
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA microfluidic device and instrument prototypes for the detection of Escherichia coli in water samples using a phage-based bioluminescence assay†
Luis F.
Alonzo‡
 *a,
Troy C.
Hinkley
a,
Andrew
Miller‡
a,
Ryan
Calderon‡
a,
Spencer
Garing‡
a,
John
Williford
a,
Nick
Clute-Reinig
a,
Ethan
Spencer‡
a,
Michael
Friend
a,
Damian
Madan‡
a,
Van T. T.
Dinh
a,
David
Bell
b,
Bernhard H.
Weigl‡
a,
Sam R.
Nugen
c,
Kevin P.
Nichols
a and
Anne-Laure M.
Le Ny‡
*a
*a,
Troy C.
Hinkley
a,
Andrew
Miller‡
a,
Ryan
Calderon‡
a,
Spencer
Garing‡
a,
John
Williford
a,
Nick
Clute-Reinig
a,
Ethan
Spencer‡
a,
Michael
Friend
a,
Damian
Madan‡
a,
Van T. T.
Dinh
a,
David
Bell
b,
Bernhard H.
Weigl‡
a,
Sam R.
Nugen
c,
Kevin P.
Nichols
a and
Anne-Laure M.
Le Ny‡
*a
aIntellectual Ventures Laboratory, 14360 SE Eastgate Way, Bellevue, WA 98007, USA
bIndependent Consultant, Issaquah, WA 98027, USA
cDepartment of Food Science, Cornell University, Ithaca, NY 14850, USA
First published on 28th January 2022
Abstract
Current quantification methods of Escherichia coli (E. coli) contamination in water samples involve long incubation, laboratory equipment and facilities, or complex processes that require specialized training for accurate operation and interpretation. To address these limitations, we have developed a microfluidic device and portable instrument prototypes capable of performing a rapid and highly sensitive bacteriophage-based assay to detect E. coli cells with detection limit comparable to traditional methods in a fraction of the time. The microfluidic device combines membrane filtration and selective enrichment using T7-NanoLuc-CBM, a genetically engineered bacteriophage, to identify 4.1 E. coli CFU in 100 mL of drinking water within 5.5 hours. The microfluidic device was designed and tested to process up to 100 mL of real-world drinking water samples with turbidities below 10 NTU. Prototypes of custom instrumentation, compatible with our valveless microfluidic device and capable of performing all of the assay's units of operation with minimal user intervention, demonstrated similar assay performance to that obtained on the benchtop assay. This research is the first step towards a faster, portable, and semi-automated, phage-based microfluidic platform for improved in-field water quality monitoring in low-resource settings.
Introduction
In 2010, safe access to clean drinking water was recognized as a basic human right by the United Nations. Many countries around the world remain committed to achieving this goal by 2030.1,2 Still, 2.2 billion people worldwide lack safely managed drinking water, and 4.2 billion people lack adequate sanitation.3 In some low- and middle-income countries, the practice of open defecation and animal excretions exacerbate the problem. Fecally contaminated water sources transmit enteric diseases estimated to result in over 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths annually.4,5
000 deaths annually.4,5
Specific commensal and pathogenic bacterial species are exclusively found in large quantities (109–1010 bacterial cells per gram) in the feces of humans and other mammals.6 Their presence reliably indicates that a water source has been contaminated with fecal matter and serves as a proxy for the presence of other disease-causing fecal-derived pathogens.7 Among these bacteria populations, Escherichia coli (E. coli) is the preferred indicator for fecal contamination in water.6,8,9 Regulatory agencies, including the United States Environmental Protection Agency (EPA) and the World Health Organization (WHO), have set a limit of zero colony-forming units (CFU) of E. coli in 100 mL for safe drinking water.8–10 Thus, the ideal limit of detection of an assay of E. coli contamination levels should be greater than 1 CFU per 100 mL.
In many remote low- and middle-income settings, monitoring for fecal contamination of drinking water is limited by the lack of laboratory facilities, financial resources, and trained staff. On-site, affordable, relatively simple, accurate, and rapid determination of E. coli contamination will enable community-led monitoring, behavior change, and emergency preparedness.11 The current gold standard used to quantify E. coli on-site includes portable kits using membrane filtration to remove bacteria from the water sample followed by a 16 to 24 hour enrichment in selective media.11 The bacteria CFUs can then be visually quantified directly on the filter. Alternatively, metabolic tests such as the hydrogen sulfide presence/absence tests are used, but they are qualitative and can lead to false positives due to the natural presence of hydrogen sulfide-producing bacteria in tropical soil.12 Overall, these methods involve long incubation periods and often require specialized training to achieve high sensitivity, thus limiting their impact.
Microfluidic platforms can enable the miniaturization of highly sensitive and rapid molecular-based identification assays for environmental monitoring applications.13–16 Dastider et al. presented an impedance microfluidic device, which relies on E. coli specific antibodies to detect 39 CFU per mL in a total assay time of 2 hours.17 Another microfluidic device detects E. coli genomic DNA equivalent to 103 CFU per mL in just 18 minutes.18 These types of devices are unable to process large volume in-field samples with complex matrices, risk the possibility of false-negative results, and rely on lab-bound instrumentation for successful operation.
Bacteriophages have been used to identify an assortment of unique bacterial populations.19–24 These viruses can rapidly infect and induce specific viable bacteria hosts into producing large quantities of new phage virions.25,26 Lytic bacteriophages can enzymatically rupture the host, leading to the release of large quantities of infectious phage virions. Researchers commonly coopt this infection process to introduce reporter genes that produce easily detectable signals when bacteria cells are infected with modified phages.27,28 We previously described a modified T7 phage engineered to produce a novel luciferase reporter fused to a cellulose-binding domain that can be used to detect E. coli with high sensitivity.29,30
While researchers continue to make impressive advances in phage-based sensors, their use remains limited to research applications and is mainly restricted to laboratory settings.31 To overcome the usability and performance challenges for rapid water quality testing in low-resource settings, we developed a portable, semi-automated, microfluidic platform. The system incorporates our previously described T7 phage-based assay into a simple-to-use, quantitative platform detecting 4.1 E. coli CFU from 100 mL of water samples within 5.5 hours.
Materials and methods
Bacteria and culture media
E. coli BL21 was obtained from ATCC (BAA-1025TM, Manassas, VA USA). Bacterial cultures were initially stored at −80 °C in 25% glycerol before use. They were grown in tryptic soy broth (TSB) and plated on Luria-Bertani (LB) agar. Overnight cultures of E. coli were prepared in 10 mL of TSB inoculated with a single bacterial colony and incubated (37 °C, 200 rpm, 18 h). Serial dilutions were performed in sterile phosphate buffer saline (PBS). Bacteria were enumerated based on the most probable number (MPN) model using Quanti-Tray/2000 system (IDEXX, Westbrook, ME) following the manufacturer's recommended protocol. MPNs and CFUs are generally used interchangeably. CFUs are the actual count from the surface of a plate, while MPN is a statistical probability of the number of organisms.Genetic engineering of T7 phage
The reporter phage T7-NanoLuc-CBM (T7-nLuc-CBM) was constructed as described previously32 (available upon request at the Nugen Lab, Cornell). Briefly, the genome of wild type bacteriophage T7 (ATCC, BAA-1025-B2) was fragmented and re-assembled with the reporter expression cassette (nLuc-CBM) containing a NanoLuc® Luciferase (Promega, Madison, WI) fused to a C-terminal cellulose-binding module (CBM2a) using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, Ipswich, MA).33 The resulting DNA was precipitated and electroporated into E. coli NEB5α cells (New England Biolabs, Ipswich, MA). Cells were recovered in Super Optimal broth with Catabolite repression (SOC) media (37 °C, 4 h, 250 rpm) until visible lysis had occurred. Cellular debris was pelleted (10000 × g, 10 min), sterile filtered (0.22 μm), and standard double overlay plaque assays were performed on serial dilutions of the supernatant. NanoGlo® (Promega, Madison, WI) substrate was applied to identify luminescent plaques resulting from NanoLuc activity. Recombinant phages were plaque purified and sequenced to confirm successful recombination before being further propagated for use in the detection assay. To reduce the background from reporter enzyme constitutively produced during propagation, microcrystalline cellulose was added to phage lysates to specifically sequester the reporter while maintaining phage titer.
