
Atomic spectrometry update: review of advances in atomic spectrometry and related techniques
E.
Hywel Evans
*a,
Jorge
Pisonero
b,
Clare M. M.
Smith
c and
Rex N.
Taylor
 d
d
aThe Open University, UK. E-mail: e.h.evans@open.ac.uk
bUniversity of Oviedo, Faculty of Science, Department of Physics, c/Federico García Lorca 18, 33006 Oviedo, Spain
cSchool of Education, College of Social Sciences, University of Glasgow, University Avenue, Glasgow G12 8QQ, UK
dOcean and Earth Science, University of Southampton, NOC, Southampton, UK SO14 3ZH
First published on 26th April 2022
Abstract
This review of 194 references covers developments in ‘Atomic Spectrometry’ published in the twelve months from November 2020 to November 2021 inclusive. It covers atomic emission, absorption, fluorescence and mass spectrometry, but excludes material on speciation and coupled techniques which is included in a separate review. It should be read in conjunction with the previous review1 and the other related reviews in the series.2–6 A critical approach to the selection of material has been adopted, with only novel developments in instrumentation, techniques and methodology being included.
1. Liquids analysis
1.1 Sample pre-treatment
One of the challenges of chip-based systems has been the problem of interfacing them with detection systems having incompatible sample introduction systems. Lackey et al.8 employed a simple interface consisting of a fused silica capillary glued into a microchip outlet using epoxy. Thus, fourteen lanthanide metal ions were separated on the microfluidic chip using isotacophoresis, and then, via a CEI-100 crosspiece to introduce makeup solution, into ICP-MS for detection. The resolution of the separation was maintained despite a 7 nL void volume at the capillary-chip junction.
Some of the challenges addressed in the forgoing reviews were addressed by Xing et al.13 They developed a method which used AuNPs modified with a thrombin aptamer, which were adsorbed onto the surface of graphene oxide (GO). In the presence of thrombin conformational changes caused desorption of the AuNPs in proportion to the amount of thrombin. Determination by spICP-MS resulted in an LOD of 4.5 fM of thrombin. Lin et al.14 used ssDNA conjugated AuNPs to label GO in order to determine the biodistribution of GO in relation to its potential use for drug delivery. The main point was to label the GO post-administration so as not to interfere with its initial biodistribution. This was possible because of the strong affinity of the ssDNA for GO. An A20R20 DNA sequence (random DNA sequence including A, T, C, and G) was found to bind most strongly to the GO. After separation of bound and unbound DNA-AuNPs, the GO-DNA-AuNP was determined using ICP-MS to achieve a method LOD of 9.3 ag L−1.
Multiplexing is another cited advantage of the elemental tagging approach. Zhang et al.15 used ssDNA probes labelled with Ag-, Au- and Pt-NPs to target three separate micro-RNA (miRNA) molecules, namely miR-486-5p, miR-221, and miR-21, in serum. These hybridised molecules were then immobilised using biotinylated capture probes on streptavidin microporous plates. Excess reagent could then be washed away and the targets released and determined using ICP-MS. LODs of 0.18 pM for miR-221, 0.23 pM for miR-486-5p, and 0.22 pM for miR-21 were achieved. Kang et al.16 developed a related but slightly different approach. They immobilised Ln labelled (with 159Tb, 165Ho, 175Lu) DNA strands on streptavidin-modified magnetic beads. Target miRNAs (miRNA-21, miRNA-155, and miRNA-10b) were hybridised with complementary MNAzymes. These activated MNAzymes then cleaved the immobilised Ln-labelled DNA strands, thus releasing the fragment with lanthanide tags which were collected after magnetic separation and analysed using ICP-MS. LODs for the three miRNAs were between 11 and 20 pmol L−1. Zhang et al.17 developed an amplification approach for the assay of the tumor biomarkers alpha-fetoprotein (AFP) and carcinoembryonic antigen (CEA). The targets were captured using biotinylated antibodies immobilised on the surface of magnetic beads. These were then bound with a secondary antibody conjugated with a rolling circular amplification (RCA) primer. Hence, signal amplification was achieved by up addition of RCA-template and DNA polymerase to produce multiple DNA sequences which were then hybridised with DNA-conjugated AuNPs for ICP-MS measurement. The method could detect AFP and CEA in 20% serum at 2 pg mL−1 and 5 pg mL−1, respectively.
Analysis of solid samples was the focus of a paper by Gao et al.18 who imaged amyloid beta peptide (Aβ) in Alzheimer’s brain tissue. They used AuNPs conjugated with antibodies to selectively bind to the Aβ sites. They then used LA-ICP-MS to quantitatively image homogenised brain slices from mice, after establishing that there was a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Anti-Aβ to Aβ (1:1), using a matrix-matched standard.
:1 ratio of Anti-Aβ to Aβ (1:1), using a matrix-matched standard.
In a slight departure from the elemental tagging approach, Chen et al.19 developed a label-free method for fluorescence and ICP-MS detection of DNA using [Ru(bpy)2dppz]2+ (bpy = 2,2′-bipyridine, dppz = dipyrido[3,2-a:2′,3′-c]phenazine) as a dual functional probe. Rather than the elemental tag being conjugated with a recognition molecule, the fluorescent [Ru(bpy)2dppz]2+ intercalates into the groove of double stranded (ds) DNA where it is not quenched by H2O molecules, leading to 100× fluorescence enhancement. Thus, it could be used as a universal dsDNA probe with no DNA sequence modification needed. The probe was added after initial amplification of hepatitis B virus (HBV) DNA using hyperbranched rolling circle amplification (HRCA). Thus, fluorescence detection was achieved directly with an LOD of 9.6 amol L−1. Also, a biotin-modified primer in HRCA allowed the dsDNA-[Ru(bpy)2dppz]2+ complexes to be subsequently captured in avidin-coated well plates, desorbed and determined using ICP-MS with an LOD of 1 amol L−1. Another label-free method was reported by Wang et al.20 to determine terminal deoxynucleotidyl transferase (TdT), polynucleotide kinase (PNK), H2O2, and mucin1 using Cu2+, Ag+, and Cd2+ as signal probes. Initially, ICP-MS was used for detection, however, a novel development was the replacement of the aforementioned signal probes with Cd QDs and then subsequently Cd2+ to allow CVG-AFS detection. The exact experimental protocol is difficult to elucidate from the paper, however.
1.2 Sample introduction
A new sample introduction method using aerosol dilution for MIP-OES was described.22 Signal emissions from N2+ and OH species were monitored for plasma diagnostics and solvent plasma load. N2+:OH signal intensity ratio measurements suggested that aerosol dilution was a desolvation process which could reduce the solvent load, thus increasing plasma energy. The efficiency of the system was tested using solutions containing Na (an EIE) and the plasma physics were found to be stable up to Na concentrations of 5 g L−1. The LOQs of Cr, Mn, V and Zn elements were found to be similar to those obtained using conventional nebulisation with lower analyte masses introduced to the MIP. The analysis of two CRMs (fertilizer, NIST 695 and tomato leaves, NIST 1573a) resulted in percentage recoveries between 90 and 130%.
The analysis of NPs (<10 nm) using ICP-MS requires low instrumental background and high detection efficiency. Kocic et al.23 evaluated the performance of a sector-field ICP-MS with standard and enhanced sensitivity (‘Jet’) vacuum interfaces combined with three different sample introduction setups: conventional pneumatic nebulisation (PN), desolvation nebulisation (DSN) and microdroplet generation (MDG). The effect of adding N2 gas to the dry aerosol was also studied. AuNP suspensions were analysed and evaluated for particle number concentration and size. Applying counting statistics, the size LOD of AuNPs was estimated to be 6.1 nm and 4.7 nm for PN and MDG with the standard interface, and 3.6 nm and 3.1 nm for DSN and MDG with the ‘Jet’ interface and N2 addition, respectively. Detection efficiencies obtained for the conventional PN were in the range of 10−4 to 10−2 counts per atom (low resolution) and 10−6 to 10−5 counts per atom (medium resolution). A significant improvement was obtained for the MDG setup with the ‘Jet’ interface and N2 addition, resulting in the range of 10−2 to 10−1 counts per atom (low resolution) and 10−4 to 10−3 counts per atom (medium resolution). Enhancement in detection efficiency was most pronounced for isotopes of lower m/z indicating reduced mass discrimination of the ‘Jet’ interface with N2 gas added to the sample aerosol. The corresponding LOD could thus be decreased by 10 or 2 times for example for Al- and Au-containing NPs, respectively. At the same time the use of an MDG for sample introduction resulted in 98.5% transport efficiency in the analyses of NP suspensions, whereas this was only 10% (PN) or 23% (DSN) with pneumatic nebulisers.
The transport efficiency of the ICP-MS is crucial for quantification of NPs using sp-ICP-MS. This usually requires SRMs with homogenous size distribution and a known particle number concentration. Currently, these SRMs are only available in limited sizes for a few metals. The determination of the loss of NPs and/or ions during their transport from the sample solution to the detection system, quantified as nebulisation (or transport) efficiency, can be time-consuming, costly and unreliable. Nebulisation of the NPs directly into the plasma (without a spray chamber) resulted in a transport efficiency of 100% and could be a promising strategy to avoid these calibration steps. This approach was investigated using a μ-dDIHEN introduction system, consisting of a demountable direct injection high-efficiency nebuliser (dDIHEN) hyphenated to a flow-injection valve and a gas displacement pump.25 With a continuous flow nebuliser, 100% transport efficiency was reported. Operated at low uptake rates (around 8 μL min−1), the average diameters of Au-, Ag- and Pt-NPs were determined in agreement with reference values. The approach was also tested for AuNPs in complex matrices, such as surface waters. sp-ICP-MS analyses with the μ-dDIHEN sample introduction system could be achieved using dissolved standard calibration to determine the average diameter and particle number concentration of NPs. In another approach, a dual-inlet setup for characterising NPs with sp-ICP-MS was developed to address the calibration issues.26 The system was based on a conventional introduction system consisting of a PN for NP solutions and a MDG for ionic calibration solutions. A new interface was developed to allow the coupling of MDG, PN and the ICP-MS system. Three independent analysis modes, based on different calibration principles, were used for determining particle size and number concentration. Mode I (counting) and mode III (MDG) had been used previously but mode II (sensitivity), used to determine the TE for inorganic ionic standard solutions only, provided a new approach to calibration that was independent of NP SRMs. The MDG based inlet system permitted improved analyte sensitivities and, therefore, lower LODs. The size dependent LODs achieved were found to be less than 15 nm for all NPs (Au, Ag, CeO2) investigated.
Double-viewing-position sp-ICP-OES was used to investigate the effects of temperature gradient, in the ICP central channel, on signal intensity.27 Correlation plots of the intensities of individual Yb2O3 particles at observation positions of 8.5 and 19.5 mm above the load coil showed extensive scattering. This lack of correlation was assigned to the relatively large gradient of ICP gas temperature in the radial direction at the lower observation position. This hypothesis was supported by the results of sp-ICP-OES measurements using a sheath gas device to confine the particles in either the centre or the outer region of the ICP central channel. Particles in the two regions resulted in distinct patterns in the correlation plot. Computer simulation showed that the heat required to bring the Yb2O3 particles to boiling was substantial and that the duration of the heat transfer process was not negligible and increased with particle mass. As a result, large particles had a relatively small degree of vaporisation at low observation positions where sp-ICP-OES intensity was not proportional to particle mass. The calibration curve was found to be concave in shape resulting in inaccurate particle size determination at low observation positions. A simulation of particle vaporisation with a wide range of particle sizes and boiling points was presented alongside a quick lookup graph of observation position (for a degree of vaporisation of 70%) versus particle diameter, allowing more accurate determinations at low observation positions. The influence of ICP-MS instrumental parameters (nebuliser gas flow, rf power and sampling depth) on the signal intensity of Au in sp-ICP-MS was evaluated using dispersions of AuNPs and a solution of dissolved Au.28 The interaction effects of the main instrumental factors were found to have a significant effect on the signal intensity, demonstrating that factor values should be jointly optimised instead of one at a time, in order to obtain maximum ion signal. Optimisation of instrumental parameter values was performed for both analyte forms and found to be in a good agreement, indicating similar behaviour of the particles in the plasma compared with the dissolved analyte. Some differences in the behaviour of the two analyte forms with respect to sampling depth position was observed, however. Neither particle size nor the presence of a complex sample matrix were found to influence the optimal instrumental parameter values, however, significant signal suppression (up to 50%) for Au was observed in matrices containing high levels of Na. Compared to frequently used ‘robust conditions’, a 70% increase in the ion signal intensity of Au and a 15% decrease in the particle size LOD was achieved with instrumental parameter optimisation.
New methods for the characterisation of dispersions containing small-diameter or low-mass-fraction NPs by sp-ICP-MS were reported.29 The optimisation of ion extraction, ion transport, and the operation of the quadrupole with increased mass bandwidth improved SNR significantly and decreased the size LOD for all NP dispersions investigated. As a model system, 10.9 ± 1.0 nm AuNPs were analysed to demonstrate the effects of increasing ion transmission. Increasing the mass bandwidth of the quadrupole improved the size LOD to 4.2 nm and enabled the resolution of NP signals from ionic background and noise. The methods were also used for the characterisation of La-doped upconversion NPs by sp-ICP-MS. Three different types (90 nm NaYF4: 20% Yb, 2% Er; 20 nm NaGdF4: 20% Yb, 1% Er; and 15 nm NaYF4: 20% Yb, 2% Er) were investigated. Y showed the best SNR with optimised ion extraction and transport parameters. The SNR of Gd, Er, and Yb were further improved by increasing the mass bandwidth of the quadrupole mass filter. The time scan measured when a dispersion of NPs is analysed by sp-ICP-MS is typically made of many intense short-lived spikes. On the basis of theoretical models, experimental data and statistical tests, it was shown that, at the very dilute limit, there was a one-to-one correspondence between the spikes present in a time scan and the points of discontinuity of the outcome of a homogeneous Poisson process.30 This relationship underlies the random nature of the sp-ICP-MS time scan. The stochastic processes describing the NP arrivals in the plasma and the time sequence of spikes in the time scan were found to be identical, providing confirmatory evidence for a hypothesis regarding the Poissonian character of the sp-ICP-MS time scan.
The characterisation of individual metallic NPs by sp-ICP-MS permits the analysis of small biological systems such as individual cells. Developments in single cell (SC) ICP-MS have increased significantly of late as a result of work on the introduction of cell suspensions to the ionisation source by developing nebuliser/spray chamber systems with high transport efficiencies. These developments have been accompanied with the commercial availability of ICP-MS instruments allowing better control of spectral interferences and TOF instruments with improved sensitivity. Recent developments in sample introduction systems for liquid cell suspensions were summarised in a review.31
The DBD has been most investigated because it can be used both for PIVG and as a trap for volatile species. He et al.33 used a nebulised film DBD for the generation of volatile species of Au, Ir, Pd, Pt and Rh with detection using ICP-MS. In their design, the liquid sample was nebulised into a cylindrical DBD, in which a plasma was maintained. The sample introduction efficiency, when using formic acid, was between 5- and 11-fold greater compared to conventional nebulisation. LODs for Au, Ir, Pd, Pt and Rh were between 0.1 and 0.7 ng L−1, and unaffected by the presence of a mixture of matrix ions up to approximately 1000 mg L−1 in total. In contrast, when a DBD is used as a trap it must be coupled with an additional method of VG. Liu et al.34 achieved this by using a traditionally configured system coupled to a DBD for VG of Pb and AFS detection. The PbH4 generated was trapped on the DBD quartz surface in 50 mL min−1 air with a 28 kHz, 9 kV discharge; and released in 110 mL min−1 H2 with a 12 kV discharge. Trapping enhancement was estimated to be 8-fold, resulting in an LOD of 8 pg for Pb (2 mL sample). Similarly, Zhang et al.35 developed a miniaturised HG-OES instrument coupled with an in situ DBD trap for the determination of As in seaweed. Arsine was generated and trapped in the DBD in an O2-containing atmosphere with a 1.35 mA discharge, then released in an Ar–H2 atmosphere. An MDL of 0.25 mg kg−1 in seaweed samples was achieved, the main advantage being the low power (<60 W), small instrument size (0.6 m × 0.5 m × 0.3 m), reasonable weight (15 kg), and low gas consumption (300 measurements for 4 L compressed Ar–H2 gas). However, it was still necessary to prepare the samples by microwave digestion so this is very much part of a mobile laboratory.
