
Strontium isotopic analysis of environmental microsamples by inductively coupled plasma – tandem mass spectrometry†
Stefano
Bertinetti
 a,
Eduardo
Bolea-Fernandez
b,
Mery
Malandrino
c,
Beatrice
Moroni
d,
David
Cappelletti
d,
Marco
Grotti
a and
Frank
Vanhaecke
*b
a,
Eduardo
Bolea-Fernandez
b,
Mery
Malandrino
c,
Beatrice
Moroni
d,
David
Cappelletti
d,
Marco
Grotti
a and
Frank
Vanhaecke
*b
aDepartment of Chemistry and Industrial Chemistry, University of Genoa, Via Dodecaneso 31, 16146 Genoa, Italy
bGhent University, Department of Chemistry, Atomic & Mass Spectrometry – A&MS – Research Group, Campus Sterre, Krijgslaan 281-S12, 9000, Ghent, Belgium. E-mail: Frank.Vanhaecke@UGent.be
cDepartment of Chemistry, University of Turin, Via Pietro Giuria 5, 10125, Turin, Italy
dDepartment of Chemistry, Biology and Biotechnology, University of Perugia, 06123 Perugia, Italy
First published on 26th November 2021
Abstract
In this work, a new method has been developed for accurate and precise Sr isotopic analysis of microsamples via inductively coupled plasma – tandem mass spectrometry (ICP-MS/MS). For introduction of low-volume samples, a setup consisting of a syringe-driven pump that allows to work at low and stable sample introduction flow rates was combined with a high-efficiency sample introduction system, originally designed for the introduction of single cells and other discrete entities into the ICP. The method requires 240 μL of sample only, introduced at a sample uptake rate of 20 μL min−1 and allows for the straightforward measurement of the 87Sr/86Sr isotope ratio without prior Rb/Sr separation. The isobaric overlap of the signals of 87Rb and 87Sr is overcome by the selective reaction between Sr+ and CH3F gas in the collision/reaction cell (CRC) of an ICP-MS/MS instrument. In this way, Sr+ ions are measured as their corresponding SrF+ reaction product ions in a mass-shift approach. A multivariate approach was applied to obtain optimum instrument settings (sample uptake rate, nebulizer gas flow rate, make-up gas flow rate, and plasma sampling depth) for maximum sensitivity and optimum precision of the 87Sr/86Sr and 88Sr/86Sr isotope ratios. The method thus developed was found to be accurate (mean bias = −0.001 ± 0.070%, with respect to a quality control reference value) and showed an external precision of 0.17% (RSD, n = 24) at an analytical concentration of 2 ng g−1. The method was applied to the analysis of small-volume digests of samples of Antarctic atmospheric particulates (PM10) and of soils from Australia and South America as potential source areas for atmospheric particulates reaching East Antarctica. The raw soil samples were handled using a dedicated resuspension/collection system to select the finest fraction (PM2.5), thus simulating the long-range transport of mineral dust in the atmosphere. The results obtained for these samples were found to be in good agreement with literature data, demonstrating the suitability of the procedure developed for the Sr isotopic analysis of such complex matrices with Sr concentrations as low as a few ng g−1 (<3 ng g−1 and <7 ng g−1, for the PM10 and soil samples, respectively). The method could also be useful for other environmental samples for which the low Sr concentrations and limited sample amounts are critical aspects.
1. Introduction
In various contexts, such as forensic, biological and/or clinical analysis, the sample amount available is often a limiting factor for elemental and isotopic analysis.1 Also in environmental studies, small-sized samples may occur. For instance, this can be the case with digests of solid matrices, whenever the final volume must be kept as small as possible to avoid excessive sample dilution. Sample size is particularly critical in the context of isotopic analysis since the attainable precision is theoretically limited by counting statistics and hence, by the signal intensity.When analysis of microsamples is required, there are usually a few ways to overcome the challenge posed by the low availability of material. One option is dilution of the sample to obtain a final volume sufficient for the method to be used. This is the easiest method and, moreover, it also allows mitigation of the matrix effects typical for high-matrix samples.2 However, this is possible only when the concentration of the target element is high enough, and the risk of contamination as a result of sample manipulation is very low. An alternative way is reducing the sample consumption during the analysis, by limiting the duration of the measurement to the largest acceptable extent (e.g., by reducing the number of isotopes monitored or the total integration time in ICP-MS analysis), and/or by reducing the sample uptake rate. However, these strategies may strongly affect the quality of the analysis results, as they influence the intensity of the (integrated) signals. In fact, at lower signal intensity/total integrated signal, the precision of the isotope ratio measurements gets worse according to Poisson statistics.3 To overcome this drawback, special high-efficiency sample introduction systems have been developed.1 These systems allow the use of a low sample uptake rate (tens of μL min−1), while still obtaining high signal intensities, comparable to those achieved with traditional sample introduction systems.4 In fact, they are able to transport the (smaller) droplets generated by the nebulizer into the plasma with higher efficiency than does the traditional system, thus reducing or eliminating the amount of sample that is discharged from the spray chamber into the waste.5 The development of suitable methods to appropriately handle microsamples via inductively coupled plasma – mass spectrometry (ICP-MS) is not a recent issue,6 but has gained significant interest over the years,1 also for the isotopic analysis of environmental samples at very low sample consumption rates.7,8
Strontium shows a widespread natural variation in its isotopic composition, mainly because of the beta decay of 87Rb into 87Sr (t1/2 = 4.96 × 1010 years).9 The 87Sr/86Sr isotope ratio is one of the most widely used tracers to study a variety of geological and environmental phenomena. For instance, Sr isotopic analysis has been widely used in mineral dust transport studies, provenance determination of agricultural products of plant or animal origin, and human migration studies.10–12 In all these cases, the 87Sr/86Sr isotope ratio of the samples was related to that of the (bio-available fraction of) Sr of the soil/rock body of the geographical location they originate from.
A major issue jeopardizing the accuracy of the Sr isotopic analysis via ICP-MS is the isobaric overlap of the signals of 87Rb and 87Sr. This can be overcome by prior chemical (chromatographic) separation of the two elements, but this process is laborious and time-consuming.13 An alternative approach is based on exploiting the different reactivity of the Rb+ and Sr+ ions towards various reaction gases (e.g., N2O, CH3Cl, CH3F or SF6) when using ICP-MS instrumentation equipped with collision/reaction cell (CRC) technology.14–16 These methods allow for a direct analysis of the samples without prior isolation of Sr, thus strongly reducing the time required for sample preparation, while at the same time mitigating the risk of contamination. The introduction of tandem ICP-MS instrumentation (ICP-MS/MS) has further extended this approach, as the double mass selection provided by an additional quadrupole located before the CRC (Q1-CRC-Q2) results in a better control over the in-cell chemistry and provides additional means to remove spectral interference based on the use of chemical resolution.17–19 In fact, the presence of two quadrupole mass filters enables such instrument to be operated in MS/MS mode, whereby only ions with the selected mass-to-charge (m/z) ratio are allowed to enter in the CRC, thus strongly reducing the formation of new interfering ions resulting from the reactions between concomitant matrix ions and the reaction gas. This capability has led to a further extension of the use of chemical resolution to overcome spectral overlap via the selection of highly reactive gases, such as NH3 or CH3F.18,19 This has also resulted in a change of the common trend of monitoring the analyte ion at its original mass after removal of the originally interfering ions (on-mass approach) into a mass-shift approach, in which the target species is monitored as a reaction product ion, formed upon selective reaction between the analyte ion and the reaction gas, at a different m/z ratio.20
In a previous study, a method for the direct Sr isotopic analysis was developed by using a mixture of CH3F and He as the CRC gas in ICP-MS/MS instrumentation.21 This approach relied on the selective reaction between Sr+ and CH3F (SrF+ was monitored as the best suited reaction product ion), while Rb+ does not react with CH3F. The same strategy was also used for the direct Sr isotopic analysis of solid samples via LA-ICP-MS/MS.22 Moreover, also interferences from other polyatomic ions (e.g., 40Ar46Ca+, 43Ca44Ca+ and 40Ar48Ca+) and from isobars (e.g., 106Cd+, 105,106Pd+ and 107Ag+, the signals of which could coincide with those of 86Sr19F+, 87Sr19F+ and 88Sr19F+) are avoided by operating the tandem ICP-MS instrument in MS/MS mode with CH3F as reactive gas.21
In this work, this method has been revisited and tuned for the analysis of microsamples (<1 mL). For this purpose, a novel setup consisting of a μL-autosampler that provides stable sample introduction at low liquid flow rates has been combined with a high-efficiency sample introduction system that improves the transport efficiency of the aerosol plume into the plasma source. To achieve optimum conditions for accurate and precise Sr isotopic analysis using minimum sample amounts, the method has been optimized using an experimental design approach to obtain a more profound insight into the interrelated effects of the operating parameters – plasma sampling depth (SD), sample uptake rate (UR), nebulizer gas flow rate (NG), and make-up gas flow rate (MG) – on the instrument sensitivity and Sr isotope ratio precision. After appropriate optimization and validation, the method has been applied to the Sr isotopic analysis of small-volume digests of samples of Antarctic atmospheric particulates and soil samples from Australia and South America collected in the context of environmental studies on mineral dust reaching Antarctica, characterized by low Sr concentrations (below 10 ng g−1).
