
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical recycling of bioplastics: technical opportunities to preserve chemical functionality as path towards a circular economy
Angel L.
Merchan
 d,
Thomas
Fischöder
e,
Johann
Hee
b,
Marcus S.
Lehnertz
g,
Ole
Osterthun
f,
Stefan
Pielsticker
c,
Julia
Schleier
d,
Till
Tiso
a,
Lars M.
Blank
a,
Jürgen
Klankermayer
f,
Reinhold
Kneer
c,
Peter
Quicker
b,
Grit
Walther
d and
Regina
Palkovits
*gh
d,
Thomas
Fischöder
e,
Johann
Hee
b,
Marcus S.
Lehnertz
g,
Ole
Osterthun
f,
Stefan
Pielsticker
c,
Julia
Schleier
d,
Till
Tiso
a,
Lars M.
Blank
a,
Jürgen
Klankermayer
f,
Reinhold
Kneer
c,
Peter
Quicker
b,
Grit
Walther
d and
Regina
Palkovits
*gh
aiAMB – Institute of Applied Microbiology, ABBt – Aachen Biology and Biotechnology, RWTH Aachen University, Worringer Weg 1, D-52074 Aachen, Germany
bTEER - Unit of Technology of Fuels, RWTH Aachen University, Wüllnerstraße 2, D-52062 Aachen, Germany
cWSA - Institute of Heat and Mass Transfer, RWTH Aachen University, Augustinerbach 6, D-52062 Aachen, Germany
dOM - Chair of Operations Management, RWTH Aachen University, Kackertstraße 7, D-52072 Aachen, Germany
eCMT - Center for Molecular Transformations, RWTH Aachen University, Worringerweg 1, D-52074 Aachen, Germany
fChair of Translational Molecular Catalysis, ITMC - Institute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, D-52074 Aachen, Germany
gChair of Heterogeneous Catalysis and Chemical Technology, ITMC - Institute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, D-52074 Aachen, Germany. E-mail: Palkovits@itmc.rwth-aachen.de
hMax-Planck-Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany
First published on 22nd November 2022
Abstract
Plastics have become an integral part of many areas of life. Their high chemical resistance has opened a huge range of applications. At the same time, this creates major challenges for the environment, so those plastic residues can now be found even in the remotest corners of the earth. The goal of the circular economy is to address this challenge by utilizing residual and waste streams as valuable raw materials. With suitable recycling strategies into the original or even added-value applications, driving forces for properly closed carbon cycles become available. In recent years, bioplastics possess tremendous growth rates. Biomass-based, they theoretically enable closed carbon cycles as their carbon atoms are harvested from CO2 by photosynthesis. Despite this advantage, their increasing market penetration must be accompanied by appropriate recycling technologies enabling sustainable and economic utilization of the chemically synthesized building blocks. This minireview addresses current knowledge on the (bio)chemical and thermal recycling of chemically novel bioplastics, that is biomass-based plastics not resembling the chemical structure of well-established fossil-derived plastics; major emphasis is on maintaining chemical functionality of the bioplastic building blocks. Available methodologies for thermal (gasification, pyrolysis), chemo-catalytic (homogeneous and heterogeneous catalysis) and bio-catalytic (enzymatic and whole-cell catalysis) recycling are summarized together with the available insights of LCA studies on recycling strategies for chemically novel biomass-based plastics. The literature review shows that although mechanical recycling presents currently the most attractive technology, LCA studies emphasize the potentially lower environmental impact of chemical recycling compared with other End-of-Life (EoL) solutions. Therefore, early development of viable technologies for chemical recycling in the growing field of biomass-based plastics is of utmost importance.
1. Introduction
Over the past seven decades, plastics have substantially revolutionized our lives. Owing to their excellent properties such as high chemical resistance and their possible application in a wide range of temperatures, it is hard to imagine our everyday life without them. Due to their simple and inexpensive production from fossil raw materials, the demand and production of plastics have increased rapidly.1,2 Worldwide plastic production amounted to almost 370 million tons in 2019 and is still growing, creating global problems, e.g. regarding CO2 emission during production, during conventional End-of-Life (EoL) treatment, and through uncontrolled discharge into the environment.3,4 For this reason, efficient and environmentally friendly recycling strategies for plastic waste are needed. In addition, in the context of defossilization of the energy and chemical sector, the need for biomass-based plastics is growing. Together, the use of biomass-based plastics and efficient recycling strategies allow the closure of the carbon loop (Fig. 1).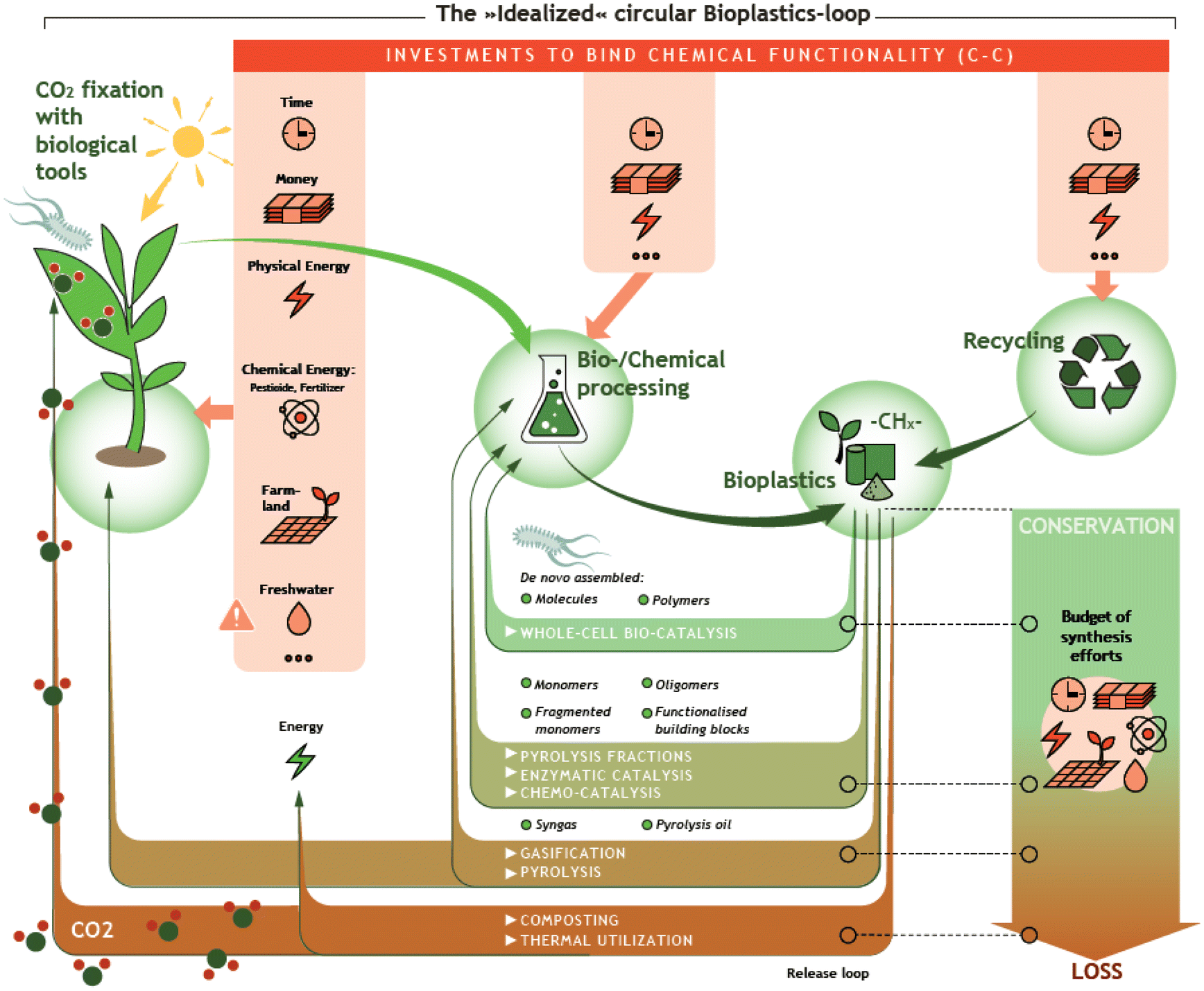 | ||
| Fig. 1 Schematic representation of the vision of an idealized circular Bioplastics loop. | ||
The term bioplastic, according to European Bioplastics, can refer to plastic which is either biomass-based, biodegradable, or features both properties. Accordingly, fossil-derived biodegradable plastics are often also called bioplastics. However, the term bioplastic is limited to biomass-based plastics in this review. Herein, biomass-based plastics (resp. bio-based) comprise a variety of materials that are derived from biomass and can be classified in different ways.
A common classification is made with respect to their biodegradability (e.g. PLA, PHA, PBS, and starch blends) or non-biodegradability (e.g. bio-based PE, PET, PA, and PTT).5 Therein, the development and use of biodegradable bio-based plastics allow for flexible recycling strategies.6 Moreover, biodegradability also presents an emergency mechanism to prevent future pollution from unintentionally released plastics.4,7 A practical alternative classification divides biomass-based plastics into two groups concerning the familiarity of their chemical structure. While drop-in polymers (e.g. bio-PA, bio-PE, bio-PET) hold the same chemical structure and properties as their fossil-based counterparts, chemical novel types (e.g. PLA, PHA) do not have such equivalents and are associated with novel properties.8 Thus, drop-ins apply to the same downstream processes, e.g. application and EoL treatment as their fossil-based counterparts.5,8 Chemical novel types though, usually cannot be integrated into the existing, circular (e.g., material conserving) EoL streams and are focus of this review.9
In 2020, the global production capacities of bioplastics added up to 2.11 Mt with shares of rounded 42% non-biodegradable and 58% biodegradable polymers.5 Yet, with a share of 61% in total, rigid and flexible packaging applications (733 kt and 538 kt, respectively) dominate other fields clearly. The largest parts of production capacity are located in Asia (55%), followed by Europe (18%) and North America (10%).5 Although until now bioplastics represent only 0.5% of the global plastic production (368 Mt), the increasing demand and diversification of the bioplastics market are reflected by increasing production capacities.3 In the upcoming years, the production capacities of bioplastics are forecasted to grow particularly in Europe, Asia, and North America by 132%, 43%, and 26%, respectively, until 2024 (compared to 2019).5 Political decisions like the ban on certain single-use plastics and requirements for utilization of biodegradable plastics in China accelerate the development of bioplastics. An increase in production capacities of 15 Mt of PBAT, 1 Mt of PBS, 3 Mt of PLA, and even some 50 kt of PHA were announced in China in 2021 alone. However, PBAT will be synthesized using fossil resources mainly. Today, bioplastics are represented in all fields of application in which fossil-based plastics are used. Forecasts predict that the trend of applying bioplastics to these short-lived applications is set to increase.5 The increasing use of chemically novel bioplastics emphasizes the urgent need to investigate and establish adequate circular EoL technologies.9
However, the EoL-treatment of plastics (bio-based as well as fossil-based) is not yet fully exploited. According to Plastics Europe, of the total 29.1 Mt of plastic post-consumer waste collected in 2018 in Europe, 42.6% were incinerated, 32.5% recycled, and 24.9% landfilled (cf.Fig. 2).3 However, for the collected post-consumer plastic packaging waste, a higher recycling share of 42% is reported. From the 9.4 Mt of plastic post-consumer waste collected for recycling, 5 Mt of recyclates were produced in Europe, of which 80% were used to produce new goods within Europe, and 20% were exported outside of Europe. However, these figures still do not match the requirements of the EU directive 2018/852, which demands that at least 55 wt% of annual plastic packaging waste needs to be recycled by the end of 2030.10
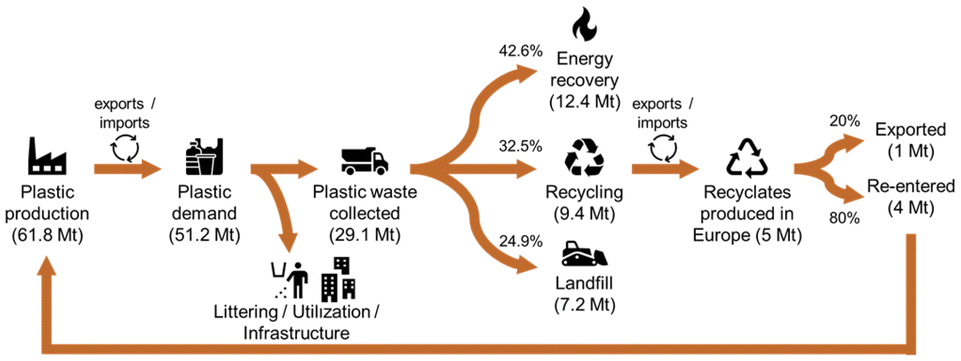 | ||
| Fig. 2 Circularity of plastics in 2018 in Europe (EU28 + NO/CH). Graphic adapted based on Plastics Europe.3 | ||
So far, the recycled share is almost exclusively achieved by mechanical recycling using physical separation processes like shredding and sorting of thermoplastics for recovery of polymers as re-granulate, which can then be used to manufacture new products. Mechanical recycling processes place high demands on the input materials, e.g. in terms of their homogeneity and low levels of contamination, which, if obtainable at all, claim high collection, sorting, and processing efforts.11 Moreover, the processes might lead to the degradation of the material's properties, resulting in recyclates unsuitable for the original application purpose. Therefore, the success of mechanical recycling is dependent on mono-fraction feedstock materials and low levels of contaminants. The recycling of PET bottles combined with a properly functioning deposit system is a rare example where these conditions are fulfilled. However, these requirements often cannot be met and the recyclates are usually used for products of lower quality instead of their original use due to the complex composition of today's composite packaging materials.12
Thus, chemical recycling could become a complementary to achieve a circular economy.13,14 Chemical recycling comprehends the chemical transformation of polymers to lower molecular weight compounds. Ideally, the products of chemical recycling present the original polymer building blocks or compounds with a high degree of chemical functionality and/or reactivity. In this way, a major part of the synthesis effort invested during preceding monomer production can be preserved. In addition, while mechanical recycling only allows a limited number of treatments due to the reduction in the quality of the recyclates, chemical recycling allows obtaining material of high quality as the derived monomers undergo a comprehensive downstream processing for purification.13,15 Keeping this in mind, a major challenge of chemical recycling relates to the need of high energy efficiency of the chemical transformation and the purification steps. Already in the past, several technologies have been developed to complement the mechanical recycling of mixed plastic waste. These technologies include for example solvolytic, catalytic, and thermochemical processes.16,17 Currently, a large number of new chemical recycling technologies are being developed aiming to be competitive. Mechanical and chemical recycling are complemented by a transformation of plastics into chemically rather inert CO2, presenting yet another EoL strategy that is potentially interesting in combination with suitable Power2X technologies.18–20 Methodologies for chemical recycling cover for example pyrolysis, chemo-catalysis, and bio-catalysis. In the ideal case, chemical recycling makes it possible to recycle plastics that cannot be treated by mechanical recycling, either because of their properties, e.g. thermosetting polymers, or because they are part of mixed waste streams, contaminated plastics, or multilayer materials. These plastic wastes are normally incinerated or landfilled. In 2018, with 0.2% and 0.1% of the recycled share of plastic packaging waste, Germany and Italy were the only countries with noticeable shares of chemical recycling as EoL treatment for plastic post-consumer waste in Europe.3 However, it must be taken into account that most chemical recycling technologies are in an early stage of development or pilot stage, and therefore the infrastructure is still limited.13,15,21 Despite the construction and operation of first industrial-scale plants to demonstrate chemical recycling technologies and identify business cases, chemical recycling still is considered limited and under development. Process reliability, sustainability in means of environmental benefits, and economic feasibility still need to be demonstrated.22,23
In politics and industry, considerable momentum but no coherent strategy can be observed regarding chemical recycling. This is highlighted by the fact, that already the definition of chemical recycling is still being discussed controversially. Also, the classification of chemical recycling in the waste hierarchy remains unclear. According to EU directive 2008/98/EC, any reprocessing of plastics into products, materials, or substances whether for the original or other purposes excluding backfilling and energy recovery is considered recycling.24,25 Overall, chemical recycling could present a viable technology to complement mechanical recycling of plastics. Especially regarding the fast-growing share of biomass-based plastics in the market, an early assessment of suitable EoL strategies appears indispensable.
