Novel thiol-functionalized covalent organic framework-enabled ICP-MS measurement of ultra-trace metals in complex matrices†
Received
5th October 2021
, Accepted 30th November 2021
First published on 1st December 2021
Abstract
The measurement of ultra-trace levels of heavy metals in complex matrices is of great concern and challenge in life and environmental science. Herein, a novel thiol-functionalized covalent organic framework (COF) was prepared, and quantitative preconcentration of trace heavy metals in environmental water samples was achieved. The material was synthesized by a two-step reaction, including a Schiff base reaction between 1,3,5-triformylphloroglucinol (Tp) and 2,5-divinyl-p-phenylenediamine (DvPa) to form a preliminary COF (TpDvPa), and a thiol–ene “click” reaction between 1,2-ethanedithiol and the vinyl groups of TpDvPa to produce thiol-functionalized TpDvPa (TpDvPa-S-SH). By packing the material into a home-made SPE micro-column, and hyphenating the micro-column with inductively coupled plasma mass spectrometry (ICP-MS), trace levels of Co2+, Ni2+ and Cd2+ in environmental waters could be successfully preconcentrated and measured, with limits of detection of 1.46, 1.38 and 0.10 ng L−1, respectively. The precisions were all less than 2.7%, with linear ranges extending from 5 to 1000 ng L−1. The proposed method was validated by the analysis of the certified reference material (GBW08608), and measurement of Co2+, Ni2+ and Cd2+ in tap water, reservoir water, effluent of a wastewater treatment plant and sea water, confirming the accuracy and feasibility of the method.
Introduction
As emerging crystalline porous polymer materials, covalent organic frameworks (COFs) have been regarded as promising adsorbents for gas storage initially.1 Owing to the notable intrinsic features, such as highly tunable porosity, ordered channels, optional building blocks, predictable control of structures, functionalization feasibility, large specific surface areas, and good thermal and chemical stability,2–4 COFs have also shown significant advantages in the adsorption and removal of organic pollutants5–8 and heavy metals.5,9–11 For instance, Sun et al.12 developed a mesoporous COF with decoration of sulfur derivatives to remove mercury from air and aqueous solutions. In addition to the superior distribution coefficient (Kd = 2.3 × 109 mL g−1), the developed material provided adsorption capacities of 1350 and 863 mg g−1 for Hg2+ and Hg0, respectively. In order to further improve the adsorption capacity and efficiency, Merí-Bofí et al.13 synthesized a novel imine-linked COF (TPB-DMTP-COF-SH). The material demonstrated extraordinary mercury uptake capacity (4395 mg g−1) and efficiency (removal of 99.98% of Hg2+ in 2 min). Although it has not been widely reported,14–16 the novel functionalized COFs could be expected to play more important roles in quantitative preconcentration of trace heavy metals in complex matrices.
At present, inductively coupled plasma mass spectrometry (ICP-MS) is regarded as the most sensitive instrument for the determination of heavy metals.17 However, the concentration of heavy metals in real samples is close to or even less than the detection limit of ICP-MS. For example, as the significant regulators of marine ecosystem dynamics and carbon cycling, cobalt, nickel and cadmium are only 0.18–18.06, 122.45–734.70 and 0.11–118.93 ng L−1 in the ocean, respectively.18 In addition, the interference of the coexisting matrices severely affects the accuracy and sensitivity of the instruments.19 The isobaric and polyatomic species originating from the sample matrix, solvent medium or plasma gas could lead to severe spectral overlap in ICP-MS.20 The high concentration of salts might also cause irreversible damage and reduce the service life of ICP-MS.21 The direct determination of trace levels of heavy metals in complex samples, such as in sea water and waste water, is still a challenge. Therefore, prior to ICP-MS analysis, the separation and preconcentration of trace heavy metals are necessary.22,23
Solid phase extraction (SPE) is suitable for on-line preconcentration of heavy metal ions, along with high repeatability and low consumption of solvents.24,25 As the essential part of SPE, selective and efficient adsorbents are of great concern in materials and separation science.14 Herein, a novel thiol-functionalized COF is designed and applied to the preconcentration of trace heavy metals in real water samples prior to ICP-MS analysis. The preliminary COFs (TpDvPa) are produced by a Schiff base reaction between 1,3,5-triformylphloroglucinol (TP) and 2,5-divinyl-p-phenylenediamine (DvPa). Then, the free vinyl groups of TpDvPa are further modified by grafting with thiol. As a β-ketoenamine linked COF, the chemical and thermal stability of the synthesized material should be adequate for SPE.26,27 In addition, the grafted thiol enables the electrostatic adherence of the material with positive metal ions. By packing the material in a micro-column, a novel online preconcentration technique (TpDvPa-S-SH based SPE-ICP-MS) for trace levels of Co2+, Ni2+ and Cd2+ is developed. Consequently, the analytical performance of ICP-MS is significantly improved.
