
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A combined photobiological–photochemical route to C10 cycloalkane jet fuels from carbon dioxide via isoprene†
Anup
Rana
 a,
Leandro
Cid Gomes
a,
João S.
Rodrigues
a,
Dalia M. M.
Yacout
b,
Hugo
Arrou-Vignod
a,
Johan
Sjölander
a,
Nathalie
Proos Vedin
a,
Ouissam
El Bakouri
a,
Karin
Stensjö
a,
Peter
Lindblad
a,
Leena
Andersson
c,
Cecilia
Sundberg
*b,
Mathias
Berglund
*c,
Pia
Lindberg
*a and
Henrik
Ottosson
*a
a,
Leandro
Cid Gomes
a,
João S.
Rodrigues
a,
Dalia M. M.
Yacout
b,
Hugo
Arrou-Vignod
a,
Johan
Sjölander
a,
Nathalie
Proos Vedin
a,
Ouissam
El Bakouri
a,
Karin
Stensjö
a,
Peter
Lindblad
a,
Leena
Andersson
c,
Cecilia
Sundberg
*b,
Mathias
Berglund
*c,
Pia
Lindberg
*a and
Henrik
Ottosson
*a
aDepartment of Chemistry – Ångström Laboratory, Uppsala University, Box 523, 751 20 Uppsala, Sweden. E-mail: henrik.ottosson@kemi.uu.se; pia.lindberg@kemi.uu.se
bDepartment of Energy and Technology, Swedish University of Agricultural Sciences (SLU), 750 07 Uppsala, Sweden. E-mail: cecilia.sundberg@slu.se
cRISE Research Institutes of Sweden, Brinellgatan 4, Box 857, 501 15 Borås, Sweden. E-mail: mathias.berglund@ri.se
First published on 7th November 2022
Abstract
The hemiterpene isoprene is a volatile C5 hydrocarbon with industrial applications. It is generated today from fossil resources, but can also be made in biological processes. We have utilized engineered photosynthetic cyanobacteria for direct, light-driven production of bio-isoprene from carbon dioxide, and show that isoprene in a subsequent photochemical step, using either near-UV or simulated or natural solar light, can be dimerized into limonene, paradiprene, and isomeric C10H16 hydrocarbons (monoterpenes) in high yields under photosensitized conditions (above 90% after 44 hours with near-UV and 61% with simulated solar light). The optimal sensitizer in our experiments is di(naphth-1-yl)methanone which we use with a loading of 0.1 mol%. It can also easily be recycled for subsequent photodimerization cycles. The isoprene dimers generated are a mixture of [2 + 2], [4 + 2] and [4 + 4] cycloadducts, and after hydrogenation this mixture is nearly ideal as a drop-in jet fuel. Importantly the photodimerization can be carried out at ambient conditions. However, the high content of hydrogenated [2 + 2] dimers in our isoprene dimer mix lowers the flash point below the threshold (38 °C); yet, these dimers can be converted thermally into [4 + 2] and [4 + 4] dimers. When hydrogenated these monoterpenoids fully satisfy the criteria for drop-in jet fuels with regard to energy density, flashpoint, kinematic viscosity, density, and freezing point. Life-cycle assessment results show a potential to produce the fuel in an environmentally sustainable way.
Introduction
In order to mitigate global warming and reach the goals of the Paris agreement, a shift towards carbon neutral fuels is necessary. For the year 2050, the International Air Transport Association (IATA) emission reduction roadmap projects a reduction in CO2 emissions from aviation by 50% compared to 2005 levels.1 This may seem modest, yet, globally air traffic increased by 4.5–8.7% per year during the period 2009–2019,2 and a low annual increase of 4% until 2050, resulting from changes in travel patterns due to COVID-19 and the installment of alternative transportation infrastructures,3 still implies more than a three-fold increase in air traffic by 2050 when compared to 2019 and approximately six-fold when compared to 2005. As the increase in air traffic is often considerably steeper in growing economies, fulfilment of the IATA goal requires prompt technological development and introduction of new sustainable aviation fuels far beyond the biofuels based on biomass cultivation and processing currently in use or at the stage to be introduced on the market.Today, there are different technologies and feedstock alternatives to conventional jet fuels.4–6 An emerging route to biofuels goes via direct production of hydrocarbons by engineered photosynthetic microorganisms, such as algae or cyanobacteria.7–10 Cyanobacteria are photosynthetic bacteria which grow on water, minerals, and CO2 from the atmosphere, using sunlight as their energy source. Many cyanobacterial strains are amenable to genetic engineering, by which new pathways may be introduced leading to generation of specific target products, and thus, they are ideal hosts for biotechnological production of sustainable fuels.11–13
Fossil-based jet fuels consist mostly of C8–C16 hydrocarbons. More explicitly, they are mixtures of n-, iso- and cyclo-alkanes, small aromatics (<25%) and alkenes (<5%).14,15 The fuel should be a proportional mixture of these compounds in order to follow the strict requirement for jet-fuels in terms of energy density, freezing point, and viscosity. In one typical jet fuel, JP-8, the proportion of C10 hydrocarbons is ∼21%.16 Hydrogenated monoterpenes (C10) and sesquiterpenes (C15) have long been considered as potential jet fuels due to their low viscosity and high energy density. Limonane (hydrogenated limonene) has been in focus among hydrogenated monoterpenes because of its availability from biomass fermentation and the low estimated cost of the resulting fuel (∼0.73 USD per L).17 Sesquiterpenes, e.g. bisabolene, farnesene and epi-isozizaene, are also molecules with potential utility.17–19
While biotechnological production of monoterpenes and sesquiterpenes has been demonstrated in various microorganisms, the toxicity of these compounds to the cells is often problematic.20 Mono- and sesquiterpenoids tend to accumulate in the biological membranes, due to their hydrophobic nature, and interfere with their integrity and function.21 On the other hand, smaller hydrocarbons, e.g., alkenes such as iso-butene and the 5-carbon-atom hemiterpenoids, are more volatile and tend to easily escape through the cell membranes.22,23 Their diffusion to the extracellular environment makes them less toxic to the cells and their harvest/capture is less costly since there is no need for cell disruption. We, therefore, suggest a two-step procedure in which these small volatile hydrocarbons (C5 and smaller) are produced photobiologically, followed by their photochemical oligomerization in a second separate step. Isoprene is a volatile five-carbon hydrocarbon and can be an ideal precursor. It contains CC double bonds which are useful as sites for (photo)oligomerization, and its production by photosynthetic engineered cyanobacteria has been demonstrated.22,24–27 Thus, even while there is at present no fully developed technology that could produce them on a commercial scale, hydrogenated isoprene oligomers could be ideal as drop-ins replacing presently used aviation fuels. To realize a production process of this type of fuel at scale will require process development to reach environmental as well as economic sustainability, both of which will be absolute requirements for commercialization of new types of sustainable aviation fuels.
There are already well-established chemical methods using heterogeneous catalysts common in industry for oligomerization of alkenes and dienes,28 which require high temperatures and pressures. Recently, Harvey and co-workers reported iron-catalyzed dimerization processes of alkenes and dienes, including isoprene, that run at ambient temperature and pressure and that produce [2 + 2] and [4 + 4] cycloadducts (Fig. 1).29,30 Interestingly, the hydrogenated [4 + 4] dimers of isoprene have better fuel properties compared to conventional jet fuels (Jet-A), and a life-cycle assessment and technoeconomic analysis showed that the process can be further improved to reduce cost and emission to compete within the sustainable aviation fuel sector.31 The [2 + 2] oligomerization of isoprene was not selective for dimers since also trimers and tetramers were formed in significant amounts. Recently, it was also reported that [4 + 2] and [2 + 2] isoprene dimers are formed upon heating at 200 °C, representing a viable route to drop-in sustainable aviation fuels.32
 | ||
| Fig. 1 (A) The two iron-based catalysts by Harvey and co-workers,29,30 and (B) the catalyzed oligomerization of isoprene. (C) Photochemical dimerization of isoprene which resulted in the formation of [2 + 2], [4 + 2] and [4 + 4] photodimers.33 Bonds formed in the reaction are marked in red. | ||
We have explored to what extent isoprene can be dimerized photochemically through triplet sensitizers using as mild conditions as possible, ultimately with solar light and in ambient conditions. The photochemical dimerization of isoprene was reported already in the 60s by Hammond, Turro and Liu using benzophenone (5 mol%) as photosensitizer (Fig. 1C), leading to 65% conversion to isoprene dimers when irradiated for five days in a sealed tube.33 Interestingly, the composition of the dimer mixtures, i.e., the distribution of [2 + 2], [4 + 2] vs. [4 + 4] cycloadducts, depended on the triplet energies of photosensitizers,34 yet importantly, trimers and longer oligomers were not formed. Combined with photosynthetic generation of isoprene from CO2, this could provide for sustainable production of hydrocarbons for jet fuels. Here it can be noted that there are only a few earlier studies on the direct production of jet fuels from CO2.35–37 An inexpensive heterogeneous Fe–Mn–K catalyst prepared by the organic combustion method was utilized for direct conversion of CO2 to jet fuel range hydrocarbons, with a yield of 17% for such hydrocarbons when run at 300 °C and 1 MPa for 20 hours,37 thus requiring a rather large energy input. Recently, a model of thermochemical solar fuel production has been demonstrated where CO2 and H2O were captured from ambient air in a process that will be suitable for fuel production in desert regions.38 Yet, we seek a process that requires as modest energy input as possible. Hence, we now report on the first formation of C10 hydrocarbons, suitable as jet fuel drop-ins after hydrogenation, in a combined two-step photobiological–photochemical approach with CO2 as carbon source and with light, either as (simulated) solar or ambient light, as the predominant energy source for the process.
To ensure a sustainable production route, a system analysis perspective is needed as it allows us to understand the different impacts of the product throughout its entire life cycle.39 Today, life cycle assessment (LCA) is employed as the main decision-support tool for implementing renewable energy technologies using a holistic framework,40–42 and several earlier studies have assessed the environmental impacts of biofuel production from microalgae using LCA.43–51 Furthermore, it has been shown that algae-derived biodiesel is the most efficient alternative in terms of land use as it avoids competition with food crops.52,53 The environmental impacts of producing cyanobacteria-based biofuels have also been assessed.42,44,54 Both Luo et al. and Quiroz-Arita et al. employed LCA to assess the life cycle energy and greenhouse gas (GHG) emissions of ethanol production via cyanobacteria,44,54 and revealed that the ethanol purification process was the main energy consumer and a significant contributor to the carbon footprint of the process. Nilsson et al. assessed the environmental impacts of photosynthetic butanol production by genetically engineered cyanobacteria,42 and found that in order to displace fossil fuels using butanol produced by cyanobacteria, significant metabolic engineering-based improvements in carbon and energy conversion efficiencies per cell are needed.
As the process reported herein is based on a volatile product which spontaneously separates from the cell culture, we can eliminate the energy requiring distillation or processing of biomass, in contrast to ethanol and larger alcohols as well as direct biodiesel production. Our combined photobiological and photochemical process thus avoids the costly and energy intensive cell disruption and organic extraction procedures required for bioproduction of non-volatile aviation fuels.55,56 The process resembles a previously envisioned strategy on catalyzed oligomerization of ethylene produced by cyanobacteria, which was explored in a technoeconomic analysis study and revealed to yield economically viable biofuels in the long term.57 We used LCA to assess the different environmental impacts of jet fuel production through the combined photobiological–photochemical route in order to identify the hot spots and improvement options. Our results should aid the further development of the novel emerging technology presented herein as it pinpoints the hurdles that need to be addressed, and thus, enable a faster realization of the technology at a large scale.
Experimental section
Cell culture and trapping of isoprene
Isoprene production in cyanobacteria was performed using an engineered Synechocystis sp. PCC 6803 strain, expressing an optimized version of isoprene synthase from Eucalyptus globulus, together with two other enzymes – DXS and IDI. Details on biological materials, seed cultures, production, extraction and trapping of isoprene can be found in the ESI.†Materials
Isoprene (99%, contains <1000 ppm p-tert-butylcatechol inhibitor as stabilizer) used for these studies was purchased from Sigma Aldrich (Merck). Inhibitor was removed by passing isoprene through a pack of activated basic alumina and used immediately. Benzophenone (99%), xanthone (97%) and thioxanthone (97%), and all common reagents used for the synthesis of other photosensitizers were purchased from Sigma Aldrich (Merck) and used for photoreaction without further purification. Detailed synthesis descriptions of the other photosensitizers can be found in the ESI.†General characterization methods
Gas chromatography-mass spectrometry (GC-MS) (Agilent 7890A GC and 5975 MSD system) was used for monitoring the photoreactions. The starting temperature of the column oven was 70 °C (0.5 min equilibration time) and the ending temperature was 320 °C, and helium was used as the carrier gas. The column used was an Agilent 19091S-433: 325 °C: 30 m × 250 μm × 0.25 μm. Mass spectrometer: source temperature: 230 °C, quad-temperature 150 °C. The 1H and 13C NMR spectra were recorded on JEOL (400YH magnet) Resonance 400 MHz spectrometer. UV-Vis absorption spectra were measured by a Varian Cary 5000 UV-Vis spectrophotometer. Further details on the general characterization methods and measurement of jet fuel properties can be found in the ESI.†Photodimerization of isoprene
RPR-100 and 200 Rayonet Photochemical Chamber Reactors were used for photoreaction. A set of 16 × 24 W UV lamps at 365 nm (purchased from Southern New England Ultraviolet Company) was used for photoirradiation. Photoreactions were performed on two different set up (a) small-scale photoreactions: 18 mL quartz cylindrical tubes (RQV-7: Rayonet; ∅ 13 mm) were used for batch reaction setup and solution was stirred during photoirradiation to homogenize the irradiation to the solution. The typical ambient temperature of the Rayonet photoreactor was ∼35–40 °C. (b) Large-scale photoreactions: Fluorinated ethylene propylene polymer (FEP) tube (O.D. × I.D.: 3.18 mm × 2.1 mm, loop volume ∼ 120 mL) and FEP tube (O.D. × I.D.: 6.35 mm × 7.94 mm, loop volume: 400 mL) coiled around the water-cooled jacketed beaker (2 L, ∅: 130 mm; height: 280 mm) were used. The distance between sample solutions and the lamps was 8.5 cm ≈ 3.3′′. A SS-F5-3A solar simulator by Enlitech, with a 300 W Xe lamp, was used in the experiments with solar simulated light. The photoreaction in solar simulator (∼10 mL loop volume) and solar irradiation (∼50 mL loop volume) were carried out in home designed flat type cell. In this setup, FEP tube (O.D. × I.D.: 3.18 mm × 2.1 mm) was coiled spirally on a flat wooden surface covered with reflective aluminum foil. Parr hydrogenation apparatus was used for hydrogenation of unsaturated isoprene dimers. More details on the photochemical dimerization procedure and photoreactors, including images, can be found in the ESI.†General details on computational methods
Geometry optimizations were performed with Gaussian 16,58 at the (U)B3LYP/6-311+G(d,p), (U)B3LYP-D3/6-311+G(d,p) and (U)M06-2X/6-311+G(d,p) levels.59–61 Stationary points were characterized as minima or transition states through frequency calculations. The enthalpy corrected energies and the Gibbs free energies were calculated at 298 K. The enthalpies of combustion are computed using the method provided by Major and co-workers62 at the M06-2X/6-31+G(d,p). Further details on the computational methods can be found in the ESI.†Results and discussion
The photobiological formation and trapping of the isoprene produced by the cyanobacteria are presented first, followed by optimization of the photoinitiated dimerization of isoprene (including bio-isoprene) to yield C10 hydrocarbons (monoterpenoids). The dimerization mechanism is analyzed through density functional theory (DFT) computations, unravelling why isoprene trimers are formed in only trace amounts. To be useful as a fuel, the monoterpenoids formed need to be hydrogenated and we determine various properties and assess the values of our hydrogenated monoterpenoids in relation to what is required for a jet fuel. We also carry out a life cycle assessment in order to pinpoint the different environmental impacts of bio-jet fuel production and to identify the related hot spots and improvement options. The results of the study will facilitate further development of the emerging technology presented.Microbial production and trapping of isoprene
Cyanobacteria, like other bacteria, are able to generate terpenoids via the methylerythritol-4-phosphate (MEP) pathway, but do not naturally produce isoprene (Fig. 2A).63 In previous work, we have established engineered strains of the unicellular cyanobacterium Synechocystis sp. PCC 6803 (hereafter Synechocystis), capable of light-driven isoprene production from CO2, via photosynthesis. This was achieved through the introduction of genes encoding an efficient isoprene synthase (IspS) and two enzymes upstream in the MEP pathway – DXS, 1-deoxy-D-xylulose-5-phosphate synthase, and IDI, isopentenyl-diphosphate isomerase (Fig. 2A).25 DXS performs the first step of the pathway by combining the two substrates pyruvate and glyceraldehyde-3-phosphate to form 1-deoxy-D-xylulose 5-phosphate (DXP). IDI performs the interconversion of isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), the substrate for the isoprene synthase to form isoprene.25,63 The reaction catalyzed by DXS includes a decarboxylation step, thereby serving as a gateway for the flux of carbon into the MEP pathway. The expression of IDI is likely necessary to maintain the balance between IPP and DMAPP, and thus enable the synthesis of essential terpenoids downstream in the terpenoid biosynthesis, when IspS expression would otherwise deplete the levels of DMAPP in the cell.25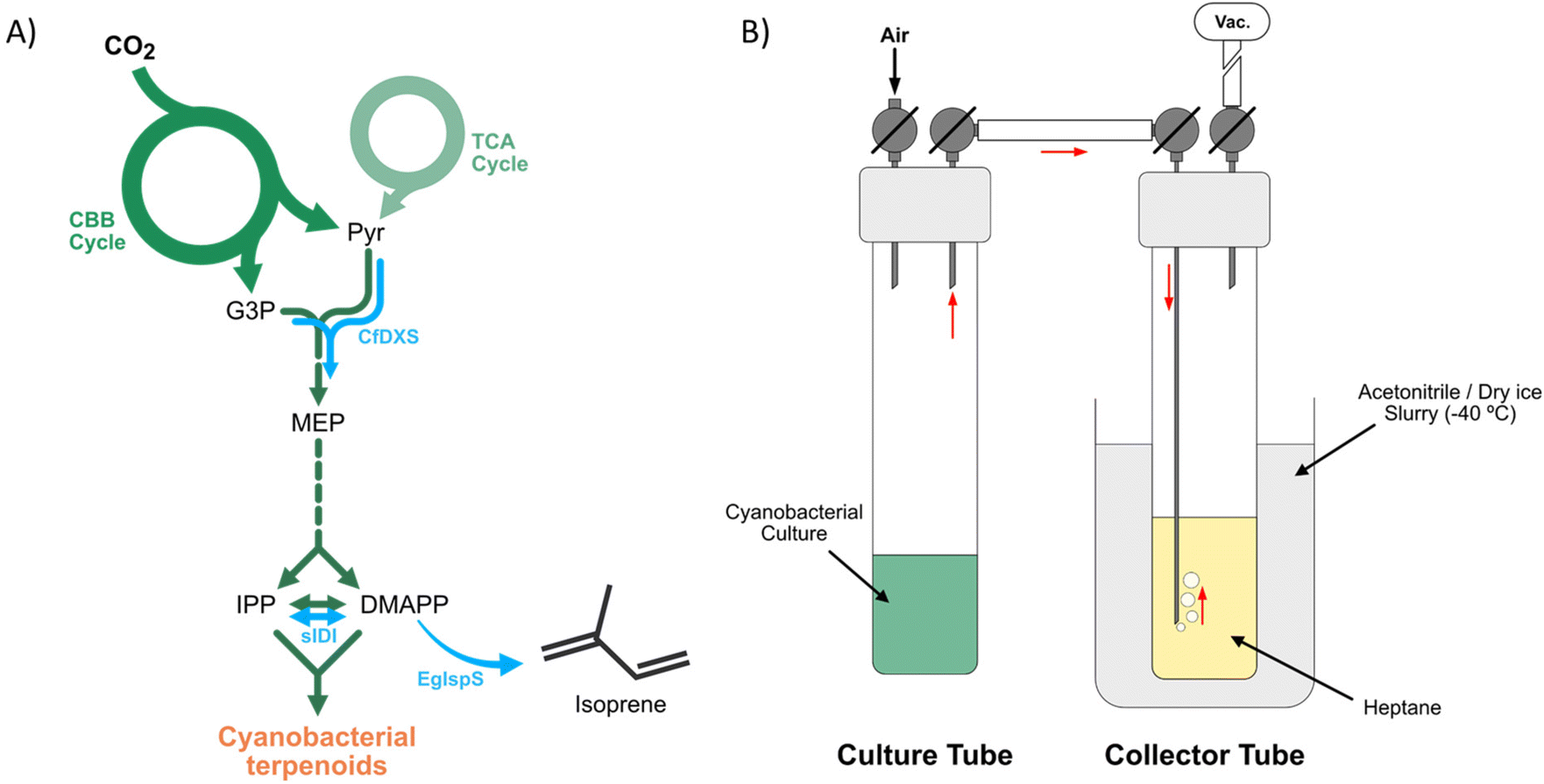 | ||
| Fig. 2 (A) Schematic representation of cyanobacterial terpenoid pathway (green) and genetic modifications (blue arrows) in the isoprene-producing strain used in this study. (B) Schematic representation of the customized isoprene capturing system. CBB – Calvin–Benson–Bassham; TCA – tricarboxylic acid; Pyr – pyruvate; G3P – glyceraldehyde-3-phosphate; MEP – methylerythritol-4-phosphate; IPP – isopentenyl-pyrophosphate; DMAPP – dimethylallyl-pyrophosphate; CfDXS – 1-deoxy-D-xylulose-5-phosphate synthase from Coleus forskohlii; sIDI – IPP/DMAPP isomerase from Synechocystis sp. PCC 6803; EgIspS – isoprene synthase from Eucalyptus globulus. Vac = vacuum line. The red arrows indicate the flow of the isoprene vapor. | ||
Here, we have used the engineered Synechocystis cells for photosynthetic production of isoprene in small-scale cultures. 20 mL of cyanobacterial culture were grown for four days in sealed 60 mL culture tubes under a constant illumination of 50 μmol photons m−2 s−1, with addition of 50 mM NaHCO3 to the culture medium. Thereafter, the headspace gas was drawn through 20 mL of cold heptane to capture produced bio-isoprene from the cultures (Fig. 2B). Isoprene concentrations in the gas phase of the cultures were determined by gas chromatography comparing to an isoprene standard, before and after capturing of the gas phase. For further experimental details, see Fig. S1, ESI.†
We achieved an isoprene titre of 1.60 mg L−1 culture after four days of cultivation under the abovementioned conditions. After capturing the isoprene in heptane in our customized trapping setup, the equivalent of 935 μg L−1 of culture remained in the cultivation tube, which translates into a capture efficiency of 41.4% (Fig. S2 and Table S1, ESI†). A second cycle of trapping resulted in the capture of ca. 490 μg L−1 culture and a higher efficiency (52.4%), for a combined trapping efficiency of ca. 70%. Additionally, we achieved higher capture efficiencies in a single trapping step for other tests, reaching as high as 89% of the isoprene produced. The bio-isoprene trapped in the heptane of the collector tubes was then used for the photochemical dimerization experiments (see section below on Photodimerization of bio-isoprene).
Throughout the experiments, we observed variability in the isoprene production by the engineered strain, likely due to genetic instability of the expression constructs. In order to improve long-term isoprene production, we therefore generated another strain of Synechocystis, where the genetic constructs conferring ability to produce isoprene are expressed from the cyanobacterial chromosome rather than from a plasmid. This was achieved by integration into the slr0168 neutral site in the genome, resulting in successful expression of EgIspS from the new site (Fig. 3A).64,65 The resulting strain, ΔNS1::2MEP-EgIspS, was evaluated for stability of isoprene production. Compared to the previous strain, ΔNS1::2MEP-EgIspS presents a consistent productivity, even when seed-cultures are inoculated from cultures that have been kept growing for several weeks (data not shown).
 | ||
| Fig. 3 Integration of isoprene expression construct into the Synechocystis genome and cultivation with intermittent collection of the headspace. (A) Genetic map showing the inserted DNA construct in strain ΔNS1::2MEP-EgIspS. Ptrc – trc promoter; BCD2 – bicistronic device 2; Strep-sIpi – codon-optimized gene encoding N-strep-tagged IPI from Synechocystis; RBS* – ribosome binding site; Strep-Cfdxs – codon-optimized gene encoding N-strep-tagged DXS from C. forskohlii; EgIspS-FLAG – codon-optimized gene encoding C-FLAG-tagged IspS from E. globulus; Ptrc2O – synthetic variant of Ptrc;66 KmR – resistance cassette against kanamycin. (B) Culture appearance during six days of cultivation with different venting regimes. (C) Cumulative isoprene production during the six-day experiment. In (B) and (C), ‘12 h’, ‘24 h’, ‘48 h’ and ‘72 h’ denote the different intervals at which the respective cultures were opened for venting the gas phase. Error bars represent standard deviations of two biological replicates. | ||
Furthermore, since the isoprene production is performed in closed vessels where isoprene accumulates in the headspace, we hypothesized that over time the concentration of isoprene and oxygen in the culture tubes may become inhibitory for cell growth and productivity. We therefore performed a set of experiments where the headspace gas was vented from the cultures at different intervals. In these experiments, closed cultures of ΔNS1::2MEP-EgIspS were grown for 6 days with sampling and removal of the gas phase at 12, 24, 48 or 72 hours intervals, and growth and isoprene production was evaluated (see Fig. S3†). In cultures with more frequent venting of the gas phase (12–48 h cycles), growth as well as productivity continued for a longer time period, and total cumulative isoprene production and rates of production were higher than in cultures in which the headspace was vented every 72 h (Fig. 3B, C and Table S2, ESI†). Regardless of the periodicity of these cycles, the cumulative amounts of isoprene were always higher than when no cycling was applied. These results are in agreement with previous reports on butanol and isobutanol production in cyanobacteria, where semi-continuous cultivation with frequent dilution resulted in prolonged and enhanced productivity of the cultures.67,68 While challenges remain regarding the details of how the downstream process for separation of product and biomass would be performed efficiently at scale, the strategy of continuous or fed-batch cultivation with frequent product removal is thus a potential avenue for developing the isoprene production process on larger scale.
Screening of triplet sensitizers
To establish a photochemical isoprene dimerization process that utilizes solar irradiation (natural or simulated) we started at the triplet sensitized diene dimerization reported by Hammond, Turro and Liu in the 60s.33 Arylketones are excellent photosensitizers due to their relatively high triplet quantum yields and exceptional photostability. The excitation wavelength of arylketones can be tuned to the visible region by extension of π-conjugation of the aryl groups. Additionally, the triplet quantum yield of ketones can be greatly improved compared to the corresponding arene chromophore.69 Such modulations push the excitation of the sensitizers toward the visible wavelength region where they can be activated by solar light (see below). In the screening of photosensitizers suitable for photodimerization of isoprene we used benzophenone (9), xanthone (10), thioxanthone (11), di(naphth-1-yl)methanone (12), naphthalen-1-yl(naphth-2-yl)methanone (13), and di(naphth-2-yl)methanone (14), see Fig. 4. The synthesis of the photosensitizers is discussed in the ESI.† The triplet energies (E(T1)) of 9–14 and isoprene, both experimentally determined and calculated using density functional theory (DFT) at the (U)B3LYP-D3/6-311+G(d,p) level,59,70–72 indicate that these ketones are suitable for effective photosensitization because their E(T1)'s are slightly higher than that of isoprene (Fig. 4 and Fig. S4, ESI†). Furthermore, the T1 states of dinapthylketones (12–14) are of ππ* character which prevents the competing H atom abstraction,73 a photoreaction that many ketones with T1 states of nπ* character initiate. In a typical photoreaction, a mixture of inhibitor-free isoprene and aryl ketone was contained in a quartz test tube under argon and irradiated with 365 nm light (Fig. S5, ESI†). The solution was stirred during the photoirradiation in order to achieve uniform light exposure. | ||
| Fig. 4 The photosensitizers used in this study as well as isoprene, and in parenthesis, their experimental triplet energies (kJ mol−1, in red) and the calculated adiabatic triplet energies (kJ mol−1, in blue) at (U)B3LYP-D3/6-311+G(d,p) level.34,73–76 | ||
The isoprene dimers formed were characterized by 1H nuclear magnetic resonance (NMR) and gas chromatography-mass spectrometry (GCMS) analysis (Fig. S6–S9, ESI†). However, we confirmed the structure of the isomers by 1H NMR as the GC chromatograms can give erroneous results on the relative product distribution due to thermal rearrangement of the dimers (see below). Seven isomeric isoprene dimers (2–8) were observed, in line with findings reported by Hammond, Turro and Liu (Fig. 1C and Fig. S9, ESI†).33 It was also proposed by Hammond and Liu that cyclooctadienes 7 and 8 might have resulted from thermal rearrangements in the GC,77 but our 1H NMR data of the isoprene dimers (purified by silica gel column by using pentane as eluent) reveals that these two dimers originate from photoinitiated dimerization and cyclization. Here it can be noted that the distribution of the various isomers depends on the E(T1)'s of the photosensitizers used. It is also noteworthy that trace amounts of isoprene trimers were formed, but not any longer oligomers (Fig. S7, ESI†).
The screening of the photosensitizers was performed by using 2 mol% loading, unless otherwise mentioned in Table 1. Depending on the photosensitizer, with the quartz tube setup (∅ 13 mm, Fig. S5, ESI†) we observed 8–41% conversion to isoprene dimers with di(naphthalen-1-yl)methanone 12 giving the highest conversion. A control experiment carried out without photosensitizer clarified its crucial role as the conversion dropped to 0.5% after 44 h of irradiation with λ = 365 nm (Fig. S10, ESI†).
|
|
||||
|---|---|---|---|---|
| Photosensitizer | Loading of photosensitizer (mol%) | Isolated yield (%) | Loading of photosensitizer (mol%) | Isolated yield (%) |
| a The actual loading was lower due to poor solubility of the sensitizer in isoprene. | ||||
| — | 0 | 0.5 | — | — |
| 9 | 2 | 36 | 0.1 | 22 |
| 10 | (0.4)a | 8 | 0.1 | 12 |
| 11 | (0.1)a | 32 | 0.1 | 32 |
| 12 | 2 | 44 | 0.1 | 42 |
| 13 | (0.3)a | 34 | 0.1 | 28 |
| 14 | (0.3)a | 21 | 0.1 | 28 |
| 15 | 2 | 11 | 0.1 | 5 |

Interestingly, the efficiency of the three dinaphthylmethanone isomers (12–14) to convert isoprene to its dimers varied from 21 to 44% due to the positional effect of naphthyl groups. Thus, the isomeric dinaphthylmethanones (12) acts as a better photosensitizer than benzophenone (9), while similar yield of isoprene dimers could be obtained with 13, and comparably the lowest yield could be obtained when 14 was used. If we compare the relative absorbance of the benzophenone (9) and the three dinaphthylmethanone isomers (12–14) at 365 nm, the maximum molar extinction coefficient is observed for 13 and minimum for 9 (Fig. S11 and S12, ESI†), and from the E(T1)'s of 12–14 (Fig. 4) it is clear that 12 is the dinaphthylmethanone with a triplet energy closest to that of isoprene. Additionally, the absorption tails of the dinapthylmethanones go beyond 400 nm, which possibly enable solar light photosensitization. As a result, the isoprene photodimerizations using dinaphthylmethanone sensitizers can be run with very low sensitizer loadings and as they absorb solar irradiation, it is apparent that particularly 12 is a suitable photosensitizer.
The yields of isoprene dimers when xanthone 10 and thioxanthone 11 were used as photosensitizers were significantly lower as compared to when benzophenone (9) was used, and we initially considered this to arise from their poor solubility in neat isoprene. To improve the solubility, we designed and synthesized 3,6-di(octyloxy)xanthenone (15) with solubilizing alkyl groups (for synthesis see ESI†). Yet, despite an improved solubility, the improvement in the isoprene-to-dimer conversion is minute (from 8 to 11%). Instead, the higher E(T1) of both 10 and 15 compared to 9 may cause less efficient triplet energy transfer to isoprene and, consequently, a less efficient isoprene dimerization. Indeed, the calculated triplet energy of 15 is higher than that of 10 by 10.9 kJ mol−1, revealing that substitution allows for further tailoring of xanthone-type sensitizers, similarly as recently reported by Booker-Milburn and co-workers.78
Optimization of dinaphthylmethanone sensitized dimerization
Having identified the most suitable photosensitizers among those selected, we determined the loading of 12 required for the optimal conversion of isoprene to its dimers. The photosensitizer loadings were screened from 0.5 down to 0.01 mol% with a similar setup as used above (see Table S3, ESI†). We could observe 21% yield of isoprene dimers in 44 h with the loading of 12 as low as 0.01 mol%. It is worth noting that the yield of the isoprene dimers does not correlate linearly with the loading of 12 as the light transmission through the solution, which is a function of the sensitizer concentration, influences the yields. We found that a loading of 12 of 0.1 mol% was adequate to get an optimized yield of the isoprene dimers. Additionally, we re-screened all photosensitizers (9–15) at 0.1 mol% concentration and the results confirmed that 12 was the most efficient photosensitizer at this concentration (Table 1).Further improvement of the photodimerization was carried out in modified reaction setups. We first used a fluorinated ethylene propylene polymer (FEP) tubing (outer diameter: 3.2 mm, ∼120 mL loop size) coiled around a water-cooled jacketed beaker (Fig. S13, ESI†). The FEP tubing setup extensively increased the surface area for the incident light, which in turn improved the light absorption. The water-cooled beaker also allows the reaction to run at ∼10 °C which, with a setup which is not fully sealed, prevents evaporation of the volatile and flammable isoprene. With this setup and with 0.1 mol% of loading of 12, we observed 89% yield of isoprene dimers (120 mL scale) when photo-irradiated for 44 h.
We further scaled up the reaction to 400 mL by using wider FEP tubing (outer diameter: 7.9 mm) coiled around the water-cooled jacketed beaker (Fig. S13, ESI†) and we observed a 48% yield of isoprene dimers when using the reaction conditions described above. Here, the lower yield can be attributed to the increased FEP tube diameter which prohibits an equal light distribution over the width of the tube. The yield of the isoprene dimers in the current larger-scale set up could be improved by employing efficient mixing and by using light source with higher intensity.
The isomer distribution between the isoprene dimers, as quantitatively determined through the 1H NMR spectrum, were found to be: 2 (16.1%), 3 (16.5%), 4 (10.7%), 5 (20.8%), 6 (21.8%), and 7 and 8 (14%) (Fig. S14, ESI†). Therefore, the major fraction of the dimers consists of [2 + 2] isoprene cycloadducts (43.3%), in accordance with the ratios observed by Liu et al.34 The lower triplet energy of dinapthylmethanone 12 than of 9 leads to preferential activation of s-cis isoprene, resulting also in high amounts of [4 + 2] cycloadducts (42.6%). The isoprene dimers and photosensitizer 12 could easily be separated by passing through a short silica gel column by using pentane as eluent or by distillation under reduced pressure (65 °C at ∼0.1 mmHg).
The isoprene dimers could be stored at 4 °C for a few months without noticeable decomposition (Fig. S15, ESI†). However, the conversion of kinetically stable [2 + 2] photodimers to the other thermodynamically more stable dimers was observed after a few months in storage (Fig. S16, ESI†) or upon heating over 100 °C in air. Also noteworthy is that under ambient conditions the photodimers tend to convert slowly over time to the corresponding immiscible epoxides and alcohols (Fig. S17 and S18, ESI†).
Now, can the photochemical formation of isoprene dimers be run under ambient atmosphere? To explore this, we analyzed the photodimerization with the aforementioned setup (120 mL) and photosensitizer content for 44 h under ambient conditions and we observed the same yield (86%) as before. The improved photosensitizing efficiency of 12 compared to benzophenone 9 is attributed to the higher absorption at 365 nm (Fig. S11, ESI†), lowest triplet energy difference as well as higher photodimerization quantum yield (ϕ = 0.91 for the dinaphthyl methanone 12versus ϕ = 0.43 for benzophenone 9, see ESI† for details). It is also noteworthy that 12 is straightforwardly synthesized in a one-pot reaction using readily available and inexpensive reagents, and after the photoirradiation it can easily be recovered (up to 95%), purified, and then used for another cycle. Finally, very low amounts of 12 as photosensitizer (0.1 mol% loading) are needed, which together with its recyclability, should significantly reduce the cost for large-scale production of isoprene dimers.
Dimerization induced by (simulated) solar irradiation
Our ultimate goal is to carry out the photodimerization of isoprene with solar irradiation (Fig. S21, ESI†). Dinaphthylmethanone 12 might be an ideal photosensitizer as its absorption tail stretches until ∼400 nm and the solar irradiation has significant light intensity at the surface of Earth at wavelengths longer than 350 nm (Fig. S22, ESI†). For this reason, we first performed the isoprene photodimerization in a solar simulator (1 sun, AM 1.5G) using a newly designed flat spiral coil made of FEP tubing for simulated solar irradiation of isoprene (Fig. 5). Now, we could obtain 61% yield of isoprene dimers (4 mL scale) when irradiated in the solar simulator for 44 h using 0.1 mol% of dinaphthylmethanone 12 as photosensitizer (Fig. S23, ESI†). | ||
| Fig. 5 (A) The custom-made setup with a coiled FEP tubing (O.D. × I.D.: 3.18 mm × 2.1 mm, 10 cm diameter) on a flat surface for solar simulator irradiation of isoprene. (B) The isoprene being photoirradiated by the solar simulator. The arrow indicates the filling level of isoprene. | ||
Using flat spiral coils (4 mL and 50 mL, Fig. 5 and S24†) and 0.1 mol% of 12, the isoprene dimerization was also tested outdoors in sunlight (Uppsala, Sweden 59°51′09.5′′N 17°39′19.9′′E, approximately 30 m above sea level on May 30–31, 2020 and September 1–2, 2021). In both experiments we observed 17% yields after a total sunlight exposure time of 20 h (Fig. S24 and S25, ESI†). These results are qualitative since reduction in light intensity due to clouds was not considered and as the solar light intensity varies over the day and locations. Thus, the experiments demonstrate that the formation of isoprene dimers under sunlight irradiation is achievable. Furthermore, the higher yield that can be estimated after 20 h in the solar simulator (28%) can be rationalized by the fact that the solar simulator has a higher relative intensity in the 350–400 nm range when compared to natural solar irradiation (see Fig. S22, ESI†).
Photodimerization of bio-isoprene
The bio-isoprene produced by the Synechocystis cells and captured in heptane was mixed with dinaphthylmethanone 12 (0.02 M), filled into the flat spiral coil and irradiated in the solar simulator (24 h, 1 sun, AM 1.5G). Despite that the concentration of bio-isoprene was low, the reaction produced bio-isoprene dimers as confirmed by GC-MS (Fig. 6), which demonstrates that we are able to generate C10 hydrocarbons from CO2. Importantly, experiments with commercially available isoprene in a similarly dilute solution (0.05 M solution in heptane) gave a comparable distribution pattern of dimers (Fig. S26 and S27, ESI†). This proof-of-principle experiment shows the possibility to turn CO2 used as carbon source into C10 cycloalkanes with our combined photobiological–photochemical approach. Bio-isoprene dimerization was also attempted under natural sunlight, yet, no dimers were detected in GCMS. This might result due to two factors; (i) the weaker intensity of the natural solar light compared to the simulated one in the 350–400 nm range (Fig. S22, ESI†), and (ii) the low concentration of the bio-isoprene in heptane. Thus, one next step is to increase the production of bio-isoprene so that a higher concentration can be achieved. This may be addressed via further metabolic engineering of the cyanobacterial strain to enhance flux of fixed carbon towards the isoprene product combined with more efficient trapping of isoprene from the culture. | ||
| Fig. 6 (A) Gas chromatogram showing peaks of bio-isoprene dimers (for the relationship between peaks and isomer types and full chromatograms, see Fig. S26, ESI†). (B) The average mass spectrum for the region RT = 2.392 to 3.623 min, for bio-isoprene solution in heptane. The reaction was photosensitized by dinaphthylmethanone 12 (0.02 M, heptane). The sample was irradiated under simulated sunlight, xenon lamp (1 sun, AM 1.5G, 24 h, flat spiral coil). | ||
Photodimerization mechanism
The reaction mechanism for light-induced formation of the isoprene dimers involves six steps (steps 1–6, Fig. 7) which we explored through DFT computations at the (U)B3LYP/6-311G(d,p) level59,70–72 (for details on the computations and for additional results at M06-2X/6-311G(d,p) level61,72 see the ESI†). The first step is the excitation and intersystem crossing (ISC) of the photosensitizer to its triplet state, followed by triplet energy transfer from the sensitizer to isoprene in the ground state, yielding isoprene in its T1 state. The T1 state isoprene can be described as a radical-pair composed of one resonance stabilized allyl radical and one methyl radical, and it exists in four different conformers with nearly equal energies, yet, separated by activation barriers of 63–67 kJ mol−1. One molecule of isoprene in its T1 state can add to an S0 state isoprene via a number of reaction paths. Among these, the addition of the methyl radical site of a T1 state isoprene molecule to an S0 state isoprene molecule proceeds over slightly lower activation barriers (step 3, lowest barrier ∼56 kJ mol−1) than the addition of the allyl radical part of T1 isoprene to an S0 state isoprene (lowest barrier ∼61 kJ mol−1). The triplet lifetime of isoprene has been determined to 27 ns,79 sufficiently long to allow the activation barrier for dimerization to be overcome. The additions, which are markedly exergonic (−90 to −70 kJ mol−1), lead to intermediate isoprene dimers that can be described as triplet state bis(allyl) radical pairs. Thus, once formed there will be no back reaction. As the two radical sites of the bis(allyl) radical pair are only weakly coupled, the singlet diradical is essentially isoenergetic with the triplet, and a rapid ISC should occur (step 5). Furthermore, the bis(allyl) radical pairs have high conformational flexibilities irrespective of electronic state because the conformer interconversions involve C–C single bond rotations (in the T1 state the rotational barriers are 10–19 kJ mol−1, step 4). Finally, when a singlet state bis(allyl) radical pair adopts a conformer with the two unpaired electrons at a sufficiently close distance they will combine into a C–C single bond (step 6), leading to the observed isoprene dimers with either cyclobutane, cyclohexene or cyclooctadiene rings (Fig. 1C). | ||
| Fig. 7 The various steps in the reaction mechanism for the formation of the cyclic isoprene dimers (steps 1 to 6) and trimers (steps 7 and 8) with the lowest activation energies at UB3LYP/6-311G(d,p) level. ISC = intersystem crossing. For further details see the ESI,† Section S6. | ||
So why is further oligomerization hampered? As the bis(allyl) radical pairs are composed of two allyl radicals which are internally stabilized through π-conjugation, they will be less reactive than triplet state isoprene which can be described as one allyl radical and one reactive methyl radical fragment. Thus, the rate for the addition of the bis(allyl) radical pair to an isoprene in its S0 state, leading to a trimer bis(allyl) radical pair, should be slow (step 7). Indeed, the lowest activation barrier for the addition of the bis(allyl) radical pair to an S0 state isoprene is 83 kJ mol−1, significantly higher than the addition of a T1 state isoprene to an S0 state isoprene (56 kJ mol−1 as seen above). A second potential route to trimers goes via addition of an T1 state isoprene to a C–C double bond of a cycloadduct (step 8), but this process should also be slow as it leads from a single carbon-centered radical to another. For this process we find a lowest calculated activation energy of 79 kJ mol−1. Together with the fact that the ring-closure of the dimer bis(allyl) radical pair is a unimolecular reaction in contrast to the bimolecular reaction to trimer bis(allyl) radical pair, this explains why the further oligomerization to trimers, tetramers, etc. is not competitive with the closure of the bis(allyl) radical pair to the cyclic dimers observed.
Finally, since the combined portions of isoprene dimers that are either [2 + 2] and [4 + 4] cycloadducts make up more than half of the dimer mix, we also tested a T1 state concerted mechanism that would involve a transition state with a cyclic array of 4n electrons stabilized by through-space Baird-aromaticity,80–82 however, we could not locate such a pathway. For further discussions, see ESI.†
Hydrogenation and fuel performance
The isoprene dimers are unsaturated, which is not ideal if they should function as a jet fuel as soot would form due to incomplete combustion when ignited. The isoprene dimers (here labelled ID-1) were therefore hydrogenated in presence of Pd/C as a catalyst at 10 atm hydrogen pressure, providing hydrogenated isoprene dimers (HID-1) in near quantitative isolated yields (see ESI† for detail procedure). These hydrogenated isoprene dimers appeared as a colorless liquid (Fig. S28, ESI†), and they were further characterized by 1H NMR and GCMS analysis (Fig. S29, ESI†). The disappearance of the alkene signals of the isoprene dimers in the 1H NMR spectrum proves a complete reduction of the C–C double bonds, leading us to the cycloalkane-based jet fuel equivalent.For this mixture of hydrogenated isoprene dimers, we determined the key fuel properties, i.e., the net heat of combustion (NHOC), kinematic viscosity, density, and flash point (Table 2). The measured density of HID-1 is 0.77 g mL−1 at 15 °C (Table S7 and Fig. S40, ESI†) which matches well with the lower required density of Jet-A. The density of the fuel is lower than that of dimethylcyclooctanes (DMCO) due to the presence of high amounts of isomers with cyclobutane rings. Moreover, the hydrogen content of the HID-1 (14.37%) is significantly higher than that of Jet-A due to the absence of aromatic and unsaturated moieties, which eventually gives a higher gravimetric NHOC value and produce clean burn without soot formation. The gravimetric NHOC is an important parameter for a jet fuel, and it should be above 42.8 MJ kg−1 according to the standard specification for jet fuels.29 Additionally, the volumetric NHOC value of HID-1 is higher than that of conventional jet fuels (Jet-A). For the two C10 hydrocarbons (18, 19, 25 and 26) in Fig. 8 which have experimentally determined NHOC,16,29 we find that computed values calculated with a DFT-based procedure by Major and co-workers62 are in good agreement (for a further description of the method see the caption Fig. 8 and the ESI†). Thus, based on the computed NHOC of the C10H20 hydrocarbons contained in HID-1 we can also conclude that their energy contents are in line with expected for aviation fuel.
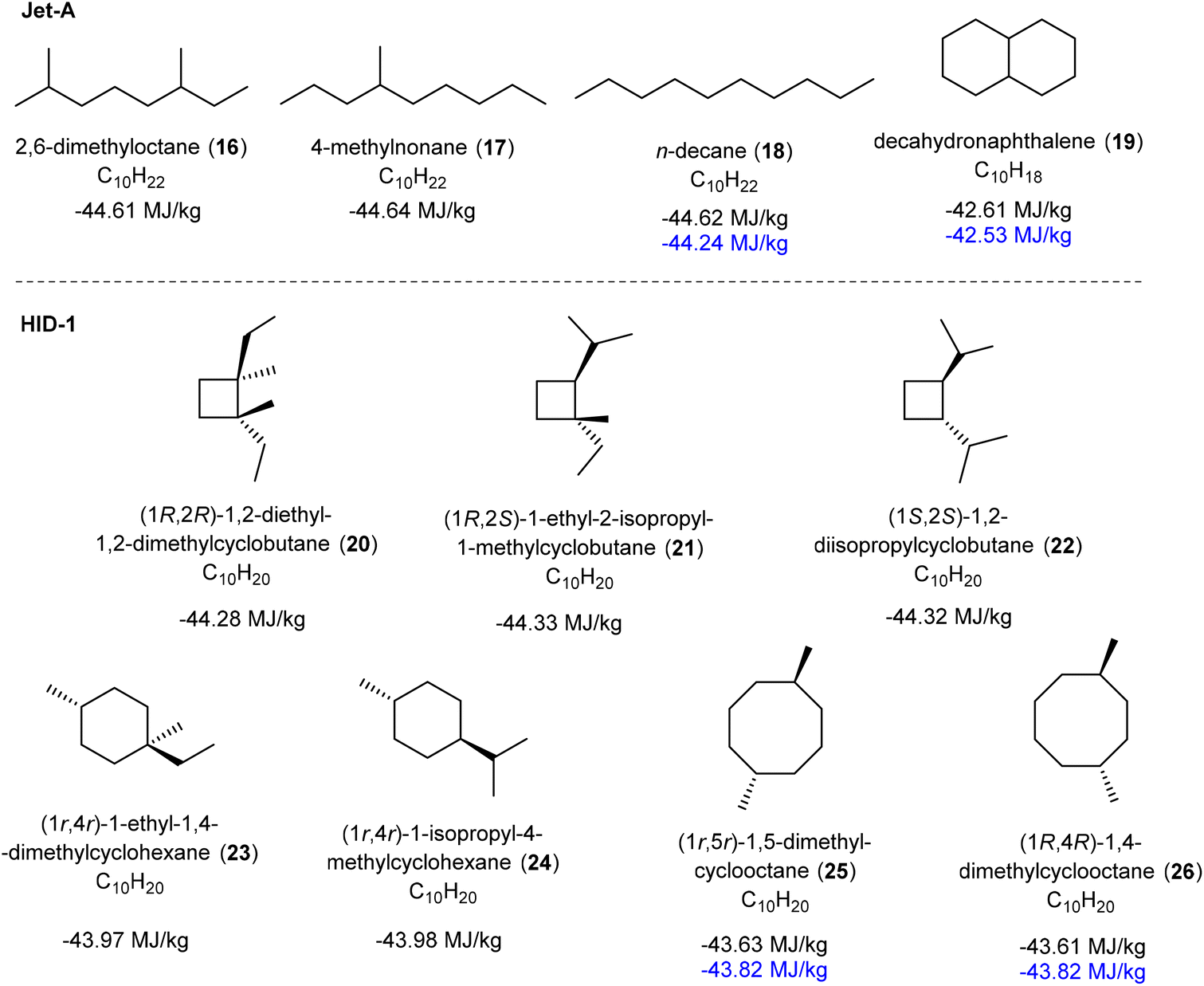 | ||
Fig. 8 Computed and experimentally determined net heat of combustion (NHOC) values for a few C10 hydrocarbons that exist in the Jet-A fuel and in our HID-1. Experimental values (in blue) from ref. 15 (compounds 18 and 19) and ref. 25 (compounds 25 and 26, determined as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 mixture). The NHOC values were computed following a DFT-based procedure by Major and co-workers developed for the M06-2X functional.62 These values contain two corrections which are needed to achieve accuracy; (i) a correction for the addition of the enthalpy of vaporization of terpenes and water, and (ii) a correction of the enthalpy of O2. The enthalpies of vaporizations were calculated using the SMD solvation model.83 :10 mixture). The NHOC values were computed following a DFT-based procedure by Major and co-workers developed for the M06-2X functional.62 These values contain two corrections which are needed to achieve accuracy; (i) a correction for the addition of the enthalpy of vaporization of terpenes and water, and (ii) a correction of the enthalpy of O2. The enthalpies of vaporizations were calculated using the SMD solvation model.83 | ||
| Fuel property | HID-1 | HID-2 | HID-3 | DMCO | Jet-A |
|---|---|---|---|---|---|
| a ASTM specification for Jet-A. Data taken from ref. 29. | |||||
| Gravimetric net heat of combustion (NHOC), MJ kg−1 | 44.23 | 43.57 | 43.59 | 43.82 | >42.8 |
| Density (15 °C), g mL−1 | 0.770 | 0.809 | 0.808 | 0.827 | >0.775 |
| Volumetric NHOC, MJ L−1 | 34.05 | 35.25 | 35.22 | 36.22 | >33.17 |
| Kinematic viscosity (−20 °C), mm2 s−1 | 1.71 | 3.16 | 2.92 | 4.17 | <8.00 |
| Freezing point, °C | <−78 | <−78 | <−78 | <−78 | <−40 |
| Flash point, °C | 33.5 | 38.5 | 38.5 | 50 | >38 |
| Hydrogen content, % mass | 14.37 | 14.37 | 14.37 | — | >13.4 |
Additionally, we have measured the kinematic viscosity of HID-1 from −40 °C to 20 °C as it is an important parameter in terms of safety and combustion of the fuel.84 A higher viscosity leads to a poorer atomization of the fuel which leads to incomplete combustion and formation of soot, eventually reducing fuel efficiency. To achieve proper atomization and combustion of a jet fuel it is strongly recommended to have a kinematic viscosity value below 12.00 mm2 s−1 at −40 °C. Rewardingly, the kinematic viscosity of HID-1 (1.71 mm2 s−1 at −20 °C) is more than 4.5 times lower than the recommended value for conventional fuel (8.00 mm2 s−1), and it is even 2.4 times lower than that recently reported for DMCO (4.17 mm2 s−1 at −20 °C) which is closely related to the structure of the molecule (C10). The kinematic viscosity at −40 °C is 2.60 mm2 s−1 (Table S5 and Fig. S39, ESI†), which is 4.6 and 3.1 times lower when compared to Jet-A and DMCO (7.95 mm2 s−1), respectively. The lower kinematic viscosity might result from the higher portion of alkylated cyclobutane isomers over cyclooctane isomers, and it will allow the drop-in to be blended with other conventional jet fuels at any ratio.
The freezing point of the jet fuel is also crucial for the safety and the flow of the fuel at high altitudes. We assessed the freezing properties of HID-1 by placing it in a dry ice/acetone bath (−78 °C) for 1.5 h and did not observe any cloudiness or crystallization, indicating that the freezing point of HID-1 is lower than −78 °C, i.e., it is much lower than the recommended value for conventional jet fuel (−40 °C). The low freezing point of HID-1 suggests that it is possible to use as a fuel in high altitude flight. Yet, a drawback of HID-1 is the flashpoint which was found to be 33.5 °C, lower than the specified value for conventional jet fuel (38 °C). The lower flash point may limit the use of HID-1 as jet fuel surrogate due to safety issues, although the commercially available Jet-B and TS-1 have much lower flash points (−18 and 28 °C, respectively) compared to the recommended value.85 Yet, these fuels have very low freezing points allowing them to be used in extremely cold environments. The low flash point of HID-1 can be attributed to the isomers with cyclobutane rings as these are more volatile.
Further modification of the C10 fuel
The fact that the flash point is slightly below the recommended value prompted us to consider modifications of the isoprene dimer mix ID-1 before the hydrogenation step. The boiling points of the various isomeric isoprene dimers (2–8, Fig. 1C) were earlier reported by Hammond, Turro and Liu and it was revealed that the [2 + 2] isomers have relatively lower boiling points than the others (Fig. S14, ESI†),33 with 2 having the lowest. This should also contribute to the low flash point of HID-1 as the flash point of a hydrocarbon correlates with its vapor pressure. A further modification of ID-1 could be performed through moderate heating which led to the conversion of cyclobutane-containing isomers to cyclooctadiene- and cyclohexene-containing ones through Cope and other thermal rearrangements.33 Here we probed two different temperatures, 135 and 160 °C, and subsequent hydrogenation gave the modified hydrogenated isoprene dimers HID-2 and HID-3 (see ESI† for detailed synthetic procedure, Fig. 9). The reaction mixtures were analyzed by 1H NMR and GCMS measurements (Fig. S30–S33, ESI†).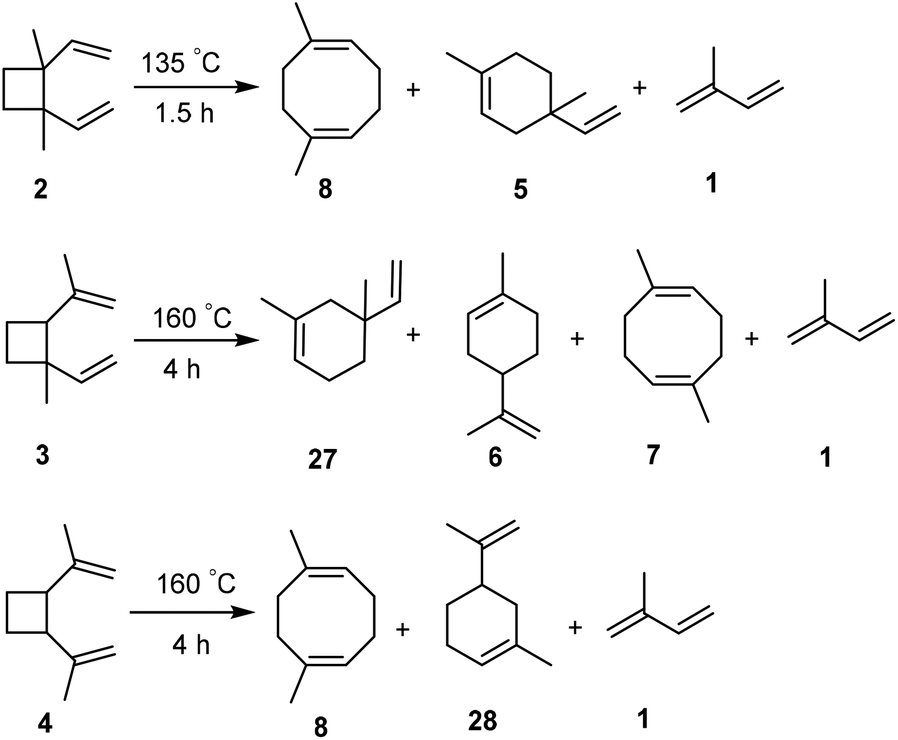 | ||
| Fig. 9 Isomerization of the cyclic [2 + 2] isoprene dimers to plausible cyclic [4 + 4] and [4 + 2] isomers through thermal Cope and other rearrangements. | ||
When ID-1 is heated at 135 °C for 1.5 h, leading to ID-2, isomer 2 rearranges to isomers 5 and 8, where isoprene is formed as a byproduct to 5% (Fig. S34, ESI†). In order to transform all [2 + 2] isoprene dimers into [4 + 2] and [4 + 4] isomers the temperature had to be elevated to 160 °C for 4 h, giving ID-3. Yet, in this case the amount of isoprene formed through a back-reaction increased to 11%, even though 3 and 4 after prolonged heating remained in the post-modified ID-3 in trace amounts of 1% and 2%, respectively (Fig. S35, ESI†). It is worth noting that the post-modification of ID-1 can be justified, as the isoprene formed as a byproduct can be recycled. After the removal of isoprene from ID-2 and ID-3, these dimer mixtures were hydrogenated using the conditions described above leading to quantitative formation of HID-2 and HID-3 (Fig. S36–S38, ESI†). Here it is noteworthy that the hydrogenation of isoprene dimers (ID-3) could be run at 1 atm H2 pressure to obtain HIDs (HID-3) in quantitative yield. However, the reaction requires longer time (48 h) to complete and 1% p-cymene is formed due to the aromatization of limonene (Fig. S69, ESI†).
After the heat treatments, the flash points of HID-2 and HID-3 increased to 38.5 °C (Table 2), i.e., above the recommended value. The identical flash point of HID-2 and HID-3 can be rationalized as they are mixtures of hydrogenated cycloalkanes with very similar boiling points. The gravimetric NHOC values of HID-2 and HID-3 decreased to 43.57 and 43.59 MJ kg−1, respectively, lower than that of HID-1 which is explained by the reduced amounts of cyclobutane isomers in the modified HID blends. Yet, the modified HID's have higher densities (both 0.809 g mL−1 at 15 °C) (Table 2 and Table S7, Fig. S40, ESI†) which leads to higher volumetric NHOC values (35.25 and 35.22 MJ L−1, respectively). The volumetric NHOC values for modified fuels are 6.3% greater compared to conventional Jet-A (>33.17 MJ L−1), which should be an added advantage. With regard to the kinematic viscosities (3.16 and 2.92 mm2 s−1 at −20 °C for HID-2 and HID-3, respectively) HID these are higher than that of HID-1 due to their lower contents of cyclobutanes (Table 2 and Table S5, Fig. S39, ESI†). Still, the values are more than 2.5 times lower than the largest recommended values, facilitating a good atomization of the HID's when used as fuels. Finally, both modified fuels have very low freezing points (<−78 °C), enabling high altitude flight (Table 2). The easy modulation of the ID-1 to ID-2 and ID-3 should be an advantage as they after hydrogenation should be ideal as drop-ins for conventional fuels for high-altitude jet engines.
There are also further favorable features of HID-1–HID-3. Conventional jet fuels contain mixtures of aromatic compounds which have added benefits as they swell the nitrile rubber elastomer valves which helps to protect the integrity of the jet engine. However, modern elastic materials do not require the aromatic content to swell the elastomers, and recent studies have shown that cycloalkane blends have similar properties as aromatics and are able to swell nitrile rubber elastomer valves.86,87 Additionally, the content of aromatic compounds in jet fuels leads to lower NHOC values as well as formation of carbon soot during the combustion which adversely affects the lifetime of the engine. Finally, aromatic compounds in jet-fuels are major health and environmental hazards. Thus, avoidance of such compounds is favorable for these reasons, and substantial interests have been focused towards development of bio-cycloalkane based fuels that mitigate the abovementioned problems.88 The very recent review by Muldoon and Harvey further highlights the potential of bio-cycloalkane based hybrid fuels for future use in military and civilian aviation fuel industries.88 In this context it can be noted that JP-10 (exo-tetrahydrodicyclopentadiene) is a synthetic C10 cycloalkane-based missile fuel.89,90 Taken together, our jet fuel mixtures (HID-1 to HID-3), which are C10 cycloalkanes, fulfil all requirements for future, less environmentally hazardous jet fuels, they are devoid of aromatic content and have high NHOC values.
System, efficiency and scale-up potentials
The emerging technology reported here is at a very early stage of development (approx. at technology readiness level 2 (TRL2)). To clarify where future research and development need to focus, we identify technological challenges by using information from recent analyses of approaches that resemble our combined photobiological–photochemical one. We also performed a life-cycle assessment (see below).In order to develop this platform into a commercial production system which is both energetically and economically sustainable, extensive improvements in performance are necessary on several levels. For the photosynthetic production of isoprene, the conversion from solar energy and CO2 to product needs to be more efficient. This will require further engineering of the host organisms, for improved photosynthetic efficiency and increased carbon fixation as well as for increased partitioning of carbon towards product formation. Furthermore, cultivation conditions need to be optimized for cell productivity. Cultivation and harvest systems also need to be further developed. While photobioreactors are commercially produced, albeit still mostly at smaller scale, efficient harvesting of a volatile product from the culture remains a challenge to solve.
A technoeconomic analysis of ethylene production by cyanobacteria has earlier been reported,57 and it was estimated that gasoline-equivalents, produced by oligomerization of the bioethylene, could be sold at a price of $28.66 per gallon in the near-term and at a price of $5.36 in the long-term. The largest cost that determined the gasoline price was the capital investment for the photobiology reactors, followed by the electricity cost for the power intense cryogenic distillation. Isoprene, contrary to ethylene, will not require cryogenic distillation as it condenses at much higher temperatures than ethylene (−104 °C vs. +34 °C). A further difference is the subsequent oligomerization which in case of ethylene uses a Ziegler catalyst, a mature technology utilized widely within the petrochemical industry. Our photochemical dimerization of isoprene is not an established technology and needs extensive process development, yet, if carried out with natural solar light it will be much less power demanding than the catalytic approach for ethylene oligomerization. The efficiency of the photochemical step is such that we can assume that all isoprene produced photobiologically within one day can be dimerized photochemically within the same amount of time. Thus, the main limiting factor for the photoproduction is the photobiological production step.
One drawback of our first strain of cyanobacteria used for isoprene generation was genetic instability of the plasmid-borne DNA construct. We successfully circumvented this by instead inserting the genes required for isoprene production into the genome of the host cyanobacterium (Fig. 3). This enables long-term stable production of isoprene, opening the possibility for continuous cultivation of the production strain for longer time periods. In a fully developed system at large scale, fed-batch or continuous cultivation combined with continuous product removal has the potential to increase productivity of the culture, while further strain engineering to enhance the productivity per cell will also be necessary.
As described above, the photochemical dimerization can be performed to very high yields (∼90%) in batch setup using thin FEP tubing, yet the yield decreases when the tube diameter increases. Process optimization in which various conditions are varied (flow rate, irradiation intensity, tubing width, laminar vs. tubular flow, and reactor design) is required. One may also search for photosensitizers with smaller S1 − T1 energy gaps than compound 12, yet still with E(T1) above that of isoprene. Such sensitizers could absorb within the visible (blue) wavelength region of the solar spectrum where the intensity is higher and still be able to transfer the triplet energy to isoprene and initiate the dimerization.
Life cycle assessment
To assess whether large scale production of photosynthetic jet fuel according to our system may become an environmentally sustainable process, we have performed a life cycle assessment (LCA) for the integrated photobiological–photochemical process, using one tonne of fuel as the functional unit (Fig. 10). For the cultivation and production of isoprene from cyanobacteria, we have used as a starting point a scenario described by Nilsson et al.,42 where the authors modeled cyanobacterial butanol production. In this system, cultivation would take place in an array of serially connected vertical flat panel photobioreactors with a total volume of 750 m3, covering 1 ha of land. We assume that the cyanobacterial cultivation would be performed in two phases. First, a pre-cultivation is performed in 10% of the whole bioreactor volume for five days to generate biomass, during which period product formation is inhibited. Second, the biomass is transferred to the whole reactor volume for a production phase of three weeks where production is induced, and 90% of fixed carbon is directed to isoprene production in the cells. Isoprene product is continuously removed and transferred to the downstream photochemical process. We make the following assumptions: (i) the carbon fixation rate is at 0.6 g L−1 day−1, based on a biomass formation rate of 1.2 g L−1 day−1 as shown by de Vree and co-workers in outdoor experiments during 77 days in the summer in the Netherlands at a latitude of approximately 52° North, and the estimate that half of the biomass is carbon;91,92 (ii) inorganic carbon is supplied from a waste resource such as biomass combustion, thus providing a carbon source at no environmental cost to our system; (iii) 80% of the water from the reactor plant is recycled after each production round; (iv) electricity needed is supplied in accordance with the Swedish energy mix based on 40% nuclear, 40% hydropower, 11% wind, 8% biofuels and waste, and 1% others (International Energy Agency and Swedish Energy Agency).93,94 In the scenario we modeled, nutrients other than CO2 are supplied based on the composition of BG11 growth medium.95 With these assumptions, the pre-cultivation phase results in a biomass concentration of 1.33 g L−1. In the production phase, with the mentioned carbon fixation rate and 90% of carbon allocated to isoprene formation, the same partitioning as modelled by Nilsson et al.,42 the time of operation of the 750 m3 reactor for generating one tonne of jet fuel at the end of the process is 2.4 days. The simultaneous biomass formation rate is 0.12 g L−1 d−1, resulting in the formation of 215 kg biomass per tonne of final fuel. This includes the effect of having day/night cycles, as the assumptions are based on outdoor experiments where no artificial light was used during the night.91 | ||
| Fig. 10 Flow chart for the envisioned process. The system boundaries for the performed LCA are marked with a red line. | ||
The bio-isoprene produced will subsequently be dimerized photochemically, and in our modeling, we utilised the input from the lab scale experiment and scaled up to produce 1 tonne of HID-2. Upon solar irradiation of isoprene (60 h) in presence of 12 as a photosensitizer to obtained isoprene dimers in 51% yield. The unreacted isoprene is distilled off to be used in the next cycle, while the isoprene dimers are separated by distillation under reduced pressure (∼70 °C, 10 mmHg pressure). The photosensitizer is easily recovered from the residue by washing with pentane and methanol (∼95% recovery, see above), and it was therefore excluded from the LCA since merely 0.1 mol% was used in the photoreaction. Further, the isoprene dimers produced will be treated thermally at 135 °C under an inert N2 atmosphere to produce ID-2 in 92% yield. The residual isoprene produced during reaction should be distilled off and used again in the photoreaction cycle. Finally, we assume that heat-treated isoprene dimers will be hydrogenated by using 10 wt% Pd/C (0.5 mol%) and H2 to obtain HID-2 in near quantitative yield, utilised as drop-in jet fuel and storable at the production site. The product could be separated by filtration of Pd/C to obtained jet fuel. The excess hydrogen used in this process would be recycled and used in the next hydrogenation cycle. The Pd/C (10 mol%) was not included in the LCA model due to low amount of loading (0.5 mol%) and reusability of the catalyst.
The process and system boundaries modeled in the LCA are shown in Fig. 10, and all inventory data summarized in Table S9, ESI.† Results from the LCA are presented in Table S10.† The climate impact was approximately 0.7 tonne CO2 eq. per tonne biofuel (Table S10, ESI†), mainly attributed to emissions from the production of sodium nitrate used in the photobiological processes (Table S11†). For further discussion on contributions to climate impact of different process parameters, see ESI 7.1 and Table S11.† The climate impact is about 20% of that of fossil jet fuel (approx. 3.8 CO2 eq. per tonne for conventional Jet-A),96 and is at the lower end of the range from 0.6–2.7 tonne CO2 eq. per tonne biofuel found in the study by Nilsson et al.,42 which investigated the environmental impacts of cyanobacteria-produced n-butanol using three different reactors. In other studies, some investigated bio-jet fuels had the best result at 0.8 tonne CO2 eq. per tonne.31
From the assessment of the overall environmental impact we see that under the assumptions made, the production of sodium nitrate completely dominates the impacts in all environmental categories (Fig. 11 and Table S11, ESI†). This nitrate is used as a nutrient for cultivation of the cyanobacteria, and amounts to 155 kg consumed per tonne of jet fuel produced. The source of sodium nitrate in our model is the global market, and it is produced using fossil fuels. Use of alternative raw materials from waste streams as a source for nitrogen instead of sodium nitrate can be a potential solution for reducing the environmental impacts. It has been shown that cyanobacteria can grow on several such waste sources of nutrients, e.g. municipal waste water and effluents of various industries.97–100 Increasing the photosynthetic efficiency of the cyanobacteria would also reduce the overall environmental impact.
 | ||
| Fig. 11 Contribution analysis showing percent of impacts originating from the different processes in the different environmental impact categories (cut-off 0.1%). The following aspects were considered: climate change (CC), human toxicity non-cancer effects (HTX), human toxicity cancer effects (CE), particulate matter (PM), photochemical ozone formation (POF), acidification potential (AP), terrestrial eutrophication (TE), freshwater eutrophication (FE), marine eutrophication (ME) and freshwater ecotoxicity (FEC). | ||
Conclusions and outlook
In this study, we demonstrated that it is possible to generate photosynthetically derived isoprene from CO2 using engineered cyanobacteria, capture the isoprene, and use it for subsequent biofuel generation via a photochemical process driven by sunlight. While further optimization of the engineered microorganisms is required for industrial applications, we were able to trap isoprene with high efficiencies relying on a simple, scalable capturing method. We could also show that repeatedly removing the product enhanced productivity from isoprene producing cyanobacterial cultures.In a subsequent photochemical step, the isoprene was dimerized into cyclic C10H20 isomers in nearly quantitative yields by usage of dinapthylmethanones as photosensitizers. The photoreaction could be run under ambient conditions, facilitating a fully renewable fuel production. Our current studies reveal that rather simple modifications of the reaction setup can greatly improve the yield of the photoreaction. Combined with a careful choice of photosensitizer this enables photodimerization of isoprene by use of solar light. The isoprene dimer mixture can be further modified by heating at moderately elevated temperatures (130–160 °C), resulting in C10 hydrocarbon mixtures which after hydrogenation fulfil all criteria to function as drop-ins for conventional jet fuels. Indeed, the modified and hydrogenated isoprene dimers have better fuel properties than the commercially available Jet-A. The very low freezing points and low viscosity should make these fuels ideal for high-altitude flights.
It is usually a challenge to compare the results and environmental impacts of an emerging technology with a mature technology due to several uncertainties such as missing data, upscaling assumptions and modeling issues.101 In case of production of photosynthetic biofuels using microalgae and cyanobacteria, the process is still in its early stages and significant productivity improvements can be expected. The results of the current LCA study will assist in further improving our novel two-step technology for bio-jet fuel production from cyanobacteria. Our LCA showed an overall positive result on the environmental sustainability of our system. It was noted that the production of nutrients, in particular nitrate, dominates the environmental impact categories. Cyanobacteria can also conceivably grow well on municipal or other wastewaters as a source of nutrients, including nitrogen,97–100 something we have not included in the above model and which would likely increase sustainability.
Hence, our results described are the very first steps toward a completely renewable jet fuel generated from CO2, water and solar light, provided that cultivation is carried out outdoors and that the hydrogenation and thermal rearrangement steps also utilize renewable energy. We report on the first proof-of-principle study of a combined photobiological–photochemical approach for jet fuel production. Extensive future research and development along various lines are needed, and several different short alkenes and dienes could be useful for similar processes. At this point it is noteworthy that emerging photobiological as well as photochemical outdoor plants utilizing solar light for direct fuel production (hydrogen) exist, although there are many challenges that remain to be addressed and solved.102,103
Author contributions
The photobiological production of isoprene has been carried out by JR, the trapping by JR and JS, the photochemical dimerization, optimization of reaction conditions, spectroscopic and chemical analysis, and hydrogenation by AR, LCG and HAV, quantum chemical calculations by NPV, OEB and AR, and determination of fuel properties by LA and MB. Life cycle assessment was conducted by DY and CS. The two-step combined photobiological–photochemical approach was conceived by HO, KS, PiL and PeL, and the application to isoprene dimerization by PiL and HO. All authors have contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts of interests.Acknowledgements
First, we are grateful to Dr Per Wiklund for helping out in establishing the contact between HO and MB, which enabled us to determine the fuel characteristics, Dr Wangchuk Rabten for design and construction of the flat tubing reactor, Dr Luke Odell for proving access to GC-MS, Prof. Johannes Messinger for access to the solar simulator, and Prof. Máté Erdélyi and Dr Ruisheng Xiong for access to their Parr hydrogenation apparatus. Financial support from the Swedish Energy Agency (grants 44728-1 and 52576-1 (HO) and 38334-3 (PiL)), Formas (grants 2017-00862 (HO) and 2020-00879 (DY)) and the NordForsk NCoE Program “NordAqua” (project number 82845) are greatly acknowledged. The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Center (NSC), Linköping, partially funded by the Swedish Research Council through grant agreement number 2018-05973.References
- International Air Transport Association (IATA), Aircraft Technology Roadmap to 2050, 2019.
- W. Walsh, International Air Transport Association Annual Review, n.d., 2021, 2021 Search PubMed.
- S. Nižetić, Int. J. Energy Res., 2020, 44, 10953–10961 CrossRef.
- A. O'Connell, M. Kousoulidou, L. Lonza and W. Weindorf, Renewable Sustainable Energy Rev., 2019, 101, 504–515 CrossRef.
- N. S. Mat Aron, K. S. Khoo, K. W. Chew, P. L. Show, W. H. Chen and T. H. P. Nguyen, Int. J. Energy Res., 2020, 44, 9266 CrossRef CAS.
- M. Wang, R. Dewil, K. Maniatis, J. Wheeldon, T. Tan, J. Baeyens and Y. Fang, Prog. Energy Combust. Sci., 2019, 74, 31–49 CrossRef.
- N. E. Nozzi, J. W. K. Oliver and S. Atsumi, Front. Bioeng. Biotechnol., 2013, 1, 7 Search PubMed.
- I. M. P. Machado and S. Atsumi, J. Biotechnol., 2012, 162, 50–56 CrossRef CAS.
- J. C. Liao, L. Mi, S. Pontrelli and S. Luo, Nat. Rev. Microbiol., 2016, 14, 288–304 CrossRef.
- C. J. Knoot, J. Ungerer, P. P. Wangikar and H. B. Pakrasi, J. Biol. Chem., 2018, 293, 5044–5052 CrossRef PubMed.
- X. Liu, H. Xie, S. Roussou and P. Lindblad, Curr. Opin. Biotechnol, 2022, 73, 143–150 CrossRef.
- J. Wichmann, K. J. Lauersen, N. Biondi, M. Christensen, T. Guerra, K. Hellgardt, S. Kühner, M. Kuronen, P. Lindberg, C. Rösch, I. S. Yunus, P. Jones, P. Lindblad and O. Kruse, Trends Biotechnol., 2021, 39, 323–327 CrossRef.
- N. Pattharaprachayakul, J. i. Choi, A. Incharoensakdi and H. M. Woo, Biotechnol. Bioprocess Eng., 2020, 25, 829–847 CrossRef CAS.
- Air BP, Handbook of Products, 2011.
- Coordinating Research Council (CRC), Handbook of Aviation Fuel Properties CRC Report No. 635, Alpharetta, 2004 Search PubMed.
- B. L. Smith and T. J. Bruno, Energy Fuels, 2007, 21, 2853–2862 CrossRef CAS.
- N. R. Baral, O. Kavvada, D. Mendez-Perez, A. Mukhopadhyay, T. S. Lee, B. A. Simmons and C. D. Scown, Energy Environ. Sci., 2019, 12, 807–824 RSC.
- P. P. Peralta-Yahya, M. Ouellet, R. Chan, A. Mukhopadhyay, J. D. Keasling and T. S. Lee, Nat. Commun., 2011, 2, 483 CrossRef.
- C. L. Liu, T. Tian, J. Alonso-Gutierrez, B. Garabedian, S. Wang, E. E. K. Baidoo, V. Benites, Y. Chen, C. J. Petzold, P. D. Adams, J. D. Keasling, T. Tan and T. S. Lee, Biotechnol. Biofuels, 2018, 11, 285 CrossRef CAS.
- J. Keasling, H. Garcia Martin, T. S. Lee, A. Mukhopadhyay, S. W. Singer and E. Sundstrom, Nat. Rev. Microbiol., 2021, 19, 701–715 CrossRef CAS.
- E. Jongedijk, K. Cankar, M. Buchhaupt, J. Schrader, H. Bouwmeester and J. Beekwilder, Appl. Microbiol. Biotechnol., 2016, 100, 2927–2938 CrossRef CAS PubMed.
- P. Lindberg, S. Park and A. Melis, Metab. Eng., 2010, 12, 70–79 CrossRef CAS PubMed.
- H. Mustila, A. Kugler and K. Stensjö, Metab. Eng. Commun., 2021, 12, e00163 CrossRef.
- X. Gao, F. Gao, D. Liu, H. Zhang, X. Nie and C. Yang, Energy Environ. Sci., 2016, 9, 1400–1411 RSC.
- E. Englund, K. Shabestary, E. P. Hudson and P. Lindberg, Metab. Eng., 2018, 49, 164–177 CrossRef PubMed.
- J. E. Chaves, P. Rueda-Romero, H. Kirst and A. Melis, ACS Synth. Biol., 2017, 6, 2281–2292 CrossRef PubMed.
- J. E. Chaves and A. Melis, Appl. Microbiol. Biotechnol., 2018, 102, 6451–6458 CrossRef CAS PubMed.
- C. P. Nicholas, Appl. Catal., A, 2017, 543, 82–97 CrossRef CAS.
- K. E. Rosenkoetter, C. R. Kennedy, P. J. Chirik and B. G. Harvey, Green Chem., 2019, 21, 5616–5623 RSC.
- D. M. Morris, R. L. Quintana and B. G. Harvey, ChemSusChem, 2019, 12, 1646–1652 CrossRef CAS PubMed.
- N. R. Baral, M. Yang, B. G. Harvey, B. A. Simmons, A. Mukhopadhyay, T. S. Lee and C. D. Scown, ACS Sustainable Chem. Eng., 2021, 9, 11872–11882 CrossRef CAS.
- J.-D. Woodroffe and B. G. Harvey, Energy Adv., 2022, 1, 338–343 RSC.
- G. S. Hammond, N. J. Turro and R. S. H. Liu, J. Org. Chem., 1963, 28, 3297–3303 CrossRef CAS.
- R. S. H. Liu, N. J. Turro and G. S. Hammond, J. Am. Chem. Soc., 1965, 87, 3406–3412 CrossRef CAS.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, J. CO2 Util., 2013, 3–4, 102–106 CrossRef CAS.
- C. Vogt, M. Monai, G. J. Kramer and B. M. Weckhuysen, Nat. Catal., 2019, 2, 188–197 CrossRef.
- B. Yao, T. Xiao, O. A. Makgae, X. Jie, S. Gonzalez-Cortes, S. Guan, A. I. Kirkland, J. R. Dilworth, H. A. Al-Megren, S. M. Alshihri, P. Dobson, G. P. Owen, J. M. Thomas and P. P. Edwards, Nat. Commun., 2020, 11, 6395 CrossRef PubMed.
- R. Schäppi, D. Rutz, F. Dähler, A. Muroyama, P. Haueter, J. Lilliestam, A. Patt, P. Furler and A. Steinfeld, Nature, 2022, 601, 63–68 CrossRef.
- International Organization for Standardization, ISO 14044 Environmental management: life cycle assessment; requirements and guidelines, ISO Geneva, 2006.
- H. Chum, A. Faaij, J. Moreira, G. Berndes, P. Dhamija, H. Dong, B. Gabrielle, A. G. Eng, W. Lucht, M. Mapako, O. M. Cerutti, T. McIntyre, T. Minowa, K. Pingoud, R. Bain, R. Chiang, D. Dawe, G. Heath, M. Junginger, M. Patel, J. Yang, E. Warner, D. Paré and S. K. Ribeiro, in Renewable Energy Sources and Climate Change Mitigation, ed. O. Edenhofer, R. Pichs-Madruga, Y. Sokona, K. Seyboth, P. Matschoss, S. Kadner, T. Zwickel, P. Eickemeier, G. Hansen, S. Schlomer and C. von Stechow, Cambridge University Press, Cambridge, 2011, pp. 209–332 Search PubMed.
- M. A. Curran, Life cycle assessment student handbook, John Wiley & Sons, 2015 Search PubMed.
- A. Nilsson, K. Shabestary, M. Brandão and E. P. Hudson, J. Ind. Ecol., 2020, 24, 205–216 CrossRef.
- L. Batan, J. Quinn, B. Willson and T. Bradley, Environ. Sci. Technol., 2010, 44, 7975–7980 CrossRef CAS.
- D. Luo, Z. Hu, D. G. Choi, V. M. Thomas, M. J. Realff and R. R. Chance, Environ. Sci. Technol., 2010, 44, 8670–8677 CrossRef CAS.
- P. K. Campbell, T. Beer and D. Batten, Bioresour. Technol., 2011, 102, 50–56 CrossRef CAS.
- P. Collet, L. Lardon, A. Hélias, S. Bricout, I. Lombaert-Valot, B. Perrier, O. Lépine, J.-P. Steyer and O. Bernard, Renewable Energy, 2014, 71, 525–533 CrossRef CAS.
- E. D. Frank, J. Han, I. Palou-Rivera, A. Elgowainy and M. Q. Wang, Center for Transportation Research, Energy Systems Division, Argonne National Laboratory, Oak Ridge, 2011, pp. 11–15 Search PubMed.
- R. M. Handler, D. R. Shonnard, T. N. Kalnes and F. S. Lupton, Algal Res., 2014, 4, 105–115 CrossRef.
- S. Ponnusamy, H. K. Reddy, T. Muppaneni, C. M. Downes and S. Deng, Bioresour. Technol., 2014, 170, 454–461 CrossRef CAS PubMed.
- J. C. Quinn, T. G. Smith, C. M. Downes and C. Quinn, Algal Res., 2014, 4, 116–122 CrossRef.
- S. C. Togarcheti, M. k. Mediboyina, V. S. Chauhan, S. Mukherji, S. Ravi and S. N. Mudliar, Resour., Conserv. Recycl., 2017, 122, 286–294 CrossRef.
- A. F. Clarens, E. P. Resurreccion, M. A. White and L. M. Colosi, Environ. Sci. Technol., 2010, 44, 1813–1819 CrossRef PubMed.
- M. L. N. M. Carneiro, F. Pradelle, S. L. Braga, M. S. P. Gomes, A. R. F. A. Martins, F. Turkovics and R. N. C. Pradelle, Renewable Sustainable Energy Rev., 2017, 73, 632–653 CrossRef.
- C. Quiroz-Arita, J. J. Sheehan and T. H. Bradley, Algal Res., 2017, 26, 445–452 CrossRef.
- J. H. K. Lim, Y. Y. Gan, H. C. Ong, B. F. Lau, W. H. Chen, C. T. Chong, T. C. Ling and J. J. Klemeš, Renewable Sustainable Energy Rev., 2021, 149, 111396 CrossRef CAS.
- A. G. Romero-Izquierdo, C. Gutiérrez-Antonio, F. I. Gómez-Castro and S. Hernández, Sustainable Alternatives for Aviation Fuels, Elsevier, 2022, pp. 103–124 Search PubMed.
- J. N. Markham, L. Tao, R. Davis, N. Voulis, L. T. Angenent, J. Ungerer and J. Yu, Green Chem., 2016, 18, 6266–6281 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian Inc., Wallingford CT Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- E. Pahima, S. Hoz, M. Ben-Tzion and D. T. Major, Sustainable Energy Fuels, 2019, 3, 457–466 RSC.
- J. S. Rodrigues and P. Lindberg, in Cyanobacteria Biotechnology, ed. P. Hudson, Wiley, 1st edn, 2021 Search PubMed.
- A. Kunert, M. Hagemann and N. Erdmann, J. Microbiol. Methods, 2000, 41, 185–194 CrossRef CAS.
- E. Englund, J. Andersen-Ranberg, R. Miao, B. Hamberger and P. Lindberg, ACS Synth. Biol., 2015, 4, 1270–1278 CrossRef PubMed.
- H.-H. Huang, D. Camsund, P. Lindblad and T. Heidorn, Nucleic Acids Res., 2010, 38, 2577–2593 CrossRef PubMed.
- X. Liu, R. Miao, P. Lindberg and P. Lindblad, Energy Environ. Sci., 2019, 12, 2765–2777 RSC.
- R. Miao, H. Xie and P. Lindblad, Biotechnol. Biofuels, 2018, 11, 267 CrossRef.
- S. K. Rajagopal, K. Nagaraj, S. Deb, V. Bhat, D. Sasikumar, E. Sebastian and M. Hariharan, Phys. Chem. Chem. Phys., 2018, 20, 19120–19128 RSC.
- A. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- S. V. Jovanovic, D. G. Morris, C. N. Pliva and J. C. Scaiano, J. Photochem. Photobiol., A, 1997, 107, 153–158 CrossRef CAS.
- S. L. Murov, PhD thesis, Univ. Chicago, Chicago, IL, 1967 Search PubMed.
- J. P. Fouassier, D. J. Lougnot, F. Wieder and J. Faure, J. Photochem., 1977, 7, 17–28 CrossRef.
- K. Meier and H. Zweifel, J. Photochem., 1986, 35, 353–366 CrossRef.
- G. S. Hammond and R. S. H. Liu, J. Am. Chem. Soc., 1963, 85, 477–478 CrossRef.
- L. D. Elliott, S. Kayal, M. W. George and K. Booker-Milburn, J. Am. Chem. Soc., 2020, 142, 14947–14956 CrossRef PubMed.
- R. A. Caldwell and M. Singh, J. Am. Chem. Soc., 1982, 104, 12152–12172 Search PubMed.
- N. C. Baird, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef.
- K. Jorner, B. O. Jahn, P. Bultinck and H. Ottosson, Chem. Sci., 2018, 9, 3165–3176 RSC.
- V. Vijay, M. Madhu, R. Ramakrishnan, A. Benny and M. Hariharan, Chem. Commun., 2020, 56, 225–228 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef PubMed.
- G. R. Wilson, T. Edwards, E. Corporan and R. L. Freerks, Energy Fuels, 2013, 27, 962–966 CrossRef.
- D. J. L. Prak, G. R. Simms, M. Hamilton and J. S. Cowart, Fuel, 2021, 286, 119389 CrossRef.
- J. L. Graham, T. F. Rahmes, M. C. Kay, J. P. Belières, J. D. Kinder, S. A. Millett, J. Ray, W. L. Vannice and J. A. Trela, Impact of alternative jet fuel and fuel blends on non-metallic materials used in commercial aircraft fuel systems. Federal Aviation Administration Report DOT, FAA/AEE/2014-10, 2014.
- Y. Liu and C. W. Wilson, Adv. Mech. Eng., 2012, 4, 127430 CrossRef.
- J. A. Muldoon and B. G. Harvey, ChemSusChem, 2020, 13, 5777–5807 CrossRef.
- T. Bruno, M. Huber, A. Laesecke, E. Lemmon and R. Perkins, Adv. Sci. Technol., 2006, 45, 1–67 Search PubMed.
- H. S. Chung, G. S. H. Chen, R. A. Kremer, J. R. Boulton and G. W. Burdette, Energy Fuels, 1999, 13, 641–649 CrossRef.
- J. H. de Vree, R. Bosma, M. Janssen, M. J. Barbosa and R. H. Wijffels, Biotechnol. Biofuels, 2015, 8, 1–12 CrossRef.
- A. A. Shastri and J. A. Morgan, Biotechnol. Prog., 2005, 21, 1617–1626 CrossRef PubMed.
- International Energy Agency (IEA), Energy Policies of IEA Countries: Sweden 2019 Review, 2019.
- Swedish Energy Agency, Energy in Sweden 2022 – An overview, 2022.
- R. Y. Stanier, R. Kunisawa, M. Mandel and G. Cohen-Bazire, Bacteriol. Rev., 1971, 35, 171–205 CrossRef.
- J. I. Hileman, D. S. Ortiz, J. T. Bartis, H. M. Wong, P. E. Donohoo, M. A. Weiss and I. A. Waitz, Near-term feasibility of alternative jet fuels, Rand Corporation, 2009 Search PubMed.
- A. R. Hughes, A. Sulesky, B. Andersson and G. Peers, Algal Res., 2018, 32, 30–37 CrossRef.
- S. Canizales, M. Sliwszcinka, A. Russo, S. Bentvelzen, H. Temmink, A. M. Verschoor, R. H. Wijffels and M. Janssen, J. Appl. Phycol., 2021, 33, 3565–3577 CrossRef.
- T. Beyl, T. M. Louw and R. W. M. Pott, Microorganisms, 2019, 7, 428 CrossRef.
- K. P. Papadopoulos, C. N. Economou, A. G. Tekerlekopoulou and D. v. Vayenas, J. Environ. Manage., 2020, 265, 110543 CrossRef.
- C. Seidel, Int. J. Life Cycle Assess., 2016, 21, 337–348 CrossRef.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef.
- Y. Chen, Energy Environ. Sci., 2022, 15, 2843–2857 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03272d |
| This journal is © The Royal Society of Chemistry 2022 |