This review provides an overview of current knowledge on the (bio)chemical and thermal recycling of chemically novel bioplastics, that is biomass-based plastics not resembling the chemical structure of well-established fossil-derived plastics; major emphasis is on maintaining chemical functionality of the bioplastic building blocks. Available methodologies for thermal (gasification, pyrolysis), chemo-catalytic (homogeneous and heterogeneous catalysis), and bio-catalytic (enzymatic and whole-cell catalysis) recycling are summarized together with the available insights of LCA studies on recycling strategies for bio-based biodegradable plastics.
2. Approaches for chemical recycling
The scope of this review will cover the following chemically novel biomass-based and biodegradable plastics, namely polyhydroxy-alkanoates (PHA), polylactic acid (PLA), thermoplastic starch (TPS), polybutylene adipate terephthalate (PBAT), polybutylene succinate (PBS) and polycaprolactone (PCL) (Fig. 3).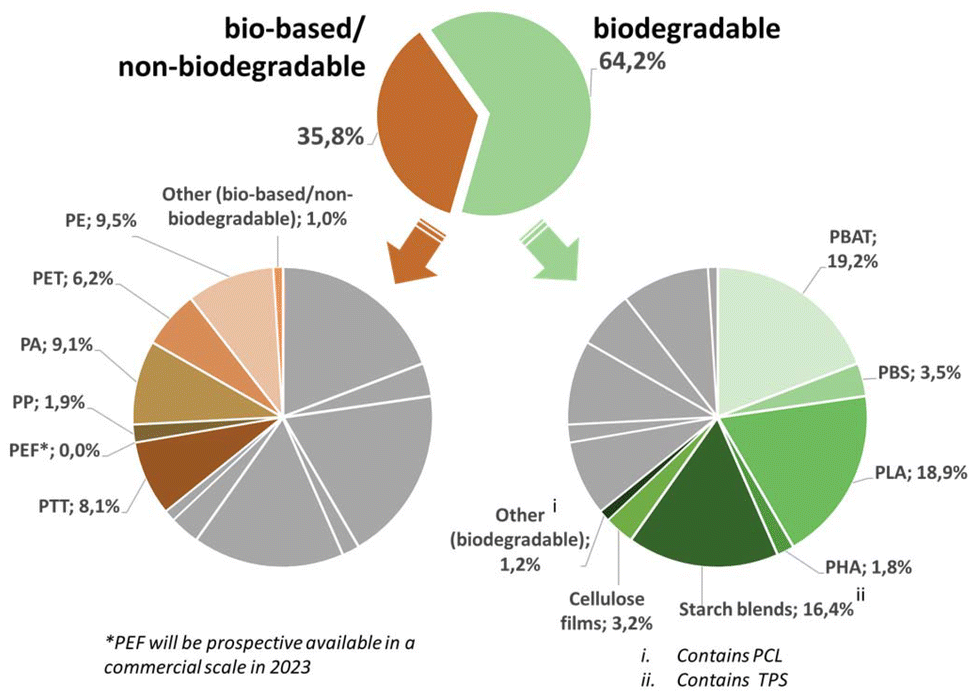 | ||
| Fig. 3 Overview of the global production capacities of bioplastics in 2021. The graph is based on data from Bioplastics Market Development Update 2021 and reworked with kind permission of European Bioplastics (european-bioplastics.org).175 | ||
Polyhydroxyalkanoates (PHA) are biopolymers synthesized as internal carbon and energy storage polymers by different bacteria. Although being researched for decades, the total production capacity of PHA worldwide is still below 50 kt per annum. However, due to available technology, the necessary CO2 neutrality, and demand for degradable plastics, a significant increase in production is expected and can already be seen in raised industrial interest, this is in particular true for polyhydroxybutyrate (PHB).26 As a side remark: carbon negative bioplastics with an EoL in landfills would be active carbon sequestration, however, risks such as land use, mismanaged storage or even sudden CO2 release due to open fires require consideration.
Polylactic acid (PLA) can also be directly synthesized by bacteria,27,28 however, the commonly used route is the microbial synthesis of the monomers L-lactic acid or D-lactic acid, with L-lactic acid being by far the prominent monomer, and subsequent chemical polymerization.29
Thermoplastic starch (TPS) is the most commonly used biopolymer and consists of restructured starch. Pure starch-based bioplastic is usually brittle. To counteract this and to facilitate thermo-plastic processing, plasticizers such as glycerol, glycol, and sorbitol are added.30 Not all plasticizers added to TPS are biocompatible and can thus affect biodegradability.31 In many cases, starch-based bioplastics are blended with biodegradable polyesters, e.g. PLA,32 PCL33 or PBAT34 to obtain plastics for industrial applications.
The co-polymer polybutylene adipate terephthalate (PBAT) is synthesized from 1,4-butanediol, adipic acid, and terephthalic acid, and thus consists of the repeating units 1,4-butanediol/adipic acid (BA) and 1,4-butanediol/terephthalic acid (BT). While for 1,4-butanediol and adipic acid biochemical synthesis routes exist and 1,4-butanediol from renewable feedstocks is commercially available, all PBAT to date originates from crude oil.35
Polybutylene succinate (PBS) can replace conventional PP for some applications. As is the case for PBAT, the monomers of PBS are usually synthesized based on fossil resources. However, microbially produced 1,4-butanediol36 and succinic acid37 from renewable carbon sources are commercially available. PBS is a linear aliphatic polyester. It is compostable, features high flexibility and excellent thermal stability. However, PBS is rather stiff and its melt viscosity for processing is often insufficient for various end-use applications.31
Polycaprolactone (PCL) is synthesized chemically by ring-opening polymerization of ε-caprolactone. ε-Caprolactone is currently produced from fossil benzene. PCL is compatible with a range of other materials and can thus be mixed for example with starch to produce TPS-type plastics. PCL can be easily degraded by hydrolysis of its ester bonds in mild conditions and is therefore used as an implantable biomaterial.38
2.1. Chemical recycling strategies
The methodologies for chemical recycling of these bio-based plastics discussed in the following comprehend enzyme catalysis, pyrolysis, chemo-catalysis using homogeneous and heterogeneous catalysis and whole-cell bio-catalysis. The aim of the chemical recycling methodologies is maximum preservation of the synthesis effort invested during preceding monomer production. This relates to maximum integrity of the chemical structure and functionality. Ideally, chemical recycling delivers compounds that can be used as monomers for subsequent polymerization, directly for other fields of application (e.g., solvents), or as substrate for further chemical conversion. Table 1 summarizes the chemical structures of selected potential products available from the different methodologies according to recent literature.
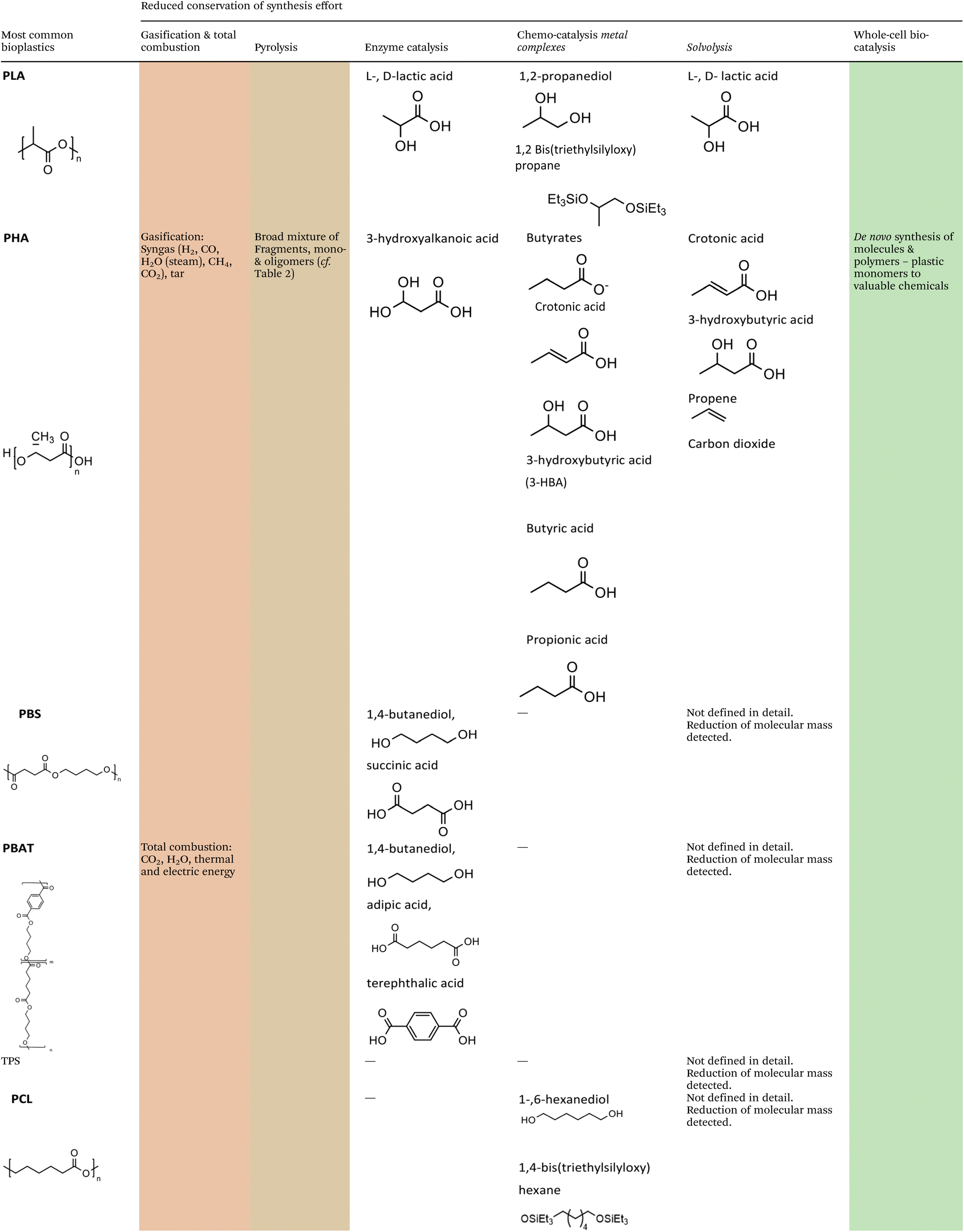
|
For most methodologies only a few literature examples on the chemical recycling of biomass-based polymers exist, currently hampering a comprehensive comparison in terms of performance, economic and application potential. In addition, investigations under industrially relevant conditions have not been carried out yet. Nevertheless, factors such as (1) costs and availability of the utilized catalysts, (2) benign reaction conditions, (3) the use of auxiliaries and additional solvents as well as (4) selectivity of the transformation to a limited set of valuable products will certainly play a crucial role for industrial viability.