2. Experimental
2.1 Instrumentation
All measurements were performed using an Agilent 8800 ICP-MS/MS instrument (Agilent Technologies, Japan). This instrument is equipped with two quadrupole mass units (Q1 and Q2) and a third generation octopole-based collision/reaction cell system (ORS) located in-between (Q1-CRC-Q2). In this work, the ICP-MS/MS instrument was always operated in MS/MS mode, whereby both quadrupoles were used as 1 amu mass filters. A high-efficiency single-cell sample introduction system (SC-SIS, Glass Expansion, Australia) was used. This SIS was originally designed for the introduction of single cells and other discrete entities into the ICP, and it was selected in this work for its high performance at low sample uptake rates.23,24 This sample introduction system comprises a high-efficiency, low-sample-consumption concentric nebulizer, mounted onto a low-volume, on-axis laminar-flow spray chamber. A secondary make-up Ar flow is introduced axially into the spray chamber to improve the transport efficiency by better shaping of the aerosol plume to reduce the impact of droplets on the spray chamber walls. An MVX-7100 μL Workstation (Teledyne CETAC Technologies, USA) was used for sample uptake instead of the conventional peristaltic pump. This setup consists of a syringe-driven pump that allows to work at low and stable sample introduction flow rates (down to 5 μL min−1) and to obtain transient signals lasting for approx. 12 min for a sample solution volume of 240 μL. For each sample, a subset of data corresponding to the central 10 minutes of the signal profile was extracted and divided into ten data fragments, from which the raw data were obtained.The CRC of the ICP-MS/MS instrument was pressurized with a mixture of CH3F/He (10% CH3F in He, “Certified Master Class”, Air Liquide, Belgium), introduced via the H2 inlet at the reported flow rate of 2.5 mL min−1. The optimum values for the sample uptake rate, nebulizer gas flow rate, make-up gas flow rate, and plasma sampling depth were obtained using an experimental design approach, as discussed below. The ICP-MS/MS instrument was operated in MS/MS mode, with Q1 set to m/z = 86, 87 and 88 and Q2 set to m/z = Q1 + 19, for the monitoring of the 86SrF+, 87SrF+, and 88SrF+ reaction product ions. The relative abundances of 86Sr, 87Sr and 88Sr are 9.86%, 7.00% and 82.58%, respectively.25 To handle these high differences, the integration time selected for each isotope was inversely proportional to its relative abundance, while keeping the total integration time (0.300 s) constant. In this way, the time dedicated to acquiring the lower-intensity signals was maximized, possibly improving the precision of the isotope ratio measurements. The Sr isotope ratios were corrected for mass bias as discussed in Section 2.5.
Instrument settings and data acquisition parameters are summarized in Table 1.
| a (*) optimized using an experimental design approach. | |
|---|---|
| Reaction gas type | CH3F/He (10/90) |
| Reaction gas flow rate (mL min−1) | 2.5 |
| Scan type | MS/MS |
| Q1 → Q2 | 86 → 105 |
| 87 → 106 | |
| 88 → 107 | |
| Plasma mode | Low matrix |
| RF power (W) | 1550 |
| Plasma sampling depth (mm) (*) | 4 |
| Sample uptake rate (μL min−1) (*) | 20 |
| Nebulizer gas flow rate (L min−1) (*) | 0.57 |
| Make-up gas flow rate (L min−1) (*) | 0.50 |
| Extract 1 (V) | −3.0 |
| Q1 bias (V) | 0 |
| Octopole bias (V) | −4.7 |
| Energy discrimination (V) | −8.4 |
| Extract 2 (V) | −175.0 |
| Integration time (s): | |
| 86SrF | 0.119 |
| 87SrF | 0.167 |
| 88SrF | 0.014 |
| Time/sweep (s) | 0.300 |
| Sweep/replicate | 198 |
| Number of replicates | 10 |
| Total analysis time/sample (min) | 14 |
2.2. Reagents and material
Only high-purity reagents were used during this work. Ultra-pure water (resistivity > 18.2 MΩ cm) was supplied by a Milli-Q Element water purification system (Millipore, France). Pro-analysis grade 14 M HNO3 (ChemLab, Belgium), further purified by sub-boiling distillation, and 9.8 M (w/w) H2O2 for ultra-trace analysis (Sigma Aldrich, USA) were used for the microwave-assisted sample digestion. Appropriate dilutions of a single-element standard solution of Sr (1 g L−1, Instrument Solutions, the Netherlands) were used for method development. The isotopic reference material NIST SRM 987 strontium carbonate (87Sr/86Sr = 0.710248 ± 0.000012)26 and an in-house standard solution of Sr (Instrument solutions, the Netherlands), previously characterized by multi-collector (MC) ICP-MS instrumentation (87Sr/86Sr = 0.70753 ± 0.00006),21 were used for mass bias correction and quality assurance and quality control (QA/QC) during sample analysis. Polycarbonate membranes (Isopore, 0.4 μm pore size) were obtained from Millipore (France).2.3. Samples and sample preparation
The method developed was applied to 26 Antarctic atmospheric particulate (PM10) samples, 11 soil samples from Australia, and 10 soil samples from South America, collected from different locations (Fig. S1†). These samples were collected in the context of the SIDDARTA project of the Italian National Program of Research in Antarctica (PNRA).The PM10 samples were collected onto PTFE hydrophilic filters (Advantec MFS, USA; H100A090C, 90 mm diameter, efficiency > 99% for 0.3 μm particles) by an Echo HiVol sampler (TCR Tecora, Italy; 200 L min−1) at the Italian-French research station Concordia (75°06′S; 123°20′E; elevation: 3233 m a.s.l.), on the East Antarctic plateau, during 2018 with monthly resolution. Half of each filter was digested via microwave-assisted acid digestion using 2.0 mL of 14 M HNO3 and 0.5 mL of 9.8 M H2O2, according to the UNI EN 14902 procedure.27 The second half of the particulate was resuspended in Milli-Q water using an ultrasonic bath for 60 minutes, after which the insoluble fraction was collected onto a polycarbonate membrane by filtration,28 thus virtually removing possible Sr contribution from marine aerosol.29 The polycarbonate filters were then digested with the same procedure used for the PM10 samples.
The 87Sr/86Sr isotope ratios of these samples were compared with those obtained for soil samples collected from potential source areas (Australia and South America), aiming at revealing the geographical origin of the mineral dust reaching the inner region of Antarctica and its temporal variation. To better mimic the natural situation, the raw soil samples were processed to select the finest fraction only, as discussed below. In fact, during the long-range transport of the mineral dust from the source region to Antarctica, the coarser particles can sediment, thus altering the size distribution of the mineral dust. Typically a mean diameter of about 2 μm, with a maximum value of approx. 5 μm is observed.30 This is a crucial aspect, because Sr shows isotope fractionation as a function of the grain size.28 Therefore, the soil samples were resuspended inside a home-made resuspension/collection chamber, developed at the University of Perugia. This device is a ∼0.2 m3 squared box made of anti-static plastic, which was designed to be very easy to clean. A gentle flow of nitrogen and a set-up with four electrically driven fans is exploited to resuspend dry soil samples directly deposited onto a small plastic plate inside the chamber. The suspended material is subsequently size-segregated by a low-volume aerosol sampler, equipped with PM10, PM2.5, and PM1.0 impactor heads, directly connected to the chamber using a Teflon joint tube. Typically, 2–3 grams of raw material are sufficient to collect a few milligrams of PM deposited on 47 mm filters in about 1 minute. The impactor head can be easily and quickly cleaned after every PM filter collection. For the present work, the air-suspended material was collected on PTFE filters using a PM2.5 impactor head. For each soil sample, both grinded and not-grinded aliquots were processed. Finally, the filters with the PM2.5 fraction of the soil samples were digested using the same procedure as for the atmospheric particulate samples.
Scanning Electron Microscopy has been used to check the size-segregation performance of this resuspension device using polycarbonate filters. An example is shown in Fig. S2,† where the non-grinded Australian dust (AUS5) sample has been deposited on polycarbonate filters using either a PM10 or a PM2.5 impactor head. For this application, we exposed the filters for 15 seconds only, to avoid undesired particle packing.
