
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid microfluidic platform for screening and enrichment of cells secreting virus neutralizing antibodies†
Weikang Nicholas
Lin‡
a,
Matthew Zirui
Tay‡
b,
Joel Xu En
Wong
b,
Chia Yin
Lee
c,
Siew-Wai
Fong
b,
Cheng-I
Wang
c,
Lisa Fong Poh
Ng
bdef,
Laurent
Renia
bgh,
Chia-Hung
Chen
 h and
Lih Feng
Cheow
*aij
h and
Lih Feng
Cheow
*aij
aDepartment of Biomedical Engineering, National University of Singapore, Singapore. E-mail: lihfeng.cheow@nus.edu.sg
bA*STAR Infectious Diseases Labs, Agency for Science, Technology and Research (A*STAR), Singapore
cSingapore Immunology Network, A*STAR, Singapore
dDepartment of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
eNational Institute of Health Research, Health Protection Research Unit in Emerging and Zoonotic Infections, University of Liverpool, Liverpool, UK
fInstitute of Infection, Veterinary and Ecological Sciences, University of Liverpool, Liverpool, UK
gLee Kong Chian School of Medicine, Nanyang Technological University, Singapore
hSchool of Biological Sciences, Nanyang Technological University, Singapore
iDepartment of Biomedical Engineering, City University of Hong Kong, Hong Kong
jInstitute for Health Innovation & Technology (iHealthtech), Singapore
First published on 13th June 2022
Abstract
As part of the body's immune response, antibodies (Abs) have the ability to neutralize pathogenic viruses to prevent infection. To screen for neutralizing Abs (nAbs) from the immune repertoire, multiple screening techniques have been developed. However, conventional methods have a trade-off between screening throughput and the ability to screen for nAbs via their functional efficacy. Although droplet microfluidic platforms have the ability to bridge this disparity, the majority of such reported platforms still rely on Ab-binding assays as a proxy for function, which results in irrelevant hits. Herein, we report the multi-module Droplet-based Platform for Effective Antibody RetrievaL (DROP-PEARL) platform, which can achieve high-throughput enrichment of Ab-secreting cells (ASCs) based on the neutralizing activity of secreted nAbs against the a target virus. In this study, in-droplet Chikungunya virus (CHIKV) infection of host cells and neutralization was demonstrated via sequential delivery of viruses and host cells via picoinjection. In addition, we demonstrate the ability of the sorting system to accurately discriminate and isolate uninfected droplets from a mixed population of droplets at a rate of 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per hour. As a proof of concept, a single-cell neutralization assay was performed on two populations of cells (nAb-producing and non-Ab producing cells), and up to 2.75-fold enrichment of ASCs was demonstrated. Finally, we demonstrated that DROP-PEARL is able to achieve similar enrichment for low frequency (∼2%) functional nAb-producing cells in a background of excess cells secreting irrelevant antibodies, highlighting its potential prospect as a first round enrichment platform for functional ASCs. We envision that the DROP-PEARL platform could potentially be used to accelerate the discovery of nAbs against other pathogenic viral targets, and we believe it will be a useful in the ongoing fight against biological threats.
000 cells per hour. As a proof of concept, a single-cell neutralization assay was performed on two populations of cells (nAb-producing and non-Ab producing cells), and up to 2.75-fold enrichment of ASCs was demonstrated. Finally, we demonstrated that DROP-PEARL is able to achieve similar enrichment for low frequency (∼2%) functional nAb-producing cells in a background of excess cells secreting irrelevant antibodies, highlighting its potential prospect as a first round enrichment platform for functional ASCs. We envision that the DROP-PEARL platform could potentially be used to accelerate the discovery of nAbs against other pathogenic viral targets, and we believe it will be a useful in the ongoing fight against biological threats.
Introduction
Pathogenic viruses are responsible for a multitude of infectious diseases that plagued humanity. As part of our adaptive immune response, neutralizing antibodies (nAbs) play an integral role in conveying protection against viruses.1 Indeed, in vivo nAb titers are strongly correlated with protection for a multitude of viral infections, including dengue,2 SARS-CoV-23 and respiratory syncytial virus (RSV).4 However, the slow onset of natural nAb production after virus infection (from a few days5 to weeks6) could leave elderly or immunocompromised individuals vulnerable to severe health consequences. In such situations, administration of exogenously produced nAbs can often prevent infected individuals from progression to severe stages of disease.Currently, there are tremendous pressures on nAb discovery efforts to keep up with the rapid mutation of existing virus strains and the emergence of novel viral threats. Although fluorescence-activated sorting (FACS) and display systems can screen a large numbers of antibody-secreting cells (ASCs) for their binding to a particular protein antigen, binding affinity is often not reflective of the true functional efficacy of a nAb candidate.7 Many high affinity mAbs bind to non-neutralizing epitopes on the viral antigen, rendering them unsuitable for therapeutic applications (Fig. 1a). In the worst case, administration of non-neutralizing Abs could contribute to antibody-dependent enhancement (ADE) effects that increase the severity of multiple viral infections.8 Meanwhile, cells producing effective nAbs can be directly screened through hybridoma generation9 and single B cell activation/expansion,10 but the laborious nature of these procedures results in a long workflow. The majority of the immune repertoire11 is overlooked due to the low throughput (102–103 candidates) nature of existing virus neutralization assays. As such, there is an unmet need for an integrated system that can rapidly perform functional Ab neutralization assay on a large population of ASCs, with the ability to not just identify but also to isolate promising candidates.
 | ||
| Fig. 1 A comparison between bulk and single-cell virus neutralization assays. (a) nAbs must bind in a manner to block virus infection. Many Abs selected solely based on their ability to bind to virus lack a neutralizing function. (b) Top: Typical workflow and timeframe required to produce clonal populations of ASCs and screen them for virus neutralizing activity. Bottom: Significant acceleration of functional Ab discovery due to the ability to perform virus neutralization assay in droplets from single cells. Sorting of droplets with high virus neutralizing activity enables retrieval of functional ASCs. | ||
Droplet microfluidics platforms present several key advantages that make them ideal for the functional screening of ASCs, which include: 1) high operating throughputs at 10–104 droplets per second,12 2) well-established toolkit for droplet manipulation such as merging,13 splitting14 and sorting15 enable complex multi-step assays to be performed, 3) the ability to accommodate a variety of assay reagents type via co-encapsulation,16 particularly reporter or effector cells needed in most Ab functional assays. While droplet-based ASC screening via Ab binding affinity has been well established,17–19 the much more clinically relevant ASC screening via a true virus neutralization assay is still lacking due to the technical challenges of performing the complex multi-step assay. A recent work describes a platform for visualization of virus neutralization by ASCs in microfluidic droplets.20 Nonetheless, this method is limited to evaluating virus neutralizing activities from 100–1000 droplets in the field-of-view and lacks the critical capability of sorting and retrieving potent nAb secreting cells for downstream analysis or expansion.
