
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Label-free 1D microfluidic dipstick counting of microbial colonies and bacteriophage plaques†
Sultan İlayda
Dönmez
 *a,
Sarah H.
Needs
a,
Helen M. I.
Osborn
a,
Nuno M.
Reis
bc and
Alexander D.
Edwards
*ac
*a,
Sarah H.
Needs
a,
Helen M. I.
Osborn
a,
Nuno M.
Reis
bc and
Alexander D.
Edwards
*ac
aReading School of Pharmacy, University of Reading, Whiteknights, RG6 6AD, UK. E-mail: s.ilaydadnmez@gmail.com; a.d.edwards@reading.ac.uk
bDepartment of Chemical Engineering and Centre for Biosensors, Biodevices and Bioelectronics (C3Bio), University of Bath, Claverton Down, Bath BA2 7AY, UK
cCapillary Film Technology Ltd, Daux Road, Billingshurst, West Sussex RH14 9SJ, UK
First published on 6th July 2022
Abstract
Counting viable bacterial cells and functional bacteriophage is fundamental to microbiology underpinning research, surveillance, biopharmaceuticals and diagnostics. Colony forming unit (CFU) and plaque forming unit (PFU) counting still requires slow and laborious solid culture on agar in Petri dishes or plates. Here, we show that dip-stick microfluidic strips can be used without growth indicator dye for rapid and simple CFU ml−1 and PFU ml−1 measurement. We demonstrate for the first time that fluoropolymer microcapillaries combined with digital imaging allow bacteriophage plaques to be counted rapidly in a dip-and-test format. The microfluidic length scales offer a linear 1-dimensional alternative to a 2D solid agar medium surface, with colonies or plaques clearly visible as “dashes” or “gaps”. An inexpensive open source darkfield biosensor system using Raspberry Pi imaging permits label-free detection and counting of colonies or plaques within 4–8 hours in a linear, liquid matrix within ∼200 μm inner diameter microcapillaries. We obtained full quantitative agreement between 1D microfluidic colony counting in dipsticks versus conventional 2D solid agar Petri dish plates for S. aureus and E. coli, and for T2 phage and phage K, but up to 6 times faster. Time-lapse darkfield imaging permitted detailed kinetic analysis of colony growth in the microcapillaries, providing new insight into microfluidic microbiology and colony growth, not possible with Petri dishes. Surprisingly, whilst E. coli colonies appeared earlier, subsequent colony expansion was faster along the microcapillaries for S. aureus. This may be explained by the microenvironment offered for 1D colony growth within microcapillaries, linked to a mass balance between nutrient (glucose) diffusion and bacterial growth kinetics. Counting individual colonies in liquid medium was not possible for motile strains that spread rapidly along the capillary, however inclusion of soft agar inhibited spreading, making this new simple dip-and-test counting method applicable to both motile and non-motile bacteria. Label-free dipstick colony and plaque counting has potential for many analytical microbial tasks, and the innovation of 1D colony counting has relevance to other microfluidic microbiology.
1. Introduction
Viable cell or virus counting is a central method in microbiology and remains vital from fundamental research to clinical and public health. Measuring microbial cell concentrations may aid diagnosis of infection or treatment decisions, and can allow detection and prevent contamination, controlling the safety of food products and water sources.1–3 This method involves the spreading of serial sample dilutions onto solid agar medium in Petri dishes and counting colony-forming units (CFU). Bacteriophage enumeration requires the addition of host bacteria cells to the solid media alongside sample of phage to allow lytic growth leading to a clear patch that allows counting of plaque-forming units (PFU).4,5 Colony counting on solid media measures viable cells that can form colonies specifically, which is why it remains a standard reference method for quantifying viable bacteria. Although minimal equipment is needed, and plates can be scanned or counted with the naked eye, it requires some skill plus sufficient incubation space for large numbers of Petri dishes, and is limited to microorganisms able to form colonies on agar. Counts are only made after overnight incubation, yet viable cell numbers can change very quickly, severely delaying knowledge of true viable cell count until after other experiments are completed.Turbidimetry, microscopy or flow cytometry can be used to rapidly estimate cell concentration. Although quick and convenient, turbidimetry measurements (typically with a spectrophotometer) are only suitable for non-cloudy samples6 and have a limited range (∼108 to 1010 CFU mL−1); estimating cell number simply by light scattering means it cannot distinguish between live and dead cells, and requires calibrating for particular species, instrument, and growth conditions. Flow cytometers are quantitative and sensitive for counting individual cells, where instrumentation is available. Microscopy can be conducted by trained staff or using digital microscopy plus image analysis, but care is needed in sample preparation. For both microscopy and cytometry, fluorescent dyes can be used to distinguish live from dead cells.7
Accurate counting of viable viral particles is significantly harder than cell counting. Viral particles are smaller than bacterial cells, making standard cytometry and microscopy techniques challenging. Molecular methods such as real-time quantitative PCR for sequence-specific measurement or using dyes for bulk viral nucleic acid concentration can be used.8 However, non-viable particles can also contain nucleic acid, so such molecular measures do not fully correlate with viable infectious particle counts. Culture based methods add further methodological complications. For bacteriophage enumeration, host bacteria must not only be available to permit viral replication but must be retained in place to permit individual replicating viral particles to be identified through plaque formation. Typically, a solid media matrix is used to retain host cells plus viral particles in a 2D plane, allowing visualisation of lysed host cells as an empty space. The double agar layer (DAL) method for bacteriophage enumeration developed by Felix d'Herelle in 1917 allows formation of visible lysis plaques in a uniform lawn of host bacteria, to detect and enumerate phages.9 But although DAL remains the standard tool for viable phage detection and measurement, plaque counting methods are restricted to skilled staff in the laboratory and remain challenging to automate or standardise to increase efficiency. To respond to antibiotic resistance using biological therapeutics such as therapeutic phage,10,11 and to simplify pathogen detection using phage,12 improved methods for phage enumeration are therefore vital. Viruses of eukaryotic hosts are likewise counted as plaque forming units on a solid culture surface coated with a monolayer of host cells. Among alternatives to the classical DAL method, one promising method for phage enumeration uses compartmentalisation into monodisperse droplets, as an alternative to a 2D solid medium surface. Droplet enzymatic testing is used to detect phage amplified in a simple droplet. Thus, phage in the emulsion culture can be visualized and counted. For example, reporter phages were detected using a reporter enzyme. However, in addition to requiring a fluorescent dye substrate, the requirement for reporter enzyme limits this example method to only certain samples.13
Microfluidic devices are increasingly used by many disciplines to overcome the limitations of traditional “millifluidic” laboratory methods. By simplifying and/or automating fluidic processing steps, speeding up diffusion-limited processes, and cutting sample volume, microfluidics provide opportunities to make faster, easier and more cost-effective biological analyses. Biosensors add monitoring and time-resolved measurements, with the potential to study microbial growth alongside enumeration.14 The speed of detection of microbes by growth may not be limited by diffusion to the same degree as other bioassays that depend on the analyte needing to diffuse to the site of a binding agent; instead miniaturisation may offer a different benefit: by confining growing cells within a smaller space, earlier detection may be possible for example by retaining converted dye in a smaller volume, increasing local concentration and thus signal intensity.15 However, timescales for bacteria counting methods that rely on growth will not necessary yield a reduction in detection times through miniaturisation alone.16
Microfluidic devices can be used to detect and characterize cell number/density with a digital growth readout within microdroplets or compartments which can be converted into viable cell counts using simple statistical distribution models.17,18 The compartment size and number affects analytical dynamic range so can be adjusted depending on the analytical need.16 Microdroplets have been used extensively for single cell characterisation studies but can also be used for accurate density calculations. A laser-induced fluorescence (LIF) based method has been developed to count the dispersion of bacterial and eukaryotic cells encapsulated in droplets. By tracking the peaks within the droplets, cell counts were accurately calculated.19 Another study used 4 pL microdroplets to determine cell counts for urinary tract infections, in which cell count is a diagnostic marker. Similarly, detection of bacteria was determined by fluorescence readout from a molecular probe for bacterial 16S.20 In another study, the concentration of live bacteria was determined by fluorescent imaging of bacteria under magnetic fields using magnetically assisted microfluidic method. Capture to antibody modified magnetic-microbeads detected ∼5 × 103 CFU ml−1E. coli within 1 hour.21 Fewer methods have been demonstrated for enumerating phage.