2.4. Multivariate optimization
The effects of the sample introduction settings (sample uptake rate, nebulizer gas flow rate and make-up gas flow rate), and of the plasma sampling depth (i.e., the distance between the tip of the sample cone and the front end of the torch) on the intensity and internal precision (RSD%) of the 86SrF+, 87SrF+ and 88SrF+ reaction product ions signals and on the internal precision of the 87Sr/86Sr and 88Sr/86Sr isotope ratios were studied using an experimental design approach,31 applying a face-centered central composite design (CCD, Table S1†). The signals were studied under the various conditions while analyzing a 10 ng g−1 Sr standard solution. The experiments were performed randomly to minimize the effect of systematic variability (e.g., instrumental drift). In contrast, the measurement corresponding to the central point (Table S1,† experiments 25–30) was regularly repeated throughout the sequence to estimate the experimental variance, necessary to calculate the significance of the models' coefficients. These were calculated by multiple linear regression analysis using the open-source statistical environment R with the software CAT (Chemometric Agile Tool).32,332.5. Mass bias correction
The method used in this work for mass bias correction has been described in detail elsewhere.21 Briefly, a combination of (i) internal correction using a 88Sr/86Sr isotope ratio of 8.3752 and Russell's exponential law (eqn (1) and (2)), and (ii) external correction (eqn (3)) using NIST SRM 987 (87Sr/86Sr = 0.710248 ± 0.000012) as an external standard in a sample standard bracketing (SSB) approach, was applied.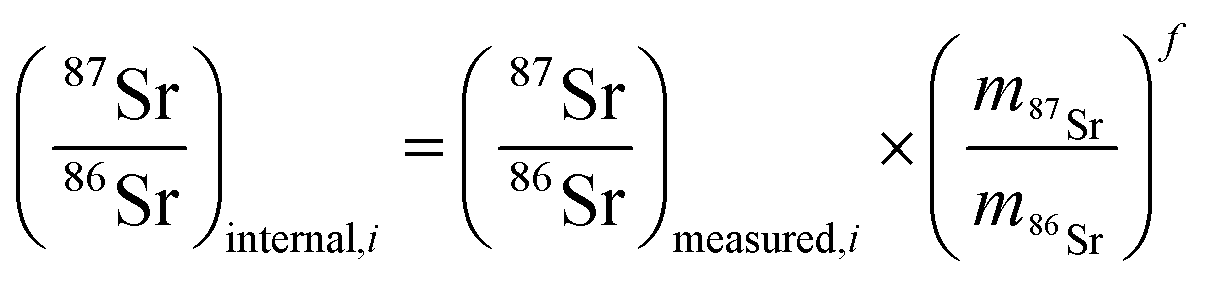 | (1) |
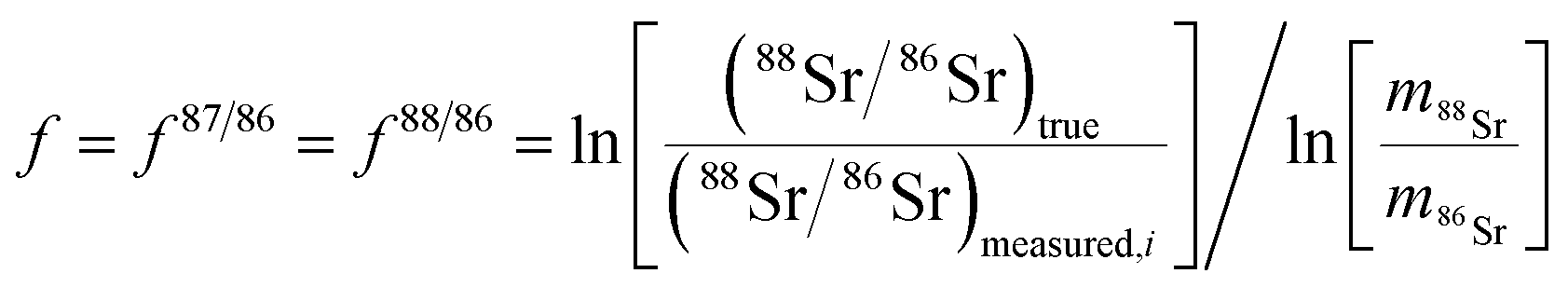 | (2) |
 | (3) |
During each analysis sequence, a standard solution of NIST SRM 987 Sr isotopic reference material, at a similar Sr concentration as in the samples, was analyzed immediately before and after each sample (sample-standard bracketing approach – SSB), to minimize the effect of the instrumental drift on the isotope ratio measurement results.
3. Results and discussion
In previous works, it was demonstrated that the use of CRC technology in ICP-MS/MS instrumentation allows for the direct determination of the 87Sr/86Sr isotope ratio without prior chromatographic Rb/Sr separation.21,22,34 An interesting approach is the use of selective fluorine transfer between CH3F and Sr+. In this way, the measurement is based on the monitoring of the signals of the 86SrF+ (m/z = 105), 87SrF+ (m/z = 106) and 88SrF+ (m/z = 107) reaction product ions, free from spectral interferences.21 However, the use of this method for the Sr isotopic analysis of environmental microsamples still remains a challenging task. In this work, this method has been revised to significantly scale down the sample uptake rate, which can affect the instrumental sensitivity, and thus, the precision of the isotope ratio measurements. This issue was particularly critical given the application to Antarctic samples, in which the expected Sr concentration is quite low (<10 ng g−1, and generally around 1 ng g−1 in the solutions resulting from the sample preparation step; see Section 2.3).Hence, an optimization study has been performed to achieve a fit-for-purpose performance for the isotopic analysis of Sr at a very low sample consumption rate and low Sr concentrations, so that the method is compatible with the analysis of environmental microsamples.
3.1. Optimization of the instrument settings