Experimental
Chemicals and materials
All chemicals used were at least of analytical grade. Deionized (DI) water (18.2 MΩ cm, obtained from a Milli-Q water purification system, Billerica, USA) was used to prepare/dilute the solutions and water samples. The element stock standard solutions of Co2+, Ni2+ and Cd2+ (1000 mg L−1) were purchased from Beijing Weiye Research Institute of Metrology and Technology (Beijing, China). Other metal solutions used for investigation of the potential interfering effects of various cationic species, i.e., K+, Na+, Ca2+, Mg2+, Zn2+, Cu2+, Mn2+, Pb2+, Ba2+, Sr2+, Al3+ and Fe3+, were prepared from corresponding salts of these species (Sinopharm chemical reagent Co., Ltd, Shanghai, China). Nitric acid (HNO3, Sigma-Aldrich trading Co., Ltd, Shanghai, China) with appropriate dilution was employed as the eluent. Nitric acid and ammonium hydroxide (NH3·H2O, Sinopharm chemical reagents Co., Ltd, Shanghai, China) were used to adjust the pH of the sample solutions. 1,3,5-Trimethylphloroglucinol (Tp, Tokyo chemical industry Co., Ltd, Shanghai, China) and 2,5-divinyl-p-phenylenediamine (DvPa, Jilin Chinese academy of sciences-yanshen technology Co., Ltd, Jilin, China) were used to synthesize the preliminary COF. 1,3,5-Trimethylbenzene (mesitylene), 1,4-dioxane, acetic acid (AcOH), acetone, azobisisobutyronitrile (AIBN), benzotrifluoride, 1,2-ethanedithiol and other solvents were obtained from Sinopharm chemical reagent Co., Ltd (Shanghai, China). Polytetrafluoroethylene (PTFE) tubes and joints of the flow injection system were purchased from Nanjing runze fluid control equipment Co., Ltd (Jiangsu, China).
Instrumentation
The target metal ions (Co2+, Ni2+ and Cd2+) were determined using an Agilent 7900 ICP-MS (Agilent, USA) with the parameters as listed in Table S1.† The synthesized material was systematically characterized by field emission scanning electron microscopy (FE-SEM, Hitachi S-4800, Hitachi, Japan), powder X-ray diffraction (PXRD, Bruker D8 AVANCE, Bruker, Germany), Fourier transform infrared spectroscopy (FT-IR, PerkinElmer Frontier, PerkinElmer, USA), 13C cross-polarization with magic-angle spinning (CP/MAS) solid-state nuclear magnetic resonance (NMR) spectroscopy (13C CP/MAS NMR, Agilent 600M, Agilent, USA), and X-ray photoelectron spectroscopy (XPS, Thermo ESCALAB XI+, Thermo, USA). In addition, the nitrogen adsorption and desorption isotherms of the synthesized material were measured at 77 K using an ASAP 2460 (Micromeritics, USA). The material was outgassed in a vacuum at 120 °C for 10 h before the measurement. Its specific surface area was calculated by using the Brunauer–Emmett–Teller (BET) model. The pore volume and pore size distribution were derived from the adsorption branches of isotherms by using the nonlocal density functional theory (NLDFT). The pH values of the sample solutions were measured using a PHS-25 pH meter (INESA Scientific Instrument Co., Ltd, Shanghai, China). The solutions were delivered using a LabM1 peristaltic pump (PP, Baoding shenchen precision pump Co., Ltd, Hebei, China). A homemade online filter with a 0.22 μm mixed cellulose ester membrane (Tianjin jinteng experiment equipment Co., Ltd, Tianjin, China) was used for online filtration of the fluids before ICP-MS detection.
Synthesis of TpDvPa-S-SH
TpDvPa-S-SH was prepared according to a reported method12,26 with some modifications (Fig. 1). Briefly, 21 mg of Tp, 24 mg of DvPa, 4.5 mL of 1,3,5-trimethylbenzene (mesitylene), 4.5 mL of 1,4-dioxane and 0.5 mL of acetic acid (AcOH) (9 mol L−1) were added into a Teflon-lined stainless steel autoclave, and heated for 72 hours at 120 °C to produce the preliminary COF (TpDvPa). The precipitated products of TpDvPa were collected by filtration and washed with acetone at least 3 times. Then, the obtained TpDvPa was dried at 60 °C for 12 h. The thiol–ene “click” reaction was carried out by mixing of 845 mg of 1,2-ethanedithiol, 50 mg of TpDvPa, 10 mg of azobisisobutyronitrile (AIBN) and 4.0 mL of benzotrifluoride at 80 °C for 48 hours under the protection of nitrogen. The produced thiol-functionalized covalent organic framework (TpDvPa-S-SH) was washed with acetone at least 3 times, and then collected by filtration (a yield of c.a. 68% was obtained).
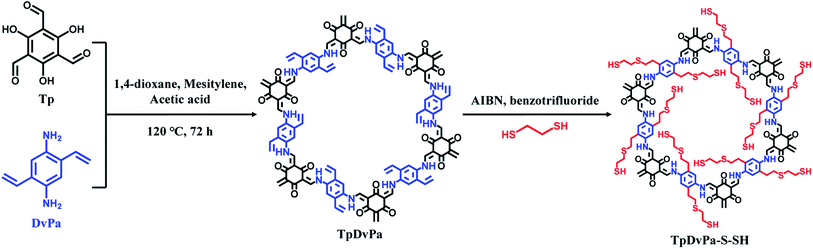 |
| | Fig. 1 Schematic illustration of the synthesis of TpDvPa-S-SH. | |
Device construction
The flow injection preconcentration system was assembled using a peristaltic pump (PP), a SPE micro-column and an online filter (Fig. 2). The PP was employed for the delivery of the sample and eluent passing through the SPE micro-column and the online filter. The eluate was injected into the ICP-MS (operational parameters are shown in Table S1†) for measurement of the metal ions. The SPE micro-column was fabricated with a piece of PTFE tube (O.D.: 2.6 mm, I.D.: 1.8 mm, length: 4 mm). The adsorbents (2 mg) were packed in the center of the PTFE tube, with two ends blocked by some silica wool. The SPE micro-column was connected with the PP and online filter using another two pieces of PTFE tubes (O.D.: 2.0 mm, I.D.: 1.0 mm). The online filter was homemade by referring to a previous study.28 The used membrane in the online filter was a 0.22 μm mixed cellulose ester membrane. Before use, the SPE micro-column was thoroughly cleaned by passing through 5 mL of acetone, HNO3 (1 mol L−1) and deionized (DI) water successively to eliminate potential matrices of the micro-column.