2.2. Gasification
Thermochemical gasification describes the conversion of carbonaceous materials into gaseous products creating added value in comparison to the raw material. In a first step, the feedstock is brought into contact at high temperatures with an externally supplied gasification agent, which carries O2, H2O or CO2 into the process and the volatile components vaporize.39,40 The thermal degassing products subsequently are converted via homogenous gas-phase reactions into mainly H2, CO, H2O (steam), CH4, and CO2 since the amount of gasification agent is lower than required for stoichiometric oxidation. In the second step, the remaining char is gasified utilizing different gas–solid reactions. The gaseous products are accompanied by high-boiling (polycyclic aromatic) hydrocarbons, gas pollutants like chlorine, sulfur and nitrogenous compounds, and dust.39,41 These by-products must be removed from the raw gas by extensive gas purification to gain a synthesis gas (syngas) suitable for further processing. The syngas is an intermediate to be used in a wide application range in the chemical industry depending on the CO/H2 ratio. In addition, to simply substituting fossil fuel in gas turbines to provide electric power and thermal energy, it can be processed downstream into substances like alcohols, ammonia, or hydrocarbon fractions using established industrial processes (e.g. Fischer–Tropsch, Haber–Bosch).42 Although being considered an important contribution to chemical recycling, the gasification of bioplastics seems, best to our knowledge, to be a technology still in the early research stage. Although not yet proven on an industrial scale, the advantage of high-temperature gasification is probably its ability to treat mixed fractions of different bioplastics, as expected for example in pre-sorted waste streams. On the other hand, the synthesis effort invested during monomer production is hardly retained and complex product mixtures of small molecular weight compounds are being formed. Alternatively, the resulting syngas mixtures of lower quality and changing composition can be used as a substrate for microbes, as commercialized by Lanzatech using syngas from steel mills to produce ethanol and in the future other bulk chemicals.432.3. Pyrolysis
Pyrolysis describes the thermal decomposition of solid material in the absence of oxygen. Typical products are CO, H2, CO2, H2O and simple hydrocarbons, condensable vapor designated as pyrolysis oil and solid residue. The product distribution strongly depends on the reaction temperature, pressure, heating rate, residence time, and the chemical structure of the raw material used. Studies dealing with the pyrolysis of polymers mostly focus on ideal parameters for depolymerization reactions to recover fractions of the monomer in the pyrolysis oil. Lamberti et al. provide an overview of recycling routes for different degradable and non-degradable bioplastics including pyrolysis as a potential treatment process.44 Most studies have focused on the pyrolysis of PLA including effects caused by mixtures with other (bio)polymers and biomass.Products of PLA pyrolysis have been analyzed using either FT-IR gas analysis or GC-MS.45,49–51 The monomer lactide, its oligomers, acetaldehyde, acetic acid, and carbon monoxide have been identified as the most prominent reaction products. Carbon dioxide,49,50 propionic acid,51 carbonyl compounds, and water were found sporadically.50,51 While most studies were carried out under isothermal conditions or with thermogravimetric analysis (TGA) at heating rates of 1–15 K min−1 with some mg of sample mass, Undri et al.51 used a microwave reactor with sample masses of up to 150 g, allowing for quantifying mass balances. The share of gas, liquid, and solid strongly depends on the reactor configuration (microwave power and absorbing additive). The highest recovery of lactide (27.7%) came along with the highest obtained liquid yield, which could be achieved with high microwave power (3 kW), comparably low residence time (25 min), and carbon as a microwave absorber. Longer residence times promoted the production of gases while other polymer absorbers (tires) led to co-pyrolysis effects with aromatic compounds formed, hindering the lactide formation.51
To selectively promote the formation of lactide monomers, the influence of catalysts on the pyrolysis process was investigated. Already Kopinke et al. postulated a reaction mechanism catalyzed by tin to exclusively form lactide.46 Nishida et al.52 observed selective lactide formation only for high Sn impurities (607 ppm), while low impurities led to the production of cyclic oligomers. Feng et al. extended the work to different end-groups (hydroxyl, carboxyl, or none) to identify the reaction pathways of Sn-catalyzed lactide formation.47
Further studies focus on the pyrolysis of PLA blends. In contrast to the previous studies dealing with pure PLA, the focus shifts from the extraction of monomers to the thermal stability properties of the polymer.53,54 Only a few studies concentrate on the product site: Wang and Li studied the thermal decomposition of PLA in the presence of different biomass to simulate waste mixtures.55 Based on their results, the interaction effect between both components regarding the decomposition kinetics and the overall mass loss seems to be small. The same holds for a mixture of PLA and PBAT.56 It should be noted, however, that in both studies no precise conclusions are drawn about the actual product distribution and it, therefore, remains open whether mixtures of different components can also be cleaved into the individual monomer building blocks with the same efficiency as the pure educt.44,45
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 57 to 87%.58 By using Mg(OH)2 as a catalyst, yields of up to 99.7% crotonic acid have been obtained.59 At higher temperatures, CO2 and propene dominate the product composition as a result of decarboxylation reactions of the crotonic acid.58,60 Concerning the degradation mechanism, there is a consensus in the literature that it almost exclusively involves a random chain scission (cis-elimination), whereby crotonic acid and its oligomers are formed.45,57,61
57 to 87%.58 By using Mg(OH)2 as a catalyst, yields of up to 99.7% crotonic acid have been obtained.59 At higher temperatures, CO2 and propene dominate the product composition as a result of decarboxylation reactions of the crotonic acid.58,60 Concerning the degradation mechanism, there is a consensus in the literature that it almost exclusively involves a random chain scission (cis-elimination), whereby crotonic acid and its oligomers are formed.45,57,61
| Substrate | Reactor | Analysis | Temperature | Products (mass yield) | Reference |
|---|---|---|---|---|---|
| PLA | TGA | FTIR | Const. heat-up until 500 °C | Lactide monomer and oligomers, acetaldehyde, CO2, CO, H2O | Zou et al. (2009)49 |
| TGA | FTIR | Const. heat-up until 750 °C | Lactide monomer and oligomers, acetaldehyde, CO2, CO, H2O, acetic acid | Badia et al. (2012)50 | |
| TGA | GC-MS | Isothermal and const. heat-up | Lactide monomer and oligomers, acetaldehyde | Aoyagi et al. (2002)45 | |
| Microwave | GC-MS | uncertain | Lactide monomer (27.7%) | Undri et al. (2014)51 | |
| PHA | TGA | GC-MS | Isothermal and const. heat-up | Crotonic acid, crotonic acid oligomers | Aoyagi et al. (2002)45 |
| TGA | GC-MS | 280 °C, no catalyst | Crotonic acid (60.7%) | Ariffin et al. (2010)59 | |
| TGA | GC-MS | 240 °C, MgO as catalyst | Crotonic acid (98.2%) | Ariffin et al. (2010)59 | |
| TGA | GC-MS | 240 °C, Mg(OH)2 as cat. | Crotonic acid (98.3%) | Ariffin et al. (2010)59 | |
| TGA | FTIR | Const. heat-up until 500 °C | Crotonic acid (35.3%), crotonic acid oligomers (57.5%), CO2, H2O, propene, ketene, acetaldehyde, β-butyrolactone, 3-butenoic acid | Grassie et al. (1984)57 | |
| PBAT | TGA | FTIR | Const. heat-up until 600 °C | CO2 (25%), CO (17%), CH (8%), aromatic compounds (2.5%), alcohols, aldehyde, ketone, ester, carboxylic compounds | Li et al. (2020)66 |
| PBS(A) | Microfurnace | GC-MS | 500 °C | 1,3-Butadiene, ester, dimers, tetrahydrofuran, cyclopentanone, hexamethylene diisocyanate | Sato et al. (2001)64 |
Overall, compared to biotechnological processes, pyrolysis is carried out at high temperatures and usually results in the formation of complex aliphatic and aromatic product mixtures. However, catalyst addition has proven successful in significantly enhancing the selectivity of individual compounds, but feedstock impurities due to the origin of waste are considered to have a certain influence on product yields and qualities.
2.4 Enzyme catalysis
With the recent developments in protein engineering and industrial protein production, enzyme catalysis for tailored plastic depolymerization is an upcoming possibility that is much discussed.7,67 Indeed, biotechnological plastic degradation inspired ideas of plastic “eating” microbes that clean the environment from plastic waste.68 To date, under confined conditions, only competitive degradation rates were reported for ester bonds, which fits well with many of the current bioplastics (PLA, PHA, PBS, PBAT). The often reiterated advantages of enzyme catalysis, in general, are high selectivity, low energy-requiring reaction conditions, water as main solvent, and potentially low environmental impact. For a particular example, one must however benchmark enzyme catalysis against other options,69,70 as low rate, low yield or high purification efforts reduce the environmental benefit.Enzymatic plastic depolymerization is the furthest development for PET, and although far from being a bioplastic, is here presented as prime example for enzyme catalysis of plastics, with a commercial plant announced just very recently (https://www.carbios.com/en/carbios-to-build-in-france-its-plant/). Indeed, enzymatic PET degradation got much attention, as not only a microbe, Ideonella sakaiensis, equipped with two enzymes to degrade PET and a biochemical pathway to degrade terephthalic acid was reported,71 also engineered esterase/cutinase enzymes with very high activity on amorphous PET inspired application ideas.72 These include the removal of broken yarn during washing of cloth73 and the recycling of PET bottles.74 The PET drinking bottle is now synonymously used for the plastic crisis we face, although in some countries with appropriate waste collection infrastructure in place, PET bottles are the prime example for successful mechanical plastic recycling. Nevertheless, in packaging in general, and food packaging specifically, many challenges exist to recycle or upcycle PET and in the future other polyesters, including bioplastics. Enzyme catalysis of plastics, mainly focusing on PET, is reviewed extensively.72,75,76 Here, we briefly summarize the information as a proxy for the technological possibilities for bioplastics. Some of the developments can be seen in analogy to cellulose/cellulase research.77 This includes fields like extensive bioprospecting, protein engineering, (e.g., catalytic rate, binding modules), and bioprocess engineering. As the interest “exploded” just recently with the prominent showcase of PET hydrolysis and repolymerization and bottle manufacturing,74 the research field is pushing all aspects at the same time. Enzymatic PET hydrolysates as a substrate for PHA production is a prominent example.78 Importantly, the life cycle analysis for enzymatic catalysis of PET is favorable under the conditions evaluated.69 The incorporation of polyester hydrolyzing enzymes into plastics is another application of enzyme catalysis that attracts a lot of attention.79 For all these examples, highly active enzymes are required that ideally have their catalytic optimum at conditions, which are compatible with the plastic properties. Indeed, for PET, hydrolyzing temperatures above the glass transition temperature is aimed at, and hence enzymes able to withstand 70 °C and higher are used for rapid PET degradation.
Prominent examples for PET degrading enzymes are esterases that have as natural product the plant material cutin, hence are cutinases. Besides leaf and branch compost cutinases (LCC), used for example by Carbios, a cutinase originating from a compost metagenome and used for example by Carbios, TfCut2 from Thermobifida fusca exhibits favorable catalytic properties. Much information on the structure of these and related enzymes exist, especially on the PETase of I. sakaiensis,80,81 and many successful protein engineering efforts are published.74,80,82 In enzyme catalysis of solids, the conditions used are key for reproducibility and comparability, however, the research field should mature as these aspects are rather poorly documented.77 While early reports discussed surface property changes, engineered esterases under optimized conditions are catalyzing, e.g. full PET degradation in under 10 hours.74 With all these successes in hand, bioprospecting seems still a powerful approach to identify enzymes with truly high activity for the different plastic polymers.83,84 As a note, although some plastics are now for five to seven decades available in the environment, no clear evidence of adaptive evolution of any enzyme activity towards plastic degradation exists, explaining the still very long availability of many plastics in the environment. It either needs more time or some of the recalcitrant chemical bonds, especially of vinyl plastics, are just not specifically attackable by enzymes.
For bioplastics, many studies exist investigating their biodegradability under many different conditions. Prominent are biodegradation tests in industrial composting, but also tests in aquatic systems including oceans exist. Indeed, some of the bioplastics such as PBAT, which is used in mulch films as PBAT-PLA blends, might be so quickly degraded that the mulch film can be left in the environment; the environmental impact of this approach is still discussed. Notably, the enzymes involved are hardly investigated. Polymers including plastics can be produced using enzyme catalysis in vitro and in vivo.85 Again, the specificity of the enzyme(s) is of outstanding importance and may be an argument for this catalytic scenario. To highlight the recent development with a long-term perspective, direct PLA synthesis by microbes is mentioned here,28,86,87 circumventing demanding lactic acid purification for chemocatalytic PLA synthesis.
2.5 Chemo-catalysis
In chemo-catalysis, molecular or solid catalysts are used to transform (bio)plastics into either the original monomers or alternative products which are potentially applicable in other value chains (cf.Fig. 1). The explored transformations majorly rely on hydrolysis of the ester bonds using catalysts bearing an acidic functionality or a reductive cleavage over tailored metal catalysts. For the field of homogeneous catalysis, solvolysis using molecular acids has to be mentioned as well. In addition, already some examples of capable metal-complex catalysts exist, while only very few studies address solid catalysts. A strength of the chemo-catalytic approaches is the often high selectivity towards the target products, thus monomers or valuable substrates. Disadvantages comprehend, dependent on the specific reaction system, the need for organic solvents, soluble acids or bases or other auxiliaries, the need for noble metal containing catalysts as well as elevated temperatures and pressures. In addition, handling of mixed waste steams can be challenging and impurities in the feed may interfere with the utilized catalyst system. Accordingly, studies under industrially relevant conditions including insight into the feasibility of catalyst recycling are indispensable to evaluate the application potential but are hardly available to date.The previously mentioned examples describe solvent and pH-value dependencies between room temperature and 70 °C. However, hydrolysis at higher temperatures (>120 °C) is also a promising approach. Fujie et al. studied a temperature range of 220–350 °C, yielding up to 90% of lactic acid at 250 °C. A further increase in temperature led to racemization and decomposition of the desired compounds and is thus detrimental.107 Lower temperatures between 120 and 190 °C further improved the lactic acid yield to >95%, though, at a reduced reaction rate.108
Mohanty et al. conducted hydrolysis experiments at 50 °C and a high relative humidity of 90% for up to 30 days. The elongation at break as well as the tensile strength of PBAT and a PBS/PBAT composite were found to be reduced. Interestingly, the impact strength of pure PBAT was not affected by the hydrolysis, whereas the composite one showed a reduced strength upon impact.114
If hydrolysis shall be inhibited, the application of nanoparticles inside the polymer matrix may be a possibility. Even if the study by Yang et al. focuses on degradation behavior in human blood with a pH value of 7 at 37 °C, the results point out that incorporated nanoparticles slightly reduce the hydrolysis rate of PBAT in the early phase.115
The application of PBS and biopolymers, in general, is often limited when elevated temperatures occur in combination with increased air humidity. The usage of anti-hydrolysis agents or polyfunctional monomer additives can increase the resistance to such adverse circumstances as demonstrated by Kim et al. for 50 °C and 90% relative humidity applied for up to 30 days.117
Polymer blends made of PLA incorporated with PBS were found to degrade much faster than the individual polymers since the macroscopic structure of the co-polymer exhibits a higher surface area and an increased hydrophilicity is present. This trend was found for degradation approaches over days118 up to several months.119 Thereby, the hydrolysis in alkaline solutions follows the surface-erosion mechanism and predominantly takes place at PLA-PBS interfaces.118
Comparable results have been obtained for Starch/PBAT blends by Yamashita et al. when applying malic, tartaric and citric acid. A higher concentration of the respective acid leads to an increased weight loss.121
A further example for alkaline hydrolysis was given by Vidaurre et al. in 5 M NaOH, focusing on the differences between linear PCL and cross-linked PCL. The latter one hydrolyzed significantly faster, most likely reasoned by a lower initial crystallinity and a higher hydrophilicity. Thus, the diffusion of NaOH into the matrix of the polymer is facilitated. In opposite to many mentions beforehand, no increase in crystallinity was observed upon hydrolysis, indicating the equal hydrolysis of both, amorphous and crystalline parts.123
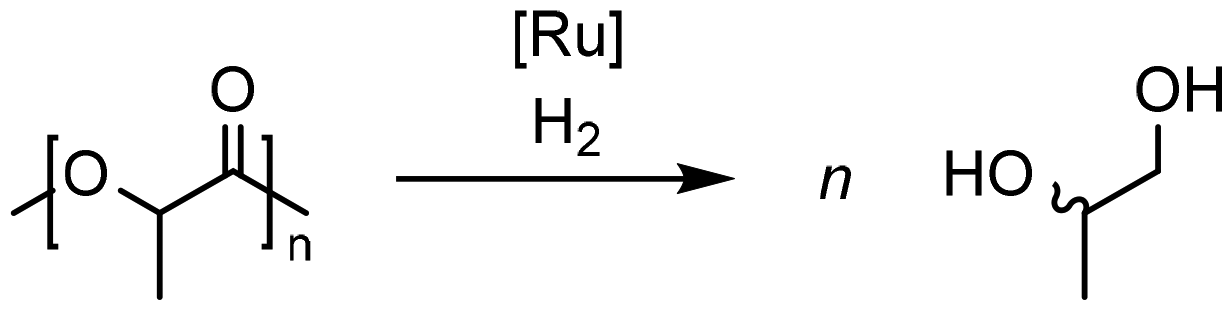 | ||
| Scheme 1 Hydrogenolysis of PLA with ruthenium-based catalysts. | ||
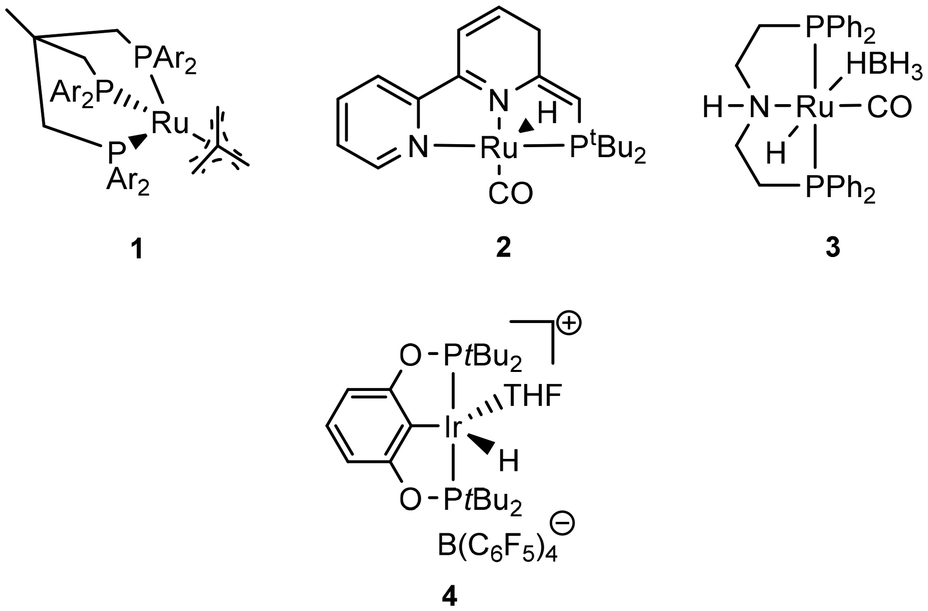 | ||
| Scheme 2 Transition-metal catalysts for the depolymerization of bioplastics. | ||
Krall showed the use of a ruthenium PNN complex (2, Scheme 2), developed initially by Milstein, as catalyst for the reductive depolymerization of PLA to 1,2-propanediol in the presence of potassium tert-butoxide.125 In their investigation defined high-purity PLA with 50 repeating units was used as substrate. In addition, an anisole-THF solvent mixture had to be used to ensure high solubility of the PLA.
Kindler developed an additive-free protocol for the reductive depolymerization of PLA by using the Ru-MACHO-BH (3, Scheme 2) catalyst.126 The reductive depolymerization could be carried out in 3 h at 140 °C, with 45 bar H2 to full conversion with 0.5 mol% catalyst loading. Analog to the work by Westhues, this catalytic system was also successfully tested on EoL PLA samples such as beverage cups, Sushi box containers, drinking straws, and PLA cutlery.
Catalytic solvolysis in methanol has been recently explored by the group of Herres-Pawlis. Therein, different Zn guanidine complexes were demonstrated to allow methanolysis of polylactide to methyl lactate at mild conditions. For the best performer, full methyl lactate yield was achieved in PLA melt within 1 h. Further studies emphasized successful catalyst recycling and stability towards mixed plastics feed. Considering the non-toxic nature and availability of these complexes, further development towards industrial application and techno-economic analysis should be carried out.
Reductive depolymerization of PLA was also achieved in presence of Zn(OAc)2 and silanes under mild reaction conditions by Fernandes.130 In particular, with 10 mol% Zn(OAc)2 and 30 mol% silane EoL-PLA samples could be converted at 110 °C within 48 h with up to 71% yield of 1,2-propane diol.
Feghali presented the depolymerization of PLA by hydrosilylation in the presence of tris(pentafluorophenyl)borane (B(C6F5)3) and a hydrosilane. Interestingly, only the use of triethylsilane (Et3SiH) has resulted in substantial yields of the silylated propylene glycol. However, up to 5 mol% of the borane catalyst had to be used. The use of TMDS or PMHS led to full conversion of PLA to propane.132
The group of Cantat used Brookhart's catalyst (4, cf. Scheme 2) in the presence of a hydrosilane to efficiently depolymerize PLA with 0.5 mol% 4 at 65 °C in chlorobenzene to the silylated propylene glycol in 64% yield (cf.Scheme 3). Commercial PLA and colored 3D printer PLA filaments were successfully converted under these conditions.131
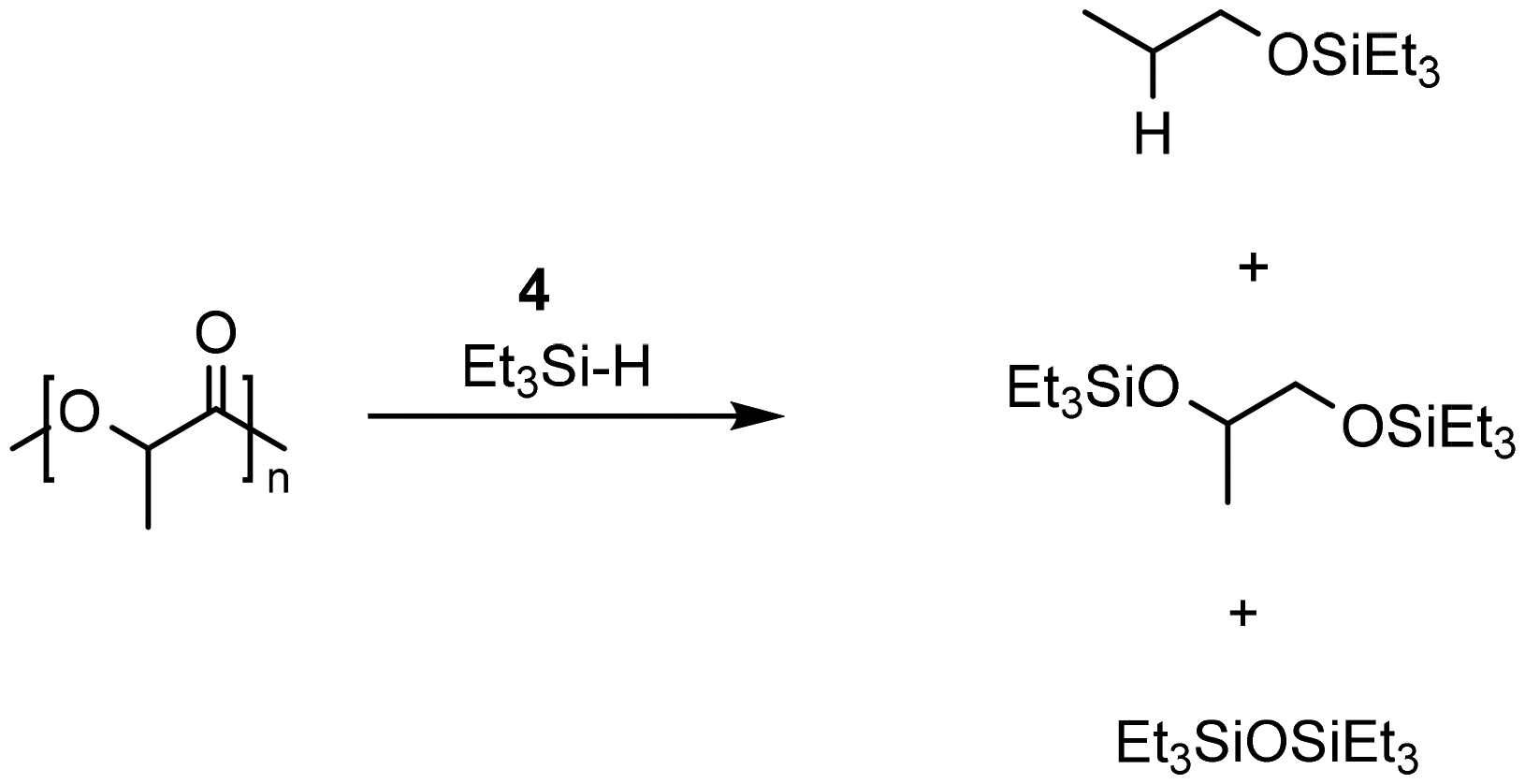 | ||
| Scheme 3 Reduction of PLA into silylated alcohols with Et3SiH and 4. | ||
Taken together, the conversion of PLA with homogeneous catalysts has been under rapid development in recent years and already EoL PLA samples can be converted with high yields to valuable diols which can be subsequently used for the production of new polymers, solvents, fuels, or specialty chemicals (Table 3).
| Substrate | Catalyst | Temperature [°C] | Pressure [bar] | Reaction time [h] | Products | Yield | Reference |
|---|---|---|---|---|---|---|---|
| PLA | 1 | 140 | 90 | 16 | 1,2-Propanediol | >99% | Westhues et al. (2018)124 |
| 2 | 160 | 54.4 | 48 | 1,2-Propanediol | >99% | Krall et al. (2014)125 | |
| 3 | 140 | 45 | 3 | 1,2-Propanediol | >99% | Kindler et al. (2020)126 | |
| Zn guanidine complexes | 60 | — | 1 | Methyl lactate | >99% | Hermann et al. (2022)127 | |
| Fuchs et al. (2022)128 | |||||||
| Stewart et al. (2022)129 | |||||||
| Zn(OAc)2 | 110 | — | 48 | 1,2-Propanediol | 60–71% | Fernandes et al. (2021)130 | |
| 4 | 65 | — | 60 |
|
64% | Monsigny et al. (2018)131 | |
| PCL | 1 | 140 | 100 | 16 | 1,6-Hexanediol | >99% | Westhues et al. (2018)124 |
| Zn(OAc)2 | 65 | — | 24 | 1,6-Hexanediol | 98% | Fernandes et al. (2021)130 | |
| 4 | r.t. | — | 2 |
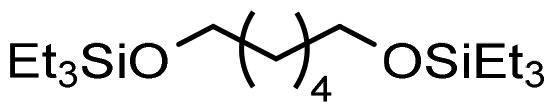
|
99% | Monsigny et al. (2018)131 | |
| PHB | 2 | 160 | 54.4 | 48 | Butyric acid | 88% | Krall et al. (2014)125 |
| P3HP | 2 | 160 | 54.4 | 48 | Propionic acid | 90% | Krall et al. (2014)125 |
 | ||
| Scheme 4 Hydrogenolysis of PCL. | ||
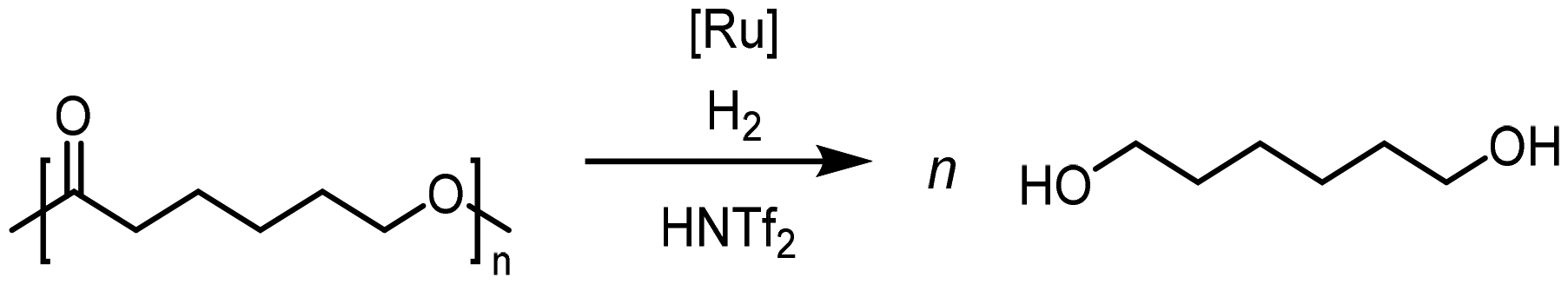
Krall used the ruthenium-PNN complex (2) not specifically for the conversion of PCL but showed successful conversion of caprolactone to 1,6-hexanediol with 47.6 bar H2 at 120 °C in 48 h.125 The zinc acetate/hydrosilane catalytic system of Fernandes allowed high conversions and yields for the reductive depolymerization of PCL with 10 mol% catalyst, and methyldiethoxysilane ((EtO)2MeSiH) as hydrosilane, to 1,6-hexanediol within 24 h at 65 °C with 98% yield. This reaction could also be performed in gram scale.130 Hydrosilylation of PCL could also be achieved in Cantat's group with Brookhart's catalyst (4) within 2 hours at room temperature and 0.3 mol% catalyst and Et3SiH as silylating agent.131
For Pd/C, under optimum reaction conditions at 200 °C and 40 bar H2 close to full conversion (∼96 wt% liquid and 0.5 wt% gaseous products) could be reached. The liquid products covered butyrates, butanol, and butyric acid with respective yields of 46, 24, and 8 wt%, while the monomer 3-hydroxybutyric acid (3-HBA) was not present in the reaction mixture. For temperatures below Tm, no conversion occurred. Higher temperatures and longer reaction times caused a decreasing butyric acid yield, while butanol yield increases. Furthermore, Sun et al. investigated the products in the absence of hydrogen. In this case, crotonic acid was the only product, reaching yields of 73 wt% after 16 h.134
Also, Cu/Zn/Al catalysts, typically employed in methanol synthesis, were investigated under reductive conditions mainly leading to butyric acid and other liquids, whereas butyrates and butanol could not be found. The butyric acid yield increased for a rising ratio of Cu to Zn from 55:35 to 65:25. This trend could be related to the preferential hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds compared to carbonyl bonds over the Cu surface sites.134
C bonds compared to carbonyl bonds over the Cu surface sites.134
In a recent study, the Palkovits group screened supported ruthenium catalysts in the hydrolysis of PHB.136 Ruthenium was selected due to the high activity previously observed in the hydrogenolysis of cellulose and polyols.137–141 Therein, the selection of the support material proved to be crucial for an efficient transformation. The best results were achieved with Ru/CeO2 allowing full conversion and a yield of 79% 3-hydroxy butyric acid, 20% butyric acid and 1% crotonic acid (200 °C, 100 bar H2, 40 min). Interestingly, for a hydrotalcite support, the product distribution changed significantly in favor of butyric acid with 53% yield, 14% hydroxybutyric acid and 10% isopropanol. Again, the reaction temperature presented the decisive parameter, with yields below 20% for a reaction at 150 °C rising to around 60% at 175 and 100% at 200 °C. The study also demonstrated the transformation of mixed waste streams consisting of PHB and PLA. Indeed, Ru/CeO2 enabled an efficient depolymerization with only minor decrease in yield compared to reactions with the pure compounds.9
Patent JPH07309863A describes the degradation of PLA to lactide in a screw-type extruder and a temperature between 200 and 400 °C. Water is added in low amounts of 0.1 to 5.0 wt% and among others, zinc oxide (ZnO) is named as a possible catalyst to be added with 0.1–1.0 wt%. It was found that the presence of ZnO promotes the reaction and inhibits the formation of undesired side products such as lactic acid ether or lactic acid anhydride (Table 4).144
| Substrate | Catalyst | Temperature [°C] | Pressure [bar] | Reaction time [h] | Products | Yield | Reference |
|---|---|---|---|---|---|---|---|
| PHB | Pd/C | 200 | 400 H2 | 12 | Butyrates | 45% | Zhang et al. (2021)134 |
| Pd/C | 200 | N2 | 16 | Crotonic acid | 73% | Zhang et al. (2021)134 | |
| Cu/Zn/Al | 200 | 400 H2 | 24 | Butyric acid | >75% | Zhang et al. (2021)134 | |
| Ru/CeO2 | 200 | 100 | 0.66 | 3-HBA | 69% | Palkovits et al. (2022)136 | |
| Ru/hydrotalcite | 200 | 100 | 0.66 | Butyric acid | 53% | Palkovits et al. (2022)136 | |
| PLA | ZnO | 200–400 | — | — | Lactic acid | — | Shirai et al. (1994)144 |
2.6. Whole-cell bio-catalysis
For the last five years or so,145 plastic waste upcycling via biotechnology has been much discussed. Here, two steps in the EoL of plastics can be defined. In the first step, enzymes can attack and cleave selectively chemical bonds present in plastic polymers (section 2.3). The second step is the upcycling of plastic hydrolysates by whole-cell biocatalysts. As mentioned, microbes exist that can utilize plastic monomers as carbon and energy source. Examples are manifold, including the monomers of bioplastics, e.g., lactic acid, 1,4-butandiol, adipic acid, and terephthalic acid. Microbes can be engineered to selectively utilize monomers of choice while not modifying other molecules. This option might simplify future recycling efforts of mixed plastic waste. Indeed, a combination of thermic, chemical, and/or biotechnological plastic hydrolysis with the conversion of plastic hydrolysates to products of value increases the option for EoL plastic treatments.Using whole-cell biocatalysts entails a fundamental distinction from chemical, enzymatic or pyrolytic polymer degradation: the employed microorganisms will metabolize the generated mono- and oligomers and eventually form biomass and CO2. This process can thus hardly be described as recycling. However, the microbes can be engineered to instead produce molecules of choice, which can be as valuable or even of higher value than the monomers from the applied plastics. If higher value products are generated, the term upcycling is used.
The vision is to exploit the metabolic funnel of microbes to convert cheap carbon sources, including monomers from bioplastic to products of value (cf.Fig. 4). In an integrated biorefinery, the synthesized CO2 is converted by microbes with green hydrogen to, e.g., ethanol. In nature, the metabolic funnel of many microbes allows the organisms to grow under ever-changing environmental conditions. Even such complex monomer mixtures as degraded lignin are completely used. In analogy, the plastic monomers are converted to central carbon metabolites and used to further produce the desired product. Recent examples for upcycling PET hydrolysates by biocatalysis78 or chemocatalysis146 are PHA/bio-PU and β-ketoadipic acid production, respectively. The use of PET pyrolysis oil for the synthesis of the bioplastic PHA was reported earlier.147 With a multitude of substrates (plastic monomers) available, the question arises which substrate allows efficient production of which chemical. In a recent analysis,148 this question was answered for many types of plastic monomers, including monomers derived from bioplastics. In short, replacing chemical synthesis routes that require a lot of resources, like the synthesis of adipic acid, are the prime targets for whole-cell biocatalysis from bioplastic monomers.148
 | ||
| Fig. 4 Concept of the metabolic funnel: polymers are hydrolyzed and the arising monomers are via catabolic pathways metabolized and converted into valuable products. (Concept borrowed from Sudarsan et al.174 | ||
As an outlook for biotechnological plastic valorization, one may want to exploit the selectivity of the enzymes used to break chemical bonds in plastics. In multi-bond materials or mixed plastic wastes, it can be advantageous if only one or selected bonds are broken, as purification and reuse might be simplified. Such materials are, for example, many PU, for which a selective approach would open possibilities for isocyanate-free PU synthesis.149 Up to now, only the ester bond is efficiently cleaved, while reports exist for enzymatic cleavage of other bonds.76,150 The biodegradability of some bioplastics potentially allows in the future consolidated bioprocessing, in which a single reactor configuration for plastic degradation and product synthesis from the resulting monomer is aimed at, in analogy to efforts in lignocellulosic ethanol production.
In the following, examples of biocatalytic degradation of bioplastics are described (i.e., the left side of the metabolic funnel from Fig. 4). Integrating these approaches in the circular bioeconomy required coupling with production pathways (right side of the bow tie from Fig. 4). In a recent study, this coupling has shown to be successful for the synthesis of an intermediate molecule from PET monomers.78
Commercial products relying on the biodegradability are for example mulch films and other films in the agricultural industry.
3. Evaluation of the different approaches for chemical recycling by Life Cycle Assessment
In the field of waste management, Life Cycle Assessment (LCA) is used in a multitude of studies to quantify the environmental impacts of products at their EoL and the underlying waste management systems.160,161 Herein, an environmental analysis of alternative EoL treatment options for plastics can complement the assessment of the technological feasibility and allow for conclusions on the contribution towards a circular economy and the reduction of greenhouse gas (GHG) emissions of these treatment options. While LCAs of chemical recycling have already been conducted for fossil-based plastic waste, studies on the chemical recycling of chemically novel biomass-based plastics, especially regarding different technologies, are rare and of limited scope.8,13,703.1. LCA methodology
LCA is a consolidated methodological framework, based on ISO14040162 and ISO14044163 allowing for quantitative analysis of the environmental impacts of a product throughout its life cycle stages (i.e. from the extraction and processing of raw materials through production, distribution, and use of the product until the final disposal or recycling of waste). Also, alternative processes within this life cycle can be evaluated concerning environmental issues, e.g. different options to treat a product at the end of its life. This enables comparing the environmental performance of different products and process alternatives and identifying the hotspots that contribute most to the environmental impact.
Moreover, the life cycle perspective of the LCA methodology enables the identification and prevention of potential environmental burden shifting within the life cycle stages of a product and between different geographic regions involved in its supply chain. Furthermore, the inclusion of a multitude of environmental impact categories in the LCA also makes it possible to recognize possible pollution transfers between environmental impacts. For example, one of the main arguments for using bioplastics instead of their fossil counterparts is their potential to reduce the carbon footprint of products. However, a greater impact on land use can potentially result from biomass production. Therefore, a burden-shifting can occur to the forest or agricultural area where the biomass is produced, and from the environmental impact category climate change to land use.
The life cycle of biomass-based plastic starts in the agriculture or forestry system where the biomass is produced. Then, through conversion processes such as fermentation, the biomass is transformed into chemicals, which are in turn converted into plastics by polymerization and plastic formulation processes. The bioplastics obtained are processed to be used in a wide range of applications such as packaging or building construction for example. After their use, these bioplastics can take different EoL pathways, including chemical and mechanical recycling but also anaerobic digestion or composting when they are biodegradables.8 Note that no EoL treatment is perfectly circular, thus there is always a loss of material from the system.13
In terms of the environmental assessment of EoL treatment options, there are specifics that need to be considered in the assessment. As pointed out by Maga et al.164 a comparison of waste management options is difficult due to different required input qualities of the waste streams, the regional conditions, and the different output products and qualities. For certain types of bioplastics, the range of EoL treatment options differs from the ones of the fossil-based materials (e.g. due to the bio-degradability). This needs to be considered sufficiently in the scope of the analyses. Besides, different technology readiness levels (TRL), accompanied by diverging levels of accessibility and quality of data for the evaluation complicate the assessment. Fig. 5 highlights the possible lifecycle of bio-based plastics.
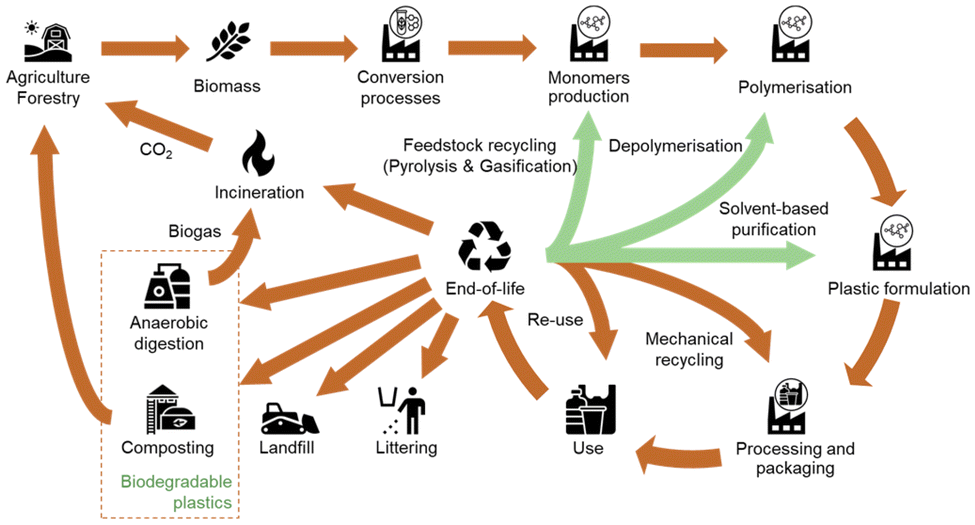 | ||
| Fig. 5 Overview of the lifecycle of bio-based plastics. Based on: Spierling et al. (2020), Koopmans et al. (2019).8,14 | ||
If bioplastics are introduced as a substitute for fossil-based plastics, Bishop et al.165 point to the importance of conducting comprehensive LCA, e.g. taking all relevant processes along the value chain into account to allow for a complete comparative evaluation of the environmental efficiency of the bioplastics against their petrochemical benchmarks. This fact is also stressed by the few existing comparative reviews on LCAs including the EoL treatment of bioplastics (cf.Table 5). The studies find deviations in the methodological approaches, such as the selection of impact categories, the accounting of credits for secondary material, or differences in the calculation of the substitutability of recyclates.8,166,167 Only two of the reviews include the EoL treatment (cradle-to-grave), due to a lack of more comparable studies that regard this lifecycle stage. While Spierling et al. aim to compare different EoL treatment options for bioplastics, they demonstrate that the focus of EoL assessment only allows for a comparison of PLA.8 Hence, the comparison of different EoL treatment options for multiple fossil- and biomass-based plastic types remains difficult.
| Reference | Content | Scope (# articles) | Quantitative comparison | Critical observations |
|---|---|---|---|---|
| Spierling et al. (2018)167 | Review of available data from LCA, S-LCA, and LCC studies for bio-based plastics | 29 (LCA) | Results for 7 fossil- and 8 bio-based polymers on a cradle-to-gate basis for 1 impact category (GWP) | • EoL not assessed |
| • Missing guidelines prevent comparability of studies | ||||
| • Comparison of one impact category only possible with restrictions | ||||
| Spierling et al. (2020)8 | Review of LCA studies on the EoL treatment of biomass-based plastics | 12 | GWP (global warming potential) for PLA | • No sufficient information for comparison of all EoL treatment options available |
| • Deviations in the results due to inconsistent assumptions, databases, etc. | ||||
| Walker and Rothman (2020)168 | Review of comparative LCA studies for fossil-based and bio-based plastics | 56 | Results for 7 fossil- and bio-based plastics (across 7 impact categories) | • Differences in impact between all plastics (fossil-based and bio-based) |
| • Much of this variation is related to the LCA methodology applied (especially for EoL treatment) |
3.2. Chemical recycling of bioplastics
The environmental performance of chemical recycling varies depending on the technology (i.e. solvent-based purification, depolymerization, pyrolysis, and gasification) and the plastic waste treated.13,21 On the one hand, the different chemical recycling technologies tend to have a lower impact on climate change than other EoL options such as incineration with energy recovery.8,13,21,169 Moreover, recyclates obtained by chemical recycling can have a lower impact on climate change than virgin plastics.169 On the other hand, mechanical recycling tends to have a better environmental performance than chemical recycling.8,13,21,169If we compare the different chemical recycling options, a study performed in the Netherlands found that pyrolysis and gasification performed better than incineration with energy recovery, but resulted in higher GHG emissions than depolymerization and dissolution technologies, the latter showing an impact on climate change in the same range as mechanical recycling.21 However, technologies with a lower impact on climate change such as depolymerization and dissolution show a greater specificity of plastic waste, while technologies with a higher impact such as pyrolysis and gasification allow treating a greater range of plastic waste streams.21 On the other hand, Davidson et al.13 reviewed nine studies on LCA of chemical recycling and identified that pyrolysis tends to have the best environmental performance among chemical recycling methods. However, the authors also highlight that the pyrolysis technology is the most investigated and therefore presents a higher quality of data, which affects the results. For this reason, more studies on the other chemical recycling technologies are necessary to have comparable results.13 This holds even more for recycling processes for biomass-based plastics.
| Reference | Polymer (waste stream)a | Processes | Indicatorsb | Findings |
|---|---|---|---|---|
| a PC: Post Consumer, PI: Post Industry. b GWP: Global Warming Impact, CC: Climate Change, HT: Human Toxicity, FD: Fossil Depletion, ALO: Agricultural Land Occupation, GWI: Global Warming Impact, OD: Ozone Depletion, POF: Photochemical Ozone Formation, A: Acidification, FEU: Freshwater Eutrophication, MEU: Freshwater Eutrophication, TEU: Freshwater Eutrophication, PM: Particulate Matter, CED: Cumulated Energy Demand. | ||||
| Piemonte et al. (2013)171 | PLA (PC bottles) | Mechanical recycling, chemical recycling (depolymerization) | Eco-Indicator99 | • Depolymerization is favorable over virgin production |
| • Mechanical recycling > chemical recycling (if substitution coefficients omitted) | ||||
| Papong et al. (2014)172 | PLA (drinking water bottles, Thailand) | Chemical recycling (depolymerization by hydrolysis), incineration with energy recovery, landfilling (with and without energy recovery), composting | GWP | • Lower cradle-to-grave emissions for biomass-based material compared to the fossil-based counterpart with all treatment options except landfilling (with and without energy recovery) |
| • Lowest contribution in GHG emissions from incineration with energy recovery followed by chemical recycling and composting | ||||
| Yano et al. (2014)173 | PLA, mix of PLA + PBSA (Household plastic packaging, Japan) | 5 treatment scenarios including: Mechanical recycling, chemical recycling, energy recovery, incineration without energy recovery, landfilling | GHG emissions | • Biomass-based materials could reduce lifecycle GHG emissions by 14–20% by replacing fossil-based material |
| • Separate collection, enabling 100%, rates of replacement, increases reduction potential further | ||||
| Cosate de Andrade et al. (2016)170 | PLA | Mechanical recycling, chemical recycling (depolymerization), composting | CC, HT, FD | • Mechanical recycling: lowest environmental impacts followed by chemical recycling |
| • Composting: worst treatment option in line with the indicators | ||||
| • Electricity consumption is decisive | ||||
| • Higher benefits of recycling technologies due to credits for the substitution of fossil-based polymers | ||||
| Maga et al. (2019)164 | PLA (PI and PC PLA waste, Germany) | Mechanical recycling, solvent-based recycling, chemical recycling, thermal treatment | FD, ALO, GWI, OD, POF, A, FEU, MEU, TEU, PM, CED | • Recycling processes lead to higher environmental benefits compared to thermal treatment |
| • Quality of waste streams influences results | ||||
| • Credits for substitution of virgin PLA by recyclates have a strong influence on the results (e.g. avoidance of impact in the categories ALO, GWI, and A) | ||||
Maga et al.164 use LCA to compare potential environmental impacts of mechanical recycling, solvent-based purification, and depolymerization with thermal treatment, e.g. incineration with energy recovery of post-consumer PLA in Germany. Their results show the environmental benefits of all recycling technologies in comparison to thermal treatment in several impact categories. The benefits are credited to the substitution of virgin PLA with recycled PLA in subsequent applications, hence the avoidance of environmental impacts. However, a correction of the resulting environmental credits (GWI) for the PLA regranulates is proposed to take decreasing quality levels into consideration as a one-to-one substitution of virgin material is not possible.
To compare mechanical recycling, depolymerization, and composting of PLA, Cosate de Andrade et al.170 use LCA, deriving data from lab-scale experiments and computer simulation. They find that mechanical recycling is associated with the lowest environmental impact in the categories of climate change, human toxicity, and fossil depletion. The authors identify industrial composting as the worst treatment option within the assessed impact categories. Also, the authors point to the fact that electricity consumption is a decisive factor in the evaluation of a recycling process. It is also highlighted that even higher levels of benefits can be achieved when considering that recycling can be repeated many times.170
In earlier studies, LCA is used to compare mechanical recycling and chemical recycling by depolymerization of PLA.171 The studies showed that depolymerization of PLA leading to lactic acid is favorable in comparison to the conventional production route of lactic acid by glucose fermentation. The results indicated that mechanical recycling is superior to depolymerization from an environmental point of view if substitution coefficients are omitted.171 Papong et al.172 use LCA to compare depolymerization by hydrolysis, incineration with energy recovery, landfilling (with and without energy recovery), and composting of PLA. Incineration is found to dominate depolymerization and composting in terms of GWP and fossil energy demand.172 Yano et al.173 use LCA in a biomass replacement case for household packaging material to compare chemical recycling in the form of superheated steam treatment and ring-opening polymerization, hyperthermal hydrolysis and anaerobic digestion, incineration (with and without energy recovery), and landfilling of PLA and a mixture of PLA and PBSA. The results indicate that chemical recycling of bioplastic packaging has a reduced environmental impact in terms of GHG emissions. The findings also emphasize the importance of source separation, which could lead to an even stronger reduction in GHG emissions for bioplastics by enabling recycling to a larger extent.173
4. Conclusion
In the broad field of strategies for recycling plastics, approaches that allow maximum preservation of chemical integrity appear particularly interesting. However, reuse in identical applications or mechanical recycling for high-value applications is often only possible with high quality and purity of the recycling streams. Chemical recycling can preserve the synthesis effort of monomer production to the greatest possible extent and at the same time can also be used for mixed and potentially contaminated material streams. A drawback of chemical recycling is the need for additional downstream processing and polymerization when compared to mechanical recycling. Nevertheless, especially in the case of mixed and/or contaminant-containing waste streams, the additional effort could be justified. Decisive for the technical implementation of chemical recycling technologies will be their economic potential and arising boundary conditions induced by e.g. recycling rate quota and CO2 footprint.Biotechnology processes like whole-cell bio-catalysis and enzymatic catalysis can deliver monomers and polymers from renewable carbon sources (i.e., biomass, CO2, waste), and thus can help to selectively break chemical bonds in plastics and to valorize the plastic hydrolysates. With these contributions, biotechnology can substantially contribute to the development of the envisioned sustainable circular plastic economy.7 The challenges in contributing to the EoL of bioplastics are in the detail, again, with an analogy to lignocellulosic biomass. Compared to the other technologies described in this review, solubility of the polymer is crucial for biotechnological transformations. This is a true challenge, as crystalline material is hardly attacked or at a very low rate.77 The production of a single product from a mixture of carbon sources is shown in industry for methane and ethanol, the largest industrial biotechnology products today. The combination of rapid hydrolysis and efficient use of hydrolysate mixtures for the synthesis of valuable chemicals might hence ask for hybrid processes, such as chemo/bio catalysis. The competition will be very high, but the effort to reduce not only plastic pollution but also the CO2 footprint is worth these efforts.
In the case of pyrolysis, the review showed that recycling of bioplastics can be achieved through pyrolysis. The observed product spectrum mainly includes oligomers with up to four repeating monomer units, the extracted monomer unit or monomer fragments with functional groups. In addition, smaller fragments can be found as well. Most of the available experimental investigations are based on thermogravimetric analysis with low heating rates and small sample mass on a laboratory scale. The focus of these studies was the identification of reaction mechanisms and potential generable products. The mostly qualitative product analysis and the low level of linkage to industrial applications (e.g., considering flash pyrolysis conditions or mixed waste streams) make an environmental evaluation (e.g., LCA) difficult.
Related to chemocatalytic strategies for chemical recycling of bioplastics, homogeneous and heterogeneous catalysis have to be named. In homogeneous catalysis, already several studies confirmed the potential of solvolysis with and without addition of molecular acids. In case of metal complexes, effective catalysts were developed and tested both for pure and real bio-plastics. In heterogeneous catalysis, only a few studies present the general feasibility of bioplastic depolymerization with solid catalysts. First examples comprehend supported noble metal catalysts known for their hydrogenation ability, such as Pd/C or Ru-based catalysts. Vital process parameters are the temperature and the gas atmosphere. A temperature above the polymers’ Tm, in most cases higher than 180 °C, is favored, since mass transfer between the liquid polymer and the solid catalyst is significantly increased.
In the broader context of all types of plastics, LCA studies have demonstrated that the environmental performance of chemical recycling varies depending on the technology and the plastic waste material treated. While chemical recycling technologies tend to have a lower impact on climate change than other EoL options, e.g. incineration with energy recovery, mechanical recycling still dominates chemical recycling in terms of environmental performance. At the same time, recyclates obtained by chemical recycling can have a lower impact on climate change than virgin plastics. In addition, regarding the LCA studies for chemical recycling of biomass-based plastics specifically, the bandwidth of results points to a variety of issues that need to be resolved for comprehensibility and comparability of future studies. From a methodological perspective, most studies account for one or two indicators only, mostly Global Warming Potential (GWP).8 However, for the identification of hot-spots and the trade-off between the impact categories for one or more waste management options, comprehensive evaluations are necessary that account for several indicators. Further, although there are studies that aim at comparing the environmental impact of fossil and bioplastics. However, these studies often neglect EoL processes and do not explicitly account for the second life of the output from the recycling processes and the dynamic nature of the technical substitutability.9 Referring to the technical perspective on the assessment, in most studies only one type of bioplastic or one form of chemical recycling is assessed. Herein, most studies on biomass-based plastics focus on PLA, while the assessment of other bioplastics remains open. To evaluate the environmental performance of the broad range of recycling technologies and materials regarding existing and prospective plastic waste streams, a broad system perspective needs to be adopted, in which the required source separation, sorting and pre-processing is integrated. For the comparison of chemical recycling technologies, different TRL levels pose a challenge for a comprehensive evaluation.
5. Outlook
The available literature clearly illustrates the high potential of chemical recycling technologies. However, many studies are still at an early stage of development. Data on the applicability for real material flows, possibilities of continuous reaction control and the design of a real overall process including downstream operation are missing for a techno-economic assessment and viable LCAs compared to other EoL strategies. Accordingly, research efforts should focus on addressing these aspects in interdisciplinary research approaches. This is necessary to build up the required database. Furthermore, beyond laboratory studies, early efforts with respect to demonstration projects are essential for a robust assessment of the promising technologies. Ideally, these efforts should be carried out in close cooperation between industrial and academic partners, taking regional conditions into account.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank funding from the Exploratory Research Space – ERS - RWTH Aachen under grant agreement PFKA008. Furthermore, the authors acknowledge funding by the Cluster of Excellence Fuel Science Center (EXC 2186, ID: 390919832) funded by the Excellence Initiative by the German federal and state governments to promote science and research at German universities. L. M. B. and J. K. acknowledge funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 870294 for the project MIX-UP. T. T. and L. M. B. acknowledge the German Federal Ministry of Education and Research for funding under the grant agreement No. 031B0899 in the project MIPLACE within the framework of the ERA-CoBioTech program of the EU. M. S. L thanks the networking program ‘Sustainable Chemical Synthesis 2.0’ (SusChemSys 2.0) for the support and fruitful discussions across disciplines. Last but not least, the authors also like to thank European Bioplastics for their valuable contribution.References
- A. L. Andrady and M. A. Neal, Philos. Trans. R. Soc., B, 2009, 364, 1977–1984 CrossRef CAS
.
- R. C. Thompson, S. H. Swan, C. J. Moore and F. S. vom Saal, Philos. Trans. R. Soc. Lond., B, 2009, 364, 1973–1976 CrossRef
-
Plastics Europe, Plastics - The facts 2020: An analysis of European plastics production, demand and waste data, Frankfurt, 2020 Search PubMed
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed
-
Institute for Bioplastics, Biocomposites, Biopolymers: facts and statistics, Hannover, 2019 Search PubMed
- T. Narancic, S. Verstichel, S. Reddy Chaganti, L. Morales-Gamez, S. T. Kenny, B. De Wilde, R. Babu Padamati and K. E. O'Connor, Environ. Sci. Technol., 2018, 52, 10441–10452 CrossRef CAS PubMed
- R. Wei, T. Tiso, J. Bertling, K. O'Connor, L. M. Blank and U. T. Bornscheuer, Nat. Catal., 2020, 3, 867–871 CrossRef CAS
- S. Spierling, V. Venkatachalam, M. Mudersbach, N. Becker, C. Herrmann and H.-J. Endres, Resources, 2020, 9, 90 CrossRef
- S. Spierling, C. Röttger, V. Venkatachalam, M. Mudersbach, C. Herrmann and H.-J. Endres, Procedia CIRP, 2018, 69, 573–578 CrossRef
- Directive (EU) 2018/852 of the European Parliament and of the Council of 30 May 2018 amending Directive 94/62/EC on packaging and packaging waste, 2018.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 1–11 CrossRef
-
P. Quicker, Waste, 3. Waste Management, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2020, vol. 6, pp. 1–15 Search PubMed
- M. G. Davidson, R. A. Furlong and M. C. McManus, J. Cleaner Prod., 2021, 293, 126163 CrossRef CAS
-
European Commission, Directorate-General for Research and Innovation, R. Koopmans, K. Doorsselaer, C. Velis, B. De Wilde, A. Ritschkoff, M. Crippa, J. Leyssens, M. Wagner and J. Muncke, A circular economy for plastics : insights from research and innovation to inform policy and funding decisions, Publications Office, 2019 Search PubMed
-
Zero Waste Europe, El Dorado of Chemical Recycling: State of play and policy challenges, Brussels, 2019 Search PubMed
-
A. Feil and T. Pretz, in Plastic Waste and Recycling, Elsevier, 2020, pp. 283–319, DOI:10.1016/b978-0-12-817880-5.00011-6
-
M. Niaounakis, in Recycling of Flexible Plastic Packaging, Elsevier, 2020, pp. 343–368, DOI:10.1016/b978-0-12-816335-1.00009-8
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC
- N. von der Assen, J. Jung and A. Bardow, Energy Environ. Sci., 2013, 6, 2721–2734 RSC
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed
-
CE Delft, Exploration chemical
recycling – Extended summary: What is the potential contribution of chemical recycling to Dutch climate policy?, Delft, 2020 Search PubMed
-
R. Geyer, in Plastic Waste and Recycling, Elsevier, 2020, pp. 13–32, DOI:10.1016/b978-0-12-817880-5.00002-5
-
M. Pohjakallio, in Plastic Waste and Recycling, Elsevier, 2020, pp. 467–479, DOI:10.1016/b978-0-12-817880-5.00018-9
- Directive 2008/98/EC of the European Parliament and of the Council of 19 November 2008 on waste and repealing certain Directives, 2018.
-
M. Pohjakallio, T. Vuorinen and A. Oasmaa, in Plastic Waste and Recycling, Elsevier, 2020, pp. 359–384, DOI:10.1016/b978-0-12-817880-5.00013-x
-
D. Tan, J. Yin and G. Q. Chen, in Current Developments in Biotechnology and Bioengineering, ed. A. Pandey, S. Negi and C. R. Soccol, Elsevier, 2017, pp. 655–692, DOI:10.1016/B978-0-444-63662-1.00029-4
- Y. K. Jung, T. Y. Kim, S. J. Park and S. Y. Lee, Biotechnol. Bioeng., 2010, 105, 161–171 CrossRef CAS PubMed
- Y. K. Jung and S. Y. Lee, J. Biotechnol., 2011, 151, 94–101 CrossRef CAS PubMed
-
K. Masutani and Y. Kimura, in Poly(lactic acid) Science and Technology: Processing, Properties, Additives and Applications, The Royal Society of Chemistry, 2015, pp. 1–36, 10.1039/9781782624806-00001
- H. Liu, F. Xie, L. Yu, L. Chen and L. Li, Prog. Polym. Sci., 2009, 34, 1348–1368 CrossRef CAS
- N. F. Zaaba and M. Jaafar, Polym. Eng. Sci., 2020, 60, 2061–2075 CrossRef CAS
- S. Khalid, L. Yu, L. Meng, H. Liu, A. Ali and L. Chen, J. Appl. Polym. Sci., 2017, 134, 45504 CrossRef
- L. Averous, L. Moro, P. Dole and C. Fringant, Polymer, 2000, 41, 4157–4167 CrossRef CAS
- V. Arunyagasemsuke, S. Suttiruengwong and M. Seadan, Adv. Mater. Res., 2012, 488–489, 57–61 CAS
- J. Jian, Z. Xiangbin and H. Xianbo, Adv. Ind. Eng. Polym. Res., 2020, 3, 19–26 Search PubMed
- A. Burgard, M. J. Burk, R. Osterhout, S. Van Dien and H. Yim, Curr. Opin. Biotechnol., 2016, 42, 118–125 CrossRef CAS
- T. U. Chae, J. H. Ahn, Y.-S. Ko, J. W. Kim, J. A. Lee, E. H. Lee and S. Y. Lee, Metab. Eng., 2020, 58, 2–16 CrossRef CAS PubMed
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC
-
M. Kaltschmitt, in Energy from Organic Materials (Biomass), ed. M. Kaltschmitt, Springer New York, New York, NY, 2019, vol. 92, ch. 244, pp. 353–389 Search PubMed
-
P. Quicker, Waste, 7. Thermal Treatment, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2020, vol. 6, pp. 1–54 Search PubMed
-
C. Heermann, F. Schwager and K. Whiting, The market for pyrolysis & gasification of solid wastes: A worldwide technology & business review, Juniper Consultancy Services Ltd, Uley, Gloucestershire, England, 2000 Search PubMed
-
C. Higman and M. van der Burgt, Gasification, Elsevier/GPP Gulf Professional Publ, Amsterdam, 2. edn, 2008 Search PubMed
- N. Fackler, B. D. Heijstra, B. J. Rasor, H. Brown, J. Martin, Z. Ni, K. M. Shebek, R. R. Rosin, S. D. Simpson, K. E. Tyo, R. J. Giannone, R. L. Hettich, T. J. Tschaplinski, C. Leang, S. D. Brown, M. C. Jewett and M. Köpke, Annu. Rev. Chem. Biomol. Eng., 2021, 12, 439–470 CrossRef PubMed
- F. M. Lamberti, L. A. Román-Ramírez and J. Wood, J. Polym. Environ., 2020, 28, 2551–2571 CrossRef CAS
- Y. Aoyagi, K. Yamashita and Y. Doi, Polym. Degrad. Stab., 2002, 76, 53–59 CrossRef CAS
- F.-D. Kopinke, M. Remmler, K. Mackenzie, M. Möder and O. Wachsen, Polym. Degrad. Stab., 1996, 53, 329–342 CrossRef CAS
- L. Feng, S. Feng, X. Bian, G. Li and X. Chen, Polym. Degrad. Stab., 2018, 157, 212–223 CrossRef CAS
- A. Babanalbandi, D. J. T. Hill, D. S. Hunter and L. Kettle, Polym. Int., 1999, 48, 980–984 CrossRef CAS
- H. Zou, C. Yi, L. Wang, H. Liu and W. Xu, J. Therm. Anal. Calorim., 2009, 97, 929–935 CrossRef CAS
- J. D. Badia, L. Santonja-Blasco, A. Martínez-Felipe and A. Ribes-Greus, Bioresour. Technol., 2012, 111, 468–475 CrossRef CAS PubMed
- A. Undri, L. Rosi, M. Frediani and P. Frediani, J. Anal. Appl. Pyrolysis, 2014, 110, 55–65 CrossRef CAS
- H. Nishida, T. Mori, S. Hoshihara, Y. Fan, Y. Shirai and T. Endo, Polym. Degrad. Stab., 2003, 81, 515–523 CrossRef CAS
- S. Zhang, Y. Liang, X. Qian, D. Hui and K. Sheng, Nanotechnol. Rev., 2020, 9, 524–533 CrossRef CAS
- E. Ozdemir, T. Tinçer and J. Hacaloglu, J. Anal. Appl. Pyrolysis, 2016, 122, 315–322 CrossRef CAS
- G. Wang and A. Li, Chin. J. Chem. Eng., 2008, 16, 929–933 CrossRef CAS
- S. Xiang, L. Feng, X. Bian, G. Li and X. Chen, Polym. Test., 2020, 81, 106211 CrossRef CAS
- N. Grassie, E. J. Murray and P. A. Holmes, Polym. Degrad. Stab., 1984, 6, 47–61 CrossRef CAS
- A. Gonzalez, L. Irusta, M. J. Fernández-Berridi, M. Iriarte and J. J. Iruin, Polym. Degrad. Stab., 2005, 87, 347–354 CrossRef CAS
- H. Ariffin, H. Nishida, Y. Shirai and M. A. Hassan, Polym. Degrad. Stab., 2010, 95, 1375–1381 CrossRef CAS
- J. M. Clark, H. M. Pilath, A. Mittal, W. E. Michener, D. J. Robichaud and D. K. Johnson, J. Phys. Chem. A, 2016, 120, 332–345 CrossRef CAS PubMed
- F.-D. Kopinke, M. Remmler and K. Mackenzie, Polym. Degrad. Stab., 1996, 52, 25–38 CrossRef CAS
- K. Chrissafis, K. M. Paraskevopoulos and D. N. Bikiaris, Thermochim. Acta, 2005, 435, 142–150 CrossRef CAS
- Y.-F. Shih and Y.-C. Chieh, Macromol. Theory Simul., 2007, 16, 101–110 CrossRef CAS
- H. Sato, M. Furuhashi, D. Yang, H. Ohtani, S. Tsuge, M. Okada, K. Tsunoda and K. Aoi, Polym. Degrad. Stab., 2001, 73, 327–334 CrossRef CAS
- J. Fojt, J. David, R. Přikryl, V. Řezáčová and J. Kučerík, Sci. Total Environ., 2020, 745, 140975 CrossRef CAS PubMed
- X. Li, Z. Cai, X. Wang, Z. Zhang, D. Tan, L. Xie, H. Sun and G. Zhong, Appl. Clay Sci., 2020, 196, 105762 CrossRef CAS
- J. C. S. Sales, A. G. Santos, A. M. de Castro and M. A. Z. Coelho, World J. Microbiol. Biotechnol., 2021, 37, 116 CrossRef CAS
- V. de Lorenzo, Environ. Microbiol., 2018, 20, 1910–1916 CrossRef PubMed
- A. Singh, N. A. Rorrer, S. R. Nicholson, E. Erickson, J. S. DesVeaux, A. F. T. Avelino, P. Lamers, A. Bhatt, Y. Zhang, G. Avery, L. Tao, A. R. Pickford, A. C. Carpenter, J. E. McGeehan and G. T. Beckham, Joule, 2021, 5, 2479–2503 CrossRef CAS
- R. Meys, F. Frick, S. Westhues, A. Sternberg, J. Klankermayer and A. Bardow, Resour., Conserv. Recycl., 2020, 162, 105010 CrossRef
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed
- R. Wei and W. Zimmermann, Microb. Biotechnol., 2017, 10, 1308–1322 CrossRef CAS PubMed
- T. Sooksai, H. Punnapayak, S. Prasongsuk and U. Sangwatanaroj, Int. J. Chem. Eng. Appl., 2017, 8, 367–370 CAS
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS PubMed
- M. Salvador, U. Abdulmutalib, J. Gonzalez, J. Kim, A. A. Smith, J.-L. Faulon, R. Wei, W. Zimmermann and J. I. Jimenez, Genes, 2019, 10, 373 CrossRef PubMed
- J. Liu, J. He, R. Xue, B. Xu, X. Qian, F. Xin, L. M. Blank, J. Zhou, R. Wei, W. Dong and M. Jiang, Biotechnol. Adv., 2021, 48, 107730 CrossRef CAS PubMed
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS
- T. Tiso, T. Narancic, R. Wei, E. Pollet, N. Beagan, K. Schröder, A. Honak, M. Jiang, S. T. Kenny, N. Wierckx, R. Perrin, L. Avérous, W. Zimmermann, K. O'Connor and L. M. Blank, Metab. Eng., 2021, 66, 167–178 CrossRef CAS
- Q. Huang, M. Hiyama, T. Kabe, S. Kimura and T. Iwata, Biomacromolecules, 2020, 21, 3301–3307 CrossRef CAS
- H. P. Austin, M. D. Allen, B. S. Donohoe, N. A. Rorrer, F. L. Kearns, R. L. Silveira, B. C. Pollard, G. Dominick, R. Duman, K. El Omari, V. Mykhaylyk, A. Wagner, W. E. Michener, A. Amore, M. S. Skaf, M. F. Crowley, A. W. Thorne, C. W. Johnson, H. L. Woodcock, J. E. McGeehan and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4350–E4357 CrossRef CAS PubMed
- S. Joo, I. J. Cho, H. Seo, H. F. Son, H.-Y. Sagong, T. J. Shin, S. Y. Choi, S. Y. Lee and K.-J. Kim, Nat. Commun., 2018, 9, 382 CrossRef PubMed
- R. Wei, T. Oeser, J. Schmidt, R. Meier, M. Barth, J. Then and W. Zimmermann, Biotechnol. Bioeng., 2016, 113, 1658–1665 CrossRef CAS
- D. Danso, C. Schmeisser, J. Chow, W. Zimmermann, R. Wei, C. Leggewie, X. Li, T. Hazen, W. R. Streit and R. E. Parales, Appl. Environ. Microbiol., 2018, 84, e02773 CrossRef CAS PubMed
- S. Francis CF, S. Rebello, M. E. Aneesh, R. Sindhu, P. Binod, S. Singh and A. Pandey, Bioengineered, 2021, 12, 1040–1053 CrossRef
- J.-I. Kadokawa and S. Kobayashi, Curr. Opin. Chem. Biol., 2010, 14, 145–153 CrossRef CAS PubMed
- S. Lajus, S. Dusséaux, J. Verbeke, C. Rigouin, Z. Guo, M. Fatarova, F. Bellvert, V. Borsenberger, M. Bressy, J. M. Nicaud, A. Marty and F. Bordes, Front. Bioeng. Biotechnol., 2020, 8, 954 CrossRef PubMed
- S. Y. Choi, M. N. Rhie, H. T. Kim, J. C. Joo, I. J. Cho, J. Son, S. Y. Jo, Y. J. Sohn, K. A. Baritugo, J. Pyo, Y. Lee, S. Y. Lee and S. J. Park, Metab. Eng., 2020, 58, 47–81 CrossRef CAS
- D. Jendrossek and R. Handrick, Annu. Rev. Microbiol., 2002, 56, 403–432 CrossRef CAS
- S. Y. Lee and J.-I. Choi, Waste Manage., 1999, 19, 133–139 CrossRef CAS
- V. Martínez, F. D. L. Peña, J. García-Hidalgo, I. D. L. Mata, J. L. García and M. A. Prieto, Appl. Environ. Microbiol., 2012, 78, 6017–6026 CrossRef
- A. A. Chowdhury, Arch. Mikrobiol., 1963, 47, 167–200 CAS
- H. S. Azevedo, F. M. Gama and R. L. Reis, Biomacromolecules, 2003, 4, 1703–1712 CrossRef CAS PubMed
- P. W. Wallace, K. Haernvall, D. Ribitsch, S. Zitzenbacher, M. Schittmayer, G. Steinkellner, K. Gruber, G. M. Guebitz and R. Birner-Gruenberger, Appl. Microbiol. Biotechnol., 2017, 101, 2291–2303 CrossRef CAS
- P. Soulenthone, Y. Tachibana, M. Suzuki, T. Mizuno, Y. Ohta and K.-I. Kasuya, Polym. Degrad. Stab., 2021, 184, 109481 CrossRef CAS
- R. N. C. Utomo, W.-J. Li, T. Tiso, C. Eberlein, M. Doeker, H. J. Heipieper, A. Jupke, N. Wierckx and L. M. Blank, ACS Sustainable Chem. Eng., 2020, 8, 17466–17474 CrossRef
- Y. Tokiwa, B. P. Calabia, C. U. Ugwu and S. Aiba, Int. J. Mol. Sci., 2009, 10, 3722–3742 CrossRef CAS
- A. Jarerat and Y. Tokiwa, Biotechnol. Lett., 2001, 23, 647–651 CrossRef CAS
- B. C. Almeida, P. Figueiredo and A. T. P. Carvalho, ACS Omega, 2019, 4, 6769–6774 CrossRef CAS
- P. Muller, Pure Appl. Chem., 1994, 66, 1077–1184 CrossRef
- S. Lazzari, F. Codari, G. Storti, M. Morbidelli and D. Moscatelli, Polym. Degrad. Stab., 2014, 110, 80–90 CrossRef CAS
- C. Y. Tham, Z. A. Abdul Hamid, Z. Ahmad and H. Ismail, Adv. Mater. Res., 2014, 970, 324–327 Search PubMed
- T. Ivanova, I. Panaiotov, F. Boury, J. E. Proust, J. P. Benoit and R. Verger, Colloids Surf., B, 1997, 8, 217–225 CrossRef CAS
- C. S. Proikakis, N. J. Mamouzelos, P. A. Tarantili and A. G. Andreopoulos, Polym. Degrad. Stab., 2006, 91, 614–619 CrossRef CAS
- G. L. Siparsky, K. J. Voorhees and F. Miao, J. Environ. Polym. Degrad., 1998, 6, 31–41 CrossRef CAS
- J. R. Rocca-Smith, N. Chau, D. Champion, C. H. Brachais, E. Marcuzzo, A. Sensidoni, F. Piasente, T. Karbowiak and F. Debeaufort, Food Chem., 2017, 236, 109–119 CrossRef CAS PubMed
- I. Osaka, M. Watanabe, M. Takama, M. Murakami and R. Arakawa, J. Mass Spectrom., 2006, 41, 1369–1377 CrossRef CAS
- H. Tsuji, H. Daimon and K. Fujie, Biomacromolecules, 2003, 4, 835–840 CrossRef CAS
- H. Tsuji, T. Saeki, T. Tsukegi, H. Daimon and K. Fujie, Polym. Degrad. Stab., 2008, 93, 1956–1963 CrossRef CAS
- G.-E. Yu and R. H. Marchessault, Polymer, 2000, 41, 1087–1098 CrossRef CAS
- S. Yoshioka, A. Kishida, S. Izumikawa, Y. Aso and Y. Takeda, J. Controlled Release, 1991, 16, 341–348 CrossRef CAS
- Y. Li and T. J. Strathmann, Green Chem., 2019, 21, 5586–5597 RSC
- H. Mitomo, Transaction, 1992, 48, 595–601 CAS
- T. Kijchavengkul, R. Auras, M. Rubino, S. Selke, M. Ngouajio and R. T. Fernandez, Polym. Degrad. Stab., 2010, 95, 2641–2647 CrossRef CAS
- R. Muthuraj, M. Misra and A. K. Mohanty, J. Appl. Polym. Sci., 2015, 132, 42189 CrossRef
- K. Fukushima, A. Rasyida and M.-C. Yang, Appl. Clay Sci., 2013, 80–81, 291–298 CrossRef CAS
- K. Cho, J. Lee and K. Kwon, J. Appl. Polym. Sci., 2001, 79, 1025–1033 CrossRef CAS
- H.-S. Kim, H.-J. Kim and D. Cho, J. Therm. Anal. Calorim., 2009, 96, 211–218 CrossRef CAS
- Y.-P. Wang, Y.-J. Xiao, J. Duan, J.-H. Yang, Y. Wang and C.-L. Zhang, Polym. Bull., 2015, 73, 1067–1083 CrossRef
- J. Zhou, X. Wang, K. Hua, C. E. Duan, W. Zhang, J. Ji and X. Yang, Iran. Polym. J., 2013, 22, 267–275 CrossRef CAS
- A. Carvalho, M. Zambon, A. Dasilvacurvelo and A. Gandini, Carbohydr. Polym., 2005, 62, 387–390 CrossRef CAS
- J. B. Olivato, M. V. Grossmann, A. P. Bilck and F. Yamashita, Carbohydr. Polym., 2012, 90, 159–164 CrossRef CAS PubMed
- H. Tsuji and T. Ishizaka, J. Appl. Polym. Sci., 2001, 80, 2281–2291 CrossRef CAS
- J. M. Meseguer-Duenas, J. Mas-Estelles, I. Castilla-Cortazar, J. L. Escobar Ivirico and A. Vidaurre, J. Mater. Sci.: Mater. Med., 2011, 22, 11–18 CrossRef CAS
- S. Westhues, J. Idel and J. Klankermayer, Sci. Adv., 2018, 4, eaat9669 CrossRef CAS PubMed
- E. M. Krall, T. W. Klein, R. J. Andersen, A. J. Nett, R. W. Glasgow, D. S. Reader, B. C. Dauphinais, S. P. Mc Ilrath, A. A. Fischer, M. J. Carney, D. J. Hudson and N. J. Robertson, Chem. Commun., 2014, 50, 4884–4887 RSC
- T.-O. Kindler, C. Alberti, E. Fedorenko, N. Santangelo and S. Enthaler, ChemistryOpen, 2020, 9, 401–404 CrossRef CAS
- A. Hermann, T. Becker, M. A. Schäfer, A. Hoffmann and S. Herres-Pawlis, ChemSusChem, 2022, 11–18 Search PubMed
- M. Fuchs, M. Walbeck, E. Jagla, A. Hoffmann and S. Herres-Pawlis, ChemPlusChem, 2022, 87, e202200029 CAS
- J. Stewart, M. Fuchs, J. Payne, O. Driscoll, G. Kociok-Köhn, B. D. Ward, S. Herres-Pawlis and M. D. Jones, RSC Adv., 2022, 12, 1416–1424 RSC
- A. C. Fernandes, ChemSusChem, 2021, 14, 4228–4233 CrossRef CAS PubMed
- L. Monsigny, J.-C. Berthet and T. Cantat, ACS Sustainable Chem. Eng., 2018, 6, 10481–10488 CrossRef CAS
- E. Feghali and T. Cantat, ChemSusChem, 2015, 8, 980–984 CrossRef CAS
- H. Zhang, S. Kang, J. Li, Z. Li, J. Chang, Y. Xu, G. Max Lu and C. Sun, Fuel, 2021, 286, 119405 CrossRef CAS
- H. Zhang, S. Kang, J. Li, Z. Li, J. Chang, Y. Xu, G. M. Lu and C. Sun, Fuel, 2021, 286, 119405 CrossRef CAS
- S. J. Organ and P. J. Barham, Polymer, 1993, 34, 2169–2174 CrossRef CAS
- M. S. Lehnertz, J. B. Mensah and R. Palkovits, Green Chem., 2022, 24, 3957–3963 RSC
- R. Palkovits, K. Tajvidi, J. Procelewska, R. Rinaldi and A. Ruppert, Green Chem., 2010, 12, 972–978 RSC
- R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576–578 RSC
- P. J. C. Hausoul, L. Negahdar, K. Schute and R. Palkovits, ChemSusChem, 2015, 8, 3323–3330 CrossRef CAS
- P. J. C. Hausoul, A. K. Beine, L. Neghadar and R. Palkovits, Catal. Sci. Technol., 2017, 7, 56–63 RSC
- A. K. Beine, A. J. D. Krüger, J. Artz, C. Weidenthaler, C. Glotzbach, P. J. C. Hausoul and R. Palkovits, Green Chem., 2018, 20, 1316–1322 RSC
- M. Niaounakis, Eur. Polym. J., 2019, 114, 464–475 CrossRef CAS
-
T. Ito, J. Tanabe and T. Taguchi, Lactide recovery apparatus and recovery method, 2010126490A, 2008 Search PubMed
-
H. Nishida, Y. Fan and Y. Shirai, Method for recovering lactide from polylactic acid product, 07309863A, 1994 Search PubMed
- N. Wierckx, M. A. Prieto, P. Pomposiello, V. de Lorenzo, K. O'Connor and L. M. Blank, Microb. Biotechnol., 2015, 8, 900–903 CrossRef
- A. Z. Werner, R. Clare, T. D. Mand, I. Pardo, K. J. Ramirez, S. J. Haugen, F. Bratti, G. N. Dexter, J. R. Elmore, J. D. Huenemann, G. L. T. Peabody, C. W. Johnson, N. A. Rorrer, D. Salvachúa, A. M. Guss and G. T. Beckham, Metab. Eng., 2021, 67, 250–261 CrossRef CAS
- S. T. Kenny, J. N. Runic, W. Kaminsky, T. Woods, R. P. Babu, C. M. Keely, W. Blau and K. E. O'Connor, Environ. Sci. Technol., 2008, 42, 7696–7701 CrossRef CAS PubMed
- T. Tiso, B. Winter, R. Wei, J. Hee, J. de Witt, N. Wierckx, P. Quicker, U. T. Bornscheuer, A. Bardow, J. Nogales and L. M. Blank, Metab. Eng., 2021, 71, 77–98 CrossRef PubMed
- A. Magnin, L. Entzmann, A. Bazin, E. Pollet and L. Averous, ChemSusChem, 2021, 14, 4234–4241 CrossRef CAS
- A. Magnin, L. Entzmann, E. Pollet and L. Avérous, Waste Manage., 2021, 132, 23–30 CrossRef CAS PubMed
- K. Beydoun and J. Klankermayer, Chem. – Eur. J., 2019, 25, 11412–11415 CrossRef CAS
- J. B. Mensah, A. H. Hergesell, S. Brosch, C. Golchert, J. Artz and R. Palkovits, Green Chem., 2020, 22, 3522–3531 RSC
- J. Meyers, J. B. Mensah, F. J. Holzhäuser, A. Omari, C. C. Blesken, T. Tiso, S. Palkovits, L. M. Blank, S. Pischinger and R. Palkovits, Energy Environ. Sci., 2019, 12, 2406–2411 RSC
- X. Qi, Y. Ren and X. Wang, Int. Biodeterior. Biodegrad., 2017, 117, 215–223 CrossRef CAS
- K.-L. G. Ho, A. L. Pometto, A. Gadea-Rivas, J. A. Briceño and A. Rojas, J. Environ. Polym. Degrad., 1999, 7, 173–177 CrossRef CAS
- I. E. Meyer-Cifuentes, J. Werner, N. Jehmlich, S. E. Will, M. Neumann-Schaal and B. Öztürk, Nat. Commun., 2020, 11, 5790 CrossRef CAS
- V. Perz, A. Hromic, A. Baumschlager, G. Steinkellner, T. Pavkov-Keller, K. Gruber, K. Bleymaier, S. Zitzenbacher, A. Zankel, C. Mayrhofer, C. Sinkel, U. Kueper, K. Schlegel, D. Ribitsch and G. M. Guebitz, Environ. Sci. Technol., 2016, 50, 2899–2907 CrossRef CAS PubMed
- T. Kijchavengkul, R. Auras, M. Rubino, M. Ngouajio and R. T. Fernandez, Chemosphere, 2008, 71, 1607–1616 CrossRef CAS PubMed
- R.-J. Müller, I. Kleeberg and W.-D. Deckwer, J. Biotechnol., 2001, 86, 87–95 CrossRef
- T. F. Astrup, D. Tonini, R. Turconi and A. Boldrin, Waste Manage., 2015, 37, 104–115 CrossRef CAS
- A. Laurent, I. Bakas, J. Clavreul, A. Bernstad, M. Niero, E. Gentil, M. Z. Hauschild and T. H. Christensen, Waste Manage., 2014, 34, 573–588 CrossRef
-
ISO, 14040: Environmental management - Life cycle assessment – Principles and framework, 2006 Search PubMed
-
ISO, 14044: Environmental management – Life cycle assessment – Requirements and guidelines, 2006 Search PubMed
- D. Maga, M. Hiebel and V. Aryan, Sustainability, 2019, 11, 5324 CrossRef CAS
- G. Bishop, D. Styles and P. N. L. Lens, Resour., Conserv. Recycl., 2021, 168, 105451 CrossRef CAS
- L. Rigamonti, A. Falbo, L. Zampori and S. Sala, Int. J. Life Cycle Assess., 2017, 22, 1278–1287 CrossRef
- S. Spierling, E. Knüpffer, H. Behnsen, M. Mudersbach, H. Krieg, S. Springer, S. Albrecht, C. Herrmann and H.-J. Endres, J. Cleaner Prod., 2018, 185, 476–491 CrossRef
- S. Walker and R. Rothman, J. Cleaner Prod., 2020, 261, 121158 CrossRef CAS
- H. Jeswani, C. Krüger, M. Russ, M. Horlacher, F. Antony, S. Hann and A. Azapagic, Sci. Total Environ., 2021, 769, 144483 CrossRef CAS
- M. F. Cosate de Andrade, P. M. S. Souza, O. Cavalett and A. R. Morales, J. Polym. Environ., 2016, 24, 372–384 CrossRef CAS
- V. Piemonte, S. Sabatini and F. Gironi, J. Polym. Environ., 2013, 21, 640–647 CrossRef CAS
- S. Papong, P. Malakul, R. Trungkavashirakun, P. Wenunun, T. Chom-in, M. Nithitanakul and E. Sarobol, J. Cleaner Prod., 2014, 65, 539–550 CrossRef CAS
- J. Yano, Y. Hirai, S.-I. Sakai and J. Tsubota, Waste Manage. Res., 2014, 32, 304–316 CrossRef CAS PubMed
- S. Sudarsan, S. Dethlefsen, L. M. Blank, M. Siemann-Herzberg and A. Schmid, Appl. Environ. Microbiol., 2014, 80(17), 5292–5303 CrossRef
- European Bioplastics, Bioplastics Market Development Update 2021, 2021 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |