
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMaterials from waste plastics for CO2 capture and utilisation
Jerald Y. Q.
Teo†
a,
Albert
Ong†
a,
Tristan T. Y.
Tan†
 a,
Xin
Li†
a,
Xian Jun
Loh
ab and
Jason Y. C.
Lim
*ab
a,
Xin
Li†
a,
Xian Jun
Loh
ab and
Jason Y. C.
Lim
*ab
aInstitute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Singapore 138634. E-mail: jason_lim@imre.a-star.edu.sg
bDepartment of Materials Science and Engineering, National University of Singapore (NUS), 9 Engineering Drive 1, Singapore 117576
First published on 16th July 2022
Abstract
Anthropogenic activities have resulted in a sustained increase in atmospheric CO2 levels over the past 200 years. To mitigate this, materials capable of capturing or chemically transforming CO2 have received tremendous attention recently. However, porous materials such as polymeric adsorbents and metal–organic frameworks (MOFs), which have demonstrated considerable success in carbon capture and utilisation, are traditionally synthesized from non-renewable petrochemical feedstock. In line with recent trends towards a circular materials economy, there have been greater emphases on functional materials design and production from societal waste. Therefore in this Perspectives article, we highlight and discuss the possibility of using waste commodity plastics as hitherto-overlooked low-cost alternative resources for the synthesis of functional materials for CO2 capture and utilisation. This not only reduces the quantity of waste plastics disposed, but also offers a second lease-of-life to these polymers by keeping them in continuous use. Herein, we examine how the polymer structures of commodity plastics have influenced the design of the resulting carbon nanomaterials, polymeric adsorbents and MOFs for effective CO2 capture and conversion into useful chemicals, such as cyclic carbonates. With this field currently only in its infancy, we also discuss the limitations of the current work, our opinions on further exploiting the potential of waste commodity plastics for CCU applications, and the essential considerations involved in this multidisciplinary endeavour.
 Jerald Y. Q. Teo | Jerald Y. Q. Teo received his B.Sc. (Hons) degree in Chemistry from the National University of Singapore in 2020. He then joined the Institute of Materials Research and Engineering (IMRE) as a research specialist working alongside Asst. Prof. Jason Lim on the upcycling of plastic waste. Currently, his work focuses on the transformation of waste plastics into functional polymeric materials. |
 Albert Ong | Albert Ong received his B.S. degree (2017) in chemistry from the National University of Singapore (NUS). He received his Ph.D. degree (2021) in Chemistry from NUS under Assoc. Prof. Chi Chunyan. Currently, He is a research scientist at IMRE, working alongside Asst. Prof. Jason Lim on the upcycling of plastic waste. |
 Tristan T. Y. Tan | Dr Tristan T. Y. Tan received his bachelor's degree in chemistry from the National University of Singapore, and his Dr rer.nat. degree from the University of Münster under the supervision of Prof. F. Ekkehardt Hahn. He has returned to Singapore and works in the Sustainable Supramolecular Materials group in IMRE on metal–organic frameworks. |
 Xin Li | Xin Li received her BA and MSc degrees in chemistry from the University of Cambridge, where she worked on metal–organic cages. She is currently working as a research specialist in IMRE with Asst. Prof. Jason Lim, where she is focused on developing MOF-polymer hybrids for biomedical and sustainability applications. |
 Xian Jun Loh | Professor Xian Jun Loh is a polymer chemist who obtained his Ph.D. degree from the National University of Singapore. He is a Fellow of Fitzwilliam College, University of Cambridge, and the Royal Society of Chemistry. Renowned internationally as a pioneer in biodegradable thermogels for biomedical, personal care and food applications, he currently sits on the editorial boards of several international journals. |
 Jason Y. C. Lim | Asst. Prof. Jason Y. C. Lim obtained his DPhil in Inorganic Chemistry from the University of Oxford, U.K., and now leads the Sustainable Supramolecular Materials Laboratory at IMRE. His current research focuses on upcycling of waste plastics into functional polymeric materials and commodity chemicals, biodegradable supramolecular biomaterials, as well as metal–organic frameworks for green chemistry applications. |
1. Introduction
Since the mid-18th century, anthropogenic activities have cumulatively emitted an estimated 1.5 trillion tonnes of CO2, with emissions continuing to grow unabated.1 With more than 34 billion tonnes of CO2 emitted annually, rising CO2 concentration in the atmosphere is the primary driver of global climate change, which could severely disrupt ecosystems and human lives. Carbon capture, storage and utilisation (CCSU) is now recognised to be amongst the most promising technologies to mitigate climate change,2 in order to limit the global average temperature increase to 1.5 °C above pre-industrial levels as ratified in the 2015 Paris Agreement.3 Carbon capture refers to the sorption of CO2 from gas streams, traditionally with aqueous amine solutions. Despite their technological maturity, such amine-based absorbent solutions are disadvantageous due to their energy-intensive and ineffective amine regeneration processes, and corrosive nature.4,5 Moreover, a large volume of liquid sorbents is required for carbon capture, where current production is insufficient to support the scale of global CO2 emission.6 Thus, recent years have witnessed increased research interest in solid adsorbent materials, which are easy to operate and regenerate with lower energy demands (30–50% reduction compared with alkanolamine solutions theoretically),7 and are easy to retrofit.8,9 On the other hand, CO2 utilisation refers to its conversion into other products, such as catalytic conversion to CO2-derived chemicals (e.g. urea and cyclic carbonates), polymers (e.g. polycarbonates and polyurethane), fuels (e.g. methanol and formic acid), mineralisation to carbonated aggregates or concrete products, and biological utilisation.10–15 A number of solid materials have shown great promise for these applications, such as activated carbons, polymeric adsorbents and metal–organic frameworks (MOFs), the last of which is especially useful for their potential for CO2 uptake, separation and conversion.16–18 Whilst these materials are traditionally produced from non-renewable petrochemical feedstock, many of them can also be produced from societal waste products such as post-use plastics. Despite their potential in addressing both pressing environmental issues of high-volume unsustainable waste plastic production and mitigating CO2 emissions, valorising waste plastics into functional materials for carbon capture and utilisation (CCU) has been surprisingly overlooked.Of the ∼6300 million metric tonnes of plastic waste produced cumulatively up to 2015, the bulk of them (∼91%) are not recycled and simply disposed after use or incinerated.19 The US Environmental Protection Agency has classified waste plastics into six major categories (SPI codes 1–6 in Fig. 1), accounting for approximately 75% of all plastic waste produced. Of these classes of plastics, only polyethylene terephthalate (PET) and high-density polyethylene (HDPE) are recovered and recycled to appreciable extents.20 Although traditionally possessing highly unsustainable linear cradle-to-grave life cycles, waste commodity plastics are now increasingly viewed as untapped resources for new materials production.21,22 Different methods of valorising waste plastics have been developed: other than carbonisation,23 they can also be subjected to post-synthetic chemical functionalisation to form functional polymers,24,25 or broken down to form small molecules which can be used as commodity chemicals (e.g. carboxylic acids),26,27 fuels, lubricants and waxes, and monomers for repolymerisation.28 Tapping on waste plastics not only reduces societal reliance on petrochemical feedstock for new materials production, saving up to an estimated 3.5 billion barrels of oil, but also contributes significantly to economic growth and brings about considerable cost savings.20 Although degradable bioplastic alternatives such as poly(hydroxyalkanoates) and poly(lactic acid) are becoming increasingly popular, their market share remains small (<1%) compared to their petroleum-derived counterparts,29 and they have not yet met the vast application scope of existing commodity plastics.30 With the global plastic waste production projected to continue unabated driven by increasing demand, potentially tripling current production rates by 2060,31 innovative methods to give waste commodity plastics new lease-of-life as functional materials will be increasingly sought-after.
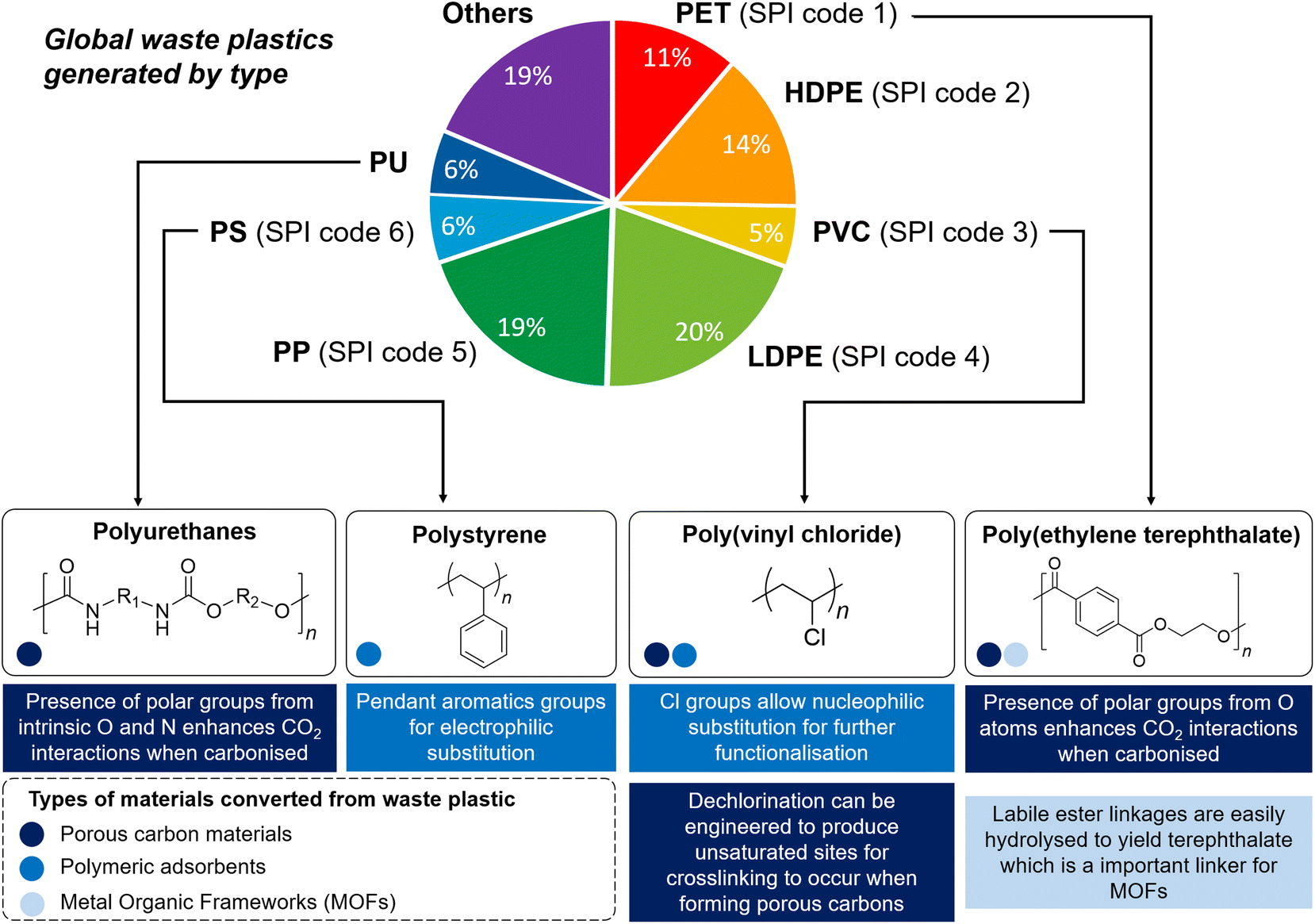 | ||
| Fig. 1 Schematic illustration showing the global primary waste plastic generation in 2015,19 the majority taken up by the six major categories of waste plastics (SPI codes 1–6). The unique chemical functionalities of PET, PUs, PVC and PS enable them to be exploited for CCU and their individual properties suitable for valorising into different types of CCU materials (differentiated by colour) are described. | ||
1.1 Designing materials for CCU from waste plastics
Plastics are carbon-rich organic polymers. Hence, of the different porous materials available for CCU, they are most suitable for valorisation into carbon-containing (1) polymeric adsorbents; (2) porous activated carbons and (3) MOFs. CO2 can be adsorbed both by physisorption and chemisorption, and a good understanding of the properties of CO2 and how it can interact with the adsorbents will be essential for designing materials from waste plastics capable of high CO2 uptake.Although CO2 is a linear centrosymmetric molecule bearing no net dipole moment, the polarisation of individual C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds due to the greater electronegativity of the terminal oxygen atoms dictates its interactions with adsorbent surfaces and reactivity. Physisorption of CO2 arises from non-covalent interactions such as ion-dipole (e.g. M+⋯OCO), dipole–dipole and van der Waal interactions with aromatic moieties.32 Chemisorption of CO2 can also occur, most often from the nucleophilic attack of amine groups on adsorbents on the electron-deficient central carbon atom of CO2. Unsurprisingly, the presence of heteroatoms (e.g. N, O, S) which confers polar and nucleophilic active sites on adsorbents can improve CO2 uptake. In addition, there is a need for sufficient pore space and volume (∼tens of nanometres) to facilitate rapid gas transport and allow unhindered gas–surface interactions. Finally, hydrophobic adsorbent surfaces can also enhance CO2 uptake by reducing the competitive binding of the active sites with water.33
O bonds due to the greater electronegativity of the terminal oxygen atoms dictates its interactions with adsorbent surfaces and reactivity. Physisorption of CO2 arises from non-covalent interactions such as ion-dipole (e.g. M+⋯OCO), dipole–dipole and van der Waal interactions with aromatic moieties.32 Chemisorption of CO2 can also occur, most often from the nucleophilic attack of amine groups on adsorbents on the electron-deficient central carbon atom of CO2. Unsurprisingly, the presence of heteroatoms (e.g. N, O, S) which confers polar and nucleophilic active sites on adsorbents can improve CO2 uptake. In addition, there is a need for sufficient pore space and volume (∼tens of nanometres) to facilitate rapid gas transport and allow unhindered gas–surface interactions. Finally, hydrophobic adsorbent surfaces can also enhance CO2 uptake by reducing the competitive binding of the active sites with water.33
The thermodynamic stability of CO2, as the most oxidised form of carbon, poses considerable challenges to its chemical utilisation. Other than the need to overcome the highly exothermic enthalpy of formation of CO2,34 the conversion of CO2 gas into a liquid or solid invokes considerable entropic cost. Thus, reactions of CO2 require highly reactive and energy-rich substrates such as aziridines and epoxides, as well as strong nucleophiles such as alkoxides, phenoxides and organometallic reagents (e.g. organolithiums and Grignard reagents).12 Currently, CO2 conversion reactions can be categorised into redox reactions forming reduced products such as methanol and formic acid, or carboxylation reactions where the whole CO2 molecule is incorporated with another reactant forming products such as cyclic carbonates.35 For CO2 to be a practical C1 feedstock, mild reaction conditions are needed, which in turn also reduces the energy demands and carbon footprint of the processes involved. Amongst various plastic-valorised porous materials, MOFs currently offer the greatest potential for CO2 utilisation due to their possibility for high CO2 uptake, selectivity and chemical activation of CO2 by design.
The structural diversity of the six major categories of waste plastics (SPI codes 1–6) offers different and unique opportunities for valorisation into materials for CCU (Fig. 1). At the time of writing, only PET, polyurethanes (PUs), poly(vinyl chloride) (PVC) and polystyrene (PS), estimated up to 30% of current plastic production, have been exploited predominantly. With its high carbon and oxygen content, PET is suitable for transformation into porous carbons that contain polar oxygenated functionalities which aid in CO2 adsorption. Furthermore, PET's susceptibility to solvolysis36 enables it to be chemically converted into terephthalic acid, an important MOF building block. Polyurethanes are useful precursors for porous carbon adsorbents as their intrinsic nitrogen content can enhance CO2 uptake. Although PVC and PS possess non-biodegradable and unreactive saturated C–C polymer backbones, the appended chlorine atoms and aromatic groups make these polymers susceptible towards nucleophilic attack and electrophilic substitution reactions, respectively, offering the possibility of post-synthetic chemical functionalisation into polymeric adsorbents. In this perspectives article, we will first consider various examples of transforming waste plastics into (1) polymeric adsorbents; (2) porous activated carbons or (3) MOFs, before we discuss potential future directions and possibilities of exploiting other types of plastics, such as the currently largely-overlooked polyolefins for CCU.
2. Upcycling waste plastics for CO2 capture and utilisation
2.1 Polymers for CO2 capture and utilisation
Porous polymeric adsorbents are light weight, possess high surface areas for CO2 uptake with porosity at the nanoscale, and offer opportunities for bottom-up design to engineer structural porosity and gas selectivity. There is ample scope for post-synthetic functionalisation of commodity plastics for the production of porous polymeric adsorbent materials, which leverages the high-volume and low-cost production of commodity plastics. This upcycling approach is especially pertinent to commodity polymers that are known to be cost-prohibitive and/or problematic to recycle, particularly expanded polystyrene (EPS) foam and PVC.37–41 Notably, EPS occupies a high volume per unit mass (27 to 40 kg m−3),42 making it costly to transport to recycling facilities, while PVC's high chlorine content can generate highly-corrosive hydrogen chloride gas and chlorinated molecules during chemical processing, and releases hazardous plasticisers which are used in significant quantities in commercial PVC formulations.43PS is useful for conversion to polymeric adsorbents due to the susceptibility of its aromatic groups to electrophilic functionalisation. Additionally, the π-electron-rich phenyl groups in PS allow induced-dipole interactions with quadrupolar CO2, which can lead to increased selectivity over N2 and CH4.44–47 Hypercrosslinked polymers (HCPs) and supported amine-functionalised polymers are two main classes of porous materials currently under development that have demonstrated good adsorption capacity and selectivity for CO2.5 Based on the same working mechanism of alkanolamines, the introduction of CO2-philic amine groups into polymers like PS and PVC has been shown to be efficient in CO2 capture.48–51 Fu et al. prepared HCPs via a one-pot Friedel–Crafts alkylation of waste EPS with a 1,2-dichloroethane crosslinker (Fig. 2A).45 Among the HCPs obtained, the highest observed CO2 adsorption capacity and CO2/N2 selectivity were 1.987 mmol g−1 and 23.4, respectively, at 273 K. Later, Fu et al. explored the use of four other crosslinkers to enhance the porosity of polymeric adsorbents derived from waste PS.46 Carbon tetrachloride-crosslinked PS (Fig. 2B) was observed to give the best CO2 adsorption capacity and selectivity over N2, with a value of 2.521 mmol g−1 and 37.8, respectively, at 273 K. Even after six adsorption–desorption cycles, no appreciable decrease in the adsorption capacity was observed, thereby demonstrating excellent reusability. On the other hand, Wu et al. developed a series of hybrid HCPs from commercial PS and octavinylsilsesquioxane (OVS) crosslinker (Fig. 2C).52 The porosity of the resulting HCPs can be fine-tuned by varying the PS/OVS ratio, and sorption experiments under a CO2/N2 gas mixture at 1 atm revealed the maximum CO2 uptake of 1.12 mmol g−1 at 298 K.
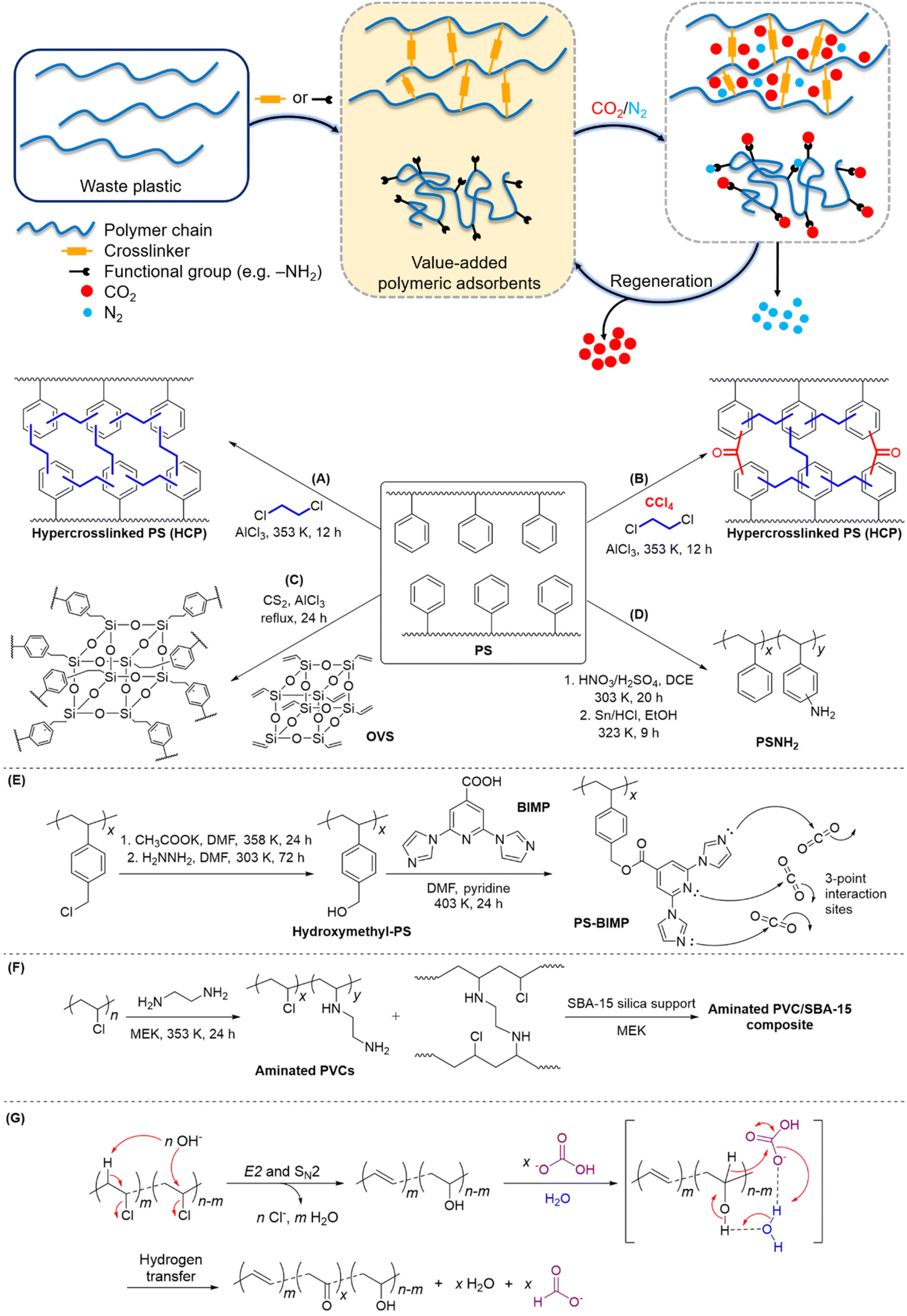 | ||
| Fig. 2 Upcycling commodity plastics into polymeric adsorbents for CCU by: (A–D) modification of PS via aromatic substitution reactions, (E) functionalisation of PS with BIMP to form multiple Lewis basic sites, (F) PVC amination and formation of composites with SBA-15 mesoporous silica, and (G) simultaneous PVC dechlorination and reduction of HCO3− to formate. | ||
Very recently, Merchán-Arenas et al. reported a two-step synthesis of an amine-tethered PS adsorbent (PSNH2) involving the nitration of waste EPS, followed by HCl/Sn-catalysed reduction of NO2 to NH2 groups (Fig. 2D).51 When applied as a solid support in the CO2 capture process, PSNH2 showed a moderate adsorption capacity of 1.05 mmol g−1 at 273 K. This could likely be improved by optimising the degree of PS amination. Gaikar and co-workers synthesized PS-based adsorbents by functionalising chloromethylated PS with different N-heterocyclic compounds.4 Adsorption studies of CO2, N2 and CH4 revealed that 2,6-bis-imidazo-1-yl-pyridine-4-carboxylic acid (BIMP)-functionalized PS (Fig. 2E) resulted in the highest CO2 uptake and selectivity; the selectivities of CO2/N2 and CO2/CH4 were 83 and 17, respectively. Although the equilibrium adsorption constant, K, for all the adsorbates had increased compared with the hydroxymethyl-PS control, the increase in K for CO2 was the greatest (i.e. higher selectivity for CO2 over N2 and CH4). Notably, an energy penalty of 38 kJ molCO2−1 for adsorbent regeneration is significantly lower than the current state-of-the-art CCU systems (172 kJ molCO2−1 for monoethanolamine),53 thereby further reinforcing its potential in CO2 sequestration applications.
Besides PS, waste PVCs are also a potential source of raw materials for the development of polymeric adsorbents for CCU. Sneddon et al. prepared a range of PVC-based/silica composites by supporting varying amounts of aminated PVC on mesoporous silica.48 In particular, the ethylenediamine-treated PVC/SBA-15 composite (Fig. 2F) comprising a 4 wt% polymer gave the highest adsorption efficiency of 0.5 mmol g−1 at 298 K. The adsorption–desorption kinetics study of this composite estimated an energy consumption of 60 kJ molCO2−1 for regeneration, which is a marked improvement from monoethanolamine.54 Moreover, the increased hydrophobicity of the composites compared with pristine silica suggests their suitability to operate under humid conditions, viz. they will not be easily deactivated by moisture present in flue gases.
Other than CO2 capture, a recent study demonstrated the non-intuitive possibility of using PVC for CO2 reduction, while achieving simultaneous de-chlorination of PVC.55 Using aqueous HCO3− to simulate the CO2 source when captured in alkaline solution, PVC was able to reduce hydrogen carbonate to formate at 300 °C in a batch tubular reactor. Mechanistic studies suggest that under alkaline conditions, PVC will undergo de-chlorination via elimination and substitution, forming a nucleophilic intermediate that could engage in redox reaction with hydrogen carbonate in water (Fig. 2G). With conversion to formate of up to 16% yield and dechlorination efficiency close to 100% achieved, the system shows promise as a green waste plastic treatment and carbon fixation tool.
2.2 Porous activated carbon adsorbents from waste plastics
Porous activated carbons are amongst the most popular materials for CO2 adsorption due to their easy synthesis and activation, affordability and ease of porosity manipulation.56 Amongst the waste commodity plastics, PET has most frequently been used to produce porous activated carbon adsorbents,57–59 and this is facilitated by the well-established collection and recycling infrastructure already in place for post-consumer PET drink bottles. PET possesses high carbon content for carbonisation, and the presence of oxygen atoms confers polar sites within the porous carbon materials to facilitate CO2 physisorption. PET typically needs to be activated by potassium hydroxide (KOH) to generate micro and mesopores, which are useful in CO2 adsorption under ambient conditions.60 The activation mechanism first involves the decomposition of PET at high activation temperature, releasing CO2 and CO. These gases can then react with KOH to give K2CO3, which in turn further reacts with carbon precursors to release more gases, thus developing and enlarging the pores within the carbon materials (Fig. 3A).57 Other than pore development, KOH activation also functionalizes the carbon precursors with additional oxygen-containing groups (e.g. carbonyl and hydroxyls) that improves CO2 binding. Elemental analysis of activated PET adsorbents shows an increase in oxygen content by weight from 11.71% to 34.33% when the KOH to carbon mass ratio increases from 0 to 3, with a corresponding increase in CO2 uptake capacity. Indeed, porous carbons formed by carbonising PET at 700 °C, and activated with a KOH to carbon mass ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, have a CO2 adsorption capacity of 1.31 mmol g−1 at 30 °C and allow complete regenerability up to four times with easy desorption.57
:1, have a CO2 adsorption capacity of 1.31 mmol g−1 at 30 °C and allow complete regenerability up to four times with easy desorption.57
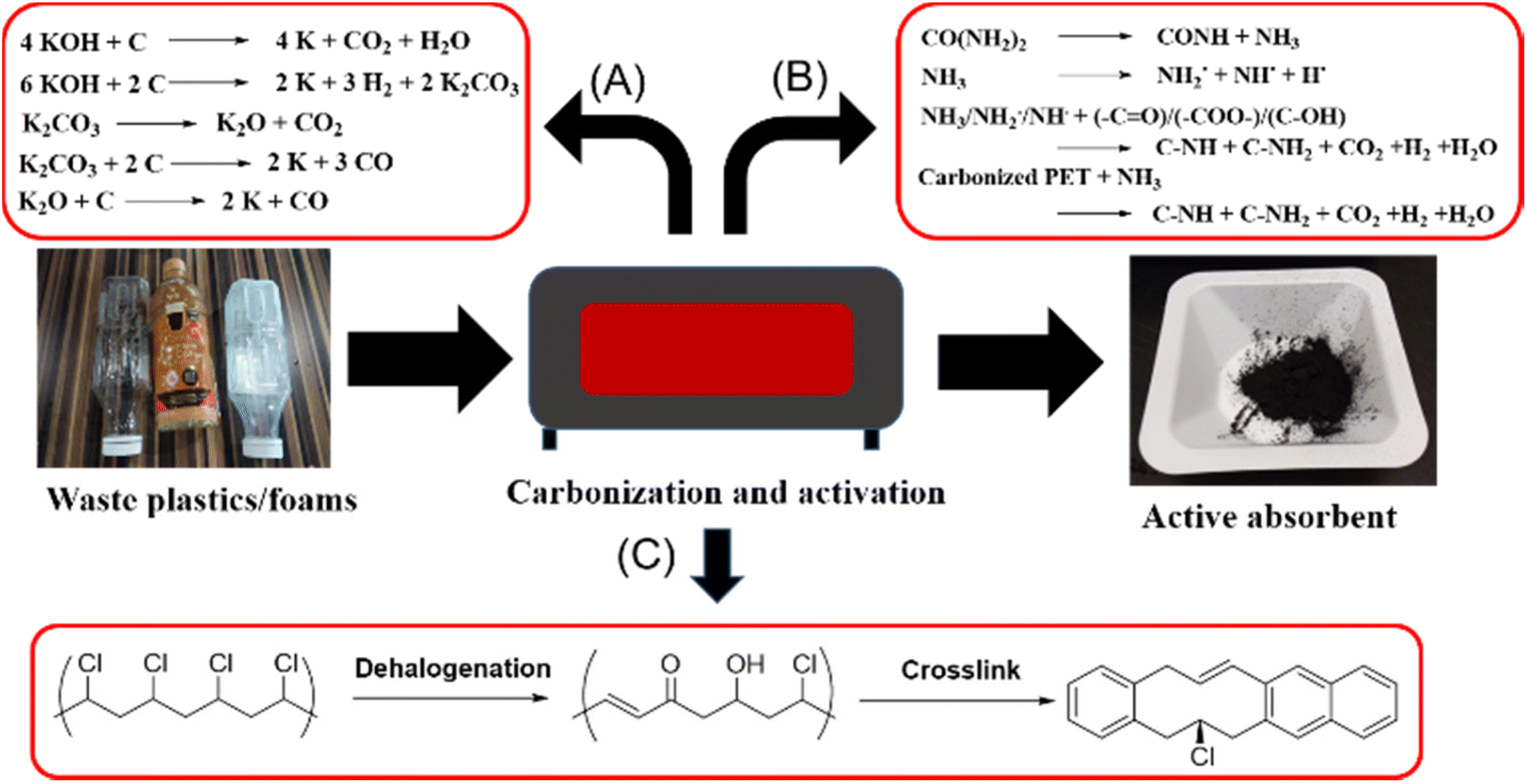 | ||
| Fig. 3 Conversion of waste plastics into porous carbon nanomaterials for CO2 absorbents: (A) KOH activation process for PET.57 (B) KOH/urea activation process for PET or PU foam.64,67 (C) Dehalogenation and crosslinking process for PVC.69 | ||
Besides oxygen-containing groups, the incorporation of nitrogen-containing groups into porous carbons can effectively enhance CO2 adsorption uptake and selectivity.61–63 Lee and co-workers64 devised a way to incorporate nitrogen-containing groups by the inclusion of urea during the KOH activation process of carbonized PET. At high temperatures, urea and NH3 reacts with carbonized PET to generate H˙, NH2˙ and NH˙ radicals. H˙ radicals could recombine to produce H2 gas which aids in pore formation, whilst the other radicals reacted with oxygen-containing groups formed from KOH activation to produce pyridinic-N and pyrrolic-N along with H2, CO2 and H2O gases (Fig. 3B).65,66 After KOH and urea activation, the sample displayed a nitrogen content of 3.23% and an oxygen content of 18.80%. Compared to absorbents without nitrogen doping, these microporous materials showed a significantly higher CO2 uptake (up to 4.58 mmol g−1 at 25 °C) under ambient conditions. Notably, nitrogen doping allowed a higher CO2 to N2 selectivity, which is of utmost importance for CO2 adsorption from flue gases containing N2 as a major component.
To further assess the environmental impact of transforming waste PET bottles into activated carbons for CO2 adsorption, a life cycle assessment was performed by Li and co-workers.58 According to the analysis, PET absorbents require a large amount of energy (5481 MJ kg−1 of material) and water resources (1261 kg/kg of material) during their lifecycle. CO2 desorption is mainly responsible for high energy consumption. For water resource consumption, adsorbent preparation and virgin PET preparation accounted for 50% and 30% respectively. Despite the high energy consumption and water consumption, the CO2 capture potential of PET absorbents still imposed a negative “Global Warming Potential”, capturing over 2100 kg of CO2 per kilogram of material with the production of only 338 kg of CO2 per kilogram of material throughout its lifecycle.
Other than PET, PU foam is a useful precursor for microporous materials for CO2 capture. The resulting activated carbon products possessed both oxygen and nitrogen functionalities due to their presence in the original PU structure (Fig. 1). This resulted in the formation of pyridine, pyrrole, quaternary N, and pyridine N-oxide species after KOH activation even without the use of urea. According to Fan and coworkers,67 CO2 adsorption due to chemical interactions with these basic nitrogen groups accounted for 20% of the total CO2 adsorption. The sample prepared under optimum conditions showed CO2 adsorption capacities of 6.67 mmol g−1 and 4.33 mmol g−1 at 0 °C and 25 °C under 1 bar, respectively, which was of comparable or higher efficiency than the aforementioned PET-based activated carbons.
Considering that PVC is amongst the world's most produced plastics,68 using PVC as a precursor for the production of microporous CO2 adsorbents is attractive. Zhang and coworkers69 developed a simple yet effective method to form porous carbon spheres for CO2 capture. PVC spheres were first dechlorinated using sodium hydroxide in the presence of tetrabutylammonium bromide as a phase transfer catalyst. Dehalogenation proceeded concurrently with oxidative cross-linking within the sphere70 to transform PVC from a thermoplastic to thermoset (Fig. 3C). Thereafter, pore creation occurred by KOH activation. Because of the spherical nature as compared to previous materials, the final sample prepared possessed a high surface area of up to 1738 m2 g−1 with a well-developed microporous structure, resulting in outstanding CO2 adsorption capacities of 8.93 and 5.47 mmol g−1 at 0 and 25 °C respectively. Compared to 5A zeolites,71 these PVC-derived activated microporous spheres can be regenerated at much lower temperatures of 180 °C.
In contrast to the aforementioned plastics, it is challenging to produce porous carbons from polyolefins due to their low-fixed-carbon content from thermochemical processes such as pyrolysis, which tend to produce more low molecular weight volatile compounds. Nonetheless, very recently, Tour and coworkers demonstrated that pyrolysis of HDPE, LDPE and PP in the presence of potassium acetate salt could yield porous carbons with pore widths of 0.7–1.4 nm for CO2 capture, instead of valueless carbon char.72 Single or mixed polyolefin feedstock could afford sorbents with CO2 capacities of 3.80 mmol g−1 (1 bar CO2, 25 °C), and could be easily regenerated at approximately 75 °C.
2.3 Plastic-derived metal–organic frameworks (MOFs)
MOFs are a class of porous materials built from metal nodes bridged by organic linkers.73 Many MOFs feature enormous internal surface areas from 1000 to 10000 m2 g−1 and permanent nanoporosity,74 and have been intensely studied as materials for CO2 adsorption and separation.75,76 In addition, the functional variability of both the inorganic and organic components also allows for the design of MOFs as catalysts for the activation and conversion of CO2.76,77 The transformation of waste plastics into materials that can be used for the organic component of MOFs would thus be an attractive way to upcycle plastics into materials for CO2 utilization.
The core organic component of many prototypical MOFs is terephthalic acid, which is also a major component of PET. The ability to extract terephthalic acid from PET via simple hydrolysis, which can then be used as an organic linker in MOFs, allows for the use of waste PET as an abundant and accessible MOF feedstock. Indeed, several reports have already demonstrated that several MOFs can be synthesized using PET as a starting material (Fig. 4A).78–82 The synthesis details of making MOFs out of PET derived terephthalate have already been well reviewed by El-Sayed and Yuan,78 and we will therefore focus on reports with specific applications towards CO2 capture and utilization.
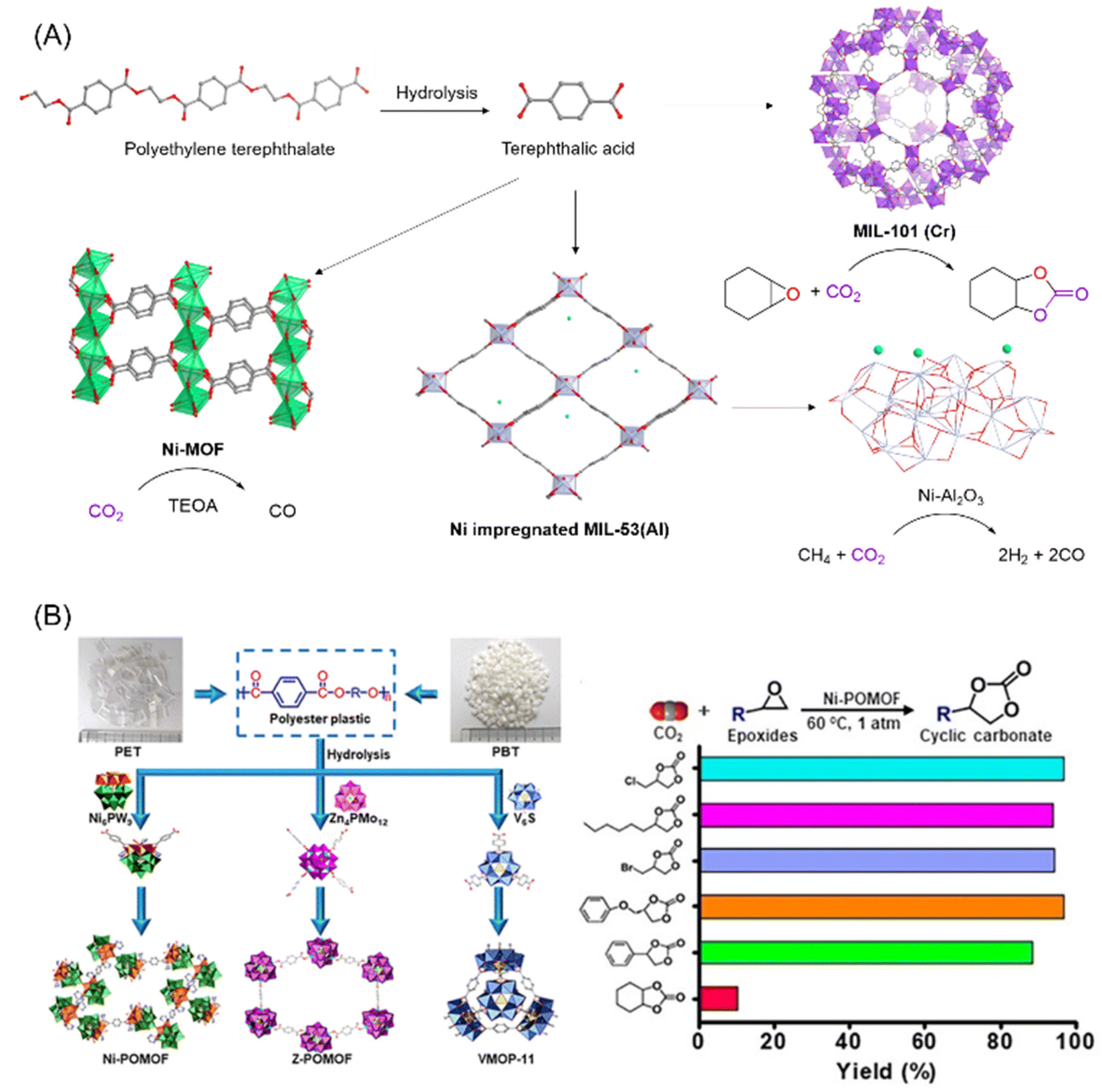 | ||
| Fig. 4 (A) Schematic illustration of the various routes for transforming PET into MOFs, and the catalytic activity of the MOFs for the utilization of CO2, figures generated from published CIF files.84–86 (B) Conversion of PET/PBT into POMOFs and catalytic performance of the Ni-POMOF for CO2 utilisation into cyclic carbonates. This figure has been published in CCS Chemistry 2019; “Polyoxometalate-Induced Efficient Recycling of Waste Polyester Plastics into Metal–Organic Frameworks”,83 and is available online at 10.31635/ccschem.019.20190028 (https://www.chinesechemsoc.org/doi/10.31635/ccschem.019.20190028). | ||
In 2016, Lo and coworkers demonstrated that waste PET pieces could be directly used as a terephthalate source for various MOFs known to adsorb CO2, namely MIL-47, MIL-53 with different metals (Al, Cr and Ga) and MIL-101(Cr).81 Using their reported synthetic procedures, waste PET could be hydrolysed in situ and thus transformed directly into the MOF in the presence of a metal precursor in good yield, with quantitative utilization of the PET in the cases of MIL-47, MIL-53(Ga) and MIL-101(Cr). CO2 adsorption of the PET derived MIL-53(Cr) and MIL-101(Cr) was measured and found to be comparable to CO2 adsorption by MOFs synthesized from commercial terephthalic acid. In addition, the PET derived MIL-47 and MIL-101(Cr) demonstrated good catalytic activity for the synthesis of cyclic carbonates from CO2 and epoxides, using tetrabutylammonium bromide as a cocatalyst.
More recently, Song and coworkers reported the upcycling of waste materials into Ni MOFs which were effective photocatalysts for the reduction of CO2.79 Impressively, both the metal and the organic component were derived from waste materials, the metal source being electroplating sludge and the terephthalate again coming from PET. The authors then compared catalytic activity of the waste material derived MOFs to those synthesized using materials from commercial sources. The photocatalytic reduction of CO2 to CO, using [Ru(bpy)3]2+ as a photosensitizer and triethanol amine as an electron donor, was investigated and it was found that the Ni MOFs derived from waste materials performed just as well as the Ni MOFs derived from commercial sources, and neither using terephthalate derived from PET nor nickel from electroplating sludge appeared to have any detrimental effect on catalytic activity.
Terephthalic acid obtained from polyesters such as PET or poly(butylene terephthalate) (PBT) can also be combined with polyoxometalates to form polyoxometalate-based metal–organic frameworks (POMOF) (Fig. 4B).83 These POMOFs, which exhibited high thermal and chemical stability, could be synthesized in one pot directly from PET/PBT in the presence of POMs under hydrothermal conditions. One of the nickel based POMOF, PET-Ni-POMOF, was tested as the catalyst for the cycloaddition of epoxides and CO2 into cyclic carbonates. Unlike most MOFs which require a co-catalyst to activate CO2, POMOF alone is sufficient to catalyse the reaction for a wide range of epoxides with conversions mostly above 95% (Fig. 4B). The POMOF catalyst was found to be structurally robust enough to withstand 10 cycles of repeated recycling, remaining structurally intact with negligible loss of catalytic performance. Other than pure CO2, using an N2/CO2 mixture which simulated flue gas from coal-powered plants could still afford high cyclic carbonate yields of up to 82.5%, showing the prospect to be used in industry for CO2 capture and utilisation.
Beyond using the MOFs directly, materials derived from MOFs have also been demonstrated to be useful in the utilization of CO2. Recently, Karam and coworkers reported the synthesis of MIL-53(Al) using terephthalic acid from PET waste, after which the MOF was impregnated with a NiII solution, calcined in air to convert the MIL-53(Al) to porous Al2O3, and treated with H2 at 800 °C to reduce NiII to Ni0.80 A similar procedure was carried out with MIL-53(Al) synthesized from commercial terephthalic acid. The obtained materials were then tested for the catalytic conversion of CO2 and CH4 to H2 and CO, also known as methane dry reforming. Notably, although the terephthalate itself is not part of the final material used in the catalysis, its initial use to make MIL-53(Al) is important to give the final Al2O3 a porous morphology, as the authors demonstrate that using commercial Al2O3 directly as a substrate lead to poor catalytic activity. Furthermore, a comparison of the MIL-53(Al) derived materials showed that the materials obtained from PET-derived MIL-53(Al) showed similar reactivity to the materials derived from commercial sources.
3. Conclusions and perspectives
Our aforementioned survey of the literature has revealed considerable potential in upcycling existing commodity plastics into materials for CCU, which is arguably only currently in its infancy. With rising concentrations of anthropogenic CO2 in the atmosphere, new and diverse strategies are urgently needed to reduce its resulting environmental and societal impacts. Although various porous materials are known to function effectively as CO2 adsorbents, materials which can also convert this useful C1 feedstock into useful chemicals represent an attractive means for CO2 valorisation by keeping them within circular chemical loops. Indeed, giving used commodity plastics a new lease of life as CCU materials could represent an attractive means to mitigate the global waste plastic problem, whilst simultaneously contributing to CO2 removal as alternative materials to potentially replace the non-ideal incumbent alkanolamine solutions. While polymeric adsorbents from waste plastics allow for a high degree of bottom-up designability for selective and high CO2 uptake, their often relatively complicated synthesis can make scaling-up difficult. Porous carbon materials generally possess high physicochemical stability, but their CO2 selectivity and adsorption capacities are often poor. Although MOFs can allow CO2 selectivity, high adsorption capacities and are versatile in applications (i.e. allowing CO2 utilisation to be also engineered), their large-scale production can be challenging and costly. Indeed, there is much room for further exploration with only PET, PVC, PU and PS amenable for conversion to materials for CCU now, up to 70% by mass of the available commodity waste plastics remain to be exploited. This section will outline some prospective future research directions in this burgeoning and highly-interdisciplinary field.Of all the waste plastics produced today, polyolefins (comprising polyethylenes and polypropylene) are the most abundant, making up close to 50% of the world's plastic waste (Fig. 1).19 Despite this, their upcycling into materials for CCU is heavily underexplored. As they comprise solely of highly-unreactive C–C or C–H bonds, post-synthetic functionalisation with CO2-philic amine groups to form polymeric adsorbents is highly challenging. Due to their low fixed carbon content, they are often unsuitable for conversion to porous carbon nanomaterials. Moreover, they also lack electronegative heteroatoms that confer polar sites to aid in CO2 adsorption. With recent advances in post-synthetic polyolefin C–H activation chemistry, however, it is now possible to incorporate hydroxyls, carbonyls87–90 and amines91,92 directly onto these polyolefins (Fig. 5). Other than allowing easy grafting of other functional units (e.g. polyamines) through these versatile groups to form polymeric adsorbents, these functionalised polyolefins could be further activated with KOH to form activated carbon materials for CO2 capture. Indeed, considering the vast scale of polyolefin production, lack of feasible alternatives to replace them in the foreseeable future and their popularity as single-use packaging materials,30 effective utilisation of polyolefins may be poised to bring about the greatest impact in waste plastic valorisation efforts.
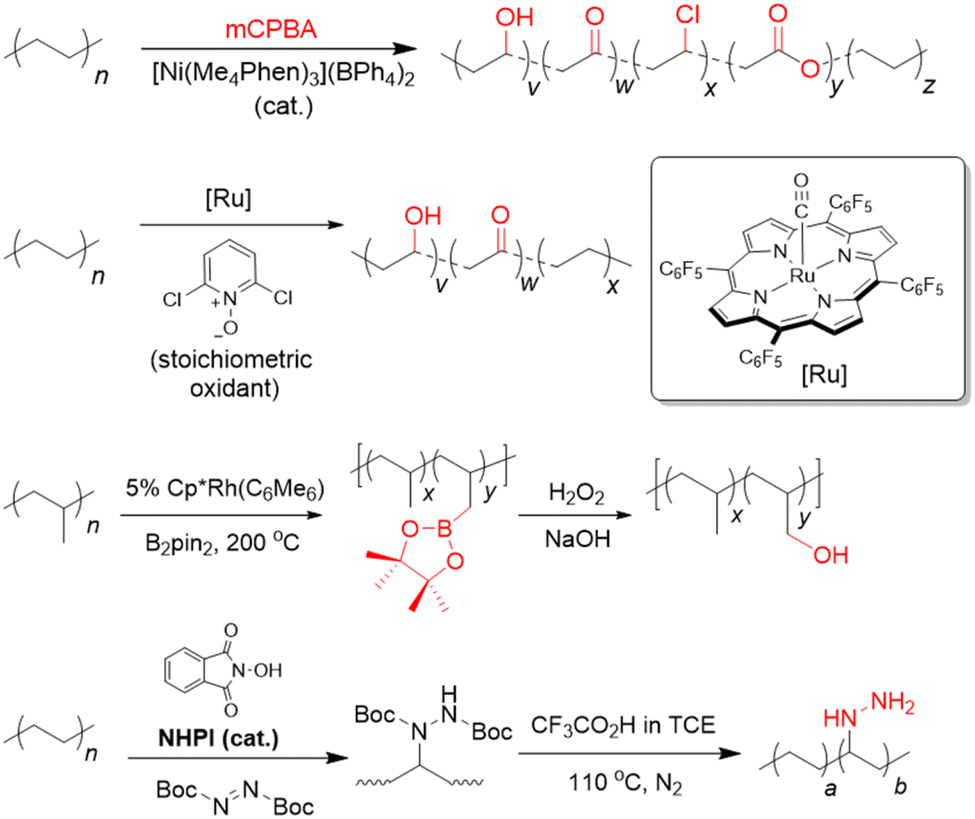 | ||
| Fig. 5 Strategies for post-synthetic C–H functionalisation of polyolefins with oxygen- or nitrogen-containing groups.90,91,93,94 | ||
For PET-derived MOFs, it is clear that sourcing terephthalic acid directly from PET had no detrimental effect on the performance of the resulting materials, thus making PET upcycling into MOFs indeed a promising route for using waste plastics in CO2 utilization. However, an inherent limitation is that only MOFs built from terephthalic acid are accessible, while one of the aspects of MOFs that make them versatile is the ability to vary and tune the functional groups on the organic linker. For instance, the simple inclusion of an amino group on the linker has been shown to greatly improve the adsorption of CO2 by MIL-5395 and UiO-66.96,97 Furthermore, the addition of an amine group can also be used to tune the bandgap of MOFs,98 and thus tune their photocatalytic activities for CO2 utilisation.99 We thus envision that the next step in the upcycling of plastics into functional MOFs would require further chemical functionalisation of the resulting terephthalic acid. Other than PET, MOFs may also be synthesized from other types of plastics. Poly(ethylene 2,5-furandicarboxylate) (PEF), which is growing in importance as a biomass-derived replacement for PET,100 can be a source of 2,5-furandicarboxylic acid that can be used to synthesize analogous MOFs. Indeed, MOFs derived from 2,5-furandicarboxylic acid such as MIL-160 have already been demonstrated to be promising materials for CO2 adsorption and separation.101,102 Although only the aromatic dicarboxylic acid component of PET and PEF are useful for MOF synthesis, the ethylene glycol component can conceivably also be upcycled into other chemical feedstocks, for instance, diformates through electro-reforming.103 In addition, a mixed-linker MOF with terephthalate and lactate ligands was synthesized directly from waste poly(lactic acid) (PLA) and terephthalic acid recently.104 Although not demonstrated in reported work, the resulting lactate-containing MOFs could be exploited for CO2 capture. Furthermore, the use of chiral lactate linkers derived from PLAs of different tacticities could in principle also afford enantioselective CO2 conversion to chiral cyclic carbonates. Although the global market for these chemicals is small, one should not undermine their contributions, as their high value can help monetise CCU and still contribute towards CO2 utilisation. In order to meet negative CO2 emission targets and turn CCU into a financially-lucrative endeavour, many large volume CO2 sinks and products need to be prioritised in the grand scheme of things.
There are other possible applications of plastic-derived materials for CCU other than the aforementioned that can and should be explored. Terephthalate-based MOFs, which can be synthesized directly from waste PET, can also be studied for electrocatalysis. For instance, MIL-53 (Al) (not PET-derived) was shown to electrochemically reduce CO2 to CO and formic acid up to a total of 40% faradaic efficiency.105 Two-dimensional porous structures could also be derived from waste plastics that allow CO2 utilisation-one may take inspiration from a 2D Cu-terephthalate material that could reduce CO2 to ethylene with up to 50% faradaic efficiency.106 The use of PET-derived terephthalate-based MOFs for photocatalytic CO2 reduction is also ready for further development, with more one-pot synthesis protocols of diverse MOF photocatalysts directly from PET necessary to be developed. Other than Song's PET-derived example of photocatalytic reduction of CO2 to CO,79 numerous other terephthalate-based MOFs such as MIL-125(Ti), MIL-88(Fe), MIL-53(Fe), MIL-101(Fe), and their amine functionalised counterparts, can be studied, as these have displayed photocatalytic activity in reducing CO2 to formate.107 Not only is waste plastic cheaper than pure ligand feedstock, the formation of MOF in situ effectively bypasses the purification of products from waste plastic depolymerisation, further saving costs in energy.
Although waste-plastic-derived porous carbons are effective CO2 adsorbents, their use in CO2 utilisation has thus far not been explored. The interested reader may take inspiration from porous carbons derived from biomass sources. For instance, Yang et al. very recently demonstrated the preparation of N-doped porous carbons from chitosan and NaNH2 under solvent-free conditions, with the NaNH2 functioning both as a nitriding agent and porogen.108 Other than showing above-average CO2 adsorption capacity, the resulting porous carbons could be used as a promoter for the N-formylation of a range of amines with CO2 (10 bar) in the presence of a hydrosilane. These N-doped porous carbons functioned both as a CO2 adsorbent, which increased the gas’ accessible reactive concentration in the liquid phase, and as CO2 activator at the Lewis basic N-sites within the material. Conceivably, the chitosan used in this work could be replaced by N-containing plastics such as polyurethanes or polyamides (e.g. nylons), with nitrogen content further increased using NaNH2 in the resulting porous materials. The usage of plastics-derived porous carbons for CO2 utilisation would vastly improve the application space for these versatile materials, ideally under benign reaction conditions and low CO2 pressures (i.e. 1 bar).
One must also consider the end of life for these waste plastic derived structures, as difficult recyclability ultimately undermines the goal of material circulation. It is our view that the post-synthesis functionalisation of commodity plastics actually facilitates their chemical degradation, as this introduces more reactive functional groups which could aid in catalyst coordination for catalytic polymer cleavage. Ultimately, these functional polymers could be broken down into small molecule carboxylic acids which can be fed back into the chemical industry through processes such as photoreforming109 or Fenton oxidation.110 MOFs, on the other hand, could in principle be recycled by first breaking them down to their constituent metal ions and ligands, then re-synthesizing them again to form pristine MOFs for CCU.111 While porous activated carbons cannot be recycled, they are in essence fly ash that could be buried with existing waste management protocols.
Other than the three main classes of plastic-derived materials that we have discussed, new avenues for CCU may be envisaged using waste plastics. One possibility is their usage as scaffolds for the biofixation of CO2. For instance, modified polystyrene (PS) could enhance the activity of bacteria Actinobacillus succinogenes when added to the culture broth.112 Owing to the high absorbance of amine-functionalized PS microspheres, CO2 supplied to the bacteria was increased, enhancing the bioproduction of succinate from CO2 by 1.6 times. Various algae–plastic composites were also developed in recent years, allowing algae produced from CO2 biofixation to be utilised effectively.113–115
Thus far, the synthesis of each class of plastic-derived CCU materials utilises single-component plastic feedstock. This is inevitable for the synthesis of polymeric adsorbents and MOFs, as the chemistry involved is inherently specific to each class of plastics used. For instance, the electrophilic substitution chemistry for PS upcycling is unsuitable for PVC, and vice versa. For MOFs, the presence of other contaminants within the PET plastic feedstock could negatively impact the crystallinity and accessible pore space of the derived MOFs. A well-established collection and sorting infrastructure exists predominantly for PET,116 one of the most recycled types of plastic worldwide mainly from used beverage bottles, and is lacking for other plastic types, such as PS, which forms a major component of municipal waste. Waste plastic sorting is currently a bottleneck of plastic valorisation efforts around the world, and requires concerted and dedicated efforts from legislators, companies and individuals. Nevertheless, we view efforts to valorise waste plastics for different applications, including for CCU described herein, as providing the necessary incentives to drive further advances in waste plastic sorting technologies. A feasible way to bypass the arduous sorting is to use mixed plastic feedstock for generating porous carbon materials for CO2 capture, though rarely demonstrated at present. Recently, porous carbon nanosheets synthesized by catalytic carbonization of a mixture of PE, PP, PS, PET and PVC on organically-modified montmorillonite have been reported to show excellent CO2 adsorption behaviour.117 More emphasis on the use of mixed plastic feedstock for producing porous carbon nanomaterials should take place.
Although giving waste plastics a second life for CCU has obvious merits from a sustainability standpoint, it is also important to consider the environmental trade-offs holistically, especially across the life cycles of the resulting CCU materials. In this regard, performing life cycle assessments (LCA) will be very helpful in evaluating their environmental impacts, and identify the key areas of improvement for greater sustainability.118 Important processes which need to be considered include sorting and transportation of waste plastics, the actual material synthesis parameters (i.e. temperature, duration, use of solvents), energy required for material regeneration (i.e. CO2 desorption), as well as additional wastes generated during waste plastic conversion, which include solvents and side products from reactions. The waste generated from these processes can be substantial, especially if multiple steps are required for sorbent material production from waste plastics, and can outweigh the benefits of plastics upcycling. To the best of our knowledge, other than activated carbons from waste PET,58 comprehensive environmental assessments have not yet been performed for other CCU materials such as polymeric adsorbents and MOFs from waste plastics. Indeed, only materials which can achieve negative CO2 emissions should be considered. As outlined by Ramírez, to qualify for negative emissions, gases removed from the atmosphere which are permanently stored should be greater than the amount of emissions associated with the removal and storage process.119 To minimise the environmental impact of such materials, environmentally-hazardous solvents (e.g. chlorinated solvents and aromatic hydrocarbons)120 should be avoided, or even better, solvent-free upcycling methods should be developed, processes should be streamlined to as few steps as practically achievable, and usage of distributed renewable sources of energy (e.g. solar power or solar heating) should be considered wherever possible.
The aforementioned plastic-derived CCU materials have thus far been demonstrated to work effectively on the lab-scale. However, for any practical implementation to be possible, a number of considerations need to be factored in, which include performance in the presence of moisture, long-term physicochemical stability and tolerance to impurities (which can deactivate catalysts for CO2 utilisation), cost of material synthesis and fabrication into the correct form-factor for application, ease of scalability and ease of materials regeneration. In order to justifiably replace the incumbent commercial amine-based solutions for carbon capture, alternatives need to offer superior practical performances with minimal differences in cost. Despite preliminary research showing enhanced performance for porous materials, they currently lack validation at large practical scales, unlike amine-based solutions (e.g. the Shell CANSOLV® CO2 Capture System).121 While plastic-derived porous carbons may be potentially cost-competitive, polymeric adsorbents and MOFs are disadvantageous in this respect, despite their often superior CO2 uptake capacities and selectivity. However, these economics may soon change with new capabilities being developed for large-scale MOF synthesis.122 Production prices are slated to drop as well, and prices as low as $10 per kg have been estimated for MIL-160(Al),123 a 2,5-furan-dicarboxylate-based MOF which could in principle be produced from PEF.
Upcycling waste plastics into materials for CCU offers new opportunities to amalgamate these apparently disparate fields of research. As an extremely low-cost resource, the use of waste plastic feedstock could substantially lower the cost of materials production, whilst simultaneously reducing our reliance on petroleum-derived chemicals for their production. Indeed, the structural diversity of waste commodity plastics offers a vast palette for innovative materials design for CCU. Other than the most common commodity plastics, one may also consider niche polymers, for instance engineering thermoplastics such as polyether ether ketone (PEEK) as starting materials, which provide excellent mechanical strength. Even next-generation bio-based plastics such as polyhydroxyalkanoates may be conceivably exploited. Other than CCU from post-combustion point sources, it is our hope that ultimately, some of these plastic-derived materials will be able to achieve the ambitious target of direct air capture of CO2, which will be a significant breakthrough both for achieving both a circular materials economy and a zero-carbon society.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the A*STAR Central Research Fund (CRF) for generous funding in support of this work.Notes and references
- H. Ritchie, M. Roser and P. Rosado, CO2 and Greenhouse Gas Emissions, https://ourworldindata.org/co2-and-other-greenhouse-gas-emissions, (accessed 23 Apr 2022, 2022).
- J. Skea, P. R. Shukla, A. Reisinger, R. l. Slade, M. Pathak, A. Al Khourdajie, R. van Diemen, A. Abdulla, K. Akimoto, M. Babiker, Q. Bai, I. Bashmakov, C. Bataille, G. Berndes, G. Blanco, K. Blok, M. Bustamante, E. Byers, L. F. Cabeza, K. Calvin, C. Carraro, L. Clarke, A. Cowie, F. Creutzig, D. K. Dadi, D. Dasgupta, H. de Coninck, F. Denton, S. Dhakal, N. K. Dubash, O. Geden, M. Grubb, C. Guivarch, S. Gupta, A. Hahmann, K. Halsnaes, P. Jaramillo, K. Jiang, F. Jotzo, T. Y. Jung, S. K. Ribeiro, S. Khennas, Ş. Kılkış, S. Kreibiehl, V. Krey, E. Kriegler, W. Lamb, F. Lecocq, S. Lwasa, N. Mahmoud, C. Mbow, D. McCollum, J. C. Minx, C. Mitchell, R. Mrabet, Y. Mulugetta, G.-J. Nabuurs, G. Nemet, P. Newman, L. Niamir, L. J. Nilsson, S. B. Nugroho, C. Okereke, S. Pachauri, A. Patt, R. Pichs-Madruga, J. P. Pereira, L. Rajamani, K. Riahi, J. Roy, Y. Saheb, R. Schaeffer, K. Seto, S. Some, L. Steg, F. L. Toth, D. Ürge-Vorsatz, D. van Vuuren, E. Verdolini, P. Vyas, Y.-M. Wei, M. Williams and H. Winkler, Climate Change 2022: Mitigation of Climate Change, Intergovernmental Panel on Climate Change (IPCC), 2022, https://report.ipcc.ch/ar6wg3/pdf/IPCC_AR6_WGIII_FinalDraft_FullReport.pdf Search PubMed.
- IPCC, Global Warming of 1.5 °C.An IPCC Special Report on the impacts of global warming of 1.5 °C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty, ed. V. Masson-Delmotte, P. Zhai, H.-O. Pörtner, D. Roberts, J. Skea, P.R. Shukla, A. Pirani, W. Moufouma-Okia, C. Péan, R. Pidcock, S. Connors, J. B. R. Matthews, Y. Chen, X. Zhou, M. I. Gomis, E. Lonnoy, T. Maycock, M. Tignor and T. Waterfield, Cambridge University Press, Cambridge, UK and New York, NY, USA, 2018, p. 616, https://doi.org/10.1017/9781009157940 Search PubMed.
- P. K. K. S. Heer, K. M. Khot and V. G. Gaikar, Sep. Purif. Technol., 2016, 158, 212–222 CrossRef CAS.
- T. C. Drage, C. E. Snape, L. A. Stevens, J. Wood, J. Wang, A. I. Cooper, R. Dawson, X. Guo, C. Satterley and R. Irons, J. Mater. Chem., 2012, 22, 2815–2823 RSC.
- J. E. Bara, Greenhouse Gases: Sci. Technol., 2012, 2, 162–171 CrossRef CAS.
- M. L. Gray, K. J. Champagne, D. Fauth, J. P. Baltrus and H. Pennline, Int. J. Greenhouse Gas Control, 2008, 2, 3–8 CrossRef CAS.
- L. Wang, Y. Yang, W. Shen, X. Kong, P. Li, J. Yu and A. E. Rodrigues, Chem. Eng. Sci., 2013, 101, 615–619 CrossRef CAS.
- S.-Y. Lee and S.-J. Park, J. Ind. Eng. Chem., 2015, 23, 1–11 CrossRef CAS.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- P. Gabrielli, M. Gazzani and M. Mazzotti, Ind. Eng. Chem. Res., 2020, 59, 7033–7045 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- A. D. N. Kamkeng, M. Wang, J. Hu, W. Du and F. Qian, Chem. Eng. J., 2021, 409, 128138 CrossRef CAS.
- H.-C. J. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- P. Wu, Y. Li, J.-J. Zheng, N. Hosono, K.-i. Otake, J. Wang, Y. Liu, L. Xia, M. Jiang, S. Sakaki and S. Kitagawa, Nat. Commun., 2019, 10, 4362 CrossRef PubMed.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- X. Zhao, B. Boruah, K. F. Chin, M. Đokić, J. M. Modak and H. S. Soo, Adv. Mater., 2021, 2100843 Search PubMed , n/a.
- C. W. S. Yeung, J. Y. Q. Teo, X. J. Loh and J. Y. C. Lim, ACS Mater. Lett., 2021, 3, 1660–1676 CrossRef CAS.
- C. Zhuo and Y. Levendis, J. Appl. Polym. Sci., 2014, 131 DOI:10.1002/app.39931.
- C. W. S. Yeung, W. W. Loh, H. H. Lau, X. J. Loh and J. Y. C. Lim, Mater. Today Chem., 2021, 21, 100524 CrossRef CAS.
- J. B. Williamson, S. E. Lewis, R. R. Johnson III, I. M. Manning and F. A. Leibfarth, Angew. Chem., Int. Ed., 2019, 58, 8654–8668 CrossRef CAS PubMed.
- E. Bäckström, K. Odelius and M. Hakkarainen, ACS Sustainable Chem. Eng., 2019, 7, 11004–11013 CrossRef.
- S. Gazi, M. Đokić, K. F. Chin, P. R. Ng and H. S. Soo, Adv. Sci., 2019, 6, 1902020 CrossRef CAS PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- . A New Industrial Revolution for Plastics, https://www.usda.gov/media/blog/2018/09/19/new-industrial-revolution-plastics, (accessed 16 Jun 2022, 2022).
- World Economic Forum, Ellen MacArthur Foundation and McKinsey & Company, The New Plastics Economy: Rethinking the future of plastics, Ellen MacArthur Foundation, 2016, http://www.ellenmacarthurfoundation.org/publications Search PubMed.
- L. Lebreton and A. Andrady, Palgrave Commun., 2019, 5, 6 CrossRef.
- H. M. Lee, I. S. Youn, M. Saleh, J. W. Lee and K. S. Kim, Phys. Chem. Chem. Phys., 2015, 17, 10925–10933 RSC.
- N. T. T. Nguyen, H. Furukawa, F. Gándara, H. T. Nguyen, K. E. Cordova and O. M. Yaghi, Angew. Chem., Int. Ed., 2014, 53, 10645–10648 CrossRef CAS PubMed.
- S. Valluri, V. Claremboux and S. Kawatra, J. Environ. Sci., 2022, 113, 322–344 CrossRef PubMed.
- E. Alper and O. Yuksel Orhan, Petroleum, 2017, 3, 109–126 CrossRef.
- A. M. Al-Sabagh, F. Z. Yehia, G. Eshaq, A. M. Rabie and A. E. ElMetwally, Egypt. J. Pet., 2016, 25, 53–64 CrossRef.
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- A. Yasuhara, T. Katami and T. Shibamoto, Bull. Environ. Contam. Toxicol., 2005, 74, 899–903 CrossRef CAS PubMed.
- J. M. García, Chem, 2016, 1, 813–815 Search PubMed.
- S. Kumagai, S. Hirahashi, G. Grause, T. Kameda, H. Toyoda and T. Yoshioka, J. Mater. Cycles Waste Manage., 2018, 20, 439–449 CrossRef CAS.
- Y. Takeshita, K. Kato, K. Takahashi, Y. Sato and S. Nishi, J. Supercrit. Fluids, 2004, 31, 185–193 CrossRef CAS.
- B. Ellis and R. Smith, Polymers: A Property Database, CRC Press, Florida, U.S.A., 2nd edn, 2009 Search PubMed.
- Y. Takeshita, K. Kato, K. Takahashi, Y. Sato and S. Nishi, J. Supercrit. Fluids, 2004, 31, 185–193 CrossRef CAS.
- M. Kaliva, G. S. Armatas and M. Vamvakaki, Langmuir, 2012, 28, 2690–2695 CrossRef CAS PubMed.
- Z. Fu, J. Jia, J. Li and C. Liu, Chem. Eng. J., 2017, 323, 557–564 CrossRef CAS.
- Z. Fu, I. M. A. Mohamed, J. Li and C. Liu, J. Taiwan Inst. Chem. Eng., 2019, 97, 381–388 CrossRef CAS.
- C. Flouraki, M. Kaliva, I. T. Papadas, G. S. Armatas and M. Vamvakaki, Polym. Chem., 2016, 7, 3026–3033 RSC.
- G. Sneddon, J. C. McGlynn, M. S. Neumann, H. M. Aydin, H. H. P. Yiu and A. Y. Ganin, J. Mater. Chem. A, 2017, 5, 11864–11872 RSC.
- W. R. Alesi and J. R. Kitchin, Ind. Eng. Chem. Res., 2012, 51, 6907–6915 CrossRef CAS.
- D. H. Jo, C. H. Lee, H. Jung, S. Jeon and S. H. Kim, Bull. Chem. Soc. Jpn., 2015, 88, 1317–1322 CrossRef CAS.
- D. R. Merchán-Arenas and A. F. Murcia-Patiño, Int. J. Environ. Sci. Technol., 2021, 18, 2519–2532 CrossRef.
- Y. Wu, L. Li, W. Yang, S. Feng and H. Liu, RSC Adv., 2015, 5, 12987–12993 RSC.
- G. Sneddon, J. C. McGlynn, M. S. Neumann, H. M. Aydin, H. H. P. Yiu and A. Y. Ganin, J. Mater. Chem. A, 2017, 5, 11864–11872 RSC.
- F. Zeman, Environ. Sci. Technol., 2007, 41, 7558–7563 CrossRef CAS PubMed.
- L. Lu, H. Zhong, T. Wang, J. Wu, F. Jin and T. Yoshioka, Green Chem., 2020, 22, 352–358 RSC.
- M. Inagaki, in New Carbons - Control of Structure and Functions, ed. M. Inagaki, Elsevier Science, Oxford, 2000, pp. 124–145, DOI:10.1016/B978-008043713-2/50005-4.
- B. Kaur, R. K. Gupta and H. Bhunia, Microporous Mesoporous Mater., 2019, 282, 146–158 CrossRef CAS.
- J. Wang, X. Yuan, S. Deng, X. Zeng, Z. Yu, S. Li and K. Li, Green Chem., 2020, 22, 6836–6845 RSC.
- A. Arenillas, F. Rubiera, J. B. Parra, C. O. Ania and J. J. Pis, Appl. Surf. Sci., 2005, 252, 619–624 CrossRef CAS.
- M. Adibfar, T. Kaghazchi, N. Asasian and M. Soleimani, Chem. Eng. Technol., 2014, 37, 979–986 CrossRef CAS.
- D. Tiwari, C. Goel, H. Bhunia and P. K. Bajpai, J. Environ. Manage., 2017, 197, 415–427 CrossRef CAS PubMed.
- R. Bai, M. Yang, G. Hu, L. Xu, X. Hu, Z. Li, S. Wang, W. Dai and M. Fan, Carbon, 2015, 81, 465–473 CrossRef CAS.
- T. J. Bandosz, M. Seredych, E. Rodríguez-Castellón, Y. Cheng, L. L. Daemen and A. J. Ramírez-Cuesta, Carbon, 2016, 96, 856–863 CrossRef CAS.
- X. Yuan, S. Li, S. Jeon, S. Deng, L. Zhao and K. B. Lee, J. Hazard. Mater., 2020, 399, 123010 CrossRef CAS PubMed.
- W. Chen, H. Yang, Y. Chen, X. Chen, Y. Fang and H. Chen, J. Anal. Appl. Pyrolysis, 2016, 120, 186–193 CrossRef CAS.
- K. Li, W. Chen, H. Yang, Y. Chen, S. Xia, M. Xia, X. Tu and H. Chen, Bioresour. Technol., 2019, 280, 260–268 CrossRef CAS PubMed.
- C. Ge, J. Song, Z. Qin, J. Wang and W. Fan, ACS Appl. Mater. Interfaces, 2016, 8, 18849–18859 CrossRef CAS PubMed.
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef CAS PubMed.
- J. Wang, F. Wang, H. Duan, Y. Li, J. Xu, Y. Huang, B. Liu and T. Zhang, ChemSusChem, 2020, 13, 6426–6432 CAS.
- B. Saethre and M. Gilbert, Polymer, 1996, 37, 3379–3386 CrossRef CAS.
- Z. Song, Q. Dong, W. L. Xu, F. Zhou, X. Liang and M. Yu, ACS Appl. Mater. Interfaces, 2018, 10, 769–775 CrossRef CAS PubMed.
- W. A. Algozeeb, P. E. Savas, Z. Yuan, Z. Wang, C. Kittrell, J. N. Hall, W. Chen, P. Bollini and J. M. Tour, ACS Nano, 2022, 16, 7284–7290 CrossRef CAS PubMed.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- H. Furukawa, E. Cordova Kyle, M. O'Keeffe and M. Yaghi Omar, Science, 2013, 341, 1230444 CrossRef PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- M. Ding, R. W. Flaig, H. L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- I. I. Alkhatib, C. Garlisi, M. Pagliaro, K. Al-Ali and G. Palmisano, Catal. Today, 2020, 340, 209–224 CrossRef CAS.
- E.-S. M. El-Sayed and D. Yuan, Green Chem., 2020, 22, 4082–4104 RSC.
- K. Song, X. Qiu, B. Han, S. Liang and Z. Lin, Environ. Sci.: Nano, 2021, 8, 390–398 RSC.
- L. Karam, A. Miglio, S. Specchia, N. El Hassan, P. Massiani and J. Reboul, Mater. Adv., 2021, 2, 2750–2758 RSC.
- S. H. Lo, D. Senthil Raja, C. W. Chen, Y. H. Kang, J. J. Chen and C. H. Lin, Dalton Trans., 2016, 45, 9565–9573 RSC.
- D. Panda, S. Patra, M. K. Awasthi and S. K. Singh, J. Chem. Educ., 2020, 97, 1101–1108 CrossRef CAS.
- Y.-J. Chen, X. Huang, Y. Chen, Y.-R. Wang, H. Zhang, C.-X. Li, L. Zhang, H. Zhu, R. Yang, Y.-H. Kan, S.-L. Li and Y.-Q. Lan, CCS Chem., 2019, 1, 561–570 CrossRef CAS.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- O. I. Lebedev, F. Millange, C. Serre, G. Van Tendeloo and G. Férey, Chem. Mater., 2005, 17, 6525–6527 CrossRef CAS.
- A. Mesbah, P. Rabu, R. Sibille, S. Lebègue, T. Mazet, B. Malaman and M. François, Inorg. Chem., 2014, 53, 872–881 CrossRef CAS PubMed.
- A. Bunescu, S. Lee, Q. Li and J. F. Hartwig, ACS Cent. Sci., 2017, 3, 895–903 CrossRef CAS PubMed.
- D. Kong, L. Wang, X. Chen, W. Xia, L. Su, F. Zuo, Z. Yan, S. Chen and J. Wu, Green Chem., 2022, 24, 2203–2211 RSC.
- C. Bae, J. F. Hartwig, H. Chung, N. K. Harris, K. A. Switek and M. A. Hillmyer, Angew. Chem., Int. Ed., 2005, 44, 6410–6413 CrossRef CAS PubMed.
- C. Bae, J. F. Hartwig, N. K. Boaen Harris, R. O. Long, K. S. Anderson and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 767–776 CrossRef CAS PubMed.
- H. Zhou, S. Wang, H. Huang, Z. Li, C. M. Plummer, S. Wang, W.-H. Sun and Y. Chen, Macromolecules, 2017, 50, 3510–3515 CrossRef CAS.
- H. Zhou, C. M. Plummer, H. Li, H. Huang, P. Ma, L. Li, L. Liu and Y. Chen, Polym. Chem., 2019, 10, 619–626 RSC.
- A. Bunescu, S. Lee, Q. Li and J. F. Hartwig, ACS Cent. Sci., 2017, 3, 895–903 CrossRef CAS PubMed.
- L. Chen, K. G. Malollari, A. Uliana, D. Sanchez, P. B. Messersmith and J. F. Hartwig, Chem, 2021, 7, 137–145 CAS.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS PubMed.
- Z. H. Rada, H. R. Abid, H. Sun and S. Wang, J. Chem. Eng. Data, 2015, 60, 2152–2161 CrossRef CAS.
- J. Ethiraj, E. Albanese, B. Civalleri, J. G. Vitillo, F. Bonino, S. Chavan, G. C. Shearer, K. P. Lillerud and S. Bordiga, ChemSusChem, 2014, 7, 3382–3388 CrossRef CAS PubMed.
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D'Arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- A. F. Sousa, R. Patrício, Z. Terzopoulou, D. N. Bikiaris, T. Stern, J. Wenger, K. Loos, N. Lotti, V. Siracusa, A. Szymczyk, S. Paszkiewicz, K. S. Triantafyllidis, A. Zamboulis, M. S. Nikolic, P. Spasojevic, S. Thiyagarajan, D. S. van Es and N. Guigo, Green Chem., 2021, 23, 8795–8820 RSC.
- W. Fan, Y. Ying, S. B. Peh, H. Yuan, Z. Yang, Y. D. Yuan, D. Shi, X. Yu, C. Kang and D. Zhao, J. Am. Chem. Soc., 2021, 143, 17716–17723 CrossRef CAS PubMed.
- D. Damasceno Borges, P. Normand, A. Permiakova, R. Babarao, N. Heymans, D. S. Galvao, C. Serre, G. De Weireld and G. Maurin, J. Phys. Chem. C, 2017, 121, 26822–26832 CrossRef CAS.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed.
- B. Slater, S.-O. Wong, A. Duckworth, A. J. P. White, M. R. Hill and B. P. Ladewig, Chem. Commun., 2019, 55, 7319–7322 RSC.
- M. Lee, A. De Riccardis, R. V. Kazantsev, J. K. Cooper, A. K. Buckley, P. W. W. Burroughs, D. M. Larson, G. Mele and F. M. Toma, ACS Appl. Energy Mater., 2020, 3, 1286–1291 CrossRef CAS.
- Y. Zhang, Y. Li, Q. Tan, S. Hong and Z. Sun, J. Exp. Nanosci., 2021, 16, 246–254 CrossRef CAS.
- J. W. Maina, C. Pozo-Gonzalo, L. Kong, J. Schütz, M. Hill and L. F. Dumée, Mater. Horiz., 2017, 4, 345–361 RSC.
- C. Yang, T. Zhao, H. Pan, F. Liu, J. Cao and Q. Lin, Chem. Eng. J., 2022, 432, 134347 CrossRef CAS.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- C.-F. Chow, W.-L. Wong, K. Y.-F. Ho, C.-S. Chan and C.-B. Gong, Chem. – Eur. J., 2016, 22, 9513–9518 CrossRef CAS PubMed.
- J. Chu, F.-S. Ke, Y. Wang, X. Feng, W. Chen, X. Ai, H. Yang and Y. Cao, Chem. Commun., 2020, 3, 5 CrossRef CAS.
- W. Zhu, Q. Li and N. Dai, Appl. Biochem. Biotechnol., 2017, 181, 584–592 CrossRef CAS PubMed.
- T. Hirotsu and T. Otsuki, in Algae Based Polymers, Blends, and Composites, ed. K. M. Zia, M. Zuber and M. Ali, Elsevier, 2017, pp. 531–564, DOI:10.1016/B978-0-12-812360-7.00014-8.
- T. Otsuki, F. Zhang, H. Kabeya and T. Hirotsu, J. Appl. Polym. Sci., 2004, 92, 812–816 CrossRef CAS.
- F. Zhang, H. Kabeya, R. Kitagawa, T. Hirotsu, M. Yamashita and T. Otsuki, J. Mater. Sci., 2000, 35, 2603–2609 CrossRef CAS.
- Plastics – the Facts 2021: An analysis of European plastics production, demand and waste data, PlasticsEurope, Brussels, Belgium, 2020 Search PubMed.
- J. Gong, B. Michalkiewicz, X. Chen, E. Mijowska, J. Liu, Z. Jiang, X. Wen and T. Tang, ACS Sustainable Chem. Eng., 2014, 2, 2837–2844 CrossRef CAS.
- ISO 14040:2006 Environmental management—Life cycle assessment—Principles and framework, International Organization for Standardization, 2006 Search PubMed.
- S. E. Tanzer and A. Ramírez, Energy Environ. Sci., 2019, 12, 1210–1218 RSC.
- M. Tobiszewski, J. Namieśnik and F. Pena-Pereira, Green Chem., 2017, 19, 1034–1042 RSC.
- . Chell CANSOLV CO2 Capture System, https://www.shell.com/business-customers/catalysts-technologies/licensed-technologies/emissions-standards/tail-gas-treatment-unit/cansolv-co2.html, (accessed 16 Jun 2022, 2022).
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, Chem. Soc. Rev., 2017, 46, 3453–3480 RSC.
- M. I. Severino, E. Gkaniatsou, F. Nouar, M. L. Pinto and C. Serre, Faraday Discuss., 2021, 231, 326–341 RSC.
Footnote |
| † These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2022 |