Photochemical vapour generation (PVG) is similar to PIVG in that some of the same mechanisms of volatile species generation may occur in both. Hu et al.36 investigated PVG of Br, Cl and F using different Cu salts to facilitate the generation of volatile species using a 15 W UV lamp. They used UV-vis, GC-MS and AFS to identify 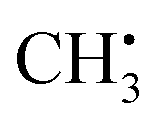 , OH˙, and Cu+ as primary reaction intermediates when CuAc2 (100 to 200 μg mL−1) was used to facilitate the formation of methyl halides. Ligand-to-metal charge transfer between acetate and Cu, then subsequent charge transfer excitation of halides was thought to account for the generation of methyl halides in organic-acid-free media. LODs of 0.03 and 3 μg L−1 for Br− and Cl− respectively were achieved using ICP-MS detection. Very similar conclusions were drawn by de Oliveira et al.37 using 2% acetic acid and 10 mg L−1 Cu2+ to generate CH3Br from Br− and BrO3−, using both 19 W (185 nm) and 15 W (254 nm) UV lamps. Volatile Br species were generated from both Br− and BrO3− at 185 nm but only from Br− at 254 nm, making Br speciation possible if both lamps were used in parallel. They found that Cu2+ ions enhanced the Br signal by 230-fold resulting in LODs of 0.01 and 0.04 ng mL−1 in the 19 W and 15 W photoreactors, respectively, using ICP-MS detection. They postulated that Cu2+ acted as a photocatalyst, with ˙CH3 and Br˙ free radical atom transfer from a copper bromide complex accounting for the high PVG efficiency. This research highlights the fact that the efficiency of PVG is affected by the addition of transition metal ions and organic compounds which was highlighted in three further studies. Re PVG was enhanced by a combination of Cd2+, Co2+, formic acid and acetic acid;38 Os with acetic acid;39 and I with ethanol.40
, OH˙, and Cu+ as primary reaction intermediates when CuAc2 (100 to 200 μg mL−1) was used to facilitate the formation of methyl halides. Ligand-to-metal charge transfer between acetate and Cu, then subsequent charge transfer excitation of halides was thought to account for the generation of methyl halides in organic-acid-free media. LODs of 0.03 and 3 μg L−1 for Br− and Cl− respectively were achieved using ICP-MS detection. Very similar conclusions were drawn by de Oliveira et al.37 using 2% acetic acid and 10 mg L−1 Cu2+ to generate CH3Br from Br− and BrO3−, using both 19 W (185 nm) and 15 W (254 nm) UV lamps. Volatile Br species were generated from both Br− and BrO3− at 185 nm but only from Br− at 254 nm, making Br speciation possible if both lamps were used in parallel. They found that Cu2+ ions enhanced the Br signal by 230-fold resulting in LODs of 0.01 and 0.04 ng mL−1 in the 19 W and 15 W photoreactors, respectively, using ICP-MS detection. They postulated that Cu2+ acted as a photocatalyst, with ˙CH3 and Br˙ free radical atom transfer from a copper bromide complex accounting for the high PVG efficiency. This research highlights the fact that the efficiency of PVG is affected by the addition of transition metal ions and organic compounds which was highlighted in three further studies. Re PVG was enhanced by a combination of Cd2+, Co2+, formic acid and acetic acid;38 Os with acetic acid;39 and I with ethanol.40
A different focus was provided by Yu et al.,41 who investigated the gas–liquid interface by using a segmented flow system. They found no difference in the PVG of As, Sn, Ir, Os, Cd, Ni, Hg, and Mo regardless of whether air or Ar was used before/after the solution slug. From this they somewhat speculatively excluded the influence of O2 on photo-generated O3. However, they did identify the volatile species (CH3)3As, Mo(CO)6 and (CH3)3Sb by GC-MS. Further experiments using EPR identified the presence of the free radical ˙CO2−. They postulated that these radicals, arising from deep UV photolysis of formic acid in the sample medium, promoted reduction of Mo to lower valence states, thus enhancing the PVG of Mo. However, while the CO2–˙CO2− couple was capable of reducing AsIII and SbIII to As0 and Sb0, they found that e(aq)− and ˙H generated could possibly play a greater role, together with ˙CH3 generation of As(CH3)3 and Sb(CH3)3. It was not always clear how the conclusions were arrived at, but the paper is worth careful analysis by other workers in the field.
1.3 Direct methods
The LS-AP-GD has the inherent capability to operate as a combined atomic and molecular (CAM) ionisation source. The plasma is sustained by placement of a high voltage (∼500 V, dc) onto an electrolytic solution through which the analyte is generally delivered to the discharge. Solvent selection provides the means of obtaining atomic/elemental and/or molecular mass spectra. Marcus et al.46 provided an overview of the varied modes of sample introduction and MS platforms to which the LS-AP-GD has been interfaced. Representative spectra and analytical figures of merit for elemental and IR measurements were presented, in addition to descriptions of the application to small organic molecules, organometallic complexes, and intact proteins. There is evidence to suggest that the diversity of analytical applications and ready implementation across MS platforms provided a versatility beyond that of other ionisation sources. In other work, the LS-AP-GD was operated as a CAM ionisation source for the analysis of elemental species, small polar molecules and proteins.47 Operation using 2% HNO3 as the electrolytic carrier solution was used for elemental analysis, while changing to a MeOH–H2O solution resulted in mass spectra indicative of the protonation of polar molecular species. The determination of low-polarity PAHs was investigated to demonstrate the versatility of the LS-AP-GD system. These molecules contain none of the common polar sites for protonation, typically undergoing ionisation via charge transfer or electron ionisation, forming radical cations. While LS-AP-GD ionisation is believed to involve both electron and Penning ionisation, as well as solvent-derived gas phase proton transfer reactions, it was expected that a radical cation formation would be the sole product for PAHs. In this study, both radical cation and protonated molecular ions were observed for each of the PAHs studied, suggesting concurrent electron/Penning ionisation and atmospheric pressure chemical ionisation. LODs of 270 pg mL−1 (5.4 pg, absolute) were obtained, demonstrating the ability of LS-AP-GD for this useful application.
2 Solids analysis
2.1 Direct methods
2.2 Indirect methods
The changes in aerosol physical properties were studied in NE-LA-ICP-MS and compared with those observed in conventional LA-ICP-MS.54 Analyte signal enhancement was found to be related to the particle number concentration of the aerosol for three sizes of AuNPs applied as droplets on the metal sample surface. The dependence of the number of generated particles on the laser fluence in the range from 0.5 to 10 J cm−2 was also studied and a different shape of particle size distribution was observed for the two modes of sample preparation. The aerosol structure was studied on filters using SEM. Compared with LA-ICP-MS, NE-LA-ICP-MS produced a larger proportion of small particles (<30 nm), approaching an optimal monodisperse aerosol introduced to the ICP. The measurements demonstrated that NPs on the sample surface could influence the evaporation, condensation and coagulation processes in aerosol formation in addition to the analyte signal in ICP-MS.
Elemental imaging by LA-ICP-MS has advanced rapidly in recent years, due to hardware development associated with fast aerosol transport technologies and a deeper understanding of the influence of operational parameters on the image quality. The effect of dosage, i.e., the number of laser pulses per pixel, on the image quality attainable by LA-ICP-QMS when mapping a biological (murine brain tissue) and a mineralogical (asbestos fibers) sample was investigated by Šala et al.55 Increasing the dosage resulted in better SNR and was crucial when elements were present at lower concentration levels, or if mapping of more than one element is required. Although this increased the mapping time, the quality of the mapping images was improved. The influence of sample surface topography on the LA process and, in turn, the analytical response of the LA-ICP-MS was studied by Salajkova et al.56 Six different surface topographies were prepared on an Al alloy sample BAM 311 and SRM NIST 610 to investigate the phenomenon. The samples were repetitively measured by LA-ICP-MS using a spot by spot analysis and the effect of laser fluence in the range of 1 to 13 J cm−2 was studied. For the majority of measured isotopes, the ICP-MS signal was amplified by roughening the sample surface. The biggest effect was observed on the Al alloy sample, with a 60× enhancement of signal compared with ablating the polished surface of the sample. This significant increase in sensitivity can suppress matrix effects. Since the effect of surface topography is different for each analyte, it was concluded that surface properties do not only affect the ICP-MS response, but also elemental fractionation in LA. These results demonstrated that varied surface topographies may lead to misleading data interpretation because even when applying ablation preshots, the signal obtained for individual elements changes.
Characterisation and investigation into complex nano-fraction particle samples have traditionally been carried out using nebuliser sp-ICP-MS. Holbrook et al.57 proposed a method for the direct analysis of multi-elemental particles in sediment samples using LA-sp-ICP-TOF-MS. A comparison between nebuliser sp-ICP-TOF-MS and LA-sp-ICP-TOF-MS was made using standard AgAu core–shell particles and environmental samples obtained via cloud point extraction procedures of a road runoff sedimentation basin. Using the results obtained, comparison metrics were then used to test the applicability of LA-sp-ICP-TOF-MS for analysis of unextracted sediment. Three main groups of signals were identified, overly abundant signals which could not be used for single-particle analysis at the chosen measurement parameters, highly abundant signals that when compared with the two previous methods produced comparable results for elemental ratios and single-particle fingerprinting. Lastly, low abundant well-defined elements such as the PGEs were found to be well suited for measurement from unextracted sediment.
Insufficient sampling of periodic signals results in an unwanted phenomenon known as ‘aliasing’. When using LA-ICP-MS it is observed that aliasing between laser pulse rates and sampling by sequential ICP-MS instruments creates erroneous variations in the measured element concentrations in the sample. Smoothing the sample flow to the ICP-MS can largely eliminate this variation but reduces spatial resolution of the time-resolved signal and is thus detrimental for imaging with fast response sample cells. A fire control circuit that fires the laser in alignment with the measurement cycle of the mass spectrometer to lessen or eliminate aliasing was described by Norris et al.58 Using the fire control circuit with a conventional ICP-QMS, the technique was able to maintain measurement precision when the extent of mixing between individual laser pulses was reduced by an order of magnitude. For high-speed and high-resolution elemental LA-ICP-QMS mapping, it is important to select the optimal operational conditions to avoid aliasing, minimise blur and maximise the SNR. Fine-tuning of operating conditions was achieved using the principle of divisors.59 This involved finding instrumental conditions to satisfy the anti-aliasing criterion, i.e., repetition rate x acquisition time = Z+. The mathematics involved was developed as an app (https://laicpms-apps.ki.si/webapps/home/). The app generates a list of optimised conditions from which the user can select the optimal set, based on the requirements for single or multiple pulse LA-ICP-QMS mapping of elements in ‘thick’ samples.
Analytical figures of merit for a low-dispersion aerosol transport system for high-throughput bulk and spatially resolved analysis via LA-ICP-MS were reported by van Acker et al.60 The device maximised the collection of aerosol particles generated during the ablation process, improving transport efficiency and minimising aerosol dispersion during transport of entrained particles into the ICP torch injector. The compression of the aerosol particles in an optimised space and time window resulted in an increase of the mass flux into the ICP, accompanied by an enhancement in sensitivity and throughput. In combination with a low-dispersion ablation cell housed in a LA-unit equipped with a nanosecond 1 kHz lasing system, a highly linear response of the integrated 238U+ signal intensity was observed upon ablation of a NIST SRM 610 glass as a function of the laser repetition rate up to 1 kHz. Single pulse responses with a full peak width at 50%, 10% and 1% of the maximum peak height of only 0.3 ± 0.1 (FWHM), 0.5 ± 0.1 (FW0.1M) and 1.2 ± 0.1 ms (FW0.01M) were achieved. The peak profiles closely resemble the profiles generated in sp-ICP-MS for individual metallic NPs and microplastics.
Low analytical throughput and lack of commercial SRMs are cited as the main obstacles to the successful analysis of metallic NPs by LA-ICP-MS. Zheng et al.61 proposed an approach termed ‘single-cell isotope dilution analysis’ (SCIDA) to overcome these obstacles. For proof of concept, macrophage cells were chosen as a model to study the uptake of AgNPs at the single-cell level. Single cells exposed to AgNPs were placed in an array by a microfluidic technique; each cell in the array was precisely dispensed with a known picoliter droplet of an enriched isotope solution using a commercial inkjet printer; accurate quantification of AgNPs in single cells was obtained using isotope dilution LA-ICP-MS. The average Ag mass of 1100 single cells, 396 ± 219 fg Ag per cell, was in good agreement with the average of the population of cells determined by solution ICP-MS analysis. The LOD was 0.2 fg Ag per cell.
3. Instrumentation and fundamentals
3.1 Instrumentation
Balaram63 reviewed (177 references) the development of MIP-OES over the period from 2012 to 2020. The review mainly covered applications of the technique for both liquid, vapour and gaseous sample introduction. Niu et al.64 reviewed the use of DBDs as sources in analytical atomic spectrometry (124 references). The different types of DBD (planar, cylindrical, surface and capillary) and the mechanism of generation of the plasma were described. Applications in OES, AAS and AFS were covered and also the use of the DBD as a vapour generation source. They concluded that the direct analysis of samples with a complex matrix is the next challenge – a common enough problem with miniaturised sources. Such impediments to miniaturisation are well documented in a review by Buckley et al.65 They examined the requirements for sources, sample introduction systems and spectrometers necessary to make a truly portable instrument, with particular focus on plasma sources and their attendant power requirements. They concluded that some of ‘cold’ plasma sources are suitable for use with portable OES but there are still some hurdles to overcome to make them energetic enough for elemental analysis.The types of sources used in analytical atomic spectrometry have not fundamentally changed since the introduction of plasmas, be they induced in an electromagnetic field or discharges. However, new variations still appear, often in the development of a miniaturised instrument. One such example is the cross double point discharge developed by He et al.66 wherein the discharge was confined between four (rather than two as in a regular point discharge) tungsten electrodes which protruded radially into a quartz tube. A micro-plasma was generated in Ar with a 70 V AC current, and had higher Trot, Texc and ne than a single point discharge. Sample introduction was via HG resulting in LODs of 2.4, 0.15 and 1.9 μg L−1 for As, Hg and Pb respectively. A single point discharge was coupled with tungsten-coil ETV sample introduction by Deng et al.67 In order to minimise sample losses the tungsten-coil was integrated into the quartz discharge tube (10 mm i.d., 12 mm o.d., 65 mm in length), within 12.5 mm of the discharge. Additional modules included power supplies for the discharge (HV) and tungsten-coil, a fibre-optic CCD spectrometer and supply of He gas. So, while portable, the instrument itself could not be said to be miniature. LODs ranged between 0.08 μg L−1 for Cd to 45 μg L−1 for As in a 10 μL sample. A similar set-up was described by Liu et al.,68 except that a DBD was used as the source and an activated carbon electrode tip doubled as both the sample collection device and the DBD inner electrode. Metals were preconcentrated from water samples onto the activated carbon tip, dried for 5 min, then fast released to produce OES during the DBD excitation process. LODs of 0.03 and 0.6 μg L−1 for 50 mL samples were obtained for Cd and Pb, respectively.
Another example of a miniaturised source is a helium flexible micro-tube plasma (FμTP) reported by Gazeli et al.69 A quartz dielectric capillary was placed axially around a 100 μm diameter tungsten electrode. A square wave voltage 1.7 kV at 20 kHz was applied and the plasma generated in a He flow of 50 mL min−1. The paper focused on the mechanisms of formation and propagation of the plasma during the positive and negative cycles of the applied voltage (50 μs full cycle). The authors found that plasma evolution occurred in the same direction during the positive and negative cycles, implying that soft ionisation could occur throughout. The plasma propagated faster during the negative half cycle because neutralisation of positive charges by the high electron concentration left from the positive half cycle occurred very fast, moving the plasma forward (from the electrode end towards the exit) at a very high speed compared to the positive half cycle. The authors noted that this may depend on the radius of the capillary, with a decrease in speed for larger radius capillaries, resulting in lower charge density.
Low power, reduced-pressure sources have been used for OES and MS for a number of years. Farcy et al.70 characterised just such an MIP source for potential use in spaceflight applications. Langmuir probe experiments were conducted to characterise the plasma and results obtained using the Saha equation were used to predict ionisation efficiencies for organic molecules and elements with high IPs. However, given that such plasmas are far from thermodynamic equilibrium it is questionable whether Langmuir probe measurements and the Saha equation are entirely applicable.
Well established sources can also be modified to improve performance. Grindlay et al.71 evaluated the performance of a conical torch for axial ICP-OES when introducing methanol and 1-propanol solutions. The conical torch differed from the Fassel torch in that it had a flared end and only carrier and outer gas flows. A plasma could be formed with 4× higher power density while consuming 50 to 70% less Ar and up to 800 W less power, compared to a Fassel torch. Operation of the ICP in the presence of organic solvents has always been a challenge, though less so since improvements in RF coupling. However, the conical torch was able to accept 100% 1-propanol or methanol when operated at 900 W and a plasma gas flow of 7 L min−1, without either needing to cool the spray chamber nor with apparent instability or ignition problems. Figures of merit were not any better than when using a Fassel torch because spectral interferences were still present, but purely based on the operating stability it would be worth evaluating for anyone working with LC-ICP methods or in the petroleum industry.
Developments in spectrometer design often occur to accommodate a modification to another part of the instrument that seems obvious once it has been attempted. Vonderach et al.72 turned an ICP-MS instrument through 90° to facilitate a downward pointing sample introduction system. The logic being that gravity now became an advantage as larger droplets no longer settled out in the spray chamber, so aerosol transport was no longer size-dependent. Droplets of up to 70 μm initial diameter were transported into the plasma, thereby increasing sample transport and improving LODs, provided that a desolvation device was used. Craig et al.73 developed a pre-cell mass analyser, comprising two Wien filters and a selection aperture and a hexapole collision/reaction cell, for use with MC-ICP-MS. The performance of the instrument, named ‘Vienna’, was unaffected and Ar-based interferences were eliminated. The abundance sensitivity at 237.05 m/z was 17 ± 3 ppb, which is equivalent to TIMS.
3.2 Fundamentals
800 ± 630 K.
Li et al.77 used step-wise resonance laser ionisation to study the photoionisation spectra of Se. Using the Rydberg series 4s24p3(4S)np3P2 with n = 15–49, the IP of Se was determined to be 76658.15(2)stat(4)sys cm−1. The authors state that this was different from the currently accepted value of 78658.35(12) cm−1 but agreed with the value of 78658.12(10) cm−1 measured by other workers.
3.2.2.1 Plasmas. Jacobs and Houk78determined collision cross sections of 89Y+, 95Mo+, 98Mo+, 89Y16O+, 107Ag+ and 109Ag+ ions in the collision cell of an ICP-MS. These were estimated from ion-stopping curves generated by making the quadrupole bias voltage progressively more positive, at He flow rates of 0, 0.5, 1, 2.5 and 5 mL min−1. Collision cross sections of ∼10−15 cm2 were determined and followed the periodic trend in atomic radius. Values for Mo+ and Ag+ were 2× to 3× smaller than other published data. They also determined the degree of kinetic energy loss caused by the addition of He into a collision reaction interface (CRI) using ion-stopping curves. They made several assumptions (probably the most significant being that collisions took place within ∼2 mm of the skimmer) and hence calculated the gas density (using the ideal gas law) in the collision region to be between ∼2.2 × 1015 cm−3 and ∼9.0 × 1015 cm−3 with increasing He flow rate from 30 mL min−1 to 100 mL min−1. They concluded that this allowed for adequate collisions to occur in the CRI while still allowing ions from the plasma to pass into the mass spectrometer. They also made the point there may be merit in redesigning the first extraction lens, and possibly the skimmer, in order to optimise ion extraction from the CRI.
Nurubeyli79 calculated the effect of ne on the degree of ion signal suppression in ICP-MS and compared this to experimental results. They seemed to conclude that ionisation suppression is more prevalent when there is high ne, caused by recombination of free electrons with Ar+ and analyte ions. This is analogous to the case in flames, necessitating the addition of an EIE to sample solutions and standards for matrix matching in FAES.
Sánchez et al.80 studied spatial ion profiles in ethanolic samples in ICP-MS with a total sample consumption system (hTISIS). Various combinations of injector ID, hTISIS temperature and extraction lens voltage were studied; the outcome being that a combination of conditions could be found where matrix suppression caused by ethanol could be minimised, often at the expense of sensitivity. When the hTISIS was set at 300 °C, a 2.5 mm i.d. injector and 5 V lens voltage resulted in lowest matrix effects. When a narrower injector was used (∼1.0 mm) there was narrower radial and axial plasma sampling zone compared to a 2.5 mm injector, so optimisation was more critical.
Modelling is a useful approach used to gain rapid insight into analytical plasmas. Javid et al.81 developed a computational fluid dynamics model of the ICP torch using energy and Saha equations. The model was incorporated into a ‘torch simulator’, run in MATLAB, which required inputs of gas and sample uptake flow rates, power, injector size, element concentration, and spray chamber efficiency as input parameters. It was then used to predict axial Texc, ne, nion, αion and Mg(II):Mg(I) intensity ratio. Good agreement between the simulator, more complex CFD models and experimental data was observed.
Chai and Kwon82 used a collisional-radiative (CR) model to measure Te and ne in a He ICP. They used a non-Maxwellian electron energy distribution and determined Tgas using N2+ rotational spectra after addition of a small amount of N2 gas. The model included electron impact excitation/de-excitation and ionisation, radiative decay with radiation trapping, heavy particle collisional ionisation between two excited atoms, and metastable diffusion. Results were compared to those obtained using a Langmuir probe, with broad agreement in trends and absolute values found for He ICPs with Te between 2 and 5 eV and ne between 108 and 1010 cm−3, gas pressure from 200 to 800 mTorr and Tgas = 500 K. Horita et al.83 also used a CR model to describe nexc as a function of ne and Te in an Ar helicon plasma. They used pairs of nexc contours, using two excitation energy levels, drawn on an ne–Te plane, to determine ne and Te from the intersection point of two contours whose values were taken from the experimental intensities. The model agreed well with results obtained using a probe method.
Zhu et al.84 characterised the fundamental parameters of an LEP by determining Texc, Trot, αion and ne. The LEP under investigation was operated in pulsed mode so a gated detector was synchronised with the LEP pulse to enable temporal resolution. They found that a long off-time interval of at least 64 ms (preferably ≥ 150 ms) between successive discharge pulses was necessary to obtain a reproducible pulse-to-pulse discharge-current. This also resulted in a higher active discharge power and a more robust plasma (based on MgII and MgI intensity measurements). The authors ascribed this to the detrimental effect of the sample solution after exposure to the LEP, such that a long off-time and high solution flow rate were required to flush out used sample solution. Hence, solution-flowing mode was preferred over static mode. The discharge current was observed to initially spike, when no analyte or background H and OH emission was observed, followed by a steady active phase when analyte emission and ionisation were observed to correlate with the current. It took between 0.7 and 0.9 ms for analyte emission to reach a maximum, thus favouring long pulse duration.
Gonzalez-Fernandez et al.85 compared experimentally measured E-field distributions with theoretical predictions of two classical models in a H GD. The E-field distributions were determined using Stark shifting and splitting of the 2S level of H, with optogalvanic detection. The length of the cathode fall region, determined using the ‘Rickards’ model, was systematically slightly shorter due to limitation of the model near the cathode region. Average ion mean free paths and energy in the cathode fall region determined using the ‘Wroński’ model were in good agreement with other workers and near ideal gas conditions. The results showed that ions originated at the cathode with high energy which was lost collisionally during transit to the centre of the discharge. Wagatsuma86 investigated ionisation and excitation of Pd in He, Ar, Ne and Kr GDs. The Pd II emission intensity correlated with the excitation energy of the plasma gases, with no intense emission observed in the Kr GD. This was attributed to an asymmetric charge transfer collision with the plasma gas ion. The excitation scheme for the Pd II lines involved the triplet metastable levels of the (4s95s 3D3,2,1), rather than the singlet ground-state level (4s10 1S0) of other elements.
Miniature DBDs have received some attention as soft ionisation sources for MS and ion mobility spectrometry. Shi et al.87 characterised an atmospheric pressure He DBD using radially resolved OES maps of He I, N2+, N2, He2, O and I species. The maps indicated that energy transfer occurred mainly via Penning ionisation of N2 with He metastable species, to produce N2+, as the plasma plume exited the capillary into open air. Charge transfer with He2+ was dominant further downstream and near the periphery of the plume. When the DBD plume was directed onto a sample surface, this had an effect on the energy transfer mechanisms. For LDPE the spatial distribution of excited species was similar but disappeared into the LDPE surface. In comparison, for a floating Cu surface there was a peak in intensity at the surface for most species. The authors proposed that higher Te and ne near the surface of Cu favoured electron impact excitation. Tian et al.88 also characterised a miniature DBD. A key finding was that a dissociative plasma did not form, unlike a DBD used for OES, thus making it suitable for soft ionisation. Badal89 used OES and MS to characterise a mixed-gas d.c. GD afterglow (FAPA) as a potential ambient desorption/ionisation source. Addition of small amounts of O2 to the He-FAPA caused an increase in the protonated water cluster ion signal and analyte-ion signals for polar analytes such as acetone and methanol. Addition of H2 produced a cleaner analyte mass spectrum with reduced total ion signal. N2 addition caused an increase in the NO3− ion signal. Spatially resolved OES observations of the anode- and negative-glow regions revealed a decrease in He and OH emission intensities on addition of molecular gases (∼0.1%). This suggested the quenching of metastable He atoms which are mainly responsible for the formation of reagent ions in the afterglow.
There has been considerable research into spICP-MS over the last few years, with some useful insights into particle atomisation and ionisation processes in the plasma. Chan and Hieftje90 further contributed to this with a study of the effect of hydrogen isotopes on plasma impedance and thermal pinching induced by single aerosol droplets. Microdroplets (50 μm diameter) were introduced vertically into an Ar ICP and two-dimensional images recorded using monochromatic imaging spectrometer at selected wavelengths. Separate introduction of H2O and D2O droplets was performed and images were obtained for Ba II at 455.4 nm, H I at 656.3 nm. D I at 656.1 nm and Ar I at 696.5 nm. The local cooling effect caused by a vapourising aerosol droplet was found to be similar for H2O and D2O. However, D2O droplets induced significantly less plasma shrinkage (between 52% and to 80%) and a smaller shift in plasma impedance compared to H2O. The authors noted that the thermal conductivity of D is about 70% of H, which is believed to play an important role in the thermal-pinch effect in the plasma. A further finding was that the decay in these effects was similar for H2O and D2O, taking ∼10 ms for plasma impedance to return to a steady state. Chun and Chan27 also used OES to investigate the effects of the temperature gradient in the ICP central channel on Yb2O3 particles. A sheath gas was used to confine particles in the centre or outer regions of the ICP central channel so that the effects of the radial temperature gradient in the central channel could be studied. Experiments were performed at 8.5 and 19.5 mm above the load coil (ALC). At the lower observation position the temperature in the inner central channel was 400 K lower than the outer region, resulting in a difference of 102 in spICP-OES emission intensity for Yb2O3 particle which have a relatively high boiling point. Hence, there was poor correlation between particle size and emission intensity. What this meant in practice is that NPs (∼<100 nm) with a boiling point of 3000 K, such as Au and Ag, should be measured at 4 mm ALC, but high-boiling or larger particles such as Yb2O3 should be measured at 10 mm ALC or above. The authors also proposed increasing the gas temperature by increasing power or adding 50% He into the Ar carrier gas.
3.2.2.2 Graphite furnaces. GF-AAS was proposed as tool to differentiate metal ions from NPs and estimate the size of these particles. Brandt and co-workers91 determined the atomisation mechanisms for ionic Au and Au NPs and used the data to discriminate between the species. Ionic Au was found to be governed by a two-precursor mechanism whereas the atomisation of Au NPs followed a one precursor atomisation mechanism. The work was extended by converting the experimental results to a computational model, producing simulations of the processes to be obtained.92 This approach reduced the calibration time by up to 80% for the quantification and sizing of AuNPs. The GF-based approach was applied to the characterisation of non-spherical Ag and AuNPs and found that these forms could be distinguished from spherical NPs.93 In addition, zero-valent Fe NPs could be distinguished from ionic Fe. However, when the technique was used for Pd and Pt, neither differentiation between ions and NPs nor size discrimination of NPs was possible.
Marrocos et al.94 used the molecular absorption of CS to investigate chemical modification for S determination using HR-CS-GF. Without chemical modification, S species were lost in the pyrolysis step, especially when using organic standards. The combination of a mixture of modifiers Pd and Mg (in solution) and W as a permanent modifier allowed the accurate determination of S in hair samples using inorganic S calibration standards.
Spectroscopic interferences were identified as a potential problem in the early days of the development of ICP-MS, with research articles dedicated to documenting them. It is possible to theoretically predict a polyatomic ion interference that might result from any given combination of elements in the sample matrix and plasma gas, but this does not necessarily tell the analyst how severe the interference will be. In order to remedy this, Lomax-Vogt et al.98 published a searchable Excel database of 2000 ions which were either experimentally identified by the authors or other workers (and cited in the literature). The database includes elemental, doubly charged elemental, oxide, hydroxide, hydride, dioxide, trioxide, polyatomic ions etc. However, as the authors conceded, it still does not contain information about the severity of interference and work is ongoing to update the database with this information.
Recent approaches for reducing spectroscopic interferences in ICP-MS have focused primarily on the use of CRC technology. Instrument manufacturers implement this technology in different ways, so some degree of caution is advised when transferring methods from one instrument to another, and research into finding alternative routes to a solution with a particular technology is useful. Sugiyama99 developed a method for reducing doubly charged REE ion interferences on As and Se using CRC-ICP-MS. The normal He collision gas was replaced with H2 and, in conjunction with kinetic energy discrimination (KED), it was possible to determine 1 μg L−1 of As and Se in the presence of 0.5 mg L−1 each of 16 REEs. This compared to a false positive result for As and Se of more than 10 μg L−1 achieved using He as the collision gas. This worked because the collision cross section of H2 is much higher than He, so it was more effective as a collision gas for doubly charged REEs, which have masses double that of the analytes. Bolea-Fernandez et al.100 investigated the role of the internal standard when using mass-shift interference correction in ICP-MS. Seventeen elements were subjected to mass-shift reactions and monitored before (as their atomic ion) and after (as a reaction product ion) and the effectiveness of the internal standard was evaluated. The found differences in behaviour between atomic ions and reaction product ions but could not say if this was due to insufficient reaction stabilisation time, reactivity or changes in the tuning conditions. Statistical evaluation did not provide evidence that closeness in mass number and/or ionisation was significant.
