activation of Hedgehog signaling pathways induce adipocyte dedifferentiation
Here, we investigated whether the concept illustrated in Fig. 2e could also be applied to signaling pathways that regulate adipogenesis. In particular, the Hippo signaling pathway has been identified as a key regulator that promotes adipogenesis,31,32 whereas the Hedgehog signaling pathway inhibits adipogenesis.33Thus, we sought to evaluate whether the suppression of the Hippo or activation of the Hedgehog signaling pathway would promote adipocyte dedifferentiation. To this end, we isolated mature adipocytes from the scWAT of Adipoq-Cre; mT/mG mice and treated them with XMU-MP1, a Hippo signaling inhibitor, or SAG21k, a Hedgehog signaling activator (Fig. 3a). Based on our findings in the preceding section, we chose Matrigel as the ECM material to eliminate the influence of ECM-induced dedifferentiation. Indeed, both XMU-MP1 and SAG21k treatment potently induced adipocyte dedifferentiation even under Matrigel culture conditions which were non-permissive for dedifferentiation in vehicle-treated control adipocytes (Fig. 3b and c).
 | ||
| Fig. 3 Inhibitors of adipogenesis and deletion of PPARγ are molecular cues that induce adipocyte dedifferentiation. a Experimental scheme for isolating primary adipocytes from Adipoq-Cre; mT/mG mice and their analysis 4 days after culture. b Confocal images of adipocytes isolated from Adipoq-Cre; mT/mG mice and treated with 10 μM XMU-MP1 or 1 μM SAG21k for 4 days. Vehicle (DMSO) is used as a control. c The shape indices of XMU-MP1 or SAG21k treated cells were significantly lower than those of vehicle-treated cells. p-Values from one-way ANOVA with Fisher's LSD post hoc test. d Schematic diagram for inducible knockout of PPARγ in adipocytes labeled with EGFP using Pparg iAKO (Adipoq-CreERT2; Ppargflox/flox; mT/mG) mice. e Experimental scheme for isolation of primary adipocytes from control (Adipoq-CreERT2; mT/mG) or Pparg iAKO mice after tamoxifen administration and their analysis 4 days after culture. f Confocal images of adipocytes isolated from control or Pparg iAKO mice 4 days after culture. Nuclei are stained with Hoechst (blue). Scale bar, 50 μm. g The shape indices of Pparg iAKO cells were significantly lower than those from control cells. p-Value from two-tailed t-test with Welch correction. | ||
Deletion of PPARγ in mature adipocytes induces dedifferentiation
Peroxisome proliferator-activated receptor γ (PPARγ) is well-known as the master regulator of adipocyte differentiation and function.34–37 Therefore, we speculated that deletion of PPARγ would also promote the dedifferentiation of adipocytes. To test this notion, we established a mouse model in which both deletion of PPARγ and fluorescent labeling of mature adipocytes could be simultaneously induced by tamoxifen administration. This model was developed by crossing three different transgenic mice (Adipoq-CreERT2; Ppargflox/flox; mT/mG, herein, Pparg iAKO) (Fig. 3d). Following tamoxifen administration to Pparg iAKO mice, primary adipocytes from scWAT were isolated and embedded in Matrigel (Fig. 3e). As anticipated, PPARγ-deleted adipocytes labeled with EGFP displayed numerous small LDs and enlarged, irregular cytoplasmic regions (Fig. 3f and g), which are a characteristic feature of dedifferentiated adipocytes.15 Altogether, these observations highlight the crucial roles of Hippo/Hedgehog signaling pathways and PPARγ in maintaining mature adipocyte identity in addition to their roles during adipogenesis. This result also emphasizes how the direct activation or suppression of downstream regulators can override Matrigel's effect in inhibiting adipocyte dedifferentiation.Dynamic cellular deformations accompany LD secretion during adipocyte dedifferentiation
Next, we took advantage of our ceiling culture chip to perform time-lapse imaging with high-spatiotemporal resolution while adipocytes were undergoing adipocyte dedifferentiation. Here, we used adipocytes derived from mT/mG mice without the adiponectin-Cre transgene, such that all cells express a membrane-targeted tdTomato signal. Adipocytes were then labeled with BODIPY FL to allow the simultaneous analysis of LD dynamics. In the early stage of the dedifferentiation, adipocytes featured a large LD and several tiny LDs around the large one. Interestingly, some cells exhibited protrusive motion with well-developed pseudopodia (ESI† Video S1), which is atypical behavior for fully differentiated adipocytes. In mature adipocytes that come into contact with the ceiling, the large central LD, which was initially spherical in shape, flattened to conform to the ceiling surface, generating many pseudopodia (Fig. 4a, 1st column). The temporal changes in the aspect ratio of lipid droplets shown in Fig. 4b implied that these LDs were likely subject to intracellular compressive stress (Fig. 4a, 2nd–3rd column). The vibrational motion is shown in ESI† Fig. S3a in detail. As soon as it is extruded out of the cell, the LD restored its initial spherical shape, as shown in Fig. 4a, 4th column, and b, suggesting the immediate release of compressive stress upon extrusion. Prior to extrusion, the LDs exhibited vigorous vibrations, quantified as the periodic fluctuations in the angle θ between the x-axis and the major axis of the LD (ESI† Video S2, Fig. 4b). As the aspect ratio becomes 1, neither the major axis nor the angle θ could be defined further (Fig. 4b and c). Some cells even displayed a rope-like structure, shown as a crescent-shape band, squeezing the LD to suddenly extrude it from the cell (ESI† Video S3). This structure appeared to be reminiscent of the contractile ring typically appearing during the telophase of the cell division. Subsequently, the rim through which the LD exited gradually shrank to finally close like a purse-string, completely isolating the extruded droplet from the parental body (Fig. 4a, 5th–9th column). The black dotted line in the last row of Fig. 4 indicates the contracting rim structure, which was traced in the phase image by following the interface of the extruded LD in contact with the ceiling. After the rim structure was closed, the extruded LD departed from the cell body and was still wrapped by the plasma membrane, evidenced by the purple tdTomato signal (Fig. 4a, 10th–14th column).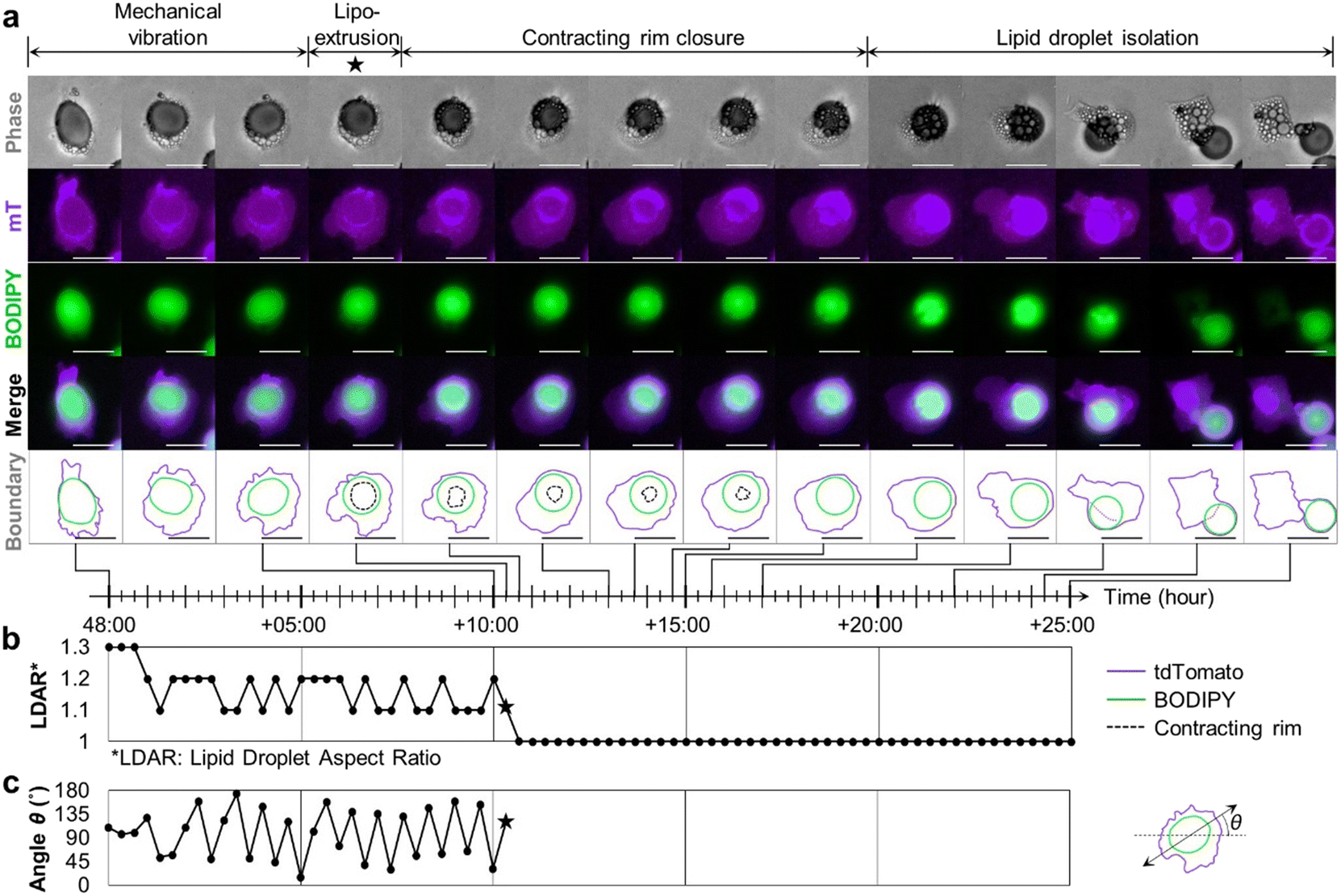 | ||
| Fig. 4 LD secretion accompanies dynamic deformation of LD. a Time-lapse image shows the entire process of LD secretion where the LD encapsulated in the dedifferentiating cell escapes from the cell body. The shape of the LD, which was initially flattened and trembling, suddenly restored its original spherical morphology. In phase images, a rim-like structure seemed to contract and then close completely. After the closure, the LD could move freely. The secreted LD seemed to be still enclosed in the cell membrane (scale bar = 100 μm, relative time). b The abrupt change in the aspect ratio of an LD upon LD extrusion at +10:20 h (marked by a star) indicates immediate release of compressive stress. c The angle θ between the x-axis and the major axis of the LD displays regular fluctuations. | ||
We analyzed 32 more liposecretion events between 24 and 144 hours after the mature adipocytes were seeded in the fibronectin-coated chip. Based on the common characteristics of the liposecretion, different modes of liposecretion can be categorized as follows (ESI† Fig. S3): (1) secretion involving mechanical vibration (Fig. 4), (2) secretion involving crescent-shaped band structure (ESI† Video S3), and (3) secretion involving sudden cellular elongation. Regardless of the mode of liposecretion, lipid extrusion seems to be made possible by intracellular mechanical contractility.