Microfluidic device design and fabrication
The commercially available microfluidic device (Fig. 1A) was designed in-house and outsourced for large-scale manufacturing (HC 1101-001, Hochuen Medical Technology, Shenzhen, China). The device was made of two 1.1 mm thick injection-molded polycarbonate enclosures (PC-110, Chi Mei Corporation, Taiwan), that sandwiched a 7 mil (177.8 μm) thick laminate polycarbonate spacer (Lexan 8010, SABIC Innovative Plastics, Shanghai, China), which was lined with 2 mil (50.8 μm) thick acrylic-based pressure-sensitive adhesive (3 M, Saint Paul, MN) on both sides. The laminate polycarbonate spacer was laser cut to allow for the internal placement of a 24.1 × 19.0 mm polyvinylidene difluoride (PVDF) membrane (0.45 μm pores, Membrane Solutions LLC., Shanghai, China) for filtering E. coli cells from water samples, a Ø 12 mm nitrocellulose (NT) membrane (0.2 μm pores, Pall Corporation, Port Washington, NY) for concentrating reporter, and a Ø 4 mm hydrophobic membrane (Versapor 450RC, Pall Corporation, Port Washington, NY) for venting the device channels.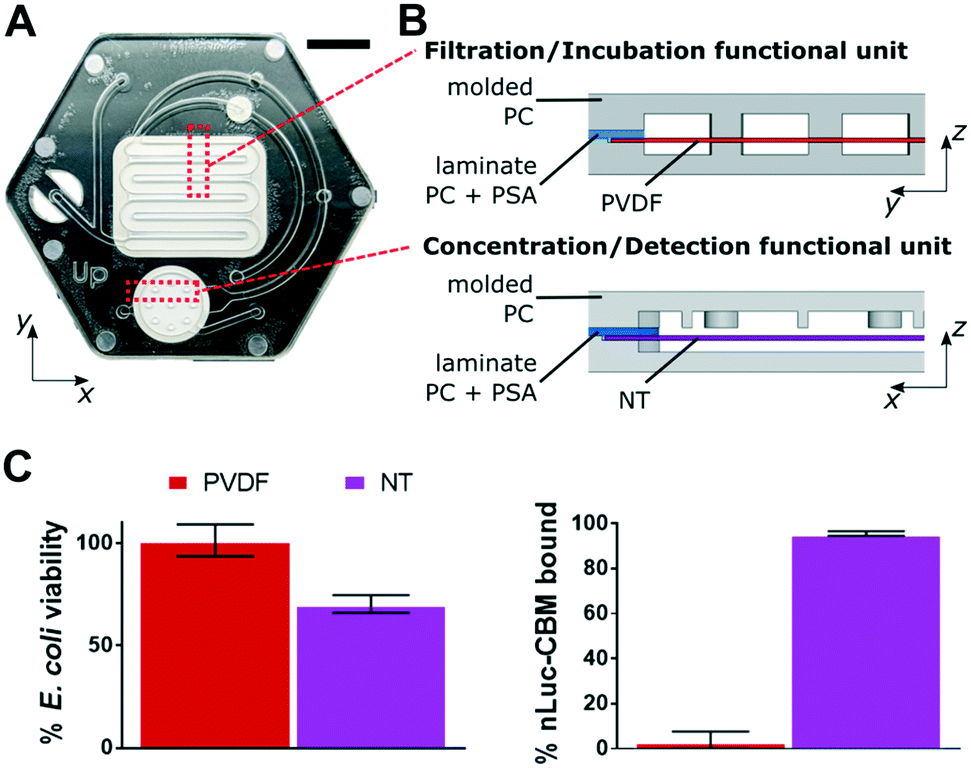 | ||
| Fig. 1 (A) Valveless microfluidic device for E. coli detection, scale bar: 10 mm. (B) Cross-sectional views of the functional dead-end filtration subunits within the microfluidic device. Gray = injection-molded polycarbonate, blue = laminate polycarbonate with double sided acrylic based PSA, red = PVDF membrane, purple = NT membrane. (C) Validation results of membrane materials for assay compatibility. | ||
A set of semi-circular features (Ø 8 mm, 3 mm apart) – present on all layers of the device – were designed around the sample input via-hole to be compatible with a custom-built, reusable world-to-chip interface. Serpentine channels (2 mm × 500 μm) were molded into the polycarbonate enclosures directly above and below the location of the PVDF membrane within the polycarbonate spacer. Similarly, circular-shaped chambers (Ø 10 mm × 500 μm) were molded into the polycarbonate enclosures directly above and below the location of the NT membrane within the polycarbonate spacer. The laser-cut spacer provided alignment of the membranes, the adhesive linings on the spacer provided seals to the enclosures around the membranes, and the walls of the serpentine channel and Ø 1 mm pillars provided structural support for the PVDF ad NT membranes, respectively. The PVDF and NT membranes acted as subunit dead-end filtration systems within the device and were connected to each other, to the sample and reagent input ports, and outlet ports by a series of 1 mm × 500 μm microfluidic channels.
The valveless microfluidic device consisted of 3 inlet and 4 outlet via-holes. Inlet and outlet ports were paired, without the need for on-device valves, by sealing unused ports, which allowed for the access of fluidic circuits corresponding to specific assay steps. A total of 5 inlet–outlet pair configurations were used for this assay, as illustrated in Fig. 2.