To exploit microfluidics for analytical microbiology, a more detailed understanding of similarities and differences in bacterial growth between conventional systems and microsystems is needed. Digital imaging has become a major tool for biosensing, with inexpensive digital microscopy and miniaturised cameras becoming particularly relevant for microbial biosensing. Smartphone cameras or the CMOS image sensors mass-produced for consumer smartphones can be adapted, for example using Raspberry Pi camera platform. Biosensing and digital imaging has the potential to provide essential insight into the engineering science of microfluidic microbiology.22 In a phage lysis monitoring by smartphone emulsion-based application, a single phage in a droplet is highly replicated in a short time by amplifying the bacteriophage in monodisperse droplets. An increase in the number of phages via β-galactosidase-activated fluorescent probes results in a fluorescent signal, and the droplet receiving the signal is considered phage positive.13 Increasing interest in open-source hardware has increased the development of inexpensive, customizable personal or analysis equipment. A fluorescence microscope device consisting of blue LEDs, a Raspberry Pi camera, and a filter allows imaging of bacteria.23 For scaled-up analytical microbiology screening, a low-cost and high-performance imaging system was developed adding the Raspberry Pi camera to a 3D printed robotic motion system allowing time-resolved automated imaging of hundreds of colorimetric and fluorescence microbiology tests.24 We believe that such biosensing platforms that combine microfluidics with digital imaging will allow us to better understand microbial growth in microfluidic culture volumes. We therefore developed a darkfield biosensing platform that detects bacteria and bacteriophage by light scattering,25 built from inexpensive, open-source hardware, for sensing bacterial growth within multiplex dip-stick microfluidic devices made from microcapillary film.26
We assessed whether the confinement of sub-mm channels was sufficient to retain bacteria and bacteriophage hosts in place to detect colonies and phage without any further labelling or plating. We demonstrate for the first time that microfluidic dip-sticks can be used as an easy-to-use “dip-and-test 1D liquid microbial media” for rapid counting of viable bacterial colony-forming units and bacteriophage plaque-forming units. Darkfield imaging allows label-free detection with simple LED plus inexpensive CMOS digital camera, and the simple microcapillary dipsticks avoid the challenge of agar plating. We explored how quickly plaques and colonies could be detected using an automated image capture system permitting detailed analysis of microfluidic microbial growth kinetics. We show these microbial dip-sticks represent a compact, accurate, rapid, simple, and inexpensive way to count colonies and plaques in liquid media, thereby replacing 140 year old agar Petri dishes with microfluidics.
2. Materials and methods
2.1 Materials and bacterial strains
Microcapillary film (MCF) produced from Teflon® fluorinated ethylene propylene (FEP) was sourced from Lamina Dielectrics Ltd (Billingshurst, West Sussex, UK) with external dimensions of 4.3 mm wide and 0.4 mm thick, containing 10 microcapillaries each with an internal diameter of approximately 200 μm.26 Non-motile E. coli B strain (a host for T2 phage) was obtained from the National Centre for Biotechnology Education, University of Reading (Reading, UK) and bacteriophage T2 was obtained from Mojgan Rabiey, School of Biological Sciences, University of Reading (Reading UK). S. aureus ATCC 19685 and S. aureus phage K ATCC 1985-B1 (family Myoviridae) were obtained from Danish Malik, Biotechnology and Biomedical Engineering, Loughborough University. Motile E. coli strain ATCC 25922 was purchased from LGC Standards (Middlesex, UK).2.2 Darkfield imaging sensor system design
A Raspberry Pi model 4 with HQ Camera (Farnell, UK) was mounted with an 8MP 50 mm 14.5 × 10.9° angle of view C-Mount lens (Cool Components, UK) focussed closely to capture macro images with an approximate field of view of 50 mm. White LED strip (RS Components, UK, product code: 153-3639) powered with a 12 V power supply (Amazon UK) were used as light source. To achieve darkfield imaging and allow dye- or label-free direct detection of bacteria inside MCF through light scattering from bacterial cells, a simple box enclosing key components comprising: light source (LED), samples of up to 8 MCF test strips spaced 9 mm apart in a 3D printed holder, 3D printed blockers to create the darkfield behind the samples (carefully aligned to eliminate direct and reflected light from the LED around the sample) and Raspberry Pi HQ camera. A 3D printed base plate to mount all components, with the camera mounted on aluminium extrusion, and added the Raspberry Pi computer and blockers to this base (Fig. 1 and S1B†). A touchscreen mounted on the side of the box helped run python scripts to take photos every 10 minutes. The photographs were subsequently analysed on an external PC using ImageJ (NIH, USA) software.27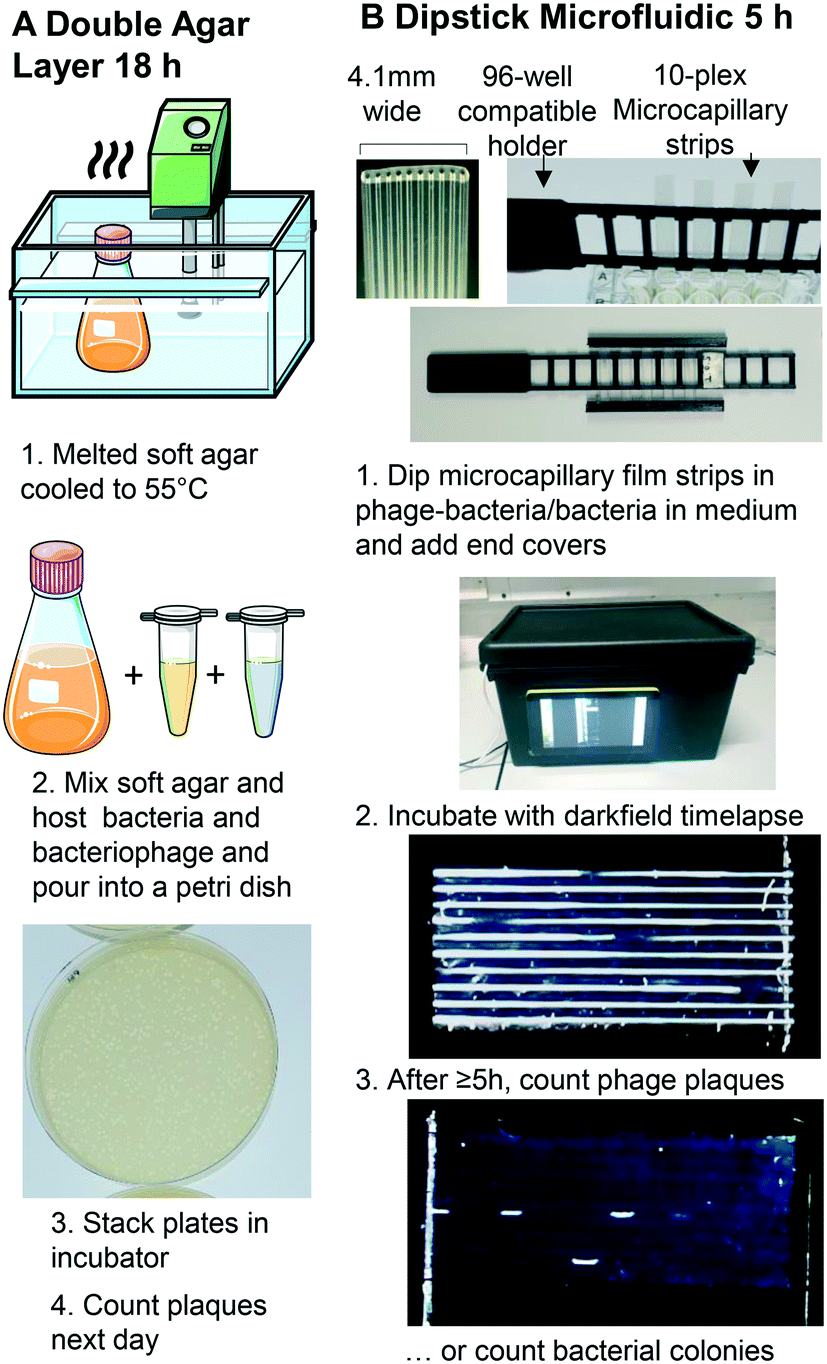 | ||
| Fig. 1 Double layer agar method vs. miniaturised 1D liquid colony and plaque counting method. A. Double layer agar method. B. 1: Sample preparation and uptake into 10× ∼1 μL microcapillary array per strip. Steps 1–5 illustrate key steps for analysis of bacterial growth kinetics within microcapillaries. A 3D printed holder permits simple uptake of up to 8 samples from a 96-well plate, and 3D printed end covers containing silicone grease seal ends to avoid air bubbles from entering or sample drying out. 2: A darkfield illuminated imaging box monitors growth by timelapse imaging with Raspberry Pi and HQ camera, operated via touchscreen. 3: The small portable imaging box fits into incubator for culture at 37 °C to capture colony or plaque growth kinetics. After incubation, bacteriophage plaques or bacterial colonies are clearly visible, from as early as 5 h. | ||
2.3 1D microcapillary dip-strip assay to measure bacterial CFU
Hydrophilic test strips were produced in 1 m long batches of MCF. MCF lengths were filled with 5 g L−1 polyvinyl alcohol (PVOH) solution in ultrapure water, as previously described.26E. coli B strain was routinely cultured on LB agar media at 37 °C overnight, a single colony was re-suspended into lysogeny broth (LB) to 0.2 OD600. S. aureus was routinely cultured on BHI agar at 37 °C overnight, a single colony was re-suspended into brain heart infusion (BHI) broth to 0.2 OD600. Bacterial suspensions were diluted a further 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 to obtain approximately 105 CFU mL−1. E. coli and S. aureus strains were prepared in 96-well plates in LB or nutrient broth (NB) respectively and serially diluted to the range of 1–3 × 103 CFU mL−1. Bacteria counts were confirmed by overnight colony counting on LB agar using the spread plate method (ESI† Methodology).