Fully differentiated mature adipocytes do not proliferate. However, a number of reports have suggested that adipocytes regain proliferative capability during the dedifferentiation process.11,15 Similarly, our EGFP+Adipoq-Cre; mT/mG adipocytes undergoing the LD secretion process were shown to proliferate, passing down the LDs to the daughter cells during cell division (ESI† Video S4).
Dynamic actin remodeling mediates LD secretion in dedifferentiating adipocytes
As is the case for many dynamic cellular processes, the whole LD secretion process that features oscillation and extrusion likely involves intracellular actin-mediated force generation. To determine how the F-actin structures of mature adipocytes are remodeled to extrude the largest organelle, i.e., the central LD of a unilocular adipocyte, out of the cell, we examined the structural organization of F-actin. First, wild-type adipocytes undergoing dedifferentiation were fixed and stained in the ceiling culture chip. Then, we arranged the images of fixed cells along a virtual time axis based on the extent to which the LDs had emerged from the cell. This arrangement allowed us to estimate the dynamic remodeling of the actin structures at each designated step of LD secretion (Fig. 4). Here, we divided the LD secretion phenomenon into three phases: the pre-secretion, secretion, and post-secretion phases. Each phase was again divided into two sub-phases (Fig. 5a). At the pre-secretion phase, cells formed pseudopodia on the ceiling. The cells at the first sub-phase featured flattened LDs with actin filaments distributed everywhere except the inside of the LD, what we called an actin shell (Fig. 5a and b). Occasionally, the actin shell developed holes of varying sizes along the surface of the LDs (Fig. 5a and c). | ||
| Fig. 5 Rapid LD secretion is accompanied by dynamic actin cytoskeleton remodeling. a Schematic representation showing each phase of rapid LD secretion. b Actin structure of representative cell at phase I-(1). c Cell at phase I-(2) having actin holes at the downside. d Cell at phase II-(1) showing actin band that is squeezing the LD. e At phase II-(2), a large LD squeezed out of the main cell body restores its original spherical shape. The actin band is rolled into a thick ring-like structure. f At phase III-(1), the actin band remains where the LD has gone (yellow arrowhead). g LDs are completely secreted at phase III-(2). Stress fibers are developed after LD secretion. Holes in the actin mesh still exist (subset). h Orthogonal y–z slices of the cell at phase II-(1). A large hole in the actin shell is clearly observed at the top view (white arrow) and the orthogonal slices (white arrowheads) (scale bar = 100 μm). | ||
At the beginning of the main secretion phase, the small actin rims merged into one or two larger ones (Fig. 5a and d). The large holes in the actin shell were also observed by the orthogonal views of 3D-reconstructed F-actin images obtained with confocal microscopy (Fig. 5h). Despite the presence of the holes, some cells still contained the LDs deformed by the actin band. Based on the morphological clues from Fig. 4, the subsequent process would be the extrusion of LDs through the opening, now identified as the rim structure made of actin. The deformed LDs in Fig. 5d were consistent with what was shown in the trembling cells shown in Fig. 4, ESI† Video S2. Therefore, we hypothesize that the vibrational movements of LDs were caused by the interactive forces between the contracting actin band and the resistance of the plasma membrane. Following the extrusion, three-dimensional actin bands rolled up to condense onto a ring structure on the ceiling (Fig. 5a and e).
After being pushed out, the LD eventually departed from the site of exit, leaving behind a hollow actin ring structure (Fig. 5a and f). Those voids, the consequences of LD secretion, are unique signatures of dedifferentiated adipocytes that are not present in normal fibroblasts. Cells that have secreted most LDs finally began to develop stress fibers, placing themselves in a different state (Fig. 5a and g). In summary, we identified the existence and the role of rearranging actin structures during rapid LD secretion by mapping the snapshots of stained cells in selected essential phases on the virtual time axis of real-time images.
Inhibition of non-muscle myosin suppresses adipocyte dedifferentiation
Actomyosin structures are critical in various intra- and inter-cellular activities that require mechanical forces, including cell division, migration, tissue morphogenesis, and muscle contraction. Actomyosin bundles can generate force through the sliding motion between actin filament and myosin units, represented by the closure of the contractile ring in cytokinesis and the rear contraction of a migrating cell. Based on the observations that cells displayed mechanical vibrations and that the actin structures were remodeled during the LD secretion process, we hypothesized that the LD secretion is induced by the contractility of non-sarcomeric actomyosin. To investigate the involvement of myosin in the squeezing motion during the extrusion process, blebbistatin treatment was used to specifically inhibit the ATPase activity of myosin II. Mature adipocytes derived from scWAT of Rosa26-mT/mG mice were isolated and loaded in a fibronectin-coated ceiling culture chip. The control group was treated with BODIPY FL, and the blebbistatin treated group was treated with BODIPY FL and 50 μM blebbistatin. Over 5 days, the cells of the control group underwent dedifferentiation, featuring prominent pseudopods and active migration, while some of them completed LD secretion (ESI† Fig. S4a, cell α, β). However, those of the blebbistatin-treated group failed to secrete LDs and exhibited negligible movements or deformations (ESI† Fig. S4b). While there were no differences between the shape indices of the two groups on day 1, the blebbistatin-treated group showed significantly higher shape index values than the control group on day 5 (ESI† Fig. S4c), implying that the dedifferentiation was suppressed by inhibition of myosin II. These results indicate that LD secretion and dedifferentiation are mediated by actomyosin-dependent contractility.Lipid droplet contents can be transported to adjacent adipocytes