Non-spectroscopic interferences caused by the matrix are the other bane of the analyst. Carter et al.101 investigated supervised and unsupervised machine learning methods to evaluate matrix effects in ICP-OES. For unsupervised learning, signal bias due to matrix effects were observed for ionic lines and low-energy atomic lines. For supervised learning, the most accurate models were those predicting analytical error for high-energy ionic lines. Signal bias correction using plasma species as internal standards was most effective for minimising EIE matrix effects on low-energy atomic lines. Predictive models, including partial least squares regression and generalised linear models were successfully applied for the determination of analytes in CRMs when analyte levels were at least 1-fold higher than the LOQs, and for concentrations >20 μg g−1 in the original sample. However, signal bias correction by internal standardisation provided no accuracy improvement.
Serrano102 investigated the effect of carbon matrix effects on atomic lines in ICP-OES. The paper included a discussion of the theoretical basis for the suppression effects. Experimental data suggested that carbon in the sample matrix affected both analyte ion and atom populations by a combination of charge transfer reactions, collisional ionisation and collisional excitation. Furthermore, they concluded that: the use of Mg(II):Mg(I) to evaluate plasma robustness is highly sensitive to carbon non-spectral interference; the source of carbon affects the intensity of matrix effect; and careful selection of analyte lines was necessary to avoid carbon-based matrix effects, particularly for Se and alkali elements. Matrix effects in nitrogen MIP-OES with a Hammer cavity were investigated by Polyakova.103 They studied the effect of matrix elements with IPs in the range from 5.72 eV to 10.48 eV on atom and ion excitation temperatures, Mg(II):Mg(I) ratio, plasma molecular species and analytical signals of 30 elements. Atomic and ionic lines of analytes behaved similarly in the presence of matrix elements with medium and low IP. They also found correlations between Mg(II):Mg(I), N2+:N2 ratios, matrix element IP and the degree of matrix effect.
4 Laser-based atomic spectrometry
Key fundamental studies and instrumental developments (published in 2021 and at the end of 2020) in laser-based atomic spectrometry are highlighted in this section. Atomic spectrometry techniques where the laser is used as either an intense energy source or a source of precise wavelength (e.g. LIBS or LIF) are here considered. However, studies related to LA-ICP-MS and -OES, and to the use of lasers for fundamental studies of the properties of atoms or for thin film deposition are not reviewed.4.1 Laser induced breakdown spectroscopy (LIBS)
This section describes the latest instrumental developments and fundamental studies related to LIBS, but it does not cover application papers in great detail. However, there are other recent reviews on LIBS applications. Applications and methodologies of elemental mapping using LIBS were evaluated by Limbeck et al.,104 highlighting the necessary instrumentation, data processing and data-reduction for imaging. An historical review of field-portable and handheld LIBS, current status and future prospects, was written by Senesi et al.105 Fundamentals and applications of underwater LIBS were reviewed by Matsumoto et al.106 The development of LIBS technology and applications, especially for Chinese LIBS research community, was reviewed by Guo et al.,107 who summarised and evaluated the research status and latest progress of the main research groups in coal, metallurgy, and water. Several international conferences were dedicated to discuss the recent progress in LIBS. In particular, the 11th Euro-Mediterranean Symposium on LIBS held in Gijón, Spain from November 29th to December 2nd (http://www.emslibs2021spain.com/), and the 4th Asian Symposium on LIBS held in Qingdao, China, from October 16 to 20th 2021 (http://www.aslibs2021.com).Spatially and temporally resolved measurements of LIBS in gases was reviewed by Parigger,110 with emphasis on LIBS spectra of H, cyanide and hydroxyl groups. Additionally, the author discussed the applicability of LIBS for analysis of astrophysical plasma (e.g. from white dwarfs). Shadowgraphy was used by Buday et al.111 to calculate the initial energy of the laser-induced shockwave expansion on 4 different matrices (steel, glass, bronze and soft tissues). The measured shockwave size was fitted using the Sedov–Taylor model during the first μs after laser ablation. This allowed the authors to compute the initial energy of the shockwave, thus enabling them to define the energy spent on ablation. Results showed that the amount of energy required to ablate steel and glass was similar, and about double the energy required to ablate bronze and soft tissues.
Cai et al.112 investigated laser–material interactions and plasma evolution of coals, with different volatile matter contents, using temporally and spatially resolved LIBS in Ar. The authors observed that volatile matter vaporised earlier and was pushed to the plasma frontier, exhibiting more intense atomic O and N emissions in this region. A plasma plume with a higher frontal particle concentration was thought to increase volatile matter content in the coal. As a result, a higher signal due to molecular emission of C species was observed in this region, and at an earlier delay, in the proximity of the target. This was thought to be caused by an increased amount of C2 molecules generated by dissociation of heavy carbon clusters and thermal removal. A conceptual model of plasma generation and evolution was proposed by the authors to explain these effects, creating a better understanding of the laser–coal interaction. Spatiotemporal spectroscopic characterisation of plasmas induced by non-orthogonal LA was investigated by Kepes et al.113 and compared to plasmas induced by orthogonal ablation. A division of the LIP into two parts was observed, one in the direction of the LA, which mainly emitted continuous radiation, and another expanding along the sample normal, which was composed of the sample material. Plasma parameters were very similar for the different ablation angles, but the emissivity of ionic species was significantly affected.
Combining emission and absorption spectroscopy can be a useful way of gaining deeper insight. LaHaye et al.114 compared the early- and late-time dynamics of LIPs using both OES and LAS. The study included the evaluation of resonance and near-resonance electronic transitions of Ca II and Al I lines. The authors considered that optical emission spectral features were initially dominated by Stark broadening, followed by instrumental broadening. The absorption spectra resulted in very narrow lines (∼2–4 pm). The Ca number density decayed quickly (∼70 μs) compared to Al because of a higher susceptibility to recombination as the plasma cooled over time. The Tkin determined from the Ca II transition (using absorption methods) appeared to follow the same trend as the Texc. The authors claimed that existence of LTE in the later evolution period of an LIP was difficult to verify in their study and required further research.
Laser trapping-LIBS and machine learning was investigated by Niu et al.115 for the determination of 4 elements absorbed in a single micro-carbon black particle in air. The authors employed a hollow laser beam for trapping, which was proposed as a possible method for online single aerosol particle analysis. A method of optical trapping as a morphologically selective tool for in situ, elemental characterisation by LIBS was developed by Purohit et al.116 Single NPs generated by LA of bulk targets in air were analysed. Results showed that the use of high laser fluence produced an aerosol with particle sizes distributed in the range between 100 and 500 nm, resulting in variability in the LIBS spectra. Lower laser fluence resulted in more large particles, which better resembled the bulk material. The dual role played by air plasmas as atomisation and excitation sources in single-particle LIBS (spLIBS) was investigated by Fortes et al.117 Both plasma imaging and time resolved spectroscopy were performed on isolated copper NPs. The Te was calculated using N II lines in an air plasma. Lower values were observed in the presence of NPs, indicating that heat transfer into the NP occurred.
Delgado et al.118 investigated the routes leading to the formation of LIBS emitting species, such as CN, C2 and NH, derived from organic compounds (such as nitrogen containing compounds, aromatic compounds and amines) in a simulated Mars atmosphere. Results showed that atmospheric C might react with organic nitrogen to yield an intense CN signal, which could be used to identify the presence of N-containing organic compounds. Nevertheless, in most of the cases, neither the atmospheric components nor the inorganic carbon, were clearly detected. The authors highlighted that LIBS may provide a clear indication of the presence of an organic compound in places where the organic compound may appear in large concentrations, such as inclusions in fracture systems or precipitates in hydrothermal deposits. Gaft et al.119 described recent developments related to the emission of halogen and rare-earth-containing molecules, and to the selective excitation of molecules by molecular LIF. In particular, the authors discussed the improved capabilities to detect some isotopic molecular signals in comparison to LA molecular isotopic spectrometry. This was primarily because molecular LIF provides strong resonance excitation of only targeted isotopes.
Galbács et al.120reviewed recent developments in methods and applications utilising NPs. Connections between nanoparticles and laser and plasma spectroscopy were discussed in three major areas, including the monitoring of NP synthesis, characterisation and plasmonic signal enhancement. The role of the NPs, which are deposited on the sample surface before the laser irradiation in nanoparticle enhanced (NE) LIBS, was investigated by Salajková et al.121 The effect of coupling the NP plasmon with the laser pulse, which improves the analytical signal, was studied for spherical AuNPs with diameters between 10 and 100 nm. Results indicated that NE-LIBS occurred at a specific NP surface concentration and that this critical concentration depended on the NP size. In another study, the shape-effect of AgNPs on NE-LIBS was evaluated for different target materials by Abdelhamid et al.122 Nanospheres, nanocubes and nanowires were tested for aluminium, zinc and silicon. The results showed that the clusters formed in the silver nanowires and nanocubes played an essential role in the laser pulse absorption mechanism, and consequently, enhanced the LIBS signal. Additionally, it was shown that plasma temperature and ne were not significantly affected by the nano-shape change. In related work, Li et al.123 developed a novel method based on AgNP preaggregation, by adding Cl−, that substantially improved sensitivity compared to conventional NE-LIBS. The Mg(I) emission lines at 383.23 nm and 383.83 nm were measured as a function of the concentration of Cl−. The authors claimed that the LOD obtained with this method was significantly improved (30.6 ppb for Mg) compared with ∼ ppm levels in the traditional LIBS method.
Liu et al.127 developed a method based on the application of two CMOS cameras, or two fibre spectrometers, to make time-resolved measurements, including the possibility to obtain both plasma images or LIBS spectra, respectively. Several drawbacks of the method, including poor signal subtraction when spectral backgrounds were dissimilar and electronic jitter preventing short timeinterval (∼10 ns) spectral acquisition. Nevertheless, the authors believed that this method would be a practical and cheap way to conduct time-resolved measurements for field or in situ detection to some degree.
Plasma emission was significantly enhanced using a solid target in conventional reheating orthogonal double-pulse LIBS in a study published by Jiang et al.128 They used an optimised inter-pulse delay of ∼4 μs. An increase in the plasma temperature was observed, together with an increase in emission intensity caused by collisional processes involving ablated particles. This resulted in improved LODs for the determination of metals in water.
A novel method to automatically control the laser focus position on the sample was developed by Fugane et al.129 They used a technique based on fast variation in the focal length a tunable planoconvex lens controlled by a function generator. This allowed the analysis of samples with tilted or irregular surfaces, e.g. analysis of a piece of scrapped stainless steel, having an about 20° tilted surface.
3D elemental maps with high spatial resolution were obtained by Huang et al.130 using a novel confocal controlled (CC) LIBS microscope. This technology provided a lateral resolution of about 700 nm for morphological imaging and about 10 μm for elemental mapping. The authors considered that the imaging process of CC-LIBS was not affected by system drifts and ambient temperature changes and thus could be used for the detection of unknown substances in complex environments.
Interference of two noncolinear fs filaments was employed by Hu et al.131 to generate a plasma grating, which was able to provide a higher local electron density. The authors observed that the resultant plasma grating induced breakdown spectroscopy system resulted in enhanced atomic emission signals compared to single filament induced breakdown spectroscopy, with reduced matrix effects. The authors considered this to be a promising tool to detect hard samples and/or those with a complex matrix.
A specially designed micro-gas column assisted LIBS system was developed by Jiang et al.132 to generate a stable micro-gas column underwater (with a diameter of about 1 mm). Laser pulses were focused on the gas–liquid interface of the column. This novel method improved LIBS signals of metal ion lines and improved LODs. The authors considered that this had a great potential to be employed in seawater investigation or water environmental monitoring.
The performance of a multi-modal laser-spectroscopy system, which combined LIBS, Raman spectroscopy and LIF was evaluated by Dhanada et al.133 The experimental set-up was based on the use of a single laser and a single detector, which consisted of a CCD-coupled echelle spectrograph.
Ewusi-Annan et al.137 developed a general approach that enables the fast automatic identification and fitting of large numbers of emission lines. Voigt profile parameters were initialised by fitting identified peaks using the second derivative of the spectrum. The methodology was employed to successfully obtain optimum parameters of the Voigt profiles that faithfully described all peaks identified in the spectrum. For instance, it was possible to achieve a Martian LIBS spectrum containing 625 identified lines in less than 30 min with fit residuals that were globally negligible. It was claimed that this work, in combination with previous studies on automatic preprocessing of LIBS spectra, might contribute to significant improvements in the analysis of large numbers and line-dense LIBS spectra.
A deep spectral convolutional neural network was proposed by Castorena et al.138 to learn to disentangle spectral signals from sources of sensor uncertainty, and to estimate qualitative and quantitative chemical content. This methodology improved on the standard pre-processing used by the Mars Science Lab, and eliminated the requirement for dark current measures, instrument response function availability and side environment information such as temperature and emitter-to-target range automatic adjustments. Different approaches for the quantification of K in unknown polymers were investigated by Brunnbauer et al.139 Two different multivariate approaches, random decision forest classification combined with conventional univariate calibration and a PLS model, were used for calibration for 8 different polymer samples. The authors claimed that using their approach it was possible to determine K in the low μg g−1 range in unknown polymer types, with a relative error typically between 30% and 90%, which is a significant improvement compared to non-matrix-matched quantification. LIBS spectra are considered to exhibit high dimensionality, redundancy, and sparsity. In this context, Képeš et al.140 proposed the use of sparse PCA for the analysis of high-dimensional sparse data, significantly improving the interpretability of loading spectra. The randomised algorithm was demonstrated to offer a 20-fold increase in computation speed compared to PCA based on singular value decomposition.
A spectral intensity correction method via probability distribution was proposed by Chang et al.141 to improve precision compared to single shot analysis. The method focused on establishing the relationship between the averaged spectral intensity of multiple measurements and any one-shot measurement. Uncertainties obtained from the use of a normal distribution were less than those for the Poisson distribution when the number of measured spectra for building the weighted correction matrix was larger than 10. The authors claimed that the proposed method provided a simple but feasible way to improve the repeatability of LIBS quantitative analysis and could greatly expand the application of LIBS in industrial scenarios, especially in the cases where multiple measurements are not available or suitable.
The broad range of excitation resulting from a LIP was investigated by Clavé et al.142 to enable simultaneous observation of multiple luminescence emission bands. They generated a LIP excitation source on a separate ablation target that was adjacent to the luminescent sample. Plasma photons, from the continuum of the plasma emission, over the whole spectral range were demonstrated to contribute to the resultant plasma induced luminescence (PIL) excitation. Side-PIL and remote-PIL (placing the luminescent samples several tens of cm away) were evaluated. In the latter case the insertion of an optical fiber between the imaging lenses filtered out the atomic emission lines from plasma radiation, which overlapped with the short decay time luminescence emission bands. The method was validated using a natural Durango apatite sample, and an artificial crystal of fluorite enriched in Eu2+, which is known to decay in less than 1 μs.
Gaft et al.143 investigated the use of BeO molecular emission signals to quantify the amount of Be at elevated concentrations, where atomic resonant emission signals suffer from auto-absorption. The authors determined LODs of ∼0.05% for BeO with a single pulse and ∼0.01% with a double pulse, which are appropriate for minerals and alloys analysis.