To address the current research gaps, we established the DROPlet-based Platform for Effective Antibody RetrievaL (DROP-PEARL) platform, a high-throughput droplet microfluidic system capable of selection and retrieval of ASCs based on the neutralizing function of secreted Abs from single cells. As a proof-of-concept, we demonstrated the use of the DROP-PEARL platform to enrich for cells secreting nAbs against Chikungunya virus (CHIKV). High-throughput screening of functional ASCs with droplet microfluidics could be a new paradigm for the rapid discovery of potent and functional biologics.
Results
Working principles of the DROP-PEARL platform
The conventional workflow for screening and isolation of ASCs with virus neutralization capability is shown in Fig. 1b (top). Due to the limited proliferation capability of patient-derived Ab-secreting B cells, they must first be immortalized (e.g. via hybridoma formation) – a process that is both time-consuming and low-yield. Following this, single immortalized ASCs are isolated in well plates (e.g. 96 or 384 well plates) and clonally expanded. This expansion step, which could take many weeks to months, is necessary to obtain sufficient Ab concentrations within the large volumes (10–100 μL) of well plates. Subsequently, secreted Abs are recovered from each well, mixed with a pre-determined amount of target virus particles for binding to occur, and finally incubated with susceptible host cells to assess the degree of virus infection. A very important final step is the specific retrieval of ASCs based on their neutralizing activities for downstream sequencing or expansion. This conventional workflow is inadequate in meeting the needs of rapid nAb discovery against novel viruses and their variants as it requires months of processing, and is limited to screening a limited number of ASCs.We reasoned that the most time-consuming step of the current workflow (i.e. cell immortalization and clonal expansion) can be eliminated if secreted Abs from a single ASC can approach concentration levels needed for effective virus neutralization. Confinement of single ASC in small volumes would allow rapid accumulation of Abs as diffusion is prevented. In the DROP-PEARL platform, single ASCs are encapsulated within picoliter droplets to facilitate rapid Ab accumulation (Fig. 1b, bottom). This is followed by delivery of live virus to each droplet to allow for binding, and finally delivery of susceptible host cells into the same droplets for virus infection to occur. Most importantly, our platform allows high throughput interrogation (at a rate of 700000 droplets per hour) of virus infection and integrated sorting and recovery of droplets with low virus infection where neutralization has occurred.
We used a Chikungunya virus (CHIKV) infection model to validate the principles and performance of the DROP-PEARL platform. Chikungunya virus is a re-emerging pathogen that is endemic in Africa and many parts of Asia, with massive outbreaks with case numbers in the millions in recent decades.21,22 There are currently no clinically licensed vaccines or treatments available for Chikungunya infection, but a monoclonal nAb treatment has shown positive results in a phase I clinical trial.23 The ability to rapidly screen for monoclonal nAbs is pertinent to the development of better therapy regimens for Chikungunya infections.
Bulk infection and neutralization assay
We first characterized the bulk infection behaviour of CHIKV. The CHIKV-ZSGreen strain encodes a fluorescent ZSGreen protein under the control of a subgenomic promoter.24 Infection and replication of CHIKV-ZSGreen in host cell would produce a green fluorescence signal. As expected, CHIKV-ZSGreen infection of HEK 293T cells in 96 well plates resulted in an increase of cells expressing green fluorescence over time, plateauing at 22 hours with 73% of infected cells at a multiplicity of infection (MOI) of 10. Additionally, to investigate the infection behavior of CHIKV in the presence of nAbs, a previously reported anti-CHIKV monoclonal nAb, 8B1025 is allowed to bind to CHIKV before susceptible cells are added. Addition of 8B10 nAb resulted in dose-dependent reduction of infection in HEK 293T cells (Fig. 2C). When 20 μg mL−1 8B10 is added, only 9.75% of cells are infected (Fig. 2b). | ||
| Fig. 2 Infection of HEK 293T cells with CHIKV-ZSGreen. (a) Bulk infection of HEK 293T cells (stained red) by CHIKV over a period of 44 hours. Green fluorescence signal increases over time due to replication of CHIKV within the cells. (b) Flow cytometry analysis enables enumeration of CHIKV virus infected cells. (c) Degree of infection observed HEK 293T cells were co-incubated with CHIKV virus for 22 hours in the presence of varying amounts of 8B10 neutralizing mAb. | ||
A staged approach to performing an in-droplet virus neutralization assay
A conventional bulk virus neutralization assay typically comprises of four steps: 1. single-cell isolation of ASCs, followed by clonal expansion for Ab accumulation over time in the supernatant. 2. Mixing of Ab-containing supernatant and virus to allow binding to occur. 3. Addition of Ab-virus mixture onto host cells for infection to occur. 4. Measurement of virus infection and retrieval of ASC candidates that have virus neutralization activities.26 Similarly, to adapt such an assay into a droplet-based setting, multiple microfluidic operations are required as follows; 1. single-cell isolation of ASCs (without requiring clonal expansion) via droplet encapsulation and accumulation of Abs in droplets. 2. Virus delivery into droplets via picoinjection. 3. Host cells delivery into droplets via picoinjection. 4. Measurement of in-droplet virus neutralization and ASC isolation via droplet sorting.We developed a strategy using picoinjection to deliver virus and host cells into ASC-containing droplets. Since picoinjection delivers only a fraction of the incoming droplet's volume, contents can be delivered successfully without significantly increasing droplet size.28 The self-triggering mechanism of picoinjection also results in robust performance even when droplet periodicity may change over the course of the experiment.29 This ensures that reagents are delivered to all droplets to maximise the proportion of droplets where CHIKV infection occurs. After each round of picoinjection, the droplet diameters were observed to increase by approximately 10 μm, or a corresponding volume increase of 50.9% and 35.8% respectively (ESI† Table S2).
In the DROP-PEARL workflow, a picoinjector chip30 is first used to deliver CHIKV into fully-formed 70 μm diameter droplets containing single ASC. Subsequently, a second picoinjector chip delivers host cells into droplets at a rate of 300 droplets per second. High cell densities ranging from 50 to 200 million cells per mL were used to ensure that multiple host cells can be delivered to each droplet. Fig. 3d showed that the number of host cells delivered into each droplet can be controlled by varying the cell densities. Finally, we investigated the viability of host cells that were delivered via picoinjection. Cell viability remained high at 94.6% at 24 hours (Fig. 3e), indicating that the process was suitable for in-droplet infection to occur. Likewise, the excellent viability of multiple cells (88.4%) over 41 hours of incubation in droplets clearly shows that sufficient nutrients are available to sustain both the ASCs and host cells throughout the DROP-PEARL workflow. The much-enhanced viability of cells in droplet could in part be attributed to the optimized nutrient-rich culture media in droplets (ESI† Fig. S2) and improved gas-exchange of droplets during incubation in 12-well plates (Methods).
 | ||
| Fig. 3 Characterization of picoinjectors. (a) 70 μm diameter droplets generated using a standard flow-focusing chip design, (b) droplets after picoinjection of CHIKV into ≈80 μm diameter droplets and (c) after subsequent cell picoinjection into ≈90 μm diameter droplets. Scale bar represents 100 μm. (d) Number distribution of picoinjected host HEK 293T cells at various cell densities. (e) In-droplet cell viability over time for droplets picoinjected with 200 million per mL host HEK 293T. | ||
In-droplet infection and purified nAb neutralization
In-droplet CHIKV infection was performed by adding 37.5 kPFU μL−1 CHIKV into 70 μm diameter droplets containing cell culture media, followed by the addition of host cells by picoinjection. For all in-droplet infection experiments, an average of 8 host cells were delivered to each droplet during picoinjection (100 million cells per mL at injection port). Twenty hours after host cells addition, detectable green fluorescence signals are observed in a majority of droplets, indicating successful in-droplet virus infection (Fig. 4a). | ||
| Fig. 4 In-droplet CHIKV infection and signal readout. (a) Droplets containing 20 μg mL−1 of 8B10 nAbs or no nAbs were picoinjected with 37.5 kPFU μL−1 CHIKV and host HEK 293T cells. Images show droplets at 0 h and 20 h from the onset of HEK 293T picoinjection. Scale bar represents 100μm. (b) Average CHIKV signal intensity of said droplets at the 20 h time point, quantified via imaging and (c) PMT measurement. (d) The percentage of infected droplets over time at varying picoinjected CHIKV viral titers, as well as in the presence of 8B10 nAbs | ||
To investigate the effect of nAbs on CHIKV infection, droplets containing 20 μg mL−1 of purified 8810 nAbs were injected with 37.5 kPFU μL−1 of CHIKV, and in-droplet infection was compared against a virus-only control (Fig. 4a). The droplet signal intensities (average of 2D image) were analysed via imaging (Fig. 4b) and compared against a continuous-flow PMT detection system (peak fluorescence of each droplet) (Fig. 4c). A significant reduction in the average CHIKV infection signal was observed in the majority of the nAb containing droplets, suggesting partial inhibition of CHIKV infection amongst the infected droplets. Partial suppression of infection is a biological phenomenon and is consistent with observations in bulk virus neutralization assay (Fig. 2b). Furthermore, a broad distribution of infection signals were observed for both nAb-containing droplets and virus-only droplets, reflecting the biological heterogeneity of the CHIKV and 293T cells.
To determine the optimal viral titer to be used in the subsequent single-cell neutralization experiments, two conditions need to be considered. Firstly, the CHIKV viral titer to be used should achieve a sufficiently high infection rate needed to reduce the number of false positives during the downstream droplet sorting step. Secondly, to reduce the probability of false negatives which stem from infection even in the presence of nAbs, the viral titer should be kept as low as possible. As such, there is a delicate balance between the infection duration and viral titer to define a window of specificity. The infection rate of droplets was studied for different viral titers over a period of 48 hours after host cell injection (Fig. 4d). In general, the higher the picoinjected viral titer, the greater the percentage of infected droplets at all time points. Additionally, the infection rate appeared to plateau after 24 hours regardless of viral titer used. Based on the results, a viral titer of 75 kPFU μL−1 was selected to be used for the single-cell neutralization assay as it provided a high droplet infection rate at 93.2% beyond 20 hours. As a positive control for the presence of nAbs, droplets containing 9 μg mL−1 of 8B10 nAbs (to simulate the average concentrations of Abs accumulated in droplets after 24 hours) are injected with 75 kPFU μL−1 CHIKV virus, followed by host cell injection for infection. We observed that the percentage of droplets with infected cells is consistently lower at all times when droplets contain nAbs, demonstrating their protective effects against CHIKV infection in this droplet-based assay. In this way, collection of droplets containing cells exhibiting a lower level of infection is expected to enrich for nAbs.
Droplet sorter characterization
The previously reported droplet-based viral neutralization assay does not allow downstream retrieval of ASCs secreting nAbs as it lacks the key enabling step for isolation of nAb-containing droplets. To address this gap, the DROP-PEARL workflow integrates a 100 μm height droplet dielectrophoretic sorters (ESI† Fig. S2) that is capable of isolating droplets based on the CHIKV infection signal. To characterize the performance of this sorter, a mixed pool containing 1:1 ratio of droplets with CHIKV-infected host cells and droplets with uninfected host cells were sorted based on the detected CHIKV fluorescence signal. The high fluorescence of droplets containing infected cells triggers a series of sorting pulses to redirect them into a waste reservoir (Fig. 5a). We defined a positive droplet as one that contains uninfected cell (an indication of presence of nAb). The false positive and true positive rates of the sorted droplets were then assessed over a range of sorting thresholds ranging from 0.12 to 3.24 V (Fig. 5e). The images of the droplets before sorting (Fig. 5b), at the collection reservoir (Fig. 5c) and in the waste reservoir (Fig. 5d) at a sorting threshold of 0.48 V demonstrates the highly accurate performance of the integrated sorter in the DROP-PEARL platform.