Most dedifferentiating cells discharged their LD contents via the dynamic actin cytoskeleton remodeling processes (Fig. 5). However, a few cells were spotted to inject their LD contents directly into their neighboring cells, evidenced by the size change of the cells in contact with others. As the diameter of the donor cell decreased, that of the receiver cell increased, supporting the transfer of LD droplet content. Cells participating in this transfer process were usually round in shape, indicating that only minimal dedifferentiation had occurred. Once the donor cells passed over their LD to the receiver cells, they either departed or shrunk on the spot (Fig. 6a and b, ESI† Video S5).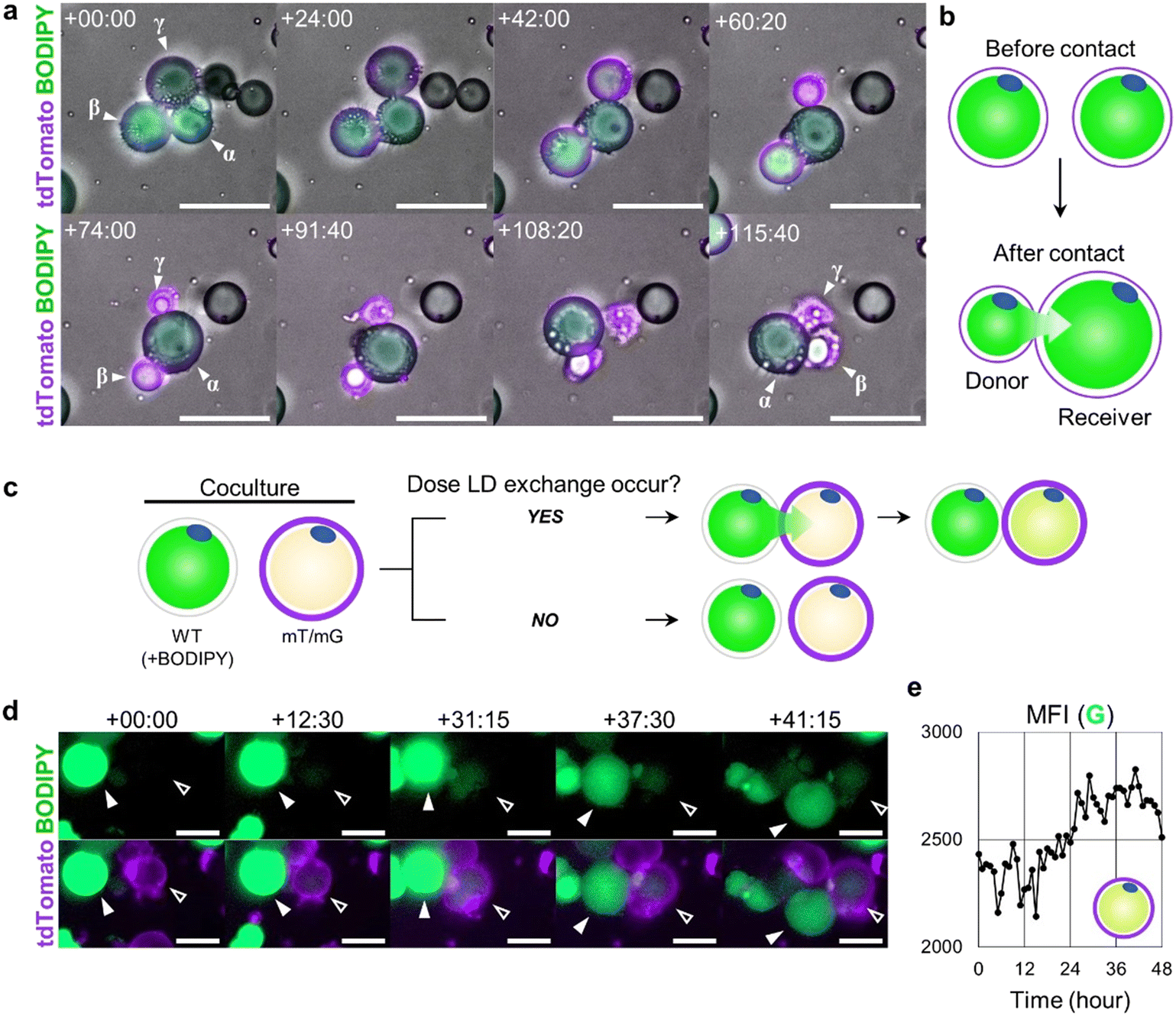 | ||
| Fig. 6 LD secretion by intercellular LD injection. a Time-lapse image showing the LD injection phenomenon (scale bar = 100 μm, relative time from 19 h:40 m). Cells β and γ transfer their LD to cell α. b Schematic representation of LD injection between two different cells. c Schematic representation of experimental setting. WT cells with LDs dyed with BODIPY and mT/mG cells were co-cultured in the ceiling culture chip. d The LD in the mT/mG cell did not express GFP at first, but over time, it gradually expressed GFP. This is evidence that mT/mG cells (open arrowhead, receiver) have received the BODIPY labeled LDs from neighboring WT BODIPY+ cells (solid arrowhead, donor) (scale bar = 50 μm, relative time from 1 d:4 h:30 m). e The mean fluorescence intensity (MFI) of the green channel measured within the boundary of the mT/mG cell (open arrowhead, receiver) demonstrates elevation over time. | ||
To confirm that the observed phenomenon was the LD transfer between two adjacent cells, not the simultaneous lipogenesis and lipolysis in two neighboring cells, we co-cultured WT mice cells and mT/mG mice cells, with only the LD contents of WT mice cells stained with BODIPY prior to the co-culture. In this setup, the mT/mG mice cells expressed membrane-targeted tdTomato signal, and WT mice cells expressed BODIPY FL signal on their LDs (Fig. 6c). Over time, it was observed that the WT donor cells appeared to inject some of their BODIPY+ LD content into mT/mG receiver cells, making the LD of the receiver mT/mG cell fluoresce green (Fig. 6d and e, ESI† Video S6).