:1000 to obtain approximately 105 CFU mL−1. E. coli and S. aureus strains were prepared in 96-well plates in LB or nutrient broth (NB) respectively and serially diluted to the range of 1–3 × 103 CFU mL−1. Bacteria counts were confirmed by overnight colony counting on LB agar using the spread plate method (ESI† Methodology).
Individual hydrophilic 33 mm MCF strips were clipped into the 3D printed rack compatible with a 96 well plate (Fig. 1B and ref. 16). One end of the microcapillary test strips were dipped into each well allowing the sample to fill the 10 microcapillaries by capillary action. Endcaps filled with silicone grease cover the end of the strips to eliminate evaporation. The test rack was put into the darkfield imaging system such that each capillary ran horizontal and incubated at 37 °C (Fig. 1B).
2.4 1D microcapillary dip-strip assay to measure bacteriophage PFU
Detailed methods of bacteriophage amplifications, storage and standard DAL can be found in the ESI† (S1 Methodology); the basic methodology is outlined in Fig. 1A. To measure PFU in Petri tubes, serial dilutions of phage K (103, 2 × 103, 4 × 103 PFU mL−1) were prepared and mixed with 4 × 108 CFU mL−1 host bacteria. A volume of 100 μL of the mix was added to wells in a microplate and MCF test strips were used as before. Bacteriophage number was confirmed using DAL method and incubated at 37 °C overnight (S1† Methodology).2.5 Image analysis
Time-lapse images from the full incubation period were stacked in the ImageJ software for analysis, including counting the number of colonies and plaques, estimating the length of the colony along the individual capillaries over time, and estimating microbial growth through analysis of “Normalised Intensity” for individual colonies. To determine the normalised intensity, identical rectangular area was selected around each bacterial colony identified at a late timepoint, and a set of 6 timepoints at regular intervals defined from before the colony first became visible – considered the baseline – to the late timepoint. The mean grey scale pixel intensity in this rectangular area was taken for each single colony at the selected time points using ImageJ (NIH, USA).27 The mean intensity for each timepoint was then normalised by firstly subtracting the mean intensity at baseline before the colony appeared, and secondly dividing by the end mean intensity at the latest time measured. Thus 100% was defined at the last time the colony measured, and 0% the starting point before the colony was visible. This analysis was conducted with the full 8-bit RGB colour image, as no significant difference in scatter intensity was seen between red, green and blue channels.3. Results and discussion
3.1 Microfluidic 1D liquid matrix counting of individual bacterial colonies using microcapillary dip-sticks
We show here for the first time that ∼200-micron diameter capillaries can be used as a 1-dimensional (1D) liquid matrix in which it is simple to detect and count bacterial colonies and phage plaques, when imaged with darkfield illumination. We termed this method 1D culture to reflect the replacement of classical 2D surface of solid agar medium in Petri dishes with liquid media in microcapillaries that constrain the growing capillaries through microfluidic dimensions.We first assessed if accurate measurement of bacterial colony forming units was possible. When growing bacteria in situ within microcapillaries, we observed bacterial colonies becoming visible after a short incubation allowing us to make absolute counts of bacterial CFU down to very low concentrations (around 250 CFU mL−1), with (Fig. 2). Instead of spreading on the surface of a 2D solid hydrogel medium, the long narrow microcapillaries allowed enumeration of colonies in a 1D liquid matrix. Each individual viable bacterial cell that could grow into a colony appeared after incubation as a clear white area of bright light scatter, enabling clear label-free counting of individual bacterial colonies (Fig. 1). 1D colony counts were compared with gold-standard, solid agar plate counts, with E. coli and S. aureus diluted to multiple cell densities. A starting target inoculum of 5 × 103 CFU mL−1 was used, a concentration at which only a few bacteria are present in each capillary (10−3 mL per ∼33 mm length). After sample loading into microcapillary dip-strips simply through capillary action and incubation, colonies became clearly visible after ∼5.5 h and ∼8.5 h for E. coli and S. aureus respectively (Fig. 2A and B) versus overnight incubation for Petri dishes. Individual colonies were easily distinguishable within microcapillaries at these cell densities and timepoints. When multiple colonies grew in a single microcapillary, as long as they were far enough apart to not overlap they could be clearly distinguished, meaning the technique is fully quantitative and not binary/digital (yes vs. no) in terms of initial number of cells per capillary, and could tolerate operation over a broader range of concentrations than simple limiting dilution counting.16 When CFU mL−1 values were calculated by 1D colony enumeration in microcapillaries, near-identical counts were obtained to gold standard 2D agar plate count (r2 = 0.99 for both E. coli and S. aureus, Fig. 2C and D). Although E. coli and S. aureus both showed close agreement between microcapillary and agar plate counts, a later timepoint was imaged for S. aureus because these colonies appeared later. Surprisingly we found the 1D microcapillary method counts had smaller standard deviations than agar plate counts (compare size of the error bars Fig. 2C and D) when each was measured in triplicate experiments, suggesting the 1D colony microcapillary counting may be more reproducible.
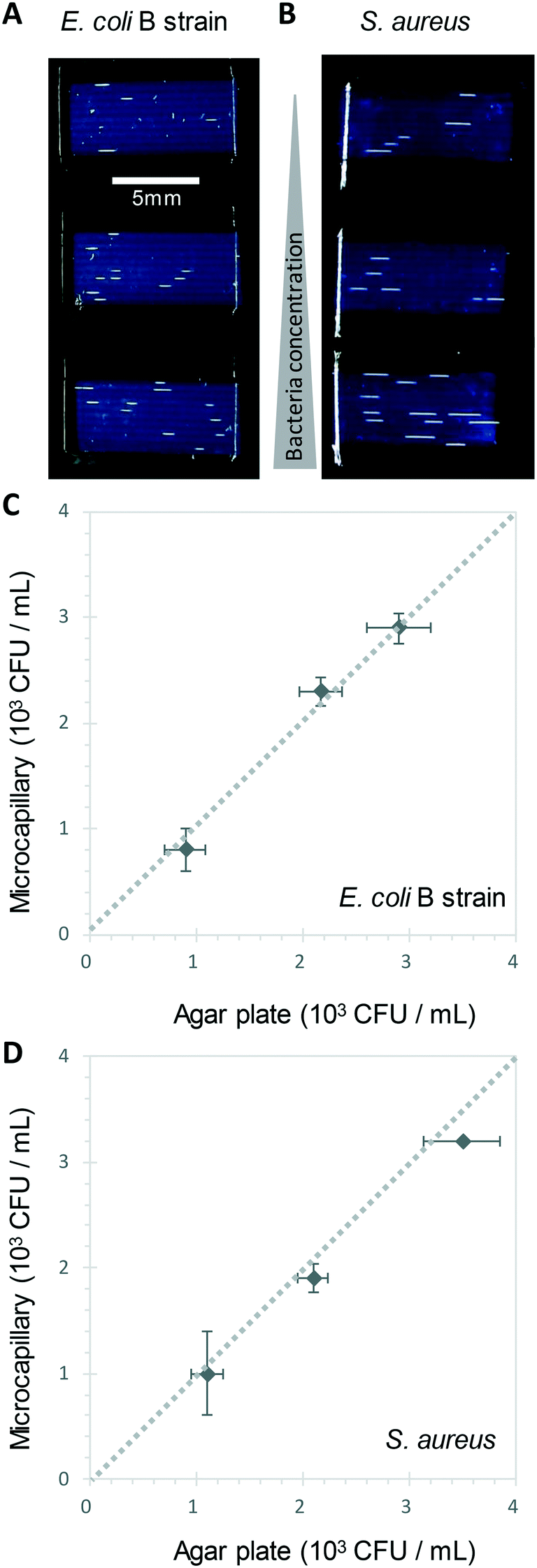 | ||
| Fig. 2 1D counting of single bacterial CFU within microcapillaries in liquid broth. A and B Image at 6 h of 3 different E. coli B strain (A) and S. aureus (B) concentrations grown in 4 μL broth per strip distributed within 10 individual microcapillaries, with individual colonies visible as short white horizontal lines along the horizontal microcapillaries. The test strips appear dark blue through light scatter by the plastic surrounding microcapillaries. C) and D) Comparison of colony forming unit (CFU) counts for E. coli (C) and S. aureus (D) measured in liquid in microcapillaries vs. solid agar plate counts. Error bars represent ± standard deviation from 3 independent replicate experiments. Dashed line indicates 1:1 relationship between the two counting methods. | ||
The imaging area of each test strip was approximately 10 mm long corresponding to a test volume of ∼4 μl in the area visible in the darkfield imaging setup for 10 capillaries, resulting in a lower limit of detection (LOD) of 2.5 × 102 CFU mL−1, assuming detection of a minimum of 1 colony forming unit. At that point the technique becomes digital and only able to differentiate between growth versus no growth. For higher concentrations there is an increasing probability of two or more CFU on the same microcapillary, potentially overlapping, similar to when plating higher cell concentrations onto agar. As the size of the colonies grows with time it becomes more probable that nearby colonies will overlap, and thus the exact upper limit of detection will vary depending on species, growth condition, and timepoint imaged. This is discussed in greater depth in section 3.2. Here, we found counts of ∼3 × 103 CFU mL−1 remained accurate (Fig. 2C and D).