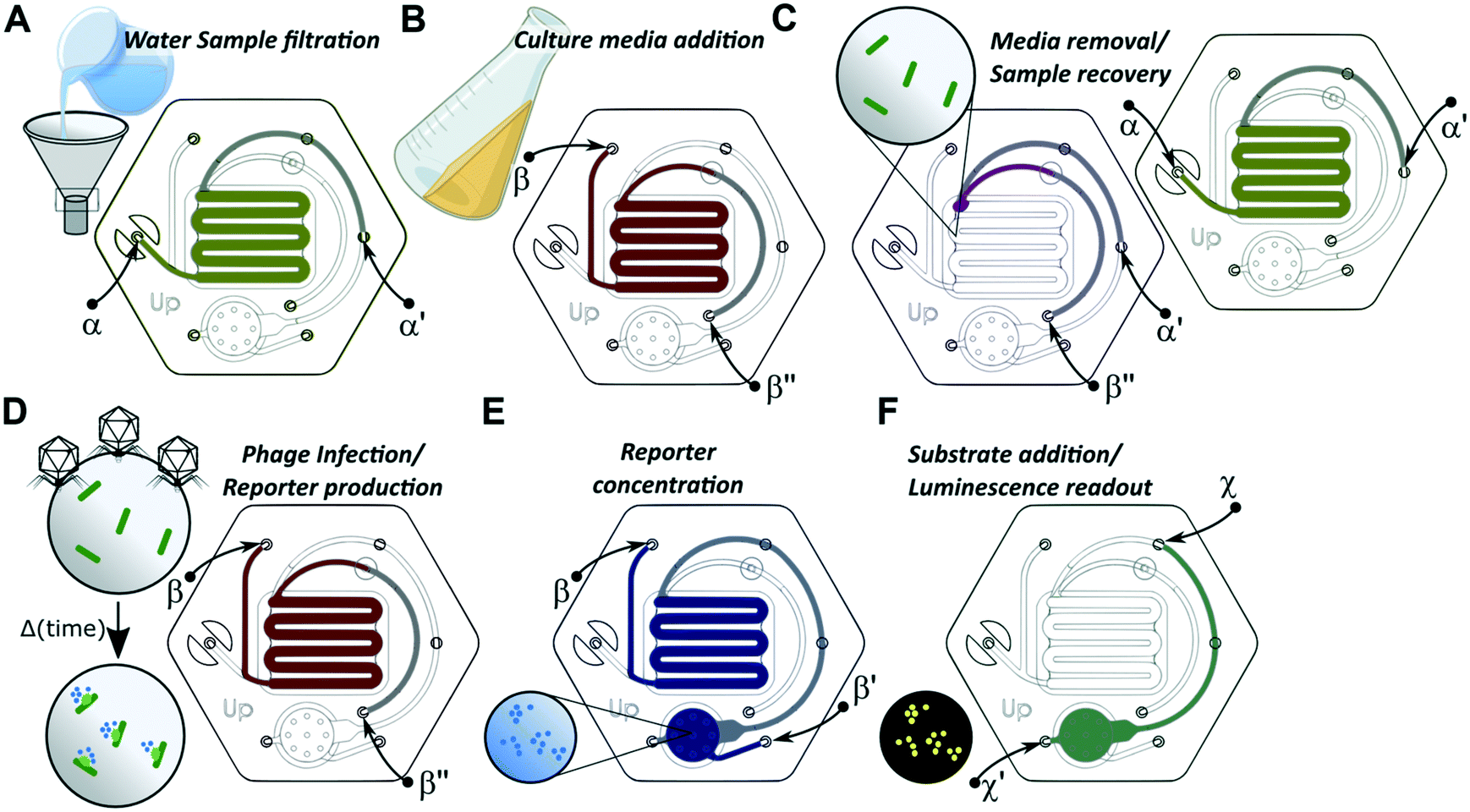 | ||
| Fig. 2 Phage-based E. coli detection assay workflow in a valveless microfluidic device. Inlet ports are connected to reagent reservoirs and outlet ports are connected to waste container, while unused ports are sealed off, restricting fluidic flow to specific areas of the microfluidic device without the need for on-board valves. (A) Step 1 – sample filtration to isolate E. coli cells from the water on the PVDF filter membrane; green line denotes the fluid pathway from inlet α to outlet α′; (B) step 2 – media addition to recover and ready cells for protein expression; red line denotes the fluid pathway from inlet β to outlet β′′; C) step 3 – media removal prior to phage addition; violet and yellow lines denote the fluid pathways from inlet β′′ to outlet α′ and from inlet α to outlet α′, respectively. This step requires two operations for the complete removal of the growth media within the microfluidic device; D) step 4 – bacteriophage addition to infect the cells and express the reporter; red line denotes the fluid pathway from inlet β to outlet β′′; E) step 5 – lysate concentration to collect and concentrate the reporter on nitrocellulose membrane; blue line denotes the fluid pathway from inlet β to outlet β′; F) step 6 – substrate addition to produce luminescence signal; green line denotes the fluid pathway from inlet χ to outlet χ′. In all drawings, the grey color denotes that the channel is on a different plane/layer than the rest of the channels with which it forms a continuous fluidic path. | ||
Instrumentation prototypes
Three custom-built instrument prototypes (Fig. S1†), one for each of the unique processes required for this assay (filtration, liquid-handling, and detection), were designed and manufactured in-house. The computer-aided designs (CAD) files were included in the ESI.†The filtration instrument was a 3D-printed clamp assembly used to dead-end filter bacteria from water samples on the PVDF membrane in the microfluidic device. The instrument base featured a hexagonal cavity for the microfluid device, a hold-down clamp, and a stainless-steel world-to-chip interface compatible with Luer-Lock connectors. The instrument lid featured an integrated orifice for the syringe (JS-S00L, JMS Co., Japan) used to introduce sample. In addition, the instrument lid featured a laser-cut silicone gasket seal for unused via-holes on the microfluidic device and a hose barb connector for a vacuum line.
The liquid-handling instrument was used to move the microfluidic device via rotation and translation. The instrument stage held the microfluidic device within a hexagonal cavity attached to a threaded shaft. The male-threaded shaft was mated with a female-threaded timing belt pulley and attached to the shaft of a permanent magnet direct current (PMDC) gearmotors, which provided combined rotational and translational motive force in isolation. The threaded timing belt pulley was coupled via timing belt at a 5:1 reduction to a second PMDC gearmotor, which provided translational motive force in isolation. When the first and second gearmotors were operated at angular velocities with a 5:1 ratio, the translational motive forces canceled out and the stage rotated without translation.
The detection instrument was a light-tight reading chamber that measured luminescent signal via a photomultiplier tube (PMT) (PDM03-9107, ET Enterprises Ltd, Uxbridge, UK). The instrument featured a hexagonal cavity to hold the microfluidic device and a Ø 25 mm cut-through, which aligned with the NT membrane within the device. The PMT was fixed underneath the base of the platform, in-line with the cut-through and directly under the NT membrane. A custom-built shutter mechanism to protect the PMT from ambient light when the microfluidic device was placed on the platform into the instrument was actuated via the opening and closing of the instrument lid. The PMT was connected to a laptop and operated with software provided by the manufacturer of the PMT.
Detection of log phase E. coli in microfluidic device
A log-phase E. coli BL21 culture was obtained by adding 200 μL of overnight culture to 4 mL of fresh TSB media. The culture was incubated until an OD600 (optical density at 600 nm) value of 0.6 was reached. At this point, serial dilutions of the culture were prepared in PBS and the dilutions were used immediately in tests. Test samples were prepared by mixing 100 μL of a dilution with 10 mL of sterile water, in triplicate, for each dilution in the series.Microfluidic devices were blocked to reduce non-specific adsorption of the phage-induced reporter protein by adding 250 μL of TSB media containing 0.1% Tween 20 to the serpentine channel above the PVDF membrane. Following a 2 h incubation period at 37 °C, the blocking solution was evacuated from the microfluidic device. Next, the filtration instrument was used to introduce test samples via syringe to the PVDF membrane in the microfluidic device. Sample volumes of 10 mL were used instead of 100 mL to expedite the filtration process and increase throughput, with no significant difference in assay results (Fig. S2†). Immediately after sample filtration, 250 μL of T7 phage solution (5 × 108 PFU per mL) was added to cover the entirety of the PVDF membrane, and the microfluidic device was moved to an incubator set to 37 °C for infection. Incubation times were varied from 1 to 4 hours to determine phage infection kinetics within the microfluidic device. After phage infection, the resulting lysate was moved within the microfluidic device from the PVDF membrane to the NT membrane, allowing for the concentration of T7-nLuc-CBM reporter. Finally, 75 μL of NanoGlo substrate was added to the chamber under the NT membrane. Bioluminescence was read immediately on the detection instrument.