Neighboring adipocytes exchanges their lipid droplet contents
As previously shown in Fig. 3b, pharmacological suppression of the Hippo pathway by XMU-MP1 induced adipocyte dedifferentiation. To further investigate the Hippo pathway function in maintaining the mature adipocyte state, we genetically blocked the Hippo pathway in adipocytes by creating Adipoq-CreERT2; Lats1/2flox/flox; tdTomato mice (referred to as Lats1/2 iAKO) (ESI† Fig. S6a), with Adipoq-CreERT2; tdTomato mice as control. Adipocytes from both Lats1/2 iAKO and control mice were loaded to the non-coated ceiling culture chip and treated with 2 μM of 4-hydroxytamoxifen to induce Cre recombination (ESI† Fig. S6b). On day 5, the shape indices of Lats1/2 iAKO cells were significantly lower than that of control cells (ESI† Fig. S5). Using this Lats1/2 iAKO model, we report another novel cellular activity revealed by the use of our ceiling culture chip, which is the intercellular LD exchange in granular form (ESI† Fig. S6c). This is distinct from the LD content injection shown in Fig. 6, where the LD contents were simply injected into the neighboring cell in a unidirectional manner. In the intercellular lipid exchange, LD contents were delivered in several small vesicles. This type of LD transfer was bi-directional and occurred while maintaining their sizes (ESI† Video S7). The mechanism governing such bi-directional LD transfer between two neighboring adipocytes is an intriguing subject that merits further investigation.Conclusion
Obesity and related metabolic diseases continue to increase, demanding further efforts to better understand adipocyte biology and pathology. In particular, to gain more complete knowledge of the regulatory mechanisms of adipocytes, it is important to study not only adipogenic differentiation but also the dedifferentiation of mature adipocytes. One reason the latter has been understudied is due to the inherent characteristics of mature adipocytes as buoyant and fragile, making it difficult to culture and observe them in vitro. As a workaround, many studies begin with preadipocytes that will be differentiated into adipocytes in vitro rather than with primary mature adipocytes since the induced adipocytes are adherent, making them easier to handle. However, those induced cells display relatively large cytoplasm with multiple droplets, not sharing key characteristics with the primary adipocytes.17 To address these challenges, our novel ceiling culture chip provides a platform for easy trapping of buoyant primary adipocytes for better imaging.Furthermore, cultured adipocytes that have undergone dedifferentiation are indistinguishable from remnant stromal cell types after primary adipocyte isolation. Thus, the impurity of stromal fibroblasts can be misinterpreted as dedifferentiated adipocytes. In this regard, the origin of the fibroblast-like cells has always been of question. The use of the Adipoq-Cre; mT/mG system, however, successfully resolved the issue by differentially labeling dedifferentiated adipocytes and stromal fibroblasts.
Equipped with the ceiling culture chip and Adipoq-Cre; mT/mG system, we established specific conditions that either promoted or inhibited adipocyte dedifferentiation. Based on well-documented studies of adipogenesis, a condition that would inhibit adipogenesis was hypothesized to promote adipocyte dedifferentiation and vice versa. We then validated our hypothesis using four specific conditions known to suppress differentiation from progenitor cell to mature adipocyte (use of fibronectin, suppression of Hippo or activation of Hedgehog signaling pathway, and the deletion of PPARγ) also promoted dedifferentiation.
Furthermore, by taking full advantage of the ceiling culture chip for high-resolution live imaging of adipocyte dedifferentiation, we uncover novel aspects of the LD secretion process. Based on real-time observation, images of fixed and immuno-stained cells could be sorted in order according to the extent of LD secretion. Here, actin cytoskeletal structures were shown to be dynamically remodeled, featuring the development of holes in the actin shell functioning as an exit for LDs. To the best of our knowledge, this study is the first report of such holes produced as a result of actin remodeling.
Another phenomenon observed with our ceiling culture chip was the transfer of the LD content between adipocytes in contact with each other. Previously, Hu et al. have reported the direct communication between cells of eWAT explant via gap-junctional couplings,38 that aligns with the LD content exchange between neighboring adipocytes observed in our study. The transfer events were shown to occur over the course of 20 h either unidirectionally or bidirectionally. Interestingly, during the unidirectional transport, donors shrank dramatically following the transfer. This phenomenon may be interpreted as a cell's effort to preserve nutrient resources by transferring the lipid stored in the unhealthy cells to healthy neighbors. On the other hand, the latter events of mutual exchange were extremely rarely observed, specifically in the Lats1/2 iAKO strain. Based on morphological analysis, the process seems to be associated with the dedifferentiation process. In addition, we developed and applied the shape index in order to quantitatively indicate the degree of dedifferentiation of adipocytes. Conventionally, the expression levels of preadipocyte markers, such as Sca1, Cd34, and Pref1, were profiled to identify the extent of dedifferentiation. Instead, the shape index can be measured without any antibody or agent, serving as a dynamic metric to characterize cells that are undergoing dedifferentiation. Despite the general perception of mature adipocytes as being rather static, we confirmed that adipocytes were, in fact, very dynamic, involving the active generation of mechanical stresses during the pathophysiological events. Furthermore, two fascinating phenomena of LD extrusion and LD transfer lead to new perspectives and opportunities for further studies in identifying the key regulators of accumulation and release of mechanical stresses through cytoskeletal remodeling during the dedifferentiation process.