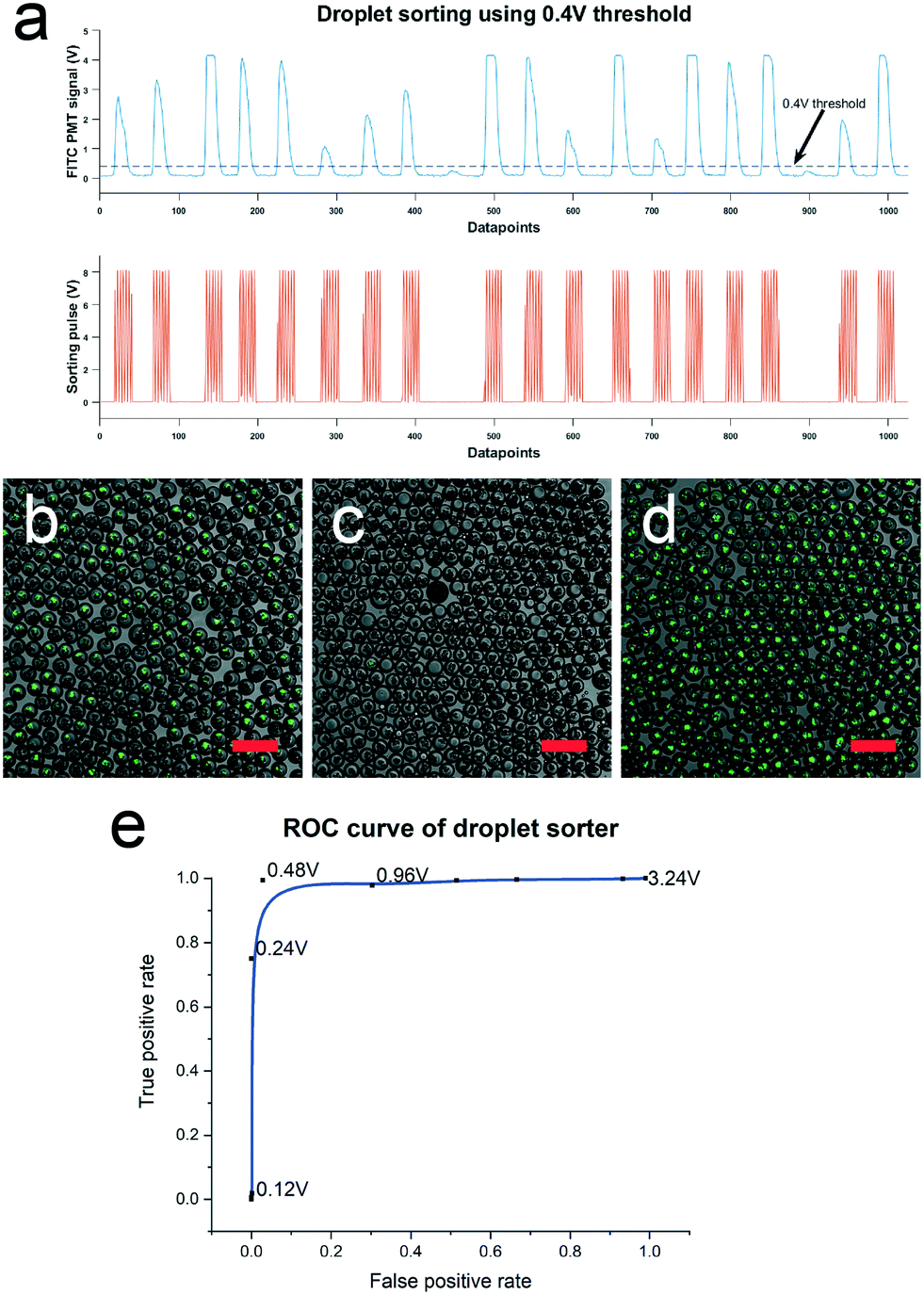 | ||
| Fig. 5 Characterization of sorters based on CHIKV infection signal. (a) PMT signal of droplets containing CHIKV infected HEK 293T cells. A sorting pulse is triggered when the PMT signal exceeds the 0.4 V threshold in this example. (b) Droplet population containing 50% infected, 50% non-infected droplets before sorting. Image of droplets retrieved at (c) collection and (d) waste outlet after sorting at 0.48 V threshold. Scale bar represents 250 μm. (e) ROC curve of sorter obtained by sorting droplets at a range of signal thresholds from 0.12 to 3.24 V. | ||
Application of DROP-PEARL to enrich for cells secreting nAbs against CHIKV infection
Finally, we validate the performance of DROP-PEARL for recovery of cells secreting nAbs against CHIKV infection. We prepared an initial population of ASCs that consists of a mixture of cells that secrete 8B10 nAbs (stained red) and cells that do not secrete nAbs (stained blue) at a 1:2 ratio. Note that the cell staining is not used for cell sorting, but rather for the downstream identification of cell types. Single cells from this mixed population are encapsulation in 70 μm diameter droplets and incubated for 24 hours to allow nAb accumulation in the droplets. Subsequently, CHIKV virus and host cells are sequentially added into the droplets and allowed to incubate for 18–22 hours for infection to occur (Fig. 6a). Droplet sorting was then performed using sorting thresholds of 0.4 V and 0.6 V, in accordance to the ROC curve established previously (Fig. 6b). The sorted droplets were then demulsified and the recovered cells were analyzed using flow cytometry to determine the percentages of ASCs (stained red) and non-secreting cells (stained blue) (Fig. 6C). While flow cytometry showed that the percentage of nAb secreting cells is 26–33% in the initial population, in experiments done over three separate occasions, the percentage of nAb secreting cells is higher in the collected set of droplets (42–57%). To reflect the efficiency of enriching nAb secreting cells, we adopted the “enrichment ratio” measure reported previously,15 defined as the percentage ratio of ASC to non-secreting cells after and before sorting. An ASC enrichment ratio of 1.90–2.75 was obtained using the DROP-PEARL platform, in a proof of concept for selecting ASCs secreting nAb against CHIKV.
 | ||
| Fig. 6 The complete in-droplet single-cell neutralization assay workflow. (a) Single-cell neutralization assay workflow. ASCs (stained red) and mock-transfected cells (stained blue) are first encapsulated in 70 μm diameter droplets and allowed to accumulate nAbs over 24 h. The droplets were then injected with CHIKV and host HEK 293T cells and incubated to allow infection to take place before they were sorted. (b) Sorted droplets retrieved from the top collection and bottom waste channels respectively when sorted using a 0.4 V threshold. Scale bar represents 100 μm. (C) Distribution of ASCs and mock transfected cells before and after sorting using a 0.4 V and 0.6 V threshold for 3 independent experimental runs. | ||
The relatively modest enrichment ratio of virus neutralization assay compared to antibody affinity assays is well within expectations. Unlike antigen-binding assay involves simple molecular interactions between antigen and antibody, virus neutralization assay is much more complicated involving the binding of antibody to the right epitope, and requires complex cellular mechanisms that determines virus infectivity and host cell permissiveness. Incomplete permissiveness of the host cells (also seen in bulk neutralization assay, where ∼30% of cells are uninfected even when there is no neutralizing Ab (Fig. 2C)) could result in false positive droplets being selected, thereby reducing the enrichment ratio. Nevertheless, this is the first demonstration of high-throughput virus neutralizing antibody enrichment in a semiautomatic microfluidics platform, and could already be used as a first enrichment step to provide a coarse selection of nAb secreting cells. We believe that this demonstration sets the foundation to its future use for a variety of viral diseases.
Finally, in view of the encouraging results of ASC enrichment from non-secreting cells using DROP-PEARL, we asked if similar enrichment of specific neutralizing antibody secreting cells can be achieved in more complex scenarios that better resembles physiological samples. We focus on two important aspects: 1) the proportion of neutralizing antibody secreting cells are low (0.1–2%) in convalescent/immunized patients, 2) there is an excess of B cells secreting unrelated antibodies in patient samples. In order to simulate these conditions, we performed an experiment where we prepared two kinds of cells secreting different monoclonal antibodies (8B10 is CHIKV neutralizing antibody, and 5A6 is a non-relevant SARS-COV-2 neutralizing antibody). ASCs secreting 8B10 (labelled red) and 5A6 (labelled blue) were mixed at a 1:50 ratio and the DROP-PEARL sorting experiment was performed as above. Representative images of the droplets before sorting, as well as sorted droplets from the top collection and bottom waste channel are shown in Fig. 7a–c. As before, the unsorted droplets consist of mostly virus infected cells and a mixture of specific (red) and non-relevant (blue) antibody secreting cells. Droplets recovered from the top collection channel contains predominantly uninfected cells, and an observable enrichment of specific CHIKV neutralizing antibodies (red). Upon FACS analysis of the recovered cells, we observed that the DROP-PEARL sorted cells contains 6.27% of CHIKV neutralizing antibody secreting cells, up from 2.69% in the unsorted population (Fig. 7d). This enrichment ratio of 2.42 at sorting threshold of 0.4 V is similar to what we obtained in the earlier proof-of-concept experiment. Our results showed that DROP-PEARL retain the ability to enrich for neutralizing antibody secreting cells even when they are present at low frequencies, and that the enrichment was not negatively impacted by the presence of large number of cells secreting non-relevant antibodies. The demonstration of successful application of DROP-PEARL in a scenario that better simulates physiological condition is an important milestone towards clinical implementation of this platform.
 | ||
| Fig. 7 Droplets retrieved from a single-cell neutralization assay performed using a mixed population of ASCs secreting 8B10 (CHIKV nAbs) and 5A6 (non-relevant SARS-COV-2 nAbs). 8B10 ASCs (stained red) and 5A6 ASCs (stained blue) are subject to the full single neutralization workflow as described in the main text (a) droplets prior to the sorting process. CHIKV-infected cells exhibit a green fluorescence signal. Sorted droplets retrieved from the (b) top collection and (c) bottom waste channels respectively when sorted using a 0.4 V green fluorescence signal threshold. Scale bar represents 100 μm. (d) Distribution of CHIKV-specific ASCs (8B10) and ASCs secreting irrelevant antibodies (5A6) before and after sorting on the DROP-PEARL platform. | ||
Discussion
The emergence of novel viral threats and rapid mutation of virus strains have posed considerable challenges for conventional screening methods to rapidly discover effective nAbs for treating infected individuals. In this manuscript, we report on DROP-PEARL, a rapid (<3 days), high throughput (150000 cells per hour) integrated microfluidic platform capable of detecting and sorting ASC based on their virus neutralization activity. The key novelties of DROP-PEARL demonstrated are: 1) rapid workflow that can be completed in <3 days, 2) high throughput screening of nAb-secreting cells (150000 cells per h) compared to existing imaging-based microfluidics method20 (∼1000 cells), 3) ability to sort and enrich for cells that can secrete neutralizing antibodies. Sorted live ASCs could be amenable to downstream expansion for large-scale production of nAb, or characterized with techniques such as single cell RNA sequencing to identify Ab-encoding gene. This platform could be a new paradigm for significantly shortening the workflow for virus neutralizing Ab discovery and production.
This is the first time an in-droplet virus neutralization assay has been demonstrated in the context of an infectious human disease where there is clinical evidence for the therapeutic benefits of exogenously administered nAb.23 Previous reports on in-droplet virus neutralization assay are only limited to characterizing the functional activities of ASCs, as it lack droplet sorting ability. In this report, we successfully developed and optimized the DROP-PEARL platform with functional ASC retrieval as an end goal, in order to realize its true potential for accelerating high throughput discovery of virus nAbs. Cells secreting monoclonal antibodies are used to validate the DROP-PEARL platform to statistically evaluate the in-droplet virus neutralizing activities. As virus neutralizing activity is performed at the single antibody-secreting cell level, it is compatible with rapid method to generating antibody-secreting cells (e.g. transient transfection) that does not require lengthy cell immortalization. On the other hand, there is also great potential to apply this method for enrichment of functional population from polyclonal antibody secreting cells, such as what would be typically found from convalescent patients.
In this proof of concept, we demonstrate that a single round of DROP-PEARL workflow enriches functional ASCs relative to non-Ab-secreting cells by 1.90–2.75 fold. Similar enrichment of specific ASC was also achieved when they were at a low proportion (∼2%) within large excess of cells secreting a irrelevant antibody, indicating a promising prospect of applying this method for enriching functional antibodies from primary samples. The relatively modest enrichment factor is consistent with a partially permissive host cell that we have used in this proof of concept. Various literature have shown the choice of host cells and reporter virus could have a significant impact on the sensitivities of virus neutralization assay.31 We believe there are significant opportunities for host cell and reporter virus engineering that could improve their permissiveness and infectivity for droplet based virus neutralization assay. For example, knocking out antiviral genes in host cells (e.g. STAT1) could drastically improve virus infection rates, as shown in dengue virus.32 Overexpression of viral receptors in host cells (e.g. ACE2 and TMPRSS2 for SARS-CoV-2,33 MXRA8 for CHIKV34) could also significantly increase the efficiency of virus entry into host cells. Engineering approaches to improve virus capsid stability and improved virus manufacturing process to reduce empty capsids35 would also increase the infection rates of virus. These approaches would be expected to significantly increase the specificity of DROP-PEARL for selection of virus-neutralizing ASCs by reducing the false positives.
DROP-PEARL is not intended to be a one-size-fits-all solution for neutralizing antibody discovery in all diseases. Due to the very different infection models and varied considerations for different viruses, the parameters in DROP-PEARL should be optimized for specific applications. Nonetheless, we expect enrichments to be achieved in majority of cases. Notwithstanding the additional specificities that could be gained from host cells and reporter virus engineering, application of DROP-PEARL to enrich for functional ASCs from convalescent individuals or immunized animals already represents a significant advance on yielding a subpopulation of ASCs secreting more potent polyclonal nAbs for immediate applications. We note that despite our best efforts to follow the conventional virus neutralizing assay steps in the single cell droplet workflow, there are unavoidable differences. Among them, the variabilities between the number of target cells, as well as heterogeneity among the antibody-secreting cells and virus, will be accentuated when they are present in small numbers within a droplet. As such, we envision DROP-PEARL as the first enrichment step to provide a coarse selection of neutralizing antibody producing cells. If desired, this enriched population could be used to generate polyclonal antibodies with better functionalities compared to the unsorted populations. A second round of DROP-PEARL workflow on the collected cells can also be performed to improve on the enrichment of ASCs. Furthermore, we envision that the DROP-PEARL workflow can be performed with ASCs enriched from previously reported Ab binding affinity assays,17,18,36 in order to obtain nAbs with high affinity. Finally, RNA sequencing of the antibody-coding genes can be performed on cells that are enriched using DROP-PEARL workflow. Antibody sequences that are enriched relative to the initial population can be identified. Such approaches have been very successfully used to identify therapeutic antibodies even when the specific ASCs constitute a small fraction of the circulating cells (e.g. from convalescence patient).37 A combination of DROP-PEARL enrichment and RNA sequencing is expected to enable rapid identification of functional antibodies from a polyclonal mixture.
In summary, we present a complete platform for the rapid discovery and retrieval of functional ASCs. We successfully demonstrated a proof of concept of the DROP-PEARL workflow to enrich for cells secreting nAbs against the CHIKV virus. In reality, the DROP-PEARL platform is versatile for rapid screening of biologics for treatment of various viral infections. We envision that it will be of great interest to the scientific community seeking to characterize nAbs against particular viruses, and biotechnology companies interested in adopting a new paradigm for rapid functional biologics discovery.
Methods
Device fabrication and operation
The polydimethylsiloxane (PDMS) microfluidic chips used in this study were made using well-established soft lithography fabrication techniques.38 Three types of microfluidic chips – droplet generator, picoinjector, droplet sorter – were used to establish the in-droplet viral neutralization assay. Flow control of both aqueous and oil phases in all processes was performed by syringe pumps (Pump 11 Elite, Harvard Apparatus, Holliston, MA) running at infusion mode. Oil phase used in the study is made up of fluorocarbon oil Novec™ HFE-7500 (3M, Singapore) containing 1% (w/w) Picosurf-1™ surfactant (Sphere Fluidics, Cambridge, UK) to stabilize droplets.A typical 60 μm height, 3-inlet flow-focusing channel design was used for droplet generation and single-cell encapsulation processes. The aqueous and oil channel widths were designed to be at 50 μm, which then constricted to 40 μm at the outlet to facilitate droplet breakup. 70 μm diameter droplets were generated by infusion of aqueous and oil. Next, to deliver CHIKV and host cells into droplets, 45 μm height/width picoinjectors with a 40 μm picoinjector nozzle width were used (ESI† Fig. S1). To generate the electric field required to disrupt the stable interface of reinjected droplets for picoinjection to occur, a 1 Vpp 20 kHz sinusoidal wave was amplified 100-fold and passed into the electrodes of the picoinjector. Lastly, to sort droplets after CHIKV infection, 100 μm height droplet sorters modified from Gielen et al.39 were used (ESI† Fig. S2). To ensure that droplets preferentially enter the top outlet channel in the absence of an electric field, the bottom outlet channel was lengthened such that its resistance is approximately 2 times the top channel. Additional shielding electrodes were included around the device to prevent undesired coalescence of droplets from the sorting electric pulses.
To fabricate the master moulds needed to create the microfluidic devices, SU-8 2050 negative photoresist (Kayaku Advanced Materials, Westborough, MA) was first spin coated onto silicon wafers. Subsequent UV exposure via a mask aligner (MJB4, SUSS MicroTec, Germany) and development was performed according to the SU-8 product sheet's recommended settings. The retrieved moulds were then surface-treated with trichloro-(1H,1H,2H,2H-perfluorooctyl)silane (Sigma-Aldrich, St. Louis, MO) in a dessicator overnight. PDMS (Sylgard 184™, Dow Corning Inc, Midland, MI) was then added over the moulds, degassed, and cured. The cured PDMS microchannels are then removed from the moulds using a scalpel, followed by creation of inlets and outlets using a 1 mm biopsy punch. The microchannels were then cleaned by ultrasonication for 10 minutes, dried, and irreversibly bonded to a substrate using a plasma cleaner (PDC-32G, Harrick Plasma, Ithaca, NY). Glass slides which were spin-coated with PDMS were used as the substrate for droplet generators, whereas uncoated glass slides were used as the substrate for picoinjectors and sorters. After plasma bonding, to create the electrodes required for the picoinjectors and sorters, a low-melting point indium alloy wire (WIREBN-52189, Indium Corporation, Clinton, NY) was melted into the electrode inlet channels, then connected to wires and secured with UV-curable glue (Uni-Seal™ 6322, Incure, Asheville, SC).
CHIKV-ZSGreen virus production
CHIKV (LR2006 OPY1)-ZsGreen infectious molecular clones were used to make viral stocks,24 which were then passaged up to two times in Vero E6 cells to amplify viruses. Virus stocks were concentrated by ultracentrifuging at 28000g for 4 hours with a sucrose cushion. Viral stock titres were quantified via plaque assay with serial dilution on Vero E6 cells.
Bulk infection and neutralization assay
For infection assays in bulk conditions, HEK 293T cells (American Type Culture Collection, ATCC) were plated in 96-well plates at 30000 cells per well, and incubated for 5 hours to allow cell adhesion. Meanwhile, antibody stocks were mixed with CHIKV-ZSGreen virus stocks (6000 pfu μl−1) for 2 h at 37 °C to allow immune complexes to form. 100 μl of virus + antibody mix were then added to cells, and incubated at 5% CO2, 37 °C for 22 hours. Cells were then trypsinized and fixed for readout by flow cytometry. Flow cytometry was performed on a BD LSR II flow cytometer.
Generation of monoclonal antibody-secreting cells
A single plasmid (encoding for 8B10 or 5A6) was constructed to express both the heavy and light chain genes of the specific antibody with the human ferritin heavy and light chain promoters respectively. HEK 293T cells were plated in a 6-well plate overnight, then transfected at around 80% confluency with the 8B10 plasmid using Lipofectamine 3000, following the manufacturer's protocol. Cells were harvested via trypsinization 24 hours post-transfection for use in assays.In-droplet CHIKV infection and purified nAb neutralization assay
In-droplet infection assays were performed at three different CHIKV viral titers (18.75/37.5/75 kPFU μL−1) to identify the optimal titer to be used in subsequent neutralization experiments. 70 μm diameter droplets were first generated using two aqueous phases – the first comprised growth media only (DMEM/F12 containing 20% FBS and 1% Penstrep) while the second comprised growth media containing 15.5% (v/v) Optiprep (Sigma-Aldrich, St. Louis, MO). The droplets were collected into syringes and incubated for 24 hours in an incubator with 5% CO2 at 37 °C. The droplets were then picoinjected with CHIKV, collected into another syringe and returned into the incubator for 3 hours before being subjected to a round of host cell picoinjection. For the host cell picoinjection process, host cells were resuspended with 15.5% (v/v) Optiprep to achieve a cell density of 100 million per mL and loaded into a syringe containing a magnetic stir bar. During the picoinjection process, the syringe was kept on ice and the stir bar is continuously agitated to ensure homogeneity of the exiting cell suspension. After the second round of picoinjection, the droplets were transferred into a 12-well plate to improve gas exchange (Corning® Costar® TC treated 12-well flat-bottom plate, Sigma-Aldrich, St. Louis, MO) containing 2 mL of HFE-7500 added with 1% (wt/wt) Picosurf-1™, sealed with parafilm and left to incubate in incubator with 5% CO2 at 37 °C. At different time points, the droplets were sampled and loaded into a disposable hemocytometer (EVETM Cell counting slides, NanoEntek, Seoul), followed by imaging via fluorescence microscopy. Infection rates were then quantified based on the CHIKV green fluorescence signal using ImageJ. Infected droplets were defined as droplets containing at least one host cell with a fluorescence intensity exceeding the background.In the purified nAb in-droplet neutralization experiment, purified 8B10 nAbs diluted with growth media to obtain a concentration of 18 μg mL−1 was used as the first aqueous phase. After dilution with the secondary aqueous phase, a final in-droplet nAb concentration of 9 μg mL−1 was obtained. The droplets were then subjected to the above mentioned in-droplet infection workflow to determine the infection rate of nAb-containing droplets over time.