Detection of stationary phase E. coli in microfluidic device
Lake water was collected from Lake Sammamish (WA, USA) and used as a representative sample matrix for environmental drinking water. The lake water was filter-sterilized (0.22 μm), inoculated with an overnight culture of E. coli BL21, and left at room temperature for 2 days. The lake-water culture was then serially diluted in PBS and the dilutions were used immediately for tests. Test samples were prepared by mixing 100 μL of a dilution with 10 mL of sterile water, in triplicate, for each dilution in the series. The filtration instrument was used to introduce samples via syringe to the PVDF membrane in the microfluidic device. 250 μL of TSB media with 0.1% Tween 20 was added over the PVDF membrane, and the device was moved to an incubator set to 37 °C. Incubation times were varied from 0 to 2 hours to evaluate bacterial growth dynamics within the microfluidic chip. Following the incubation period, the media was discarded, and T7 phage solution was added. The microfluidic device was moved back into the incubator at 37 °C for 3 h. After phage infection, the resulting lysate was fully transferred to the NT membrane. NanoGlo® substrate was added, and luminescence was read immediately on the detection instrument.Testing performance of microfluidic device with turbid samples
Stablcal® turbidity standards (HACH, Loveland, CO) were diluted with sterile filtered water to produce 100 mL turbidity samples ranging from 0 to 1000 nephelometric turbidity units (NTU). Real-world water samples were collected from different sites, including hand-dug wells, boreholes, lakes, and rivers. Turbidity values were validated using a portable turbidity meter. The filtration instrument was used to introduce 100 mL of turbidity samples via syringe to the PVDF membrane in the microfluidic device. Time-to-filtration measurements were collected at 10 mL increments until all the sample was successfully filtered or the PVDF membrane was completely fouled.Validation of automated liquid handling instrumentation prototype
Log-phase E. coli BL21 dilutions were prepared and introduced into the microfluidic device using the filtration instrument. The microfluidic device was then transferred to the liquid-handling instrument. A user interface (available in ESI†) was designed on LabVIEW to program the rotational/translational stage mechanism to execute the assay steps. Assay reagents (e.g., T7-nLuc-CBM solution and NanoGlo substrate) were prepared in vials and connected to the inlet ports of the manifold on the lid using Tygon® tubing. The central laboratory vacuum system was connected to the outlet ports of the manifold to induce fluid flow when the microfluidic device was sealed in place. Temperature was regulated using a Peltier cell set at 37 °C for 3 hours during the phage incubation period. Following phage incubation and substrate addition, the microfluidic device was transferred to the detection instrument to measure the resulting luminescence.Results and discussion
Microfluidic device design
The microfluidic device was designed to consolidate a phage-based E. coli detection assay,29,30 which consists of multiple steps: 1) filtration of 100 mL of liquid sample, 2) in situ culture and metabolic recovery of isolated E. coli cells, 3) in situ infection of E. coli with T7 phage, 4) concentration of phage-induced luminescence-generating, enzymatic reporter onto a cellulose membrane, and 5) measurement of luminescence output signal (Fig. 2). These assay steps are accomplished with two functional subunits combined within an injection-molded microfluidic device (Fig. 1B). Polycarbonate, a thermoplastic material, was selected as the primary device material due to its low enzyme adsorption, thermal stability during incubations, optical transparency, and compatibility with scale-up manufacturing processes34,35 to reduce fabrication costs. An acrylic-based adhesive was used to assemble the three polycarbonate layers of the device and was shown to not affect assay performance (Fig. S3†).Steps 1–3 of the assay ran within the filtration/incubation functional subunit of the device (Fig. 1B). A dead-end filtration architecture with a 0.45 μm filter membrane concentrated bacteria cells from 100 mL of water. A serpentine channel design provided fast sample filtration times. Preliminary channel dimensions were optimized through a series of empirical tests (Fig. S3†), with final adjustments to channel dimensions made during preparation of the device for manufacturing. After filtration of the sample left concentrated bacteria cells on the filter membrane, cell metabolic recovery followed by cell phage infection occurred directly on the membrane. PVDF was selected as the filtration membrane because it demonstrated E. coli viability (101.4 ± 7.8%) and negligible nLuc-CBM adsorption (3.3 ± 4.5%) (Fig. 1C and S3†).
Steps 4 and 5 of the assay ran within the concentration/detection functional subunit (Fig. 1B). A dead-end filtration architecture with a capture membrane concentrated the reporter which was expressed in the phage-infected E. coli as a recombinant fusion of NanoLuc enzyme and a cellulose-binding module. The NT membrane was selected as the capture membrane because it demonstrated near complete binding of the reporter (95.6 ± 1.0%) (Fig. 1C and S3†). The dimensions of this functional subunit were determined empirically (Fig. S3†). The NT membrane was tested for the ability to handle steps 1–5 of the assay, but the NT membrane reduced cell viability in steps 1–3 compared to the PVDF membrane (Fig. 1C). In addition, the need to minimize the area of detection over the PMT in step 5 was at odds with the desire to maximize filtration rate of large sample volumes.
Further optimization of the microfluidic device design improved performance of each functional subunit over early-stage designs (Fig. S4†). Improvements to the manufacturability of the device included replacement of disposable Luer Lock® components with the novel fluidic interconnect on the filtration instrument (Fig. S1†), transition from onboard pneumatic valves to a valveless device regulated via a novel stage mechanism on the liquid-handling instrument. These changes to the design unlocked an injection-molded manufacturing process, which considerably reduced the overall cost to less than $1 at a production scale of 250000 devices per year (Fig. S6†).
Validation of T7 phage infection in microfluidic device
To evaluate phage infection kinetics in this unique microfluidic system,36 several durations of phage infection of E. coli were tested in the device by measuring luminescence at specific time intervals (Fig. 3). Phage infection duration can depend on the specific phage strain used, so allowing time for complete infection of the bacteria population is critical for increased assay sensitivity.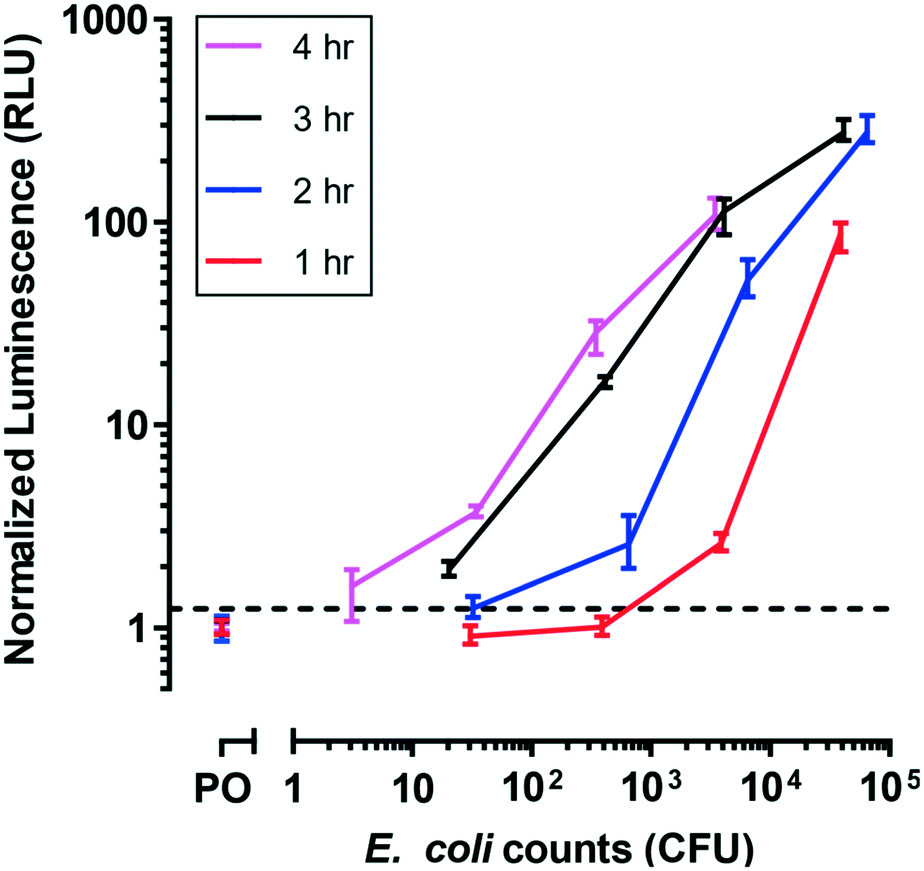 | ||
| Fig. 3 T7-nLuc-CBM phage infection dynamics on filter membrane within the microfluidic device. Luminescence (RLU: relative light unit) correlated with bacteria concentrations, with increased phage incubation time leading to limit of detection improvements. The limit of detection (dash line) was defined as the negative control (PO: phage only) plus three standard deviations. Data points represent the average of three replicates and error bars represent standard deviations. | ||
For these experiments, E. coli BL21 was cultured off-device to minimize growth within the device prior to infection with phage. For each of the infection durations tested, luminescence signal increased with bacterial cell counts as expected. Signal also increased with the infection duration, such that the limit of detection improved with increased phage infection. The data show we can measure 20.3 MPN after 3 h of phage incubation. However, the similarities of the response curves for infection duration of 3 h and 4 h suggested that no appreciable advantage was derived from extending phage incubation periods beyond 3 h for the cell densities tested, because most E. coli cells were infected. Thus, for all following experiments, this infection duration was used.
Phage-based assay in microfluidic device
To evaluate E. coli incubation kinetics in the microfluidic device, BL21 E. coli cells were conditioned in filtered lake water until a stationary growth phase was reached, to simulate their state in natural environments.37 Several durations of pre-infection incubations of sample derived from those cells were tested in the device by measuring luminescence after the full assay. Sample incubation duration can impact bacterial growth rates, so allowing time to obtain actively growing host cells is critical for maximal phage infection.38The sample incubation duration was varied to allow injured cells to recover from a metabolically inefficient state (Fig. 4). For each of the incubation durations tested, luminescence signal increased with bacterial cell counts as expected. Signal also increased with the incubation duration, such that the limit of detection improved with increased sample incubation duration. A 1 h incubation interval allowed for the detection of less than 13.4 CFU in approximately 4 h (i.e., 1 h sample incubation plus 3 h phage infection). The luminescence output for stationary cells incubated in media for 1 h closely matched the response observed in log-phase cells infected with T7-nLuc-CBM for 3 h (Fig. 3), and additional incubation duration (t = 2 h) yields a higher luminescent signal. This suggests that additional media incubation (t > 1 h) resulted in considerable pre-enrichment prior to phage infection, allowing for the detection of 4.1 E. coli CFU.
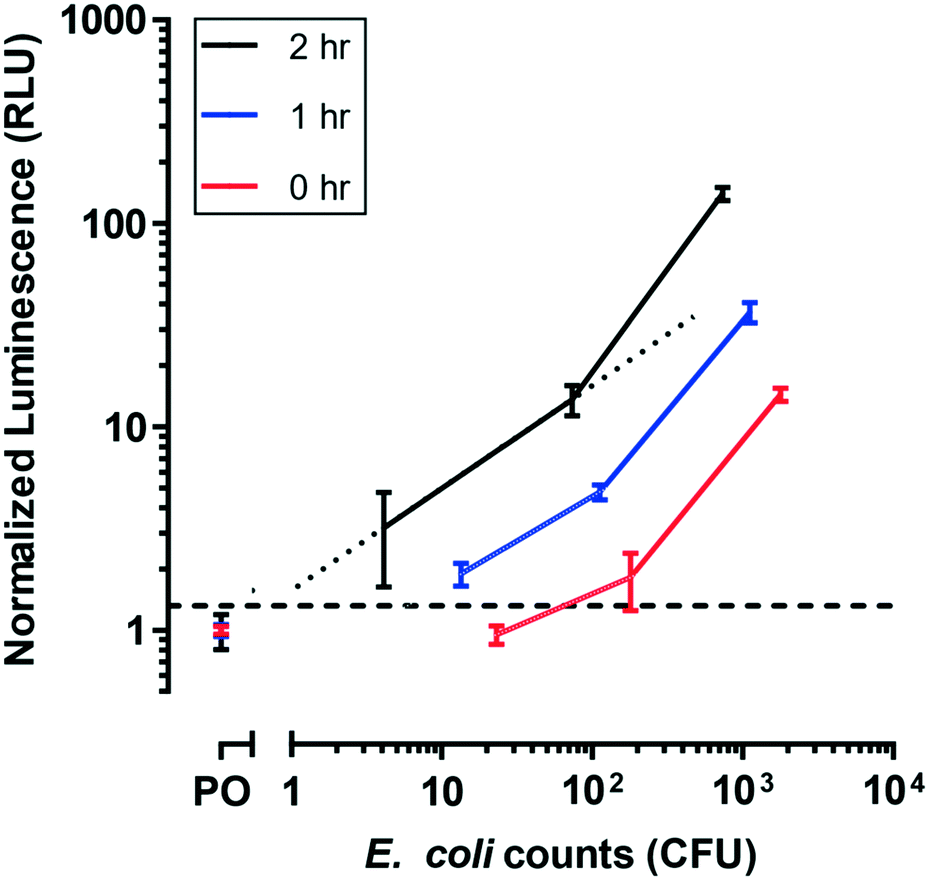 | ||
| Fig. 4 E. coli metabolic recovery from lake water treated samples. Increased incubation periods in nutrient rich media leads to limit of detection improvements. For BL21 E. coli cells, a 2 h media incubation, followed by a 3 h phage incubation, allows for the detection of <10 MPN. The limit of detection (dash line) was defined as the negative control (PO: phage only) plus three standard deviations. Data points represent the average of three replicates and error bars represent standard deviations. Dotted line represents linear interpolation used to apply a semi-quantitative interpretation of the results. | ||
Our data can be analyzed to provide semi-quantitative results in relevant concentration ranges. A linear interpolation was performed on a subset of the data obtained after 2 h of media incubation (dotted line in Fig. 4), which contained MPN counts between 1 and 100. Altogether, the following cut-off values were determined: <1 MPN corresponded to relative light unit (RLU) < 1.3, 1–10 MPN corresponded to 1.3 < RLU < 5, 10–100 MPN corresponded to 5 < RLU < 15, and >100 MPN corresponded to RLU > 15. Note that the calibration curve determined here is specific to BL21 upon T7 phage infection. Additional calibration curves would have to be generated to determine appropriate values for other samples (e.g., for other reporter phages, specific bacteria strains, or unknown bacteria populations). However, these results show that this assay, using the microfluidic device, is conducive to a semi-quantitative method to determine levels of bacterial contamination.
Influence of sample turbidity on device performance
We challenged our device with several lab-made turbidity standards and real-world samples, because highly turbid samples are known to generate membrane fouling issues in dead-end filtration devices39 (Fig. 5). Environmental samples isolated from borehole and hand-dug drinking wells (0.18 and 0.22 NTU) appeared visibly clear, while river and lake samples (16.35 and 19.93 NTU) contained large particle contaminants visible by eye.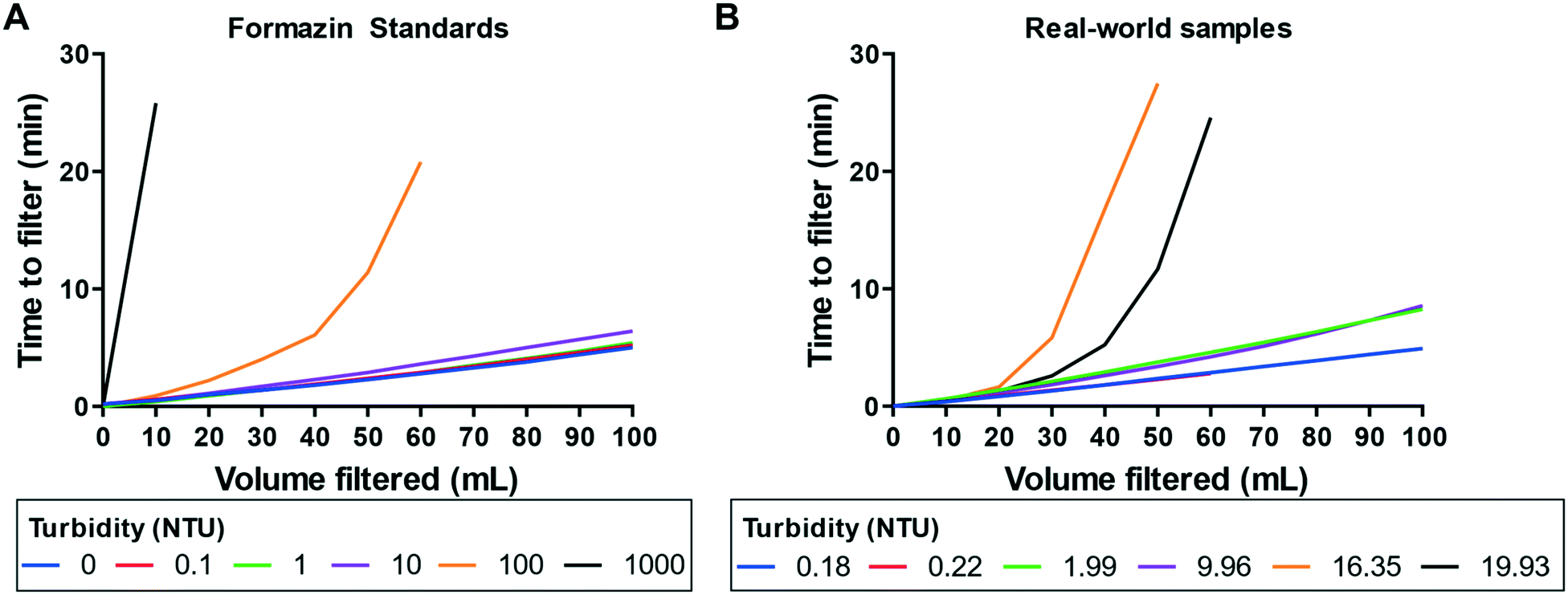 | ||
| Fig. 5 Performance validation of microfluidic device against water samples with variable turbidity. (A) Time required to filter formazin turbidity standards (0–1 NTU data points are clustered together). (B) Time required to filter real world water samples (1.99 and 9.96 NTU samples are dilutions of the 19.93 NTU sample). | ||
We observed a direct correlation between filtration rates and sample turbidity of ≤10 NTU (Fig. 5A). With our current microfluidic device architecture and material selection, we can filter 100 mL of ≤10 NTU formazin samples in less than 10 min without any sample manipulation. For formazin samples with turbidity >10 NTU, only a portion of the 100 mL could be filtered in the device before membrane fouling was observed. Formazin is an accepted turbidity standard, but appreciable differences were observed between the formazin and the river and lake samples (Fig. 5B). For borehole and hand-dug drinking well water samples (<10 NTU), we observed <10 min filtration in the device, as observed with formazin samples (<10 NTU). However, river and lake samples (10–20 NTU) were only partially filtered in the device, an outcome most similar to 100 NTU formazin samples. Still, simple dilution of the river and lake samples with sterile water to reduce the turbidity below 10 NTU allowed for the complete filtration of these difficult-to-process samples.
Performance of prototype instrumentation for portable microfluidic device use
Our microfluidic phage-based assay comprised five steps, three of which required user interaction and two of which required the incubation durations established above. The three steps that required user interaction with the device were: 1) sample filtration, 2) assay reagent delivery, and 3) bioluminescent signal readout. As a proof of concept for a field-operable set of equipment, we constructed three custom, portable instrument prototypes to assist with the operation of the microfluidic device. These designs – for filtration, liquid-handling, and detection instruments – could be adapted to be field deployable.The filtration instrument was designed to rapidly but safely introduce large sample volumes to the microfluidic device. A unique feature of the filtration instrument design was the reusable world-to-chip interface (Fig. S1A†), which worked similarly to a standard Luer Lock connector. This interface was continuously tested and showed no signs of sample leaking when 100 mL was introduced to the sample cup (i.e., 100 mL syringe with Luer Lock). Additionally, no cross-contamination was observed between samples.
The liquid-handling instrument was designed to deliver a series of liquid reagents and incubate the microfluidic device at accurately controlled temperatures (Fig. S1B†). Rotation and translation of the microfluidic device on a custom, dual-gearmotor-drive stage enabled the removal of on-device valves, by aligning inlet and outlet via holes with reagent input and waste while sealing unused holes. The assay timing steps and corresponding stage coordinates were verified empirically and pre-programmed onto the microcontroller within the instrument, completely automating the assay. The assay performance on-instrument was compared to assay performance off-instrument, as established in the other experiments within this study, and they showed equivalent performance (Fig. 6).
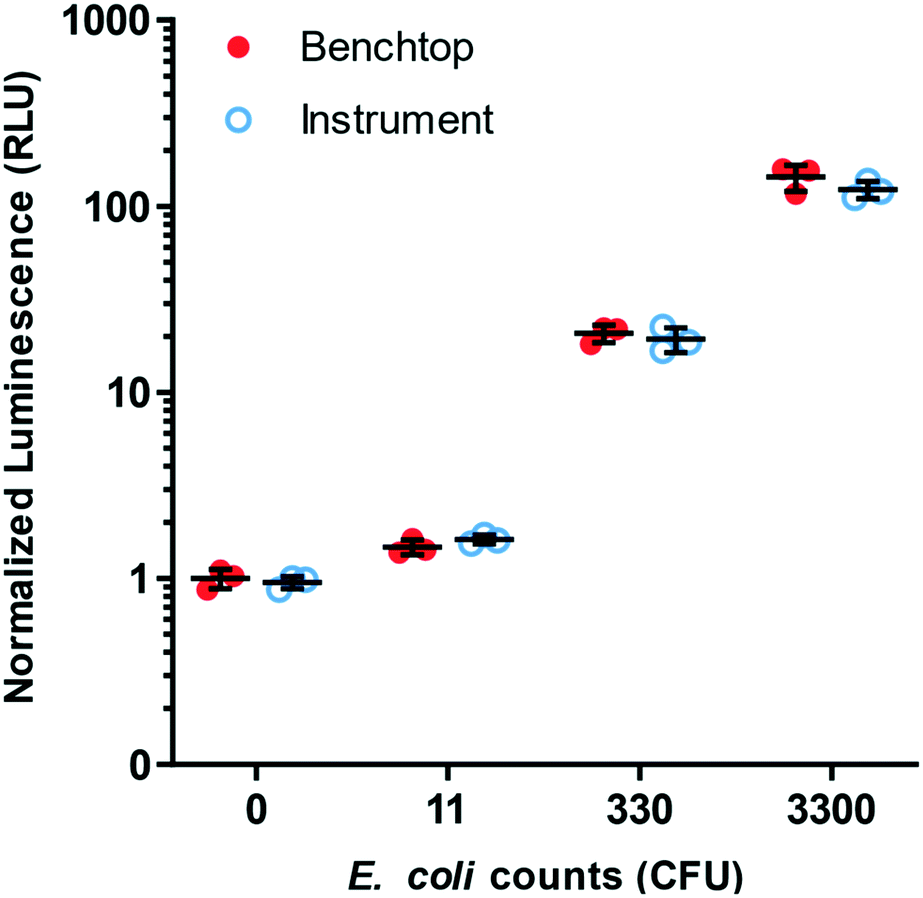 | ||
| Fig. 6 Validation of instrumentation prototypes for portable and semi-automated operation of the microfluidic device. Data showing equivalent performance of the microfluidic phage-based E. coli detection assay performed on the prototype instrumentation versus on the laboratory benchtop. For each E. coli concentration tested, there is no significant difference (n = 3, p > 0.05) between benchtop and instrument operation. | ||
The detection instrument was designed to sensitively detect luminescence signal from the captured reporter on the NT membrane in the microfluidic device using a PMT, but alternative detector technologies can be considered.40–45 A simple mechanical shutter mechanism (Fig. S1C†) was designed to shield the PMT's detector from excess ambient light when the platform's cover was open during sample introduction. Validation experiments showed that while we had some light contamination during the opening of the cover, the effects were reversible upon closure, and the PMT's dark counts were recovered in approximately 30 s (Fig. S1C†).
Conclusions
There is a great need for improved bacterial detection technologies in drinking water systems, especially in low-resource settings where sanitation infrastructure is insufficient. Interested stakeholders have expressed a desire for alternative diagnostic tools that are rapid, sensitive, specific, portable, and affordable.11 Our results demonstrate a portable and scalable microfluidic platform solution to process complex environmental matrices with minimal user interventions. The successful application of our rapid and highly sensitive phage-based assay on a microfluidic platform achieves the detection of single-cell levels of E. coli in large volumes of liquid samples within a third of the time of conventional testing methods. A single E. coli strain was used to demonstrate the semi-quantitative detection capabilities of this phage-based platform, but the system has the potential to be used as a broad tool for detection of other relevant strains and pathogens. Future optimization efforts will be focused on increasing test throughput and automatization of the instrumentation to allow for timely surveillance and ease of use.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Bill and Melinda Gates Foundation Trust for their sponsorship through Intellectual Venture's Global Good Fund I, LLC (https://www.globalgood.com). We would also like to thank Joshua Bishop for their help editing the manuscript.References
- The human right to water and sanitation, GA Res 64/292, UN GAOR, 64th sess, 108th plen mtg, Agenda Item 48, UN Doc A/RES/64/292 (adopted 28 July 2010) Search PubMed.
- Transforming our world: the 2030 Agenda for Sustainable Development, GA Res 70/1, UN GAOR, 70th sess, 4th plen mtg, Agenda Item 15 and 116, UN Doc A/RES/70/1 (adopted 25 September 2015) Search PubMed.
- UNICEF and WHO, Progress on household drinking water, sanitation and hygiene 2000–2017: Special focus on inequalities, 2019 Search PubMed.
- A. Prüss-Ustün, J. Bartram, T. Clasen, J. M. Colford, O. Cumming, V. Curtis, S. Bonjour, A. D. Dangour, J. De France, L. Fewtrell, M. C. Freeman, B. Gordon, P. R. Hunter, R. B. Johnston, C. Mathers, D. Mäusezahl, K. Medlicott, M. Neira, M. Stocks, J. Wolf and S. Cairncross, Burden of disease from inadequate water, sanitation and hygiene in low- and middle-income settings: a retrospective analysis of data from 145 countries, Trop. Med. Int. Health, 2014, 19, 894–905 CrossRef PubMed.
- WHO, Preventing diarrhoea through better water, sanitation and hygiene: exposures and impacts in low- and middle-income countries, Geneva, 2014 Search PubMed.
- R. G. Feachem, D. J. Bradley, H. Garelick and D. D. Mara, Sanitation and Disease: Health Aspects of Excreta and Wastewater Management, John Wiley & Sons, New York, 1983 Search PubMed.
- A. Dufour, Bacterial indicators/health hazards associated with water, ed. A. W. Hoadley and B. J. Dutka, 1977, pp. 48–58 Search PubMed.
- S. C. Edberg, E. W. Rice, R. J. Karlin and M. J. Allen, Escherichia coli: the best biological drinking water indicator for public health protection, J. Appl. Microbiol., 2000, 88, 106S–116S CrossRef PubMed.
- WHO, Guidelines for drinking-water quality, Geneva, 4th edn, 2017 Search PubMed.
- United States Environmental Protection Agency, National Primary Drinking Water Regulations, 2009 May, Report No.: EPA 816-F-09-004 Search PubMed.
- UNICEF, Target Product Profile: Rapid E.coli Detection Tests v3.0, December 2019 Search PubMed.
- H. Yang, J. A. Wright, R. E. S. Bain, S. Pedley, J. Elliott and S. W. Gundry, Accuracy of the H2S test: A systematic review of the influence of bacterial density and sample volume, J. Water Health, 2013, 11, 173–185 CrossRef PubMed.
- A. Jang, Z. Zou, K. K. Lee, C. H. Ahn and P. L. Bishop, State-of-the-art lab chip sensors for environmental water monitoring, Meas. Sci. Technol., 2011, 22(3), 1–18 CrossRef.
- S. Kou, D. Cheng, F. Sun and I. M. Hsing, Microfluidics and microbial engineering, Lab Chip, 2016, 16, 432–446 RSC.
- R. Pol, F. Céspedes, D. Gabriel and M. Baeza, Microfluidic lab-on-a-chip platforms for environmental monitoring, TrAC, Trends Anal. Chem., 2017, 95, 62–68 CrossRef CAS.
- B. C. Dhar and N. Y. Lee, Lab-on-a-Chip Technology for Environmental Monitoring of Microorganisms, BioChip J., 2018, 12, 173–183 CrossRef CAS.
- S. G. Dastider, A. Abdullah, I. Jasim, N. S. Yuksek, M. Dweik and M. Almasri, Low concentration E. coli O157:H7 bacteria sensing using microfluidic MEMS biosensor, Rev. Sci. Instrum., 2018, 89(12), 125009 CrossRef PubMed.
- H. Tachibana, M. Saito, S. Shibuya, K. Tsuji, N. Miyagawa, K. Yamanaka and E. Tamiya, On-chip quantitative detection of pathogen genes by autonomous microfluidic PCR platform, Biosens. Bioelectron., 2015, 74, 725–730 CrossRef CAS PubMed.
- N. Bhardwaj, S. K. Bhardwaj, J. Mehta, K. H. Kim and A. Deep, MOF-bacteriophage biosensor for highly sensitive and specific detection of staphylococcus aureus, ACS Appl. Mater. Interfaces, 2017, 9, 33589–33598 CrossRef CAS PubMed.
- Ł. Richter, M. Janczuk-Richter, J. Niedziółka-Jönsson, J. Paczesny and R. Hołyst, Recent advances in bacteriophage-based methods for bacteria detection, Drug Discovery Today, 2018, 23, 448–455 CrossRef PubMed.
- I. Sorokulova, E. Olsen and V. Vodyanoy, Bacteriophage biosensors for antibiotic-resistant bacteria, Expert Rev. Med. Devices, 2014, 11, 175–186 CrossRef CAS PubMed.
- S. Niyomdecha, W. Limbut, A. Numnuam, P. Kanatharana, R. Charlermroj, N. Karoonuthaisiri and P. Thavarungkul, Phage-based capacitive biosensor for Salmonella detection, Talanta, 2018, 188, 658–664 CrossRef CAS PubMed.
- M. Tolba, M. U. Ahmed, C. Tlili, F. Eichenseher, M. J. Loessner and M. Zourob, A bacteriophage endolysin-based electrochemical impedance biosensor for the rapid detection of Listeria cells, Analyst, 2012, 137, 5749–5756 RSC.
- J. Paczesny, Ł. Richter and R. Hołyst, Recent Progress in the Detection of Bacteria Using Bacteriophages: A Review, Viruses, 2020, 12, 845 CrossRef CAS PubMed.
- D. P. Pires, S. Cleto, S. Sillankorva, J. Azeredo and T. K. Lu, Genetically Engineered Phages: a Review of Advances over the Last Decade, Microbiol. Mol. Biol. Rev., 2016, 80, 523–543 CrossRef CAS PubMed.
- S. Chibani-chennoufi, A. Bruttin, H. Brüssow, M. Dillmann and H. Bru, Phage-Host Interaction : an Ecological Perspective MINIREVIEW, J. Bacteriol., 2004, 186, 3677–3686 CrossRef CAS PubMed.
- A. P. Sagona, A. M. Grigonyte, P. R. MacDonald and A. Jaramillo, Genetically modified bacteriophages, Integr. Biol., 2016, 8, 465–474 CrossRef PubMed.
- A. E. Smartt, T. Xu, P. Jegier, J. J. Carswell, S. A. Blount, G. S. Sayler and S. Ripp, Pathogen detection using engineered bacteriophages, Anal. Bioanal. Chem., 2012, 402, 3127–3146 CrossRef CAS PubMed.
- T. C. Hinkley, S. Singh, S. Garing, A. L. M. Le Ny, K. P. Nichols, J. E. Peters, J. N. Talbert and S. R. Nugen, A phage-based assay for the rapid, quantitative, and single CFU visualization of E. coli (ECOR #13) in drinking water, Sci. Rep., 2018, 8, 1–8 CAS.
- T. C. Hinkley, S. Garing, S. Singh, A. L. M. Le Ny, K. P. Nichols, J. E. Peters, J. N. Talbert and S. R. Nugen, Reporter bacteriophage T7NLC utilizes a novel NanoLuc::CBM fusion for the ultrasensitive detection of: Escherichia coli in water, Analyst, 2018, 143, 4074–4082 RSC.
- J. Xu, Y. Chau and Y. K. Lee, Phage-based electrochemical sensors: A review, Micromachines, 2019, 10, 1–20 Search PubMed.
- L. J. Marinelli, M. Piuri, Z. Swigoňová, A. Balachandran, L. M. Oldfield, J. C. van Kessel and G. F. Hatfull, BRED: A simple and powerful tool for constructing mutant and recombinant bacteriophage genomes, PLoS One, 2008, 3(12), 1–8 CrossRef PubMed.
- B. W. McLean, M. R. Bray, A. B. Boraston, N. R. Gilkes, C. A. Haynes and D. G. Kilburn, Analysis of binding of the family 2a carbohydrate-binding module from Celludomonas fimi xylanase 10a to cellulose: Specificity and identification of functionally important amino acid residues, Protein Eng., 2000, 13, 801–809 CrossRef CAS PubMed.
- D. Ogończyk, J. Węgrzyn, P. Jankowski, B. Dąbrowski and P. Garstecki, Bonding of microfluidic devices fabricated in polycarbonate, Lab Chip, 2010, 10, 1324 RSC.
- P. N. Nge, C. I. Rogers and A. T. Woolley, Advances in Microfluidic Materials, Functions, Integration, and Applications, Chem. Rev., 2013, 113, 2550–2583 CrossRef CAS PubMed.
- D. Vandenheuvel, A. Singh, K. Vandersteegen, J. Klumpp, R. Lavigne and G. Van Den Mooter, Feasibility of spray drying bacteriophages into respirable powders to combat pulmonary bacterial infections, Eur. J. Pharm. Biopharm., 2013, 84, 578–582 CrossRef CAS PubMed.
- S. E. Finkel, Long-term survival during stationary phase: Evolution and the GASP phenotype, Nat. Rev. Microbiol., 2006, 4, 113–120 CrossRef CAS PubMed.
- Bacteriophages, ed. M. R. J. Clokie and A. M. Kropinski, Humana Press, Totowa, NJ, 2009, vol. 501 Search PubMed.
- WHO, WATER QUALITY AND HEALTH - REVIEW OF TURBIDITY: Information for regulators and water suppliers, 2017 Search PubMed.
- M. M. Calabretta, L. Montali, A. Lopreside, F. Fragapane, F. Iacoangeli, A. Roda, V. Bocci, M. D'Elia and E. Michelini, Ultrasensitive On-Field Luminescence Detection Using a Low-Cost Silicon Photomultiplier Device, Anal. Chem., 2021, 93, 7388–7393 CrossRef CAS PubMed.
- A. Sevastou, S. S. Tragoulias, D. P. Kalogianni and T. K. Christopoulos, Mix-and-read method for assessment of milk pasteurization using a smartphone or a common digital camera, Anal. Bioanal. Chem., 2020, 412, 5663–5669 CrossRef CAS PubMed.
- L. Montali, M. M. Calabretta, A. Lopreside, M. D'Elia, M. Guardigli and E. Michelini, Multienzyme chemiluminescent foldable biosensor for on-site detection of acetylcholinesterase inhibitors, Biosens. Bioelectron., 2020, 162, 112232 CrossRef CAS PubMed.
- M. M. Calabretta, R. Álvarez-Diduk, E. Michelini, A. Roda and A. Merkoçi, Nano-lantern on paper for smartphone-based ATP detection, Biosens. Bioelectron., 2020, 150, 111902 CrossRef CAS PubMed.
- H. Li, N. Lopes, S. Moser, G. Sayler and S. Ripp, Silicon photomultiplier (SPM) detection of low-level bioluminescence for the development of deployable whole-cell biosensors: Possibilities and limitations, Biosens. Bioelectron., 2012, 33, 299–303 CrossRef CAS PubMed.
- Y. Jung, C. Coronel-Aguilera, I.-J. Doh, H. J. Min, T. Lim, B. M. Applegate and E. Bae, Design and application of a portable luminometer for bioluminescence detection, Appl. Opt., 2020, 59, 801 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1lc00888a |
| ‡ New laboratory name: Global Health Labs, 14360 SE Eastgate Way, Bellevue, WA 98007, USA. E-mail: luis.alonzo@ghlabs.org, annelaure.leny@ghlabs.org. |
| This journal is © The Royal Society of Chemistry 2022 |