Author contributions
Conceptualization: J. K., K. Y. P., S. C., J. M. S., and J. H. S., data curation: J. K., and K. Y. P., formal analysis: J. K., and K. Y. P., funding acquisition: J. K., K. Y. P., J. M. S., and J. H. S., investigation: J. K., K. Y. P., S. C., and U. H. K., methodology: J. K., K. Y. P., S. C., U. H. K., D. S. L., J. M. S., and J. H. S., project administration: J. M. S., and J. H. S., resources: J. K., K. Y. P., S. C., U. H. K., D. S. L., J. M. S., and J. H. S., software: J. K., and K. Y. P., supervision: J. M. S., and J. H. S., validation: J. K., K. Y. P., S. C., J. M. S., and J. H. S., visualization: J. K., K. Y. P., J. M. S., and J. H. S., writing – original draft: J. K., K. Y. P., J. M. S., and J. H. S., writing – review & editing: J. K., K. Y. P., S. C., U. H. K., D. S. L., J. M. S., and J. H. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF-2020M3A9E4039658 and 2021R1A2C200757311). This work was supported by KAIST intramural grants (N11190172 and N11200076).Notes and references
- T. Song and S. Kuang, Clin. Sci., 2019, 133, 2107–2119 CrossRef CAS PubMed.
- P. E. Scherer, Diabetologia, 2019, 62, 223–232 CrossRef PubMed.
- A. Sakers, M. K. De Siqueira, P. Seale and C. J. Villanueva, Cell, 2022, 185, 419–446 CrossRef CAS PubMed.
- A. G. Cristancho and M. A. Lazar, Nat. Rev. Mol. Cell Biol., 2011, 12, 722–734 CrossRef CAS PubMed.
- Q. A. Wang, A. Song, W. Chen, P. C. Schwalie, F. Zhang, L. Vishvanath, L. Jiang, R. Ye, M. Shao, C. Tao, R. K. Gupta, B. Deplancke and P. E. Scherer, Cell Metab., 2018, 28, 282–288 CrossRef CAS PubMed , e283.
- X. Fu, A. Zhao and T. Hu, in Cellular Dedifferentiation and Regenerative Medicine, Springer Berlin Heidelberg, Berlin, Heidelberg, 2018, pp. 175–193, DOI:10.1007/978-3-662-56179-9_9.
- Y. Oki, S. Watanabe, T. Endo and K. Kano, Cell Struct. Funct., 2008, 33, 211–222 CrossRef CAS PubMed.
- A. Poloni, G. Maurizi, F. Serrani, S. Mancini, G. Discepoli, A. L. Tranquilli, R. Bencivenga and P. Leoni, Cell Proliferation, 2012, 45, 66–75 CrossRef CAS PubMed.
- T. Sakuma, T. Matsumoto, K. Kano, N. Fukuda, D. Obinata, K. Yamaguchi, T. Yoshida, S. Takahashi and H. Mugishima, J. Urol., 2009, 182, 355–365 CrossRef PubMed.
- A. Poloni, G. Maurizi, S. Anastasi, E. Mondini, D. Mattiucci, G. Discepoli, F. Tiberi, S. Mancini, S. Partelli, A. Maurizi, S. Cinti, A. Olivieri and P. Leoni, Exp. Hematol., 2015, 43, 137–146 CrossRef CAS PubMed.
- J. A. Côté, G. Ostinelli, M.-F. Gauthier, A. Lacasse and A. Tchernof, Cell Tissue Res., 2019, 378, 385–398 CrossRef PubMed.
- J. A. Côté, M. F. Gauthier, G. Ostinelli, D. Brochu, K. Bellmann, A. Marette, F. Julien, S. Lebel and A. Tchernof, J. Cell. Physiol., 2019, 234, 10270–10280 CrossRef PubMed.
- J. H. Shin, D. W. Shin and M. Noh, Biochem. Pharmacol., 2009, 77, 1835–1844 CrossRef CAS PubMed.
- G. Maurizi, A. Poloni, D. Mattiucci, S. Santi, A. Maurizi, V. Izzi, A. Giuliani, S. Mancini, M. C. Zingaretti, J. Perugini, I. Severi, M. Falconi, M. Vivarelli, M. R. Rippo, S. Corvera, A. Giordano, P. Leoni and S. Cinti, J. Cell. Physiol., 2017, 232, 2887–2899 CrossRef CAS PubMed.
- H. Sugihara, N. Yonemitsu, S. Miyabara and K. Yun, Differentiation, 1986, 31, 42–49 CrossRef CAS PubMed.
- J. A. Côté, J. Lessard, M. Pelletier, S. Marceau, O. Lescelleur, J. Fradette and A. Tchernof, FEBS Open Bio, 2017, 7, 1092–1101 CrossRef PubMed.