4.2 Cavity ringdown spectroscopy
A novel CRDS method was developed by Bracken et al.144 to overcome resource requirements and technical challenges for N2O isotopic isomer measurements, considered a useful tool to study soil N cycling processes. A combined laser spectrometer and small sample isotope module method were used to determine N2O concentration, isotope ratios, and consequently site preference measurements using gas sample volumes <20 mL. The authors claimed that the repeatability made this approach suitable for N2O ‘isotopomer mapping’ to distinguish dominant source pathways, such as nitrification and denitrification, and required less extensive lab resources than the traditionally used GC-IRMS methods.4.3 Laser induced fluorescence (LIF)
Bueno et al.145 compared intermodulated LIF and intermodulated optogalvanic spectroscopy using the same experimental set-up. They used a homemade Er–Ar HCL, which is a well-stablished source used in isotope shift studies. Both of these sub-Doppler techniques allowed the detection of the isotope shift of two Er transitions, 576.440 nm and 582.842 nm. The transition at 585.693 nm was also observed using intermodulated LIF which presented improved discharge stability independence, being a faster and simpler way to detect spectroscopic signals in this environment, although the results were not highly precise.5 Isotope analysis
The development of more sensitive mass spectrometric and sample introduction techniques has encouraged greater development of LA systems. Primarily these have been used as a tool to investigate finer resolution of particles, crystals and concentric growth without sacrificing the precision of the target IR. Indeed, it is perhaps almost greedy to demand even more from a single laser pit, however, the use of split-stream LA to simultaneously determine both IRs and elemental abundance on separate mass spectrometers has become increasingly common. Instrumental sensitivity, low-noise detectors, efficient LA cells, and reaction/collision interference reduction are all advances that have allowed detection and precision to be achieved at progressively lower signal levels.5.1 Reviews
Bouden et al.146 provided a useful retrospective on MC-SIMS triple oxygen isotope measurements. They reviewed advances in SIMS hardware in the 1980’s and ’90s, through to current detector and resolution systematics. In particular, the paper provided an assessment of the typical variations in standard error in respect of varying count rates received in 1011 and 1012 Ω preamplifiers: a useful graphical guide as to what to expect from different sample types and spot sizes.Techniques and developments in calcium isotope measurement were reviewed by Chakrabarti et al.147 who compared the main MS instrumentation of TIMS and MC-ICP-MS, comparing the specific benefits of each. Primarily, TIMS produces the highest quality ∂44/40Ca and ∂44/42Ca due to isolation of Ca through temperature-specific ionisation, whereas MC-ICP-MS has a faster throughput. Useful tables were provided which defined the double spikes used, instrumentation, sensitivity and precision for different laboratories around the world, with representative publications detailing each laboratory’s technique. Future developments were discussed in terms of standardisation of measurements, with a particular issue being the presence of Sr in Ca isotope standards, which produces doubly charged interference on Ca isotopes. The recommendation suggested was to develop a Sr-free Ca isotope standard.
An expanding field in analytical science is the use of MS instruments to map chemical variation in geological and biological materials. Chew et al.148 reviewed LA-ICP-MS imaging, in particular focusing on the application to U–Pb geochronology. They documented examples of resolving elemental concentration variations (chiefly U and Pb) in zoned or complex growth crystals, such as zircon and titanite. The images produced by a 10 μm spot size were found to be useful in targeting suitable sectors of crystals for dating studies. Chu et al.149 evaluated Os isotope measurement and PGE determinations in geological materials. The main chemical dissolution methods, including NiS fire assay, alkali fusion, Carius tube acid digestion and high-pressure asher digestion were reviewed. There was a useful table containing a compilation of PGE concentrations and 187Os/188Os of international rock standards, including estimates of precision, and citations for data sources. The key techniques of TIMS and MC-ICP-MS were also reviewed, with details of interference corrections, spiking methodology and sample introduction. Tulej et al.150 examined the use of a LIMS-TOF system in the simultaneous determination of Mg, Ni and B isotopes in trevorite and micrometre-size inclusions within the crystal. Because of the small size of this instrument it had the potential to be deployed in space exploration. This technique demonstrated the potential for useful precision for isotopic characterisation with ∂26Mg = ±0.96 and ∂60Ni at ±1.15 in the test materials.
5.2 Radiogenic isotope ratio analysis
Fréres et al.151 examined how high levels of NdO+ formation effect instrumental mass bias for Nd isotopic measurements by MC-ICP-MS. They correlated the effects of instrumental conditions (such as wet or dry plasma, introduction methods and cone geometry) with oxide formation and Nd IRs. Thus, a mathematical model was developed to predict offsets from correct values. Recommendations were made on which instrumental parameters significantly degrade ratio precision. Reinhard et al.152 trialled a new ATONA amplifier system, fitted to a TIMS instrument, for the measurement of sub-nanogram loads of Nd for isotopic characterisation. The study examined instrument methodology to find optimal parameters, including cycle integration times and baseline length. It was found that 100 pg of Nd, from the JNdi reference material, resulted in a precision for 143Nd/144Nd of ±0.000012 using a 30 s integration time. The potential for these collectors was discussed, concluding that they were most effective when measuring high-magnitude isotope ratios, where very large and tiny masses need to be detected simultaneously.Isotope ratio analysis using LA is hampered by problems with integrating the transient signals Yamamoto et al.153 examined the measurement of U isotopes by LA-MC-ICP-MS in zircon and titanite mineral phases. When using 1012 Ω and 1013 Ω amplifiers with Faraday collectors there is the possibility of time-correction errors in response to transient signals. In order to eliminate these errors a new signal processing method was developed. This involved continuous ion-monitoring to integrate single signal events for ion currents greater than the maximum ion currents of the U isotopes. The study concluded that the reference material used showed significant deviation from previously reported 235U/238U ratios in a number of the titanite grains. In addition, the 234U/238U for these FCT titanites was found to be consistently 15% higher than secular equilibrium values. Hirata et al.154 found that shortening the dwell time erroneously increased signal intensity due to including adjacent time-slices in the count integration. Correction for the counting loss for different dwell times based on standard data produced 206Pb/238U data within 2–3% (2SD) for a range of known zircon and glass reference materials.
Zircon dating was the subject of a study by Huang et al.,155 who investigated laser focus as a source of error in U–Pb isotope measurement. Their study found that variation in laser focus by 30 μm led to systematic shift in 206Pb/238U, ranging from <1% for NIST 610 to 12% for rutile (TiO2). Notably, a similar shift was not observed between 206Pb and 207Pb, which would mean a potential false discordance of data on concordia plots. They concluded that laser focus should remain with 5 μm of the analyte surface in order to avoid bias. Iwano et al.156 examined the possibility of measuring U–Pb ages of zircons by single pulse LA-MC-ICP-MS: i.e. on a shot-by-shot basis. This was tested using young (<35 Ma) Himalayan zircons. Each shot was calculated as an age, including the surface cleaning first shot, which frequently resulted in an aberrant age. One objective of this was to enable measurement of, and derive an age for, the thin outer rim of zircons. The method was used to define the age of metamorphism and final cooling stages of zircons from the Mount Everest region. Lin et al.157 also investigated high spatial resolution analysis of zircons by LA-MC-ICP-MS. Their study used ≤10 μm spot size and examined the effects of down-hole elemental fractionation as a function of different laser settings. Trials with a number of zircon reference materials confirmed that this method was capable of providing age discrimination between core, mantle and rim zones, as well as avoiding visible imperfections in the crystals.
Schaltegger et al.158 evaluated the use of solutions used as references for U–Pb geochronology by ID-TIMS. Solution ET100 was tested with different TIMS instruments, but no significant differences were found between the results from different instruments. A key finding was that ensuring a complete homogenisation and equilibration of sample and spike was essential in producing consistent age determinations. Liu et al.159 designed a protocol to measure IRs on small quantities of Pb (∼16 ng), entitled ‘liquid spray dielectric barrier discharge plasma-induced chemical vapour generation MC-ICP-MS’. In this system, Pb was converted to a volatile species in the presence of formic acid, resulting in a seven-fold increase in sensitivity. One potential issue was the mass fractionation created by the introduction system, which was corrected by using 205Tl/203Tl. The method would be most useful for low-Pb or small volume samples, but has the potential to be extended to Pb double spiking or 2× double spiking to measure yet smaller samples, until the limits of blank contributions are reached. Even smaller quantities of Pb were isotopically characterised in a study by Nagaishi et al.160 using MC-ICP-MS with Aridus II sample introduction. A key feature of their design was to admit a Tl spike solution in line with the sample solution rather than adding directly to the analyte. The reason for adding Tl immediately before introduction to the desolvation nebuliser was to minimise Tl oxidation in the Pb solution – a state documented to induce a poor mass fractionation relationship between Pb and Tl. Results indicated that using 0.4 ng Pb produced levels of precision (±0.0032 2SD for 206Pb/204Pb), which put the technique’ precision on a par with double spike analyses.
5.3 New developments
Di et al.161 produced an elegant mathematical solution that deconvoluted the changes in relative Faraday cup efficiencies. Multi-collector TIMS and ICP-MS often use multidynamic analyses (i.e., periodically switching the mass spectrum between different collector sets) which can be used to neutralise collector differences. These authors used relative changes in static isotopic analysis within dynamic routines to track Faraday cup deterioration. Specifically, they used radiogenic Sr isotope measurement from TIMS, gathering data from routine measurements rather than specially designed experiments.Sun et al.162 developed a new method for the isolation of multiple transition metals before stable isotope measurement. They used a multi-stage ion exchange system that started with Re-resin to isolate U and Mo, followed by an anion AG MP-1M resin column to separate Cu, Fe, Zn and Cd. A further stage isolated Ni using additional cationic and Re-resin columns. The study provided a useful table of isobaric and doubly-charged interferences for each of the key isotopes in the element systems examined. This included monitored interferent isotopes, the correction ratios used, and the accepted values for these ratios. One key conclusion from this study was that where double spikes or indeed multiple double spikes are used, care must be taken to ensure that impurities in spikes used in one system do not induce analytical artefacts in another elements’ isotopes.
5.4 Geological studies
Measurement of Sr, Pb and Mg isotopes in crude oil was investigated by Henn et al.163 who trialled two methods – microwave-assisted wet digestion within an ultra-high pressure digestion cavity and solubilisation of inorganic solids using membrane filtration. The latter method did not retain Pb, possibly due to its affinity with unrecovered organic compounds, but both methods satisfactorily recovered Sr and Mg. Measurement of IRs in Brazilian crude oil samples was performed using MC-ICP-MS. Results showed a mixing relationship between seawater composition and a radiogenic Sr and Pb end member, with 206Pb/204Pb > 19.2.Li et al.164 investigated the possibility of completing the separation of K, Ca and Sr in a single ion exchange column prior to stable and radiogenic isotope measurement for these systems. The column used an AG50W-X12 (200 to 400 mesh) cation exchange resin with the sample loaded in a solution of 2 M HCl + 0.1 M HF. This resulted in a clean separation of K from Al, Mg, Fe and Na, then subsequent elutions of 2 M HCl and 3 M HCl recovered the Ca and Sr respectively. Analysis of the three isotope systems generated IRs for reference materials that were indistinguishable from accepted values.
Yim et al.165 investigated the relationship between the mass fractionation of Sr and Rb during determination of 87Sr/86Sr by MC-ICP-MS. They found that in low Rb/Sr materials, such as NIST 616, the isobaric interference from 87Rb on 87Sr was insignificant. However, in high-Rb/Sr materials such as NIST 610 and 612 correction was essential. Mass fractionation factors derived from the non-radiogenic Sr IRs were found to produce unreliable results when applied to the Rb correction, indicating that fractionation of these elements was not systematically related. The most reliable correction method was found to be using an interpolated factor based on Rb fractionation via sample-standard bracketing.
5.5 Stable isotope ratio studies
Boron isotopes are a key proxy for palaeooceanographic pH estimation. Buisson et al.166 utilised a new micro-sublimation and direct injection uptake system with MC-ICP-MS. 10B and 11B ions were detected using Faraday cups equipped with 1013 Ω amplifiers. With this system ∂11B was determined on ∼1 ng with external repeatability of 0.2‰, which is a similar level of precision achieved when B is isolated by ion exchange chromatography. Evans et al.167 advanced a technique introduced by Standish et al. (Rapid Commun. Mass Spectrom., 2019, 33(10), 959–968) to minimise the interference from multiply charged Ca ion species on ∂11B measurement. Their modification to this method was to utilise 1013 Ω Faraday amplifiers to make more precise measurement of the Ca interference by closely defining the shape of the isobaric interference. The results from this showed that 70 μm single-spot LA-MC-ICP-MS analysis of ∂11B was reproducible to 0.5‰ (2SE) and with 40 μm to 1.4‰; the latter equating to 2 pg B. Chalk et al.168 examined the use of 10B and 11B as tracers in studies of plant nutrition. This became a ‘growth’ area following the development of ICP-MS, and the ability to effectively measure B isotopes in organic materials, and within-plant mobility of B in crops. Malinovsky et al.169 designed a method to measure B isotopes by MC-ICP-MS using fractionation correction to admixed internal standards. They experimented by adding Li, C and Mg, which span the mass range and the expected mass fractionation of B. It was determined that Mg and C performed better than Li in minimising random fluctuations and temporal drift as well as accounting for matrix related changes. However, care had to be taken to accommodate the spectral interference of 12C1H+ on the 13C isotope.Potassium isotope ratios, such as 41K/39K, are susceptible to large interferences from ArH+ and Ar+. Chen et al.170 utilised He as a collision gas and H2 as a reaction gas in the CRC cell of an MC-ICP-MS to effectively eliminate these interferences. Within-run precision of ∂41K was found to be better than ±0.04‰ and long-term reproducibility better than ±0.07‰.
Moynier et al.171 also measured K isotopes using the MC-ICP-MS in seawater and other terrestrial and lunar materials, using an Apex Omega aspiration system. They used a CRC to achieve measurement reproducibility of typically 0.05‰ (2SD). A key advance was the high instrument sensitivity for K+ of ∼2000 V μg mL−1, enabling accurate measurements in solutions containing only 25 ng mL−1. They also recommended that Ca be removed to leave the K analyte at better than 99% purification, and that sample and standard concentrations and molarities be closely matched. Gu and Sun172 also determined ∂41K using MC-ICP-MS, but without a collision cell, and in a low-resolution mode. They used cold (RF power ∼600 W) and wet plasma conditions to suppress the ArH+ interference. Long-term measurements using their method revealed that precision and accuracy were significantly degraded if different acid molarities were used between bracketing standards and samples. Using the same acid, precision over a six-month period was ∂41K ± 0.08‰ 2SD. These authors also considered173 the problems of K isotope measurement on the interference-free peak shoulder of ArH+. Because of the narrowness of this shoulder even a small drift in mass stability of ∼0.002 m/z was found to induce failure of the analysis. When a continuous acquisition method was introduced, which does not load and check instrument parameters between samples and standards, significantly greater mass stability, and hence more reliable ∂41K, was achieved.
Iron isotopes were measured by Yuan et al.174 using LA-MC-ICP-MS. Problems of matrix effects influencing mass bias in dry plasma conditions were minimised by using lower fluence and inducing wet plasma conditions. The wet plasma was generated by adding water vapour and N2 after the ablation cell. This resulted in ∂56Fe reproducibility of ±0.1‰ (2SD) on pyrite, manganite, hematite and chalcopyrite. Improvements in Fe isotope measurements by SIMS was the subject of a study by Decraene et al.175 They used a new RF plasma ion source, Hyperion-II, which increased the ion beam density by an order of magnitude compared to a conventional Duoplasmatron source. The aim of the study was to enable measurement of micro-pyrites, which are less than 20 μm, and results indicated that reproducibility for ∂56Fe was ±0.25‰ (2SD) on the Balmat pyrite standard.
Measurement of Mg isotopes in cerebrospinal fluid is challenging due to the invasive nature of the procedure, but has potential uses in studies of neurodegeneration. Grigoryan et al.176 used 1013 Ω amplifiers with MC-ICP-MS to reduce errors on the micro sample determinations of small amounts of Mg (between 7 and 10 μg L−1). The ∂Mg values were found to be similar to the ratios determined for 150 μg L−1 samples. A microanalytical approach to measuring Mg isotopes was taken by Sadekov et al.,177 who used LA-MC-ICP-MS to examine the isotopic variation in biogenic carbonates. They reported precision and accuracy down to ±0.2‰ (2SE), which was achieved by careful monitoring of baseline interferences and using carbonate standards with low trace metal compositions (similar to typical biogenic carbonates). The spot size of 0.2 μm provided a remarkably high spatial resolution which would be useful where isotopic discrimination is needed on annual growth accretion.