A disadvantage of the current capillary system is that within the relatively small imaging area inherent in microfluidic systems, in contrast to a traditional 100 mm diameter Petri dish, only a few colonies or plaques are counted in each experiment. It's well known when measuring viable cell numbers that as the numbers of colonies counted falls towards low absolute numbers (e.g. 5–15 colony range), the uncertainty around the absolute CFU mL−1 increases. Some guidelines advise counting at least 25 colonies on agar plates to gain sufficient resolution for accurate counts. We conducted counts with larger numbers of replica samples (n = 6) and observed counts to be very repeatable even with low absolute numbers of colonies (Fig. S4†). Viable cell counts are typically measured over multiple orders of magnitude expressed on logarithmic scales, and note that care is required in diluting samples prior to colony counting. However, it should be remembered that higher uncertainty might be encountered in methods such as this 1D microcapillary system where a low absolute number of counts are recorded, and advise that where precise measurement of absolute viable cell numbers are important, only appropriate statistical expression of uncertainty in counts are used. For example, with low absolute colony counts, parametric statistical tests should be avoided since variation in counts would not be expected to be normally distributed. To overcome this limitation, longer strips or larger numbers of strips could be assessed, providing a larger growth volume and imaging area, thus higher absolute numbers of colonies or plaques to be counted. We also note that in common with many other viable cell or viral counting methods there are other sources of variation in measuring viable cell numbers; for example if sampling a growing population with a doubling time approaching 20 minutes, cell counts can change considerably depending on the exact timing of sample application to the strip.
3.2 Kinetics of bacterial colony growth in microcapillaries
The Raspberry Pi HQ camera darkfield imaging biosensor system used offered full control over imaging and gathering of time-resolved growth data, enabling detailed analysis 1D colony growth kinetics within microcapillaries. This setup was optimised for simultaneous capture of up to 8 microcapillary dip-strips, with total volume sampled of 8 × 4 μL tests, corresponding to 80 individual microcapillaries. From images recorded every 10 minutes, it was possible to generate time-lapse videos illustrating colony growth (Video S1 and Fig. S2†). The time-lapse image sets allowed us to clearly visualise colony growth using distance versus time plots that showed very distinct growth dynamics between the two bacterial species tested (Fig. 3). Thus, E. coli colonies appeared at earlier timepoints and yet the speed of colony expansion quickly slows (Fig. 3A). In contrast, although S. aureus colonies appeared later, they showed faster and more sustained colony expansion along the capillaries (Fig. 3B).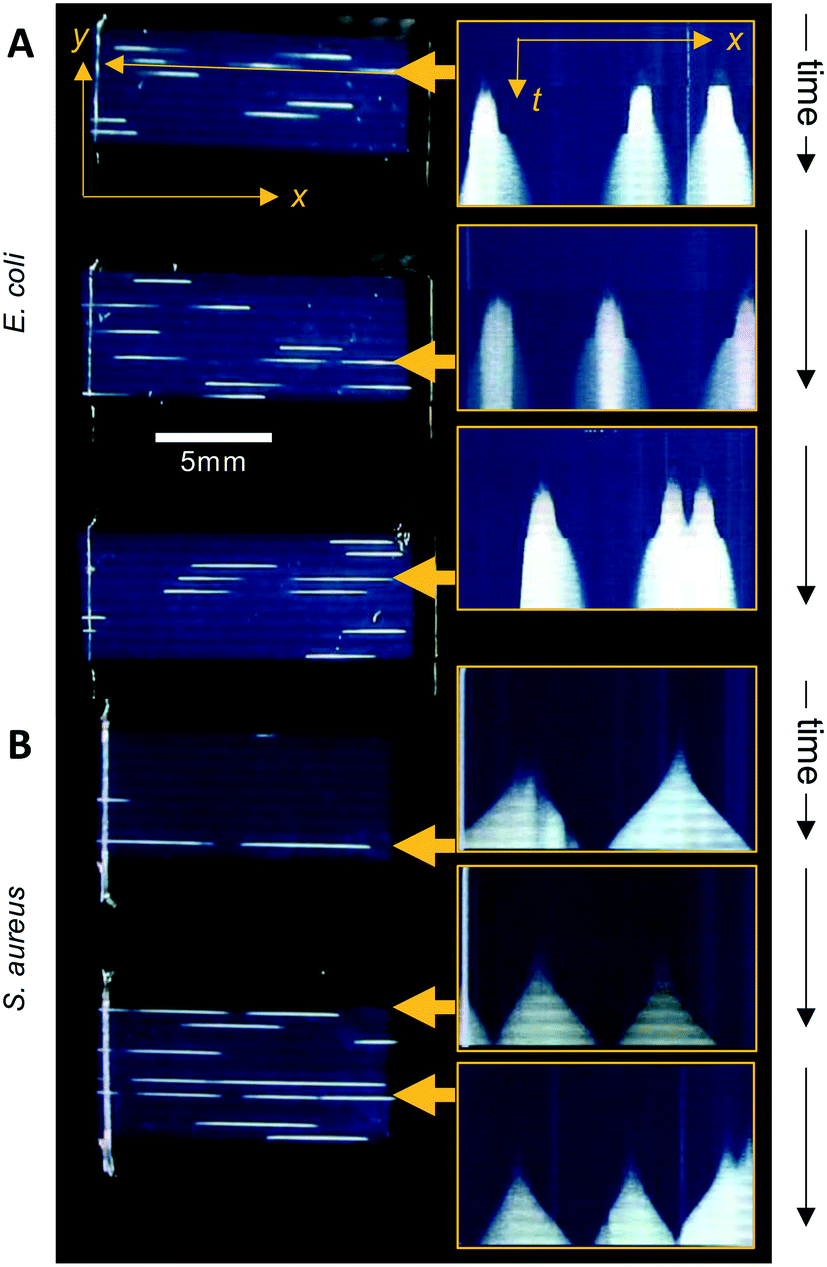 | ||
| Fig. 3 Visualising capillary growth dynamics in 1D liquid culture. On the right-hand side: intensity along the length of individual capillaries was plotted vs. time from analysis of timelapse image stacks, to illustrate the different colony growth dynamics for (A) E. coli vs. (B) S. aureus. The 6 capillaries indicated by yellow arrows were plotted on the left hand side, with the vertical axis representing time increasing downwards, with 1 pixel representing one individual image; images were captured at 10 minute intervals and are ∼80 pixels high, representing ∼13 hours total incubation. Scale measurement bar is the same for images to left as for x–t plots to right. | ||
Initially, we asked how fast colony counts could be made, by comparing growth of multiple individual capillaries in one set of strips. As expected, given the clonal nature of the suspensions tested, all individual colonies appeared at almost the same time for each sample tested. The earliest E. coli colonies to appear were visible 4.5 hours after incubation, and all colonies were visible within 50 minutes of these earliest colonies (Fig. 4A). For S. aureus, the fastest colonies to appear became visible after 8 h of incubation (Fig. 4B), again with all colonies growing at a similar rate and all becoming detectable within 1 h of the first colony detected. We conclude that confidant final counts could be determined at 6 h and 9 h for E. coli and S. aureus respectively, without the risk of missing slower-growing colonies.
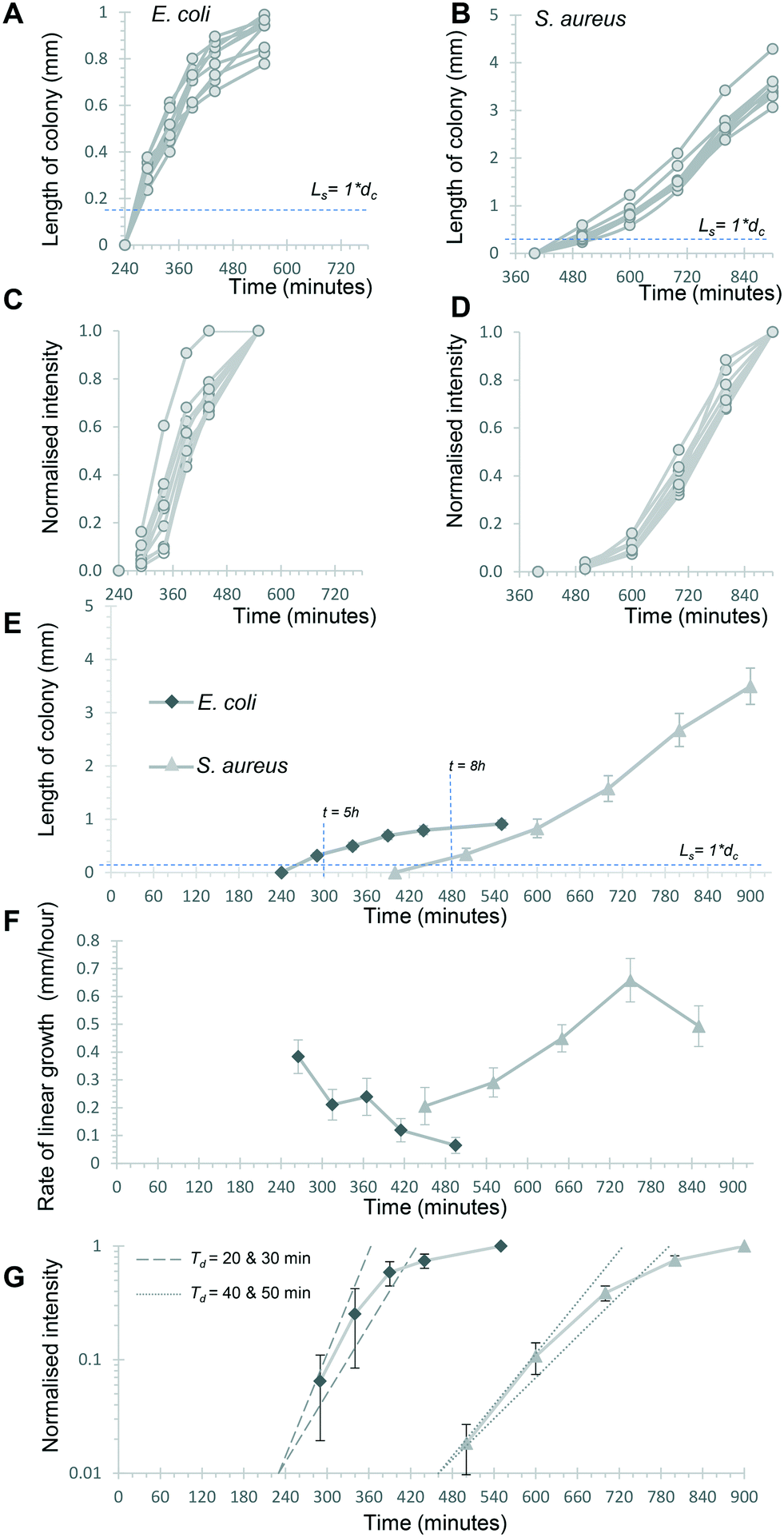 | ||
| Fig. 4 Kinetics of capillary colony growth in 1D liquid culture. Linear growth of 9 individual E. coli and S. aureus colonies was measured at the 6 timepoints indicated and all colonies plotted individually (A) and (B) to see variation in growth between colonies. Note different time and length axes are used given the different rates and sizes between these species. Bacterial growth kinetics within colonies was also estimated by calculating a normalised light scattering intensity measurement for both species (C and D) for the same 9 colonies. The mean length was plotted for both species (E) for the full experiment to show the distinct growth dynamics, with earliest time for counting at the marked timepoints. F) The rate of linear colony growth was calculated for each pair of length measurements for individual colonies and plotted at the middle timepoint. Mean rates for the 9 colonies are plotted, with error bars indicating ±1 standard deviation. G) By plotting mean normalised intensities on a logy scale, it's possible to estimate initial colony intensity rise during exponential growth. Dotted and dashed lines illustrate theoretical exponential growth for 20- and 30 minute doubling times (Td), starting from a normalised intensity of 0.01 at 240 minutes (pair of dashed lines) vs. 430 minutes (dotted pair). | ||
Notably, 1D colonies were far clearer with non-motile bacterial strains. The initial samples of E. coli B strain and S. aureus are non-motile; but when low cell concentrations of a motile strain of E. coli 25922 were incubated in microcapillaries, although at earlier time points initial bright foci appeared indicating colony growth were transiently observed, shortly after appearing became impossible to distinguish individual colonies of bacteria, presumably with their motility allowing rapid seeding and further growth along the length of the capillary (Video S2 and bottom pane in Fig. S3†). However, colonies of motile strains could be more clearly distinguished upon inclusion of a low concentration of agar in the culture medium, replicating the semi-solid agar used in traditional Petri dish methods that significantly limits the spread of the motile bacteria (top pane in Fig. S3 and Video S2†). The inclusion of low agar concentrations does add an extra step to the testing method, and semi-solid agar needs to be prepared by melting and maintained at an adequate temperature, otherwise flow of sample into capillaries by capillary action was not possible due to viscosity of solution. Thus, whilst 1D colony counting is far easier with non-motile strains, use of conventional semi-solid media additives makes this technique compatible with motile bacteria.
The time-resolved darkfield imaging of 1D colony growth allowed us to explore the rate of growth using different measurements. First, we analysed colony length as a simplified 1D measure of colony growth in liquid media, schematically represented in Fig. S6A and B.† It was clear this increased over a similar profile for multiple individual colonies over the 6 time points selected, from just before the first colony became visible (Fig. 4A and B), again confirming uniform growth of individual CFU. Secondly, we explored if quantitative measurement of growth of the bacterial suspension could be estimated from light scatter intensity. Other recent studies measuring bacterial cell density using innovative digital imaging techniques that detect light scatter have shown good correlation with colony counts.28,29 The dynamic range of darkfield scatter intensity was limited as images were optimised for early colony detection (i.e. brightest possible images at early timepoints) and for 8 test strips (i.e. lower resolution, larger number of samples); thus intensities rapidly became saturated and intensity non-linear to microbial cell density as the colony expanded. With this exact setup, the outline of each colony was hard to define automatically using methods such as particle analysis. We therefore calculated a normalised intensity to estimate growth, by defining a single fixed rectangular area of interest that fully contained the colony at a late stage of growth, to avoid artefacts from defining the colony area, and then calculating a normalised scatter intensity for this area of interest, such that the mean intensity of the final image varied from 0% (before colony appeared) to 100% (at last timepoint analysed) for the same rectangular area of interest (Fig. 4C and D). The normalised intensity and 1D length increased together but were clearly independent measurements, with length initially increasing faster than intensity (Fig. S5†). The close overlay of normalised intensity kinetics for 9 separate colonies each of E. coli and S. aureus suggests this measurement is repeatable (Fig. 4C and D). Without the normalisation step, the mean greyscale pixel intensity varied more between individual colonies (Fig. S6†) likely due to different areas of interest being selected for each colony. Small drift in microcapillary position during incubation prevented analysis of smaller areas of interest to more precisely define each colony. We therefore believe that, in spite of some limitations, this normalised intensity measurement provides a useful estimate of colony growth through increasing intensity as cell density increases (expected to correspond to turbidity measurements in conventional liquid culture), as opposed to linear expansion along the microcapillary.
3.3 Understanding bacterial colony growth dynamics in microfluidic culture
Having established that these two measurements – linear 1D colony growth and normalised intensity – were repeatable for sets of 9 individual colonies for each species, we then examined the microfluidic bacterial colony growth dynamics both in space and in cell density. This confirmed the very distinct patterns of colony growth between E. coli and S. aureus that were visible from distance-time plots (Fig. 3). We observed faster linear expansion of the latter despite a slower time to colony appearance and slower initial logarithmic growth in intensity. Whilst E. coli colonies appeared earlier, once colonies were visible the rate of linear colony growth was significantly faster for S. aureus and these colonies achieved a longer length (Fig. 4E). This could be explained by changes in colony growth speed – an initial rapid linear growth rate for E. coli of 0.4 mm h−1 declined as the colony grew, such that ∼5 h after first appearing the rate was 4× slower than S. aureus and the colony length remained under 1 mm (Fig. 4F). In contrast, although S. aureus colonies appeared ∼3 h later with a slower initial growth rate of 0.2 mm h−1, this speed steadily increased to around 0.4–0.5 mm h−1 (Fig. 4F), with colonies exceeding 3 mm in length by the end of the experiment.The plots of normalised intensity followed a typical microbial growth s-curve, suggesting an initial exponential growth period as the colony first appears, after which image intensity becomes saturated so we could no longer distinguish between image intensity saturation versus a growth plateau from nutrient depletion. We were able to estimate the rate of initial exponential growth and compare with expected liquid culture log phase growth rates for the two organisms studied. For E. coli initial exponential growth rates appear to approach a 20 minute doubling time (Fig. 4G – dashed lines indicate theoretical 20 and 30 minute doubling times). Previous studies of growth in fluoropolymer MCF using colorimetric resazurin dye agree that E. coli can grow with doubling time between 20–30 minutes.16S. aureus typically grows rapidly with similar doubling time to E. coli, yet we observed later appearance and slower initial growth for this species, suggesting the environment within the microcapillaries may become anaerobic slowing the growth rate for this facultative anaerobe. The initial exponential rate appeared closer to 40 minutes doubling time for S. aureus (Fig. 4G – dotted lines indicate theoretical 40 and 50 minute doubling times).
Previously, we reported that darkfield imaging could detect E. coli suspensions with turbidity at or above 0.34 OD600,25 corresponding to cell concentrations of 2.8 × 108 CFU mL−1. For a colony with a length with Ls equal to one microcapillary diameter dc and thus a volume of 6 nanolitres, this corresponds to ∼1700 cells. Starting from 1 CFU in this volume, this represents 1700-fold growth, requiring 11 successive doublings (211 = 2048-fold expansion). For a constant 20 minute doubling time this would take 220 minutes, not far from the 260 minutes before E. coli colonies reached this length. Similarly, at a constant 40 minute doubling time it would take 440 minutes to grow 1 CFU up to ∼2000 cells, consistent with the 480 minutes before S. aureus colonies reached this size. These estimates do not account for any initial lag prior to exponential growth, or in differences in the minimum cell density that can be detected through darkfield imaging given that we are using a slightly different configuration of camera; furthermore, light scattering intensity may differ between these species.
It was informative to contrast colony appearance times and increase in normalised intensity with linear colony growth kinetics. Surprisingly, linear colony expansion was not only faster but more sustained for S. aureus than E. coli, contrasting to the slower rate of initial intensity rise and later appearance. This discrepancy of appearing later plus slower initial intensity growth from an individual CFU, followed by faster linear colony expansion at later timepoints, could be due to differences in growth properties of the Gram-negative E. coli, vs. Gram-positive S. aureus. The later appearance of S. aureus might also be attributed to lower light scattering from these smaller cocci,30 but this would not affect the gradient of initial exponential increase in scatter intensity, that indicate that S. aureus does indeed growing slower than E. coli in this microsystem – possibly due to limited oxygen levels. However, when we evaluated growth of an obligate aerobic bacterial strain in rich broth in microcapillaries, no difference was found between growth in a shaker incubator vs. in fluoropolymer microcapillaries imaged by darkfield illumination, with similar growth curves observed in both conditions (Fig. S8†). This clearly indicates that even if oxygen may be depleted at some point by microbial metabolism within microcapillaries, there is sufficient oxygen in the system to clearly detect strong light scattering signal at microbial cell densities high enough for detecting colonies by darkfield imaging (Fig. S8†).
To understand this further, we calculated the expected rate of nutrient diffusion along a microcapillary. Assuming growth of bacteria is limited by carbon source (glucose), the diffusion time of glucose also becomes relevant to the dynamics of colony growth (Fig. S7†). We have estimated a rate of diffusion for glucose around 0.8 mm h−1 based on data from the literature.31 As the size of the colony reaches the diameter of the microcapillary, the 1D growth of the colony becomes limited by the diffusion of glucose. Also, in light of Monod's equation, the rate of growth of a microorganism is dependent on the concentration of the limiting nutrient on the interface. As the colony grows further, the carbon source fully depletes within the colony, therefore stimulating the growth of the colony along the length of the microcapillary. The rate of growth of the microorganism may also have a strong influence on the 1D rate of expansion of the colony. In our case S. aureus, with the linear rate of colony growth being smaller than rate of glucose diffusion, it is likely the glucose concentration at the interface will remain high, therefore the microorganism is able to grow near the maximum rate (Fig. S7C, bottom†). In contrast, with a faster-growing microorganism like E. coli, the glucose concentration in the interface can be entirely depleted as diffusion is not rapid enough to replenish at the interface, which forces the colony to grow at a lower growth rate (Fig. S7C, top†). This means the rate of growth of the colony becomes dependent on the rate of mass transfer of the limiting nutrient (glucose) from the bulk of the microcapillary to the interface, demonstrating 1D microcapillary counting can be manipulated by playing with the microenvironment, in particular growth conditions of the microorganism (such as temperature, different carbon and energy sources, or pH), the mass transport, or both, which is far harder to control on a standard solid medium such as agar Petri dish.
3.4 Simple, accurate, 1D microfluidic agar-free bacteriophage plaque counting
Bacteriophage enumeration is far more challenging than agar plate counting of bacterial colonies. The reference method, DAL, requires the live host organism to be seeded and movement restricted by plating soft agar on top of another agar layer. Careful addition of the correct concentration of bacteria to agar while it is still liquid requires careful temperature control to ensure the bacteria remain viable. We tested bacteriophage with E. coli and S. aureus host strains to determine whether phage count could be rapidly determined using a 1D capillary liquid medium method, a major improvement over the current method (Fig. S1†). The permissive S. aureus host bacteria were mixed with dilutions of bacteriophages and the hydrophilic MCF test strips added to the samples. As with the bacteria colony counts, an excellent agreement between phage plaque numbers and PFU concentrations for phage K were observed with 1D capillary counting compared to conventional DAL (Fig. 5). Likewise accurate phage counting of T2 phage concentration was achieved with the E. coli B strain host (r2 = 0.99 both T2 and phage K) (Fig. 6). This was also confirmed using soft agar in the capillary strips with no difference in results irrespective of whether liquid or agar was used in microcapillary dip-strips, indicating agar is not necessary for bacteriophage enumeration with these non-motile hosts (Fig. 6).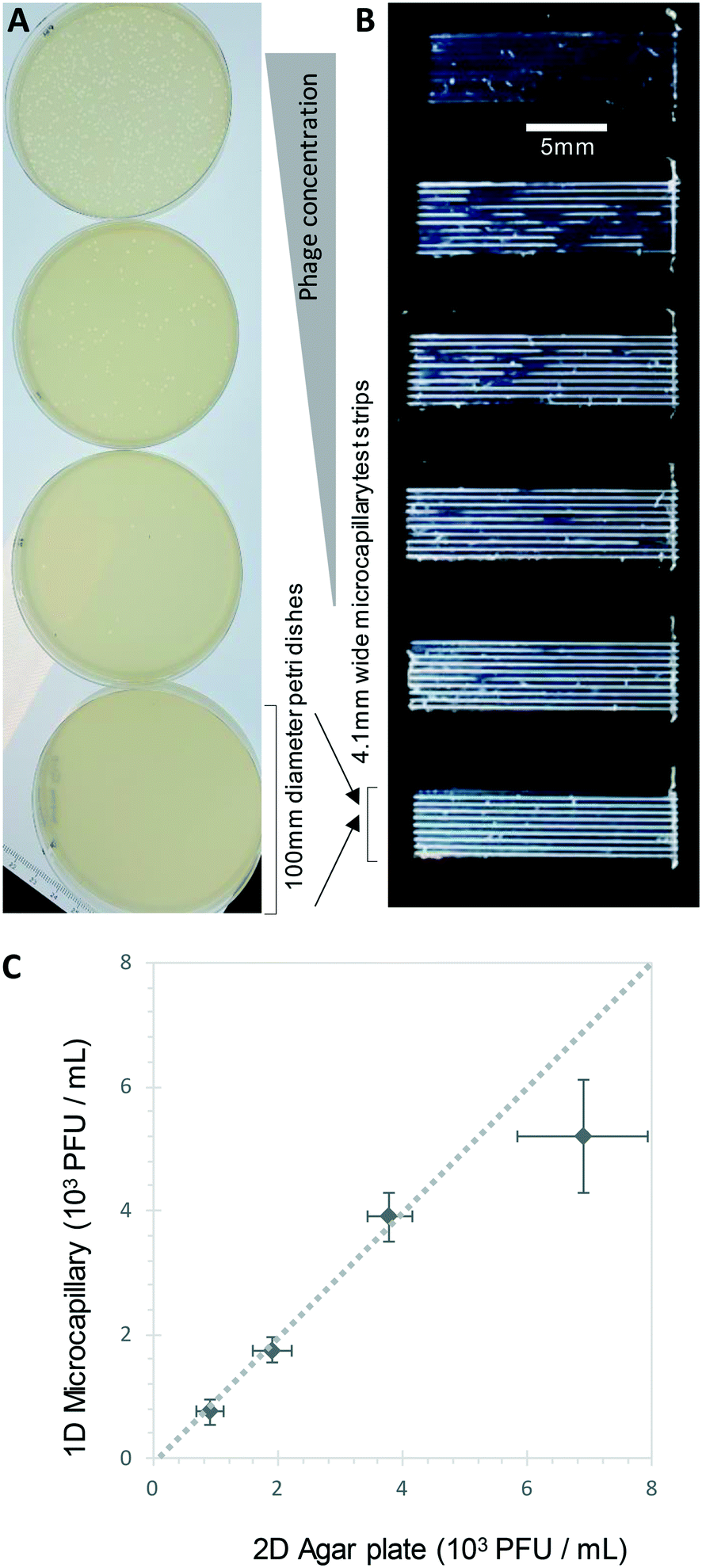 | ||
| Fig. 5 1D counting S. aureus phage K plaques in liquid broth within microcapillaries. A) Example endpoint images of five increasing phage K concentrations plated with conventional double agar layer method in Petri dishes, plus no phage control. B) The same phage K concentrations were measured within microcapillary dip-strips. C) Comparison of plaque forming units counted in microcapillaries vs. double agar layer method; it was not possible to calculate plaques for the highest concentration sample as most of the capillary was cleared hence only 4 concentrations were plotted. Mean counts of 10 replicate capillaries are plotted with error bars indicating ±1 standard deviation. Dashed line indicates 1:1 relationship between the two counting methods. | ||
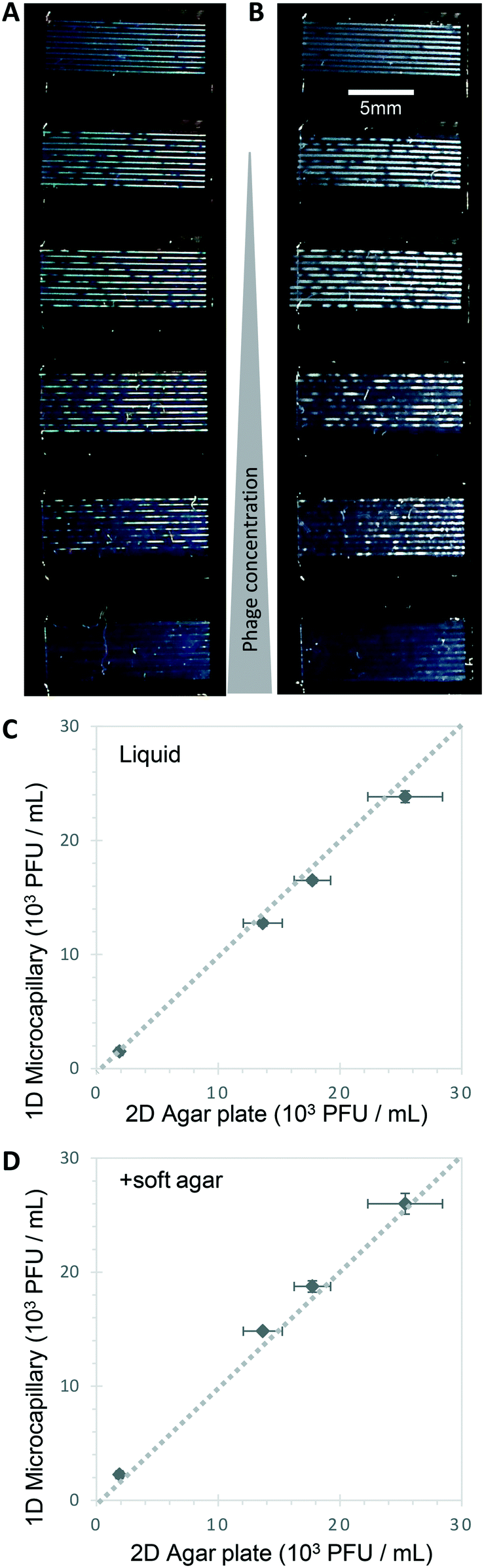 | ||
| Fig. 6 Agar is not required to detect T2 bacteriophage plaques within microcapillaries. A) and B): E. coli capillary phage counting in liquid broth was similar with or without soft agar. Endpoint images showed the same number and size of bacteriophage plaques in liquid media without agar (A), as seen with addition of 0.5% agar to broth prior to dipping and testing (B) within microcapillaries. C) Counting T2 phage concentration via DAL (double agar layer) on plate vs. microcapillaries in liquid broth. D) Counting T2 phage concentration via DAL (double agar layer) vs. microcapillaries with inclusion of soft agar. Dashed line indicates 1:1 relationship between the two counting methods. | ||
Plaques could be clearly counted after just 5 h and detected at concentrations down to 250 PFU mL−1 (Fig. 5 and 6) corresponding to the theoretical viral concentration where 1 plaque is present per 4 μL sample volume in 10 capillaries along the visible section of test strip. As with the reference method, changes in parameters like host inoculum and media viscosity can affect the assay measurements including plaque size.32 We found plaque size within 1D capillaries varied depending on host/phage system studied. Phage K had larger plaque sizes compared to T2 (Fig. 5 and 6). The smaller plaque size of T2 made it possible to count much higher numbers of bacteriophage with an upper limit approaching 3 × 104 PFU mL−1, before plaques began overlapping (Fig. 6).
Of particular note, the current gold standard DAL method for phage counting involves suspending host bacteria in warm agar, requiring care to ensure even seeding while avoiding killing host bacteria if the temperature of the melted agar is not optimal. Secondly, through miniaturisation, the storage and incubator space required for large stacks of Petri dishes is avoided alongside the refrigerated storage for prepared agar plates. Due to the small size, the test can be combined with a small portable incubating reader for timelapse imaging. This allows the development of automated image capture and analysis of more samples than traditional methods. Counting within 1D microcapillaries also significantly reduced the time needed to count CFU or PFU to between 5–9 h depending on culture characteristics of different bacteria – faster than typical overnight timepoints needed for agar plate colony or plaque counts. By permitting simple counting of individual colonies or plaques, grown from single cells or viral particles, we achieved a lower limit of detection approaching 250 CFU mL−1 or PFU mL−1, similar to agar colony or DAL plaque counting in Petri dishes. Lower concentrations can be measured simply by increasing the sample volume using more test strips or imaging longer microcapillary sections.
3.5 Benefits of open source darkfield microfluidic imaging system
Previously we combined microfluidic capillary measurements with light scatter detection to detect bacterial cell suspensions. A minimum of ∼0.5–0.1 OD600 turbidity, corresponding to ∼107–108 CFU mL−1, was needed to produce a solid line that was immediately visible with darkfield illumination geometry optimised for smartphones (Dönmez et al., 2020 (ref. 25)). Whilst this provides a microfluidic smartphone alternative to conventional cuvette plus spectrometer, our new observations move beyond estimating only high cell concentrations, to precisely measuring absolute CFU and PFU at many orders of magnitude lower concentrations. At the same time, we replaced smartphone imaging with an open source sensor system, exploiting the computing and imaging power of the Raspberry Pi HQ camera equipped with a macro lens.While bacterial colonies for rapidly dividing E. coli strains on solid agar media can be visible in as little as 7 h,24 these systems tend to be low-throughput and rarely is kinetic data collected due to the size/shape of Petri dishes which are not optically well designed for digitally recording results. However, some studies have examined the kinetics of solid agar colony growth of organisms such as S. aureus,33Vibrio spp,34E. coli, Klebsiella aerogenes, and Klebsiella pneumoniae35 by time-lapse imaging of Petri dishes. The imaging systems used are designed to examine Petri dishes, with different approaches used, including light scattering but can often only examine one Petri dish at a time, and it can be hard to define colony appearance as agar and colony appear similar. Fluorescence and dyes can be added but can further complicate the analytical system. For example, label-free bacterial detection is especially valuable for detecting bacterial lysis by phage. This cannot be easily monitored using metabolic growth dyes because the dye is likely to be irreversibly converted by bacteria before lysis occurs. The darkfield imaging system therefore adds the benefits of time-resolved darkfield imaging to microsystems; label-free detection being especially beneficial.
Counting bacterial colonies grown within capillaries has been exploited in the past, having been described as early as 1956 (ref. 36 and 37) using individual glass capillaries with colonies detected visually by eye. This confirms that the initial measurements here with a few defined strains are likely to be representative of a wider range of organisms. However, the flat, multiplex format possible using microcapillary film significantly increases throughput over individual glass capillaries. The refractive index match between aqueous culture medium and fluoropolymer device results in high optical transparency, allowing small colonies to be detected earlier. The benefits of digital camera for time resolved imaging further increases the utility of this method, especially with the inexpensive optoelectronic components now available (e.g. Raspberry Pi camera and LED illumination used here). We believe that microcapillary bacteriophage plaque counting has not previously been reported, and given the complexity of performing DAL method, we suggest this may be the most significant step forward with this method.
One limitation of the capillary method is that in contrast to the surface of solid medium on a plate, there is no straightforward way to sample bacterial colonies or phage plaques for subsequent analysis. This makes it more suitable to counting and quantitation than to identification or phenotypical characterisation. The inability to further sample colonies or plaques is shared with several other closed microfluidic systems. The melt-extrusion manufacturing method makes MCF especially suited to disposable quantitation devices, whereas other microsystems (e.g. droplet microfluidics) that are more complex and expensive may be more suitable for applications requiring downstream manipulations. Another limitation of the current method is that it was far less effective with motile organisms that could spread rapidly along the capillary, preventing clear colony or plaque detection (Fig. S3†). This again was clearly visible through time-resolved imaging, and with further optimisation it may be possible to count colonies as very transient points of bright scatter prior to spread. The inclusion of low concentration of agar in the sample medium did inhibit this spread but also reduced the simplicity of performing the test. It remains important therefore to further develop these methods of delaying or inhibiting the spread of motile organisms within the microcapillaries, for example substituting alternative hydrogel-forming polymers, or incorporating such polymers inside the hydrophilic coated capillaries. The transient detection further illustrates another advantage of time-resolved reader versus endpoint image. Time-lapse imaging also improves counting with higher concentrations of bacteria. Over time, the colony length increased making identification of several colonies per capillary more challenging.
4. Conclusion
This microfluidic method can accurately count bacterial CFU and bacteriophage PFU in liquid media, label-free. 1D counting of bacteria and plaques in Petri Tubes allows low concentrations of bacteria and phage, down to ∼250 CFU mL−1 or 250 PFU mL−1 to be counted in a simple liquid media, reducing the number of steps compared to reference methods. The 1D method was also found to reduce variability compared to the reference methods. The microfluidic geometry permits a miniaturised 1D liquid alternative to 2D distribution of bacterial cells over a solid media surface. The miniaturised test permits the application of timelapse imaging for multiple samples in parallel (80 distinct microcapillaries in 8 dip-strips; total sample volume 32 microlitres) for detailed kinetic growth analysis providing a wealth of quantitative information not easily obtained for standard Petri dish culture. The use of simple liquid microcapillary cultures may be beneficial for specific applications including environmental sampling in the field or where resources are limited. Overall, this concept of 1D liquid culture for colony and plaque counting demonstrates a novel and effective way to make use of miniaturisation to speed up and simplify analytical microbiology over conventional agar-based culture methods; this approach has the potential to deliver significant impact from research to clinical microbiology and beyond.Conflicts of interest
ADE and NMR are the inventors of patent application protecting aspects of the novel microfluidic devices tested in this study and is a director and shareholder in Capillary Film Technology Ltd, a company holding a commercial license to this patent application: WO2016012778 “Capillary assay device with internal hydrophilic coating” AD Edwards, NM Reis.Acknowledgements
We thank Danish Malik from Loughborough University for Phage K and host. We thank the Turkish Ministry of National Education, Republic of Turkey for Postgraduate Study Abroad Program to support Sultan İlayda Dönmez. This project was partially funded by Engineering and Physical Science Research Council (EPSRC) grants EP/R022410/1 and EP/S010807/1. Images from Servier Medical Art (https://smart.servier.com) were adapted and used for Fig. 1 and S1,† under a Creative Commons Attribution license.References
- A. Kumar, et al., Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock*, Crit. Care Med., 2006, 34(6), 1589–1596 CrossRef PubMed.
- F. Edition, Guidelines for drinking-water quality, WHO Chron., 2011, 38(4), 104–108 Search PubMed.
- A. H. Havelaar, et al., World Health Organization Global Estimates and Regional Comparisons of the Burden of Foodborne Disease in 2010, PLoS Med., 2015, 12(12), e1001923 CrossRef PubMed.
- J. T. P. Larry Maturin, Aerobic Plate Count, 2001 January, 27, 2022, Available from: https://www.fda.gov/food/laboratory-methods-food/bam-chapter-3-aerobic-plate-count Search PubMed.
- J. Alford and E. E. Steinle, A double layered plate method for the detection of microbial lipolysis, J. Appl. Bacteriol., 1967, 30(3), 488–494 CrossRef CAS PubMed.
- A. Zapata and S. Ramirez-Arcos, A Comparative Study of McFarland Turbidity Standards and the Densimat Photometer to Determine Bacterial Cell Density, Curr. Microbiol., 2015, 70(6), 907–909 CrossRef CAS PubMed.
- J. Robertson, C. McGoverin, F. Vanholsbeeck and S. Swift, Optimisation of the Protocol for the LIVE/DEAD® BacLightTM Bacterial Viability Kit for Rapid Determination of Bacterial Load, Front. Microbiol., 2019, 10, 801 CrossRef PubMed.
- S. Heider and C. Metzner, Quantitative real-time single particle analysis of virions, Virology, 2014, 462-463, 199–206 CrossRef CAS PubMed.
- M. d'Herelle, Sur un microbe invisible antagoniste des bacilles dysentériques, Acta Kravsi, 1961, 165, 373–375 Search PubMed.
- C. Brives and J. Pourraz, Phage therapy as a potential solution in the fight against AMR: obstacles and possible futures, Palgrave Commun., 2020, 6(1), 100 CrossRef.
- D. J. Malik, et al., Formulation, stabilisation and encapsulation of bacteriophage for phage therapy, Adv. Colloid Interface Sci., 2017, 249, 100–133 CrossRef CAS PubMed.
- J. Paczesny, Ł. Richter and R. Hołyst, Recent Progress in the Detection of Bacteria Using Bacteriophages: A Review, Viruses, 2020, 12(8), 845 CrossRef CAS PubMed.
- K. F. Tjhung, et al., Rapid Enumeration of Phage in Monodisperse Emulsions, Anal. Chem., 2014, 86(12), 5642–5648 CrossRef CAS PubMed.
- S. F. Berlanda, et al., Recent Advances in Microfluidic Technology for Bioanalysis and Diagnostics, Anal. Chem., 2021, 93(1), 311–331 CrossRef CAS PubMed.
- S. A. Byrnes, et al., Polydisperse emulsion digital assay to enhance time to detection and extend dynamic range in bacterial cultures enabled by a statistical framework, Analyst, 2018, 143(12), 2828–2836 RSC.
- S. H. Needs, H. M. I. Osborn and A. D. Edwards, Counting bacteria in microfluidic devices: Smartphone compatible ‘dip-and-test’ viable cell quantitation using resazurin amplified detection in microliter capillary arrays, J. Microbiol. Methods, 2021, 187, 106199 CrossRef CAS PubMed.
- O. Scheler, W. Postek and P. Garstecki, Recent developments of microfluidics as a tool for biotechnology and microbiology, Curr. Opin. Biotechnol., 2019, 55, 60–67 CrossRef CAS PubMed.
- T. S. Kaminski, O. Scheler and P. Garstecki, Droplet microfluidics for microbiology: techniques, applications and challenges, Lab Chip, 2016, 16(12), 2168–2187 RSC.
- H. Lu, et al., High throughput single cell counting in droplet-based microfluidics, Sci. Rep., 2017, 7(1), 1366 CrossRef PubMed.
- P. Zhang, et al., Facile syringe filter-enabled bacteria separation, enrichment, and buffer exchange for clinical isolation-free digital detection and characterization of bacterial pathogens in urine, Analyst, 2021, 146(8), 2475–2483 RSC.
- D. Rodoplu, et al., A simple magnetic-assisted microfluidic method for rapid detection and phenotypic characterization of ultralow concentrations of bacteria, Talanta, 2021, 230, 122291 CrossRef CAS PubMed.
- L. Castillo-Henríquez, et al., Biosensors for the Detection of Bacterial and Viral Clinical Pathogens, Sensors, 2020, 20(23), 6926 CrossRef PubMed.
- I. Nuñez, et al., Low cost and open source multi-fluorescence imaging system for teaching and research in biology and bioengineering, PLoS One, 2017, 12(11), e0187163 CrossRef PubMed.
- S. H. Needs, et al., Exploiting open source 3D printer architecture for laboratory robotics to automate high-throughput time-lapse imaging for analytical microbiology, PLoS One, 2019, 14(11), e0224878 CrossRef CAS PubMed.
- S. İ. Dönmez, et al., Label-free smartphone quantitation of bacteria by darkfield imaging of light scattering in fluoropolymer micro capillary film allows portable detection of bacteriophage lysis, Sens. Actuators, B, 2020, 323, 128645 CrossRef.
- N. M. Reis, et al., Lab on a stick: multi-analyte cellular assays in a microfluidic dipstick, Lab Chip, 2016, 16(15), 2891–2899 RSC.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, NIH Image to ImageJ: 25 years of image analysis, Nat. Methods, 2012, 9(7), 671–675 CrossRef CAS PubMed.
- F. Zhang, et al., Rapid Antimicrobial Susceptibility Testing on Clinical Urine Samples by Video-Based Object Scattering Intensity Detection, Anal. Chem., 2021, 93(18), 7011–7021 CrossRef CAS PubMed.
- S. Vargas, et al., Dynamic light scattering: A fast and reliable method to analyze bacterial growth during the lag phase, J. Microbiol. Methods, 2017, 137, 34–39 CrossRef CAS PubMed.
- B. K. Wilson and G. D. Vigil, Automated bacterial identification by angle resolved dark-field imaging, Biomed. Opt. Express, 2013, 4(9), 1692–1701 CrossRef PubMed.
- S. Miyamoto, et al., Estimating the Diffusion Coefficients of Sugars Using Diffusion Experiments in Agar-Gel and Computer Simulations, Chem. Pharm. Bull., 2018, 66(6), 632–636 CrossRef CAS PubMed.
- D. Rajnovic, X. Muñoz-Berbel and J. Mas, Fast phage detection and quantification: An optical density-based approach, PLoS One, 2019, 14(5), e0216292 CrossRef PubMed.
- J. Bär, et al., Efficient microbial colony growth dynamics quantification with ColTapp, an automated image analysis application, Sci. Rep., 2020, 10(1), 16084 CrossRef PubMed.
- K. Huff, et al., Light-scattering sensor for real-time identification of Vibrio parahaemolyticus, Vibrio vulnificus and Vibrio cholerae colonies on solid agar plate, Microb. Biotechnol., 2012, 5(5), 607–620 CrossRef PubMed.
- H. Wang, et al., Early detection and classification of live bacteria using time-lapse coherent imaging and deep learning, Light: Sci. Appl., 2020, 9(1), 118 CrossRef PubMed.
- T. Yanagita, Capillary tube method for counting viable bacteria, J. Bacteriol., 1956, 71(3), 381–382 CrossRef CAS PubMed.
- R. L. Bowman, P. Blume and G. G. Vurek, Capillary-Tube Scanner for Mechanized Microbiology: A photoelectric scanner measures growth in agar-filled capillaries and gives a new approach to microbiology, Science, 1967, 158(3797), 78–83 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2lc00280a |
| This journal is © The Royal Society of Chemistry 2022 |