
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzyme-based digital bioassay technology – key strategies and future perspectives
Hiroyuki
Noji
*,
Yoshihiro
Minagawa
and
Hiroshi
Ueno
Department of Applied Chemistry, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, Japan. E-mail: hnoji@g.ecc.u-tokyo.ac.jp
First published on 14th July 2022
Abstract
Digital bioassays based on single-molecule enzyme reactions represent a new class of bioanalytical methods that enable the highly sensitive detection of biomolecules in a quantitative manner. Since the first reports of these methods in the 2000s, there has been significant growth in this new bioanalytical strategy. The principal strategy of this method is to compartmentalize target molecules in micron-sized reactors at the single-molecule level and count the number of microreactors showing positive signals originating from the target molecule. A representative application of digital bioassay is the digital enzyme-linked immunosorbent assay (ELISA). Owing to their versatility, various types of digital ELISAs have been actively developed. In addition, some disease markers and viruses possess catalytic activity, and digital bioassays for such enzymes and viruses have, thus, been developed. Currently, with the emergence of new microreactor technologies, the targets of this methodology are expanding from simple enzymes to more complex systems, such as membrane transporters and cell-free gene expression. In addition, multiplex or multiparametric digital bioassays have been developed to assess precisely the heterogeneities in sample molecules/systems that are obscured by ensemble measurements. In this review, we first introduce the basic concepts of digital bioassays and introduce a range of digital bioassays. Finally, we discuss the perspectives of new classes of digital bioassays and emerging fields based on digital bioassay technology.
1. Introduction
Digital bioassays have emerged as a new class of bioanalytical method with single-molecule detection sensitivity.1–3 The main principle of digital bioassays is to micro-compartmentalize assay solutions to encapsulate individual target molecules stochastically within reactors in a “one-or-none” manner. The number, as well as the concentration, of a target molecule in the specimen is determined by counting the number of reactors showing positive signals originating from single target molecules after signal binarization into “1” or “0”.Digital bioassays can be categorized into two classes based on the type of signal amplification in the microreactor.4,5 Digital polymerase chain reaction (PCR) and other digital nucleic acid amplification assays form the first category, and these rely on exponential signal amplification.6 The second category is enzyme-based digital bioassays, and these are the subject of this review. Herein, we refer to these as “digital bioassays” for simplicity. In digital bioassays, a single enzyme molecule produces a signal in a linear fashion with respect to reaction time. Therefore, the digitalization of single-molecule enzyme-based bioassays requires smaller reactors than digital PCR.
Digital bioassay has been established, when micron-sized reactor array systems became available.7 The most prominent benefit of digital bioassays is their high sensitivity. Downsizing of reactor volume is one of the key points to achieve a high signal-to-noise ratio, that is, signal separation between positive and negative reactors. Another key aspect of digital bioassays is the use of a large number of reactors that enables the detection of the target molecule at very low concentrations. In addition, the highly parallelized nature of micron-sized reactor systems allows quantitative measurements over a wide range of target concentrations. In general, tens or hundreds of thousands of reactors can be prepared, yielding a dynamic range of over 3–4 orders of magnitude. Another benefit of digital bioassays is that they allow the study of the heterogeneity of enzymes that are masked in ensemble measurements. Single-molecule studies have revealed that the catalytic activity of enzymes differs from molecule to molecule, suggesting the individualities of each enzyme molecule. Digital bioassays enable quantitative analysis of the individualities of enzymes owing to the high-throughput nature of the method.8–11
Currently, many types of digital bioassays have been reported because many bioanalytical methods and diagnostic tests utilize enzymes for signal amplification. A representative application of the digital bioassay is the digital enzyme-linked immunosorbent assay (ELISA).12,13 Owing to the versatility of digital ELISAs, various types of digital ELISAs and related assays have been reported.12–19 Nowadays, the concept of digital bioassays has been extended to other assays, such as membrane transporter assays,20,21 nucleic acid detection22,23 with Cas12/13 proteins,24 and cell-free gene expression (digital gene expression) for the accurate screening of random mutation libraries.25
This review focuses on digital bioassays based on single-molecule enzyme reactions that show the linear amplification of signals over time. Although there are several excellent review papers1–3 in which digital bioassays are featured with an emphasis on high sensitivity, the fundamental points and technical issues are not well discussed. This review provides key points for the design of digital bioassays and introduces their expanded applications from the perspective of next-generation digital bioassays.
2. Basics of digital bioassays
2.1. Concept behind digital bioassays
A digital bioassay relies on the stochastic encapsulation of target molecules into reactors; when the concentration of the target molecule is sufficiently low to ensure that the mean number of target molecules per reactor is sufficiently lower than 1.0, the reactors show discrete fluorescence signals, that is, binary signals (0 or 1) representing none or one. Fig. 1a shows schematics of a digital bioassay and a conventional bioassay in bulk solution for comparison. Once the signal separation between positive reactors with a single enzyme molecule and negative reactors without an enzyme has been established, the absolute number of target molecules can be determined by counting the positive reactors after signal digitalization with an adequate threshold. Fig. 1b shows experimental images of a digital bioassay of the alkaline phosphatase (ALP) enzyme.26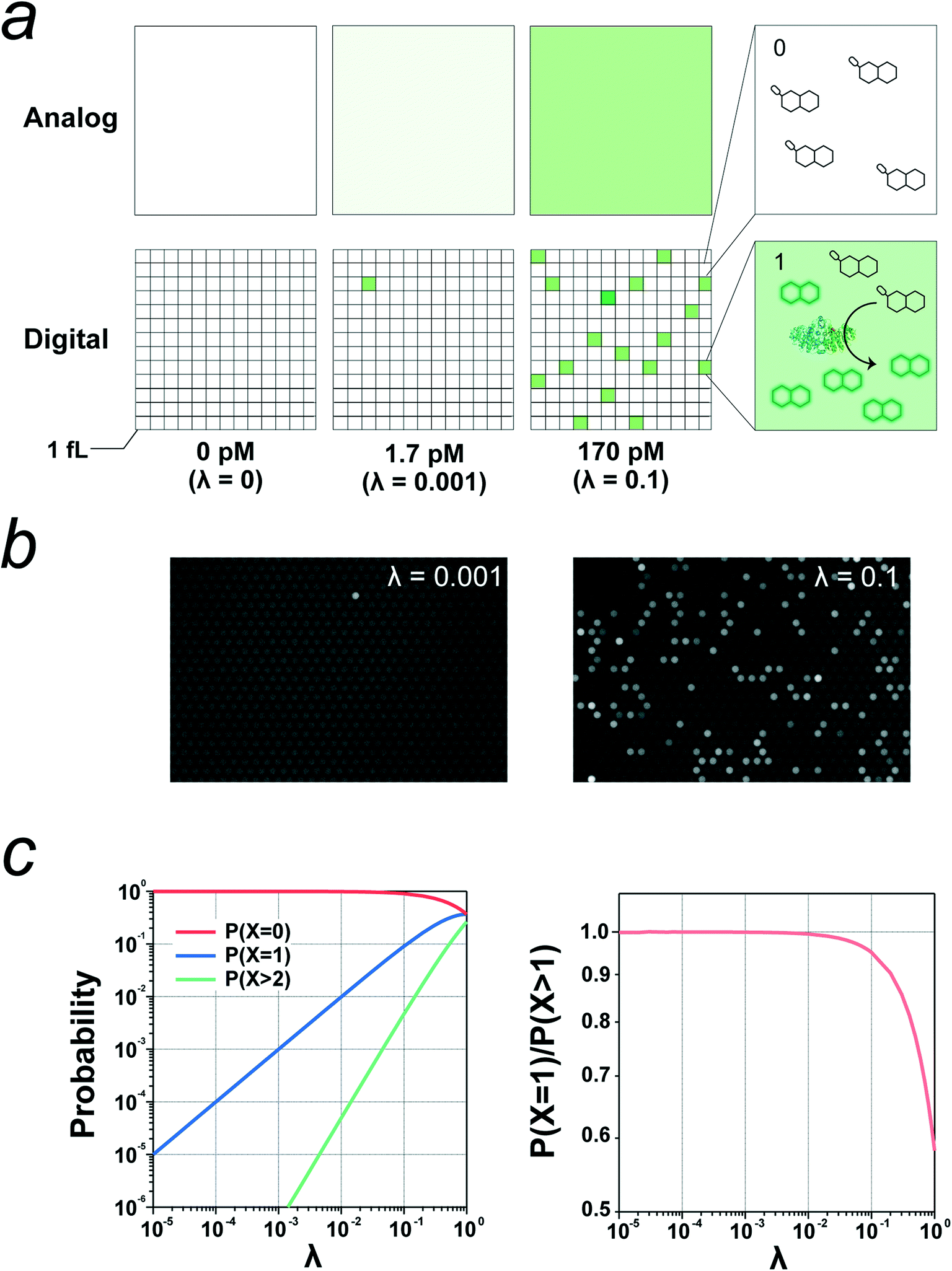 | ||
| Fig. 1 Concept of digital bioassay. a) Schematics of conventional (analog) bioassay and digital bioassay. In the digital format, assay mixture is micro-compartmentalized to stochastically encapsulate single enzyme molecules into reactors with volume of 1 fL. λ represents the mean number of enzyme molecule per reactor at the given concentrations of enzyme. b) Fluorescence images of digital bioassay of alkaline phosphatase (ALP) obtained at the indicated λ values. c) Probability of reactors with zero, one or two or more molecules of enzyme plotted against λ value (left). Probability of reactor with single enzyme molecule among all of positive reactors with one or more enzyme molecules (right). | ||
The fraction of reactors accommodating target molecules obeys Poisson distribution:
| P(n, λ) = e−λ·λn/n! (n = 0, 1, 2, 3…), |
| P(n ≥ 1, λ) = 1 − P(n = 0) = 1 − e−λ | (1) |
2.2. Three components of digital bioassays
Digital bioassay systems include three principal components: a micron-sized reactor for micro-compartmentalization, enzymes as target molecules for investigation or labelling molecules of target molecules, and fluorogenic substrates or chemical probes that provide the fluorescence signals. In addition to these components, other components or molecules are integrated in each type of assay, such as antibody-coated microbeads for digital ELISA and lipid bilayers for digital transporter assays. In the following section, the microreactors, enzymes, and fluorogenic probes are introduced.2.3. Microreactors
Various types of microreactors are currently available for digital bioassays. The earliest class of single-molecule enzyme assays was reported by Rotman et al. in 1961,28 in which water-in-oil (w/o) droplets were used for the emulsification of a fluorogenic assay solution of β-galactosidase (β-gal) (Fig. 2a). This report represents a pioneering study of single-molecule bioanalyses. However, the method is not suitable for quantitative analysis because of the high heterogeneity of w/o droplet size. Quantitative single-molecule enzymatic assays have become available when microfabrication technology has enabled the preparation of regularly shaped femtoliter reactors.7,29 The first quantitative single-molecule enzymatic assay was demonstrated using a polydimethylsiloxane (PDMS)-based femtoliter reactor array system (Fig. 2b).7 In this system, an aqueous solution was encapsulated in micron-sized reactors formed between the microcavities on a PDMS sheet and a glass coverslip. A similar method was proposed in which an optical fiber plate with micron-sized holes on the surface was pressed against a PDMS gasket sheet to prepare a femtoliter reactor array (Fig. 2c).29,30 However, PDMS/glass-based reactor systems require a mechanical compression process for sealing the microreactors, which is not suitable for reproducible and high-throughput generation of femto-reactors.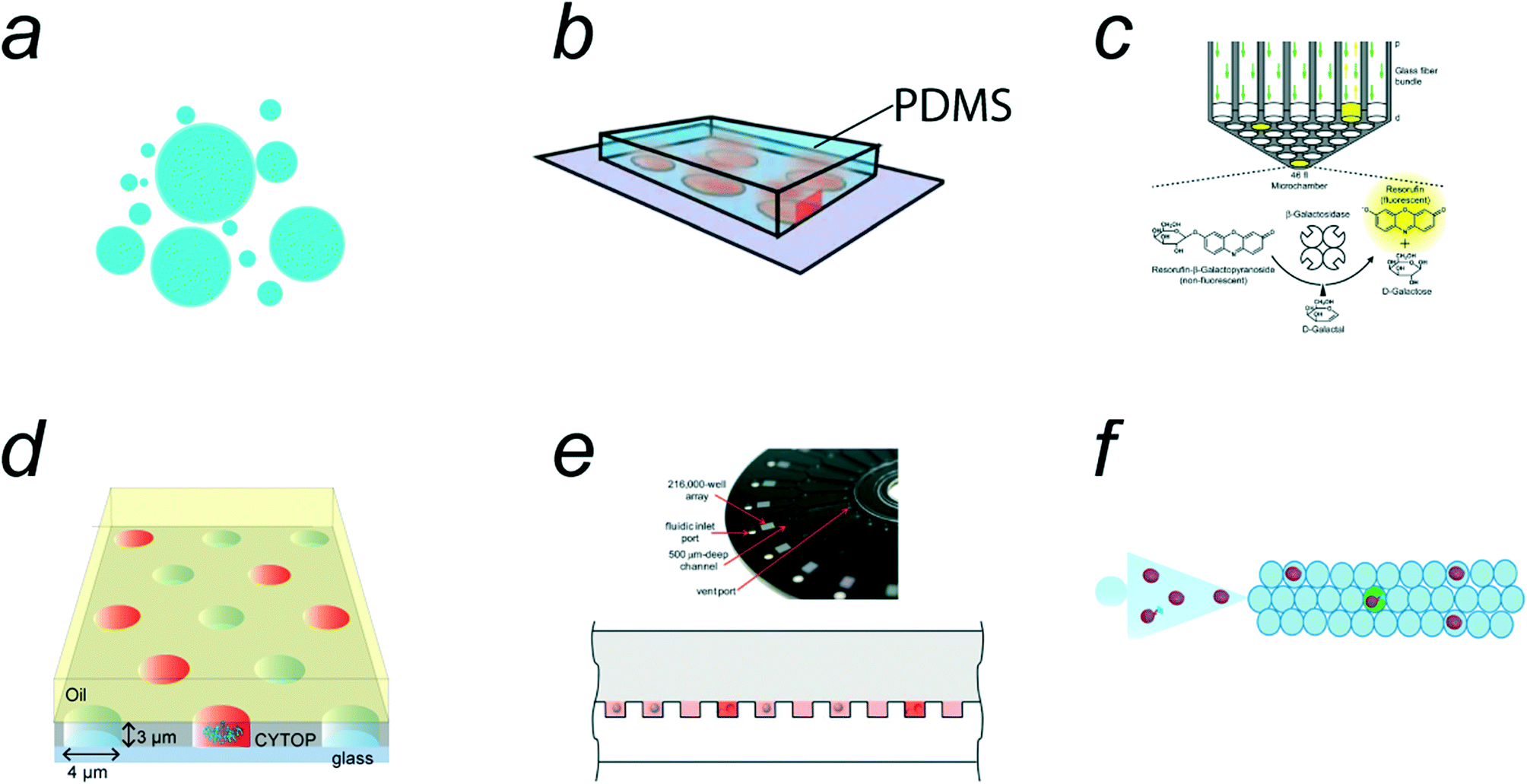 | ||
| Fig. 2 Micro-compartmentalization methods. a) Water-in-oil emulsion. b) PDMS (polydimethylsiloxane)/coverslip microreactor system. Reproduced from ref. 7, Copyright 2005 Springer Nature. c) Optic fiber plate/PDMS microreactor system. Reproduced from ref. 75, Copyright 2007 National Academy of Sciences. d) Femtoliter droplet array device (FRAD). e) Arrayed droplets on COP (cyclicolefin polymer) device, SiMOA (single molecule array). Reproduced from ref. 33, Copyright 2012 The Royal Society of Chemistry. f) Schematic of droplet microfluidic system. Reproduced from ref. 39, Copyright 2020 American Chemistry Society. | ||
Following the PDMS/glass-based systems, a w/o droplet array system was proposed; this system contains a million femtoliter-sized w/o droplets on the device surface.31 The device, termed the femtoliter reactor array device (FRAD), is composed of a coverslip coated with a fluorinated polymer where a million micron-sized cavities are displayed on the coverslip surface. The w/o droplets are formed within the cavities when they are sealed with an oil layer (Fig. 2d). Owing to the versatility and stability of this system, FRAD has been utilized in various types of digital bioassays, including single-molecule enzyme analyses10–12,26 and enzyme screening.25 In addition, a cyclic olefin polymer (COP)-based device for droplet arrays has been developed (Fig. 2e).32,33 This system, termed SiMoA (representing “single-molecule array”), is made of a COP plate having micron-sized cavities in which water droplets are held and sealed with oil, similar to FRAD. Because COP chips are suitable for mass production by the mold-injection process, SiMoAs have been commercialized for digital ELISAs.34,35 Both FRAD and SiMoA systems allow the facile and high-throughput generation of w/o droplets at rates of 105–106 droplets per device.
Free water droplets can also be used for digital bioassays (Fig. 2f). Because conventional droplet microfluidic systems generate w/o droplets with volumes over 10 pL, relatively large droplets (Φ = 10 μm) are often used for digital bioassays,24,36–39 leading to a compromise in detection time. However, there have been a few reports of digital bioassays in free w/o droplets with femtoliter volumes.16,40 Unlike arrayed microreactor systems, the total number of droplets is unlimited in principle. Droplet microfluidic systems can generate millions of droplets every minute, which exceeds the number of reactors on arrayed systems. Another beneficial feature of free-droplet systems is that the encapsulation efficiency of the sample solution is generally higher than that of array-type systems.
In addition to array and free w/o droplet systems, other technologies for micro-compartmentalization have been applied to digital bioassays. One of the distinctive microreactor technologies is an arrayed lipid bilayer chamber (ALBiC) technology, which has been applied for digital bioassays of membrane transporters.20 In this review, ALBiCs and related technologies are introduced in subsection 4.7. In addition, recently developed technologies, such as the particle-templated method and compartmentalization-free methods, are introduced as emerging technologies in subsections 4.2 and 5.2.
2.4. Enzymes
Currently, only a few types of enzymes have been applied in digital bioassays, although the variety of enzymes is expanding. This is because enzymes must meet the following two requirements. First, fluorogenic substrates must be available for the enzymes. Second, the enzyme must possess a sufficiently high catalytic power to produce a high fluorescence signal rapidly.β-gal,7,26,30,41,42 β-glucuronidase (β-gluc)8,43 and alkaline phosphatase (ALP)11,26,44,45 are often used in digital bioassays because the chemistry of their fluorogenic substrates has been well established, thus providing several types of fluorogenic substrates for each enzyme. Horseradish peroxidases (HRPs) have also been studied in digital bioassays.7,46 Fluorogenic substrates are also available as esterases. However, a digital bioassay for esters has not been reported, probably because the fluorogenic substrates are autolyzed, causing background signals. Nucleases such as Cas13a and Cas12a are new families employed for digital bioassay.24,38,47–49 The very stringent activation of the nuclease activity of Cas proteins upon target polynucleotide recognition is ideal for digital bioassays that detect single molecules of RNA or DNA derived from pathogenic entities such as viruses.
The catalytic power of enzymes is one of the most important factors in obtaining high signals in digital bioassays. Fig. 3a shows the distribution of catalytic turnover rates of several enzymes. As shown, there is log-normal distribution with a median value of 13.7 turnover per s.50 Digital bioassays employ enzymes with catalytic turnovers of 10–1000 turnover per s. As discussed in the next subsection, the catalytic turnover rate depends on the type of fluorogenic substrate.
 | ||
| Fig. 3 Enzymes and substrates of digital bioassays. a) Distribution of enzyme's kcat, modified from ref. 50, Copyright 2011 American Chemistry Society. The kcat values of representative enzymes used for digital bioassays were indicated. b) Chemical structures and spectra of three major fluorescence dyes used for fluorogenic substrates: 4-methylumbelliferone (4-MU), fluorescein, and resorufin. Dot lines and lines represent absorbance and fluorescence spectra, respectively. c) Fluorogenic substrates for ALP: 4-methylumbelliferyl phosphate (4-MUP), fluorescein diphosphate (FDP), and resorufin-phosphate. d) Autolysis of FDP found in digital bioassay of ALP. Time-lapse images and time courses with two distinctive groups: first and slow ones. The fast group corresponds to catalysis by single ALP molecule. Slow one represents autolysis of FDP. Modified from ref. 11, Copyright 2021 American Chemistry Society. Note that the original data includes the third group corresponding to enzyme molecules with halved activity due to heterodimer ALP composed of inactive monomer and active monomer. This group was omitted in this figure for clarity. | ||
In addition to high catalytic power, enzyme substrate promiscuity is required for the rapid catalysis of synthetic fluorogenic substrates. Because of the differences in the chemical structures of fluorogenic substrates compared to natural ones, the turnover rate of enzymes for fluorogenic substrates differs. Therefore, the overall fluorescence signal from the fluorogenic assay of enzymes is a combination of enzymatic and chemical properties of fluorogenic substrates.
2.5. Fluorogenic substrates
Most fluorogenic substrates used in most digital bioassays contain either of three types of fluorescent chromophores in their chemical structures: 4-methylumbelliferone (4-MU), fluorescein, and resorufin (Fig. 3b). These substrates are chemical conjugates of fluorescent chromophores and a particular chemical group for enzyme recognition and reaction.51Fig. 3c shows representative fluorogenic ALP substrates composed of a fluorophore and phosphate ester group. Upon enzymatic reaction, typically, hydrolytic cleavage of the substrate occurs, and a fluorogenic chromophore is released, thus resulting in the resume of a high fluorescence quantum yield. Fluorescein, resorufin, and coumarin are suitable base structures because their fluorescence occurs in an all-or-none manner upon enzymatic cleavage. This fluorescence switching is crucial for achieving a high fluorescence contrast between positive and negative reactors.There are other factors to achieve a high fluorescence contrast between positive and negative reactors. One is the compatibility of the fluorogenic substrates with the specificity spectrum of enzymes. Enzymes have an intrinsic substrate promiscuity to some degree, enabling recognition and reaction with nonnatural substrates. When a fluorogenic substrate is compatible with an enzyme, the enzyme can catalyze the conversion of the substrate at a high rate, thus generating a high fluorescence signal. In the case of Escherichia coli ALP (EcALP), the 4-MU derived substrate, 4-methylumbelliferyl phosphate (4-MUP) is a better substrate than the fluorescein-derived fluorescein diphosphate (FDP). Crucially, EcALP catalyzes the dephosphorylation of coumarin-based substrates at a higher rate than fluorescein-based substrates. This is attributed to the chemical difference between FDP and 4-MUP; FDP has two phosphate bonds for cleavage, whereas 4-MUP has a single phosphate group. The obligatory double digestion mechanism of fluorescein is responsible for its slower catalysis. A similar trend was observed for β-gal, which hydrolyzes resorufin-β-D-galactopyranoside (RDG) 10-times faster than fluorescein-di-β-D-galactopyranoside (FDG); the former substrate has a single galactose moiety, whereas the latter has two.
Another important property of fluorogenic substrates is the low permeability of fluorogenic substrates/products in sealing oil. Fluorescent chromophores with high fluorescence intensity generally contain aromatic rings; therefore, they are hydrophobic to some degree. As a result, fluorogenic substrates are often modified with polar groups to achieve biocompatibility. However, the leakage of the fluorogenic substrate/product can still occur, thus reducing the signal-to-noise ratio. For example, the leakage of reaction products from fluorogenic substrates was reported in a digital bioassay of EcALP with a fluorogenic 4-MUP substrate.26 The reaction product of 4-MUP is 4-methylumbelliferone (4-MU), which has an additional hydroxyl moiety on the coumarin structure that enhances its water solubility. At neutral pH, where the hydroxyl group is protonated, 4-MU is electrically neutral and leaks into the oil phase. Therefore, for digital bioassays with 4-MU-based fluorogenic substrates, the assay mixture should be at alkaline pH to retain 4-MU molecules in the w/o droplets. For example, digital bioassays for EcALP and influenza virions based on neuraminidase activity with a 4-MU-based substrate were conducted at pH 9.25 and 9.0, respectively.26,52
The chemical stability of fluorogenic substrates in reaction mixtures is also important for digital bioassays. Some fluorogenic substrates are spontaneously cleaved under ambient conditions, even in the absence of enzymes. Fig. 3d shows the time-course of fluorescence signal of digital bioassay of EcALP with FDP as a fluorogenic substrate. The base lines; the time-courses of empty reactors showing the autolysis of FDP.11 For quantitative analysis, the background signal from the autolysis of the fluorogenic substrates has to be subtracted from the signals from the positive reactors.
Quencher probes have been used in digital bioassays of nucleases. A representative digital bioassay with nuclease is CRISPR-Cas12/Cas13-based assays.24,47 Cas13 is a Cas family protein that shows high RNase activity when RNA with the correct target sequence is recognized via CRISPR RNA bound to the Cas13 protein. When activated upon binding to the target RNA, Cas13 cleaves RNA molecules in a sequence-independent manner, although each Cas13 protein has a preference for the substrate RNA sequence to some degree. When the CRISPR-Cas13 complex cleaves the quencher probe RNA labeled with a fluorescent dye and quencher dye, probes regain fluorescence, allowing digital counting with a high signal-to-noise ratio. Similar digital bioassays containing the Cas12a protein that recognize DNA and activates trans-ribonuclease activity have also been reported.38,48,49 Peptide-linked quencher probes should enable the development of digital bioassays of peptidases, although this has not been previously reported.
3. Design principles for digital bioassays
3.1. Signal digitalization
The most critical point in the design of digital bioassays is the precise discrimination of positive and negative reactors for signal digitalization with a threshold value determined from the signal histogram (Fig. 4a). Importantly, for accurate classification, the positive reactor signal should be well separated from the negative signal, i.e. the mean signal values of the positive and negative reactors should be separated sufficiently by the sum of the standard deviations of the signals, as follows:| Δμ = μP − μN ≫ σP + σN, |
| θ = μN + n·σN (n ≥ 3). |
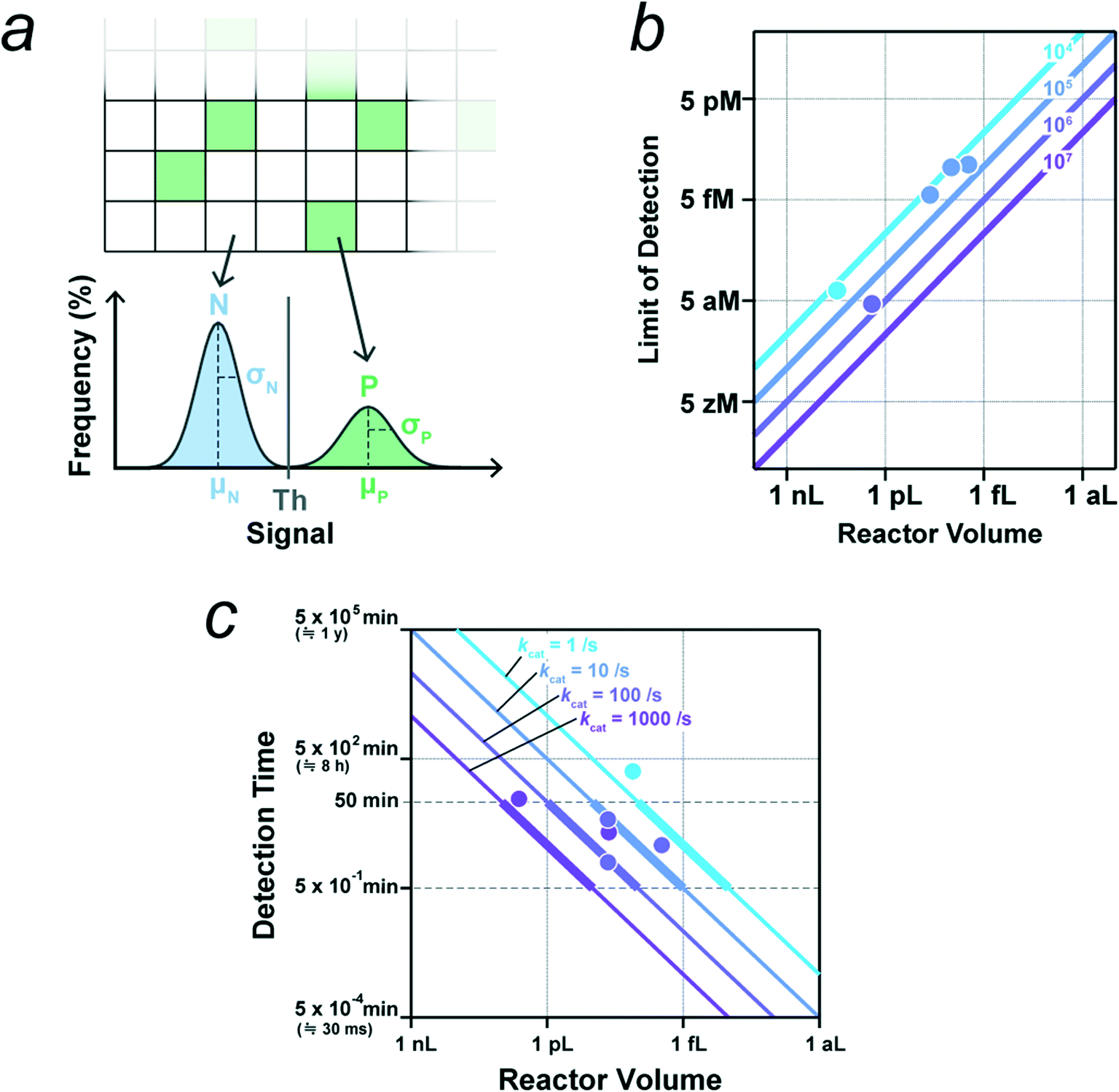 | ||
| Fig. 4 Key parameters for digital bioassay. a) Threshold for signal binarization. Schematic of distributions of signals from negative reactors without enzyme (N) and from positive reactors containing single molecule of enzyme (P). μ and σ represent the mean and standard deviation. b) Theoretical limit of detection (LOD) determined by the total reaction volume that is the reactor number multiplied by the reactor volume. Lines represent LOD versus reactor volume for systems with indicated number of reactors. Data points indicate the experimental LOD values from selected literatures (see main text, and also Table 1), and they are colored according to the corresponding number of reactors. c) Detection time required for the signal detection of single molecule of enzyme with indicated catalytic turnover rate. The detection time is estimated as the time for the accumulation of reaction product to 500 nM in a microreactor of indicated volume. Data points indicate the detection times from selected literatures (see main text, and also Table 1), and they are colored according to the corresponding catalytic turnover rate. | ||
3.2. Theoretical limit-of-detection
The LOD is determined by two factors: the total number of reactors and the fraction of pseudo-positive reactors. For simplicity, we assume that there are no pseudo-positives in this subsection. When the total reaction volume per device (i.e., the number of reactors multiplied by the reactor volume) is high, the possibility of capturing target molecules in some reactors is also high, leading to a high detection sensitivity (i.e., low LOD). When the mean number of target molecules per the total reactor volume is one per device, the probability of finding a positive reactor is estimated to be 63% according to eqn (1);| P(n ≥ 1, λ = 1) ≅ 0.63 |
| P(n ≥ 1, λ = 3) ≅ 0.95 |
| Enzyme | Substrate | k cat or turnover (s−1) | Detection time (min) | Reactor volume (v = fL) | Reactor number (n) | LODtheo (= 3/(n × v × NA))a (fM) | LODexp (fM) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a N A is the Avogadro constant. b The values were determined from heterogeneity in catalytic activity distribution of single enzyme. c The values were determined in conventional biochemical assay. d The value was determined by using three crRNA. e Digital homogeneous entropy-driven biomolecular assay (dHEBA). f The values were determined as the lowest concentration for quantification. | ||||||||
| Mutant E. coli. ALP | 4-MUP | 891 | 10 | 44 | 912![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.13 | 7 | 26 |
| Tissue nonspecific ALP | 4-MUP | 873b 1542b | 20 | 46 | 60000 |
1.8 | — | 9 |
| HRP | Amplex red | 38 | 2 | 46 | — | — | — | 46 |
| β-Gal from E. coli | RDG | 534 | 2 | 46 | — | — | — | 56 |
| β-Glucosidase from P. furiosus | FDGlu | 3.8 | 260 | 13 | — | — | — | 40 |
| Cas12a from L. bacterium | FAM-CCCCCC-BHQ1 | 4850c | 60 | 4300 | — | — | 28.7 × 10–3f | 38 |
| Cas13a from L. wadei | FAM-rUrUrUrUrU-3lABkFQ′ | 110 | <5 | 3 | 120000 |
14 | 56 (5.7d) | 47 |
| Cas13a from L. buccalis | FAM- rUrUrUrUrU-3lABkFQ′ | — | 60 | 14000 |
20000 |
18 × 10−3 | 10 × 10–3f | 24 |
| β-Gal | FDG | — | 10 | 32 | 200000 |
0.78 | 46 | 16 |
| dHEBAe | qProbe | — | 10 | 2600 | 100920 |
19 × 10−3 | 4 × 10−3 | 53 |
3.3. Dynamic range
Dynamic range is also an important performance parameter for analytical methods. As abovementioned, the lower limit for analysis in digital bioassay is three molecules per total reactor volume. On the other hand, λ has to be less than 0.1 for linear response to target concentration. Therefore, for simple digital bioassay, the order of the dynamic range is approximately 1/10 of the total number of reactors, e.g. 105 for assay with 106 reactors. The Poisson distribution also allows the estimation of target molecule concentration even when the probability of positive reactor is close to saturation; λ > 0.1. Under such experimental conditions, it could be more effective to count the number of negative reactors instead of measuring the number of positive reactors, which does not respond linearly to target concentration. In this manner, the dynamic range can be extended to the order of the total reactor number.The above argument assumes that the reactors for micro-compartmentalization are homogeneous, and that the encapsulation process simply obeys the Poisson distribution. There are several methods proposed for breaking the limit of the dynamic range determined by the number of reactors. One is the method with multivolume reactor devices that display the array of microreactors with several classes of volume.54 This method was principally developed for digital PCR that amplifies signal in exponential manner so that distinctive signal is obtained with a wide range of reaction volume. This strategy could also be effective for enzyme-based digital bioassay although it should compensate the detection time because a larger reactor requires longer incubation time to gain sufficiently high signal. Another method is also based on arrayed type reactor system, termed Brownian trapping with drift.55 In this method, target molecules are actively captured with binder molecules immobilized on reactors during the injection of the target molecule solution into reactors. Resultantly, the probability of positive reactors shows exponentially decay along the direction of solution injection. This method effectively extends the limit of dynamic range although the probability of positive reactor no longer shows linear response to target molecule concentration.
3.4. Enhancing positive signals
The above calculation assumes that all positive reactors are well separated from negative ones. For efficient signal digitalization, the mean value of the fluorescence signal from the positive reactors should be sufficiently high to avoid pseudo-positive signals. Here, we consider the lowest concentration of the fluorescent dye for signal digitalization to be at least 500 nM, based on our own measurements and an analysis of the literature.11,16,26,56 Although it is possible to obtain images of fluorescent dye solutions of 100 nM or less, high concentrations are required for quantitative imaging with an objective lens of medium or low numerical aperture (less than 0.76), i.e., that suitable for high-throughput wide-view imaging. The major factors for obtaining a high fluorescence signal are the reactor volume and reaction time, as well as the properties of enzymes and substrates such as turnover rate, oil permeability, and photo/chemical stabilities (see subsection 2.5.).The size of the microreactors is a critical factor for achieving a high fluorescence signal. In principle, smaller reactors yield higher signals owing to the higher condensation effect. Typically, digital bioassays use reactors with volumes in the femto- to picoliter range, but several studies have reported attoliter chamber systems for the effective detection of slow enzymes.45,57 Thus, reducing the reactor volume is the principal strategy for achieving a high signal intensity, in particular in the digital assay for enzymes with slow catalysis. However, it should be noted that reducing the reactor depth to less than the depth of focus of the objective lens can be less efficient and even reduce the signal intensity. This can happen particularly when the enzyme exhaust the substrate, and the fluorescent dye concentration is constant, because the total number of fluorescent dye molecules that give a fluorescence signal in imaging should be proportional to the reactor volume under these conditions. The depth of focus of the objective lens used in digital bioassays is 1–2 μm. Therefore, the size of microreactors used in digital bioassays is typically 1–10 μm.
Another strategy for signal enhancement is the extension of the reaction time to allow the greater accumulation of fluorescent dyes in the reactors. Fig. 4c shows the correlation between the reactor volume and reaction time require to reach 500 nM for enzymes with the indicated catalytic turnover rate: 1, 10, 100, or 1000 turnover per s. Preferentially, the reaction time should be within 1 h, especially considering the stability of the optical systems and their application in diagnostic tests. The data points in Fig. 4c indicate the reactor volume and reaction time from the literatures selected based on the clarity of the experimental conditions.9,26,40,46,47,56 The colors of the lines and points indicate the range of catalytic turnover rate. As shown, the data points fall on or above the corresponding lines. Although longer incubation principally results in higher fluorescence signal, the signal should gradually reach a plateau level that is theoretically limited by the concentration of fluorogenic substrate in reactors.
3.5. Suppressing negative signals
Ideally, reactors without target molecules should not cause a pseudo-positive signal. However, in reality, the complete suppression of pseudo-positive signals is quite difficult because these signals have a range of origins, including optical, device, and sample solution effects.The noise from the components of the optical imaging systems is generally low compared to other noise sources. Camera noise is typically negligible. A typical noise source in optical imaging systems is the uneven excitation light in the imaging field. Because it is difficult to suppress this completely, the effect of uneven excitation should be corrected during image processing. In some cases, spherical aberrations can cause systematic noise. Because spherical aberration is severe only in the peripheral area, this effect can be minimized by omitting the peripheral area from image analysis.
One major cause of pseudo-positive signals or high background noise in reactor array systems is the residual enzyme assay solution in the device. Because the residual solution also generates a high fluorescence signal, uniform oil sealing to wash out excess enzyme solution is crucial to maintain a low level of background noise. Sometimes, exceptionally high noise causing outliers originates from impurities in the device, such as a residual photoresist. However, pseudo-positive signals arising from impurities do not increase fluorescence signal with reaction time. Therefore, when time-lapse images are analyzed to measure the rate of increase in fluorescence, pseudo-positive signals originating from device impurities can be discriminated from active reactors by differentiating the static and time-dependent signals.
Some pseudo-positive signals originate from the assay solution or the specimen. In the case of digital ELISA, the most typical pseudo-positive result is due to the nonspecific binding of enzyme-conjugated antibody molecules on microbeads or the device surfaces.12 Because enzymes can retain catalytic activity even if they are attached to microbeads or the device surface, these nonspecifically bound enzymes cause pseudo-positive signals at the same level as the positive signal. When clinical specimens are used as samples, contaminating enzymes in the specimens can also cause pseudo-positive signals, as seen in digital influenza virus counting.52 Another pseudo-positive mechanism, which increases the signal with time, is the autolysis of fluorogenic substrates, as described in subsection 2.5. Although the rate of spontaneous cleavage is evidently lower than that of the enzyme-catalyzed reaction, it becomes more significant when the reaction volume is large, where the signal increase caused by the enzymatic reaction is relatively small.
4. Applications of digital bioassays
4.1. Digital ELISA and related assays
A representative digital bioassay expected to be a next-generation diagnostic test is digital ELISA. Since the first digital ELISA assay was reported,13 various types of digital ELISAs have been reported.4,15,16,18,19,34,36,39 Although a microreactor system composed of a PDMS gasket and fabricated optic fiber plate was used in early studies (Fig. 5a),13,58 w/o droplet array systems have been widely employed12,32 after the first w/o array system was developed (Fig. 5b).31 Furthermore, the widespread application and further development of digital ELISA were triggered by the commercialization by an array device termed single-molecule array (SiMoA) and the related analysis system59 of Quanterix. This commercialization makes digital ELISA accessible to nonexperts and has greatly contributed to the development of disease-related marker assays such as those for prostate specific antigen (PSA),60 amyloid-β (1–42) peptide,61 interleukin proteins,62 and tau proteins14 (for a more comprehensive review on the applications of SiMoA, see ref. 34). | ||
| Fig. 5 Digital ELISAs. a) The schematic of digital ELISA with arrayed microreactor system. Reproduced from ref. 13, Copyright 2010 Springer Nature. b) Digital ELISA with FRAD. Reproduced from ref. 12, Copyright 2012 Royal Society of Chemistry. c) Digital homogeneous ELISA (homogeneous entropy-driven biomolecular assay). Reproduced from ref. 53, Copyright 2016 American Chemistry Society. d) Compartmentalization-free digital ELISA by use of tyramide deposition. Reproduced from ref. 19, Copyright 2016 American Chemistry Society. | ||
The reported LOD values of digital ELISA with array systems generally range from femtomolar to attomolar levels,4,34 which are 2–4 orders of magnitude better than that of conventional ELISA and are lower than the theoretical values estimated from the total reactor volume of droplet array systems. This is because, in the general protocol of digital ELISA, target molecules are enriched in a pull-down process using antibody-coated microbeads. However, the experimentally determined LODs do not reach the theoretical value of LOD (around the zeptomolar level), when taking the enrichment process into account.12 One of the major obstacles to achieving a theoretical LOD is the nonspecific binding of enzyme-conjugated antibodies, which causes a pseudo-positive reactor.12 Nonspecific binding of the conjugate can be mitigated by decreasing the concentration of the conjugate in the reaction mixture. However, there is a trade-off between the efficiency of antigen-antibody formation and the reduction of nonspecific binding. Another technical challenge in digital ELISA is sampling efficiency: that is, how efficiently target molecules are incorporated into microreactors after being captured on antibody-coated microbeads. In the conventional format of digital ELISA with arrayed reactors, the sampling efficiency ranges from 5% to 20%.12,59 Thus, the improvement of the sampling efficiency could result in 10-times lower LOD.
A free-droplet format has also been used. The earliest class of digital ELISA based on droplet fluidics was reported by Shim et al.,16 in which an immunocomplex for the detection of PSA was encapsulated in femtoliter w/o droplets with a volume of approximately 10 fL, and 20000 droplets were analyzed. The LOD of the system was 46 fM, higher by 3–4 orders of magnitude than that for digital ELISA with arrayed reactors12,13 because an enrichment process was not implemented. Later, another type of droplet-based digital ELISA was reported, in which a large number of droplets with picoliter volumes were analyzed.39 The resultant LOD values ranged from to 20–30 aM, which are 10-times better than that of the digital ELISA with the SiMoA system.
Conventional digital ELISAs are heterogeneous immunoassays that require bound/free (B/F) separation; before being introduced into a microreactor device or emulsified with droplet fluidics, samples must be subjected to B/F separation to remove unbound antibody conjugates. Because this process is laborious and requires additional instruments, a homogeneous method of digital ELISA is highly desirable. However, very few homogeneous formats of digital ELISA have been reported. Kim et al.53 exploited an entropy-driven nucleic acid strand displacement assay as a signal amplification process for a homogeneous entropy-driven biomolecular assay (HEBA) that provides a fluorescence signal without the B/F separation process (Fig. 5c). They achieved the attomolar-level detection of influenza nucleoproteins. Akama et al.15 reported another format for homogeneous digital immunoassay, termed digital HoNon-ELISA (digital homogeneous non-enzymatic immunosorbent assay). In this method, target molecules are captured in FRAD reactors via capture antibodies that are deposited on the reactors. For the detection of target molecules, magnetic nanoparticles conjugated with secondary antibodies are actively introduced into reactors via an external magnetic field to ensure immunocomplex formation with the target molecules, and the reactors are sealed with oil. Functional immunocomplexes were detected as nanoparticles showing tethered Brownian motion. Byrnes et al. reported another type of homogeneous digital ELISA that is based on proximity ligation where target molecule enhances the ligation reaction of two oligo nucleotides conjugated with antibodies.63 The ligation product is amplified in subsequent in-droplet PCR that gives high signal for binarization. The reported LOD is still in pM range, posing technical challenges for further improvements.
In addition to disease-related marker detection, digital ELISA can also be applied for enzymatic assays. Wang et al. reported eSiMoA, a novel ultra-sensitive enzyme assay based on digital ELISA; in this process, enzymes react with substrate-conjugated beads on which enzymes deposit phosphate or nucleotides on catalytic activity.64 Subsequently, the reacted beads are counted with antibody molecules against the deposited chemical groups in digital ELISA format. They achieved highly sensitive assays for protein kinases, telomerase, and transferases.
4.2. Digital ELISA in compartmentalization-free format
Compartmentalization-free formats for digital ELISAs were developed with the aim of circumventing the laborious and costly microfabrication process.18,19 These compartmentalization-free methods utilize “reaction localization” by the use of enzyme-catalyzed fluorescence deposition,19 rolling circle amplification of DNA for localized amplification of signals,18 or enzyme localization on substrate-coated microbeads,17 instead of the physical compartmentalization of the solution.The earliest class of compartmentalization-free digital ELISAs was reported by Akama et al.,19 who exploited tyramide signal amplification chemistry (Fig. 5d). HRP was used as a reporter enzyme and conjugated with the antibody. When reacted with fluorescently labeled tyramide substrates, HRP converted the tyramide substrates into short-lived radicals that formed covalent bonds with nearby proteins. When HRP-conjugated antibody molecules were bound to the target molecules captured on the microbeads, the tyramide radical reacted with the contiguous protein molecules on the microbeads, depositing fluorescent dyes on the microbead surface. Consequently, microbeads carrying ELISA complexes were selectively labeled with fluorescent dyes, allowing digital counting with flow cytometry. This compartmentalization-free digital ELISA achieved a LOD at the sub-femtomolar level, although the detection efficiency of the fluorescent beads was approximately 70% when compared with the droplet array-based digital ELISA because of the partial fluorescent labelling of the microbeads. A similar method based on tyramide chemistry that achieved an LOD around the femtomolar level has also been reported, in which the microbeads were embedded within fibrin hydrogels after reaction for microscopic imaging.65
Wu et al.18 reported another type of compartmentalization-free digital ELISA, employing phi29 DNA polymerase as the reporter enzyme that amplifies signals as long DNA strands in concatemer form via rolling circle amplification. In this study, the sample solution was casted and evaporated on a coverslip, and, because the amplified DNA molecules labeled with fluorescent oligo-probes were sufficiently stable to emit high fluorescence after drying, this method achieved an attomolar-level LOD, a 25-fold improvement compared to digital ELISA with the SiMoA system.
A compartmentalization-free digital bioassay with T4 polynucleotide kinase phosphatase (T4 PNKP)17 is also notable, even though this method is not an immunoassay. This assay is based on the unique features of T4 PNKP; that is, this enzyme catalyzes the dephosphorylation of the 3′-PO4 of DNA molecules and then translates to the neighboring DNA molecules for the next round of catalysis. Thus, once a single molecule of T4 PNKP attaches to a DNA-labeled microbead, the enzyme dephosphorylates many DNA molecules on the microbeads before dissociation. Therefore, when dephosphorylated DNA strands were fluorescently labeled, the number of T4 PNKPs could be counted based on the number of fluorescent beads.
4.3. Digital assays for the detection of SARS-CoV-2
The recent COVID 19 pandemic has drawn unprecedented demand for simple, rapid, and highly sensitive assays for the detection of RNA and/or proteins related to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) obtained from nasal swabs or other fluid samples from patients or people to be tested. Quantitative reverse transcription PCR (RT-qPCR) is currently the gold standard method for detecting the presence of the viral RNA from samples. However, RT-qPCR has some disadvantages. For example, RT-qPCR is not rapid: the standard protocol takes several hours for full processing. In addition, RT-qPCR requires the use of expensive instruments, hampering the use of RT-PCR in small clinics and in less developed countries/areas. As a result of the acute demand for simple, rapid, and highly sensitive assays for the detection of SARS-CoV-2, the development of digital bioassays has accelerated remarkably, and, since the emergence of COVID 19, this field has made important progress.Ge et al. developed a digital ELISA for the detection of the nucleocapsid protein (N protein) from SARS-Cov-2 using the aptamer/antibody sandwich method.41 They enhanced the detection limit by a factor of 300 compared to that of conventional ELISA methods.
An emerging class of digital bioassays to meet the requirements for the detection of SARS-Cov-2 RNA is Cas13-based digital bioassays. Cas13a proteins recognize target RNA via CRISPR RNA prebound on the protein. The association with the target RNA activates the trans-RNA cleavage activity of Cas13a to digest RNA molecules almost nonspecifically.66 This enables the digital counting of target RNA using an RNA quencher probe. Tian et al. reported the method for digital counting of RNA where the Cas13a assay mix was micro-compartmentalized into w/o droplets (Fig. 6a).24 Owing to the high catalytic activity of Cas13a (103–4 turnover per s), Cas13a accumulates detectable levels of fluorescent dyes in relatively large volume droplets (1 pL) as a microcompartment for digital bioassay. Due to the large size, the incubation for 60 min was required. This method reported an LOD of 6 copies per μL for a model sample. Shinoda et al. performed digital SARS-CoV-2 RNA counting using a FRAD.47 The volume was only 3 fL, which was sufficiently small to detect signals within 5 min, and the LOD of this system was approximately 10 fM. In a recent study, they reported higher sensitivity, 6.5 aM or less by employing pull-down enrichment process.67 These digital counting methods enable simple assay procedures without a preamplification process with reverse transcription. The simplicity of the protocol is one of the remarkable advantages compared with other Cas13a-based assays, such as SHERLOCK66 which requires nucleic acid amplification.
 | ||
| Fig. 6 Applications of digital bioassay. a) Digital Cas13 assay for the detection of SARS-CoV-2 RNA. Reproduced from ref. 24, Copyright 2021 American Chemistry Society. b) Heterogeneity analysis of ALP enzyme molecules investigated with FRAD. The histogram shows that ALP has two fractions: half active and fully active ones, each corresponding to heterodimers from active and inactive monomers and homodimer of active monomers. Reproduced from ref. 11, Copyright 2021 Wiley. c) Highly accurate screening method based on cell-free gene expression from single template DNA (digital gene expression). Reproduced from ref. 25, Copyright 2019 American Association for the Advancement of Science. d) ALBiC (arrayed lipid bilayer chamber) for single-molecule analysis of active transporter protein, F0F1 ATPase. Reproduced from ref. 20, Copyright 2014 Springer Nature. | ||
The digital counting of SARS-CoV-2 RNA with Cas12a has also been reported.48 Because Cas12a recognizes DNA to activate the trans-cleavage activity against RNA, the protocol includes a reverse-transcription process. Although the assay includes additional steps, this method achieved very high detection sensitivity, a single copy per microliter, owing to the preamplification process. A similar assay using a sample prewarming protocol to avoid undesired premature target amplification by Cas12a was reported to achieve comparable sensitivity (5 copies per mL).49 A digital counting assay with the Cas12a system would be suitable for diagnostic tests to detect patients infected with DNA viruses.38
Thus, digital assay with Cas proteins has seen rapid growth. However, it should be noted that the COVID 19 pandemic has accelerated not only digital bioassays but also other bioanalytic methods. In particular, compact diagnostics test systems have been developed based on RT-PCR or strand-exchange reactions.68,69 These methods also achieved swift and highly sensitive detection of SARS-CoV-2 RNA, and these have been commercialized already. Compared with these methods, digital bioassay based on Cas proteins are not obviously superior in terms of detection sensitivity and detection time. A remarkable feature of digital bioassays with Cas proteins, which differentiates them from other methods, is that Cas protein-based assays do not require thermal cycle or incubation at higher temperature that inevitably requires an external heating system, restricting the size reduction of the total system. Therefore, digital bioassay with Cas protein should seek more compact and cost-effective systems. In addition, upon the development of swift assay methods, it would become more important to develop simple and swift methods for preparations of biofluid samples that should be compatible with digital bioassays and/or other highly sensitive bioassays.
In addition to detection of SARS-CoV-2 RNA, digital ELISAs/immunoassays have been developed for immune response analysis of patients. Norman et al. reported digital ELISAs for the highly sensitive detection of anti-SARS-CoV-2 antibodies in the plasma of patients.70 They developed a multiplex digital ELISA for the detection of three immunoglobulins (IgG, IgM, and IgA) against three SARS-CoV-2 proteins. For the detection of each immunoglobulin, color-barcoded microbeads, each modified with either of the SARS-CoV-2 proteins and a corresponding color dye, were reacted with a sample and subjected to digital counting with the SiMoA system. This method allows the high-resolution profiling of the early seroconversion of plasma samples from patients.
4.4. Molecule-to-molecule heterogeneity analysis
Digital bioassays have contributed to the elucidation of the molecular mechanisms of enzymes, uncovering the features of enzymes masked by ensemble averaging in bulk solution experiments. One of the advantageous features of digital bioassay methods for enzyme studies is that they provide direct information on the heterogeneity of the catalytic activity of enzyme molecules. In particular, with arrayed microreactor systems, digital bioassays enable time-lapse analysis, allowing quantitative analysis of the catalytic activity of single enzyme molecules in a high-throughput manner. To date, all enzymes that have been quantitatively analyzed so far show molecule-to-molecule heterogeneity in catalytic activity. Because such heterogeneity is usually observed on long time scales, even after time-averaging for minutes to hours, the heterogeneities found in digital bioassays are considered to be “static” heterogeneity, unlike the “dynamic disorder”71 that shows dynamic fluctuation in catalytic rate within a short time (typically, millisecond to seconds). Although static heterogeneities were reported in the earliest single-molecule studies,72 population analysis of molecule-to-molecule heterogeneity has become accessible with digital bioassay.Distinctive heterogeneities in enzyme activity have been found in enzymes with oligomeric structures, for example, β-gal with a homo-tetramer56 and ALP with a homodimer,11 both of which are often used in digital ELISAs or related assays. Li et al. reported a non-Gaussian distribution of the catalytic activity of engineered hetero-tetrameric β-gal molecules composed of active monomer(s) and mutated inactive monomer(s). They also measured the mistranslation frequency of a reconstituted cell-free transcription and translation system (PURE system) by counting the number of hetero-tetrameric β-gal molecules carrying a single copy of an active monomer that was occasionally expressed in DNA encoding the inactive β-gal gene. Ueno et al. reported a digital bioassay for the quantitative analysis of the intrinsic heterogeneity of the ALP enzyme,11 and revealed that ALP has two distinctive populations, half-active and fully active; the former is a heterodimer enzyme with one inactive monomer unit, whereas the latter is a homodimer composed of active monomers (Fig. 6b). The distinctive heterogeneity of ALP was found in ALPs from different species, indicating that static heterogeneity is a common feature of ALPs.
Honda et al. developed a digital bioassay platform for serial buffer exchange to measure single molecules of enzymes under multidimensional conditions, i.e., substrate and inhibitor concentrations.10 They investigated the heterogeneity of neuraminidase activity among single influenza virus particles; each virus particle exhibited not only different neuraminidase activity but also different sensitivity to the inhibitors oseltamivir and zanamivir. It was shown that the half maximum inhibitory concentration (IC50) values differed by 20% among virus particles. Interestingly, no correlation was observed between the IC50 values of the two inhibitors, suggesting that the molecular mechanisms for the sensitivity/resistance of the virus to oseltamivir and zanamivir are different.
Functional substrates showing static heterogeneity between enzyme molecules are considered to be a key for the acquisition of new functions in evolutionary processes,73 and the evolutionary optimization of enzyme function can occur by shifting the population balance between functional substrates. Leibherr et al. investigated the molecule-to-molecule diversity in the catalytic activity of an in vitro partially evolved mutant of β-gluc that has high substrate promiscuity to catalyze hydrolytic reactions against a wide range of glycoside substrates.8 In comparison with wild-type β-gluc, the promiscuous mutant enzyme showed higher diversity in catalytic activity. This is an excellent example demonstrating the power of digital bioassays to provide important implications about enzyme evolution. From the viewpoint of protein engineering, molecule-to-molecule diversity could be considered as a barometer for enzyme evolvability in directed evolution experiments. More comprehensive analyses are required to generalize these findings.
4.5. Activity dynamics analysis
In addition to static heterogeneity, dynamic heterogeneity, i.e. fluctuations in catalytic activity is also accessible using digital bioassays. However, compared to single-turnover measurments,74 this approach is not superior in terms of time resolution. Therefore, there have been a limited number of studies on dynamic heterogeneity using digital bioassays. For example, Gorris et al. reported the activity fluctuation of β-gal in the presence of the slow-binding inhibitor D-galactal.75 Because the data did not show a clear transition between the active and inactive states, they measured the autocorrelation function to estimate the kinetic features of the inhibitor association and dissociation. Rojek et al. attempted heat stimulation of the transitions among the functional substates of β-gal.76 They concluded that individual enzymes switch their functional state almost randomly, and no correlation was found before and after the heat pulse.4.6. Digital bioassays for cell-free synthetic biology
Cell-free synthetic biology is a rapidly growing field that aims to reconstitute cellular functions using molecules outside cells.77–80 This trend has contributed significantly to the elucidation of molecular functions and also to technological developments for various applications, such as enzyme screening/selection, drug discovery, metabolic engineering, and biomanufacturing. The “single DNA molecule in a single micro-compartment” concept has been employed in a cell-free method for the directed evolution of enzymes, termed “in vitro compartmentalization (IVC)”,81 in which a single molecule of DNA is compartmentalized in w/o droplets with a cell-free transcription and translation (TXTL) system. The concept of gene expression from a single DNA molecule in a single reactor (digital gene expression) is essential for IVC methods to achieve efficient genotype–phenotype coupling.Cell-free TXTL has been implemented in a system for enzyme screening based on digital gene expression (Fig. 6c).25 Because the reactors of FRAD are highly monodisperse and the arrayed system allows time-lapse imaging for highly quantitative measurements, this method has achieved the highest enrichment factors, 104, and demonstrated the highly accurate selection of catalysis-enhanced ALP enzymes. Digital gene expression has also been reported in monodisperse w/o droplets prepared with microfluidics system82 and monodisperse liposomes.83
Another example of a digital concept in cell-free synthetic biology is gene replication from a single DNA/RNA molecule in a reactor, so-called digital gene replication.84–86 Ichihashi et al. reported cell-free gene replication using Qβ phage RNA polymerase proteins expressed from cell-free TXTL in a w/o emulsion.85 They found that TXTL-coupled gene replication under digital conditions was critical for avoiding or eliminating parasite RNA molecules. Similar findings have been reported by Matsumura et al.86 Recently, over 200 kbp DNA replication in w/o droplets under digital conditions using a reconstituted replisome system from E. coli was reported,84 termed reconstituted cycled replication (RCR).87
Thus, digital gene expression and replication are essential concepts in the field of cell-free synthetic biology. It is expected that the further implementation of cell-free systems, including synthetic molecular systems, into microcompartments will realize highly functionalized cell-like systems (artificial cells) with greater autonomy.
4.7. Digital bioassays for membrane transporter
Membrane transporter proteins play pivotal roles in many cellular activities such as signal transduction, energy transduction, the active uptake of nutrient molecules, the efflux of waste products or xenobiotic molecules, and membrane quality control.88,89 Therefore, there are great demands for quantitative analytical methods for membrane transporter proteins. Among the membrane transporter proteins, ion channel proteins can generate a high flux of ions across membranes (of the order of 107 ions per s) driven by the electrochemical potential across the membrane. Therefore, even single ion channel molecules can dissipate a large amount of energy, allowing electrochemical detection at the single channel level.90 However, most transporters are slow (generally slower than 102 transport events per s). In addition, transport substrates are not always electrically charged; therefore, transporters for such neutral substrates are quite difficult to detect using electrochemical methods.Recently, there have been increasing reports on new microsystems for digital bioassays of membrane transporters. The principal idea is the accumulation/ejection of transport substrates into or from a microreactor sealed by lipid bilayers. Transport activity is generally monitored using fluorescent probes encapsulated in the reactors. Tonooka et al. reported the detection of the passive Ca2+ transporting activity of α-hemolysin with picoliter droplets covered with a lipid bilayer.21 Watanabe et al. reported another microsystem termed ALBiC (arrayed lipid bilayer chamber) comprising 105 droplets covered with a lipid bilayer (Fig. 6d). The chamber volume was only 7 fL, which is sufficiently small for the detection of slow transporters.20 They successfully detected the transporting activities of not only a passive transporter, α-hemolysin, but also an active proton transporter, F0F1 ATPase, at the single-molecule level. To enhance the detection sensitivity further, an ALBiC with attoliter volume was developed.57 Another beneficial feature of ALBiC is that it allows the formation of an asymmetric bilayer with different lipid compositions in the upper and lower lipid layers.91 Taking this advantage, Watanabe et al. reported a single-molecule kinetic study of a phospholipid scramblase protein.92 A monodisperse liposome system has also been used for digital bioassays of transporters such as α-hemolysin and F0F1 ATPase.83 These lipid bilayer microsystems are expected to pave the way for the digital bioassays of a wide variety of membrane proteins.
5. Digital bioassays: perspectives
5.1. Enzyme variety
There are restrictions on the types of enzymes that are applicable to digital bioassays. One of the main reasons for this is that fluorogenic substrates are only available for a limited types of enzymes such as phosphatases, glycolysis enzymes, and nucleases. Therefore, the expansion of the chemical variety of fluorogenic substrates is required. Another way to expand the enzyme variety in digital bioassays is to utilize detection methods other than fluorescence imaging. Zhang et al. reported an electrochemical system for a digital influenza virus counting.93 Electrochemical digital bioassays are currently inferior in detection sensitivity and time compared to digital analysis with optical imaging/detection methods. However, given that many biosensors currently in widespread use are based on electrochemical detection, the potential of this strategy is particularly notable. In addition to electrochemical detection, mass spectrometry would also be an appropriate alternative detection method, especially considering that mass spectrometric analysis has been utilized for the analysis of the secretion activity of single cells entrapped in the FRAD system.945.2. Emerging technologies
The current methodology for micro-compartmentalization is also limited to a few major methods, such as FRAD, SiMoA, and w/o droplets. However, these methods require microfabrication for device preparation, and this has hindered the widespread use of digital bioassays. In this regard, the particle-template droplet generation method has great potential as an easily accessible method for generating monodisperse droplets.95 Recently, hydrogel particle-templated droplet formation was developed, and, using this, the digital counting of β-gal has been demonstrated.425.3. Challenges facing digital ELISA
Digital bioanalytical methods represented by digital ELISA are expected to have applications as ultrasensitive clinical diagnostic agents. However, there are some challenges preventing their clinical use. One is the false-positive signals that prevents digital ELISA from reaching its theoretical detection sensitivity.12 One of the major causes of false-positive signals is the nonspecific binding of antibody–enzyme conjugates to microbeads or device surfaces. Nonspecific binding could be minimized by coating the device surface with microbeads. Another challenge for the clinical application of digital ELISA is the size reduction of the total system down to a palmtop size or the scale of lateral-flow test kits to enable digital bioassays to enter the rapid diagnostic market. A straightforward strategy for downsizing the digital ELISA system is to use a small fluorescence imaging system. In this regard, smartphone-based detection systems have already been reported.36,96 In addition to downsizing the detection system, it is also necessary to reduce the size of the sample liquid-handling system. On-chip liquid handling methods, such as digital microfluidics97,98 would be suitable for downsizing the sample handling system for digital ELISA. An alternative is to improve wetwares. For example, the application of a homogeneous ELISA in digital format would enable a very simple digital immunoassay that does not require the B/F separation process. In addition to B/F separation, the enrichment process is another hurdle for downsizing of the liquid-handling process. Thus, the bead pull-down process should be replaced with another method that enables downsizing of the process.Another challenge of digital ELISA is how to implement the power of digital bioassay to discriminate molecule-to-molecule individuality. Multiplex digital assays by use of multiple fluorogenic substrates demonstrated the discrimination of ALP isoforms at single-molecule level.44 Because in digital ELISA target molecule is recognized by antibody, it should be hard to directly detect molecular individuality of target molecules. However, it would be achievable to detect the oligomeric state of target molecules by the quantification of the number of bound antibody-conjugated enzyme molecules. The formation of hetero molecular complex state with other proteins would be also achievable by use of multiple antibody-conjugated enzymes. These could be next challenges of digital ELISA to access the individuality of target molecules.
5.4. Integration of molecular systems
Digital bioassays have seen not only sophistication but also expansion to other fields, such as cell-free synthetic biology. This is because digital bioassays offer quantitative methods to analyze biomolecular systems reconstituted in microcompartments, such as artificial cell reactors. In addition to cell-free TXTL and DNA replication systems, the integration of other cell-free systems, such as DNA computing systems or biosensing systems, would open new avenues to create microreactor systems with higher autonomy and capability for information processing. For example, Gines et al. reported a novel digital counting method for microRNA molecules by implementing a DNA computing reaction system in microdroplets.99 Such integration of molecular systems into microreactors for more intelligent information processing would represent a new trend in the field of digital bioassays.6. Conclusions
Digital bioassays have been established as novel bioanalysis methods that offer ultrahigh sensitivity and allow for the analysis of the enzyme heterogeneity that is masked in conventional ensemble averaging. Digital ELISA, a representative digital bioassay, is expected to be a next-generation diagnostic assay and has shown tremendous progress in the development of ultrasensitive antigen/antibody detection and quantification. The digital bioassay concept has been applied to realize the single-molecule analysis of membrane transporters and to build functional cell-free microsystems for enzyme screening, information processing, and the reconstitution of autonomous molecular systems. Technologies for digital bioassays are being constantly updated, and novel technologies that could revolutionize the digital bioassay field are emerging.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr. Ron Milo (The Weizmann Institute of Science, Israel) for the data of Fig. 3a. This work was supported in part by Grant-in-Aid for Scientific Research on Innovation Areas (JP18H04817, JP19H05380, JP21H00388 to H. U.), Grant-in-Aids for Scientific Research (S) (JP19H05624 to H. N.) from the Japan Society for the Promotion of Science, and JST CREST, Japan (JPMJCR19S4 to H. N.).Notes and references
- Z. Farka, M. J. Mickert, M. Pastucha, Z. Mikusova, P. Skladal and H. H. Gorris, Angew. Chem., Int. Ed., 2020, 59, 10746–10773 CrossRef CAS PubMed
.
- Y. Zhang and H. Noji, Anal. Chem., 2017, 89, 92–101 CrossRef CAS PubMed
- L. Cohen and D. R. Walt, Annu. Rev. Anal. Chem., 2017, 10, 345–363 CrossRef CAS PubMed
- A. S. Basu, SLAS Technol., 2017, 22, 387–405 CrossRef
- A. S. Basu, SLAS Technol., 2017, 22, 369–386 CrossRef PubMed
- B. Vogelstein and K. W. Kinzler, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9236–9241 CrossRef CAS PubMed
- Y. Rondelez, G. Tresset, K. V. Tabata, H. Arata, H. Fujita, S. Takeuchi and H. Noji, Nat. Biotechnol., 2005, 23, 361–365 CrossRef CAS PubMed
- R. B. Liebherr, M. Renner and H. H. Gorris, J. Am. Chem. Soc., 2014, 136, 5949–5955 CrossRef CAS PubMed
- Y. Jiang, X. Li and D. R. Walt, Angew. Chem., Int. Ed., 2020, 59, 18010–18015 CrossRef CAS PubMed
- S. Honda, Y. Minagawa, H. Noji and K. V. Tabata, Anal. Chem., 2021, 93, 5494–5502 CrossRef CAS PubMed
- H. Ueno, M. Kato, Y. Minagawa, Y. Hirose and H. Noji, Protein Sci., 2021, 30, 1628–1639 CrossRef CAS PubMed
- S. H. Kim, S. Iwai, S. Araki, S. Sakakihara, R. Iino and H. Noji, Lab Chip, 2012, 12, 4986–4991 RSC
- D. M. Rissin, C. W. Kan, T. G. Campbell, S. C. Howes, D. R. Fournier, L. Song, T. Piech, P. P. Patel, L. Chang, A. J. Rivnak, E. P. Ferrell, J. D. Randall, G. K. Provuncher, D. R. Walt and D. C. Duffy, Nat. Biotechnol., 2010, 28, 595–599 CrossRef CAS PubMed
- D. Mengel, T. H. Mok, A. Nihat, W. Liu, R. A. Rissman, D. Galasko, H. Zetterberg, S. Mead, J. Collinge and D. M. Walsh, Cell, 2021, 10, 3514 CrossRef CAS PubMed
- K. Akama, N. Iwanaga, K. Yamawaki, M. Okuda, K. Jain, H. Ueno, N. Soga, Y. Minagawa and H. Noji, ACS Nano, 2019, 13, 13116–13126 CrossRef CAS PubMed
- J. U. Shim, R. T. Ranasinghe, C. A. Smith, S. M. Ibrahim, F. Hollfelder, W. T. Huck, D. Klenerman and C. Abell, ACS Nano, 2013, 7, 5955–5964 CrossRef CAS PubMed
- L. Zhang, W. Fan, D. Jia, Q. Feng, W. Ren and C. Liu, Anal. Chem., 2021, 93, 14828–14836 CrossRef CAS PubMed
- C. Wu, P. M. Garden and D. R. Walt, J. Am. Chem. Soc., 2020, 142, 12314–12323 CrossRef CAS
- K. Akama, K. Shirai and S. Suzuki, Anal. Chem., 2016, 88, 7123–7129 CrossRef CAS PubMed
- R. Watanabe, N. Soga, D. Fujita, K. V. Tabata, L. Yamauchi, S. Hyeon Kim, D. Asanuma, M. Kamiya, Y. Urano, H. Suga and H. Noji, Nat. Commun., 2014, 5, 4519 CrossRef CAS PubMed
- T. Tonooka, K. Sato, T. Osaki, R. Kawano and S. Takeuchi, Small, 2014, 10, 3275–3282 CrossRef CAS PubMed
- L. Cohen, M. R. Hartman, A. Amardey-Wellington and D. R. Walt, Nucleic Acids Res., 2017, 45, e137 CrossRef CAS PubMed
- Z. Li, R. B. Hayman and D. R. Walt, J. Am. Chem. Soc., 2008, 130, 12622–12623 CrossRef CAS PubMed
- T. Tian, B. Shu, Y. Jiang, M. Ye, L. Liu, Z. Guo, Z. Han, Z. Wang and X. Zhou, ACS Nano, 2021, 15, 1167–1178 CrossRef CAS PubMed
- Y. Zhang, Y. Minagawa, H. Kizoe, K. Miyazaki, R. Iino, H. Ueno, K. V. Tabata, Y. Shimane and H. Noji, Sci. Adv., 2019, 5, eaav8185 CrossRef CAS PubMed
- Y. Obayashi, R. Iino and H. Noji, Analyst, 2015, 140, 5065–5073 RSC
- J. E. Kreutz, T. Munson, T. Huynh, F. Shen, W. Du and R. F. Ismagilov, Anal. Chem., 2011, 83, 8158–8168 CrossRef CAS PubMed
- B. Rotman, Proc. Natl. Acad. Sci. U. S. A., 1961, 47, 1981–1991 CrossRef CAS
- D. M. Rissin and D. R. Walt, J. Am. Chem. Soc., 2006, 128, 6286–6287 CrossRef CAS PubMed
- D. M. Rissin and D. R. Walt, Nano Lett., 2006, 6, 520–523 CrossRef CAS PubMed
- S. Sakakihara, S. Araki, R. Iino and H. Noji, Lab Chip, 2010, 10, 3355–3362 RSC
- H. Zhang, S. Nie, C. M. Etson, R. M. Wang and D. R. Walt, Lab Chip, 2012, 12, 2229–2239 RSC
- C. W. Kan, A. J. Rivnak, T. G. Campbell, T. Piech, D. M. Rissin, M. Mosl, A. Peterca, H. P. Niederberger, K. A. Minnehan, P. P. Patel, E. P. Ferrell, R. E. Meyer, L. Chang, D. H. Wilson, D. R. Fournier and D. C. Duffy, Lab Chip, 2012, 12, 977–985 RSC
- C. Wu, A. M. Maley and D. R. Walt, Crit. Rev. Clin. Lab. Sci., 2019, 270–290, DOI:10.1080/10408363.2019.1700903
- P. Mogalisetti and D. R. Walt, Methods Enzymol., 2016, 581, 541–560 CAS
- V. Yelleswarapu, J. R. Buser, M. Haber, J. Baron, E. Inapuri and D. Issadore, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4489–4495 CrossRef CAS PubMed
- Z. Guan, Y. Zou, M. Zhang, J. Lv, H. Shen, P. Yang, H. Zhang, Z. Zhu and C. J. Yang, Biomicrofluidics, 2014, 8, 014110 CrossRef PubMed
- H. Yue, B. Shu, T. Tian, E. Xiong, M. Huang, D. Zhu, J. Sun, Q. Liu, S. Wang, Y. Li and X. Zhou, Nano Lett., 2021, 21, 4643–4653 CrossRef CAS PubMed
- L. Cohen, N. Cui, Y. Cai, P. M. Garden, X. Li, D. A. Weitz and D. R. Walt, ACS Nano, 2020, 14, 9491–9501 CrossRef CAS PubMed
- R. Arayanarakool, L. Shui, S. W. Kengen, A. van den Berg and J. C. Eijkel, Lab Chip, 2013, 13, 1955–1962 RSC
- C. Ge, J. Feng, J. Zhang, K. Hu, D. Wang, L. Zha, X. Hu and R. Li, Talanta, 2022, 236, 122847 CrossRef CAS PubMed
- Y. Wang, V. Shah, A. Lu, E. Pachler, B. Cheng and D. Di Carlo, Lab Chip, 2021, 21, 3438–3448 RSC
- Y. Jiang, X. Li, B. R. Morrow, A. Pothukuchy, J. Gollihar, R. Novak, C. B. Reilly, A. D. Ellington and D. R. Walt, ACS Cent. Sci., 2019, 5, 1691–1698 CrossRef CAS PubMed
- S. Sakamoto, T. Komatsu, R. Watanabe, Y. Zhang, T. Inoue, M. Kawaguchi, H. Nakagawa, T. Ueno, T. Okusaka, K. Honda, H. Noji and Y. Urano, Sci. Adv., 2020, 6, eaay0888 CrossRef CAS PubMed
- T. Ono, T. Ichiki and H. Noji, Analyst, 2018, 143, 4923–4929 RSC
- H. H. Gorris and D. R. Walt, J. Am. Chem. Soc., 2009, 131, 6277–6282 CrossRef CAS PubMed
- H. Shinoda, Y. Taguchi, R. Nakagawa, A. Makino, S. Okazaki, M. Nakano, Y. Muramoto, C. Takahashi, I. Takahashi, J. Ando, T. Noda, O. Nureki, H. Nishimasu and R. Watanabe, Commun. Biol., 2021, 4, 476 CrossRef CAS PubMed
- J. S. Park, K. Hsieh, L. Chen, A. Kaushik, A. Y. Trick and T. H. Wang, Adv. Sci., 2021, 8, 2003564 CrossRef CAS PubMed
- X. Ding, K. Yin, Z. Y. Li, M. M. Sfeir and C. C. Liu, Biosens. Bioelectron., 2021, 184, 113218 CrossRef CAS PubMed
- A. Bar-Even, E. Noor, Y. Savir, W. Liebermeister, D. Davidi, D. S. Tawfik and R. Milo, Biochemistry, 2011, 50, 4402–4410 CrossRef CAS PubMed
- Y. Urano, Curr. Opin. Chem. Biol., 2012, 16, 602–608 CrossRef CAS PubMed
- K. V. Tabata, Y. Minagawa, Y. Kawaguchi, M. Ono, Y. Moriizumi, S. Yamayoshi, Y. Fujioka, Y. Ohba, Y. Kawaoka and H. Noji, Sci. Rep., 2019, 9, 1067 CrossRef PubMed
- D. Kim, O. B. Garner, A. Ozcan and D. Di Carlo, ACS Nano, 2016, 10, 7467–7475 CrossRef CAS PubMed
- F. Shen, B. Sun, J. E. Kreutz, E. K. Davydova, W. Du, P. L. Reddy, L. J. Joseph and R. F. Ismagilov, J. Am. Chem. Soc., 2011, 133, 17705–17712 CrossRef CAS PubMed
- S. Ge, W. Liu, T. Schlappi and R. F. Ismagilov, J. Am. Chem. Soc., 2014, 136, 14662–14665 CrossRef CAS PubMed
- X. Li, Y. Jiang, S. Chong and D. R. Walt, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8346–8351 CrossRef CAS PubMed
- N. Soga, R. Watanabe and H. Noji, Sci. Rep., 2015, 5, 11025 CrossRef PubMed
- S. Nie, E. Benito-Pena, H. Zhang, Y. Wu and D. R. Walt, Anal. Chem., 2013, 85, 9272–9280 CrossRef CAS PubMed
- D. H. Wilson, D. M. Rissin, C. W. Kan, D. R. Fournier, T. Piech, T. G. Campbell, R. E. Meyer, M. W. Fishburn, C. Cabrera, P. P. Patel, E. Frew, Y. Chen, L. Chang, E. P. Ferrell, V. von Einem, W. McGuigan, M. Reinhardt, H. Sayer, C. Vielsack and D. C. Duffy, J. Lab. Autom., 2016, 21, 533–547 CrossRef CAS PubMed
- D. H. Wilson, D. W. Hanlon, G. K. Provuncher, L. Chang, L. Song, P. P. Patel, E. P. Ferrell, H. Lepor, A. W. Partin, D. W. Chan, L. J. Sokoll, C. D. Cheli, R. P. Thiel, D. R. Fournier and D. C. Duffy, Clin. Chem., 2011, 57, 1712–1721 CrossRef CAS PubMed
- L. Song, D. R. Lachno, D. Hanlon, A. Shepro, A. Jeromin, D. Gemani, J. A. Talbot, M. M. Racke, J. L. Dage and R. A. Dean, Alzheimer's Res. Ther., 2016, 8, 58 CrossRef PubMed
- M. Gilbert, R. Livingston, J. Felberg and J. J. Bishop, Anal. Biochem., 2016, 503, 11–20 CrossRef CAS PubMed
- S. A. Byrnes, T. Huynh, T. C. Chang, C. E. Anderson, J. J. McDermott, C. I. Oncina, B. H. Weigl and K. P. Nichols, Anal. Chem., 2020, 92, 3535–3543 CrossRef CAS PubMed
- X. Wang, A. F. Ogata and D. R. Walt, J. Am. Chem. Soc., 2020, 142, 15098–15106 CrossRef CAS PubMed
- A. M. Maley, P. M. Garden and D. R. Walt, ACS Sens., 2020, 5, 3037–3042 CrossRef CAS PubMed
- C. Myhrvold, C. A. Freije, J. S. Gootenberg, O. O. Abudayyeh, H. C. Metsky, A. F. Durbin, M. J. Kellner, A. L. Tan, L. M. Paul, L. A. Parham, K. F. Garcia, K. G. Barnes, B. Chak, A. Mondini, M. L. Nogueira, S. Isern, S. F. Michael, I. Lorenzana, N. L. Yozwiak, B. L. MacInnis, I. Bosch, L. Gehrke, F. Zhang and P. C. Sabeti, Science, 2018, 360, 444–448 CrossRef CAS PubMed
- H. Shinoda, T. Iida, A. Makino, M. Yoshimura, J. Ishikawa, J. Ando, K. Murai, K. Sugiyama, Y. Muramoto, M. Nakano, K. Kiga, L. Cui, O. Nureki, H. Takeuchi, T. Noda, H. Nishimasu and R. Watanabe, Commun. Biol., 2022, 5, 473 CrossRef CAS PubMed
- M. C. Smithgall, I. Scherberkova, S. Whittier and D. A. Green, J. Clin. Virol., 2020, 128, 104428 CrossRef CAS PubMed
- J. Lee and J. U. Song, J. Med. Virol., 2021, 93, 4523–4531 CrossRef CAS PubMed
- M. Norman, T. Gilboa, A. F. Ogata, A. M. Maley, L. Cohen, E. L. Busch, R. Lazarovits, C. P. Mao, Y. Cai, J. Zhang, J. E. Feldman, B.
M. Hauser, T. M. Caradonna, B. Chen, A. G. Schmidt, G. Alter, R. C. Charles, E. T. Ryan and D. R. Walt, Nat. Biomed. Eng., 2020, 4, 1180–1187 CrossRef CAS PubMed
- S. C. Kou, B. J. Cherayil, W. Min, B. P. English and X. S. Xie, J. Phys. Chem. B, 2005, 109, 19068–19081 CrossRef CAS PubMed
- Q. Xue and E. S. Yeung, Nature, 1995, 373, 681–683 CrossRef CAS PubMed
- O. Khersonsky and D. S. Tawfik, Annu. Rev. Biochem., 2010, 79, 471–505 CrossRef CAS PubMed
- P. Turunen, A. E. Rowan and K. Blank, FEBS Lett., 2014, 588, 3553–3563 CrossRef CAS PubMed
- H. H. Gorris, D. M. Rissin and D. R. Walt, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17680–17685 CrossRef CAS PubMed
- M. J. Rojek and D. R. Walt, PLoS One, 2014, 9, e86224 CrossRef PubMed
- A. D. Silverman, A. S. Karim and M. C. Jewett, Nat. Rev. Genet., 2020, 21, 151–170 CrossRef CAS PubMed
- V. Noireaux and A. P. Liu, Annu. Rev. Biomed. Eng., 2020, 22, 51–77 CrossRef CAS PubMed
- N. Laohakunakorn, L. Grasemann, B. Lavickova, G. Michielin, A. Shahein, Z. Swank and S. J. Maerkl, Front. Bioeng. Biotechnol., 2020, 8, 213 CrossRef PubMed
- J. G. Perez, J. C. Stark and M. C. Jewett, Cold Spring Harbor Perspect. Biol., 2016, 8, a023853 CrossRef PubMed
- O. J. Miller, K. Bernath, J. J. Agresti, G. Amitai, B. T. Kelly, E. Mastrobattista, V. Taly, S. Magdassi, D. S. Tawfik and A. D. Griffiths, Nat. Methods, 2006, 3, 561–570 CrossRef CAS PubMed
- F. Courtois, L. F. Olguin, G. Whyte, D. Bratton, W. T. Huck, C. Abell and F. Hollfelder, ChemBioChem, 2008, 9, 439–446 CrossRef CAS PubMed
- N. Soga, A. Ota, K. Nakajima, R. Watanabe, H. Ueno and H. Noji, ACS Nano, 2020, 14, 11700–11711 CrossRef CAS PubMed
- H. Ueno, H. Sawada, N. Soga, M. Sano, S. Nara, K. V. Tabata, M. Su'etsugu and H. Noji, ACS Synth. Biol., 2021, 10, 2179–2186 CrossRef CAS PubMed
- N. Ichihashi, K. Usui, Y. Kazuta, T. Sunami, T. Matsuura and T. Yomo, Nat. Commun., 2013, 4, 2494 CrossRef PubMed
- S. Matsumura, A. Kun, M. Ryckelynck, F. Coldren, A. Szilagyi, F. Jossinet, C. Rick, P. Nghe, E. Szathmary and A. D. Griffiths, Science, 2016, 354, 1293–1296 CrossRef CAS PubMed
- M. Su'etsugu, H. Takada, T. Katayama and H. Tsujimoto, Nucleic Acids Res., 2017, 45, 11525–11534 CrossRef PubMed
- M. H. Saier Jr., Microbiol. Mol. Biol. Rev., 2000, 64, 354–411 CrossRef CAS PubMed
- C. International Transporter, K. M. Giacomini, S. M. Huang, D. J. Tweedie, L. Z. Benet, K. L. Brouwer, X. Chu, A. Dahlin, R. Evers, V. Fischer, K. M. Hillgren, K. A. Hoffmaster, T. Ishikawa, D. Keppler, R. B. Kim, C. A. Lee, M. Niemi, J. W. Polli, Y. Sugiyama, P. W. Swaan, J. A. Ware, S. H. Wright, S. W. Yee, M. J. Zamek-Gliszczynski and L. Zhang, Nat. Rev. Drug Discovery, 2010, 9, 215–236 CrossRef PubMed
- D. Sigg, J. Gen. Physiol., 2014, 144, 7–26 CrossRef CAS PubMed
- R. Watanabe, N. Soga, T. Yamanaka and H. Noji, Sci. Rep., 2014, 4, 7076 CrossRef PubMed
- R. Watanabe, T. Sakuragi, H. Noji and S. Nagata, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3066–3071 CrossRef CAS PubMed
- Z. Wu, C. H. Zhou, L. J. Pan, T. Zeng, L. Zhu, D. W. Pang and Z. L. Zhang, Anal. Chem., 2016, 88, 9166–9172 CrossRef CAS PubMed
- H. Fujita, T. Esaki, T. Masujima, A. Hotta, S. H. Kim, H. Noji and T. M. Watanabe, RSC Adv., 2015, 5, 16968–16971 RSC
- M. N. Hatori, S. C. Kim and A. R. Abate, Anal. Chem., 2018, 90, 9813–9820 CrossRef CAS PubMed
- Y. Minagawa, H. Ueno, K. V. Tabata and H. Noji, Lab Chip, 2019, 19, 2678–2687 RSC
- K. Choi, A. H. Ng, R. Fobel and A. R. Wheeler, Annu. Rev. Anal. Chem., 2012, 5, 413–440 CrossRef CAS PubMed
- D. Witters, K. Knez, F. Ceyssens, R. Puers and J. Lammertyn, Lab Chip, 2013, 13, 2047–2054 RSC
- G. Gines, R. Menezes, K. Nara, A. S. Kirstetter, V. Taly and Y. Rondelez, Sci. Adv., 2020, 6, eaay5952 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2022 |