
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From haemoglobin to single-site hydrogenation catalyst†
Alain Y.
Li
a,
Angus
Pedersen
 ab,
Jingyu
Feng
a,
Hui
Luo
a,
Jesús
Barrio
ab,
Julien
Roman
a,
King Kuok (Mimi)
Hii
c and
Maria-Magdalena
Titirici
*ad
ab,
Jingyu
Feng
a,
Hui
Luo
a,
Jesús
Barrio
ab,
Julien
Roman
a,
King Kuok (Mimi)
Hii
c and
Maria-Magdalena
Titirici
*ad
aDepartment of Chemical Engineering, Imperial College London, Exhibition Road, London SW7 2AZ, UK. E-mail: m.titirici@imperial.ac.uk
bDepartment of Materials, Royal School of Mines, Imperial College London, Exhibition Road, London SW7 2AZ, England, UK
cDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, 80, Wood Lane, London W12 0BZ, UK
dAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, 2-1-1 Katahira, Aobaku, Sendai, Miyagi 980-8577, Japan
First published on 6th September 2022
Abstract
Iron-based single-site catalysts hold immense potential for achieving highly selective chemical processes, with the added advantage of iron being an earth-abundant metal. They are widely explored in electrocatalysis for oxygen reduction and display promising catalytic activity for organic transformations. In particular, FeNx@C catalysts are active for the reduction of nitroarene into aromatic amines. Yet, they are difficult to mass-produce, and most preparation methods fail to avoid single site aggregation. Here we prepared FeNx@C catalysts from bio-derived compounds, xylose and haemoglobin, in a simple two-step process. Since haemoglobin naturally contains FeNx single-sites, we successfully repurposed them into hydrogenation catalytic centers and avoided their aggregation during the preparation of the material. Their single-site nature was demonstrated by aberration-corrected transmission electron microscopy and X-ray absorption techniques. They were shown to be active for transfer hydrogenation of nitroarenes into anilines, with excellent substrate selectivity and recyclability, as demonstrated by the preserved yield across seven catalytic cycles. We also showed that FeNx@C could be used to prepare 2-phenylbenzimidazole through a reduction/condensation tandem. Our work shows for the first time the viability of biomass precursors to prepare Fe single-site hydrogenation catalysts.
Introduction
Nitroarene reduction into aniline derivatives is the main method to obtain aromatic amines in the chemical industry.1 Most processes rely on stoichiometric reduction using Fe0/HCl (Béchamp process), or more commonly heterogeneous metallic hydrogenation catalysts (Pd, Ni Cu, Mn, or Fe).2 Since Corma and Serna's breakthrough using Au/TiO2,3 a wide variety of nanocatalysts have been reported to date.4 Additionally, in order to circumvent the use of flammable H2 gas, alternative hydrogen donors such as iPrOH, HCO2H, or silanes have been investigated. Indeed, many metals, ranging from precious metals (Au, Pt, Pd) to more abundant metals (Fe, Ni, Co) have been reported to be catalytically active in nitroarene transfer hydrogenation reactions.5 Among these, Fe, while standing as the second most abundant transition metal in the earth crust, has emerged as a highly efficient catalyst.6–10 Since the pioneering work of Beller and co-workers in using Fe-phenanthroline pyrolysed over carbon for nitroarene hydrogenation,11–14 many nitrogen/carbon-supported Fe catalysts have since emerged (Fig. 1A).14–18 In most reports, Fe was found as an ill-defined mixture of nanoparticulate species (metals, oxides, carbides and nitrides), without formal evidence of single sites. This mixture could lead to potential issues such as catalytic selectivity and metal leaching. | ||
| Fig. 1 Fe-Based catalysts for nitroarene reduction using (A) FexOyNz nanoclusters (B) using single-site FeNx@C, and (C) our system using single-site FeNx@C prepared from haemoglobin and using iPrOH/KCO2H reductants. | ||
Single-site catalysts, with their well-defined atomic sites, can allow maximum metal usage while achieving high activity/selectivity.19–22 There has been few examples of single-site FeNx@C-catalysed hydrogenation: Cheong et al. reported a SBA-15-templated material, using Fe(NO3)3 and glucosamine as precursors, whereas Yun and Lu et al. opted for a MOF-encapsulated Fe precursor (Fig. 1B).23–25 While all three catalysts were reported to be highly active and selective for nitroarene transfer hydrogenation reactions, the processes deployed highly carcinogenic hydrazine as a hydrogen source. In this work, we report a single-site FeNx@C catalyst for highly-selective reduction of nitroarenes to anilines, using isopropanol as a hydrogen source.