Optical and electronics setup for droplet sorting
The optical setup used in droplet sorting comprised of a fluorescence light source (SPECTRA III, Lumencor Inc., USA) and a photomultiplier tube (PMT) detection system (H9306-03, Hamamatsu Photonics K.K., Japan). Only the FITC channel was used in all droplet sorting experiments.Voltage signals obtained from the PMT were parallelized into two outputs for sorting and signal recording. For signal recording, the PMT analog signals were converted to digital signals using a data acquisition card (USB-6002, National Instruments, USA) and recorded using the in-built Analog Input Recorder application in MATLAB (R2019b, MathWorks, USA). For sorting, PMT signals were processed in real-time using an Arduino DUE microprocessor (Arduino, USA) to determine if a particular droplet signal exceeds a pre-defined threshold. The DUE microprocessor was used to control an Arduino UNO microprocessor (Arduino, USA) responsible for generating 8 Vpp, 10 kHz square waves which were amplified 100-fold through a high-voltage amplifier before they were passed into the sorter's electrodes. Upon detection of a PMT signal which exceeds the threshold, sorting waves were switched on for 1750 μs to actuate the droplet towards the bottom sorting channel.
For ROC curve characterization, a 50:50 mixed pool of infected and non-infected droplets were sorted at 11 different thresholds ranging from 0.12 to 3.24 V. 0.5 μL of droplets from each sorting condition was then sampled and fluorescently imaged to identify the number of true and false positives.
Single-cell droplet neutralization assay
A single-cell in-droplet neutralization assay was performed to verify the ability of the workflow in enriching for ASCs secreting functional nAbs against CHIKV. A cell suspension comprising a mix of CHIKV nAb-secreting ASCs and non-secreting cells in a ratio of 1:2 was first resuspended in 15.5% (v/v) Optiprep. For later identification between the two populations of cells, the ASCs were stained red (CellTracker™ Red CMTPX Dye, Invitrogen, Waltham, MA) while the non-secreting cells were stained blue (Cell Proliferation Dye eFluor™ 450, Invitrogen, Waltham, MA) beforehand. Seventy μm diameter droplets were then generated using the cell suspension as the first aqueous phase and growth media as the secondary aqueous phase. A 20% single-cell encapsulation rate was used. The droplets were collected into a syringe, placed in an incubator with 5% CO2 at 37 °C for 24 hours to allow nAb accumulation within the droplets, before they were subjected to the in-droplet infection assay workflow. Droplet sorting was performed between 18–22 hours after host cell picoinjection when an overall droplet infection rate of >90% was achieved. The resulting droplets from each sorting condition and the pre-sorting baseline were then fluorescently imaged, demulsified using 20% (v/v) 1H,1H,2H,2H-perfluoro-1-octanol (Sigma-Aldrich, St. Louis, MO) in HFE7500, and the aqueous phases were recovered. Cells retrieved from the droplets were trypsinized and fixed, then analysed by flow cytometry for percentages of ASCs and non-secreting cells.
To evaluate the performance of DROP-PEARL for enrichment of specific functional ASCs in the presence of large excess of cells secreting irrelevant antibodies, 8B10 ASCs (secreting CHIKV nAbs) and 5A6 ASCs (secreting non-relevant SARS-COV-2 nAbs) are subject to the DROP-PEARL workflow as described above. An initial cell population comprising of 8B10 and 5A6 ASCs at a ratio of 1:50 was first encapsulated in 70 μm diameter droplets to achieve a single-cell occupancy rate of 20%. The droplets were then incubated to allow the accumulation of Abs within the droplets over a period of 24 h. The droplets were then picoinjected with 75 kPFU uL−1 of CHIKV, incubated for 3 h to allow Ab neutralization of CHIKV, before a second picoinjection step to deliver 293T host cells at a cell density of 100 million per mL. The droplets were then incubated for 20 h to allow infection to take place before they were dielectrophoretically sorted at a 0.4/0.6 V signal threshold.
Author contributions
L. F. P. N. provided CHIKV expertise and virus; S. W. F., J. X. E. W. and M. Z. T. prepared the CHIKV stocks; C. Y. L., M. Z. T., J. X. E. W. and C. W. designed and prepared the 8B10 and 5A6 nAbs and ASCs; L. R. and C. C. conceptualized the project; M. Z. T., W. N. L. and L. F. C. designed the research; M. Z. T. and W. N. L. performed the microfluidic and bulk experiments; M. Z. T., W. N. L. and L. F. C. analyzed the data; M. Z. T., W. N. L. and L. F. C. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Singapore Immunology Network (SIgN) Flow Cytometry core for assistance with cytometry design, the SIgN mouse core for support in animal breeding, and Institute for Health Innovation and Technology (iHealthtech) for use of core facilities. Prof Andres Merits from University of Tartu constructed and provided the infectious clone of ZSGreen-tagged viruses used in this project. This project is funded by Agency for Science, Technology and Research (A*STAR) core grant awarded to LR and MZT, by the National Medical Research Council of Singapore (MOH-000219-00 and NMRC/OF-YIRG19nov-0059), and the Singapore Ministry of Education (MOE-000063).References
- C. D. Murin, I. A. Wilson and A. B. Ward, Nat. Microbiol., 2019, 4, 734–747 CrossRef CAS PubMed.
- L. C. Katzelnick, M. Montoya, L. Gresh, A. Balmaseda and E. Harris, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 728–733 CrossRef CAS PubMed.
- D. S. Khoury, D. Cromer, A. Reynaldi, T. E. Schlub, A. K. Wheatley, J. A. Juno, K. Subbarao, S. J. Kent, J. A. Triccas and M. P. Davenport, Nat. Med., 2021, 27, 1205–1211 CrossRef CAS PubMed.
- A. G. Buchwald, B. S. Graham, A. Traore, F. C. Haidara, M. Chen, K. Morabito, B. C. Lin, S. O. Sow, M. M. Levine, M. F. Pasetti and M. D. Tapia, Clin. Infect. Dis., 2021, 73, 4421–4427 CrossRef PubMed.
- W. M. P. B. Wahala and A. M. D. Silva, Viruses, 2011, 3, 2374–2395 CrossRef CAS PubMed.
- Y. Galipeau, M. Greig, G. Liu, M. Driedger and M.-A. Langlois, Front. Immunol., 2020, 11, 610688 CrossRef CAS PubMed.
- D. R. Burton and J. R. Mascola, Nat. Immunol., 2015, 16, 571–576 CrossRef CAS PubMed.
- S. M. Tirado and K. J. Yoon, Viral Immunol., 2003, 16, 69–86 CrossRef CAS PubMed.
- L. Olsson and H. S. Kaplan, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 5429–5431 CrossRef CAS PubMed.
- L. M. Walker, S. K. Phogat, P.-Y. Chan-Hui, D. Wagner, P. Phung, J. L. Goss, T. Wrin, M. D. Simek, S. Fling, J. L. Mitcham, J. K. Lehrman, F. H. Priddy, O. A. Olsen, S. M. Frey, P. W. Hammond, S. Kaminsky, T. Zamb, M. Moyle, W. C. Koff, P. Poignard and D. R. Burton, Science, 2009, 326, 285–289 CrossRef CAS PubMed.
- R. R. Beerli and C. Rader, mAbs, 2010, 2, 365–378 CrossRef PubMed.
- K. Matuła, F. Rivello and W. T. S. Huck, Adv. Biosyst., 2020, 4, 1900188 CrossRef PubMed.
- X. Niu, S. Gulati, J. B. Edel and A. J. deMello, Lab Chip, 2008, 8, 1837–1841 RSC.
- D. R. Link, S. L. Anna, D. A. Weitz and H. A. Stone, Phys. Rev. Lett., 2004, 92, 054503 CrossRef CAS PubMed.
- J. C. Baret, O. J. Miller, V. Taly, M. Ryckelynck, A. El-Harrak, L. Frenz, C. Rick, M. L. Samuels, J. B. Hutchison, J. J. Agresti, D. R. Link, D. A. Weitz and A. D. Griffiths, Lab Chip, 2009, 9, 1850–1858 RSC.
- Y. Yuan, J. Brouchon, J. M. Calvo-Calle, J. Xia, L. Sun, X. Zhang, K. L. Clayton, F. Ye, D. A. Weitz and J. A. Heyman, Lab Chip, 2020, 20, 1513–1520 RSC.
- A. Gérard, A. Woolfe, G. Mottet, M. Reichen, C. Castrillon, V. Menrath, S. Ellouze, A. Poitou, R. Doineau, L. Briseno-Roa, P. Canales-Herrerias, P. Mary, G. Rose, C. Ortega, M. Delincé, S. Essono, B. Jia, B. Iannascoli, O. Richard-Le Goff, R. Kumar, S. N. Stewart, Y. Pousse, B. Shen, K. Grosselin, B. Saudemont, A. Sautel-Caillé, A. Godina, S. McNamara, K. Eyer, G. A. Millot, J. Baudry, P. England, C. Nizak, A. Jensen, A. D. Griffiths, P. Bruhns and C. Brenan, Nat. Biotechnol., 2020, 38, 715–721 CrossRef PubMed.
- R. Ding, K.-C. Hung, A. Mitra, L. W. Ung, D. Lightwood, R. Tu, D. Starkie, L. Cai, L. Mazutis, S. Chong, D. A. Weitz and J. A. Heyman, RSC Adv., 2020, 10, 27006–27013 RSC.
- L. Mazutis, J. Gilbert, W. L. Ung, D. A. Weitz, A. D. Griffiths and J. A. Heyman, Nat. Protoc., 2013, 8, 870–891 CrossRef CAS PubMed.
- J. A. Wippold, H. Wang, J. Tingling, J. L. Leibowitz, P. de Figueiredo and A. Han, Lab Chip, 2020, 20, 1628–1638 RSC.
- B. Wahid, A. Ali, S. Rafique and M. Idrees, Int. J. Infect. Dis., 2017, 58, 69–76 CrossRef PubMed.
- M. S. Cunha, P. A. G. Costa, I. A. Correa, M. R. M. de Souza, P. T. Calil, G. P. D. da Silva, S. M. Costa, V. W. P. Fonseca and L. J. da Costa, Front. Microbiol., 2020, 11, 1297 CrossRef PubMed.
- Moderna, Moderna's therapeutics: Antibody against Chikungunya virus (mRNA-1944), https://investors.modernatx.com/static-files/20407a28-5283-4b70-a39d-d683bac985d2.
- A. Utt, P. K. Das, M. Varjak, V. Lulla, A. Lulla and A. Merits, J. Virol., 2015, 89, 3145–3162 CrossRef CAS PubMed.
- L. Warter, C. Y. Lee, R. Thiagarajan, M. Grandadam, S. Lebecque, R. T. P. Lin, S. Bertin-Maghit, L. F. P. Ng, J.-P. Abastado, P. Desprès, C.-I. Wang and A. Nardin, J. Immunol., 2011, 186, 3258 CrossRef CAS PubMed.
- Y. Fu, Z. Zhang, J. Sun and Q. Zhu, J. Immunol., 2015, 194, 206.232 CrossRef PubMed.
- A. Isozaki, Y. Nakagawa, M. H. Loo, Y. Shibata, N. Tanaka, D. L. Setyaningrum, J.-W. Park, Y. Shirasaki, H. Mikami, D. Huang, H. Tsoi, C. T. Riche, T. Ota, H. Miwa, Y. Kanda, T. Ito, K. Yamada, O. Iwata, K. Suzuki, S. Ohnuki, Y. Ohya, Y. Kato, T. Hasunuma, S. Matsusaka, M. Yamagishi, M. Yazawa, S. Uemura, K. Nagasawa, H. Watarai, D. D. Carlo and K. Goda, Sci. Adv., 2020, 6, eaba6712 CrossRef CAS PubMed.
- M. Rhee, Y. K. Light, S. Yilmaz, P. D. Adams, D. Saxena, R. J. Meagher and A. K. Singh, Lab Chip, 2014, 14, 4533–4539 RSC.
- D. J. Eastburn, A. Sciambi and A. R. Abate, PLoS One, 2013, 8, e62961 CrossRef CAS PubMed.
- A. R. Abate, T. Hung, P. Mary, J. J. Agresti and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19163–19166 CrossRef CAS PubMed.
- W. Zhai, D. N. Zhang, C. Mai, J. Choy, G. Jian, K. Sra and M. S. Galinski, PLoS One, 2012, 7, e52327 CrossRef CAS PubMed.
- https://www.atcc.org/services/cell-engineering/stat1-knockout .
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N. H. Wu, A. Nitsche, M. A. Müller, C. Drosten and S. Pöhlmann, Cell, 2020, 181, 271–280.e8 CrossRef CAS PubMed.
- R. Zhang, A. S. Kim, J. M. Fox, S. Nair, K. Basore, W. B. Klimstra, R. Rimkunas, R. H. Fong, H. Lin, S. Poddar, J. E. Crowe, B. J. Doranz, D. H. Fremont and M. S. Diamond, Nature, 2018, 557, 570–574 CrossRef CAS PubMed.
- P. Nestola, C. Peixoto, R. R. Silva, P. M. Alves, J. P. Mota and M. J. Carrondo, Biotechnol. Bioeng., 2015, 112, 843–857 CrossRef CAS PubMed.
- N. Shembekar, H. Hu, D. Eustace and C. A. Merten, Cell Rep., 2018, 22, 2206–2215 CrossRef CAS PubMed.
- Y. C. Tan, L. K. Blum, S. Kongpachith, C. H. Ju, X. Cai, T. M. Lindstrom, J. Sokolove and W. H. Robinson, Clin. Immunol., 2014, 151, 55–65 CrossRef CAS PubMed.
- J. C. McDonald and G. M. Whitesides, Acc. Chem. Res., 2002, 35, 491–499 CrossRef CAS PubMed.
- F. Gielen, R. Hours, S. Emond, M. Fischlechner, U. Schell and F. Hollfelder, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7383–E7389 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2lc00018k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |