
Lead determination by HG-MIP OES with nitrogen plasma after a variable optimization study
Alicia
Mollo
 *a,
Alexandra
Sixto
b,
Florencia Cora
Jofré
c,
Mariela
Pistón
a and
Marianela
Savio
c
*a,
Alexandra
Sixto
b,
Florencia Cora
Jofré
c,
Mariela
Pistón
a and
Marianela
Savio
c
aGrupo de Análisis de Elementos Traza y Desarrollo de Estrategias Simples para Preparación de Muestras (GATPREM), Facultad de Química, Universidad de la República, Montevideo, Uruguay. E-mail: amollo@fq.edu.uy
bAnalytical Chemistry, DEC, Facultad de Química, Universidad de la República, Montevideo, Uruguay
cInstituto de Ciencias de la Tierra y Ambientales de La Pampa (INCITAP), Santa Rosa, La Pampa, Argentina
First published on 30th November 2021
Abstract
A hydride generation nitrogen microwave induced plasma optical emission spectrometry (HG-MIP OES) system was developed for lead determination. Plumbane chemical generation was performed using 0.045 mol L−1 K3Fe(CN)6 as the additive in acidic medium and 2.0% w/v NaBH4 in 1% w/v NaOH as the reductant. The additivated sample and the reductant were pumped towards a cyclonic chamber where hydride generation and separation from the solution occurred. Operating conditions such as the additive and reductant concentration, sample, reductant and nitrogen flow-rate, the cyclonic chamber stabilization time and the viewing position in the torch were optimized. A meticulous study of the variation of the nitrogen plasma response as a function of the hydrochloric acid concentration was carried out. The analytical performance of the method was studied and its applicability evaluated in a Trace Element in Water Certified Reference Material and a Soil Reference Material. The method performance in terms of limits of detection and quantification (0.54 μg L−1 and 1.8 μg L−1 Pb, respectively) was found comparable to other atomic spectrometry techniques. This is to our knowledge the first research on the analytical performance of lead determination by PbH4 generation and determination by MIP OES.
Introduction
Lead occurs in the Earth's crust associated with other elements; the industrial processes release it to air, water and land. Adverse health effects are related to continuous low concentration exposure. Even if several anthropogenic sources have been eliminated, its persistence in the environment makes exposure monitoring relevant due to its toxicological concern.1 Hence, when selecting the analytical method for its determination, low detection limit techniques are required. Analytical atomic spectrometric techniques (i.e., atomic absorption spectrometry (AAS), atomic emission spectrometry (AES) and atomic fluorescence spectrometry (AFS)) coupled with chemical vapour generation (CVG) are suitable alternatives to fulfil this purpose.2CVG is a widespread sample introduction technique utilized in analytical chemistry. Hydride generation (HG) was first reported for hydride forming elements such as As, Bi, Ge, Hg, Pb, Sb, Se, Sn and Te but was later extended to noble metals such as Au, Ag, Pd, Pt, Rd, Rh, and Os and other transition metals such as Co, Cu, Cr, Fe, Ir, Mn, Ni, Rh, Ti, and Zn.3–5
Sodium tetrahydroborate (THB) has been the most common derivatization reagent used for HG. The HG mechanism depends on the reaction medium acid concentration as it determines the prevailing reagent and analyte species in the solution, and thus their mutual reactivity as well as the formed hydride stability.3,6,7
However, in the case of lead, the direct reaction efficiency between Pb(II) and THB yielding lead tetrahydride (plumbane, PbH4) is very low. Its generation needs an additive with a ligand/donor and/or redox properties which interact with the analyte and/or THB producing an analytically useful reactivity modification.6,8 D'Ulivo et al. largely studied its role in PbH4 generation, where hexacyanoferrate(III) is suitable for that purpose.6,8–10 They postulated that hexacyanoferrate(III) formed a hydridoboron intermediate that reacts with Pb(II) enhancing its formation.8,9
The volatile species once generated are driven towards the atomization device for its determination. Several detection systems have been coupled to CVG: quartz tube atomic absorption spectrometry (QT AAS), probably the most used; electrothermal atomic absorption spectrometry (ETAAS); inductively coupled plasma optical emission spectrometry (ICP OES); microwave induced plasma optical emission spectrometry (MIP OES); and inductively coupled plasma mass spectrometry (ICP MS).6,11–14
MIP OES with nitrogen plasma is an interesting alternative technique to ICP OES and is able to be hyphenated with HG.15 In 1987, Barnett16 reported the first evidence of lead tetrahydride detection by MIP OES with helium plasma after HG with sodium persulphate. However, since the commercially available high-power MIP OES with nitrogen plasma in 2012, only a few studies have determined Pb by MIP OES; nevertheless, some used conventional chambers and none others clarified if it was done with or without hydride generation.17–23
The microwave plasma (MIP) is created by nitrogen excitation through the combination of an axial magnetic field and a transverse electric one allowing a good interaction of the plasma and the sample, as well as higher plasma temperature. It reaches temperatures around 5000 K depending on the nitrogen flow-rate selected in the chamber and the plasma viewing position.24,25
For MIP OES analysis the hydride is generated in a multimode spray chamber (MSIS) which acts both as the generating compartment and gas–liquid separator. It consists of a glass cyclonic chamber fitted with two vertically opposed conical tubes where sample and reductant flows converge and mix in a thin film. A nitrogen flow strips the volatile species generated towards the torch. There are minimum transport losses as the cyclonic chamber is connected directly to the atomization device.14,26–28
To the best of our knowledge the first evaluation of MIP OES with nitrogen plasma for lead determination using hydride generation as the sample introduction technique is performed in this work. The chemical variables affecting the hydride generation and the instrumental parameters for its determination are presented as well as the analytical figures of merit for this methodology.
Experimental
Instrumentation
A microwave-induced plasma optical emission spectrometer MIP OES Agilent model 4210 (Agilent Technologies, Santa Clara, USA) equipped with a multimode spray chamber (MSIS, Agilent), and a standard torch was used for determination. An online nitrogen generator model 4107 (Agilent Technologies, Santa Clara, USA), which takes in air from the environment through an air compressor model KK70 TA-200 K (DürrTechnik, Bietigheim-Bissingen, Germany) was used.A microwave-assisted digestion system (Mars 6 CEM, North Carolina, USA) equipped with 12 Easy Prep Plus vessels was used to prepare the soil reference material for lead determination.
Materials
A Millipore water purification system (resistivity 18.2 Mohm cm) was used to obtain deionized water. Lead working solutions were prepared daily by dilution of the corresponding 1000 mg L−1 (atomic absorption standard solution, Merck, Germany). The following reagents used were of analytical grade: potassium hexacyanoferrate(III), 63% w/w nitric acid and 37% w/w hydrochloric acid. Sodium tetrahydroborate solutions were prepared from sodium tetrahydroborate (hydride generation grade, Fluka) in 1% w/v NaOH.The certified reference material, MRC.INO.101 trace elements in water, was obtained from Laboratorio Tecnológico del Uruguay (LATU), Montevideo, Uruguay (2% v/v HNO3 medium, certified value: 34.48 μg L−1 Pb).
Soil reference material: RM-Agro E2002a (Embrapa Pecuária Sudeste, São Carlos SP, Brazil). Reference value: Pb (mg kg−1) 172.41 ± 4.02 (RSD% 2.33).
No sample preparation step was required for Trace Element in Water CRM and it was measured directly.
Analytical HG procedure
Lead solutions already containing the additive 0.045 mol L−1 K3Fe(CN)6 and acid (HCl or HNO3 as appropriate) and 2.0% w/v THB were pumped towards the MSIS spray chamber at 0.90 mL min−1. Triplicate measurements were performed for 5 s after the stabilization of the spray chamber with PBH4 generation for at least 20 s. The nitrogen flow-rate towards the plasma was about 750 mL min−1 and viewing position 0. Lead determination was performed at 405.781 nm with an automatic background correction. A general scheme of the described configuration of MSIS for PbH4 generation and determination by MIP OES is shown in Fig. 1.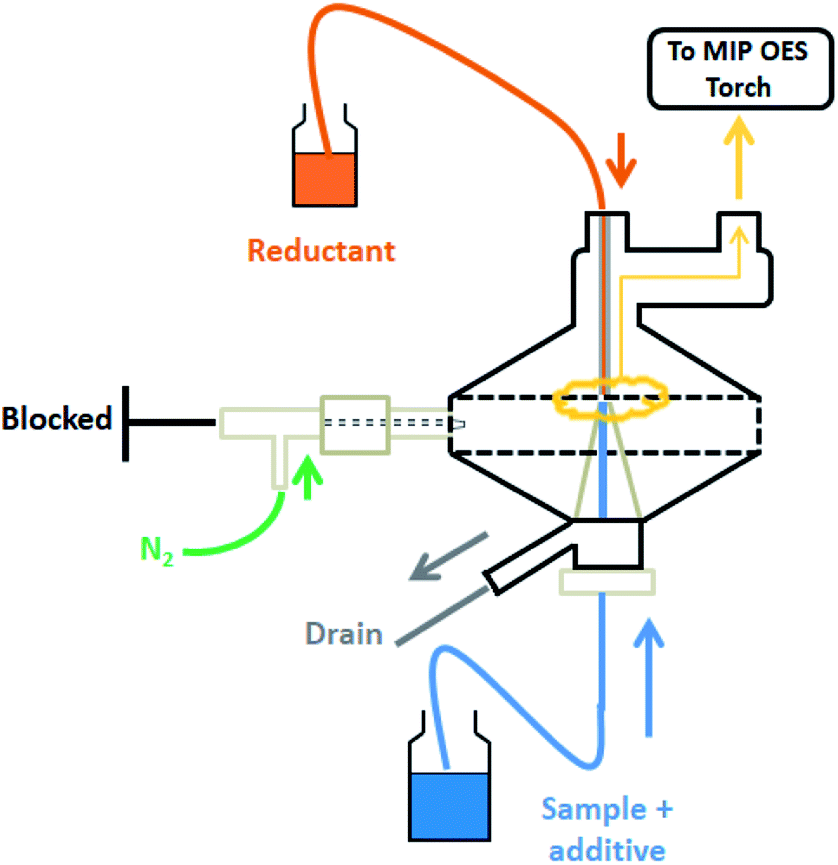 | ||
| Fig. 1 Scheme of the multimode spray chamber operating for plumbane generation. | ||
Results and discussion
Plumbane generation: preliminary optimization
Several reagents are described in the literature as additives for PbH4 generation.8,10,31–34 In this work K3Fe(CN)6 in acidic medium was chosen. The chemical variables studied were K3Fe(CN)6, HCl and NaBH4 concentrations. The reductant and sample flow-rates, and the instrumental parameters such as the stabilization time in the chamber, the nitrogen flow-rate and the viewing position in the torch were also evaluated.The variables and the determining parameters were evaluated one at a time keeping the others constant. Conditions leading to the best signal intensity for PBH4 were taken as the optimum criterion.
As a starting point, instrumental parameters and operating conditions were set as established by the manufacturer: N2 flow-rate 0.75 mL min−1, stabilization time 10 s, viewing position 0 and reagent flow-rate 0.45 mL min−1. Chemical working conditions were chosen close to those found optimum in Kratzer's work.10 Three concentration levels were chosen for K3Fe(CN)6 (0.015–0.030–0.045 mol L−1) and NaBH4 (0.4–1.0–2.0% w/v in 1% NaOH), and two concentration levels for HCl (0.12–1.0 mol L−1).
In the preliminary optimization process, THB concentration was kept fixed at 0.4% w/v while each K3Fe(CN)6 and HCl concentration combinations were run. The minor signal was found for 1.0 mol L−1 HCl at every ferricyanide level. Reagent (0.45 and 0.90 mL min−1) and N2 (0.5–0.75–1.0 mL min−1) flow-rates and the stabilization time of the cyclonic chamber (0, 10, 20 s) were sequentially assayed for each K3Fe(CN)6 concentration level at 0.12 mol L−1 HCl. Next, for the aforementioned conditions leading to the highest signals, different viewing position in the torch (−60 – −30 – 0 – 30 – 60) were tried.
Finally, the reductant concentration (0.4–1.0–2.0% w/v in 1% w/v NaOH) was optimized. Table 1 summarizes the instrumental and working conditions which lead to the maximum signals.
| a Prepared in 1% w/v NaOH. | |||
|---|---|---|---|
| Instrumental parameters | |||
| Pump speed (rpm solution flow-rate 0.90 mL min−1) | 30 | ||
| Nitrogen flow (L min−1) | 0.75 | ||
| Reading time (s) | 5 | ||
| Viewing position | 0 | ||
| Stabilization time (s) | 20 | ||
| Wavelength (nm) | 405.781 | ||
| Background correction | Automatic | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Experimental conditions | |||
| K3Fe(CN)6 | HCl | NaBH4a | |
| 0.045 mol L−1 | 0.12 mol L−1 | 2.0% w/v | |
Cheng et al.34 reached similar results using HCl, indicating that at low acid concentrations Pb does not completely convert into hydride, while at high concentrations the hydride is diluted, observing lower signal intensity in both cases.
From the preliminary optimization stage it follows that the variable of influence impacting the most in the response was the HCl concentration. The study was then focused on the response to that concentration seeking for further optimization.
Optimization of the acidic medium
To weigh up the effect of the HCl concentration on the determination of lead by HG-MIP OES with nitrogen plasma, the signals obtained at different acid concentrations were compared with the signal under the optimal working conditions found in the preliminary optimization study (0.12 mol L−1 HCl–0.045 mol L−1 K3Fe(CN)6). Solutions containing 50 μg L−1 Pb–0.045 mol L−1 K3Fe(CN)6 were prepared at increasing HCl concentrations within 0.05 and 1.15 mol L−1 (evaluated solutions) and run according to the analytical HG procedure described above together with a 50 μg L−1 Pb–0.12 mol L−1 HCl–0.045 mol L−1 K3Fe(CN)6 solution. The signal of the latter was taken as the reference. The relative response was calculated as shown in eqn (1) and plotted against the hydrochloric acid concentration (Fig. 2).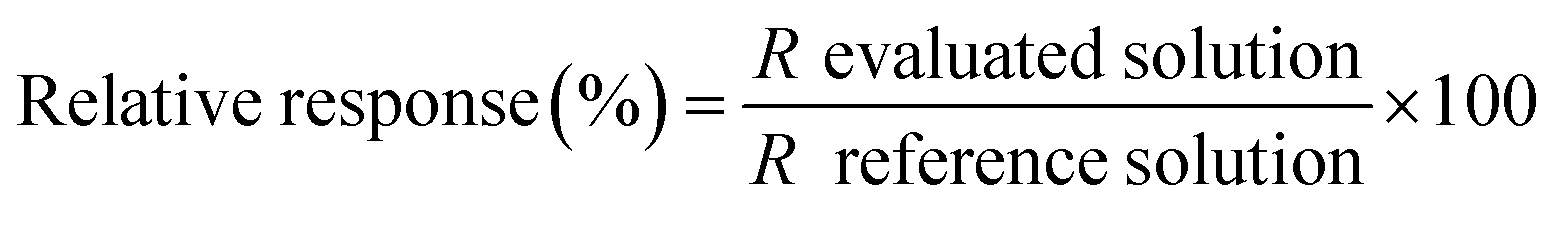 | (1) |
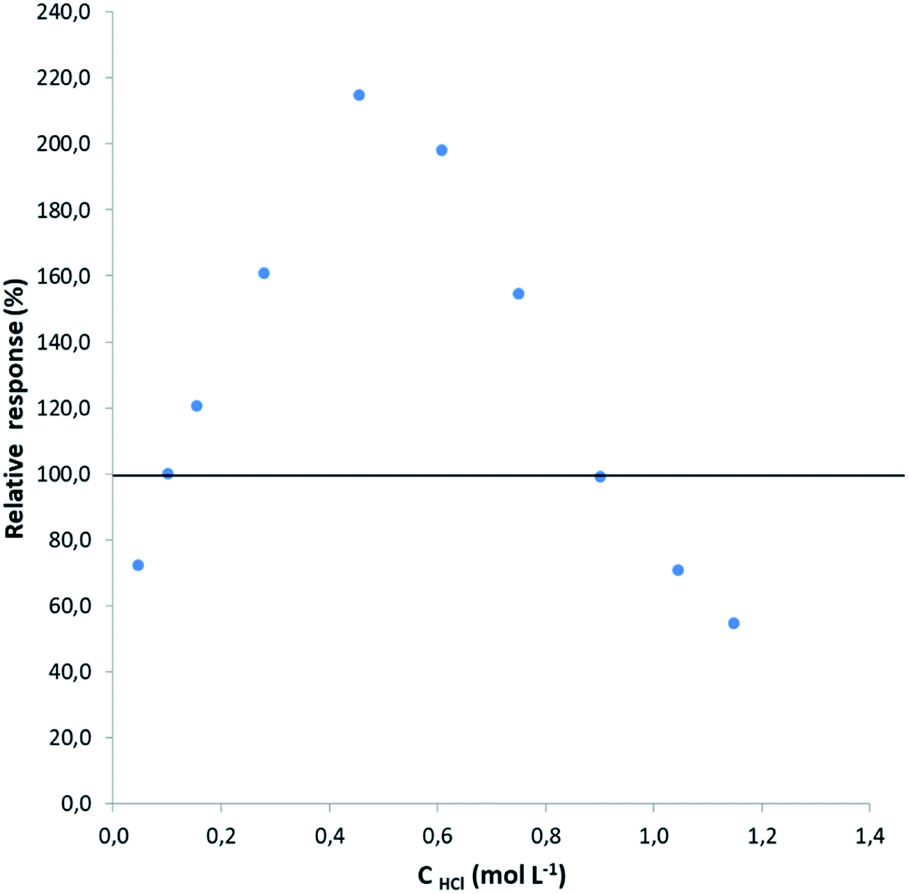 | ||
| Fig. 2 Variation of the relative response with increasing HCl concentration. The net signal of the 50 μg L−1 Pb–0.12 mol L−1 HCl–0.045 mol L−1 K3Fe(CN)6 solution was taken as the reference. 2% w/v NaBH4 in 1% w/v NaOH was used as the reductant. | ||
As can be observed, there is a signal enhancement within an acidic range of 0.12 and 0.90 mol L−1, varying quite deeply in a narrow range of hydrochloric acid concentration, reaching an optimum close to 0.5 mol L−1. At higher hydrochloric acid concentrations, the tetrahydride acid decomposition probably overcomes the HG or the increased amount of hydrogen evolution due to the acid decomposition of NaBH4 dilutes the hydride formed.
These results differ with regard to the ones met by other authors employing AAS. Elci et al.35 and Ertas et al.36 used 0.090 mol L−1 K3Fe(CN)6, using a flow injection system and a continuous flow one for the HG, respectively; both found the optimum acid concentration at 0.036 mol L−1 HCl.
Kratzer10 used a continuous flow generation system for PbH4 generation in 0.030 mol L−1 K3Fe(CN)6 and found a signal enhancement up to 0.10 mol L−1 HCl, a plateau up to 0.2 mol L−1 HCl and a signal drop for higher acid concentrations. Similar results were found by Bakırdere et al.37 using the same K3Fe(CN)6 concentration in a batch system with an optimum signal at 0.16 mol L−1 HNO3 followed by a decay which they attribute to the degradation of ferrycyanide. Nonetheless, Afonso et al.38 used more concentrated ferricyanide, 0.090 mol L−1 K3Fe(CN)6, and found the optimum acid concentration quite close, at 0.091 mol L−1 HCl for a continuous flow system by ICP OES determination.
Except for Ertas et al. who employed a quartz atom trap and made the determination by FAAS, and Afonso as previously mentioned, all the other authors accomplished the atomization in an externally heated quartz tube and the determination by AAS. It is well established that the H radical population in the quartz tube atomizer determines the atomization efficiency.6,11,39 Atomization proceeds according to a radical mechanism for which high temperatures are not needed, just enough hydrogen to supply the radicals; however, a too high hydrogen flux can result in dilution of the volatile species and thus in a signal decay. A high acidic medium results in high hydrogen evolution caused by the tetrahydroborate decomposition.
Among the literature, the work by Ikeda et al.40 was the only one which reported acidic conditions similar to this work. However, the generation system (continuous flow with two gas–liquid separators for plasma stability against hydrogen evolution), as well as the derivatizing agent (oxygen peroxide) are different, which makes the comparison difficult.
Petrov et al.41 used a similar MSIS for the generation system to that used in the present work. Oxygen peroxide was used instead of potassium ferricyanide and ICP-MS for determination, and 0.1 mol L−1 HNO3 was found as the optimum acidic medium.
Lead determination is usually performed after a wet digestion of the sample where nitric acid is frequently used either for this purpose or for liquid sample preservation. Envisaging the method applicability, the influence of the HNO3 concentration in the response was also evaluated. The response of 50 μg L−1 Pb–0.045 mol L−1 K3Fe(CN)6 solutions in (0.12–0.30–0.75–0.90–1.20) mol L−1 HNO3 was compared to the response of 50 μg L−1 Pb–0.045 mol L−1 K3Fe(CN)6 solutions in (0.12–0.30–0.75–0.90–1.20) mol L−1 HCl. For that purpose the relative response for each solution was calculated according to eqn (1). As previously mentioned, the reference solution was 50 μg L−1 Pb–0.12 mol L−1 HCl–0.045 mol L−1 K3Fe(CN)6 both for hydrochloric and nitric acid solutions.
The results are presented in Table 2. The difference in the relative response found when the acidity of the medium came from HNO3 was less than 10% that the one obtained for HCl in the acid concentration range where there is no signal detriment. Hence, using HCl or HNO3 would obtain similar results.
| HCl/HNO3 (mol L−1) | Relative response (%) HCl | Relative response (%) HNO3 |
|---|---|---|
| 0.12 | 100.0 | 101.5 |
| 0.30 | 201.3 | 194.2 |
| 0.75 | 154.6 | 146.3 |
| 0.90 | 99.1 | 104.0 |
| 1.20 | 54.7 | 47.2 |
Among 0.12 and 0.90 mol L−1 HCl or HNO3 have a positive impact on the HG which in terms of acids usually added to the samples corresponds to (1.0–7.5)% v/v HCl or (0.8–6.0)% v/v HNO3. The signal variation is wide in a quite narrow range of acid concentration; hence, the calibration standards must have the same acidity as the samples in order to prevent biased results. Whenever possible, the calibration standards and the samples should be matched in a concentration range where the signal is as high as possible.
In samples requiring high nitric acid concentration digestion (above 6.0% v/v) a neutralization and acid concentration adjustment should be performed before achieving lead determination by HG-MIP OES with nitrogen plasma.
Analytical performance
The method was characterized according to the Eurachem Guide recommendations.42 The analytical parameters of the proposed method such as linear range, limits of detection (3s) and quantification (10s), precision (repeatability) and trueness were evaluated to determine its applicability.According to the results obtained about the relevance of the acid concentration in the sample, the characterization was carried out at the same CRM acid concentration, taking advantage that it is a concentration for which the generation efficiency is high enough.
The linear range was evaluated up to 100 μg L−1. The quantification was thus accomplished using a six-point external standard calibration curve in 0.28 mol L−1 HNO3. The LOD and LOQ were estimated from the standard deviation (s) of ten replicates of the reagent blank signal (0 μg L−1 Pb–0.28 mol L−1 HNO3–0.045 mol L−1 K3Fe(CN)6).
Precision under conditions of repeatability was calculated as the relative concentration standard deviation (RSD) of 6 replicates in Trace Element in Water CRM.
Trueness was assessed by means of Pb recovery after its determination by HG-MIP OES with nitrogen plasma in 6 replicates of the Trace Element in Water CRM (2% v/v or 0.28 mol L−1 HNO3 and 34.48 μg L−1 Pb).
Results are presented in Table 3.
| a I: intensity; C concentration (μg L−1); LOD: limit of detection; LOQ: limit of quantification; s: standard deviation. | |
|---|---|
| Linear range: 1.8–100 μg L−1 | |
| Calibration function (R2 = 0.9995): I = 151.8C + 132.6 | |
| Detection and quantification limits, n = 10 | |
| LOD: 0.54 μg L−1 | LOQ: 1.8 μg L−1 |
| Precision (repeatability, n = 6): 3.3% | |
| Recovery (n = 6) ± s: (89.5 ± 2.9)% | |
The methodology was also applied to analyse a soil reference material which was previously digested as already explained, diluted 50 times and acidified up to 0.28 mol L−1 HNO3. The recovery found was (90.7 ± 4.6)% for two replicates.
A bibliographic research was carried out to compare the proposed methodology analytical performance with other frequently used ones. A comparison of Pb LODs found in the literature with those of other techniques is summarized in Table 4. The LOD found for HG-MIP OES with nitrogen plasma is comparable to those achieved by HG and other atomic spectrometries, with the benefits of considerably reducing costs per run.
| Technique | LOD (μg L−1) | Reference |
|---|---|---|
| a FAAS: flame atomic absorption spectrometry, MIP OES: microwave induced plasma atomic emission spectrometry, ETAA: electrothermal atomic absorption spectrometry, HG-ICP OES: hydride generation inductively coupled plasma atomic emission spectrometry, HG-AAS: hydride generation atomic absorption spectrometry, HG-MIP OES: hydride generation microwave induced plasma atomic emission spectrometry, HG-ICP MS: hydride generation inductively coupled plasma mass spectrometry, and ICP MS: inductively coupled plasma mass spectrometry. | ||
| FAAS | 10 | 11 |
| MIP OES | 7 | 43 |
| ETAAS | 1.7 | 44 |
| HG-ICP OES | 1.0 | 40 |
| HG-AAS | 0.56 | 37 |
| HG-MIP OES | 0.54 | Present work |
| HG-ICP MS | 0.20 | 41 |
| ICP MS | 0.18 | 41 |
Conclusions
This is the first attempt that a novel Pb determination technique involving HG-MIP OES with nitrogen plasma is presented and its analytical performance determined. Experimental conditions were optimized and the relevance of the acid concentration of the reaction medium was studied. The methodology has been successfully applied to the determination of lead in water CRM and a soil RM. The LODs achieved are suitable and in good compliance to those reported by other atomic spectrometries.The combination of HG and MIP OES with nitrogen plasma could be postulated as an advantageous and promising cost-effective alternative for Pb determination in water and soils compared with high-cost instrumental techniques that require argon or acetylene.
To the best of our knowledge this is the first research on the PbH4 generation behaviour in acidic medium for determination by the MIP OES with nitrogen plasma system.
Author contributions
Alicia Mollo: conceptualization, data curation, formal analysis, investigation, methodology, project administration, supervision, validation, writing original draft. Alexandra Sixto: data curation, formal analysis, investigation, methodology, validation, writing review & editing. Florencia Cora Jofré: resources, visualization, writing review & editing. Mariela Pistón: resources, writing review & editing. Marianela Savio: conceptualization, resources, writing review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors wish to thanks Joaquim A. Nóbrega, ANII (Agencia Nacional de Investigación e Innovación), Comisión Sectorial de Investigación Científica (CSIC) and the Programa de Desarrollo de las Ciencias Básicas (PEDECIBA – Química).Notes and references
- Agency for Toxic Substances and Disease Registry (ATSDR), Toxicological Profile for Lead, U.S. Department of Health and Human Services, Public Health Service, Atlanta, GA, 2020, https://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=96%26tid=22, last accessed 10/12/20, DOI:10.15620/cdc:95222.
- A. D'Ulivo, . Dědina, Z. Mester, R. E. Sturgeon, Q. Wang and B. Welz, Pure Appl. Chem., 2011, 83(6), 1283, DOI:10.1351/PAC-REP-09-10-03.
- J. Dědina, Encyclopedia of Analytical Chemistry, Online © 2006–2010, John Wiley & Sons, Ltd 2010, DOI:10.1002/9780470027318.a9127.
- T. Matoušek, Anal. Bioanal. Chem., 2007, 388, 763, DOI:10.1007/s00216-006-1017-x.
- P. Pohl and B. Prusisz, Anal. Bioanal. Chem., 2007, 388, 753, DOI:10.1007/s00216-006-0678-9.
- J. Dedina and D. L. Tsalev, Hydride Generation Atomic Absorption Spectrometry, Wiley, Chichester, 1995 Search PubMed.
- A. D'Ulivo, Spectrochim. Acta, Part B, 2016, 119, 91, DOI:10.1016/j.sab.2016.03.003.
- A. D’Ulivo, J. Anal. At. Spectrom., 2019, 34, 823–847, 10.1039/c9ja00039a.
- A. D'Ulivo, M. Onor, R. Spiniello and E. Pitzalis, Spectrochim. Acta, Part B, 2008, 63, 835, DOI:10.1016/j.sab.2008.02.007.
- J. Kratzer, Spectrochim. Acta, Part B, 2012, 71–72, 40, DOI:10.1016/j.sab.2012.04.005.
- B. Welz and M. Sperling, Atomic Absorption Spectroscopy, Wiley–VCH, Weinheim, Germany, 3rd edn, 1999 Search PubMed.
- J. Dědina, Spectrochim. Acta, Part B, 2007, 62, 846, DOI:10.1016/j.sab.2007.05.002.
- P. Smichowski and S. Farías, Microchem. J., 2000, 67, 147 CrossRef CAS.
- M. Ślachciński, Appl. Spectrosc. Rev., 2014, 49(4), 271, DOI:10.1080/05704928.2013.823547.
- V. Balaram, Microchem. J., 2020, 159, 105483, DOI:10.1016/j.microc.2020.105483.
- N. Barnett, Spectrochim. Acta, Part B, 1987, 42, 859 CrossRef.
- D. H. Bonemann, A. C. Beduhn Luckow, C. Correa Pereira, A. Ossanes de Souza, S. Cadore, A. M. Nunes, M. Antunes Vieira and A. Schwingel Ribeiro, J. Food Compos. Anal., 2021, 96, 103716, DOI:10.1016/j.jfca.2020.103716.
- D. Pemberthy, Y. Padilla and G. A. Peñuela, Environ. Sci. Pollut. Res., 2020 DOI:10.1007/s11356-020-11894-7.
- S. E. Gallego Ríos, C. M. Ramírez, B. E. López, S. M. Macías, J. Leal and C. M. Velásquez, Water, Air, Soil Pollut., 2018, 229, 275, DOI:10.1007/s11270-018-3933-8.
- S. E. Gallego Ríos, G. A. Peñuela and C. M. Ramírez Botero, Food Anal. Methods, 2017, 10, 3407, DOI:10.1007/s12161-017-0908-0.
- H. Matusiewicz and M. Slachcinski, Spectrosc. Lett., 2010, 43, 172, DOI:10.1080/00387010903284323.
- I. Rodriguez Pereiro and A. Carro Díaz, Anal. Bioanal. Chem., 2002, 372, 74, DOI:10.1007/s00216-001-1139-0.
- N. Ozbek, M. Koca and S. Akman, Food Anal. Methods, 2016, 9(8), 2246, DOI:10.1007/s12161-016-0421-x.
- M. Hammer, Spectrochim. Acta, Part B, 2008, 63, 456, DOI:10.1016/j.sab.2007.12.007.
- D. Goncalves, T. McSweeney and G. Donati, J. Anal. At. Spectrom., 2016, 31, 1097, 10.1039/C6JA00066E.
- R. L. J. McLaughlin and I. D. Brindle, U.S Pat., 6,891,605, 2005 Search PubMed.
- R. Mclaughlin, P. Cheese, M. Ding, A. Ptolemy, A. Conn and I. Brindle, A convenient sample introduction system for ICP, ICP-MS, and AA, American Laboratory, 2006, vol. 38 Search PubMed.
- I. D. Brindle, Vapor generation, in Sample Introduction Systems in ICPMS and ICPOES, ed. D. Beauchemin, Elsevier, 2020, p. 381, DOI:10.1016/B978-0-444-59482-2.00008-7.
- I. Machado, I. Dol, E. Rodríguez-Arce, M. V. Cecio and M. Pistón, Microchem. J., 2016, 128, 128, DOI:10.1016/j.microc.2016.04.016.
- M. Pistón, A. Suárez, V. Bühl, F. Tissot, J. Silva and L. Panizzolo, J. Food Compos. Anal., 2020, 103624, DOI:10.1016/j.jfca.2020.103624.
- M. C. Valdes-Hevia y Temprano, M. R. Fernandez de la Campa and A. Sanz-Medel, J. Anal. At. Spectrom., 1993, 8, 821, 10.1039/ja9930800821.
- C. Nerin, S. Olavide, J. Cacho and A. Garnica, Water, Air, Soil Pollut., 1989, 44, 339, DOI:10.1007/bf00279263.
- L. Elçi, Z. Arslan and J. F. Tyson, J. Hazard. Mater., 2009, 162, 880, DOI:10.1016/j.jhazmat.2008.05.113.
- J. Cheng, Q. Li, M. Zhao and Z. Wang, Anal. Chim. Acta, 2019, 1077, 107, DOI:10.1016/j.aca.2019.06.003.
- L. Elci, Z. Arslan, J. F. Tyson, J. Hazard. Mater., 2009, 996, https://scholarworks.umass.edu/chem_faculty_pubs/996 Search PubMed.
- N. Ertas, Z. Arslan and J. F. Tyson, J. Anal. At. Spectrom., 2008, 23, 223, 10.1039/b712126a.
- S. Bakırdere, D. S. Chormey, Ç. Büyükpınar, N. San and S. Keyf, Anal. Lett., 2016, 49(12), 1917, DOI:10.1080/00032719.2015.1127380.
- D. D. Afonso, S. Baytak and Z. Arslan, J. Anal. At. Spectrom., 2010, 25, 726, 10.1039/b920280c.
- P. Dvořák, M. Talába, J. Kratzer and J. Dedina, Chem. Sci., 2019, 10, 3643, 10.1039/c8sc05655b.
- M. Ikeda, J. Nishibe, S. Hamada and R. Tujinoa, Anal. Chim. Acta, 1981, 125(C), 109, DOI:10.1016/s003-2670(01)85055-8.
- P. K. Petrov, G. Wibetoe and D. L. Tsalev, Spectrochim. Acta, Part B, 2006, 61, 50, DOI:10.1016/j.sab.2005.11.001.
- B. Magnusson and U. Ornemark, Eurachem Guide: the Fitness for Purpose of Analytical Methods – a Laboratory Guide to Method Validation and Related Topics, http://www.eurachem.org, 2nd edn, 2014 Search PubMed.
- S. E. Gallego Ríos, G. A. Peñuela and C. M. Ramírez Botero, Food Anal. Methods, 2017, 10, 3407–3414, DOI:10.1007/s12161-017-0908-0.
- C. E. Dogan and H. M. Ortner, Instrum. Sci. Technol., 2008, 36, 267, DOI:10.1080/10739140801944043.
| This journal is © The Royal Society of Chemistry 2022 |