- M. J. Harms, Q. Li, S. Lee, C. Zhang, B. Kull, S. Hallen, A. Thorell, I. Alexandersson, C. E. Hagberg, X.-R. Peng, A. Mardinoglu, K. L. Spalding and J. Boucher, Cell Rep., 2019, 27, 213–225 CrossRef CAS PubMed , e215.
- A. Yokomizo, Y. Morimoto, K. Nishimura and S. Takeuchi, Micromachines, 2019, 10, 358 CrossRef PubMed.
- D. Kim, K. Kim and J. Y. Park, Lab Chip, 2021, 21, 1974–1986 RSC.
- B. D. Pope, C. R. Warren, M. O. Dahl, C. V. Pizza, D. E. Henze, N. R. Sinatra, G. M. Gonzalez, H. Chang, Q. Liu, A. L. Glieberman, J. P. Ferrier, C. A. Cowan and K. K. Parker, Lab Chip, 2020, 20, 4152–4165 RSC.
- J. Eguchi, X. Wang, S. Yu, E. E. Kershaw, P. C. Chiu, J. Dushay, J. L. Estall, U. Klein, E. Maratos-Flier and E. D. Rosen, Cell Metab., 2011, 13, 249–259 CrossRef CAS PubMed.
- A. Sassmann, S. Offermanns and N. Wettschureck, Genesis, 2010, 48, 618–625 CrossRef CAS.
- W. He, Y. Barak, A. Hevener, P. Olson, D. Liao, J. Le, M. Nelson, E. Ong, J. M. Olefsky and R. M. Evans, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15712–15717 CrossRef CAS PubMed.
- M. D. Muzumdar, B. Tasic, K. Miyamichi, L. Li and L. Luo, Genesis, 2007, 45, 593–605 CrossRef CAS PubMed.
- T. Heallen, M. Zhang, J. Wang, M. Bonilla-Claudio, E. Klysik, R. L. Johnson and J. F. Martin, Science, 2011, 332, 458–461 CrossRef CAS PubMed.
- M. Kim, M. Kim, S. Lee, S. Kuninaka, H. Saya, H. Lee, S. Lee and D.-S. Lim, EMBO J., 2013, 32, 1543–1555 CrossRef CAS PubMed.
- L. Madisen, T. A. Zwingman, S. M. Sunkin, S. W. Oh, H. A. Zariwala, H. Gu, L. L. Ng, R. D. Palmiter, M. J. Hawrylycz, A. R. Jones, E. S. Lein and H. Zeng, Nat. Neurosci., 2010, 13, 133–140 CrossRef CAS PubMed.
- D. Lin, T. H. Chun and L. Kang, Biochem. Pharmacol., 2016, 119, 8–16 CrossRef CAS PubMed.
- K. O'Connor, H. Song, N. Rosenzweig and D. Jansen, Biotechnol. Lett., 2004, 25, 1967–1972 CrossRef PubMed.
- E. C. M. Mariman and P. Wang, Cell. Mol. Life Sci., 2010, 67, 1277–1292 CrossRef CAS.
- J.-H. Hong, E. S. Hwang, M. T. McManus, A. Amsterdam, Y. Tian, R. Kalmukova, E. Mueller, T. Benjamin, B. M. Spiegelman, P. A. Sharp, N. Hopkins and M. B. Yaffe, Science, 2005, 309, 1074–1078 CrossRef CAS PubMed.
- Y. An, Q. Kang, Y. Zhao, X. Hu and N. Li, PLoS One, 2013, 8, e72042 CrossRef CAS PubMed.
- J. Chen, C. Bao, J. T. Kim, J. S. Cho, S. Qiu and H. J. Lee, J. Agric. Food Chem., 2018, 66, 11926–11934 CrossRef CAS PubMed.
- P. Tontonoz, E. Hu and B. M. Spiegelman, Cell, 1994, 79, 1147–1156 CrossRef CAS.
- Y. Barak, M. C. Nelson, E. S. Ong, Y. Z. Jones, P. Ruiz-Lozano, K. R. Chien, A. Koder and R. M. Evans, Mol. Cell, 1999, 4, 585–595 CrossRef CAS PubMed.
- E. D. Rosen, P. Sarraf, A. E. Troy, G. Bradwin, K. Moore, D. S. Milstone, B. M. Spiegelman and R. M. Mortensen, Mol. Cell, 1999, 4, 611–617 CrossRef CAS PubMed.
- F. Wang, S. E. Mullican, J. R. DiSpirito, L. C. Peed and M. A. Lazar, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18656–18661 CrossRef CAS PubMed.
- J. Hu, X. Li, R. L. Judd and C. J. Easley, Lab Chip, 2020, 20, 1503–1512 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2lc00428c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |