
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceViable protoplast formation of the coral endosymbiont alga Symbiodinium spp. in a microfluidics platform†
Faiza
Bashir
ab,
Sándor
Kovács‡
a,
Ágnes
Ábrahám
cd,
Krisztina
Nagy
c,
Ferhan
Ayaydin§
 e,
Ildikó
Valkony-Kelemen
e,
Györgyi
Ferenc
a,
Péter
Galajda
c,
Szilvia Z.
Tóth
a,
László
Sass
a,
Péter B.
Kós
af,
Imre
Vass
*a and
Milán
Szabó
*ag
e,
Ildikó
Valkony-Kelemen
e,
Györgyi
Ferenc
a,
Péter
Galajda
c,
Szilvia Z.
Tóth
a,
László
Sass
a,
Péter B.
Kós
af,
Imre
Vass
*a and
Milán
Szabó
*ag
aInstitute of Plant Biology, Biological Research Centre, Eötvös Loránd Research Network, Szeged, Hungary. E-mail: vass.imre@brc.hu; szabo.milan@brc.hu
bDoctoral School of Biology, University of Szeged, Szeged, Hungary
cInstitute of Biophysics, Biological Research Centre, Eötvös Loránd Research Network, Szeged, Hungary
dDoctoral School of Multidisciplinary Medical Sciences, University of Szeged, Szeged, Hungary
eCellular Imaging Laboratory, Biological Research Centre, Eötvös Loránd Research Network, Szeged, Hungary
fDepartment of Biotechnology, Faculty of Science and Informatics, University of Szeged, Szeged, Hungary
gClimate Change Cluster, University of Technology Sydney, Australia
First published on 19th May 2022
Abstract
Symbiodiniaceae is an important dinoflagellate family which lives in endosymbiosis with reef invertebrates, including coral polyps, making them central to the holobiont. With coral reefs currently under extreme threat from climate change, there is a pressing need to improve our understanding on the stress tolerance and stress avoidance mechanisms of Symbiodinium spp. Reactive oxygen species (ROS) such as singlet oxygen are central players in mediating various stress responses; however, the detection of ROS using specific dyes is still far from definitive in intact Symbiodinium cells due to the hindrance of uptake of certain fluorescent dyes because of the presence of the cell wall. Protoplast technology provides a promising platform for studying oxidative stress with the main advantage of removed cell wall, however the preparation of viable protoplasts remains a significant challenge. Previous studies have successfully applied cellulose-based protoplast preparation in Symbiodiniaceae; however, the protoplast formation and regeneration process was found to be suboptimal. Here, we present a microfluidics-based platform which allowed protoplast isolation from individually trapped Symbiodinium cells, by using a precisely adjusted flow of cell wall digestion enzymes (cellulase and macerozyme). Trapped single cells exhibited characteristic changes in their morphology, cessation of cell division and a slight decrease in photosynthetic activity during protoplast formation. Following digestion and transfer to regeneration medium, protoplasts remained photosynthetically active, regrew cell walls, regained motility, and entered exponential growth. Elevated flow rates in the microfluidic chambers resulted in somewhat faster protoplast formation; however, cell wall digestion at higher flow rates partially compromised photosynthetic activity. Physiologically competent protoplasts prepared from trapped cells in microfluidic chambers allowed for the first time the visualization of the intracellular localization of singlet oxygen (using Singlet Oxygen Sensor Green dye) in Symbiodiniaceae, potentially opening new avenues for studying oxidative stress.
Introduction
Coral reef ecosystems represent essential habitats for a wide variety of marine animals.1 Reef-building corals belonging to the class anthozoa (Cnidaria) are among the most biologically diverse groups. Corals harbour endosymbiotic algae Symbiodiniaceae that reside within the endodermal tissue of the anthozoan host.2Symbiodinium spp. are the crucial symbiotic component of reef-building corals and a primary producer in the aquatic ecosystem on Earth.3 In this symbiotic relationship, Symbiodinium provides organic compounds to the host (Anthozoa) and in return it receives inorganic compounds and protection from the host, which makes it pivotal for coral physiology.4Symbiodiniaceae not only drive coral productivity and reef growth,5 but their physiological, biochemical, morphological and genetic diversity can also determine the thermal bleaching thresholds of their coral host.6–10 Cultured Symbiodinium exhibits daily morphological changes between a flagellated (motile) stage during the day to a non-flagellated spherical (coccoid) stage at night.11Symbiodinium cells in the motile stage have thin thecal plates,12,13 and thus, some Symbiodinium species have been formally described with thecal plate tabulations for species criterion.14–16 However, the differences in thecal tabulations between genetically distant species are small;15 therefore, molecular grouping has been commonly used. As a result, Symbiodinium has been classified into groups (clades A–I) and each clade consists of numerous subclades or types.17 However, more recent molecular and taxonomic analyses indicated the necessity to reassign Symbiodinium clades as individual genera within the family Symbiodiniaceae.18 Therefore, a new taxonomic nomenclature applies to the previously defined clades, with only clade A being referred to as Symbiodinium, which is consistently applied in the current work.
The host-symbiont relationship is very sensitive to environmental stresses such as ocean acidification and increased temperature, but also pollutions, pathogens and changes in salinity.19 As a consequence of the ecological stressors, the beneficial interaction between the coral host and its algal symbiont may cease, prompting the expulsion of the zooxanthellae and leading to a phenomenon known as coral bleaching.20,21 The mechanism of the expulsion of Symbiodinium is still not fully known. The role of reactive oxygen species (ROS), such as the highly reactive singlet oxygen (1O2),22 the superoxide anion radical, hydrogen peroxide and the hydroxyl radical which are generated under heat and light stress, potentially causing breakdown of the symbiosis, has been extensively studied.23–29 Previous studies have shown that the increased production of 1O2 occurs primarily under high light stress conditions inside chloroplasts,30,31 which will ultimately result in the inactivation of photosystem II (PSII) and photoinhibition.32–351O2 may accumulate in Symbiodinium cells and can be transferred to its coral host.361O2 detection is important, but still largely unresolved at subcellular level. The fluorescein derivative 1O2-specific fluorescent dye, Singlet Oxygen Sensor Green (SOSG) has been applied to detect 1O2 in intact leaves37,38 and in single cells of the green alga Chlamydomonas reinhardtii.39 However, SOSG has constrained penetration into cyanobacterial cells39 and SOSG was found to have no or limited ability to penetrate into Symbiodinium cells.36,40 Therefore, for the purpose of intracellular detection of 1O2 it is crucial to employ living cells which are devoid of cell wall, such as protoplasts.
Protoplast is a term used for bacterial, fungal or plant cells whose cell wall was removed mechanically or enzymatically.41 Protoplast technology has extensive applications in plants and microalgae for nuclear transformation, somatic hybridization, in vitro cell culturing and plant regeneration.42,43 Whereas various methods and procedures have been developed and successfully applied to a range of microalgae species, isolation and regeneration of viable protoplasts in Symbiodiniaceae has just recently been established44 and attempted to be applied for transformation of the nuclear genome of Symbiodinium (to date, the nuclear transformation of the Symbiodinium genome has been unsuccessful45). The major structural element of the cell wall of Symbiodinium is cellulose associated with proteins/glycoproteins, therefore the Symbiodinium cell wall can be digested with cellulase enzymes.46 However, the cell wall organization of dinoflagellates is complex, it is composed of several pellicles and thecal plate layers,47,48 which may represent a significant hindrance in cell wall digestion using cellulase enzymes. The first successful isolation of protoplasts from Symbiodinium was carried out recently,44 but the digestion and regeneration of the cell wall was time consuming, making it cumbersome for downstream applications. Furthermore, the lack of high-resolution real time analysis of the protoplast formation process at the single-cell level hinders the understanding, efficacy and physiological features of protoplast formation. Therefore the application of protoplast technology as a biotechnology tool remains far from routine in Symbiodiniaceae.
Protoplast formation encounters the challenges of low output and loss of cell viability in traditional laboratory settings. Microfluidic methods have demonstrated their high potential in various microbial applications under controlled environments and high-throughput biological assays.49–52 Numerous microfluidic systems have been used to grow microalgae and study their growth rate at microscale.53 Microfluidic technology enables studying the growth and single-cell heterogeneity of microalgae,54–56 and the production of various compounds such as extracellular polymeric substances57 or pigments.58 Several microfluidics-based approaches have also been applied to investigate the chemoattraction and chemotaxis behavior of marine phytoplankton and bacteria59 and diseases caused by bacterial infections in corals.60 To obtain the ideal growth conditions for microalgae, microscale photobioreactors have been developed, which have a high potential for strain optimization and phenotype assays.55 Microfluidics-based methods also allowed the trapping and characterization of plant protoplasts and monitoring their regeneration.61 By manipulating the geometry of the microfluidic channels and adjusting the flow rate, it is possible to optimize the protoplast yield from plant cells.62 Recently, a microfluidics-based single-cell phenotyping assay has been developed for various Symbiodiniaceae, offering a high-throughput capability to monitor photophysiological changes at the single-cell level under heat stress.63,64 However, protoplast isolation and monitoring their physiological/photosynthetic capacity in microfluidic chambers have not been established so far. Furthermore, the ability of intracellular detection of 1O2 in Symbiodinium has remained far from definitive.
In the present work, we aimed to optimize the protoplast formation and regeneration process and determine the timescale of protoplast formation using a morphometric analysis. We investigated the morphological differences in the cell division pattern in intact Symbiodinium cells and protoplasts. The novelties of the study are 1) the establishment of a microfluidic system in which viable and physiologically competent Symbiodinium protoplasts can be obtained and regenerated into intact cells, 2) the real-time monitoring of the process, which allows the establishment of a single-cell morphometric analysis and process optimization by varying flow rates to achieve high yield of protoplasts, and 3) the application of protoplasts for intracellular detection of singlet oxygen, and for demonstrating the uptake of synthetic oligonucleotides.
Experimental
Symbiodinium cultures
Symbiodinium microadriaticum Freudenthal culture strain CCMP2467 (former clade A1) was obtained from the National Center for Marine Algae and Microbiota (NCMA), Bigelow Laboratory for Ocean Sciences USA. Symbiodinium microadriaticum CCMP2467 was originally isolated from the scleractinian coral Stylophora pistillata. Strain CCMP2467 belongs to clade A1 based on the nuclear internal transcribed region 2 (ITS2).65Cells were grown in F/2 medium at 24 °C at a light intensity of 50 μmol photons m−2 s−1, with a light:dark period of 12 h:12 h. The growth rate and number of cells were determined daily by using a LUNA automated cell counter (Biosystems). The cells were collected during their exponential growth phase by centrifugation at 2000 g for 4 min and resuspended in fresh F/2 medium for the experiments.
Microfluidics setup and cell monitoring
The microfluidic device (Fig. 1) was designed according to the characteristic cell size of about 10 μm (Fig. 3). It is composed of an array of traps with three different sizes, with the purpose of assessing the suitability of trapping and monitoring Symbiodinium cells in traps of varying volumes. All traps were found to be suitable for monitoring single cells throughout the whole process of protoplast formation and regeneration. The microfluidic devices were designed using the open source KLayout software (ref: https://www.klayout.de). The devices were fabricated using soft lithography.66 Briefly, a master mold was created from SU-82015 photoresist (Microchem corp., Westborough MA, USA) on a silicon substrate by laser direct writing using a Heidelberg μPG 101 micro pattern generator (Heidelberg Instruments GmbH, Heidelberg, Germany). The master mold was replicated by casting of Sylgard 184 polydimethylsiloxane (The Dow Chemical Company, Midland MI, USA). After punching access holes into the cured PDMS pieces, they were bound to glass coverslips by oxygen plasma treatment using a Harrick PDC-002 plasma cleaner (Harrick Plasma, Ithaca NY, USA). | ||
| Fig. 1 Schematics of the microfluidic device for single-cell protoplast preparation and analysis. (A) Overview of the microfluidic device showing the inlet, the trifurcated branches and the outlet. (B) The detailed trifurcated part containing traps of different sizes. The right side shows the inlet which leads to the open side of the traps. From the inlet, culture medium and cells are injected into the device, and after passing through the traps the medium leaves the device at the outlet. The red arrow shows the flow direction. (C) Schematic representation of single traps with three different sizes. (i) Large trap with 60 μm width, (ii) medium-sized trap with 40 μm width, (iii) small trap with 30 μm width. For single-cell monitoring, the medium-sized traps were used in most experiments. All the traps have a 10 μm wall thickness with 5 μm gaps. | ||
The setup is composed of three parts, the microfluidic device, a tubing system and a syringe pump. Cell cultures were diluted twofold (cell density was diluted from ∼105 cells per ml diluted to 5 × 104 cells per ml) and were loaded from the inlet by using a pipette. The microfluidic device was mounted on the microscope stage and a long tube was inserted in the inlet hole, while a smaller tube was inserted in the outlet hole (Fig. 1). The inlet tube was attached to a syringe (Omnifix-F Solo Luer 1 mL, B. Braun) containing the media and/or the enzyme mounted on a syringe pump (SyringeTwo-SKU 4000, New Era Pump Systems, Inc. USA). Different flow rates, 20, 40, 60, 80, 100 (μL h−1) were applied to investigate the impact of flow rate on the dynamics and efficiency of protoplast formation. The images were captured with a 25× objective (Hund Wetzlar, Helmut Hund GmbH, Wetzlar, Germany) by using a light microscope (H 600/12, Hund Wetzlar, Helmut Hund GmbH, Wetzlar, Germany) equipped with a microQ digital camera (UCMOS08000KPB, ToupTek Photonics Co., Ltd., Hangzhou, China). ToupView, a camera control software (ToupTek Photonics Co., Ltd., Hangzhou, China) was used to continuously capture the images every 10 s. The changes in the morphology and cell size throughout the process of protoplasting and cell wall regeneration were analyzed with the Matlab software (The MathWorks Inc., Natick, MA, USA). A microscopy version of a Pulse Amplitude Modulation Chlorophyll fluorescence imaging system (Imaging-PAM M-Series Chlorophyll Fluorometer, Microscopy version, Heinz Walz GmbH, Effeltrich, Germany) was attached to the microscope and single-cell photophysiology and morphology were monitored under identical conditions, on the same cells (see below).
Characterization of the fluid flow by model calculations
The characteristics of the fluid flow within the microfluidic device were calculated with the Comsol Multiphysics 4.3a software. The velocity field was calculated using the “Laminar flow” model with a time dependent study. We applied the “shallow channel” approximation to get a quasi 3D model of the flow. Calculations were done for 20 μL h−1, 60 μL h−1 and 100 μL h−1 flow rates. Results show that the average flow velocity differs by less than 10% in the three branches of the device. Fig. 2A show the flow fields (flow lines and color coded flow velocity magnitudes) for the three different trap sizes. In case of a 60 μL h−1 overall flow rate in the device maximal flow velocities are in the 350–450 μm s−1 range within the traps (Fig. 2B).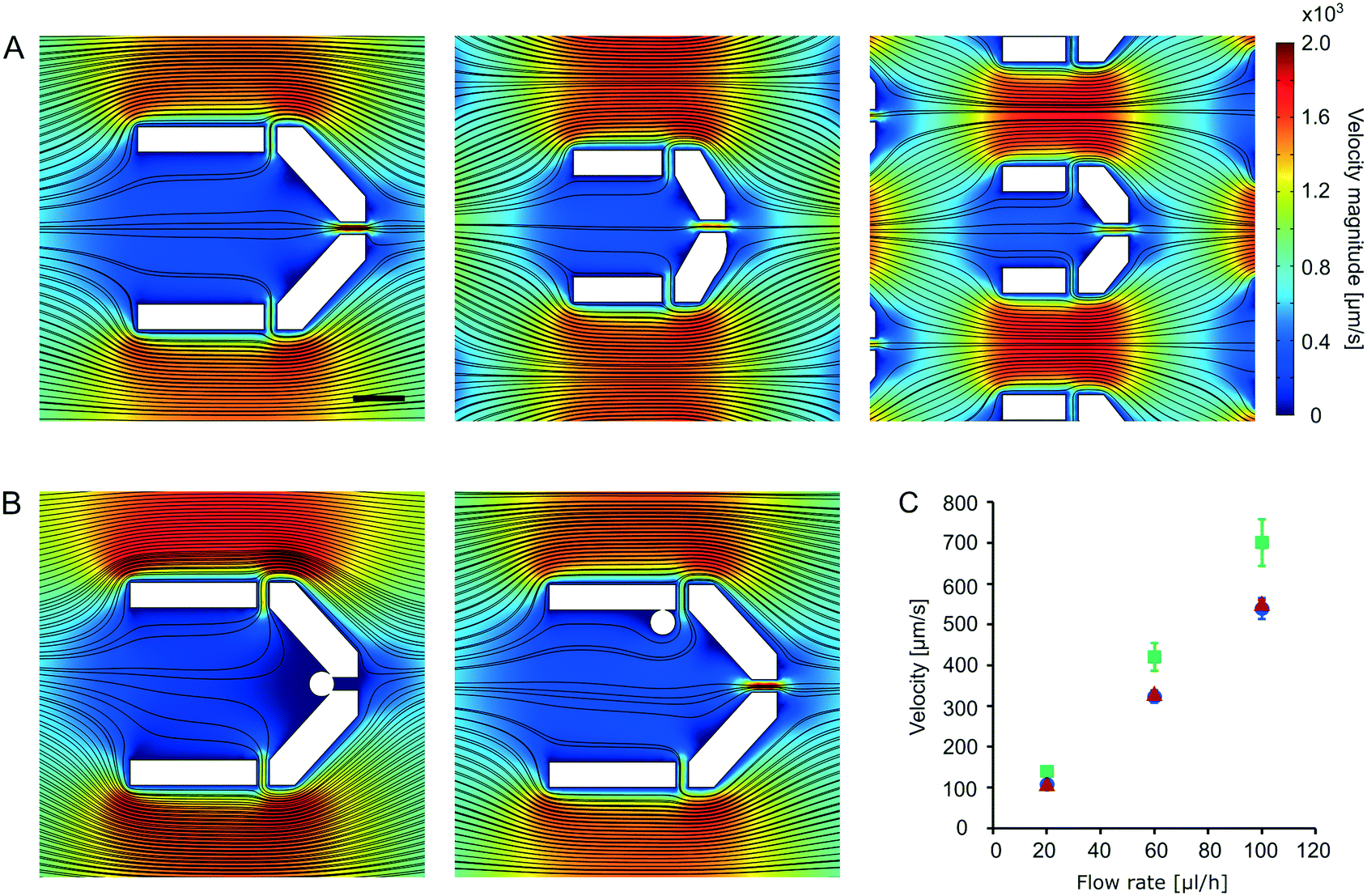 | ||
| Fig. 2 Calculated flow fields around and within the cell traps. A) Flow fields in case of three different traps sizes used in the microfluidic devices for 60 μL h−1 overall flow rate in the device. The scale bar is 20 μm. B) Flow fields for the largest trap with 10 μm diameter circular particles representing cells. Positioning of the model particles is based on typical observations as shown in Fig. 5B and 7A. Calculations were done for 60 μL h−1 overall flow rate in the device. The scale bar and color bar indicated on panel A apply here too. C) Maximal flow velocities calculated at the entrance of the traps (i.e. along the lines connecting the leftmost corners of a given trap) as a function of the overall flow rate applied to the device. Blue circles: largest traps; red triangles: mid-sized traps; green squares: small traps. The standard deviations indicated are based on data from n = 3 (large and mid-sized traps across the width of the channel) and n = 5 (small traps across the width of the channel) traps. | ||
In order to determine the shear stress on cells, calculations were performed with a 10 μm diameter model particle placed within the trap. Positioning of the model particles (as shown on Fig. 2B) represent typical scenarios such as those shown on Fig. 5B and 7A. Color coded representation of the shear stress is shown in supplementary material Fig. S1.† According to the calculations the maximal shear stress is experienced by cells positioned in the vicinity of gaps. In this case the maximal shear stress is about 6 dyn cm−2.
Optimization of protoplast isolation and regeneration procedures
In order to generate protoplasts from bulk cultures in Petri dishes, the protocol by Levin et al.44 was employed with some modifications. Symbiodinium cells were taken from a one-week-old culture (exponential phase) and 4 mL of suspension (cell density ∼105 cells ml−1) was centrifuged at 2000 g for 4 minutes. The pellet was resuspended in the digestion medium, which was prepared by mixing 4% cellulase Onozuka RS (Duchefa Biochemie, Haarlem, Netherlands) and 1% macerozyme R-10 (Duchefa Biochemie, Haarlem, Netherlands) in pre-cooled sterile F/2 medium. The enzyme solution was centrifuged at 10000 g for 10 min at 4 °C in order to remove any potential impurities of the enzyme powder, and the supernatant was filter sterilized by using a 0.22 μm sterile syringe filter (Merck Millipore Ltd.). Sterile D-sorbitol (Molar Chemicals Ltd.) was added as osmoticum in a final concentration of 0.5 M to the enzyme solution just before initiating protoplast isolation. Cells were incubated in the enzyme solution at 30 °C in the dark for 24 h in a shaking incubator at 100 rpm. After 24 h, protoplasts were centrifuged for 4 minutes at 200 g and the pellet was resuspended in the washing medium (0.5 M sucrose, 0.5 M D-sorbitol, and 25 mM CaCl2 in sterile F/2). The protoplasts in washing medium were incubated in the dark in a shaking incubator at 25 °C for 3 h. The protoplasts were pelleted and the step was repeated. To avoid microbial contamination, 100 μg mL−1 kanamycin was used during the washing step. After washing, protoplasts were resuspended in regeneration medium prepared by adding 25 mM CaCl2 and 0.5 M D-sorbitol to the culture medium (pH 7.0), and recurred to Symbiodinium growth conditions (50 μmol photons m−2 s−1, 25 °C, and 12 h light:12 h dark cycle). All experiments were performed in triplicates.Protoplast isolation and regeneration procedure in microfluidic devices
The digestion solution applied in the microfluidic experiments contained 4% cellulase Onozuka RS (Duchefa Biochemie, Haarlem, Netherlands) and 1% macerozyme R-10 (Duchefa Biochemie, Haarlem, Netherlands) in filter-sterilized F/2 medium, including 0.5 M sterile D-Sorbitol. The digestion solution was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g at 4 °C for 10 min in order to remove any potential impurities of the enzyme powder. The digestion solution was loaded into a syringe (Omnifix-F Solo Luer 1 mL, B. Braun), and the syringe containing the digestion solution was attached to the tubing of the microfluidic system and a constant flow rate was applied in order to move the enzyme solution through the microfluidic devices containing the trapped cells. After enzyme treatment, the digestion solution was replaced by washing solution and protoplasts were washed with continuous flow to remove the enzyme solution from the microfluidic devices. After 4–5 h of washing, regeneration medium was applied to the cells for 2 days, after which culture medium was added. All experiments were performed in triplicates.
000 g at 4 °C for 10 min in order to remove any potential impurities of the enzyme powder. The digestion solution was loaded into a syringe (Omnifix-F Solo Luer 1 mL, B. Braun), and the syringe containing the digestion solution was attached to the tubing of the microfluidic system and a constant flow rate was applied in order to move the enzyme solution through the microfluidic devices containing the trapped cells. After enzyme treatment, the digestion solution was replaced by washing solution and protoplasts were washed with continuous flow to remove the enzyme solution from the microfluidic devices. After 4–5 h of washing, regeneration medium was applied to the cells for 2 days, after which culture medium was added. All experiments were performed in triplicates.
Confocal microscopy
Cell wall digestion and regeneration were confirmed by Calcofluor White (CFW) (50 μM) (Sigma-Aldrich) that specifically stains cellulose in the cell wall. The permeabilization of the cell membrane and cell wall for CFW staining was improved by treating the cells with 30 μL of a mixture of 20% dimethyl sulfoxide (DMSO) and 10% potassium hydroxide for 5 minutes.44 Cells were imaged by a Leica SP5 confocal laser scanning microscope (LSM) using the HCX PL APO 63× oil immersion objective (NA: 1.4), using sequential scan with 405 nm excitation and 415–485 nm emission detection for cellulose detection and an excitation of 543 nm and 639–778 nm emission detection for chlorophyll (Chl) fluorescence detection.Single-cell chlorophyll fluorescence measurements (microscopy imaging PAM analysis)
Single-cell Chl fluorescence was determined by pulse-amplitude modulated imaging (PAM) microfluorometry (Imaging-PAM M-Series chlorophyll fluorometer, microscopy version, with a IMAG-CG control unit and IMAG-L450 measuring head, Heinz Walz GmbH, Effeltrich, Germany) equipped with an IMAG-K6 CCD camera (Allied Vision Technologies GmbH, Ahrensburg, Germany). A non-biological fluorescence standard was used to equalize the maximum fluorescence yield (Fm) to minimum fluorescence yield (F0) in the absence of variable fluorescence from biological samples, in order to avoid potential artifacts in the high sensitivity mode (this high sensitivity mode is required to obtain variable fluorescence image of cells with appropriate S/N). Once the steady state fluorescence yield (F0) stabilized, an F0 image was obtained (measuring light intensity = 4, approx. 0.3 μmol photons m−2 s−1, gain = 10, frequency = 1, damping = 5, F0 averaging n = 3) after 3 minutes of dark adaptation. A saturating pulse of blue light (460 nm, approx. 2000 μmol photons m−2 s−1, 0.8 s pulse width) was applied to obtain Fm images, and the Fv/Fm parameter, which reflects PSII activity, was calculated as Fv/Fm = (Fm − F0)/Fm.Morphometric analysis of protoplast formation
In order to analyze the morphological changes of protoplasts, an automated image analysis procedure was developed (using Matlab version 2018b, the MathWorks Inc., Natick, MA, USA). The sequential images collected during protoplast formation were segmented using RGB (red, green, blue) parameters, and the area, eccentricity, major and minor axes were calculated for each cell according to the image processing toolbox (The MathWorks Inc., Natick, MA, USA) functions. The eccentricity is the ratio of the distance between the foci of the ellipse and its major axis length. The value is between 0 and 1 (0 and 1 are degenerate cases. An ellipse whose eccentricity is 0 is actually a circle, while an ellipse whose eccentricity is 1 is a line segment). The procedure allows to display these morphological parameters over time, therefore the kinetics of the morphological changes during protoplast formation can be visualized. Where applicable, the curves of the time-dependent morphological changes were fitted with logistic function (using OriginPro 2018, OriginLab Corp. Northampton, MA, USA), according to the following equation:where A1 is the asymptote of the initial value, A2 is the asymptote of the final value, x0 is the value of the sigmoid midpoint, p is the power (sigmoidicity).

Subcellular localization of Singlet Oxygen Sensor Green (SOSG)
Intact cells and protoplasts were stained with 100 μM SOSG dye and incubated for one hour in the dark followed by illumination for 5 min under 2300 μmol photons m−2 s−1 visible light. Then the cells were washed 2 times and images were taken by using 488 nm excitation and 510–590 nm emission with a Leica SP5 Confocal microscope.36 SOSG staining was tested both in the plate protoplast experiment where cells were treated with the enzyme in a small petri plate (see details in the protoplast isolation and regeneration procedures) and the microfluidic devices. The cells were washed 2 times with washing solution to remove excess dye. First the cells were imaged without light treatment and then they were illuminated on the microscope stage to follow the changes in SOSG fluorescence intensity in the same cells after illumination. The distribution pattern of 1O2 in intact cells and protoplasts was quantified by using Leica Microsystems LAS-X software. SOSG fluorescence was quantified by drawing a transect on the cells and then signal intensity was obtained in the graphical form by using the Leica software. The experiment was performed in three biological replicates.Treatment with fluorescein-labelled oligonucleotides
For oligonucleotide uptake experiments, protoplasts were washed with washing solution by centrifugation at 200 g at 25 °C for 5 minutes with F/2 medium containing 0.5 M sorbitol. Fluorescein (FAM) labelled random nonsense oligonucleotides (ODN) composed of 53 nucleotides (5′-3′, sequence HS-C6-aatctgtaTCTATATTCATCATAGGAAACACCAAAGATGATATTTTCTTTAAT-FAM, where HS-C6 refers to a thiol group added on the 6th alkyl chain to enhance stability against exonucleases, synthesized in the Nucleic Acid Synthesis Laboratory, Biological Research Centre, Szeged, Hungary) were used for the staining of control and protoplast cells (10 μM concentration). The oligonucleotide sequence did not show any significant similarity with the Symbiodinium microadriaticum genome sequence based on BLAST® search. Samples were incubated with the ODN solution for 15 min in dark and then washed 2–3 times (by centrifugation at 200 g at 25 °C for 3 min with F/2 medium containing 0.5 M sorbitol) and imaged by confocal microscopy as described above. In order to avoid autofluorescence interfering with the green fluorescence of FAM-labelled ODN, autofluorescence was eliminated by smart gain setting before staining with FAM-ODN. Images were also acquired for control samples (protoplast washed with washing solution only). The experiment was performed in three biological replicates.Results and discussion
In order to determine the enzyme concentration and mixture required for protoplast formation in the microfluidic chambers, a preliminary cell wall digestion assay was performed in Petri plates. 3KU of cellulase treatment resulted in the partial digestion of the cell wall, and the protoplast formation process required approx. 48 h, in agreement with previous findings.44 Complete cell wall digestion was obtained by using 4% (6KU) of cellulase in combination with 1% (1.5KU) macerozyme (Fig. 3), which resulted in a significant increase in the longitudinal and transversal length of the cells (major and minor axis, respectively, in accordance with Levin et al. 2017 (ref. 44)). | ||
| Fig. 3 Light microscope images of (A) untreated (control) and (B) treated (protoplast) Symbiodinium cells from the conventional petri plate-based digestion assay experiment. (C) Statistical analysis of the changes in cell morphology (major and minor axis, ‘C.’ denotes control cells, ‘P.’ denotes protoplasts). (D) Statistical analysis of the changes in the cell shape index (major axis/minor axis). Cells were treated with 4% cellulase and 1% macerozyme for 24 h. Different letters indicate statistically different means, values sharing common letters are not significantly different from one another (p = 0.05). | ||
Protoplast formation was confirmed by the lack of Calcofluor White (CFW) staining, which gives blue fluorescence when it reacts with cellulose of intact cells (Fig. 4, and S2† displays more examples for each condition). Pronounced blue fluorescence was observed in the cell wall of control cells (Fig. 4A). The lack of CFW fluorescence in the protoplast demonstrated the digestion and removal of the cell wall (Fig. 4B). Regeneration of cell wall was confirmed by the reappearance of CFW fluorescence in the regenerated protoplasts (Fig. 4C). Protoplast formation was also confirmed by incubating protoplasts in distilled water, in which protoplasts exhibited bursts due to the decrease of turgor pressure (Fig. 4D). The same composition of the enzyme digestion solution was applied for the protoplast isolation and regeneration procedure in microfluidic chambers.
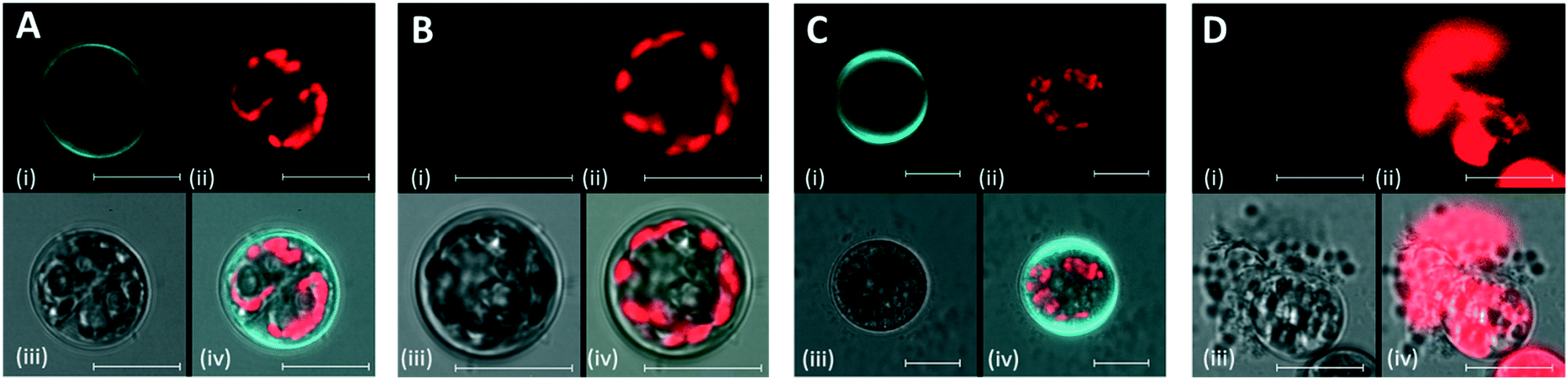 | ||
| Fig. 4 Cell wall staining of Symbiodinium cells and protoplasts from the conventional petri plate-based digestion assay experiment. (A) Control Symbiodinium cells stained with CFW, (B) protoplast stained with CFW, (C) CFW staining after regeneration of cell wall, (D) ruptured protoplast in distilled water. In each panel, the 1st channel (top left) shows CFW fluorescence, 2nd (top right) is Chl fluorescence, 3rd (bottom left) is transmission and 4th (bottom right) is the merged image. Scale bar represents 10 μm. | ||
Investigation of protoplasts in microfluidic chambers
The microfluidic system was designed and applied to investigate the efficiency and time course of protoplast formation and cell wall regeneration under precisely controlled environmental conditions, considering medium, temperature, and flow rate. Individual cells were captured in the traps, which allowed the characterization of the morphological and physiological changes of single cells throughout the loading, enzyme digestion and regeneration process.Most of the cells stopped moving after being loaded in the chamber, indicating that cells were possibly under loading stress. However, motility was regained after four hours. Cells were motile before enzyme treatment (pre-digestion) (Fig. 5A), and Symbiodinium cells exhibited regular division in the traps, indicating that the trapping procedure and keeping the cells in the traps did not compromise the physiological activity of Symbiodinium. The enzyme digestion procedure caused marked changes in the morphology of trapped Symbiodinium cells, the sequence of which could be recorded over time. The cells lost motility on day 1 post-digestion (Fig. 5A), potentially due to cell cycle arrest in the coccoid stage67,68 or mechanical loss of flagella69 during the cell wall digestion procedure. The cells were in the dividing phase and motile forms before enzyme treatment, but after 1 h of enzyme treatment most of the cells stopped moving and cell division also halted. However, some of the cells still divided in the protoplast phase (see below). The motility of cells recovered after 2 days' incubation in the regeneration medium, and the number of motile cells increased when they were incubated in the culture medium. By day 3 in the culture medium, all the cells divided into very actively moving daughter cells (it has to be noted that after cell division, many daughter cells escaped the traps due to their high motility, but since these cells were not the focus of interest of the current study, the escape of motile cells at the end of the experiment did not influence our observations).
 | ||
| Fig. 5 Motility pattern and morphological changes of Symbiodinium cells before, during and after enzyme treatment. (A) Percentage of cell motility (mean ± SE, n = 3). The black arrow on day −1 indicates when cells were loaded in the chamber and the blue arrow on day 1 marks when the enzyme solution was employed (t = 0 h), as described in the experimental section. The red arrow marks when regeneration medium was applied, and the teal arrow marks when the culture medium started to flow. (B) Cells in the trap of microfluidic device at various times after the enzyme treatment (0 h, 4 h, 8 h, 15 h, 25 h and 35 h). The flow rate was 40 μL h−1, the capture rate 60 seconds and the scale bar represents 10 μm. | ||
Changes in size and morphology were observed during the process of protoplast formation. Before enzyme treatment the cells were more or less elongated and had a spindle shape, but after a few hours they started to become round and increased in diameter. The diameter reached its maximum when the cells were fully protoplasted (Fig. 5B). The process of protoplast formation and associated changes in the size and morphology of the cells can be seen in Fig. 5B.
Enzyme treatment in microfluidic devices (Section ‘Protoplast isolation and regeneration procedure’) was started when the cells were in the actively growing phase, as during this phase dinoflagellates shed their thecal plates and external membranes, so cell wall digestion is potentially more efficient.47 Some divisions were also observed during the enzyme treatment, where protoplasts divided into two rounded non-motile daughter cells (Fig. 6B), as opposed to the cell division of non-digested control cells, where two spindle-shaped motile daughter cells are formed (Fig. 6A). Protoplast formation, i.e. the complete rounding of the daughter cells was faster in dividing vs. non-dividing cells (cf. 5B and 6B), indicating that actively growing cells are more efficiently protoplasted than non-dividing cells. The cell division pattern was different after regeneration of the cell wall as compared to untreated cells. In normal cell division, the mother cell divides into 2 or 4 daughter cells (Fig. 6A), but after regeneration more than 4 daughter cells were formed with maximum division into 8 daughter cells with cell sizes smaller than normal daughter cells (Fig. 6C). In intact cells, the division process into 2 motile daughter cells was completed in 3 minutes, whereas in the protoplast stage the division process required 3 hours to complete and it resulted in two round and non-motile cells instead of motile and spindle-shaped daughter cells (Fig. 6B). These daughter cells expanded in size after cell division.
 | ||
| Fig. 6 Pattern of cell division and morphological differences in control cells, protoplasts and after regeneration of cell wall. (A) Cell division of intact cells into two motile daughter cells shown by arrows, (B) cell division of protoplasts into two daughter cells, (C) pattern of cell division after regeneration of cell wall into more than 4 daughter cells shown by arrows (0 min in panel A is the cell before division, and 1 min, 2 min and 3 min are the time series of the cell division process after 1, 2 and 3 minutes respectively. In panel B, 0 h is the state of the cell before enzyme treatment, and 2 h, 2.3 h, 2.5 h, 2.75 h, 3 h and 3.08 h are the time points after 120, 140, 150, 165 and 185 min of enzyme treatment. In panel C, images 1 to 3 (12 h, 24 h and 48 h) show protoplasts after 12 h, 24 h and 48 h, respectively, in regeneration medium, whereas images 4 to 6 (20 h, 30 h and 40 h) show cells after 20 h, 30 h and 40 h in culture medium. Scale bar represents 10 μm. | ||
These results indicate that Symbiodinium cells that are undergoing cell wall digestion are able to divide (albeit with a low efficiency), and the daughter cells formed during the cell wall digestion procedure are prone to protoplast formation with higher efficiency than non-dividing cells. This could be important for further optimization of the protoplast preparation procedure, i.e. by means of culture synchronization when the protoplast yield could be enhanced.
In order to investigate the regeneration capacity of the protoplasts after removal of the digestion solution, changes in cell morphology were monitored under continuous flow of regeneration medium. After the addition of the digestion enzyme solution, all the moving forms were converted into non-moving forms both in the plate and the microfluidic traps. Cells were kept in the regeneration medium for two days followed by incubation in the culture medium. After one day in the culture medium, cells regained their division ability. All the cells regained motility after 2 days in the culture medium, and cell wall regeneration of the protoplast was completed in 4 days.
Time-resolved morphometric analysis of protoplast formation
In order to investigate and quantify the morphological changes over time during protoplast formation, an automated image processing and analysis procedure was applied to individually trapped cells.In a typical experiment, after addition of the digestion solution, the area (Fig. 7B), major and minor axis (Fig. 7D and E, respectively) of the cells increased, until an equilibrium state in these parameters was attained. In the case of 40 μL h−1 flow rate, the equilibrium state was attained after approx. 35 h. However the timescale of these morphological changes was dependent on the flow rate (see below). The eccentricity of cells decreased (Fig. 7C), indicating that the cells became more round over time. However, the time required to attain equilibrium state of the eccentricity parameter could not be unequivocally determined. It has to be noted that minor changes in cell morphology occurred after loading the cells in the microfluidic chambers, indicating that loading stress might have perturbed cell morphology (indicated by the time period after loading but before the start of the cell wall digestion). Cell morphology is known to vary in Symbiodinium as a result of stress conditions (e.g. elevated temperature70,71), however the characteristic increase followed by the equilibrium state of cell area and other morphological parameters during the enzymatic cell wall digestion could be clearly distinguished from other stress-induced cell morphology changes, because heat stress or bleaching conditions typically cause vacuolization, disorganization of chloroplast and other structural degradation of symbionts,70,71 which was not observed during protoplast formation. The curves of the morphological parameters could be reasonably well fitted with a logistic (sigmoid) function, and the x0 value (representing the midpoint of the timescale of the changes) can be used as a quantitative descriptor of the time scale of protoplast formation (see below).
 | ||
| Fig. 7 Morphometric analysis of protoplast formation over time in the microfluidic device. (A) Original images of traps showing the selected areas of interest of single cells selected for morphological analysis. The same cell is shown over time at the beginning (top image) and at the end (bottom image) of the experiment. (B) Area, (C) eccentricity, (D) major axis, (E) minor axis of cells. Insets show the changes in the parameters after approx. 35 h (tfinal), when the changes attain the equilibrium state (mean ± S.D., n = 10, different letters indicate significant differences (p = 0.05)). Black solid arrow indicates the phase when cells were loaded in the microfluidic traps. Blue solid arrow represents when enzyme solution was applied to the cells (t0). The red curves represent the logistic fitting of the original data. The applied flow rate was 40 μL h−1. Scale bar represents 10 μm. | ||
In order to investigate whether protoplast formation could be accelerated using the microfluidics approach, different flow rates were tested (20, 40, 60, 80 and 100 μL h−1) for the enzyme digestion process. Elevated flow rates resulted in similar trajectories of the time-dependent changes of morphological parameters during the digestion process, and the time required for protoplast formation was found to be significantly shorter only at the 100 μL h−1 flow rate (Fig. 8A; the original traces depicting the time course of the relative changes in area, eccentricity, major and minor axis at different flow rates are shown in Fig. S3†).
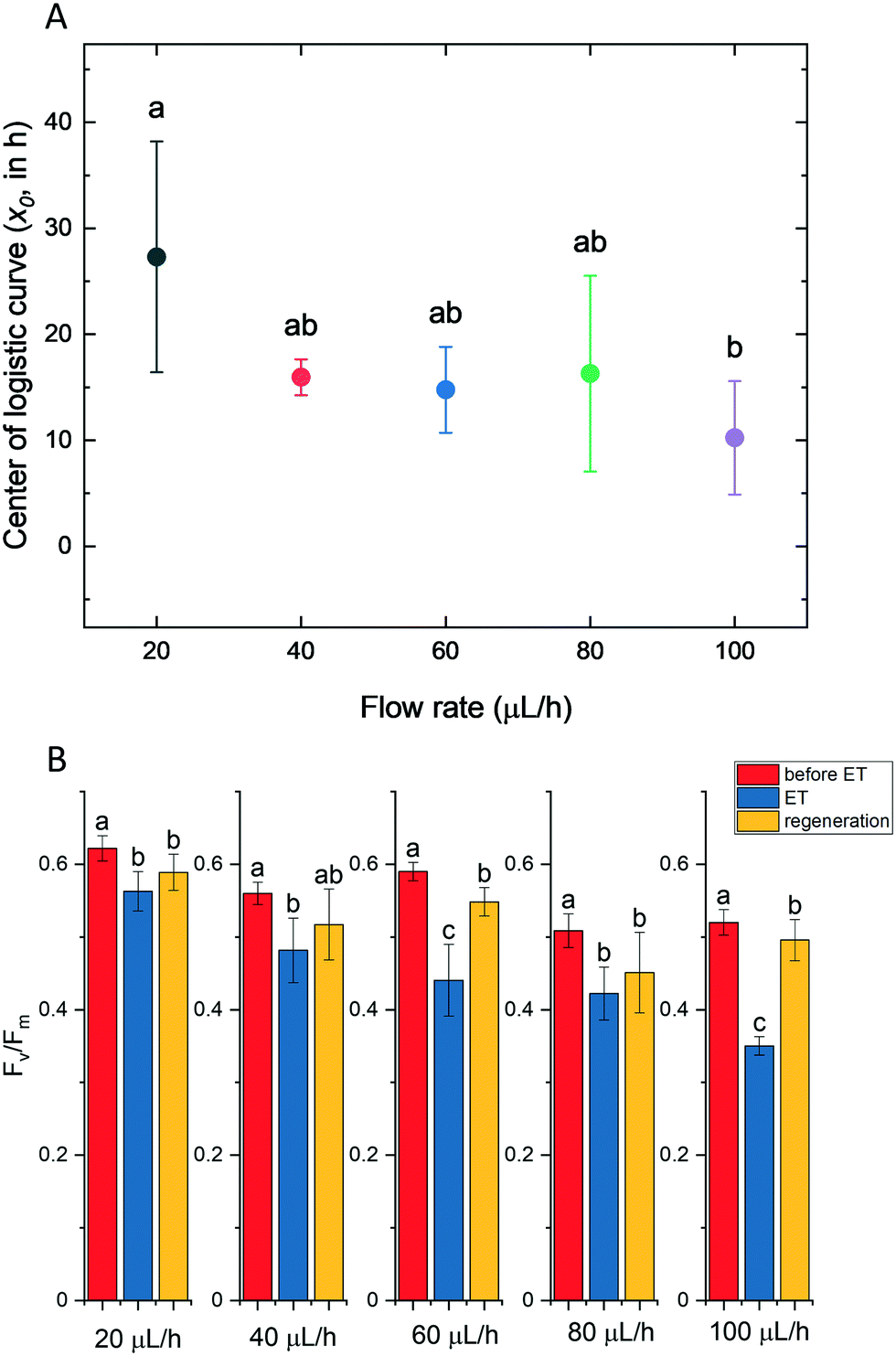 | ||
| Fig. 8 Physiological status of Symbiodinium cultures throughout protoplast generation and cell wall regeneration in the microfluidic system. (A) The time course of protoplast formation at various flow rates (μL h−1) (mean ± SD, n = 10). (B) Effect of various flow rates on the FV/Fm value during protoplast formation and after regeneration of cell wall (mean ± SD, n = 10). ET, enzyme treatment. Measurement points for 20 μL h−1, 40 μL h−1, 60 μL h−1, 80 μL h−1 and 100 μL h−1 are 40 h, 35 h, 30 h, 18 h, and 15 h respectively. Different letters indicate statistically different means, values sharing common letters are not significantly different from one another (p = 0.05). | ||
Although the timescale of the morphological changes could be monitored at different flow rates, it was also an important question to clarify whether protoplasts retained physiological or photosynthetic competency across the applied range of the flow rate, and whether flow rates influenced the activity of PSII (Fv/Fm). Fv/Fm in cells under control conditions (in F/2 medium) was not affected by the applied flow rate. Protoplasting caused very minor decrease at a low flow rate, whereas the increased flow rate has damaging effect particularly in the 80–100 μL h−1 flow range (Fig. 8B, original microscope PAM data depicting F0, Fm and Fv/Fm images of single intact cells and protoplast are shown in Fig. S4 and S5,† respectively).
F v/Fm substantially recovered in the regeneration phase, when the digestion solution was replaced with regeneration medium, indicating that the enzyme digestion procedure did not impair PSII activity. However, it has to be noted that although higher flow rates enable a faster protoplast formation process, they also cause a significant loss in photosynthetic efficiency during enzyme treatment. Therefore, based on the above results, for the optimization of the protoplast isolation procedures it is important to consider that faster protoplast isolation procedure at elevated flow rates might partially compromise photosynthetic activity, which can be particularly important when maintaining physiological competence of protoplasts is a priority.
Applications of protoplast technology for singlet oxygen labelling in Symbiodinium
Oxidative stress and oxidative damage have long been implicated as major factors in coral bleaching,72 recently reviewed in Szabó et al.73 In the past years, emphasis of investigating the impact of oxidative stress and reactive oxygen species in coral physiology is increasingly based on single-cell studies applying multiple fluorescent sensors and single-cell photosynthetic activity assays (e.g.71,74). For singlet oxygen detection the applicable fluorescent dye is Singlet Oxygen Sensor Green (SOSG). However, Rehman et al.36 found that SOSG did not cross the cell wall, therefore intracellular 1O2 detection could not be performed in Symbiodinium. Wietheger et al.40 reported that some intracellular SOSG staining could be observed in intact Symbiodinium cells, however subcellular localization of SOSG could not be assigned and no optical sectioning was performed to analyze the intracellular distribution of the dye. Therefore, it was of particular importance in the present study to investigate intracellular 1O2 formation using the SOSG dye, for which the preparation of physiologically competent protoplasts is essential. Using the advantage of the protoplast preparation method in a highly controlled environment as demonstrated in the current study, SOSG labeling was attempted in order to image the intracellular distribution of 1O2 in Symbiodinium. Intact cells did not exhibit any internal staining, whereas a strong SOSG fluorescence was observed at the perimeter when the cells were exposed to light for 5 min (Fig. 9A and B), in agreement with Rehman et al.36 in protoplasts, green fluorescence of SOSG was observed to be co-localized with the red fluorescence of Chl, indicating the localization of the SOSG dye in the chloroplast (Fig. 9D and H), however in some cases the SOSG dye was evenly distributed in the cytoplasm and did not specifically co-localize with Chl autofluorescence (more examples of the localization of Chl and SOSG fluorescence in individual protoplasts that are trapped in the devices are shown in supplementary material Fig. S6,† and the merged images and the intensity distribution of the Chl and SOSG fluorescence across defined transects of cells are shown in Fig. S7†). Thus, we demonstrated for the first time that SOSG penetrates the protoplasts of Symbiodinium and therefore protoplasts are amenable to investigation of singlet oxygen formation in Symbiodinium. | ||
| Fig. 9 Confocal microscopy imaging of Symbiodinium intact cells and protoplasts stained with Singlet Oxygen Sensor Green (SOSG). Three detection channels are represented: (i) SOSG fluorescence; (ii) Chl fluorescence; (iii) transmission. Panel (iv) shows the merged images of the three signals. (A) Laser scanning microscopy (LSM) imaging of cells without light treatment. (B) LSM imaging of cells which were exposed to light (2300 μmol photons m−2 s−1 for 5 min. (C) LSM imaging of protoplasts without light treatment. (D) LSM imaging of protoplasts which were exposed to light (2300 μmol photons m−2 s−1) for 5 min. E–H show the intensity distribution of the three detection channels at the indicated transect lines for A–D respectively (bright field, black; Chl, red; SOSG, green). Scale bar represents 10 μm. | ||
To confirm 1O2 production in protoplasts, SOSG staining was done in combination with our histidine assay.36Symbiodinium protoplasts showed strong SOSG fluorescence when illuminated with light due to the production of 1O2; however, this fluorescence disappeared when SOSG was used together with histidine, indicating 1O2 uptake by histidine (Fig. S8†) in agreement with previous results.36
Based on the current study, selective enhancement of SOSG fluorescence of trapped protoplasts upon transient strong illumination could open new possibilities for quantifying or semi-quantifying 1O2 in Symbiodinium cells, thereby potentially enabling the investigation of the intracellular distribution of 1O2. Furthermore, protoplasts could be suitable experimental subjects for other physiological assays requiring cell wall impermeable fluorophores. In this manner, various physiological processes can be investigated with fluorescent dyes whose applicability is otherwise limited due to their impermeability through the cell wall.
Applications of protoplast technology for oligonucleotide uptake in Symbiodinium
Oligonucleotide-directed mutagenesis represents a genome-editing tool which uses synthetic oligonucleotides for targeted exchange of nucleotides.75 It has been successfully applied in plant protoplasts and in vivo in intact plant tissues.76–78 Antisense oligonucleotide technology was applied in a dinoflagellate species Lingulodinium polyedrum to knock down a cellulose synthase gene.79 However, antisense oligonucleotide technology is unexploited in Symbiodiniaceae. As an important potential application of oligonucleotide-mediated mutagenesis in Symbiodinium, the uptake of short DNA oligonucleotides was tested in intact Symbiodinium cells and protoplasts. Cells and protoplasts were treated with fluorescein-labelled antisense oligonucleotides (ODN). Confocal microscopy was performed to visualize the uptake of ODNs in Symbiodinium. Intact cells and protoplasts did not show green fluorescence without ODN labelling (Fig. 10A and B, respectively). When the intact cells were incubated with ODN, no green fluorescence could be observed, indicating the inability of intact cells with cell wall for ODN uptake (Fig. 10C). FAM-ODN labelled protoplasts showed a clear green spot, which did not co-localize with Chl autofluorescence (chloroplasts) (Fig. 10D, further examples of ODN-labelled protoplasts are shown in Fig. S9†). From approx. 120 protoplasts, 85 showed the uptake of ODN, which indicates that the uptake efficiency was about 70% (examples of several FAM-ODN labelled protoplasts which exhibited or lacked the green spot of FAM fluorescence are shown in Fig. S10†). We could also demonstrate that the green fluorescence of the FAM-labeled oligonucleotides showed co-localization with the blue fluorescence of DAPI, a commonly used fluorescent dye to label nuclear DNA (Fig. S11†). This indicates that Symbiodinium protoplasts are able to take up small fluorescently labelled ODNs, which are potentially targeted to nucleus. Further studies are needed to establish whether oligonucleotide technology is applicable for genome editing and oligonucleotide-directed mutagenesis in Symbiodiniaceae.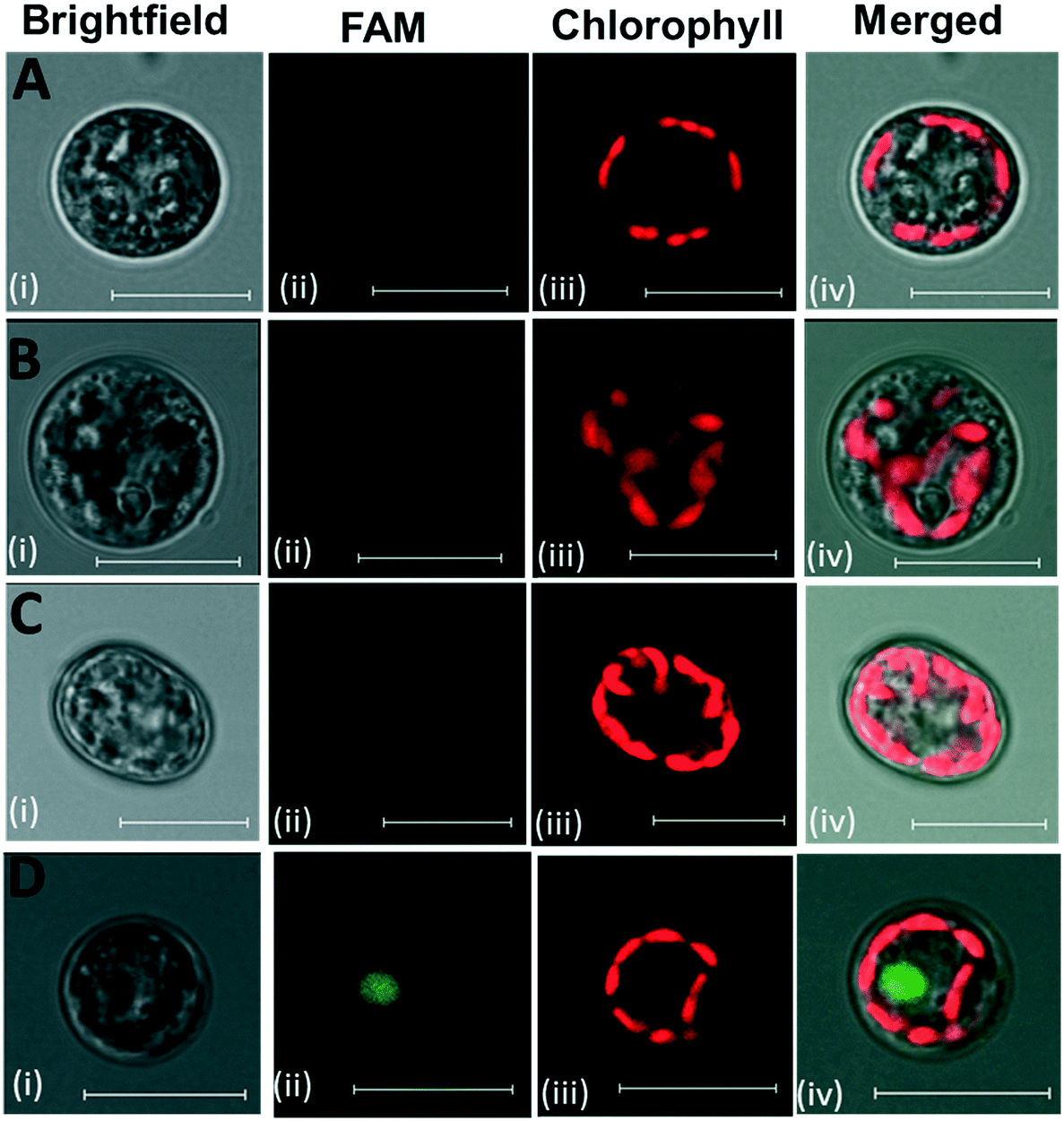 | ||
| Fig. 10 LSM imaging of FAM-ODN labelling of Symbiodinium cells. (A) Unlabelled cells, (B) unlabelled protoplasts, (C) labelled cells, (D) labelled protoplasts. (i) Brightfield; (ii) FAM (green) fluorescence; (iii) Chl fluorescence; (iv) merged images of the three channels. Scale bar represents 10 μm. | ||
Conclusions
In the current study, protoplasts were successfully isolated from Symbiodinium in a microfluidic system, which enabled morphometric analysis of single cells throughout the whole protoplast formation and regeneration time. With the presented method, the protoplast formation process and its associated morphological parameters could be monitored on the single-cell level with high time resolution, which is essential for further biotechnological and environmental applications. The trapped cells retained physiological activity and photosynthetic competence and were able to regrow their cell walls. The microfluidic method also allowed the localization of 1O2 inside Symbiodinium protoplasts using the 1O2-sensitive dye SOSG, thereby creating several possibilities for the investigation of the intracellular distribution of 1O2. Furthermore, it was demonstrated that Symbiodinium protoplasts are able to take up short (≤53 nucleotides) oligonucleotides. The protoplast technology applied in microfluidic chamber can be considered as an attractive method for the analysis of single cells, which therefore may open further possibilities for protoplast fusion and manipulations under precisely controlled conditions.Data availability statement
All results generated during and/or analyzed during the current study are included in the main manuscript and in the Electronic Supplementary Information; raw data is available from the corresponding author upon reasonable request.Author contributions
Faiza Bashir – investigation, formal analysis, visualization, writing – original draft; Sándor Kovács – methodology (microfluidics of Symbiodinium spp.), investigation, software; Ágnes Ábrahám – methodology (microfluidic devices), visualization; Krisztina Nagy – methodology (microfluidic devices), visualization; Ferhan Ayaydin – methodology (confocal fluorescence microscopy methods of protoplasts), visualization, formal analysis, writing – review & editing; Ildikó Valkony-Kelemen – methodology (confocal microscopy); Györgyi Ferenc – methodology, resources (oligonucleotides); Péter Galajda – conceptualization (design of microfluidic devices), methodology, resources, writing – review & editing; Szilvia Z. Tóth – conceptualization (single cell morphology and physiology), writing – review & editing; László Sass – software (morphological analysis), visualization; Péter Kós – methodology, data curation (molecular biology); Imre Vass – conceptualization (morphological analysis and protoplast isolation from Symbiodinium spp.), resources, funding acquisition, project administration, writing – original draft, writing – review & editing, supervision; Milán Szabó – formal analysis, supervision, validation, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the grants GINOP-2.3.2-15-2016-00026, Hungarian Academy of Sciences, MTA Premium Postdoctoral Research Program PREMIUM-2017-38 (awarded to M. S.), National Research, Development and Innovation Office grant number NKFIH FK 128977 (awarded to M. S.), NKFIH K 116016, NKFIH K 116526 (awarded to P. G.), and NKFIH PD 124889 (awarded to K. N.) K. N. was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/00463/18/8). The authors thank László Dér for valuable discussions and his help with the model calculations.References
- R. E. Steele, C. N. David and U. Technau, Trends Genet., 2011, 27, 7–13 CrossRef CAS PubMed.
- A. Bertucci, A. Moya, S. Tambutté, D. Allemand, C. T. Supuran and D. Zoccola, Bioorg. Med. Chem., 2013, 21, 1437–1450 CrossRef CAS PubMed.
- S. A. Murray, D. J. Suggett, M. A. Doblin, G. S. Kohli, J. R. Seymour, M. Fabris and P. J. Ralph, Perspect. Psychol. Sci., 2016, 3, 37–52 Search PubMed.
- D. Yellowlees, T. A. Rees and W. Leggat, Plant, Cell Environ., 2008, 31, 679–694 CrossRef CAS PubMed.
- L. Muscatine, Ecosystems of the World, Coral Reefs, 1990, vol. 25, pp. 75–87 Search PubMed.
- R. Berkelmans and M. J. Van Oppen, Proc. R. Soc. B, 2006, 273, 2305–2312 CrossRef PubMed.
- W. K. Fitt, B. E. Brown, M. E. Warner and R. P. Dunne, Coral Reefs, 2001, 20, 51–65 CrossRef.
- E. Howells, V. Beltran, N. Larsen, L. Bay, B. Willis and M. Van Oppen, Nat. Clim. Change, 2012, 2, 116–120 CrossRef.
- R. A. Levin, V. H. Beltran, R. Hill, S. Kjelleberg, D. McDougald, P. D. Steinberg and M. J. Van Oppen, Mol. Biol. Evol., 2016, 33, 2201–2215 CrossRef CAS PubMed.
- I. Yuyama, S. Harii and M. Hidaka, Mar. Environ. Res., 2012, 76, 41–47 CrossRef CAS PubMed.
- H. D. Freudenthal, J. Protozool., 1962, 9, 45–52 CrossRef.
- A. R. Loeblich and J. L. Sherley, J. Mar. Biol. Assoc. U. K., 1979, 59, 195–205 CrossRef.
- R. K. Trench and R. J. Blank, J. Phycol., 1987, 23, 469–481 CrossRef.
- G. Hansen and N. Daugbjerg, J. Phycol., 2009, 45, 251–263 CrossRef PubMed.
- H. J. Jeong, S. Y. Lee, N. S. Kang, Y. D. Yoo, A. S. Lim, M. J. Lee, H. S. Kim, W. Yih, H. Yamashita and T. C. LaJeunesse, J. Eukaryotic Microbiol., 2014, 61, 75–94 CrossRef CAS PubMed.
- S. Y. Lee, H. J. Jeong, N. S. Kang, T. Y. Jang, S. H. Jang and T. C. Lajeunesse, Eur. J. Phycol., 2015, 50, 155–172 CrossRef.
- X. Pochon and R. D. Gates, Mol. Phylogenet. Evol., 2010, 56, 492–497 CrossRef CAS PubMed.
- T. C. LaJeunesse, J. E. Parkinson, P. W. Gabrielson, H. J. Jeong, J. D. Reimer, C. R. Voolstra and S. R. Santos, Curr. Biol., 2018, 28, 2570–2580.e2576 CrossRef CAS PubMed.
- J. M. Pandolfi, S. R. Connolly, D. J. Marshall and A. L. Cohen, Science, 2011, 333, 418–422 CrossRef CAS PubMed.
- B. Brown, Coral Reefs, 1997, 16, S129–S138 CrossRef.
- O. Hoegh-Guldberg, P. J. Mumby, A. J. Hooten, R. S. Steneck, P. Greenfield, E. Gomez, C. D. Harvell, P. F. Sale, A. J. Edwards, K. Caldeira, N. Knowlton, C. M. Eakin, R. Iglesias-Prieto, N. Muthiga, R. H. Bradbury, A. Dubi and M. E. Hatziolos, Science, 2007, 318, 1737–1742 CrossRef CAS PubMed.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- M. P. Lesser, W. R. Stochaj, D. W. Tapley and J. M. Shick, Coral Reefs, 1990, 8, 225–232 CrossRef.
- M. P. Lesser, Limnol. Oceanogr., 1996, 41, 271–283 CrossRef CAS.
- B. Brown, C. Downs, R. Dunne and S. Gibb, Mar. Ecol.: Prog. Ser., 2002, 242, 119–129 CrossRef.
- C. Downs, J. E. Fauth, J. C. Halas, P. Dustan, J. Bemiss and C. M. Woodley, Free Radical Biol. Med., 2002, 33, 533–543 CrossRef CAS PubMed.
- S. Richier, P. Furla, A. Plantivaux, P.-L. Merle and D. Allemand, J. Exp. Biol., 2005, 208, 277–285 CrossRef PubMed.
- V. M. Weis, J. Exp. Biol., 2008, 211, 3059–3066 CrossRef CAS PubMed.
- T. D. Hawkins, T. Krueger, S. P. Wilkinson, P. L. Fisher and S. K. Davy, Coral Reefs, 2015, 34, 1229–1241 CrossRef.
- É. Hideg, I. Vass, T. Kálai and K. Hideg, in Methods Enzymol., Elsevier, 2000, vol. 319, pp. 77–85 Search PubMed.
- É. Hideg, K. I. Ogawa, T. Kálai and K. Hideg, Physiol. Plant., 2001, 112, 10–14 CrossRef PubMed.
- É. Hideg and U. Schreiber, Photosynth. Res., 2007, 92, 103–108 CrossRef PubMed.
- C. Triantaphylides, M. Krischke, F. A. Hoeberichts, B. Ksas, G. Gresser, M. Havaux, F. Van Breusegem and M. J. Mueller, Plant Physiol., 2008, 148, 960–968 CrossRef CAS PubMed.
- C. Triantaphylidès and M. Havaux, Trends Plant Sci., 2009, 14, 219–228 CrossRef PubMed.
- P. Pospíšil and A. Prasad, J. Photochem. Photobiol., B, 2014, 137, 39–48 CrossRef PubMed.
- A. U. Rehman, M. Szabó, Z. Deák, L. Sass, A. Larkum, P. Ralph and I. Vass, New Phytol., 2016, 212, 472–484 CrossRef CAS PubMed.
- C. Flors, M. J. Fryer, J. Waring, B. Reeder, U. Bechtold, P. M. Mullineaux, S. Nonell, M. T. Wilson and N. R. Baker, J. Exp. Bot., 2006, 57, 1725–1734 CrossRef CAS PubMed.
- S. M. Driever, M. J. Fryer, P. M. Mullineaux and N. R. Baker, in Plant Signal Transduction: Methods and Protocols, ed. T. Pfannschmidt, Humana Press, Totowa, NJ, 2009, pp. 109–116, DOI:10.1007/978-1-59745-289-2_7.
- A. Prasad, M. Sedlářová and P. Pospíšil, Sci. Rep., 2018, 8, 1–13 Search PubMed.
- A. Wietheger, D. E. Starzak, K. S. Gould and S. K. Davy, Biol. Bull., 2018, 234, 11–21 CrossRef CAS PubMed.
- E. C. Cocking, Nature, 1960, 187, 962–963 CrossRef.
- P. S. Carlson, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 598 CrossRef CAS PubMed.
- J. Noda, A. Mühlroth, L. Bučinská, J. Dean, A. M. Bones and R. Sobotka, J. Appl. Phycol., 2017, 29, 853–863 CrossRef CAS.
- R. A. Levin, D. J. Suggett, M. R. Nitschke, M. J. van Oppen and P. D. Steinberg, J. Eukaryotic Microbiol., 2017, 64, 588–597 CrossRef CAS PubMed.
- J. E. Chen, A. C. Barbrook, G. Cui, C. J. Howe and M. Aranda, PLoS One, 2019, 14, e0211936 CrossRef CAS PubMed.
- D. A. Markell, R. K. Trench and R. Iglesias-Prieto, Symbiosis, 1992, 12, 19–31 CAS.
- L. Morrill and A. Loeblich Iii, J. Plankton Res., 1981, 3, 53–65 CrossRef.
- T. S. Wakefield, M. A. Farmer and S. C. Kempf, Biol. Bull., 2000, 199, 76–84 CrossRef CAS PubMed.
- O. Scheler, W. Postek and P. Garstecki, Curr. Opin. Biotechnol., 2019, 55, 60–67 CrossRef CAS PubMed.
- K. Nagy, Á. Ábrahám, J. E. Keymer and P. Galajda, Front. Microbiol., 2018, 9, 496 CrossRef PubMed.
- D. B. Weibel, W. R. DiLuzio and G. M. Whitesides, Nat. Rev. Microbiol., 2007, 5, 209–218 CrossRef CAS PubMed.
- E. M. Payne, D. A. Holland-Moritz, S. Sun and R. T. Kennedy, Lab Chip, 2020, 20, 2247–2262 RSC.
- Y. J. Juang and J. S. Chang, Biotechnol. J., 2016, 11, 327–335 CrossRef CAS PubMed.
- C. Westerwalbesloh, C. Brehl, S. Weber, C. Probst, J. Widzgowski, A. Grünberger, C. Pfaff, L. Nedbal and D. Kohlheyer, PLoS One, 2019, 14, e0216093 CrossRef CAS PubMed.
- C. Castaldello, E. Sforza, E. Cimetta, T. Morosinotto and F. Bezzo, Ind. Eng. Chem. Res., 2019, 58, 18036–18045 CrossRef CAS.
- E. Széles, K. Nagy, Á. Ábrahám, S. Kovács, A. Podmaniczki, V. Nagy, L. Kovács, P. Galajda and S. Z. Tóth, Cells, 2022, 11, 285 CrossRef PubMed.
- M. J. Kim, J. R. Youn and Y. S. Song, Lab Chip, 2018, 18, 1017–1025 RSC.
- J. Yao, H. S. Kim, J. Y. Kim, Y.-E. Choi and J. Park, Lab Chip, 2020, 20, 647–654 RSC.
- J. R. Seymour, R. Simo, T. Ahmed and R. Stocker, Science, 2010, 329, 342–345 CrossRef CAS PubMed.
- E. Gibbin, A. Gavish, I. Domart-Coulon, E. Kramarsky-Winter, O. Shapiro, A. Meibom and A. Vardi, BMC Microbiol., 2018, 18, 1–10 CrossRef PubMed.
- K. Sakai, F. Charlot, T. Le Saux, S. Bonhomme, F. Nogué, J.-C. Palauqui and J. Fattaccioli, Plant Methods, 2019, 15, 79 CrossRef PubMed.
- M.-S. Hung and J.-H. Chang, Proc. Inst. Mech. Eng., Part N, 2012, 226, 15–22 CAS.
- L. Behrendt, M. M. Salek, E. L. Trampe, V. I. Fernandez, K. S. Lee, M. Kühl and R. Stocker, Sci. Adv., 2020, 6, eabb2754 CrossRef CAS PubMed.
- M. Andersson, S. Johansson, H. Bergman, L. Xiao, L. Behrendt and M. Tenje, Lab Chip, 2021, 21, 1694–1705 RSC.
- M. Aranda, Y. Li, Y. J. Liew, S. Baumgarten, O. Simakov, M. C. Wilson, J. Piel, H. Ashoor, S. Bougouffa and V. B. Bajic, Sci. Rep., 2016, 6, 39734 CrossRef CAS PubMed.
- D. Qin, Y. Xia and G. M. Whitesides, Nat. Protoc., 2010, 5, 491–502 CrossRef CAS PubMed.
- I. Pozdnyakov and S. Skarlato, Protistology, 2012, 7, 108–115 Search PubMed.
- L. H. Wang, Y. H. Liu, Y. M. Ju, Y. Y. Hsiao, L. S. Fang and C. S. Chen, Coral Reefs, 2008, 27, 823 CrossRef.
- G. Bricheux, D. G. Mahoney and S. P. Gibbs, Protoplasma, 1992, 168, 159–171 CrossRef.
- D. J. Franklin, O. Hoegh-Guldberg, R. J. Jones and J. A. Berges, Mar. Ecol.: Prog. Ser., 2004, 272, 117–130 CrossRef.
- K. Petrou, D. A. Nielsen and P. Heraud, Front. Mar. Sci., 2018, 5, 110 CrossRef.
- M. P. Lesser, Coral Reefs, 1997, 16, 187–192 CrossRef.
- M. Szabó, A. W. D. Larkum and I. Vass, in Photosynthesis in Algae: Biochemical and Physiological Mechanisms, ed. A. W. D. Larkum, A. R. Grossman and J. A. Raven, Springer International Publishing, Cham, 2020, pp. 459–488, DOI:10.1007/978-3-030-33397-3_17.
- D. A. Nielsen, K. Petrou and R. D. Gates, ISME J., 2018, 12, 1558–1567 CrossRef CAS PubMed.
- H. Tiricz, B. Nagy, G. Ferenc, K. Török, I. Nagy, D. Dudits and F. Ayaydin, J. Plant Res., 2018, 131, 179–189 CrossRef CAS PubMed.
- F. Rádi, B. Nagy, G. Ferenc, K. Török, I. Nagy, Z. Zombori, D. Dudits and F. Ayaydin, Acta Physiol. Plant., 2021, 43, 79 CrossRef.
- E. Dinç, S. Z. Tóth, G. Schansker, F. Ayaydin, L. Kovács, D. Dudits, G. Garab and S. Bottka, Plant Physiol., 2011, 157, 1628–1641 CrossRef PubMed.
- P. R. Beetham, P. B. Kipp, X. L. Sawycky, C. J. Arntzen and G. D. May, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8774–8778 CrossRef CAS PubMed.
- W. S. Chan, A. C. M. Kwok and J. T. Y. Wong, Front. Microbiol., 2019, 10, 546 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2lc00130f |
| ‡ Present address: Institute for Zoology, Experimental Morphology, University of Cologne, Zülpicher Strasse 47b, D-50674 Cologne, Germany. |
| § Present address: Functional Cell Biology and Immunology Advanced Core Facility, Hungarian Centre of Excellence for Molecular Medicine (HCEMM), University of Szeged, Szeged, Hungary. |
| This journal is © The Royal Society of Chemistry 2022 |