The combined effects of plasma sampling depth (SD), sample uptake rate (UR), nebulizer gas flow rate (NG), and make-up gas flow rate (MG) on the signal intensities of the 86SrF+, 87SrF+ and 88SrF+ reaction product ions and their internal precision, and on the precision of the 87Sr/86Sr and 88Sr/86Sr isotope ratios, have been studied according to an experimental design approach, as described in Section 2.4. The models' coefficients obtained with their significance are reported in Tables S2–S5,† and representative response surfaces are plotted in Fig. 1–4 and S3–S6.†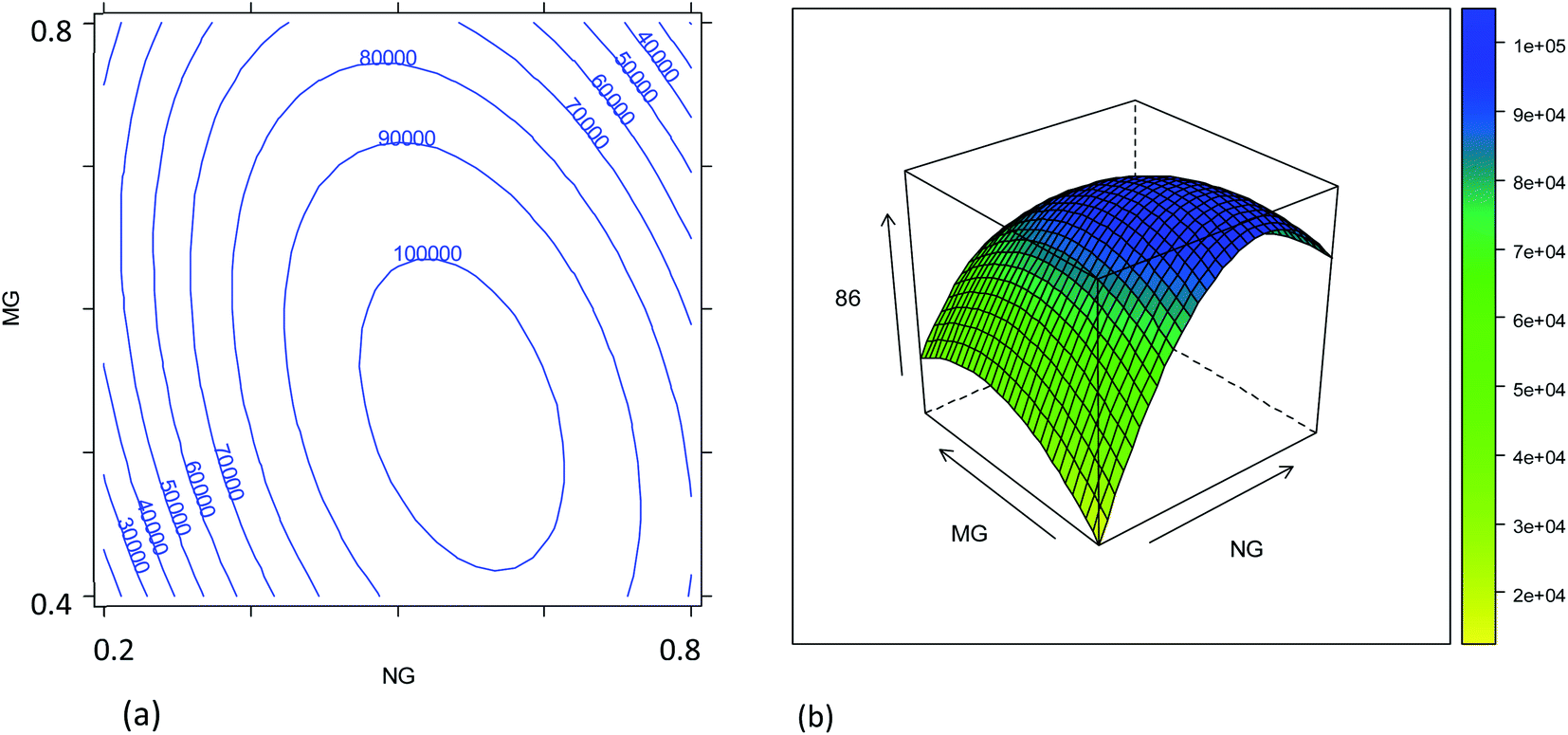 | ||
| Fig. 1 Combined effect of the nebulizer gas flow rate (NG, L min−1) and make-up gas flow rate (MG, L min−1) on the 86SrF+ signal intensity: (a) contour plot; (b) response surface (SD = 4 mm; UR = 35 μL min−1). | ||
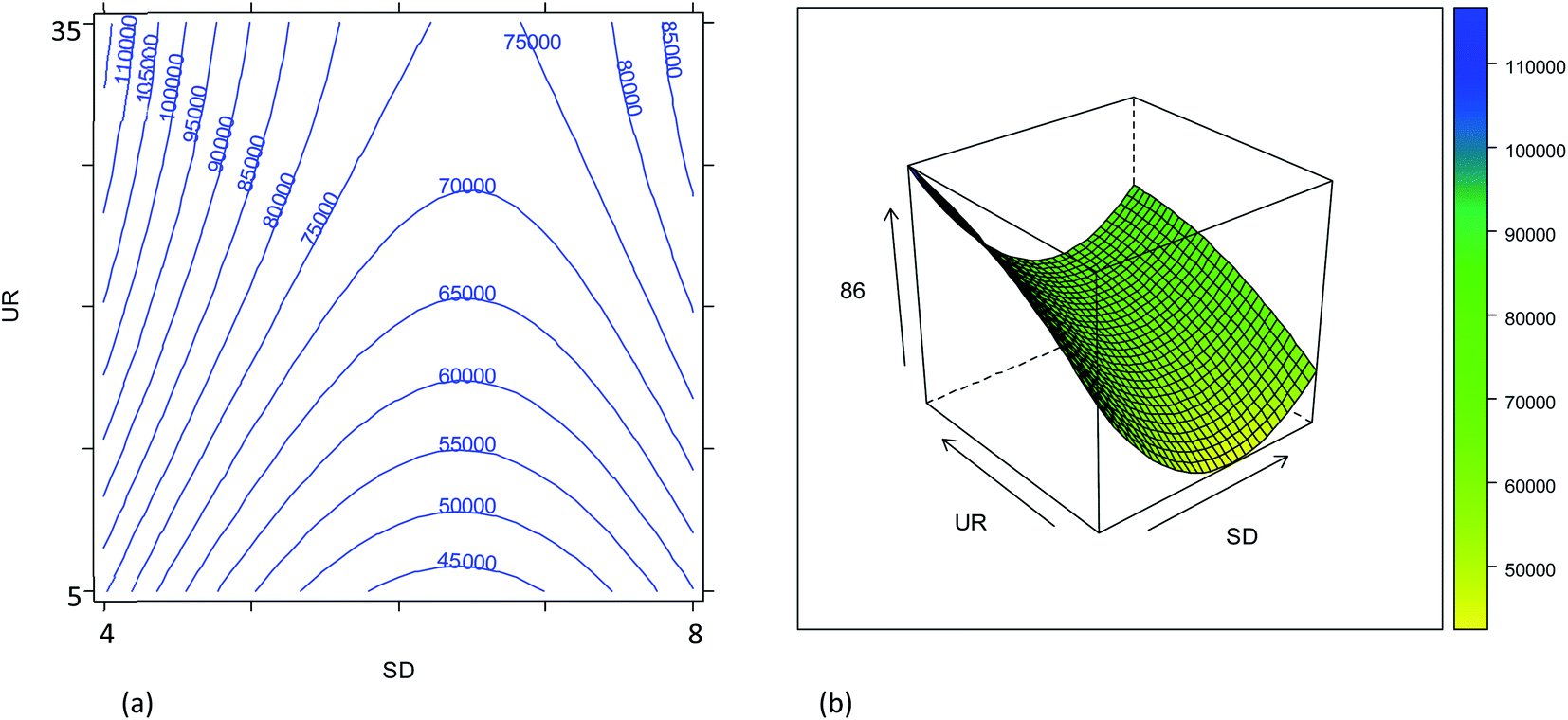 | ||
| Fig. 2 Combined effect of the plasma sampling distance (SD, mm) and sample uptake rate (UR, μL min−1) on the 86SrF+ signal intensity: (a) contour plot; (b) response surface (NG = 0.57 L min−1; MG = 0.5 L min−1). | ||
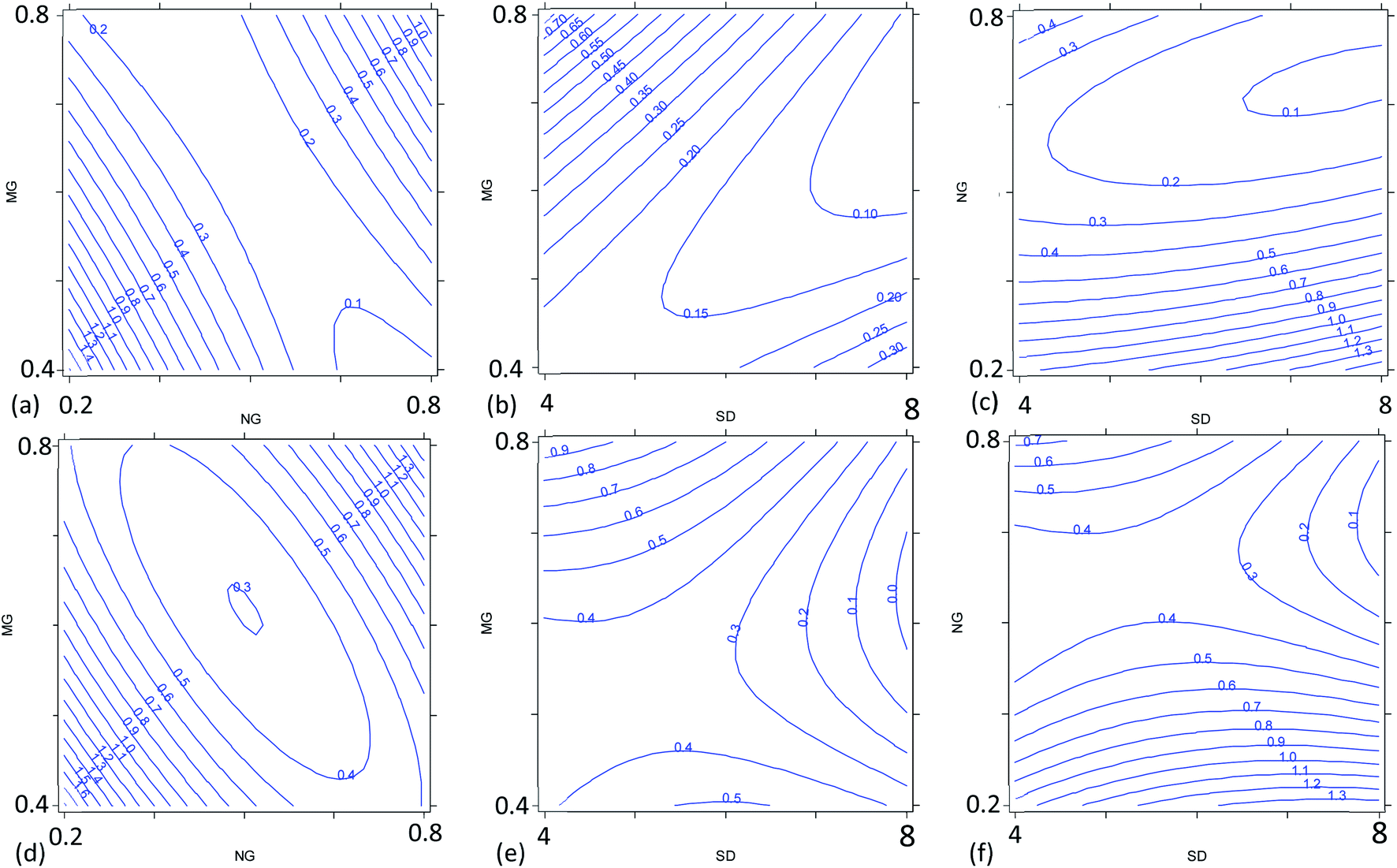 | ||
| Fig. 3 Combined effect of the nebulizer gas flow rate (NG, L min−1), plasma sampling distance (SD, mm) and make-up gas flow rate (MG, L min−1) on the precision of the 87Sr/86Sr (contour plots a–c) and 88Sr/86Sr (contour plots d–f) isotope ratios. UR = 20 μL min−1 for each graph; for (a) and (d) SD = 6 mm (similar trends were observed for SD = 4 mm and 8 mm), for (b) and (e) NG = 0.57 L min−1, and for (c) and (f) MG = 0.50 L min−1. | ||
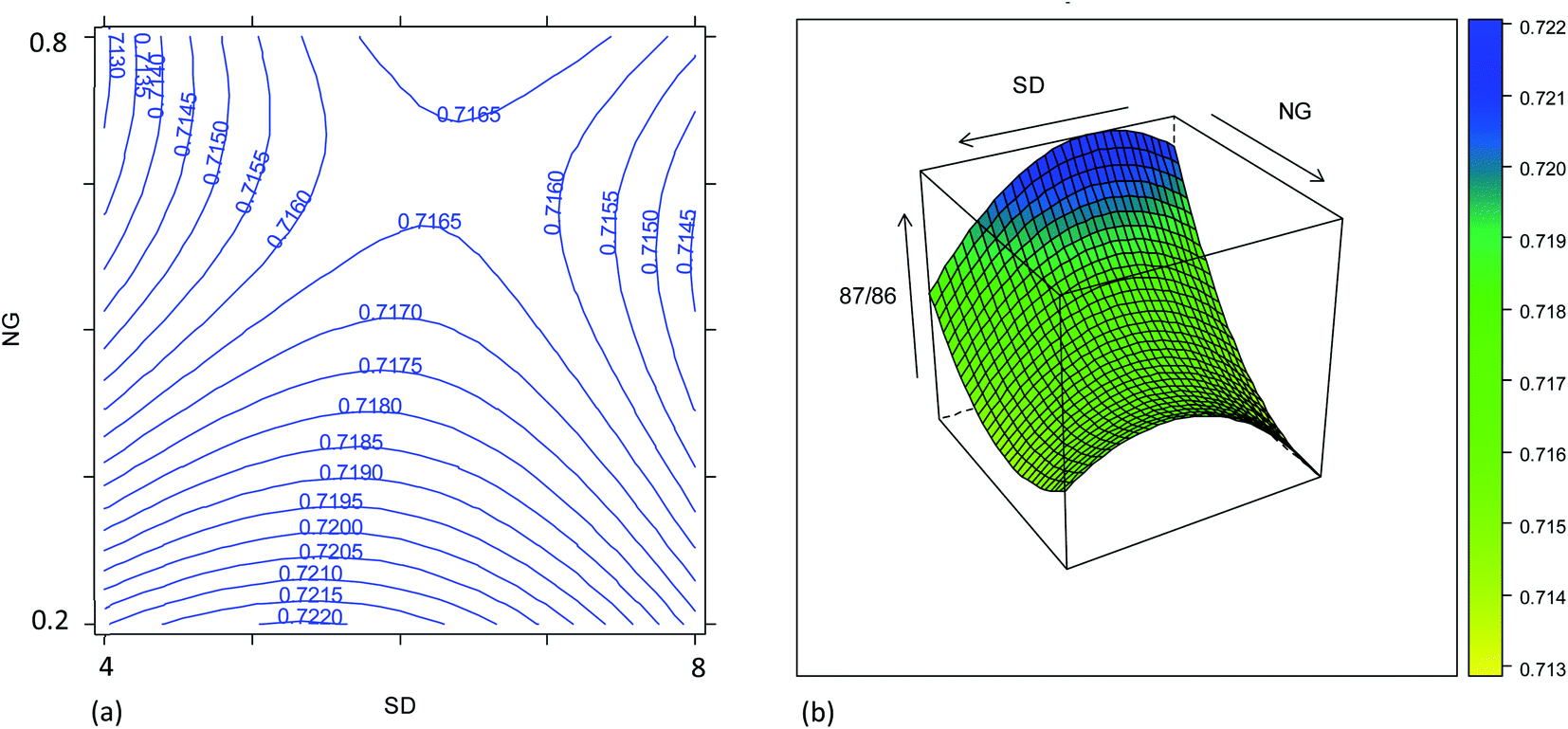 | ||
| Fig. 4 Combined effect of the plasma sampling distance (SD, mm) and nebulizer gas flow rate (NG, L min−1) on the 87Sr/86Sr isotope ratio: (a) contour plot; (b) response surface (MG = 0.50 L min−1; UR = 20 μL min−1). | ||
Due to the strong correlation between the intensities of the different reaction product ions (r > 0.9999), only the results for 86SrF+ have been reported as a measure for sensitivity (Table S2,†Fig. 1 and 2). It can be noted that the nebulizer gas flow rate (NG) and its interaction with the make-up gas flow rate (MG) affect the instrument sensitivity to the largest extent.4 In addition, a strong quadratic effect of the NG on the signal intensity was observed. In fact, the signal intensity first increases upon increase of the NG, then reaches a maximum, after which there is a reduction in sensitivity by further increasing this parameter (Fig. 1). The initial increase can be attributed to the improved aerosol formation and transport to the plasma source, and to the spatial overlay of the high-density zone of the singly charged ions with the sampling zone when increasing the NG and MG flow rates.35 On the other hand, an excessive increase of these gas flow rates leads to a decrease of the analytical signal, probably due to the reduction of the residence time of the sample aerosol in the plasma and an alteration of the ion spatial distribution with respect to the plasma sampling region. Moreover, the significant interaction between the NG and MG gas flow rates shifts the optimum conditions towards higher NG flow rates when decreasing the MG (Fig. 1). The formation of a finer aerosol benefits from a lower MG to prevent the impact of droplets against the chamber walls.4 Similar response surfaces have been obtained for other SD values (Fig. S3†).
The combined effect of the sample uptake rate (UR) and the plasma sampling depth (SD) is displayed in Fig. 2. The first parameter showed a roughly linear effect on the sensitivity (only the linear term was significant, Table S2†), but the effect turned out to be much less important than that of the other parameters. Hence, considering that a major goal was to reduce the sample consumption, an intermediate value of 20 μL min−1 (coded as 0) was selected as a good compromise. Concerning the effect of the SD, it was found that the central values provide worse results than the extreme values (Fig. S3†). These results can be related to the spatial overlay of the high-density zone for M+ with the plasma sampling zone, although the SD played a minor role compared to that of the gas flow rates. Considering the effect of all the variables evaluated, the optimum conditions for maximum instrument sensitivity were: SD = 4 mm (coded as −1), UR = 20 μL min−1 (coded as 0), NG = 0.57 L min−1 (coded as 0.25) and MG = 0.50 L min−1 (coded as −0.5). Fig. 2 shows that the conditions chosen fall in a region in which a little (random) variation in the UR or SD values causes a high variation in the ion intensity, negatively affecting the precision. However, it should be noticed that the conditions selected also fall in a quite flat region of the response surface of NG vs. MG (Fig. 1). The use of a more stable sample introduction system than the traditional peristaltic pump, makes UR a low-noise parameter, whereas the gas fluxes could have higher fluctuations, but marginally affecting the precision. Therefore, the RSD% of the 86SrF+ signal obtained under the selected conditions was not much higher than that achieved with other SD, NG and MG values (Table S3 and Fig. S5†).
The multivariate effect of these operating parameters on the internal precision of the 87Sr/86Sr and 88Sr/86Sr isotope ratios is reported in Tables S4 and S5† and shown in Fig. 3 and 4. First, a high correlation (r = 0.91) between the two ratios was found, meaning that the variation in the operating conditions affected the precision of both isotope ratios in a similar way. The coefficients of the models and their significance (Tables S4 and S5†) indicated that the precision is mainly influenced by the paired interactions between SD, NG and MG, and by the quadratic behavior of NG. These effects are illustrated for the two isotope ratios in Fig. 3. It can be seen that the values of NG and MG that maximize the instrumental sensitivity also lead to low % RSD-values (Fig. 3a–d), in agreement with the theoretical relationship between the precision attainable and the signal intensity:3
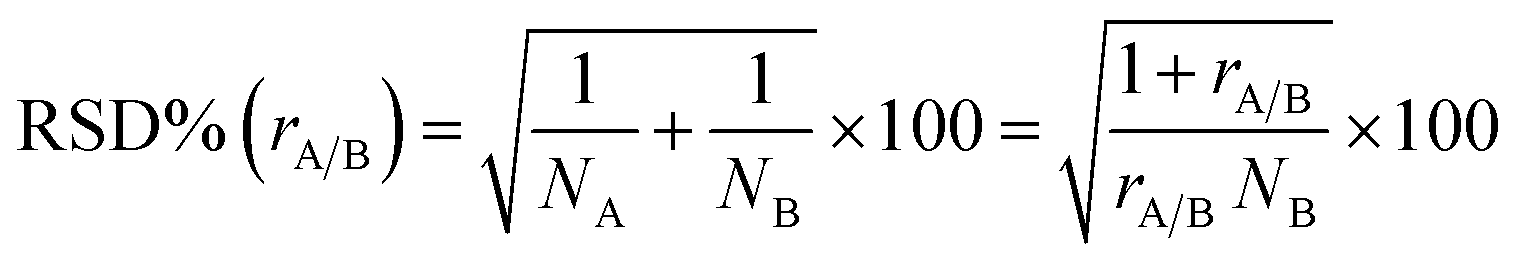 | (4) |
Despite the high correlation between the two isotope ratios, the optimal values of SD for the lowest RSD% are different: 6 mm and 8 mm for 87Sr/86Sr and 88Sr/86Sr, respectively. Moreover, the RSD% of 88Sr/86Sr is more sensitive to the variation of the SD from 6 to 8 mm (Fig. 3b–e and c–f). Therefore, to achieve high precision for both isotope ratios, the value of SD = 8 mm would be preferred. However, this value is different from that required for the highest intensity (SD = 4 mm, coded as −1), as previously also reported by Andrèn et al.36 In fact, RSD% (rA/B) is also affected by the isotope ratio value, which is strongly dependent on the SD setting (see Section 3.2). Therefore, a compromise choice for this parameter appeared to be necessary. As instrumental sensitivity could be a critical issue for the specific application considered in this work, the SD was set at 4 mm to achieve the maximum sensitivity. On the other hand, for the Sr isotopic analysis of samples with higher Sr concentration, the optimal conditions to maximize the precision (SD = 8 mm) should be selected.
Finally, the models were validated by five repetitions under optimal conditions, obtaining a good agreement between the experimental and the predicted values for 86SrF+ signal intensity, and the internal precision of 87Sr/86Sr and 88Sr/86Sr isotope ratio measurements (Table S6†).
3.2 Effect of the instrument settings on the 87Sr/86Sr isotope ratio
The effect of the instrument settings studied above on the measured 87Sr/86Sr isotope ratio was also investigated using the results from the same experimental design. The model is reported in Table S7† and plotted in Fig. 4. The raw 87Sr/86Sr isotope ratio result (i.e., not corrected for mass bias) was mostly affected by the NG and MG, and their interaction with the SD. The effect of the NG can be explained considering the higher lateral diffusion of the lighter isotopes in the plasma.35,36 In fact, by increasing the NG + MG flow rate, the residence time of the ions inside the plasma is reduced, thus reducing the loss of the lightest isotope and thus reducing the 87Sr/86Sr ratio. Also, the nozzle separation effect, that is strongly related to the nebulizer gas flow rate, introduces a mass bias in the same direction.37 Moreover, the efficiency of ionization is not uniform inside the plasma, and it is not the same for the different isotopes. According to the zone model,35 the zone of maximum ion density for the lighter isotope is closer to the torch than that for the heavier isotopes. Therefore, the 87Sr/86Sr isotope ratio should increase moving away from the torch, i.e., upon increasing the SD. This behavior can be recognized in Fig. 4, which shows the effect of the SD and NG on the 87Sr/86Sr ratio. However, a more complex dependence of the isotope ratio on the SD was evident, with a decrease in the isotope ratio by increasing the SD observed at high values. By comparing Fig. 4a and S4a,† a difference in the effect of SD on the 87Sr/86Sr ratio and 86SrF+ ion intensity can be observed. According to eqn (4), this led to a different effect of SD on the precision, as discussed above. Finally, also the model for the isotope ratio 87Sr/86Sr was validated, observing a good agreement between the predicted and experimental values (Table S6†).3.3. Precision and accuracy
To assess the precision and accuracy of the 87Sr/86Sr isotope ratio measurements under the optimal conditions, an in-house standard solution of Sr, with a concentration of 2 ng g−1, was periodically analyzed, and the 87Sr/86Sr isotope ratio thus obtained was compared to that previously determined via MC-ICP-MS (87Sr/86Sr = 0.70752 ± 0.00006). The mean measured value (87Sr/86Sr = 0.7075 ± 0.0005, mean ± 95% confidence interval, n = 24) did not differ significantly from the reference value. The internal precision of the raw data was 0.20 ± 0.03% (mean ± 95% confidence interval, n = 24), whereas the internal precision of the corrected isotope ratios, computed by applying the general error propagation law for eqn (1)–(3), was 0.58 ± 0.04%. The external precision, estimated from the dispersion of the isotope ratio values obtained in 6 consecutive days of analysis, was 0.17% (n = 24). For each day (about 10 hours of analysis), an external (daily) precision of 0.15 ± 0.07% (mean ± 95% confidence interval, n = 6) was obtained, considering the dispersion of the measurement results occurring each day. Finally, the mean bias from the reference value was −0.001 ± 0.070% (mean ± 95% confidence interval, n = 24).The precision obtained in this work was worse than that obtained in a previous work, using the same instrument, but with a traditional SIS (MicroMist nebulizer fitted onto a Peltier-cooled Scott-type spray chamber).21 In that case, internal precisions of 0.12% and 0.09% for raw and internally-corrected data, respectively, and an external precision of 0.04% were obtained. These significant differences could be hypothetically attributed to the lower sample uptake rate used in this work (20 μL min−1vs. ∼400 μL min−1) and the lower Sr concentration (2 ng g−1vs. 10 ng g−1) of the standard solution used for QA purposes, despite the high-efficiency SIS used.
3.4. Determination of Sr isotopic composition of Antarctic PM10 and PSA soil samples
The method developed was finally applied to the determination of the 87Sr/86Sr isotope ratios of soil samples collected in Australia and South America (Patagonia and Central-West Argentina), and PM10 samples collected on the Antarctic plateau. The digests (see Section 2.3) were analyzed and precise measurement of the 87Sr/86Sr ratios was achieved while consuming 240 μL of solution only. The results are reported in Tables S8 and S9† and shown in Fig. 5, also presenting literature data for comparison. The solutions analyzed had an estimated Sr concentration lower than 7 ng g−1 and than 3 ng g−1, for the soil and PM10 samples, respectively. The 87Sr/86Sr ratios in the total and insoluble fraction of the PM10 samples did not show any significant difference (t-test for paired samples, p > 0.1, n = 13) at the precision level achieved in this work. Similarly, no significant differences were found between the isotope ratios of grinded and non-grinded soil samples, although the number of paired samples was too low to apply statistical tests (n = 5 and n = 3 for Australia and South America, respectively). Most importantly, a good agreement between the Sr isotopic composition of the soil samples analyzed in this work and relevant literature data was observed (Fig. 5). Most of the soil samples from South America showed the “Patagonian” isotopic signature, and only a couple of samples resembled more the signature of Central Western (CW) Argentina. The 87Sr/86Sr ratios obtained for the Antarctic PM10 samples are spread within a range between the South Australian and Patagonian reference values, rendering the recognition of their origin, as previously reported, not straightforward.38 In fact, when using one isotope ratio only, potential overlap of the signatures of different potential source areas, such as Patagonia, New Zealand and Antarctica Dry Valley, may occur and interfere with an unequivocal provenancing.29,39 In future investigations, the Sr isotope ratio results will be combined with data on other isotopic proxies (Nd, Pb). Considering the necessity to perform different analyses on the same sample, the development of a method that consumes less than 1 mL of sample is of even greater importance.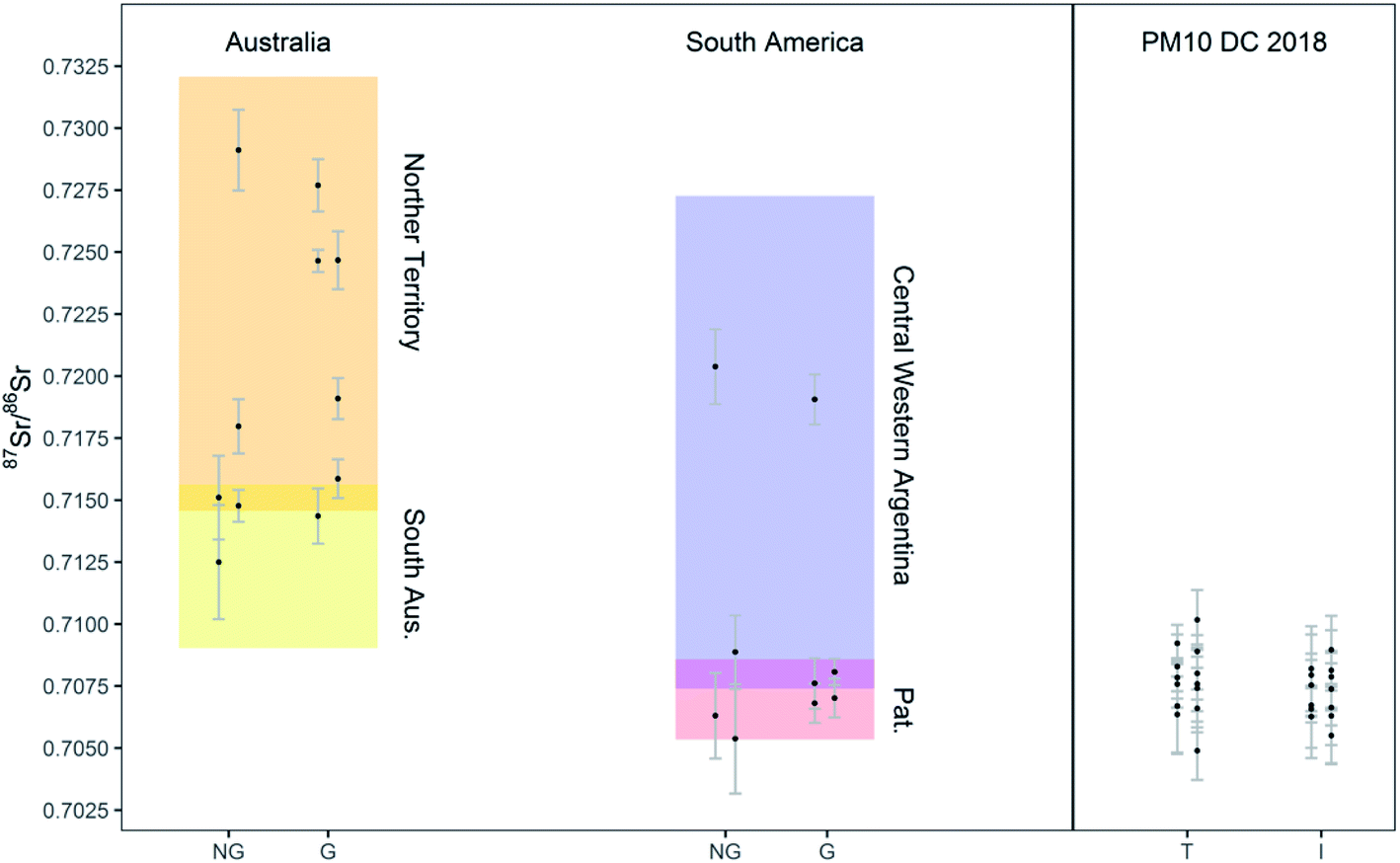 | ||
| Fig. 5 Sr isotope ratio results for soil samples and PM10 Antarctic samples. NG: non-grinded fractions of soil samples; G: grinded fractions of soil samples; T: total fraction of PM10; I: insoluble fraction of PM10. Sr isotope ratios from literature for Australian (Northern Territory and South Australia) and South American (Patagonian and Central Western Argentinian) soils are also reported for comparison as shaded areas.10,40 | ||
4. Conclusions
In this work, a new ICP-MS/MS method for Sr isotopic analysis with low sample consumption (240 μL) has been developed. The use of an MVX-7100 μL autosampler allowed a very stable micro-flow of sample (20 μL min−1) to be obtained. In addition, the expected decrease in sensitivity due to the low sample uptake rate was compensated for by using a high-efficiency sample introduction system, originally developed for the analysis of single cells. Finally, the selective reaction between Sr+ and CH3F and the better control over the in-cell chemistry provided by ICP-MS/MS instrumentation were exploited to achieve the direct determination of the 87Sr/86Sr ratio at the ng g−1 concentration level, without prior chromatographic Rb/Sr separation.The instrumental parameters related to the sample introduction system were optimized by an experimental design methodology to obtain the best instrument sensitivity and internal precision for the 87Sr/86Sr and 88Sr/86Sr isotope ratios. Under the optimal conditions, the method showed fit-for-purpose performance, as demonstrated by the analysis of digests of soil samples from Australia and South America, and Antarctic atmospheric particulate samples, with a Sr concentration around 1 ng g−1.
Although the method developed presents a lower precision than that obtained using ICP-MS/MS at a conventional sample uptake rate for samples with higher Sr concentration (around 10 ng g−1),21 it can be very useful to analyze samples for which the low volume available and/or the target element concentration are critical aspects, such as small-volume digests or pre-concentrated samples.
Author contributions
Conceptualization: Stefano Bertinetti, Eduardo Bolea-Fernandez, Marco Grotti, Frank Vanhaecke. Methodology: Stefano Bertinetti, Eduardo Bolea-Fernandez, Marco Grotti, Frank Vanhaecke. Investigation: Stefano Bertinetti, Mery Malandrino, Beatrice Moroni, David Cappelletti. Validation: Stefano Bertinetti, Eduardo Bolea-Fernandez, Marco Grotti, Frank Vanhaecke. Resources: Mery Malandrino, David Cappelletti, Marco Grotti, Frank Vanhaecke. Visualization: Stefano Bertinetti. Writing – original draft: Stefano Bertinetti. Writing – review and editing: Eduardo Bolea-Fernandez, David M. Cappelletti, Marco Grotti, Frank Vanhaecke. Supervision: Eduardo Bolea-Fernandez, Marco Grotti, Frank Vanhaecke. Funding acquisition: Marco Grotti, Frank Vanhaecke. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was financially supported by the MIUR (Italian Ministry of University and Research) and PNRA (Programma Nazionale di Ricerca in Antartide) through the PNRA 2016/AC2.04 (PNRA 16_00252-Linea A2) Project “SIDDARTA: Source IDentification of (mineral) Dust to AntaRcTicA” and PNRA 2015/AC3.04 (PNRA 14_00091-Linea A3) Project “LTCPAA: Long-Term Measurements of Chemical and Physical Properties of Atmospheric Aerosol at Dome C”. EBF thanks FWO-Vlaanderen for his postdoctoral grant (12ZA320N). FV and EBF acknowledge Teledyne Cetac Technologies and Glass Expansion for lending them the MVX-7100 μL sample introduction unit and the single-cell glassware, respectively.References
- J. L. Todolí and J. M. Mermet, Sample introduction systems for the analysis of liquid microsamples by ICP-AES and ICP-MS, Spectrochim. Acta, Part B, 2006, 61, 239–283, DOI:10.1016/j.sab.2005.12.010.
- C. Ferrer, A. Lozano, A. Agüera, A. J. Girón and A. R. Fernández-Alba, Overcoming matrix effects using the dilution approach in multiresidue methods for fruits and vegetables, J. Chromatogr. A, 2011, 1218, 7634–7639, DOI:10.1016/j.chroma.2011.07.033.
- A. Ulianov, O. Müntener and U. Schaltegger, The ICPMS signal as a Poisson process: a review of basic concepts, J. Anal. At. Spectrom., 2015, 30, 1297–1321, 10.1039/c4ja00319e.
- S. Bertinetti, F. Ardini, L. Caiazzo and M. Grotti, Determination of major elements in Antarctic snow by inductively coupled plasma optical emission spectrometry using a total-consumption sample introduction system, Spectrochim. Acta, Part B, 2021, 181, 106231, DOI:10.1016/j.sab.2021.106231.
- M. Grotti, F. Ardini and J. L. Todolì, Total introduction of microsamples in inductively coupled plasma mass spectrometry by high-temperature evaporation chamber with a sheathing gas stream, Anal. Chim. Acta, 2013, 767, 14–20, DOI:10.1016/j.aca.2013.01.017.
- E. D. Salin and R. L. A. Sing, Microsample Liquid Analysis with a Wire-Loop Direct Sample Insertion Technique in an Inductively Coupled Plasma, Anal. Chem., 1984, 56, 2596–2598, DOI:10.1021/ac00277a074.
- A. Bazzano and M. Grotti, Determination of lead isotope ratios in environmental matrices by quadrupole ICP-MS working at low sample consumption rates, J. Anal. At. Spectrom., 2014, 29, 926–933, 10.1039/c3ja50388g.
- A. Bazzano, K. Latruwe, M. Grotti and F. Vanhaecke, Lead isotopic analysis of Antarctic snow using multi-collector ICP-mass spectrometry, J. Anal. At. Spectrom., 2015, 30, 1322–1328, 10.1039/c4ja00484a.
- I. M. Villa, P. De Bièvre, N. E. Holden and P. R. Renne, IUPAC-IUGS recommendation on the half life of 87 Rb, Geochim. Cosmochim. Acta, 2015, 164, 382–385, DOI:10.1016/j.gca.2015.05.025.
- M. Revel-Rolland, P. De Deckker, B. Delmonte, P. P. Hesse, J. W. Magee and I. Basile-Doelsch, et al., Eastern Australia: A possible source of dust in East Antarctica interglacial ice, Earth Planet. Sci. Lett., 2006, 249, 1–13, DOI:10.1016/j.epsl.2006.06.028.
- M. Barbaste, M. Barbaste, K. Robinson, S. Guilfoyle, B. Medina and R. Lobinski, Precise determination of the strontium isotope ratios in wine by inductively coupled plasma sector field multicollector mass spectrometry (ICP-SF-MC-MS), J. Anal. At. Spectrom., 2002, 17, 135–137, 10.1039/b109559p.
- R. A. Bentley, Strontium isotopes from the earth to the archaeological skeleton: A review, J. Archaeol. Method Theor, 2006, 13, 135–187, DOI:10.1007/s10816-006-9009-x.
- A. Y. Kramchaninov, I. V. Chernyshev and K. N. Shatagin, Isotope analysis of strontium by multicollector inductively-coupled plasma mass spectrometry: High-precision combined measurement of 88Sr/86Sr and 87Sr/86Sr isotope ratios, J. Anal. Chem., 2012, 67, 1084–1092, DOI:10.1134/S1061934812140067.
- D. R. Bandura, V. I. Baranov and S. D. Tanner, Reaction chemistry and collisional processes in multipole devices for resolving isobaric interferences in ICP-MS, Fresenius. J. Anal. Chem., 2001, 370, 454–470, DOI:10.1007/s002160100869.
- L. J. Moens, F. F. Vanhaecke, D. R. Bandura, V. I. Baranov and S. D. Tanner, Elimination of isobaric interferences in ICP-MS, using ion-molecule reaction chemistry: Rb/Sr age determination of magmatic rocks, a case study, J. Anal. At. Spectrom., 2001, 16, 991–994, 10.1039/b103707m.
- E. Bolea-Fernandez, L. Balcaen, M. Resano and F. Vanhaecke, Overcoming spectral overlap: Via inductively coupled plasma-tandem mass spectrometry (ICP-MS/MS). A tutorial review, J. Anal. At. Spectrom., 2017, 32, 1660–1679, 10.1039/c7ja00010c.
- L. Balcaen, E. Bolea-Fernandez, M. Resano and F. Vanhaecke, Inductively coupled plasma – Tandem mass spectrometry (ICP-MS/MS): A powerful and universal tool for the interference-free determination of (ultra)trace elements – A tutorial review, Anal. Chim. Acta, 2015, 894, 7–19, DOI:10.1016/j.aca.2015.08.053.
- L. Balcaen, E. Bolea-Fernandez, M. Resano and F. Vanhaecke, Accurate determination of ultra-trace levels of Ti in blood serum using ICP-MS/MS, Anal. Chim. Acta, 2014, 809, 1–8, DOI:10.1016/j.aca.2013.10.017.
- E. Bolea-Fernandez, L. Balcaen, M. Resano and F. Vanhaecke, Potential of Methyl Fluoride as a Universal Reaction Gas to Overcome Spectral Interference in the Determination of Ultratrace Concentrations of Metals in Biofluids Using Inductively Coupled Plasma-Tandem Mass Spectrometry, Anal. Chem., 2014, 86, 7969–7977, DOI:10.1021/ac502023h.
- M. Resano, M. Aramendía, F. V Nakadi, E. García-Ruiz, C. Alvarez-Llamas and N. Bordel, et al., Breaking the boundaries in spectrometry. Molecular analysis with atomic spectrometric techniques, TrAC, Trends Anal. Chem., 2020, 129, 115955, DOI:10.1016/j.trac.2020.115955.
- E. Bolea-Fernandez, L. Balcaen, M. Resano and F. Vanhaecke, Tandem ICP-mass spectrometry for Sr isotopic analysis without prior Rb/Sr separation, J. Anal. At. Spectrom., 2016, 31, 303–310, 10.1039/c5ja00157a.
- E. Bolea-Fernandez, S. J. M. Van Malderen, L. Balcaen, M. Resano and F. Vanhaecke, Laser ablation-tandem ICP-mass spectrometry (LA-ICP-MS/MS) for direct Sr isotopic analysis of solid samples with high Rb/Sr ratios, J. Anal. At. Spectrom., 2016, 31, 464–472, 10.1039/c5ja00404g.
- E. Bolea-Fernandez, A. Rua-Ibarz, M. Velimirovic, K. Tirez and F. Vanhaecke, Detection of microplastics using inductively coupled plasma-mass spectrometry (ICP-MS) operated in single-event mode, J. Anal. At. Spectrom., 2020, 35, 455–460, 10.1039/c9ja00379g.
- T. Liu, E. Bolea-Fernandez, C. Mangodt, O. De Wever and F. Vanhaecke, Single-event tandem ICP-mass spectrometry for the quantification of chemotherapeutic drug-derived Pt and endogenous elements in individual human cells, Anal. Chim. Acta, 2021, 1177, 338797, DOI:10.1016/j.aca.2021.338797.
- N. E. Holden, T. B. Coplen, J. K. Böhlke, L. V. Tarbox, J. Benefield and J. R. De Laeter, et al., IUPAC Periodic Table of the Elements and Isotopes (IPTEI) for the Education Community (IUPAC Technical Report), Pure Appl. Chem., 2018, 90, 1833–2092, DOI:10.1515/pac-2015-0703.
- W. Zhang and Z. Hu, Estimation of isotopic reference values for pure materials and geological reference materials, At. Spectrosc., 2020, 41, 93–102, DOI:10.46770/as.2020.03.001.
- EN 14902:2005; Ambient air quality – Standard method for the measurement of Pb, Cd, As and Ni in the PM10 fraction of suspended particulate matter, 2005, available from: http://store.uni.com/catalogo/en-14902-2005 Search PubMed.
- B. Delmonte, P. S. Andersson, H. Schöberg, M. Hansson, J. R. Petit and R. Delmas, et al., Geographic provenance of aeolian dust in East Antarctica during Pleistocene glaciations: preliminary results from Talos Dome and comparison with East Antarctic and new Andean ice core data, Quat. Sci. Rev., 2010, 29, 256–264, DOI:10.1016/j.quascirev.2009.05.010.
- B. Delmonte, I. Basile-Doelsch, J. R. Petit, V. Maggi, M. Revel-Rolland and A. Michard, et al., Comparing the Epica and Vostok dust records during the last 220,000 years: Stratigraphical correlation and provenance in glacial periods, Earth-Sci. Rev., 2004, 66, 63–87, DOI:10.1016/j.earscirev.2003.10.004.
- B. Delmonte, J. R. Petit, K. K. Andersen, I. Basile-Doelsch, V. Maggi and V. Y. Lipenkov, Dust size evidence for opposite regional atmospheric circulation changes over east Antarctica during the last climatic transition, Clim. Dynam., 2004, 23, 427–438, DOI:10.1007/s00382-004-0450-9.
- B. Benedetti, V. Caponigro and F. Ardini, Experimental Design Step by Step: A Practical Guide for Beginners, Crit. Rev. Anal. Chem., 2020, 1–14 DOI:10.1080/10408347.2020.1848517.
- R Core Team. R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, 2020, [cited 2021 Jul 6]. available from: https://www.r-project.org/ Search PubMed.
- R. Leardi, C. Melzi, and G. Polotti, CAT (Chemometric Agile Tool), 2017, available from: http://www.gruppochemiometria.it/index.php/software/19-download-the-r-based-chemometric-software Search PubMed.
- K. J. Hogmalm, T. Zack, A. K. O. Karlsson, A. S. L. Sjöqvist and D. Garbe-Schönberg, In situ Rb-Sr and K-Ca dating by LA-ICP-MS/MS: An evaluation of N2O and SF6 as reaction gases, J. Anal. At. Spectrom., 2017, 32, 305–313, 10.1039/c6ja00362a.
- F. Vanhaecke, R. Dams and C. Vandecasteele, “Zone model” as an explanation for signal behaviour and non-spectral interferences in inductively coupled plasma mass spectrometry, J. Anal. At. Spectrom., 1993, 8, 433–438, 10.1039/JA9930800433.
- H. Andrén, I. Rodushkin, A. Stenberg, D. Malinovsky and D. C. Baxter, Sources of mass bias and isotope ratio variation in multi-collector ICP-MS: Optimization of instrumental parameters based on experimental observations, J. Anal. At. Spectrom., 2004, 19, 1217–1224, 10.1039/b403938f.
- K. G. Heumann, S. M. Gallus, G. Rädlinger and J. Vogl, Precision and accuracy in isotope ratio measurements by plasma source mass spectrometry, J. Anal. At. Spectrom., 1998, 13, 1001–1008, 10.1039/a801965g.
- B. Delmonte, H. Winton, M. Baroni, G. Baccolo, M. Hansson and P. Andersson, et al., Holocene dust in East Antarctica: Provenance and variability in time and space, Holocene, 2020, 30, 546–558, DOI:10.1177/0959683619875188.
- B. G. Koffman, S. L. Goldstein, G. Winckler, A. Borunda, M. R. Kaplan and L. Bolge, et al., New Zealand as a source of mineral dust to the atmosphere and ocean, Quat. Sci. Rev., 2021, 251, 106659, DOI:10.1016/j.quascirev.2020.106659.
- S. Gili, D. M. Gaiero, S. L. Goldstein, F. Chemale, J. Jweda and M. R. Kaplan, et al., Glacial/interglacial changes of Southern Hemisphere wind circulation from the geochemistry of South American dust, Earth Planet. Sci. Lett., 2017, 469, 98–109, DOI:10.1016/j.epsl.2017.04.007.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ja00329a |
| This journal is © The Royal Society of Chemistry 2022 |