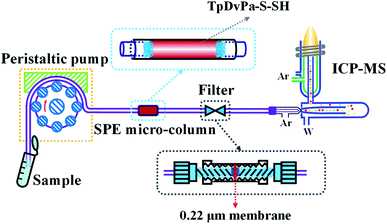 |
| | Fig. 2 Schematic representation of the TpDvPa-S-SH based SPE-ICP-MS system for the preconcentration and determination of Co2+, Ni2+ and Cd2+. | |
Procedures of the SPE preconcentration
The pretreated 5 mL of sample was pumped to flow through the SPE micro-column at a flow rate of 0.5 mL min−1. Then, 0.2 mL of HNO3 (0.5 mol L−1) was used to elute the SPE micro-column at a flow rate of 0.25 mL min−1. The eluate was delivered to flow through the online filter, and introduced into ICP-MS for measurement of the metal ions. The SPE micro-column was thoroughly flushed with 5 mL of HNO3 (1 mol L−1) to eliminate the residual metal ions and other coexisting species. Finally, the SPE micro-column was flushed to neutral using 3 mL of DI water at a flow rate of 0.5 mL min−1 for the next run of preconcentration.
Sampling and pretreatment
Tap water was obtained from the lab of Qingdao University of Technology (QUT, Qingdao, China). Reservoir water was obtained from the Shipeng Reservoir (Qingdao, China). Effluent of wastewater treatment plant was obtained from the Licun River Wastewater Treatment Plant (Qingdao, China). Sea water was obtained from the seaside of the May fourth square of Qingdao. Certified reference material, GBW08608 was purchased from Beijing Weiye Research Institute of Metrology and Technology (Beijing, China). All the water samples were filtered through a 0.22 μm mixed cellulose ester membrane filter and stored at 4 °C in a refrigerator. The sample filtrates were diluted properly with DI water and adjusted to pH = 6 before analysis.
Results and discussion
Characterization of TpDvPa-S-SH
The prepared material was characterized to be fibrous crystals by field emission scanning electron microscopy (FE-SEM, Fig. 3a) and powder X-ray diffraction (PXRD, Fig. 3b). The diameter of the fibrous crystal was approximately 0.02–0.05 μm. Similar to TpDvPa and the reported COFs,26,29 TpDvPa-S-SH exhibited a PXRD pattern with a prominent diffraction peak at 2θ = 4.7°, which was assigned to the (100) plane diffraction. In addition, other diffraction peaks at 10.2°, 11.9° and 27.1° corresponded to the (002), (210) and (001) planes, respectively. The broadened PXRD peaks of TpDvPa-S-SH could be attributed to the plenty of flexible chains in the pores of the material.30
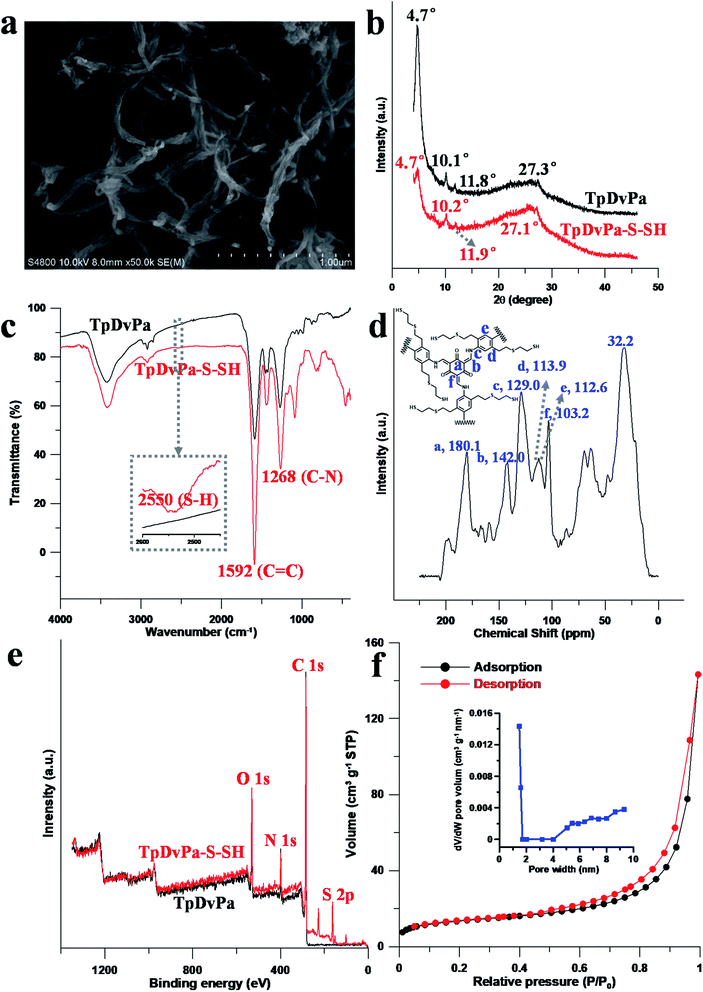 |
| | Fig. 3 (a) FE-SEM image of TpDvPa-S-SH. (b) PXRD patterns of TpDvPa (black) and TpDvPa-S-SH (red). (c) FT-IR spectra of TpDvPa (black) and TpDvPa-S-SH (red). (d) 13C CP/MAS NMR spectra of TpDvPa-S-SH. (e) Comparison of the XPS of TpDvPa (black) and TpDvPa-S-SH (red). (f) Nitrogen adsorption and desorption isotherms, and pore size distribution (inset) of TpDvPa-S-SH. | |
The synthesized TpDvPa-S-SH and TpDvPa were also characterized by Fourier transform infrared (FT-IR) spectroscopy and 13C cross-polarization/magic-angle spinning (CP/MAS) solid-state nuclear magnetic resonance (NMR) spectroscopy. Typical stretching band peaks of C–N (1268 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1592 cm−1) were obtained (Fig. 3c), indicating the β-ketoenamine structure of TpDvPa-S-SH and TpDvPa. As reported, the CO peak of TpDvPa-S-SH might merge with the CC stretching band at 1592 cm−1,26 therefore the separated CO peak was not observed in the FT-IR spectra. In addition, the characteristic stretching bands of O–H and CN of the enol form were not observed in the spectra, which also proved that the C–C and CN bonds on the skeleton were transformed into the CC and C–N bonds through tautomerism. The band of S–H (2550 cm−1) appeared in the FT-IR spectra of TpDvPa-S-SH, confirming the successful modification of TpDvPa with thiol groups. In the 13C CP/MAS NMR spectra (Fig. 3d), the characteristic peaks at 180.1, 142.0 and 103.2 ppm corresponding to the CO, C–N and CC carbon were observed (i.e., “a, b and f” carbon in the inset of Fig. 3d), which also proved the formation of the β-ketoenamine structure by tautomerization. The peaks around 32.2 ppm could be attributed to the methyl carbon of the thiol groups. The peaks at 129.0, 113.9 and 112.6 ppm could be attributed to the “c, d and e” carbon in the inset of Fig. 3d, respectively. These results all indicated the successful synthesis of TpDvPa-S-SH.
C (1592 cm−1) were obtained (Fig. 3c), indicating the β-ketoenamine structure of TpDvPa-S-SH and TpDvPa. As reported, the CO peak of TpDvPa-S-SH might merge with the CC stretching band at 1592 cm−1,26 therefore the separated CO peak was not observed in the FT-IR spectra. In addition, the characteristic stretching bands of O–H and CN of the enol form were not observed in the spectra, which also proved that the C–C and CN bonds on the skeleton were transformed into the CC and C–N bonds through tautomerism. The band of S–H (2550 cm−1) appeared in the FT-IR spectra of TpDvPa-S-SH, confirming the successful modification of TpDvPa with thiol groups. In the 13C CP/MAS NMR spectra (Fig. 3d), the characteristic peaks at 180.1, 142.0 and 103.2 ppm corresponding to the CO, C–N and CC carbon were observed (i.e., “a, b and f” carbon in the inset of Fig. 3d), which also proved the formation of the β-ketoenamine structure by tautomerization. The peaks around 32.2 ppm could be attributed to the methyl carbon of the thiol groups. The peaks at 129.0, 113.9 and 112.6 ppm could be attributed to the “c, d and e” carbon in the inset of Fig. 3d, respectively. These results all indicated the successful synthesis of TpDvPa-S-SH.
The elemental composition of TpDvPa-S-SH and TpDvPa (Fig. 3e) was determined by X-ray photoelectron spectroscopy (XPS). The results of XPS showed photoelectron lines at binding energies of 530, 398, 284 and 162 eV, which were assigned to O 1s, N 1s, C 1s and S 2p, respectively. Compared with the preliminary COF, the binding energy of S 2p at 162.5 eV was observed in the XPS of TpDvPa-S-SH, further verifying the successful modification of TpDvPa with the thiol group. The porosities of TpDvPa-S-SH were measured by the nitrogen adsorption and desorption experiment at 77 K. TpDvPa-S-SH exhibited the reversible type-I adsorption isotherms (Fig. 3f). The Brunauer–Emmett–Teller (BET) surface area and pore volume of TpDvPa-S-SH were 46 m2 g−1 and 0.19 cm3 g−1. The Langmuir surface area was 194 m2 g−1. The pore size distribution was calculated by using the nonlocal density functional theory (NLDFT), and a major pore size of 1.5 nm was characterized (Fig. 3f, inset).
Optimization of the analytical conditions
The experimental conditions used for adsorption and desorption of Co2+, Ni2+ and Cd2+ were optimized in this study. Because the pH of the solution influenced the surface property of the adsorbent (TpDvPa-S-SH), the effects of the pH of the sample were studied (Fig. 4a). At a low sample pH (i.e., pH = 2), the adsorption efficiencies of TpDvPa-S-SH for Co2+, Ni2+ and Cd2+ were extremely low. The recoveries of the target ions in the eluate were only 6.4, 12.2 and 3.5% for Co2+, Ni2+ and Cd2+, respectively. This might be attributed to the fact that the thiols of TpDvPa-S-SH are highly protonated and thus the binding sites are closely coordinated with H+. The positively charged TpDvPa-S-SH surface repels the adsorption of Co2+, Ni2+ and Cd2+, which results in low efficiencies of preconcentration. With increasing pH of the sample, the binding sites of TpDvPa-S-SH were de-protonated and facilitated the adsorption of positive metal ions. When adjusting the pH of the sample to 6, the highest recoveries of the target ions in the eluate were obtained, i.e., 91.9, 97.6 and 92.0% for Co2+, Ni2+ and Cd2+, respectively. On further increasing the pH of the sample, the metal ions would form hydroxide complexes in an alkaline medium. As a result, the metal adsorption would be supressed, i.e., the recoveries of the target ions in the eluate decreased to 53.3, 74.8 and 63.5% for Co2+, Ni2+ and Cd2+, respectively. Therefore, the sample pH of 6 was selected in this study.
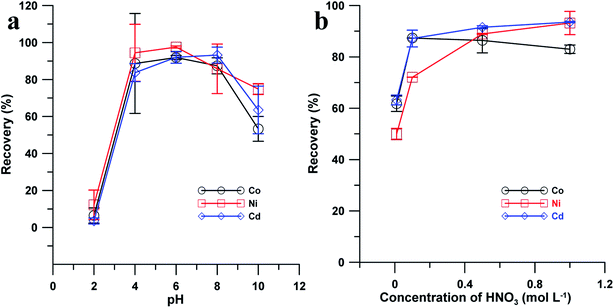 |
| | Fig. 4 The effects of (a) the sample pH and (b) the concentration of HNO3 on the recoveries of Co2+, Ni2+ and Cd2+ retained on the TpDvPa-S-SH micro-column. Sample volume: 5 mL; eluent: 0.2 mL HNO3; Co2+ and Cd2+: 0.01 μg L−1; Ni2+: 0.05 μg L−1. | |
The influences of the concentration levels of the HNO3 solution on the desorption of Co2+, Ni2+ and Cd2+ were tested and shown in Fig. 4b. By increasing the concentration of the HNO3 solution from 0.01 to 0.1 mol L−1, a steep rise of the recoveries of the target metal ions was recorded. By continuously increasing the acid concentration from 0.1 to 1.0 mol L−1, the recoveries of Co2+, Ni2+ and Cd2+ were not significantly changed. By considering the obtained desorption efficiencies and dosage of HNO3, 0.5 mol L−1 HNO3 was selected as the eluent. Other experimental conditions, including the sample flow rate, eluent flow rate, and the amount of the adsorbents were also tested in this study. As a result, the sample flow rate of 0.5 mL min−1 (Fig. S1†), eluent flow rate of 0.25 mL min−1 (Fig. S2†), and the amount of the adsorbents of 2 mg (Fig. S3†) were selected in this method.
The effects of metal ions
The influences of the commonly co-existing ions (e.g., K+, Na+, Ca2+, Mg2+, Zn2+, Cu2+, Mn2+, Pb2+, Ba2+, Sr2+, Al3+ and Fe3+) on the preconcentration of target metals were tested by gradually increasing the amount of coexisting metal ions in a standard solution (Co2+ and Cd2+: 0.01 μg L−1 and Ni2+: 0.05 μg L−1). The results showed that the co-existing ions at the concentration levels of 1000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times of the target metal ions did not interfere with the preconcentration and determination of the analyte within a 10% error range. The results are summarized in Table 1. Generally, for real environmental water samples, the concentration level of the co-existing ions in real water samples or after appropriate dilution would be not higher than the tested levels, therefore the developed SPE method can be directly used without further treatments or masking reagents.
000 times of the target metal ions did not interfere with the preconcentration and determination of the analyte within a 10% error range. The results are summarized in Table 1. Generally, for real environmental water samples, the concentration level of the co-existing ions in real water samples or after appropriate dilution would be not higher than the tested levels, therefore the developed SPE method can be directly used without further treatments or masking reagents.
Table 1 The effects of metal ions on the developed SPE-ICP-MS method
| Metal ions |
Co2+/0.01 μg L−1 |
Ni2+/0.05 μg L−1 |
Cd2+/0.01 μg L−1 |
| Spiked (μg L−1) |
Recovery (%) |
Spiked (μg L−1) |
Recovery (%) |
Spiked (μg L−1) |
Recovery (%) |
| K+ |
100.00 |
90.7 |
500.00 |
92.2 |
100.00 |
91.4 |
| Na+ |
100.00 |
90.1 |
500.00 |
95.5 |
100.00 |
99.9 |
| Ca2+ |
100.00 |
90.4 |
500.00 |
97.8 |
100.00 |
97.2 |
| Mg2+ |
10.00 |
90.1 |
500.00 |
105.4 |
10.00 |
91.3 |
| Zn2+ |
100.00 |
93.6 |
500.00 |
93.6 |
100.00 |
91.1 |
| Cu2+ |
100.00 |
93.0 |
500.00 |
90.6 |
100.00 |
96.3 |
| Mn2+ |
100.00 |
98.0 |
50.00 |
91.2 |
10.00 |
97.4 |
| Pb2+ |
100.00 |
103.0 |
500.00 |
96.9 |
100.00 |
104.1 |
| Ba2+ |
100.00 |
96.6 |
500.00 |
88.5 |
100.00 |
94.5 |
| Sr2+ |
100.00 |
104.2 |
500.00 |
93.6 |
100.00 |
104.4 |
| Al3+ |
10.00 |
90.9 |
500.00 |
92.1 |
100.00 |
90.7 |
| Fe3+ |
100.00 |
97.3 |
500.00 |
94.8 |
100.00 |
91.4 |
Analytical performances
The adsorption capacity of TpDvPa-S-SH was tested to be 131.7, 179.2, 173.3, 227.7, 34.3, 34.8, and 68.8 mg g−1 for Co2+, Ni2+, Cd2+, Pb2+, Ba2+, Sr2+ and Cu2+. Under the optimized conditions, the analytical performances of the TpDvPa-S-SH based SPE-ICP-MS system for detection of Co2+, Ni2+ and Cd2+ were assessed, including the linear ranges, coefficients of determination (R2), limits of detection (LODs), relative standard deviations (RSDs) and enrichment factors (EFs). The obtained results are summarized in Table 2. With a sample volume of 5 mL and a eluent of 0.2 mL (0.5 mol L−1 HNO3), quantitative preconcentration was achieved in linearity ranges of 0.005–0.5 μg L−1 of Co2+ and Cd2+, and 0.05–1.0 μg L−1 of Ni2+. The obtained coefficients of determination (R2) were all ≥0.9997 (Fig. 5). Upon comparison of the direct measurements by ICP-MS (Fig. S4†), the EFs (calculated by comparing the slopes of the calibration curves of this method and the direct measurement method) of 22.7 (Co2+), 24.4 (Ni2+) and 22.8 (Cd2+), along with the limits of detection of 1.46 (Co2+), 1.38 (Ni2+) and 0.10 ng L−1 (Cd2+) were obtained by the developed method. The precisions for the determination of Co2+, Cd2+ and Ni2+ were 2.7, 1.7 and 1.7% (RSD, n = 7) at a concentration of 0.01, 0.01 and 0.05 μg L−1, respectively. In addition, with more than 100 times of adsorption and desorption, the packed SPE micro-column did not show significant deterioration during this study, well indicating the stability of the SPE micro-column. Comparison of the developed SPE-ICP-MS method with other recently reported ICP-MS methods for the determination of heavy metal ions (Table 3) shows that the sensitivity (LOD) and the precision (RSD) of the developed SPE-ICP-MS method were significantly improved.
Table 2 Analytical performances of the developed SPE-ICP-MS method
| Target metal ions |
Co2+ |
Ni2+ |
Cd2+ |
|
C
Co = CCd = 0.01 μg L−1, CNi = 0.05 μg L−1, n = 7.
|
| Linear ranges (μg L−1) |
0.005–0.5 |
0.05–1 |
0.005–0.5 |
| Regression equations |
S = 408896CCo + 617.5 |
S = 88038CNi + 1096.6 |
S = 101170CCd − 267.7 |
| Coefficients of determination |
0.9999 |
0.9999 |
0.9997 |
| Limits of detection (ng L−1) |
1.46 |
1.38 |
0.10 |
| Relative standard deviationsa (%) |
2.7 |
1.7 |
1.7 |
| Enrichment factors |
22.7 |
24.4 |
22.8 |
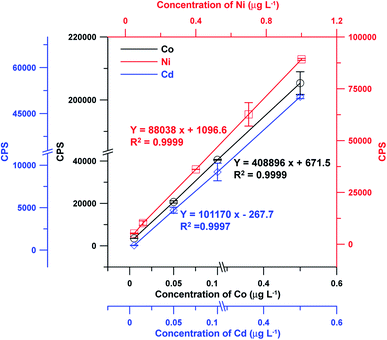 |
| | Fig. 5 The calibration curves of the developed TpDvPa-S-SH based SPE-ICP-MS system for the measurement of Co2+, Ni2+ and Cd2+. | |
Table 3 The comparison of analytical performances of the developed SPE-ICP-MS method and recently reported ICP-MS methods
| Analytes |
Adsorbent |
Method |
Eluent |
LOD (ng L−1) |
RSD (%) |
Ref. |
|
Fe3O4@SiO2@TiO2: Fe3O4 magnetic core-SiO2 and TiO2 double shell system.
MSPE: magnetic solid phase extraction.
CTpBD: carboxylated 1,3,5-triformylphloroglucinol-benzidine covalent organic frameworks.
SPE-FI: solid phase extraction-flow injection.
PPy-CS2: poly (pyrrole-N-carbodithioic acid).
UAD-μSPE: ultrasound-assisted dispersive micro solid-phase extraction.
CHPMs: carboxyl-functionalized hollow polymer microspheres.
SPE: solid phase extraction.
|
| Cu, Zn, Cd, Pb |
Fe3O4@SiO2@TiO2a |
MSPEb |
10 mL 2 mol L−1 HNO3 |
41–82 |
<10 |
31
|
| Cr, Co, Cd, Zn, Pb |
Carbon-coated Fe3O4 nanoparticles with surface amido groups |
MSPEb |
4 mL 2 mol L−1 HNO3 |
1–110 |
6.2–9.4 |
32
|
| Cr, Mn, Co, Ni, Cd, V, Cu, As, Se, Mo |
CTpBDc (COF) |
SPE-FId |
2 mL 0.7 mol L−1 HNO3 |
2.1–21.6 |
1.2–4.3 |
14
|
| Cd, Co, Cu, Ni, Pd, Zn |
PPy-CS2e |
UAD-μSPEf |
1 mL 2 mol L−1 HNO3 |
0.3–11.2 |
2.2–6.3 |
33
|
| V, Cr, Cu, Cd, Pd |
CHPMsg |
SPEh |
3 mL 1 mol L−1 HNO3 |
0.8–3.2 |
1.2–3.5 |
34
|
| Co, Ni, Cd |
TpDvPa-S-SH (COF) |
SPEh |
0.2 mL 0.5 moL L−1 HNO3 |
0.10–1.46 |
1.7–2.7 |
This work |
Validation and applications
The developed TpDvPa-S-SH based SPE-ICP-MS system was validated by the analysis of the certified reference material (GBW08608, contains 2.2 mg L−1 of K+, 23 mg L−1 of Na+, 39 mg L−1 of Ca2+ and 11 mg L−1 of Mg2+ as its matrices) and environmental water samples. The results are summarized in Table 4. It clearly showed that the detected concentration levels of Cd2+ and Ni2+ were in good agreement with the certified values. Because GBW08608 did not contain Co2+, it was spiked with 10.00 μg L−1 of Co2+ for cobalt detection. As a result, 10.12 ± 0.14 μg L−1 of Co2+ was found in the spiked GBW08608, indicating that a satisfactory recovery of 101.1% was obtained. In addition, the developed SPE-ICP-MS method was used to analyze tap water, reservoir water, effluent of a wastewater treatment plant and sea water. It can be seen that trace levels of metal ions were successively detected, i.e., 0.40 ± 0.01 to 3.09 ± 0.11 μg L−1 of Ni2+ and Cd2+ were found. Since the concentration levels of Co2+ were lower than the LOD of the present method, Co2+ was not detected in these samples except in sea water (1.15 ± 0.52 μg L−1). Moreover, acceptable spiking recoveries (87.5–95.4%) were achieved in these waters. These results well indicated the reliability and applicability of the TpDvPa-S-SH based SPE-ICP-MS method for the determination of trace levels of Co2+, Ni2+ and Cd2+ in environmental water samples.
Table 4 The determination of Co2+, Ni2+, Cd2+ in GBW08608, tap water, reservoir water, effluent of a wastewater treatment plant and sea water
| Sample |
Spiked (μg L−1) |
Found (μg L−1) |
Recovery (%) |
| Co2+ |
Ni2+ |
Cd2+ |
Co2+ |
Ni2+ |
Cd2+ |
Co2+ |
Ni2+ |
Cd2+ |
|
Certified values.
—: not spiked.
n.d.: not detected.
|
| GBW08608 |
10.00 |
62.87 ± 2.00a |
12.37 ± 0.50a |
10.12 ± 0.14 |
67.30 ± 1.04 |
12.09 ± 0.65 |
101.2 |
— |
— |
| Tap water |
—b |
— |
— |
n.d.c |
3.09 ± 0.11 |
0.44 ± 0.02 |
— |
— |
— |
| 1.00 |
5.00 |
1.00 |
0.875 ± 0.001 |
7.84 ± 0.01 |
1.38 ± 0.10 |
87.5 |
95.0 |
94.0 |
| Reservoir water |
— |
— |
— |
n.d. |
1.31 ± 0.14 |
0.411 ± 0.002 |
— |
— |
— |
| 1.00 |
5.00 |
1.00 |
0.94 ± 0.01 |
6.08 ± 0.01 |
1.360 ± 0.004 |
94.0 |
95.4 |
94.9 |
| Effluent of a wastewater treatment plant |
— |
— |
— |
n.d. |
1.17 ± 0.16 |
0.40 ± 0.01 |
— |
— |
— |
| 1.00 |
5.00 |
1.00 |
0.906 ± 0.005 |
5.75 ± 0.01 |
1.33 ± 0.03 |
90.6 |
91.6 |
93.0 |
| Sea water |
— |
— |
— |
1.15 ± 0.52 |
6.41 ± 1.04 |
1.70 ± 1.36 |
— |
— |
— |
Conclusion
In this study, a novel COF material (TpDvPa-S-SH) was synthesized and applied for the preconcentration of trace heavy metals in environmental water samples. Owning to the characteristic properties of TpDvPa-S-SH, i.e., the large specific surface area, permanent porosity and abundant thiol groups, the material was illustrated to be an excellent adsorbent for the sorption of Co2+, Ni2+ and Cd2+. In addition, the adsorbed metal ions can be easily eluted using nitric acid. By packing TpDvPa-S-SH in a micro-column and coupling with ICP-MS, trace levels of Co2+, Ni2+ and Cd2+ in complex matrices (such as sea water, tap water, reservoir water and effluent of a wastewater treatment plant) can be quantitatively preconcentrated and detected. The analytical results of the certified reference material (GBW08608) and environmental water samples well verified the accuracy and practicality of the developed method, also suggesting its promising applications.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant numbers 21876181, 21727811 and 22176106).
References
- M. G. Rabbani, A. K. Sekizkardes, Z. Kahveci, T. E. Reich, R. Ding and H. M. El-Kaderi, Chem.–Eur. J., 2013, 19, 3324–3328 CrossRef CAS PubMed.
- K. Y. Geng, T. He, R. Y. Liu, S. Dalapati, K. T. Tan, Z. P. Li, S. S. Tao, Y. F. Gong, Q. H. Jiang and D. L. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- K. Y. Geng, V. Arumugam, H. J. Xu, Y. N. Gao and D. L. Jiang, Prog. Polym. Sci., 2020, 108, 101288 CrossRef CAS.
- L. X. Chen, Q. Wu, J. Gao, H. Li, S. Q. Dong, X. F. Shi and L. Zhao, TrAC, Trends Anal. Chem., 2019, 113, 182–193 CrossRef CAS.
- X. Zhong, Z. P. Lu, W. Liang and B. W. Hu, J. Hazard. Mater., 2020, 393, 122353 CrossRef CAS.
- L. Liu, W. K. Meng, L. Li, G. J. Xu, X. Wang, L. Z. Chen, M. L. Wang, J. M. Lin and R. S. Zhao, Chem. Eng. J., 2019, 369, 920–927 CrossRef CAS.
- W. Gao, Y. Tian, H. Liu, Y. Q. Cai, A. F. Liu, Y. L. Yu, Z. S. Zhao and G. B. Jiang, Anal. Chem., 2019, 91, 772–775 CrossRef CAS PubMed.
- J. L. Liu, E. M. Zong, H. Y. Fu, S. R. Zheng, Z. Y. Xu and D. Q. Zhu, J. Colloid Interface Sci., 2012, 372, 99–107 CrossRef CAS PubMed.
- G. L. Li, J. R. Ye, Q. L. Fang and F. Liu, Chem. Eng. J., 2019, 370, 822–830 CrossRef CAS.
- Y. Z. Jiang, C. Y. Liu and A. S. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 32186–32191 CrossRef CAS PubMed.
- M. H. Zhan, H. M. Jia, J. Y. Fan, H. M. Yu, E. Amador and W. Chen, Anal. Chem., 2019, 91, 6103–6110 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. C. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Q. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS PubMed.
- L. Merí-Bofí, S. Royuela, F. Zamora, M. L. Ruiz-González, J. L. Segura, R. Muñoz-Olivas and M. J. Mancheño, J. Mater. Chem. A, 2017, 5, 17973–17981 RSC.
- J. M. Liu, X. Z. Wang, C. Y. Zhao, J. L. Hao, G. Z. Fang and S. Wang, J. Hazard. Mater., 2018, 344, 220–229 CrossRef CAS PubMed.
- R. X. Bi, F. L. Li, J. B. Chao, H. H. Dong, X. L. Zhang, Z. H. Wang, B. Li and N. Zhao, J. Chromatogr. A, 2021, 1635, 461712 CrossRef CAS PubMed.
- T. Zhang, C. W. Gao, W. Huang, Y. L. Chen, Y. Wang and J. M. Wang, Talanta, 2018, 188, 578–583 CrossRef CAS PubMed.
- Z. R. Liu, X. T. Li, G. Y. Xiao, B. B. Chen, M. He and B. Hu, TrAC, Trends Anal. Chem., 2017, 93, 78–101 CrossRef CAS.
- Y. Sohrin and K. W. Bruland, TrAC, Trends Anal. Chem., 2011, 30, 1291–1307 CrossRef CAS.
- S. H. Tan and G. Horlick, J. Anal. At. Spectrom., 1987, 2, 745–763 RSC.
- T. S. Lum and K. S. Y. Leung, J. Anal. At. Spectrom., 2016, 31, 1078–1088 RSC.
- M. R. Plantz, J. S. Fritz, F. G. Smith and R. S. Houk, Anal. Chem., 1989, 61, 149–153 CrossRef CAS.
- H. Wang, X. L. Liu, K. Nan, B. B. Chen, M. He and B. Hu, J. Anal. At. Spectrom., 2017, 32, 58–77 RSC.
- M. He, L. J. Huang, B. S. Zhao, B. B. Chen and B. Hu, Anal. Chim. Acta, 2017, 973, 1–24 CrossRef CAS PubMed.
- C. K. Su and J. Y. Lin, Anal. Chem., 2020, 92, 9640–9648 CrossRef CAS PubMed.
- L. Chen, Z. Z. Wang, J. X. Pei and X. J. Huang, Anal. Chem., 2020, 92, 2251–2257 CrossRef CAS PubMed.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed.
- W. Gao, J. W. Cheng, X. L. Yuan and Y. Tian, Talanta, 2021, 222, 121501 CrossRef CAS PubMed.
- Y. Tian, M. L. Chen, X. W. Chen, J. H. Wang, Y. Hirano, H. Sakamoto and T. Shirasaki, J. Anal. At. Spectrom., 2011, 26, 133–140 RSC.
- B. P. Biswal, S. Kandambeth, S. Chandra, D. B. Shinde, S. Bera, S. Karak, B. Garai, U. K. Kharul and R. Banerjee, J. Mater. Chem. A, 2015, 3, 23664–23669 RSC.
- F. Xu, H. Xu, X. Chen, D. C. Wu, Y. Wu, H. Liu, C. Gu, R. W. Fu and D. L. Jiang, Angew. Chem., Int. Ed., 2015, 54, 6814–6818 CrossRef CAS PubMed.
- M. A. Habila, Z. A. ALOthman, A. M. El-Toni, J. P. Labis and M. Soylak, Talanta, 2016, 154, 539–547 CrossRef CAS PubMed.
- M. A. Habila, Z. A. AlOthman, A. M. El-Toni, S. A. Al-Tamrah, M. Soylak and J. P. Labis, Microchim. Acta, 2017, 184, 2645–2651 CrossRef CAS.
- A. Rohanifar, N. Alipourasiabi, G. S. Shyam Sunder, J. G. Lawrence and J. R. Kirchhoff, Mikrochim. Acta, 2020, 187, 339 CrossRef CAS PubMed.
- J. X. Qin, Z. Su, Y. H. Mao, C. C. Liu, B. Qi, G. Z. Fang and S. Wang, Ecotoxicol. Environ. Saf., 2021, 208, 111729 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ja00341k |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Jiawen
Cheng
a,
Xiaoxuan
Han
*b,
Yutong
Li
b,
Ting
Yang
a,
Jiawen
Cheng
a,
Xiaoxuan
Han
*b,
Yutong
Li
b,
Ting
Yang