In this work, the single-site catalyst FeNx@C was prepared using haemoglobin as a bio-derived N/C precursor (Fig. 1C). As a waste product from the meat industry, haemoglobin contains a low Fe content (0.35 wt%) of FeN4 heme units, which we repurposed into catalytic sites. It has been explored previously to prepare fuel cell cathode catalysts, by direct pyrolysis26–31 or templated by MgCl2·6H2O.32,33 To the best of our knowledge, it has not been applied as a heterogeneous catalyst material. The present work will report a two-step preparation of a FeNx@C catalyst, using only biomass starting materials derived from haemoglobin and xylose as a carbon source. The latter was selected due to its capacity as a C5 sugar to form polymerisable furans, and its availability from non-edible lignocellulosic biomass.34
Results and discussion
The catalyst was prepared in a two-step fashion (Scheme 1). Firstly, an aqueous solution of haemoglobin and xylose was subjected to hydrothermal carbonisation treatment (HTC) using kayexalate (poly(sodium 4-styrenesulfonate)) as a structure-directing agent.35 HTC is a biomass carbonisation method, which entails the heating of aqueous solutions containing typically sugar-like molecules at 180–250 °C in a sealed autoclave.36 The combination of heat and the autogenous pressure triggers sugar dehydration into furanic intermediates, followed by their polymerisation into carbon microspheres (2–5 μm).37 This approach is based on a procedure developed previously within our research group, for the preparation of nitrogen-doped carbons from glucose and albumin protein.38,39 | ||
| Scheme 1 Preparation of FeNx@C. | ||
In a second step, the material obtained from HTC (named FeHTC) was thermally treated in a furnace under different atmospheres of varying oxygen/nitrogen ratios (0–10% O2 in N2). We measured the Fe concentation by ICP-MS through the catalyst preparation, showing that it increased from 580 ppm in the initial mixture to 1600 ppm after the HTC step, and to 2300–4000 ppm after pyrolysis depending on the oxygen content. The highest value (4000 ppm) was found at 6% oxygen. Throughout the HTC/pyrolysis sequence, we were able to increase the iron concentration relative to the other elements (C,N,O) from waste protein and biomass – while obtaining a nanoporous material.40
The preparation also resulted in an increase in the specific surface area of all the materials (from 40 m2 g−1 for FeHTC to 70–740 m2 g−1, Fig. 2A and Table S1†), with pores <5 nm in diameter (Fig. 2B). Conducting the synthesis of the catalyst without the template increased the specific area to 610 m2 g−1 after pyrolysis.
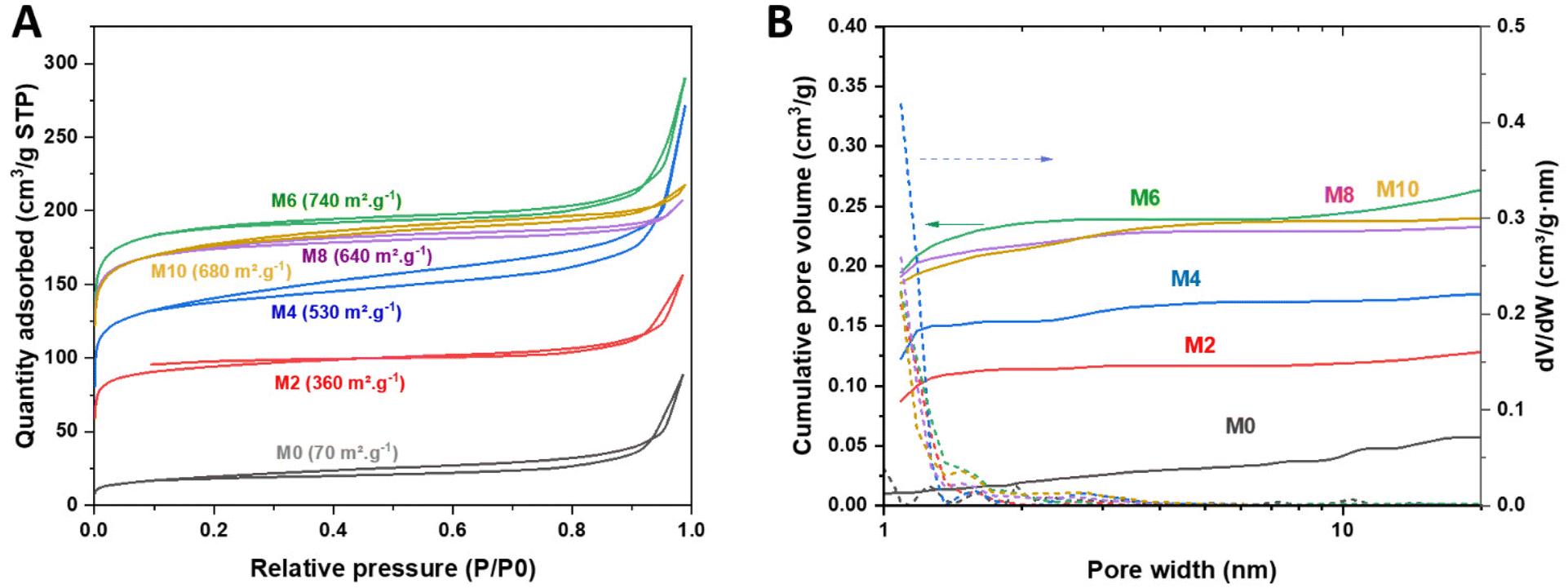 | ||
| Fig. 2 (A) N2 adsorption–desorption isotherm and (B) cumulative pore volume distributions for samples with different oxygen percentages during pyrolysis (the number after M indicates the oxygen percentage during pyrolysis). | ||
Next, the catalytic activity of all of the prepared FeNx@C samples was tested in the reduction of nitroarenes (Table 1). 4-Nitrotoluene was selected as a model substrate, using iPrOH both as a solvent and a reductant, in the presence of K2CO3 and KCO2H as additives at 150 °C. While temperature conditions are considerably higher than for other single-site iron-based systems (25–60 °C), we strived to use safer reagents and solvents compared to the previous systems (comprising hydrazine, THF, NEt3). FeHTC, and both the material treated with no template or no oxygen (Table 1, entries 1–3) were catalytically inactive (0% yield). With 2% oxygen thermal treatment, the yield increased to 7% (Table 1, entry 4), and increased to 56 and 75% yield at 4% and 6% oxygen treatment, respectively (Table 1, entries 5 and 6). Upon further increasing the oxygen content to 8% and 10%, the yield decreased to 20 and 1% respectively (Table 1, entries 7 and 8). Thus, 6% oxygen treatment proved to be the most active catalyst, with a turnover frequency of 770 × 10−3 h−1 (assuming a full utilisation of the Fe atoms as single sites), in line with its higher BET specific surface area and highest Fe loading.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | TOF (10−3 h−1) | Yield (%) |
| Reaction conditions: 4-Nitrotoluene (12.4, 88 μmol), K2CO3 (6.1 mg, 0.5 equiv.), KCO2H (3.7 mg, 0.5 equiv.), catalyst (50 mg), isopropyl alcohol (2.0 mL), Ar, 150 °C, 24 h. | |||
| 1 | FeHTC | 0 | 0 |
| 2 | Same than FeNx@C, without template | 0 | 0 |
| Oxygen % during pyrolysis | TOF (10 −3 h −1 ) | Yield (%) | |
| 3 | 0 | 0 | 0 |
| 4 | 2 | 82 | 7 |
| 5 | 4 | 719 | 56 |
| 6 | 6 | 770 | 75 |
| 7 | 8 | 249 | 20 |
| 8 | 10 | 111 | 1 |

In Fig. 3 we have summarised the various parameters explored so far. We will thus keep the material at 6% oxygen throughout the rest of the manuscript, and refer to it as FeNx@C.
 | ||
| Fig. 3 Fe ICP-MS concentration, BET specific surface area and turnover frequency for the various materials prepared. | ||
X-ray Photoelectron Spectroscopy (XPS) showed a significant amount of surface oxygen (7.57 at%), along with 2.54 at% nitrogen and 0.05 at% Fe, the rest being carbon (analysis of signals deconvolution is given Table S2 and Fig. S9 in ESI†). Raman and X-ray diffraction respectively showed that the carbon is amorphous (with an IG/ID ratio of 0.94), with no trace of crystalline iron oxide domain (Fig. S10 and S11†).
The single-site nature of FeNx@C was then confirmed by means of electron microscopy (Fig. 4). SEM showed the spherical macrostructure of the carbon (2–10 μm, Fig. 4A and S8†). With TEM, no Fe particles were detected within the amorphous carbon matrix even at high magnification, excluding the presence of Fe species aggregates (Fig. 4B). Finally, aberration-corrected high resolution HAADF-STEM, showed sparse bright spots within the material (highlighted with red arrows), indicative of Fe single sites (Fig. 4C and D, Fig. S2–S4 in ESI†). More Fe single sites are embedded in the porous carbon matrix, which are difficult to image owing to the thickness of the carbon spheres.
 | ||
| Fig. 4 (A) SEM image (B) TEM image (C) and (D) HAADF-STEM images of FeNx@C (6% oxygen treatment, single site Fe are indicated with a red arrow). | ||
Unlike for other FeNx@C examples, no acid washing was required since no Fe-based nanocluster was detected by HAADF-STEM, XRD or X-ray absorption spectroscopy (XAS, vide infra). Indeed, the catalyst showed the same catalytic activity and Fe content with or without acid washing. We surmise that both the pre-coordination of Fe in heme sites along with their very low concentration contributed synergistically to avoid metal aggregation.41
Cryo (5K) X-band EPR was used to selectively probe Fe3+ sites within the catalyst, with potential Fe2+ and Fe4+ sites typically being EPR silent (Fig. 5).42 The Fe precursor, haemoglobin, gives a clear signal at g ∼ 5.8 and g ∼ 4.3 which relates to high spin (S = 5/2) Fe in methaemoglobin and transferrin impurity, respectively. Additionally, a broad signal centered around g ∼ 2.1 derives from ferritin, with this signal more evident at higher EPR temperatures. The sharp signal at g = 2 originates from organic radicals, with some potential contribution from the EPR tube or resonator. Interestingly, only a very small signal at g ∼ 4.3 was found in the FeNx@C catalyst, assigned to non-specifically bound high spin Fe3+ with large rhombic zero field splitting, possibly due to some leftover traces transferrin.42,43 EPR sensitivity is typically in the order of μM,44 while the Fe concentration in the catalyst is in the order of mM, suggesting Fe3+ in any significant proportion should be detected. Thus, the limited EPR response indicates that Fe exists almost entirely in the Fe2+ or Fe4+ state.
 | ||
| Fig. 5 X-band EPR spectrum of haemoglobin (red), FeNx@C (blue) and empty tube (black). | ||
Having confirmed the single-site nature of FeNx@C, we then investigated the role of each reaction component by conducting a series of control experiments (Table 2), starting with the use of base additives. In previous reports of both heterogeneous and homogeneous Fe catalysis, it has also been shown that the addition of a base helps activate the metal complex.45,46 Typically, 1.0–5.0 equivalents of corrosive hydroxide salts (NaOH, KOH) are required. Only a few examples of base-free Fe catalysts have been reported for the transfer hydrogenation of nitroarenes (Fe(BF4)2/HCO2H,47 FeBr2/PhSiH3,48 Fe2O3/HCO2H49), but they all required costly phosphine ligands.
|
|
||||
|---|---|---|---|---|
| Entry | Deviation from standard conditions | Yield (%) | ||
| Standard reaction conditions: 4-nitrotoluene (12.4 mg, 88 μmol), base and additive (0.5 equiv. each), Fe source (4 mol% Fe), isopropyl alcohol (2.0 mL), Ar, 150 °C, 24 h.a 38% of 1,2-di-p-tolylhydrazine was formed. | ||||
| 1 | None | 75 | ||
| 2 | Removal | No K2CO3 | 10 | |
| 3 | No KCO2H | 34a | ||
| 4 | No catalyst | 0 | ||
| 5 | Base | Na2CO3 | 31 | |
| 6 | KOH | 36 | ||
| 7 | NEt3 | 16 | ||
| 8 | Reductant | NH 4 CO 2 H | (0.5 equiv.) | 0 |
| 9 | (1 equiv.) | 23 | ||
| 10 | (1.5 equiv.) | 25 | ||
| 11 | Iron source | Fe 3+ | FeCl3 | 0 |
| 12 | Fe(acac)3 | 0 | ||
| 13 | Fe 2+ | FeCl2 | 14 | |
| 14 | FePc | 0 | ||
| 15 | Pristine haemoglobin | 6 | ||

In our system, removing K2CO3 decreased the yield from 75% to 10% (Table 2, entry 1), showing the essential role of adding a base. Removing KCO2H led to a similar conversion of the starting material (75%), but with half of the product being the hydrazine intermediate (34% of amine, 38% of 1,2-di-p-tolylhydrazine were formed, Table 2, entry 3). This shows that KCO2H helps cleave the N–N bond of the hydrazine intermediate. Indeed, at no other point of the investigation this intermediate was detected when KCO2H was present in the mixture. The indicates that the reduction occurs probably through the indirect nitroarene reduction pathway (see further details in ESI, Fig. S17 and S18†). As expected, the reaction did not proceed in the absence of catalyst, confirming the catalyst-base-reductant synergy (Table 2, entry 4). Substituting K2CO3 with Na2CO3 decreased the yield to 31%, in consistence with other reports on nitroarene transfer hydrogenation, with K2CO3 having a better solubility in alcohols (Table 2, entry 5).50 KOH or NEt3 decreased the yield to 36 and 16%, respectively (Table 2, entries 6 and 7). Replacing KCO2H with NH4CO2H gave no yield (Table 2, entry 8), although some product was detected upon increasing the amount of NH4CO2H to 1 and 1.5 equivalents (23 and 25% yield, Table 2, entries 9 and 10). Finally, we used different Fe salts directly as catalysts in our optimal conditions. Fe3+ sources, such as Fe(acac)3 and FeCl3, were inactive for the reaction (Table 2, entries 11 and 12), while FeCl2 gave a 14% yield, hinting at Fe2+ as the active species (Table 2, entry 13). Iron(II) phthalocyanine (FePc, Table 2, entry 14) was catalytically inactive, although this could be attributed to it being insoluble in iPrOH.51 In comparison, a low level of catalytic activity was observed using the pure haemoglobin precursor (6% yield, Table 2, entry 15). Due to the chemical complexity of the precursor, we cannot exclude that its catalytic activity comes from the residual ferritin or the transferrin impurity in the sample (vide supra for the EPR analysis).
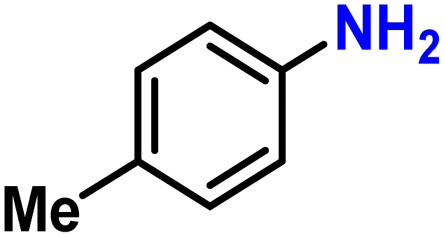
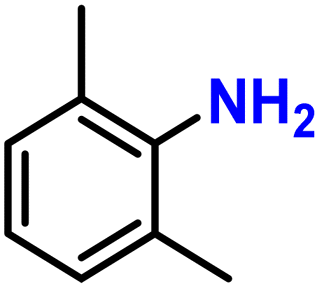
Next, the catalytic study was extended to other nitroarene substrates (Table 3). We first investigated steric hindrance surrounding the – nitro group. In comparison with the 85% yield obtained in the reduction of nitrobenzene (Table 3, entry 2), more sterically hindered substrates such as 2-ethylnitrobenzene or 1,3-dimethyl-2-nitrobenzene required a slightly increased Fe loading (5 mol%) to maintain high yields (69 and 77%, Table 3, entries 3 and 4). This is consistent with the predominantly nanoporous nature of the catalyst, making the catalytic site more difficult to access for bulkier substrates.
|
|
|||
|---|---|---|---|
| Entry | Product | Yield (%) | |
| Standard reaction conditions: nitroarene (88 μmol), K2CO3 (6.1 mg, 0.5 equiv.), KCO2H (3.7 mg, 0.5 equiv.), FeNx@C (50 mg, 4 mol% Fe), isopropyl alcohol (2.0 mL), Ar, 150 °C, 24 h.a 5 mol% Fe was used by adjusting substrate amount to 70 μmol.b KCO2H was not added. | |||
| 1 |
|
75 | |
| 2 |

|
85 | |
| 3 |

|
69a | |
| 4 |
|
77 | |
| 5 |

|
84 | |
| 6 |

|
58 | |
| 7 |

|
46 | |
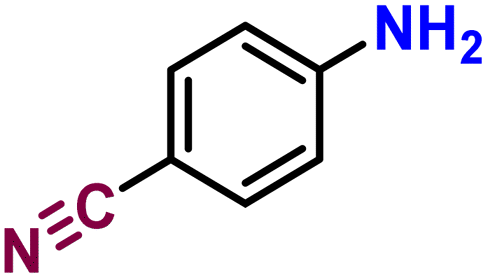
| 8 |

|
Ortho- | 97a |
| 9 | Meta- | 93a | |
| 10 | Para- | 57a | |
| 11 |

|
71 | |
| 12 |
|
41b | |
| 13 |

|
37a | |

Chemoselectivity is a major challenge in nitroarene reduction,52 since hydrogenation catalysts can also cleave weak bonds (such as carbon–halogen bonds), or reduce unsaturated bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO). Therefore, bifunctional nitroarenes bearing cleavable bonds or reducible groups were also included in the substrate study. The C–O ether bond in 1,2-(methylenedioxy)-4-nitrobenzene was preserved while keeping a high yield (85%, Table 3, entry 5). Although yields were lower, 4-iodonitrobenzene and 1,2,3-trichloro-5-nitrobenzene were successfully reduced while fully preserving the C–I and C–Cl bonds (58 and 46%, Table 3, entries 6 and 7). As for nitroanilines, the three isomers were reduced in high yields (97, 93 and 53%, Table 3, entries 8–10). Similarly, CC, C
C, CO). Therefore, bifunctional nitroarenes bearing cleavable bonds or reducible groups were also included in the substrate study. The C–O ether bond in 1,2-(methylenedioxy)-4-nitrobenzene was preserved while keeping a high yield (85%, Table 3, entry 5). Although yields were lower, 4-iodonitrobenzene and 1,2,3-trichloro-5-nitrobenzene were successfully reduced while fully preserving the C–I and C–Cl bonds (58 and 46%, Table 3, entries 6 and 7). As for nitroanilines, the three isomers were reduced in high yields (97, 93 and 53%, Table 3, entries 8–10). Similarly, CC, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, CN bonds remained unaffected: 3-nitrostyrene gave a 71% yield (Table 3, entry 11), whereas 4-nitrobenzonitrile and 1-ethynyl-4-nitrobenzene gave only 41 and 37% yields respectively (Table 3, entries 12 and 13). In the case of 4-nitrobenzonitrile, standard conditions gave a mixture of 4-aminobenzonitrile and 4-nitrobenzylamine (26% and 10% respectively), while only the former was produced when KCO2H was removed. Overall, we showed the excellent chemoselectivity of our catalytic system can be achieved, with the exception of nitrobenzaldehydes and nitrovinyls (see ESI for details, Fig. S16†).
C, CN bonds remained unaffected: 3-nitrostyrene gave a 71% yield (Table 3, entry 11), whereas 4-nitrobenzonitrile and 1-ethynyl-4-nitrobenzene gave only 41 and 37% yields respectively (Table 3, entries 12 and 13). In the case of 4-nitrobenzonitrile, standard conditions gave a mixture of 4-aminobenzonitrile and 4-nitrobenzylamine (26% and 10% respectively), while only the former was produced when KCO2H was removed. Overall, we showed the excellent chemoselectivity of our catalytic system can be achieved, with the exception of nitrobenzaldehydes and nitrovinyls (see ESI for details, Fig. S16†).
With this methodology, we were also able to prepare 2-phenylbenzimidazole, an important scaffold involved in anti-cancer drugs, in a one-pot reduction/cyclisation tandem reaction from 2-nitroaniline.53 For this, we replaced isopropanol with benzyl alcohol, which generates an equivalent of benzaldehyde byproduct in the reduction of 2-nitroaniline into ortho-phenylenediamine. The two products can then undergo condensation into the desired heterocycle with 64% yield (Fig. 6).
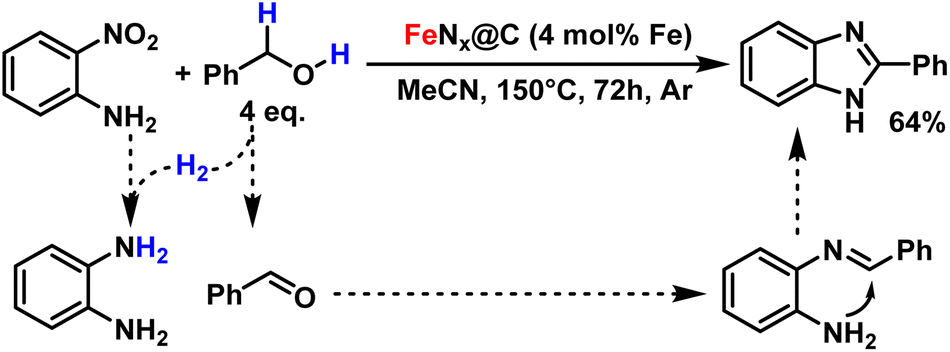 | ||
| Fig. 6 One-pot preparation of 2-phenylbenzimidazole. | ||
The catalyst was easily recycled by centrifugation, and retained its activity for up to 7 cycles without a significant decrease in yield for the reduction of 4-nitrobenzene (Fig. 7).
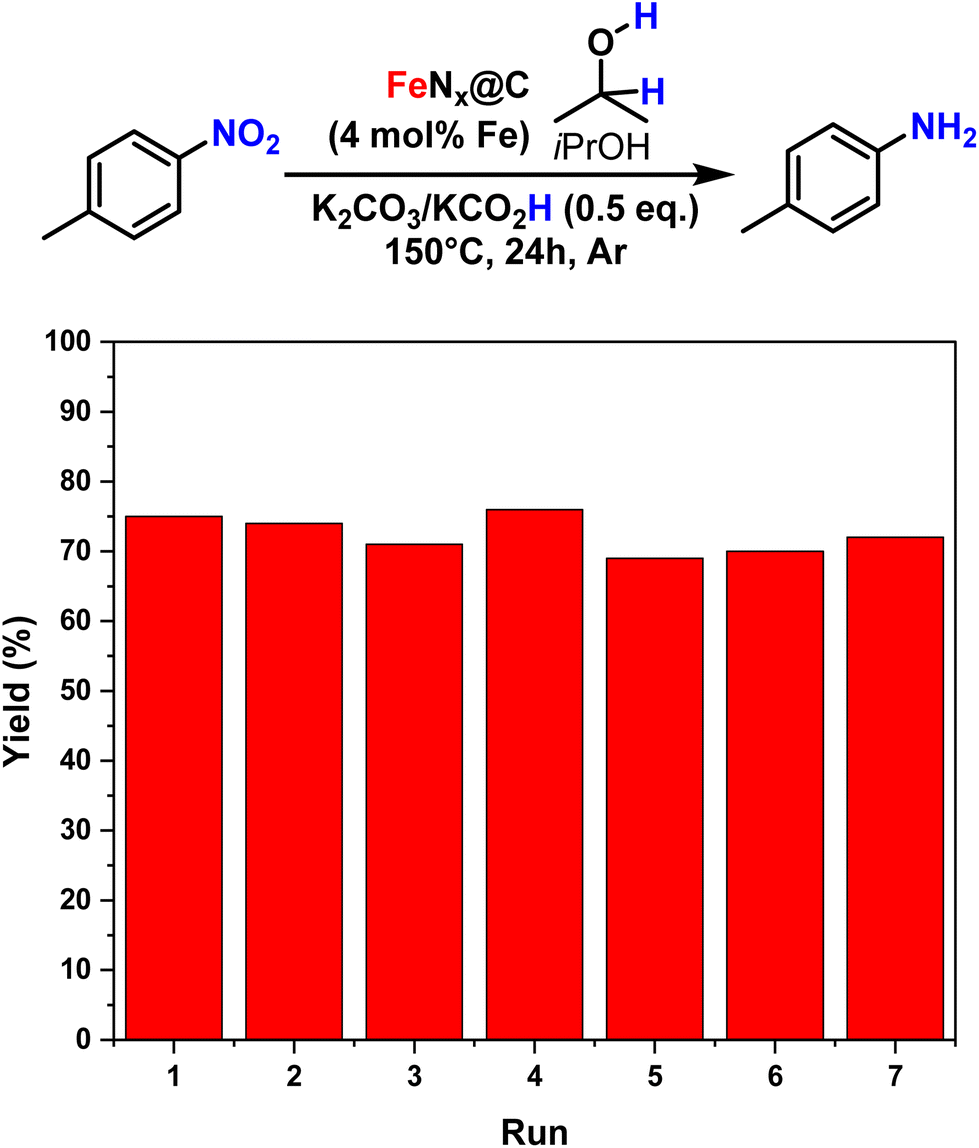 | ||
| Fig. 7 Recycling of the catalyst. | ||
The recovered material (after one catalytic cyle) was subjected to HAADF-STEM analysis, showing no appearance of FexOy aggregates, and we were still able to identify single-site Fe bright dots (Fig. 8, and Fig. S5–S6†), implying that the catalytic sites were retained.
 | ||
| Fig. 8 (A) and (B) Post-reaction HAADF-STEM image of FeNx@CAR (single site Fe are indicated with a red arrow). | ||
Information on FeNx coordination before and after reaction was also obtained by XAS. As shown in Fig. S12,† the Fe–K-edge X-ray absorption near-edge spectroscopy (XANES) for the catalyst before and after the reaction (FeNx@C and FeNx@CAR respectively) is similar to that of the FePc reference sample. FeNx@CAR showed an increased pre-edge intensity (at 7113 eV), which indicates that Fe has a more symmetric coordination pattern than in FeNx@C.54 The Fourier transform (FT) of the extended X-ray absorption fine structure (EXAFS) spectrum for Fe-before and Fe-after samples showed a main peak at 1.5 Å and 1.4 Å (phase uncorrected), corresponding to the first shell Fe–N or Fe–O (Fig. S12E†). The shorter first shell bond distance for FeNx@CAR suggests a change in the local coordination environment. A comparison with Zitolo et al.'s spectra (Fig. S14†) indicates the similarities of our samples (FeNx@C and FeNx@CAR) with their FeN4–O and FeN4–O2 samples. Together with the pre-edge features, we propose a FeN4–O structure for FeNx@C and FeN4–O2 for FeNx@CAR, respectively.55 Wavelet transform (WT) EXAFS was employed to visualise the nearby atoms by providing the radial distance and k space resolutions. As shown in Fig. S15,† WT of FeNx@C and FeNx@CAR showed one prominent peak at ∼4.7 Å−1 and 4.2 Å−1, respectively, which is very close to that in the reference FePc (∼4.2 Å−1). Moreover, they are different from the FeO, Fe2O3, and Fe foil reference samples, suggesting the dominant iron species in both samples is FeN4 sites, at least at the resolution of this experiment. Due to the similar bond distance, it is difficult to distinguish the difference between Fe–N and Fe–O in single sites. However, together with XPS, EPR, and STEM, we propose that Fe–N is the main species on the first shell.
EXAFS fittings were performed to get more insight on the coordination environment (Fig. S12†). All fittings are in good consistency with experimental data. The best fit values (Table S3†) of FeNx@C gave an average coordination number of 4.85 for Fe–N at 2.04 Å, 2.10 for Fe–C at 2.35 Å, while the coordination number for FeNx@CAR was 5.95 for Fe–N at 1.91 Å, 2.63 for Fe–C at 2.28 Å, and 0.77 for Fe–Fe at 2.52 Å. A minor Fe–Fe peak arising from FeNx@CAR sample might originate from trace amount Fe2O3 from Celite used for filtration or traces Fe in K2CO3 (see notes in ESI†).56 As shown in Fig. S8d,† an increased coordination number and decreased bond length could be seen in the FeNx@CAR. Together with XANES pre-edge features, we propose a FeN4 structure coordinated to one axial ligand before reaction, while FeNx@CAR exhibits a FeN4 structure coordinated to 2 axial ligands with a more symmetric structure. The nature of the axial ligand is still uncertain, although at least one is most likely an oxygen molecule, or some leftover nitrogen-containing product from the reaction.
Conclusions
In summary, we have shown for the first time the potential of haemoglobin-derived Fe catalysts for organic transformations. Unlike most reports, only biomass precursors were used (xylose and haemoglobin) and no Fe aggregation was observed, avoiding a commonly-used washing step in the preparation. Its single-site nature was demonstrated by high-end techniques (XAS, HAADF-STEM), and remained stable throughout the catalytic cycles. We have shown it to be highly selective towards a number of substrates, and active at low Fe loading (4–5 mol%), with isopropanol, K2CO3 and KCO2H. We believe that this work will pave a way for pure biomass-based catalysts applied to organic reactions, while setting a high standard for single-site material characterisation for thermal catalysis.Author contributions
A. Y. L. designed the project, prepared the materials and performed all the catalytic tests, the ICP-MS and N2 adsorption experiments. H. L. performed the TEM/SEM images and assisted for HAADF-STEM analysis. J. F. and H. L. performed the XAS analysis. A. P. performed the EPR analysis. J. B. performed the XPS analysis. J. R. performed the Raman and the XRD analysis and helped for N2 adsorption result interpretation. M. M. T. and K. K. H. supervised the study. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Monica Amboage from the I20-EDE (SP28663) at Diamond Light Source to support and assist the X-Ray measurements, and Dr Thomas Slater from the electron Physical Science Imaging Centre (ePSIC) at Diamond Light Source to support and assist the HAADF-STEM imaging (mg28698). The authors thank as well Patricia Carry and Kaho Cheung for the use and support of the Analytical Laboratory in the Department of Chemical Engineering at Imperial College London and Gwilherm Kerherve for the use and support of the XPS suite in the Department of Materials at Imperial College London. A. P. thanks the EPSRC Centre for Doctoral Training in the Advanced Characterization of Materials (grant number EP/L015277/1) and the Imperial College London SPIN-Lab (equipment grant number: EP/P030548/1). A. Y. L. acknowledges Marie Skłodowska-Curie Fellowship H2020-MSCA-IF-2019 (892614) through the project HAEMOGLOBIN. M. M. T. acknowledges the Royal Academy of Engineering Chair in Emerging Technologies Fellowship. We thank Dr Servann Herou for his priceless advice and training, and Mengnan Wang for handling the elemental analysis of the catalyst.References
-
B. Amini and S. Lowenkron, in Kirk–Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Ltd, 2003 Search PubMed
.
-
K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, 3rd, Completely Revised Edition, 2008 Search PubMed
- A. Corma and P. Serna, Science, 2006, 313, 332 LP–332334 CrossRef PubMed
- P. Serna and A. Corma, ACS Catal., 2015, 5, 7114–7121 CrossRef CAS
- J. Song, Z.-F. Huang, L. Pan, K. Li, X. Zhang, L. Wang and J.-J. Zou, Appl. Catal., B, 2018, 227, 386–408 CrossRef CAS
- D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS PubMed
- H. Niu, J. Lu, J. Song, L. Pan, X. Zhang, L. Wang and J.-J. Zou, Ind. Eng. Chem. Res., 2016, 55, 8527–8533 CrossRef CAS
- G. Wienhöfer, M. Baseda-Krüger, C. Ziebart, F. A. Westerhaus, W. Baumann, R. Jackstell, K. Junge and M. Beller, Chem. Commun., 2013, 49, 9089–9091 RSC
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed
- A. Li, S. A. Nicolae, M. Qiao, K. Preuss, P. A. Szilágyi, A. Moores and M.-M. Titirici, ChemCatChem, 2019, 11, 3602–3625 CrossRef CAS
- R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed
- R. V. Jagadeesh, T. Stemmler, A.-E. Surkus, H. Junge, K. Junge and M. Beller, Nat. Protoc., 2015, 10, 548 CrossRef CAS PubMed
- R. v. Jagadeesh, G. Wienhöfer, F. A. Westerhaus, A.-E. Surkus, M.-M. Pohl, H. Junge, K. Junge and M. Beller, Chem. Commun., 2011, 47, 10972–10974 RSC
- L. Liu, B. Wang, R. Gao, D. Zhang, W. Xu, L. Chen, X. Yan and Y. Li, RSC Adv., 2020, 10, 10689–10694 RSC
- R. v. Jagadeesh, K. Natte, H. Junge and M. Beller, ACS Catal., 2015, 5, 1526–1529 CrossRef CAS
- P. Veerakumar, I. Panneer Muthuselvam, C.-T. Hung, K.-C. Lin, F.-C. Chou and S.-B. Liu, ACS Sustainable Chem. Eng., 2016, 4, 6772–6782 CrossRef CAS
- O. Beswick, I. Yuranov, D. T. L. Alexander and L. Kiwi-Minsker, Catal. Today, 2015, 249, 45–51 CrossRef CAS
- X. Cui, Q. Zhang, M. Tian and Z. Dong, New J. Chem., 2017, 41, 10165–10173 RSC
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed
- H. Jin, P. Li, P. Cui, J. Shi, W. Zhou, X. Yu, W. Song and C. Cao, Nat. Commun., 2022, 13, 723 CrossRef CAS PubMed
- H.-Y. Zhuo, X. Yu, Q. Yu, H. Xiao, X. Zhang and J. Li, Sci. China Mater., 2020, 63, 1741–1749 CrossRef CAS
- Q. Shen, H. Jin, P. Li, X. Yu, L. Zheng, W. Song and C. Cao, Nano Res., 2022, 15, 5024–5031 CrossRef CAS
- G. Lu, K. Sun, Y. Lin, Q. Du, J. Zhang, K. Wang and P. Wang, Nano Res., 2022, 15, 603–611 CrossRef CAS
- W.-C. Cheong, W. Yang, J. Zhang, Y. Li, D. Zhao, S. Liu, K. Wu, Q. Liu, C. Zhang, D. Wang, Q. Peng, C. Chen and Y. Li, ACS Appl. Mater. Interfaces, 2019, 11, 33819–33824 CrossRef CAS PubMed
- R. Yun, F. Zhan, N. Li, B. Zhang, W. Ma, L. Hong, T. Sheng, L. Du, B. Zheng and S. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 34122–34129 CrossRef CAS PubMed
- J. Maruyama and I. Abe, Chem. Mater., 2005, 17, 4660–4667 CrossRef CAS
- J. Maruyama and I. Abe, Chem. Mater., 2006, 18, 1303–1311 CrossRef CAS
- J. Maruyama, J. Okamura, K. Miyazaki and I. Abe, J. Phys. Chem. C, 2007, 111, 6597–6600 CrossRef CAS
- J. Maruyama, J. Okamura, K. Miyazaki, Y. Uchimoto and I. Abe, J. Phys. Chem. C, 2008, 112, 2784–2790 CrossRef CAS
- W. Chen, X. Luo, S. Ling, Y. Zhou, B. Shen, T. J. A. Slater, J. A. Fernandes, T. Lin, J. Wang and Y. Shen, Carbon, 2020, 168, 588–596 CrossRef CAS
- C.-Z. Guo, C.-G. Chen and Z.-L. Luo, J. Power Sources, 2014, 245, 841–845 CrossRef CAS
- J. Maruyama, T. Hasegawa, T. Amano, Y. Muramatsu, E. M. Gullikson, Y. Orikasa and Y. Uchimoto, ACS Appl. Mater. Interfaces, 2011, 3, 4837–4843 CrossRef CAS PubMed
- J. Maruyama, T. Hasegawa, S. Iwasaki, H. Kanda and H. Kishimoto, ACS Sustainable Chem. Eng., 2014, 2, 493–499 CrossRef CAS
- X. Li, Y. Chen and J. Nielsen, Curr. Opin. Biotechnol, 2019, 57, 56–65 CrossRef CAS PubMed
- Y. Gong, Z. Wei, J. Wang, P. Zhang, H. Li and Y. Wang, Sci. Rep., 2014, 4, 6349 CrossRef CAS PubMed
- M.-M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC
- S. A. Nicolae, H. Au, P. Modugno, H. Luo, A. E. Szego, M. Qiao, L. Li, W. Yin, H. J. Heeres, N. Berge and M.-M. Titirici, Green Chem., 2020, 22, 4747–4800 RSC
- K. Preuss, L. C. Tănase, C. M. Teodorescu, I. Abrahams and M.-M. Titirici, J. Mater. Chem. A, 2017, 5, 16336–16343 RSC
- N. Baccile, M. Antonietti and M.-M. Titirici, ChemSusChem, 2010, 3, 246–253 CrossRef CAS
- M. Paneque, J. M. de la Rosa, J. Kern, M. T. Reza and H. Knicker, J. Anal. Appl. Pyrolysis, 2017, 128, 314–323 CrossRef CAS
- M. C. Martins Alves and G. Tourillon, J. Phys. Chem., 1996, 100, 7566–7572 CrossRef
- W. R. Hagen, Dalton Trans., 2006, 4415–4434 RSC
- L. Ni, C. Gallenkamp, S. Paul, M. Kübler, P. Theis, S. Chabbra, K. Hofmann, E. Bill, A. Schnegg, B. Albert, V. Krewald and U. I. Kramm, Adv. EnergySustain. Res., 2021, 2, 2000064 Search PubMed
- M. M. Roessler and E. Salvadori, Chem. Soc. Rev., 2018, 47, 2534–2553 RSC
- S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC
- P. E. Sues, K. Z. Demmans and R. H. Morris, Dalton Trans., 2014, 43, 7650–7667 RSC
- G. Wienhöfer, I. Sorribes, A. Boddien, F. Westerhaus, K. Junge, H. Junge, R. Llusar and M. Beller, J. Am. Chem. Soc., 2011, 133, 12875–12879 CrossRef PubMed
- K. Junge, B. Wendt, N. Shaikh and M. Beller, Chem. Commun., 2010, 46, 1769–1771 RSC
- K. J. Datta, A. K. Rathi, M. B. Gawande, V. Ranc, G. Zoppellaro, R. S. Varma and R. Zboril, ChemCatChem, 2016, 8, 2351–2355 CrossRef CAS
- H. Wiener, J. Blum and Y. Sasson, J. Org. Chem., 1991, 56, 4481–4486 CrossRef CAS
- F. Ghani, J. Kristen and H. Riegler, J. Chem. Eng. Data, 2012, 57, 439–449 CrossRef CAS
- H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS
- S. Yadav, B. Narasimhan and H. kaur, Adv. Anticancer Agents Med. Chem., 2016, 16, 1403–1425 CrossRef CAS
- X. Xie, C. He, B. Li, Y. He, D. A. Cullen, E. C. Wegener, A. J. Kropf, U. Martinez, Y. Cheng, M. H. Engelhard, M. E. Bowden, M. Song, T. Lemmon, X. S. Li, Z. Nie, J. Liu, D. J. Myers, P. Zelenay, G. Wang, G. Wu, V. Ramani and Y. Shao, Nat. Catal., 2020, 3, 1044–1054 CrossRef CAS
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed
- Imerys filtration, Celite specification sheet, https://nika-iv.com/en/diatomite/tds/FR_C_574.pdf, (accessed 2 December 2021).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02344j |
| This journal is © The Royal Society of Chemistry 2022 |