Selenium isotopes were determined by Karasinski et al.178 who developed a method using wet plasma MC-ICP-MS without hydride generation or Se separation. This utilised three mass bias correction protocols, combining sample-standard bracketing, internal standardisation and optimised regression models. The latter method was found to result in the best precision and elimination of matrix effects, and was significantly better than a stand-alone sample-standard bracketing correction.
Measurement of the 121Sb![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :123Sb IR is an emerging method used to investigate pollution related to mining and the potential for isotopic fractionation in response to biological processes and anthropogenic smelting. Kaufmann et al.179 determined ∂123Sb using solution and LA MC-ICP-MS of Sb minerals and found precision and accuracy to be better than 0.1‰. An approach using precise LA analysis of Sb-rich minerals (e.g. stibnite) was favoured because it was thought most likely to detect micrometre-scale processes reflected in the Sb isotopes. A new purification technique for Sb isotopes was designed by Li et al.180 They pre-reduced the sample using KI and ascorbic acid before a thiol silica column, eluted with HCl, was used to separate Sb from matrix elements. The approach did not appear to cause any isotope fractionation.
:123Sb IR is an emerging method used to investigate pollution related to mining and the potential for isotopic fractionation in response to biological processes and anthropogenic smelting. Kaufmann et al.179 determined ∂123Sb using solution and LA MC-ICP-MS of Sb minerals and found precision and accuracy to be better than 0.1‰. An approach using precise LA analysis of Sb-rich minerals (e.g. stibnite) was favoured because it was thought most likely to detect micrometre-scale processes reflected in the Sb isotopes. A new purification technique for Sb isotopes was designed by Li et al.180 They pre-reduced the sample using KI and ascorbic acid before a thiol silica column, eluted with HCl, was used to separate Sb from matrix elements. The approach did not appear to cause any isotope fractionation.
Li et al.181 described a new Zr(HPO4)2 emitter to enhance the ionisation efficiency of Ni in TIMS. It was found to inhibit Fe and Zn signals which are the main potential isobaric interferences on Ni isotope measurement. However, the key advance was a reduction in the amount of Ni required for analysis to 200 ng compared to the ∼1000 ng required by the traditional H3BO3–Al and H3PO4–Al emitters.
Liu et al.182 explored natural Sr isotopic variation, measured using MC-ICP-MS. This method combined the use of a 84Sr–87Sr double spike for Sr fractionation correction with a Zr external normalisation to correct for mass fractionation of the isobaric interferences. Kr and Rb interferences would normally be a limitation, but the method significantly improved accuracy and internal precision to better than ±0.01‰ (2SE), which is comparable with that achieved by TIMS.
Sulfur isotopes were measured by Lu et al.183 using LA-MC-ICP-MS. Their study targeted sulfate minerals, such as barite (BaSO4) and anhydrite (CaSO4). The results suggested that minimal fractionation was generated during the formation of these minerals in the natural environment. Any observed instrument-induced fractionation was minimised by lowering laser repetition rates and fluence output. Oelze et al.184 presented an analytical routine to simultaneously measure S isotope ratios and trace element chemistry. This method employed the increasingly common technique of splitting the laser ablation stream between an MC-ICP-MS, to measure the S isotopes and ICP-QMS to quantify the element. The study found it was possible to achieve good precision for S ratios while maintaining the trace element signals above background levels. An application of this split stream technique was to analyse individual pyrite crystals, with the isotopic and trace element data feeding into knowledge of the sulphide mineral formation histories.
Peng et al.185 used a double spike technique to conduct high-precision Cd isotope analyses of geological RMs. They used an MC-ICP-MS in conjunction with a 111Cd–113Cd double spike, proportioned 54.52%:45.48%. The paper provided a useful analysis of the error propagation for all of the Cd double spike options, demonstrating that the error minimum extends from ∼30% to 80% spike in the mixture for most double spike combinations, with the optimum at around 50:50. Following corrections for the interfering elements Sn, Pb and In, the ∂114/110Cd was determined with a precision of ∼0.05‰ for the RMs.
5.6 Nuclear forensics
Triple quadrupole ICP-MS is becoming a key tool in environmental isotopic analysis and Bu et al.186 utilised this technique to determine Pu isotopes in low-level soil RMs. A key benefit of this instrumentation is the gas reaction cell, and in this study mixed NH3–He (proportioned 1:9) was used to remove UH+ interference on 239Pu while retaining suitable sensitivity of Pu. Lindahl et al.187 used ICP-TQMS with a gas-free collision/reaction cell to assess the precision and accuracy of U isotope measurement. Drift in the mass calibration of the two quadrupole filters in conjunction with mass resolution settings were found to significantly affect measured U isotopes. A recommendation to the manufacturer was made to further investigate the impact of the second mass filter on mass discrimination. Lin et al.188 determined the high-magnitude isotope ratio of 236U/238U using MC-ICP-MS. Previously, AMS has been used to measure 236U/238U to 10−12, but SF-MS analysis suffers from tailing of 238U on 236U. The technique in this study was to adjust the retarding potential quadrupole filter to lower the ion yield to ∼20%, which resulted in a clear separation of 236U from the 238U tail. They quantified 236U/238U in the reference material NBL-112A to be 1.8 × 10−10. The methodology opens the way for measurement of environmental samples with 236U/238U at 10−9 or 10−10.
Improving 241Pu/239Pu measurement by TIMS was the subject of a study by Hernández et al.189 This ratio is affected by an isobaric interference from 241Am, so the authors utilised a two-part correction. First, a theoretical correction for the Am in-growth in the time elapsed between Pu purification and measurement; and second, an estimation of the Am signal at m/z 241 based on the ionisation efficiency of the two elements. Results indicated that 241Pu/239Pu could be reproduced to within ±1.5% of RM accepted values.
Gao et al.190 tackled the measurement of U IRs in individual uranium particles. Particle sizes of ∼1 μm meant that high-efficiency ionisation was required during TIMS measurement to achieve acceptable precision (±0.5% RSD on 235U/238U). This study examined the effects of different Re filament emitters, including loading the particles with glucose, collodion and high-vacuum grease prior to filament heating. Of these, the experiments determined that the vacuum grease provided the lowest errors and the highest ionisation efficiency.
Ito et al.191 devised a method to measure radioactive 90Sr using total evaporation TIMS. It utilised the differential ionisation properties of Sr and isobaric Zr to effectively eliminate 90Zr from the measurement, despite a potentially 108 times greater signal. Using ID it was possible to measure the quantity of 90Sr down to 0.029 fg. High-precision analysis of Sm isotopes in nuclear materials was the subject of a study by Louis-Jean et al.192 who used a TIMS instrument with Pt activator on a Re filament and a total evaporation analysis. One aim of the study was to detect the short-lived fission product 147Nd which beta decays to 147Sm. The purpose was to detect perturbations in Sm IRs, which through correction for half-life may enable the reconstruction of original 147Nd concentrations. Their method managed to identify deviations in 147Sm/148Sm and 149Sm/148Sm consistent with short-lived fission fragments of masses 147 and 149, meaning that the method could potentially be used in a range of nuclear and geological materials.
Mathew et al.193 prepared gravimetric mixtures of 241Am/243Am to calibrate TIMS for Am isotopic assay measurements. These were produced at nominal ratios of 1:1, 20:1 and 200:1. A total evaporation technique was deployed to characterise batches of AmO2 from plutonium materials at the Los Alamos Laboratory, with expected accuracy within ±0.03%.
The long-lived radionuclide 129I accumulates in the environment from spent nuclear fuel reprocessing and nuclear accidents, and determining its concentration is important in assessing the safety of waste repositories. A summary paper by Kim et al.194 documented the key mass spectrometric techniques used to measure 129I relative to 127I: TIMS, AMS and ICP-MS. The paper covered the purification methodology and the required loading chemical substrates for each technique. AMS was defined as the most sensitive technique with LoD down to 10−15 for 129I/127I. In comparison, SFMS techniques were capable of measurement to only 10−8, but were considered to represent a significant analytical cost and time saving compared with AMS.
6. Glossary of abbreviations
Whenever suitable, elements may be referred to by their chemical symbols and compounds by their formulae. The following abbreviations may be used without definition. Abbreviations also cover the plural form.| AAS | atomic absorption spectrometry |
| ac | alternating current |
| AES | atomic emission spectrometry |
| AFS | atomic fluorescence spectrometry |
| ALC | above load coil |
| AMS | accelerator mass spectrometry |
| ANN | artificial neural network |
| CAM | combined atomic and molecular |
| CCD | charge coupled detector |
| CR | collisional radiative |
| CRC | collision/reaction cell |
| CRDS | cavity ringdown spectroscopy |
| CRI | collision/reaction interface |
| CRM | certified reference material |
| CVG | chemical vapour generation |
| DBD | dielectric barrier discharge |
| DIHEN | direct injection high efficiency nebuliser |
| DSN | desolvation nebulisation |
| EIE | easily ionisable element |
| EPR | electron paramagnetic resonance |
| ETV | electrothermal vaporisation |
| FAES | flame atomic emission spectrometry |
| FIB | fast ion bombardment |
| FLA | flowing liquid afterglow |
| FLC | flowing liquid cathode |
| FWHM | full width at half maximum |
| GC | gas chromatography |
| GD | glow discharge |
| GF | graphite furnace |
| GIS | gas injection system |
| GO | graphene oxide |
| HCL | hollow cathode lamp |
| HG | hydride generation |
| HR | high resolution |
| HV | high voltage |
| ICP | inductively coupled plasma |
| IDMS | isotope dilution mass spectrometry |
| IP | ionisation potential |
| IR | isotope ratio |
| IRMS | isotope ratio mass spectrometry |
| KED | kinetic energy discrimination |
| LA | laser ablation |
| LAMIS | laser ablation molecular isotopic spectrometry |
| LC | liquid chromatography |
| LDPE | low density polyethylene |
| LEP | liquid electrode plasma |
| LIBS | laser induced breakdown spectroscopy |
| LIF | laser induced fluorescence |
| LIP | laser induced plasma |
| LIPS | laser induced plasma spectroscopy |
| LLE | liquid–liquid extraction |
| LOD | limit of detection |
| LOQ | limit of quantitation |
| LPE | liquid plasma electrode |
| LTE | local thermodynamic equilibrium |
| MC | multicollector |
| MDG | microdroplet generation |
| MDL | method detection limit |
| MIP | microwave induced plasma |
| MS | mass spectrometry |
| n e | electron number density |
| n ion | ion number density |
| NE | nanoparticle enhanced |
| NIST | National Institute of Standards and Technology |
| NP | nanoparticle |
| OES | optical emission spectroscopy |
| PAH | polyaromatic hydrocarbon |
| PCA | principal components analysis |
| PGE | platinum group element |
| PIVG | plasma induced vapour generation |
| PMMA | polymethyl methacrylate |
| PN | pneumatic nebulisation |
| PTFE | polytetrafluoroethyleme |
| PVG | photochemical vapour generation |
| QD | quantum dot |
| QMS | quadrupole mass spectrometry |
| REE | rare earth element |
| RF | radiofrequency |
| RM | reference material |
| SC | single cell |
| SD | standard deviation |
| SE | standard error |
| SEM | scanning electron microscopy |
| SF | sector field |
| SFMS | sector field mass spectrometry |
| SIMS | secondary ion mass spectrometry |
| SNR | signal-to-noise ratio |
| sp | single particle |
| SRM | standard reference material |
| TE | transport efficiency |
| T e | electron temperature |
| T exc | excitation temperature |
| TIMS | thermal ionisation mass spectrometry |
| T ion | ionisation temperature |
| TISIS | total sample consumption system |
| TOF | time-of-flight |
| TOFMS | time-of-flight mass spectrometry |
| TQ | triple quadrupole |
| TQMS | triple quadrupole mass spectrometry |
| T rot | rotational temperature |
| UV | ultra violet |
| VG | vapour generation |
| α ion | degree of ionisation |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Jorge Pisonero acknowledges support from Ministerio de Ciencia e Innovación (Spain) through the I+D+i project referenced PID2020-113951GB-100/AEI/10.13039/501100011033.References
- E. H. Evans, J. Pisonero, C. M. M. Smith and R. N. Taylor, J. Anal. At. Spectrom., 2020, 35(5), 830–851 RSC.
- C. Vanhoof, J. R. Bacon, U. E. A. Fittschen and L. Vincze, J. Anal. At. Spectrom., 2021, 36(9), 1797–1812 RSC.
- J. R. Bacon, O. T. Butler, W. R. L. Cairns, O. Cavoura, J. M. Cook, C. M. Davidson and R. Mertz-Kraus, J. Anal. At. Spectrom., 2021, 36(1), 10–55 RSC.
- M. Patriarca, N. Barlow, A. Cross, S. Hill, A. Robson, A. Taylor and J. Tyson, J. Anal. At. Spectrom., 2021, 36(3), 452–511 RSC.
- E. H. Evans, J. Pisonero, C. M. M. Smith and R. N. Taylor, J. Anal. At. Spectrom., 2021, 36(5), 868–891 RSC.
- R. Clough, C. F. Harrington, S. J. Hill, Y. Madrid and J. F. Tyson, J. Anal. At. Spectrom., 2021, 36(7), 1326–1373 RSC.
- T. Wang, W. Wang, Q. Zheng, Y. Ma and C. Xu, Anal. Chem., 2021, 93(32), 11233–11242 CrossRef CAS PubMed.
- H. Lackey, D. Bottenus, M. Liezers, S. Shen, S. Branch, J. Katalenich and A. Lines, Anal. Chim. Acta, 2020, 1137, 11–18 CrossRef CAS PubMed.
- K. G. Stevens and T. L. Pukala, Trends Anal. Chem., 2020, 132, 116064 CrossRef CAS PubMed.
- A. Delgado-Gonzalez and R. M. Sanchez-Martin, Anal. Chem., 2021, 93(2), 657–664 CrossRef CAS PubMed.
- L. Feng, At. Spectrosc., 2021, 42(3), 262–270 CrossRef.
- D. Torregrosa, G. Grindlay, L. Gras and J. Mora, Microchem. J., 2021, 166, 106200 CrossRef CAS.
- Y. Xing, J. Han, X. Wu, D. T. Pierce and J. X. Zhao, Analyst, 2021, 145(24), 7932–7940 RSC.
- J. Y. Lin, P. X. Lai, Y. C. Sun, C. C. Huang and C. K. Su, Anal. Chem., 2020, 92(20), 13997–14005 CrossRef CAS PubMed.
- F. Zhang, L. Yong, X. Hua, F. You, B. Wang, Y. L. Feng and L. Mao, Analyst, 2021, 146(6), 2074–2082 RSC.
- Q. Kang, M. He, B. Chen, G. Xiao and B. Hu, Anal. Chem., 2021, 93(2), 737–744 CrossRef CAS PubMed.
- J. Zhang, R. Zhou, Y. Jin and N. Cheng, Microchem. J., 2021, 160, 105541 CrossRef CAS.
- X. Gao, H. Pan, Y. Han, L. Feng, J. Xiong, S. Luo and H. Li, Anal. Chim. Acta, 2021, 1148, 238197 CrossRef CAS PubMed.
- B. Chen, G. Xiao, M. He and B. Hu, Anal. Chem., 2021, 93(27), 9454–9461 CrossRef PubMed.
- X. Wang, X. Chen, R. Zhou, P. Hu, K. Huang and P. Chen, Anal. Chem., 2021, 93(8), 3889–3897 CrossRef CAS PubMed.
- J. W. Olesik, J. A. Hartshorne, N. Casey, E. Linard and J. R. Dettman, Spectrochim. Acta, Part B, 2021, 176, 106038 CrossRef CAS.
- A. A. Rocha, L. da Costa, C. Duyck, J. R. Barbosa and R. Lorencatto, Anal. Chim. Acta, 2020, 1140, 41–49 CrossRef CAS PubMed.
- J. Kocic, D. Günther and B. Hattendorf, J. Anal. At. Spectrom., 2021, 36(1), 233–242 RSC.
- E. Bolea, M. S. Jimenez, J. Perez-Arantegui, J. C. Vidal, M. Bakir, K. Ben-Jeddou, A. C. Gimenez-Ingalaturre, D. Ojeda, C. Trujillo and F. Laborda, Anal. Methods, 2021, 13(25), 2742–2795 RSC.
- M. Tharaud, P. Louvat and M. F. Benedetti, Anal. Bioanal. Chem., 2021, 413(3), 923–933 CrossRef CAS PubMed.
- D. Rosenkranz, F. L. Kriegel, E. Mavrakis, S. A. Pergantis, P. Reichardt, J. Tentschert, N. Jakubowski, P. Laux, U. Panne and A. Luch, J. Visualized Exp., 2020,(163), e61653 Search PubMed.
- K.-H. Chun and W.-T. Chan, J. Anal. At. Spectrom., 2021, 36(6), 1261–1271 RSC.
- V. Kinnunen, S. Perämäki and R. Matilainen, Spectrochim. Acta, Part B, 2021, 177, 106104 CrossRef CAS.
- S. Meyer, R. Gonzalez de Vega, X. Xu, Z. Du, P. A. Doble and D. Clases, Anal. Chem., 2020, 92(22), 15007–15016 CrossRef CAS PubMed.
- P.-E. Peyneau, Spectrochim. Acta, Part B, 2021, 178, 106126 CrossRef CAS.
- M. Corte-Rodríguez, R. Álvarez-Fernández, P. García-Cancela, M. Montes-Bayón and J. Bettmer, TrAC, Trends Anal. Chem., 2020, 132, 116042 CrossRef.
- X. Pan, Y. Lin, Y. Su, J. Yang, L. He, Y. Deng, X. Hou and C. Zheng, Anal. Chem., 2021, 93(23), 8257–8264 CrossRef CAS PubMed.
- Q. He, X. Wang, H. He and J. Zhang, J. Anal. At. Spectrom., 2020, 35(11), 2704–2711 RSC.
- M. Liu, L. Ding, J. Liu, X. Mao, X. Na and Y. Shao, Anal. Sci., 2021, 37(2), 321–327 CrossRef CAS PubMed.
- Y. Zhang, X. Mao, D. Tian, J. Liu and C. Li, Anal. Methods, 2021, 13(36), 4079–4089 RSC.
- J. Hu, H. Chen, X. Jiang and X. Hou, Anal. Chem., 2021, 93(32), 11151–11158 CrossRef CAS PubMed.
- R. M. de Oliveira, D. L. G. Borges, P. Grinberg, Z. Mester and R. E. Sturgeon, J. Anal. At. Spectrom., 2021, 36(6), 1235–1243 RSC.
- Y. Zhen, H. Chen, M. Zhang, J. Hu and X. Hou, Appl. Spectrosc. Rev., 2021, 1–20 Search PubMed.
- R. M. de Oliveira, D. L. G. Borges, P. Grinberg and R. E. Sturgeon, J. Anal. At. Spectrom., 2021, 36(10), 2097–2106 RSC.
- R. M. Oliveira, B. S. Soares and D. L. G. Borges, J. Food Compos. Anal., 2021, 95, 103655 CrossRef CAS.
- Y. Yu, H. Chen, Q. Zhao, Q. Mou, L. Dong, R. Wang, Z. Shi and Y. Gao, Anal. Chem., 2021, 93(7), 3343–3352 CrossRef CAS PubMed.
- P. Maung and D. Beauchemin, J. Anal. At. Spectrom., 2021, 36(6), 1104–1111 RSC.
- P. Pohl, P. Jamroz, K. Greda, M. Gorska, A. Dzimitrowicz, M. Welna and A. Szymczycha-Madeja, Anal. Chim. Acta, 2021, 1169, 338399 CrossRef CAS PubMed.
- P. Zheng, Y. Luo, J. Wang, Q. Hu, Y. Yang, X. Mao and C. Lai, J. Anal. At. Spectrom., 2021, 36(6), 1228–1234 RSC.
- T. J. Williams and R. K. Marcus, Spectrochim. Acta, Part B, 2021, 179, 105994 CrossRef CAS.
- R. Kenneth Marcus, E. D. Hoegg, K. A. Hall, T. J. Williams and D. W. Koppenaal, Mass Spectrom. Rev., 2021 DOI:10.1002/mas.21720.
- T. J. Williams, J. R. Bills and R. K. Marcus, J. Anal. At. Spectrom., 2020, 35(11), 2475–2478 RSC.
- A. V. Kuptsov, A. V. Volzhenin, V. A. Labusov and A. I. Saprykin, J. Anal. At. Spectrom., 2020, 35(11), 2600–2605 RSC.
- A. V. Kuptsov, A. V. Volzhenin, V. A. Labusov and A. I. Saprykin, J. Anal. At. Spectrom., 2021, 36(4), 829–835 RSC.
- A. V. Kuptsov, A. V. Volzhenin, V. A. Labusov and A. I. Saprykin, Spectrochim. Acta, Part B, 2021, 177, 106047 CrossRef CAS.
- G. Fröhlich, J. Anal. Chem., 2021, 76(1), 112–128 CrossRef.
- A. Priebe, T. Xie, L. Pethö and J. Michler, J. Anal. At. Spectrom., 2020, 35(12), 2997–3006 RSC.
- K. J. Suski, D. M. Bell, M. K. Newburn, M. L. Alexander, D. Imre, D. W. Koppenaal and A. Zelenyuk, Spectrochim. Acta, Part B, 2021, 179, 106092 CrossRef CAS.
- M. Holá, Z. Salajková, A. Hrdlička, J. Ondráček, K. Novotný, D. Pavliňák, M. Vojtíšek-Lom, L. Čelko, P. Pořízka, V. Kanický, D. Prochazka, J. Novotný and J. Kaiser, J. Anal. At. Spectrom., 2020, 35(12), 2893–2900 RSC.
- M. Šala, V. S. Šelih, C. C. Stremtan, T. Tămaş and J. T. van Elteren, J. Anal. At. Spectrom., 2021, 36(1), 75–79 RSC.
- Z. Salajkova, M. Hola, D. Prochazka, J. Ondracek, D. Pavlinak, L. Celko, F. Gregar, P. Sperka, P. Porizka, V. Kanicky, A. De Giacomo and J. Kaiser, Talanta, 2021, 222, 121512 CrossRef CAS PubMed.
- T. R. Holbrook, D. Gallot-Duval, T. Reemtsma and S. Wagner, J. Anal. At. Spectrom., 2021, 36(10), 2107–2115 RSC.
- C. A. Norris, L. Danyushevsky, P. Olin and N. R. West, J. Anal. At. Spectrom., 2021, 36(4), 733–739 RSC.
- J. T. van Elteren, D. Metarapi, M. Šala, V. S. Šelih and C. C. Stremtan, J. Anal. At. Spectrom., 2020, 35(11), 2494–2497 RSC.
- T. Van Acker, S. J. M. Van Malderen, T. Van Helden, C. Stremtan, M. Šala, J. T. van Elteren and F. Vanhaecke, J. Anal. At. Spectrom., 2021, 36(6), 1201–1209 RSC.
- L. N. Zheng, L. X. Feng, J. W. Shi, H. Q. Chen, B. Wang, M. Wang, H. F. Wang and W. Y. Feng, Anal. Chem., 2020, 92(21), 14339–14345 CrossRef CAS PubMed.
- L. Qiao, Z. Wu, Y. Li and Y. Xu, Microchem. J., 2020, 159, 105474 CrossRef CAS.
- V. Balaram, Microchem. J., 2020, 159, 105483 CrossRef CAS.
- G. Niu, A. Knodel, S. Burhenn, S. Brandt and J. Franzke, Anal. Chim. Acta, 2021, 1147, 211–239 CrossRef CAS PubMed.
- B. T. Buckley, R. Buckley and C. L. Doherty, Molecules, 2021, 26(16), 4761 CrossRef CAS PubMed.
- L. He, P. Li, K. Li, T. Lin, J. Luo, X. Hou and X. Jiang, J. Anal. At. Spectrom., 2021, 36(6), 1193–1200 RSC.
- Y. Deng, J. Hu, M. Li, L. He, K. Li, X. Hou and X. Jiang, Anal. Chim. Acta, 2021, 1163, 338502 CrossRef CAS PubMed.
- S. Liu, X. X. Xue, Y. L. Yu and J. H. Wang, Anal. Chem., 2021, 93(15), 6262–6269 CrossRef CAS PubMed.
- O. Gazeli, C. Lazarou, G. Niu, C. Anastassiou, G. E. Georghiou and J. Franzke, Spectrochim. Acta, Part B, 2021, 182, 106248 CrossRef CAS.
- B. J. Farcy, R. D. Arevalo, M. Taghioskoui, W. F. McDonough, M. Benna and W. B. Brinckerhoff, J. Anal. At. Spectrom., 2020, 35(11), 2740–2747 RSC.
- G. Grindlay, S. Alavi and J. Mostaghimi, J. Anal. At. Spectrom., 2020, 35(12), 2956–2963 RSC.
- T. Vonderach, B. Hattendorf and D. Gunther, Anal. Chem., 2021, 93(2), 1001–1008 CrossRef CAS PubMed.
- G. Craig, H. Wehrs, D. G. Bevan, M. Pfeifer, J. Lewis, C. D. Coath, T. Elliott, C. Huang, N. S. Lloyd and J. B. Schwieters, Anal. Chem., 2021, 93(30), 10519–10527 CrossRef CAS PubMed.
- A. Safi, S. M. Aberkane, A. Botto, B. Campanella, S. Legnaioli, F. Poggialini, S. Raneri, F. Rezaei and V. Palleschi, Appl. Spectrosc., 2021, 75(6), 654–660 CrossRef CAS PubMed.
- P. V. Vashchenko, V. A. Labusov, V. G. Garanin and A. V. Borisov, Inorg. Mater., 2020, 56(14), 1441–1445 CrossRef CAS.
- M. Fikry, I. A. Alhijry, A. M. Aboulfotouh and A. M. El Sherbini, Appl. Spectrosc., 2021, 75(10), 1288–1295 CrossRef CAS PubMed.
- R. Li, Y. Liu, M. Mostamand and J. Lassen, Spectrochim. Acta, Part B, 2021, 175, 106017 CrossRef CAS.
- J. L. Jacobs and R. S. Houk, Spectrochim. Acta, Part B, 2021, 176, 106042 CrossRef CAS.
- T. K. Nurubeyli, Tech. Phys., 2020, 65(12), 1963–1968 CrossRef CAS.
- C. Sánchez, R. Sánchez, C.-P. Lienemann and J.-L. Todolí, J. Anal. At. Spectrom., 2021, 36(10), 2085–2096 RSC.
- S. M. Javid, S. Alavi, X. Guo, A. Zabihihesari and J. Mostaghimi, Spectrochim. Acta, Part B, 2021, 180, 106182 CrossRef CAS.
- K.-B. Chai and D.-H. Kwon, Spectrochim. Acta, Part B, 2021, 183, 106269 CrossRef CAS.
- H. Horita, D. Kuwahara, H. Akatsuka and S. Shinohara, AIP Adv., 2021, 11(7), 075226 CrossRef CAS.
- Y. Zhu, R. E. Russo and G. C. Y. Chan, Spectrochim. Acta, Part B, 2021, 179, 106089 CrossRef CAS.
- V. Gonzalez-Fernandez, A. Steiger, K. Grützmacher and M. I. de la Rosa, Spectrochim. Acta, Part B, 2020, 172, 105972 CrossRef CAS.
- K. Wagatsuma, Spectrochim. Acta, Part B, 2021, 175, 106018 CrossRef CAS.
- S. Shi, K. Finch and G. Gamez, J. Anal. At. Spectrom., 2021, 36(5), 1055–1073 RSC.
- C. Tian, N. Ahlmann, S. Brandt, J. Franzke and G. Niu, Spectrochim. Acta, Part B, 2021, 181, 106222 CrossRef CAS.
- S. P. Badal, P. B. Farnsworth, G. C. Y. Chan, B. T. Molnar, J. R. Hellinger and J. T. Shelley, Spectrochim. Acta, Part B, 2021, 176, 106043 CrossRef CAS.
- G. C. Y. Chan and G. M. Hieftje, Spectrochim. Acta, Part B, 2021, 176, 106035 CrossRef CAS.
- A. Brandt, B. Gómez-Nieto, J. Friedland, R. Güttel and K. Leopold, Spectrochim. Acta, Part B, 2020, 173, 105976 CrossRef CAS.
- J. Friedland, A. Brandt, K. Leopold and R. Güttel, Spectrochim. Acta, Part B, 2021, 182, 106249 CrossRef CAS.
- A. Brandt, K. Kees and K. Leopold, J. Anal. At. Spectrom., 2020, 35(11), 2536–2544 RSC.
- V. C. P. Marrocos, R. A. Gonçalves, F. G. Lepri and T. D. Saint’Pierre, Spectrochim. Acta, Part B, 2020, 174, 106008 CrossRef CAS.
- S. Liu, Z. Han, X. Kong, J. Zhang, Z. Lv and G. Yuan, Appl. Spectrosc. Rev., 2021, 1–29 CrossRef.
- S. Diez-Fernández, H. Isnard, A. Nonell, C. Bresson and F. Chartier, J. Anal. At. Spectrom., 2020, 35(12), 2793–2819 RSC.
- V. Balaram, Rapid Commun. Mass Spectrom., 2021, 35(10), e9065 CrossRef CAS PubMed.
- M. C. Lomax-Vogt, F. Liu and J. W. Olesik, Spectrochim. Acta, Part B, 2021, 179, 106098 CrossRef CAS.
- N. Sugiyama, J. Anal. At. Spectrom., 2021, 36(2), 294–302 RSC.
- E. Bolea-Fernandez, A. Rua-Ibarz, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2021, 36(6), 1135–1149 RSC.
- J. A. Carter, L. M. O’Brien, T. Harville, B. T. Jones and G. L. Donati, Talanta, 2021, 223(Pt 2), 121665 CrossRef CAS PubMed.
- R. Serrano, G. Grindlay, L. Gras and J. Mora, Spectrochim. Acta, Part B, 2021, 177, 106070 CrossRef CAS.
- E. V. Polyakova and O. V. Pelipasov, Spectrochim. Acta, Part B, 2020, 173, 105988 CrossRef CAS.
- A. Limbeck, L. Brunnbauer, H. Lohninger, P. Porizka, P. Modlitbova, J. Kaiser, P. Janovszky, A. Keri and G. Galbacs, Anal. Chim. Acta, 2021, 1147, 72–98 CrossRef CAS PubMed.
- G. S. Senesi, R. S. Harmon and R. R. Hark, Spectrochim. Acta, Part B, 2021, 175, 106013 CrossRef CAS.
- A. Matsumoto and T. Sakka, Anal. Sci., 2021, 37(8), 1061–1072 CrossRef CAS PubMed.
- L.-B. Guo, D. Zhang, L.-X. Sun, S.-C. Yao, L. Zhang, Z.-Z. Wang, Q.-Q. Wang, H.-B. Ding, Y. Lu, Z.-Y. Hou and Z. Wang, Frontiers in Physics, 2021, 16(2), 22500 CrossRef.
- Y.-T. Fu, W.-L. Gu, Z.-Y. Hou, S. A. Muhammed, T.-Q. Li, Y. Wang and Z. Wang, Frontiers in Physics, 2021, 16(2), 22502 CrossRef.
- M. S. Afgan, S. Sheta, Y. Song, W. Gu and Z. Wang, J. Anal. At. Spectrom., 2020, 35(11), 2649–2655 RSC.
- C. G. Parigger, Spectrochim. Acta, Part B, 2021, 179, 106122 CrossRef CAS.
- J. Buday, P. Pořízka, M. Buchtová and J. Kaiser, Spectrochim. Acta, Part B, 2021, 182, 106254 CrossRef CAS.
- J. Cai, M. Dong, Y. Zhang, Y. Chen, Y. Liang and J. Lu, Spectrochim. Acta, Part B, 2021, 180, 106195 CrossRef CAS.
- E. Kepes, I. Gornushkin, P. Porizka and J. Kaiser, Analyst, 2021, 146(3), 920–929 RSC.
- N. L. LaHaye, S. S. Harilal and M. C. Phillips, Spectrochim. Acta, Part B, 2021, 179, 106096 CrossRef CAS.
- C. Niu, X. Cheng, T. Zhang, X. Wang, B. He, W. Zhang, Y. Feng, J. Bai and H. Li, Anal. Chem., 2021, 93(4), 2281–2290 CrossRef CAS PubMed.
- P. Purohit, F. J. Fortes and J. J. Laserna, Anal. Chem., 2021, 93(4), 2635–2643 CrossRef CAS PubMed.
- F. J. Fortes, P. Purohit and J. J. Laserna, Spectrochim. Acta, Part B, 2021, 180, 106193 CrossRef CAS.
- T. Delgado, L. García-Gómez, L. M. Cabalín and J. J. Laserna, Spectrochim. Acta, Part B, 2021, 179, 106114 CrossRef CAS.
- M. Gaft, L. Nagli, I. Gornushkin and Y. Raichlin, Spectrochim. Acta, Part B, 2020, 173, 105989 CrossRef CAS.
- G. Galbács, A. Kéri, A. Kohut, M. Veres and Z. Geretovszky, J. Anal. At. Spectrom., 2021, 36(9), 1826–1872 RSC.
- Z. Salajková, V. Gardette, J. Kaiser, M. Dell’Aglio and A. De Giacomo, Spectrochim. Acta, Part B, 2021, 179, 106105 CrossRef.
- M. Abdelhamid, Y. A. Attia and M. Abdel-Harith, J. Anal. At. Spectrom., 2020, 35(12), 2982–2989 RSC.
- Z. Li, K. Wu, J. Shen, W. Zhang, C. Zhou and B. Hu, J. Anal. At. Spectrom., 2021, 36(11), 2346–2352 RSC.
- C. Liu, J. Guo, Y. Tian, C. Zhang, K. Cheng, W. Ye and R. Zheng, Sensors, 2020, 20(24), 7341 CrossRef CAS PubMed.
- Y. Zhang, Y. Tian, Y. Lu, L. Guo, Y. Li, J. Guo and R. Zheng, J. Anal. At. Spectrom., 2021, 36(3), 644–653 RSC.
- F. Huang, Y. Tian, Y. Li, W. Ye, Y. Lu, J. Guo and R. Zheng, Appl. Opt., 2021, 60(6), 1595–1602 CrossRef PubMed.
- Z. Liu, Y. Tian, Y. Lu, J. Guo, Y. Li, W. Ye and R. Zheng, Spectrochim. Acta, Part B, 2021, 180, 106202 CrossRef CAS.
- Y. Jiang, Z. Lin, R. Li and Y. Chen, Spectrochim. Acta, Part B, 2021, 181, 106221 CrossRef CAS.
- Y. Fugane, S. Kashiwakura and K. Wagatsuma, ISIJ Int., 2020, 60(12), 2845–2850 CrossRef CAS.
- W. Huang, C. He, Y. Wang, W. Zhao and L. Qiu, J. Anal. At. Spectrom., 2020, 35(11), 2530–2535 RSC.
- M. Hu, J. Peng, S. Niu and H. Zeng, Adv. Photonics, 2020, 2(06), 065001 CAS.
- L. Jiang, M. Sui, Y. Fan, H. Su, Y. Xue and S. Zhong, Spectrochim. Acta, Part B, 2021, 177, 106065 CrossRef CAS.
- V. S. Dhanada, K. S. Choudhari, S. D. George, V. B. Kartha, S. Chidangil and V. K. Unnikrishnan, J. Anal. At. Spectrom., 2021, 36(11), 2391–2403 RSC.
- V. Costa, D. Babos, J. Castro, D. Andrade, R. Gamela, R. Machado, M. Sperança, A. Araújo, J. Garcia and E. Pereira-Filho, J. Braz. Chem. Soc., 2021, 31(12), 2439–2451 Search PubMed.
- L.-N. Li, X.-F. Liu, F. Yang, W.-M. Xu, J.-Y. Wang and R. Shu, Spectrochim. Acta, Part B, 2021, 180, 106183 CrossRef CAS.
- D. Zhang, H. Zhang, Y. Zhao, Y. Chen, C. Ke, T. Xu and Y. He, Appl. Spectrosc. Rev., 2020, 57(2), 89–111 CrossRef.
- E. Ewusi-Annan and N. Melikechi, Spectrochim. Acta, Part B, 2021, 177, 106109 CrossRef CAS.
- J. Castorena, D. Oyen, A. Ollila, C. Legget and N. Lanza, Spectrochim. Acta, Part B, 2021, 178, 106125 CrossRef CAS.
- L. Brunnbauer, J. Gonzalez, H. Lohninger, J. Bode, C. Vogt, M. Nelhiebel, S. Larisegger and A. Limbeck, Spectrochim. Acta, Part B, 2021, 183, 106272 CrossRef CAS.
- E. Képeš, J. Vrábel, P. Pořízka and J. Kaiser, J. Anal. At. Spectrom., 2021, 36(7), 1410–1421 RSC.
- F. Chang, J. Yang, H. Lu and H. Li, J. Anal. At. Spectrom., 2021, 36(8), 1712–1723 RSC.
- E. Clavé, M. Gaft, V. Motto-Ros, C. Fabre, O. Forni, O. Beyssac, S. Maurice, R. C. Wiens and B. Bousquet, Spectrochim. Acta, Part B, 2021, 177, 106111 CrossRef.
- M. Gaft, L. Nagli, A. Gorychev and Y. Raichlin, Spectrochim. Acta, Part B, 2021, 182, 106233 CrossRef CAS.
- C. J. Bracken, G. J. Lanigan, K. G. Richards, C. Muller, S. R. Tracy, R. Well, R. Carolan and P. N. C. Murphy, Rapid Commun. Mass Spectrom., 2021, 35(8), e9049 CrossRef CAS PubMed.
- P. Bueno, M. E. Sbampato, J. W. Neri, J. J. Neto, L. F. N. Barreta, J. R. dos Santos and M. G. Destro, Spectrochim. Acta, Part B, 2021, 181, 106220 CrossRef CAS.
- N. Bouden, J. Villeneuve, Y. Marrocchi, E. Deloule, E. Füri, A. Gurenko, L. Piani, E. Thomassot, P. Peres and F. Fernandes, Frontiers in Earth Science, 2021, 8, 10 CrossRef.
- R. Chakrabarti, S. Mondal, A. D. Jacobson, M. Mills, S. J. Romaniello and H. Vollstaedt, Chem. Geol., 2021, 581, 120398 CrossRef CAS.
- D. Chew, K. Drost, J. H. Marsh and J. A. Petrus, Chem. Geol., 2021, 559, 119917 CrossRef CAS.
- Z. Chu, Front. Chem., 2020, 8, 615839 CrossRef CAS PubMed.
- M. Tulej, A. Neubeck, A. Riedo, R. Lukmanov, V. Grimaudo, N. F. W. Ligterink, M. Ivarsson, W. Bach, C. de Koning and P. Wurz, J. Mass Spectrom., 2020, 55(12), e4660 CrossRef CAS PubMed.
- E. Frères, D. Weis, K. Newman, M. Amini and K. Gordon, Geostand. Geoanal. Res., 2021, 45(3), 501–523 CrossRef.
- A. Reinhard, J. Inglis, R. Steiner, S. LaMont and Z. Palacz, Rapid Commun. Mass Spectrom., 2021, 35(7), e9032 CrossRef CAS PubMed.
- K. Yamamoto, H. Asanuma, H. Takahashi and T. Hirata, J. Anal. At. Spectrom., 2021, 36(3), 668–675 RSC.
- T. Hirata, S. Niki, S. Yamashita, H. Asanuma and H. Iwano, J. Anal. At. Spectrom., 2021, 36(1), 70–74 RSC.
- H.-Q. Huang, M. Guillong, Y. Hu and C. Spandler, J. Anal. At. Spectrom., 2021, 36(4), 836–844 RSC.
- H. Iwano, T. Hirata, J. Hosoi, H. Sakai, Y. Orihashi and T. Danhara, Chem. Geol., 2021, 559, 119903 CrossRef CAS.
- M. Lin, G. Zhang, N. Li, H. Li and J. Wang, Geostand. Geoanal. Res., 2021, 45(2), 265–285 CrossRef CAS.
- U. Schaltegger, M. Ovtcharova, S. P. Gaynor, B. Schoene, J. F. Wotzlaw, J. Davies, F. Farina, N. D. Greber, D. Szymanowski and C. Chelle-Michou, J. Anal. At. Spectrom., 2021, 36(7), 1466–1477 RSC.
- X. Liu, Z. Zhu, C. Yang, G. Wang, H. Zheng and S. Hu, ACS Earth Space Chem., 2021, 5(7), 1762–1771 CrossRef CAS.
- K. Nagaishi, R. Nakada and T. Ishikawa, Geochem. J., 2021, 55(1), 1–9 CrossRef CAS.
- Y. Di, Z. Li and Y. Amelin, J. Anal. At. Spectrom., 2021, 36(7), 1489–1502 RSC.
- M. Sun, C. Archer and D. Vance, Geostand. Geoanal. Res., 2021, 45(4), 643–658 CrossRef CAS.
- A. S. Henn, S. M. Chernonozhkin, F. Vanhaecke and E. M. M. Flores, J. Anal. At. Spectrom., 2021, 36(7), 1478–1488 RSC.
- X. Li and G. Han, J. Anal. At. Spectrom., 2021, 36(3), 676–684 RSC.
- S.-G. Yim, M.-J. Jung, Y.-J. Jeong, Y. Kim and A. C.-s. Cheong, J. Anal. Sci. Technol., 2021, 12(1), 10 CrossRef CAS.
- M. Buisson, P. Louvat, C. Thaler and C. Rollion-Bard, J. Anal. At. Spectrom., 2021, 36(10), 2116–2131 RSC.
- D. Evans, A. Gerdes, D. Coenen, H. R. Marschall and W. Müller, J. Anal. At. Spectrom., 2021, 36(8), 1607–1617 RSC.
- P. Chalk, C. J. Smith, D. Chen and J.-Z. He, Arch. Agron. Soil Sci., 2020, 68(4), 561–578 CrossRef.
- D. Malinovsky, P. J. H. Dunn and H. Goenaga-Infante, J. Anal. At. Spectrom., 2020, 35(11), 2723–2731 RSC.
- H. Chen, N. J. Saunders, M. Jerram and A. N. Halliday, Chem. Geol., 2021, 578, 120281 CrossRef CAS.
- F. Moynier, Y. Hu, K. Wang, Y. Zhao, Y. Gérard, Z. Deng, J. Moureau, W. Li, J. I. Simon and F.-Z. Teng, Chem. Geol., 2021, 571, 120144 CrossRef CAS.
- H.-O. Gu and H. Sun, J. Anal. At. Spectrom., 2021, 36(11), 2545–2552 RSC.
- H. O. Gu, H. Sun, C. Huang, F. Wang and C. Ge, Rapid Commun. Mass Spectrom., 2021, 35(13), e9105 CrossRef CAS PubMed.
- H. Yuan, At. Spectrosc., 2021, 42(5), 282–293 CrossRef.
- M. N. Decraene, J. Marin-Carbonne, A. S. Bouvier, J. Villeneuve, N. Bouden, B. Luais and E. Deloule, Rapid Commun. Mass Spectrom., 2021, 35(3), e8986 CrossRef CAS PubMed.
- R. Grigoryan, M. Costas-Rodriguez, P. Santens and F. Vanhaecke, Anal. Chem., 2020, 92(24), 15975–15981 CrossRef CAS PubMed.
- A. Sadekov, N. S. Lloyd, S. Misra, J. P. D’Olivo and M. McCulloch, Rapid Commun. Mass Spectrom., 2020, 34(23), e8918 CrossRef CAS PubMed.
- J. Karasinski, A. Tupys, L. Yang, Z. Mester, L. Halicz and E. Bulska, Anal. Chem., 2020, 92(24), 16097–16104 CrossRef CAS PubMed.
- A. B. Kaufmann, M. Lazarov, S. Kiefer, J. Majzlan and S. Weyer, J. Anal. At. Spectrom., 2021, 36(7), 1554–1567 RSC.
- S. Li, Y. Deng, H. Zheng, X. Liu, P. Tang, J. Zhou and Z. Zhu, J. Anal. At. Spectrom., 2021, 36(1), 157–164 RSC.
- C.-F. Li, Z.-Y. Chu, X.-C. Wang, L.-J. Feng and J.-H. Guo, At. Spectrosc., 2020, 41(6), 249–255 CAS.
- H.-C. Liu, Y.-H. Chen, C.-F. You and C.-H. Chung, J. Anal. At. Spectrom., 2021, 36(11), 2322–2329 RSC.
- J. Lu and W. Chen, At. Spectrosc., 2020, 41(6), 223–233 CrossRef CAS.
- M. Oelze, D. A. Frick and S. A. Gleeson, J. Anal. At. Spectrom., 2021, 36(6), 1118–1124 RSC.
- H. Peng, D. He, R. Guo, X. Liu, N. S. Belshaw, H. Zheng, S. Hu and Z. Zhu, J. Anal. At. Spectrom., 2021, 36(2), 390–398 RSC.
- W. Bu, M. Gu, X. Ding, Y. Ni, X. Shao, X. Liu, C. Yang and S. Hu, J. Anal. At. Spectrom., 2021, 36(11), 2330–2337 RSC.
- P. Lindahl, G. Olszewski and M. Eriksson, J. Anal. At. Spectrom., 2021, 36(10), 2164–2172 RSC.
- H. T. Lin, H. W. Chiang, T. L. Yu, M. Christl, J. Liu, K. DeLong and C. C. Shen, Anal. Chem., 2021, 93(24), 8442–8449 CrossRef CAS PubMed.
- A. M. Sánchez Hernández, J. Horta Domenech, D. Wojnowski and E. Zuleger, Int. J. Mass Spectrom., 2021, 468, 116660 CrossRef.
- J. Gao, C. Xu and Y. Zhao, Spectrochim. Acta, Part B, 2021, 182, 106252 CrossRef CAS.
- C. Ito, R. Shimode, T. Miyazaki, S. Wakaki, K. Suzuki and Y. Takagai, Anal. Chem., 2020, 92(24), 16058–16065 CrossRef CAS PubMed.
- J. Louis-Jean, J. D. Inglis, S. Hanson, A. Pollington, D. Meininger, S. Reilly and R. Steiner, J. Radioanal. Nucl. Chem., 2021, 327(1), 317–327 CrossRef CAS.
- K. J. Mathew, C. F. Ottenfeld, R. C. Keller, K. J. Kuhn and J. B. Fulwyler, Int. J. Mass Spectrom., 2020, 458, 116430 CrossRef CAS.
- J. Kim, J.-Y. Kim, S.-E. Bae, K. Song and J.-H. Park, Microchem. J., 2021, 169, 106476 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |