
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hemicellulose content affects the properties of cellulose nanofibrils produced from softwood pulp fibres by LPMO†
Salla
Koskela
 ab,
Li
Zha
a,
Shennan
Wang
a,
Max
Yan
c and
Qi
Zhou
*ab
ab,
Li
Zha
a,
Shennan
Wang
a,
Max
Yan
c and
Qi
Zhou
*ab
aDivision of Glycoscience, Department of Chemistry, KTH Royal Institute of Technology, AlbaNova University Centre, SE-106 91 Stockholm, Sweden. E-mail: qi@kth.se
bWallenberg Wood Science Center, Department of Fiber and Polymer Technology, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden
cDepartment of Applied Physics, KTH Royal Institute of Technology, SE-114 19 Stockholm, Sweden
First published on 15th August 2022
Abstract
Lytic polysaccharide monooxygenase (LPMO)-catalysed oxidation of cellulose has emerged as a green alternative to chemical modifications in the production of cellulose nanofibrils (CNFs) from wood pulp fibres. The effect of the hemicellulose content of the starting pulp fibres on the oxidation capabilities of cellulose-active LPMO is important and has not been investigated previously. In this study, the production of LPMO-oxidised CNFs was evaluated on two commercial softwood pulp fibres with different hemicellulose contents. Thin and colloidally stable CNFs were readily obtained from kraft pulp with a hemicellulose content of 16%. The preserved hemicellulose fraction in the kraft pulp enhanced the access of LPMO into the fibre cell wall, enabling the production of homogeneous CNFs with a thin width of 3.7 ± 1.7 nm. By contrast, the LPMO-oxidised dissolving pulp with a lower hemicellulose content of 4% could only be partially disintegrated into thin CNFs, leaving a large amount of cellulose microfibril aggregates with widths of around 50 to 100 nm. CNFs disintegrated from the LPMO-oxidised kraft pulp could be processed into nanopapers with excellent properties including an optical transmittance of 86%, tensile strength of 260 MPa, and Young's modulus of 16.9 GPa. Such CNFs also showed acid-triggered nanofibril gelation owing to the introduced carboxyl groups on cellulose microfibril surfaces. These results indicate that the inherent hemicelluloses present in the wood cell wall are essential for LPMO-mediated CNF production from wood pulp fibres.
Introduction
Renewable origin, biodegradability, light weight, and suitability for an astonishingly wide variety of advanced materials have made CNFs particularly popular in the development of renewable materials and credible rivals to synthetic polymers.1,2 Processing of wood pulp fibres into CNFs has become industrially appealing, and several pilot plants for commercial production of CNFs have already been established.3 In a typical process to produce CNFs, wood pulp fibres are chemically treated to become more susceptible to disintegration. Subsequently, the fibre slurry is passed through a microfluidizer that disintegrates the fibre cell wall into thin, nanoscale filaments. However, to produce truly sustainable nanocellulose-based materials, the environmental impact of the manufacturing process should also be considered. Life-cycle assessments have indicated that nanocellulose has a much larger environmental footprint than its raw material, pulp fibre. In addition to the mechanical disintegration step, a large part of this environmental burden results from the chemical pre-treatment, emphasising the urgent need to seek more sustainable solutions.4Enzymatic pre-treatment has long been recognised as an environmentally friendly alternative to chemical modifications and has been successfully used to alter various properties of wood pulp fibres. By working under mild, aqueous reaction conditions, enzymes can potentiate considerable reductions in the environmental impact of industrial processes.5 In CNF production, a classical approach has been the pre-treatment of the pulp fibres with monocomponent endoglucanases which impair the integrity of the fibre cell wall and facilitate subsequent mechanical disintegration.6–8 As the degree of polymerisation of cellulose rapidly decreases while its surface chemistry is not altered by the hydrolytic endoglucanase reaction,9 multiple passes of endoglucanase hydrolysed pulp fibres through the microfluidizer are still required in order to produce CNFs.7 In addition, the lack of stabilising surface charges makes the obtained CNFs prone to aggregate, forming bundles of filaments.
The recent discovery of lytic polysaccharide monooxygenases (LPMOs) has provided a unique opportunity for the oxidative modification of cellulose without the use of harsh chemicals.10 In particular, cellulose-active C1-oxidising LPMOs have piqued interest in the green production of CNFs, as they can introduce charged carboxyl groups on cellulose microfibril surfaces.11 This resembles one of the common chemical pre-treatment methods, 2,2,6,6-tetramethylpyperidine-1-oxy radical (TEMPO)-mediated oxidation, which selectively oxidises the C6 primary hydroxyl groups of cellulose to carboxyl groups.12 Compared to other cellulose-active enzymes, the reaction mechanism of LPMOs is rather different. LPMOs contain catalytic copper in their active site.13 Binding onto cellulose, LPMOs employ a radical oxygen species to catalyse selective hydroxylation of the glucose moiety. This results in destabilization and cleavage of the β-1,4 glycosidic bond and formation of two new cellulose chain ends, one native and the other modified.14 In the case of C1-oxidising LPMOs, the modified reducing end contains an aldonic acid, i.e. gluconic acid.15 Therefore, the requirements for the reaction to occur include an electron donor, typically ascorbic or gallic acid, for the reduction of the catalytic copper on the enzyme's active site, and an oxygen species for the formation of a copper–oxyl radical.16 Depending on the available oxygen species, which can be either O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14,17 or H2O2,18,19 LPMOs demonstrate either oxygenase or peroxygenase activity, respectively. Of these two possible co-substrates, H2O2 has been shown to result in higher reaction rates.20,21
14,17 or H2O2,18,19 LPMOs demonstrate either oxygenase or peroxygenase activity, respectively. Of these two possible co-substrates, H2O2 has been shown to result in higher reaction rates.20,21
Owing to the newly-formed cellulose chain ends with an aldonic acid functionality, the pulp fibres oxidised by C1-oxidising LPMOs can be mechanically disintegrated by using mild disintegration methods and the produced CNFs show colloidal stability in aqueous suspension.22–25 The ability of C1-oxidising LPMOs to boost cellulose nanofibrillation has already been demonstrated on both softwood22,23 and hardwood pulp fibres.24,25 Particularly, in our previous work, the nanofibrillation extent of LPMO-treated wood holocellulose fibres has even reached a similar level to that of chemical pre-treatments, i.e. individualisation of nanofibrils with a width of ca. 4 nm.23 By contrast, the typical width of nanofibrils produced by hydrolytic endoglucanase pre-treatment is much larger, 10–30 nm.6–8
Although the advantages of LPMO treatment over endoglucanases have been rather well demonstrated, many aspects of the oxidation capabilities of LPMOs are still unknown, including their ability to oxidise different types of wood pulp fibres. Two of the most common commercial wood pulps, dissolving pulp and kraft pulp, have different chemical compositions. Dissolving pulp is produced by an acid pre-hydrolysis step to remove hemicelluloses, yielding fibres comprising 90–92% cellulose and 3–6% hemicellulose.26 Kraft pulp retains a substantial amount of hemicellulose, approximately 17%.27 This hemicellulose fraction has been shown to reduce hornification in the pulp fibre, increase its water retention, and promote re-swelling of dry pulp in water.28–30 Interestingly, numerous studies have indicated that hemicellulose also has a significant impact on both chemical modifications and the performance of cellulose-active enzymes on pulp fibres. Kraft pulp fibres rich in hemicellulose are generally regarded as highly accessible for chemical modifications, such as TEMPO-oxidation.12 Conversely, pure cellulosic substrates, including bacterial and tunicate cellulose, often show low chemical modification yields.31 By contrast, the effect of hemicellulose has been the opposite for enzymatic modification. Substrates enriched in hemicellulose have shown lower hydrolysis yields by cellulases than pure cellulose, indicating that hemicellulose might form steric hindrance on cellulose microfibril surfaces preventing enzyme binding.32,33 In addition, a recent study found that LPMO released more soluble products from bacterial cellulose than hemicellulose-containing pulp fibres.34 Therefore, it could be expected that the presence of hemicellulose has a profound impact on the accessibility of wood pulp fibres toward LPMOs.
In this work, we aimed to clarify the significance of the starting material composition in the production of CNFs by enzymatic pre-treatment with LPMO. For this, a C1-oxidising TtLPMO9E originating from the ascomycetous fungus Thermothielavioides terrestris was produced in Pichia pastoris, and its optimal reaction conditions were determined. LPMO was used to oxidise both dissolving pulp and kraft pulp fibres. After the LPMO treatment, the pulp fibres were homogenised in a microfluidizer, and the morphology and properties of the resulting CNFs were characterised. In addition, the obtained CNFs were further used to prepare cellulose nanopapers and hydrogels, and their mechanical and rheological properties were studied.
Results
Optimal reaction conditions for TtLPMO9E
TtLPMO9E was obtained from a heterologous host P. pastoris in a good yield, approximately 125 mg L−1 of the culture supernatant as determined after purification. The apparent molecular weight of the recombinant enzyme containing a C-terminal (His)6 tag was 30 kDa (Fig. S1†). In addition to the oxidase or peroxygenase activities demonstrated by LPMOs in the presence of cellulose, LPMOs also show peroxidase activity in the presence of H2O2. The peroxidase activity has been utilised as a reporter for LPMO catalytic integrity in a spectrophotometric assay where a soluble substrate 2,6-dimethoxyphenol (2,6-DMP) is converted by LPMO into a chromogenic product at the expense of H2O2.35 This assay confirmed that the recombinantly produced TtLPMO9E was active. Even though 2,6-DMP is not a native substrate for LPMOs, the detected activity on this substrate confirmed the presence of the catalytic copper on the enzyme's active site and indicated that TtLPMO9E was correctly processed by the host.TtLPMO9E has been previously shown to be cellulose-active and C1-specific,36 while its thermostability and optimal reaction pH have not been reported. To evaluate the stability of TtLPMO9E, the enzyme was incubated for up to 72 h at various temperatures ranging from 30 to 90 °C, after which the residual activity was assayed against 2,6-DMP. The results indicated a good ability to resist heat induced inactivation. During the whole incubation time of 72 h, TtLPMO9E retained over 38% of its initial activity at temperatures up to 60 °C, while being not completely inactivated even at 90 °C (Fig. 1a). The most favourable temperature for TtLPMO9E appeared to be 40 °C, as it retained 74% of the initial activity, the highest among all the tested temperatures. In addition, a moderate increase in the initial activity (24%) was also observed after 6 to 24 h of incubation at 40 °C. It is not surprising that TtLPMO9E demonstrated good thermostability, as T. terrestris has proven to be an excellent source of thermostable enzymes,37,38 and LPMOs in general have been found to be thermally resistant.39
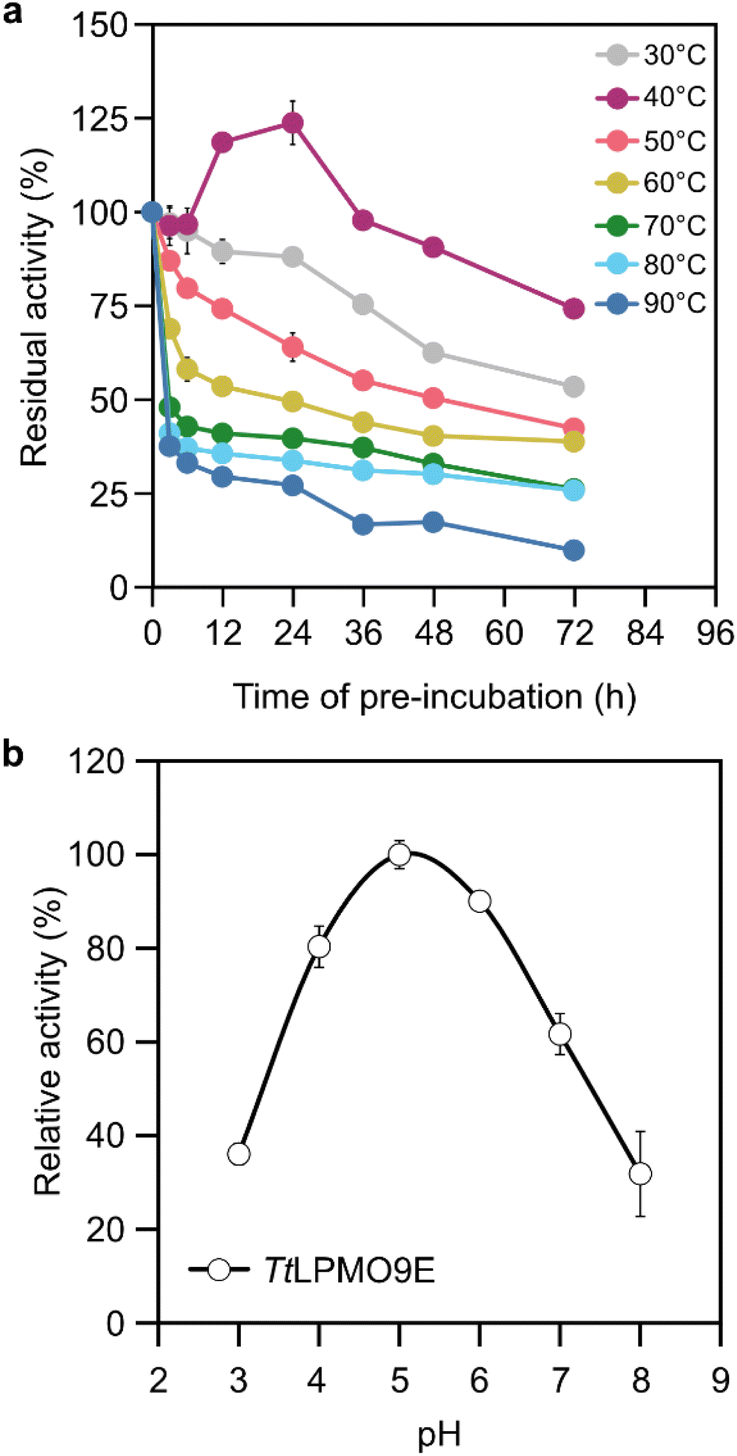 | ||
| Fig. 1 (a) Stability of TtLPMO9E after pre-incubation at various temperatures. The residual activities were measured against 2,6-DMP at pH 6.5 and 25 °C. (b) Optimal reaction pH for TtLPMO9E in a series of 50 mM sodium malonate buffers at 40 °C. The relative activities were determined as the release of glucose equivalents from microcrystalline cellulose (MCC). | ||
A model cellulosic substrate, microcrystalline cellulose (MCC) was used to probe the optimal reaction pH. Quantification of the released soluble sugars, i.e. cello-oligosaccharides during TtLPMO9E oxidation of MCC, revealed a bell-shaped profile over the studied pH range (Fig. 1b). It should be noted that the pH profile describes the overall reaction of cellulose chain cleavages, as pH has an impact to both LPMO activity and the ability of the reductant to donate an electron. By using gallic acid as the electron donor, the highest amount of soluble sugars released by TtLPMO9E was observed at pH 5.0. These optimised reaction conditions (40 °C and pH 5.0) were employed for the following reactions on pulp fibres.
Nanofibrillation of pulp fibres treated with TtLPMO9E
The commercial dissolving pulp and kraft pulp fibres originated from softwood and had hemicellulose contents of 4 and 16%, respectively, as determined by carbohydrate analysis (Table S1†). The pulp fibres were treated with TtLPMO9E in reactions containing gallic acid as an electron donor for the enzyme. After the enzymatic reactions, the solid contents were adjusted to 1% (w/v) and water slurries of the pulp fibres were disintegrated using a microfluidizer M-110EH (Microfluidics Ind., USA). After one pass through the 200 and 100 μm microfluidic chambers at a pressure of 1600 bar, the water slurries of TtLPMO9E-oxidised pulp fibres were transformed into gel-like suspensions. The CNFs disintegrated from the TtLPMO9E-oxidised dissolving pulp and kraft pulp fibres are hereafter termed D-CNFs and K-CNFs, respectively. The successful oxidation of the two pulps was confirmed by FTIR analysis (Fig. S2†). The FTIR spectra of K-CNFs and D-CNFs showed a new peak at 1595 cm−1 corresponding to the deprotonated carboxyl group in the LPMO reaction product aldonic acid, suggesting that TtLPMO9E was able to introduce carboxyl groups on both types of pulp fibres. The carboxyl contents of K-CNFs and D-CNFs were 0.22 and 0.08 mmol g−1, respectively, as measured by conductometric titration.The crystallinity index (CrI) values of the pulp fibres and the obtained CNFs were deduced by the peak height method from their XRD patterns (Fig. S3†). The CrI values for D-CNFs and K-CNFs were 58.3% and 58.2%, respectively. The CrI values for the original dissolving pulp and kraft pulp fibres were 66.6% and 63.7%, respectively. These results indicated that TtLPMO9E-oxidation combined with mechanical disintegration reduced the crystallinity of the dissolving pulp by 8.3% while the crystallinity of the kraft pulp fibres was reduced by 5.5%. The observed reduction in crystallinity suggested that TtLPMO9E mainly targeted and disrupted the crystalline regions of cellulose as previously discovered.11 However, no significant difference in crystallinity was found between the obtained D-CNFs and K-CNFs.
The yields of the CNFs were assessed as a mass recovery from the starting pulp fibres, which indicated yields of 68% and 84% for D-CNFs and K-CNFs, respectively. Carbohydrate analysis showed that the enzymatic oxidation did not significantly alter the hemicellulose content of the two pulp fibres (Table S1†). The degree of polymerisation (DP) values of the starting pulp fibres and the obtained CNFs were determined by viscometric analysis. The DP values for D-CNFs and K-CNFs were 720 and 820, respectively, while the DP values of the starting dissolving pulp and kraft pulp were 1240 and 1870, respectively. This result indicated that TtLPMO9E catalysed a relatively higher number of cellulose chain cleavages on kraft pulp than on dissolving pulp.
In addition to introducing carboxyl groups to the cellulose microfibril surfaces, the cellulose chain cleavages catalysed by LPMO should also result in the release of soluble cello-oligosaccharides.40 The release of soluble cello-oligosaccharides during the LPMO reaction can to some extent indicate the number of available LPMO binding sites, which relates to the accessibility of the pulp fibre cell wall. The initial release of soluble sugars by TtLPMO9E from the two wood pulp fibres was found to be similar (Fig. 2). However, after 12 h, the liberation of soluble sugars from the dissolving pulp slowed down and after 36 h it halted completely, signifying that the accessible surfaces had been exhausted. By contrast, soluble products were released from the kraft pulp during the whole reaction time of 72 h, which could indicate that kraft pulp has more accessible surfaces for the LPMO. This observation agreed with the observed higher relative loss of DP in the kraft pulp than in the dissolving pulp. The final soluble sugar yield was 54 mg g−1 for kraft pulp, which was 80% higher than that (30 mg g−1) for the dissolving pulp.
 | ||
| Fig. 2 Releases of soluble glucose equivalents by TtLPMO9E during the oxidation of dissolving pulp and kraft pulp fibres. The soluble sugars were detected using a phenol–sulphuric acid assay. The standard deviations represent three replicate reactions. | ||
The obtained CNF suspensions were further examined by light microscopy and atomic force microscopy (AFM) (Fig. 3). Light microscopy revealed that both pulp fibres were extensively disintegrated by one pass through a microfluidizer after LPMO pre-treatment. The starting pulp fibre cells were several millimetres long and tens of micrometres wide (Fig. 3a and b). By contrast, the gel-like CNF suspensions no longer contained any micrometre-sized particles (Fig. 3c and d). AFM analysis showed that K-CNF nanofibrils were well-individualised and had a uniform thin width (Fig. 3f). D-CNF fibrils formed a mass of entangled threads with varying widths in the range of 10 to 50 nm, and D-CNFs also contained large aggregates of 50 to 100 nm formed by nanofibril coagulation (Fig. 3e). The thinner width of K-CNFs was related to their higher carboxyl content, similar to the CNFs produced by TEMPO-mediated oxidation.41 The widths of K-CNFs were further analysed from the AFM images and a histogram of the width distribution is shown in Fig. S4.† The widths of D-CNFs could not be analysed, as they were not apparent in the AFM images due to extensive aggregation. The results showed that K-CNFs had a narrow width distribution ranging from 1 to 10 nm with an average width of 3.7 ± 1.7 nm. Such a width profile is similar to that of the nanofibrils obtained from wood pulp fibres using TEMPO-mediated oxidation (2–3 nm),42 carboxymethylation (2.3–3.5 nm),43 or quaternisation (1.6–2.1 nm).44
 | ||
| Fig. 3 Light microscopy images of the starting (a) dissolving pulp and (b) kraft pulp fibres, and corresponding nanofibrils (c) D-CNFs and (d) K-CNFs prepared by TtLPMO9E-oxidation and subsequent mechanical disintegration. AFM height images of (e) D-CNFs and (f) K-CNFs. (g–j) Photographs of vials containing 0.1% (w/v) CNF suspensions demonstrate the stability of the dispersions before and after 7 days of incubation. | ||
The colloidal stability of the CNF water suspensions was investigated by a sedimentation test. The solid contents of the CNF suspensions were first adjusted to 0.1% (w/v), after which the suspensions were left to stand at room temperature. The aqueous suspension of K-CNFs appeared rather transparent (Fig. 3i) while the suspension of D-CNFs was opaque white (Fig. 3g). The D-CNFs were observed to sediment after being incubated for 7 days (Fig. 3h), while the K-CNFs remained as a stable colloidal suspension (Fig. 3j). Analysis of the charge density revealed the underlying reason for the differences between the two CNFs in colloidal stability. The obtained ζ-potential for K-CNFs was −41 ± 1.7 mV. The ζ-potential of D-CNFs was −29 ± 1.4 mV, which was deemed insufficient to stabilise the CNF suspension as a ζ-potential value higher than −30 mV is generally regarded unstable.
Properties of the nanopapers
A thinner nanofibril width is generally preferred in various material applications due to enhancements in the optical- and mechanical properties of the bulk material.45,46 To compare their performance in bulk materials, D-CNFs and K-CNFs were processed into nanopapers by vacuum filtration from their water suspensions followed by drying under compression. Field emission scanning electron microscopy (FE-SEM) micrographs of the nanopaper surfaces and cross-sections are presented in Fig. 4. The surfaces of the nanopapers showed typical network structures of random-in-plane oriented CNFs (Fig. 4a and b). The surface of the K-CNF nanopaper appeared slightly smoother than that of the D-CNF nanopaper. In addition, the cross-section of the K-CNF nanopaper revealed a uniform layered structure of evenly deposited nanofibrils, formed during vacuum filtration of a stable colloidal suspension (Fig. 4d). By contrast, the layered structure of the D-CNF nanopaper contained a few large fibril aggregates formed by CNF agglomeration in the unstable suspension of nanofibrils with a lower carboxyl content (Fig. 4c). | ||
| Fig. 4 FE-SEM micrographs for the surfaces (a and b) and freeze-fractured cross-sections (c and d) of nanopapers prepared from D-CNFs and K-CNFs. | ||
Photographs of the obtained nanopapers are presented in Fig. 5. The D-CNF nanopaper was white and translucent (Fig. 5a). The K-CNF nanopaper with a smooth surface and a neat, layered structure was transparent (Fig. 5b). Such transparent feature is typically observed for nanopapers prepared from thin, well-individualised nanofibrils. The optical properties of the nanopapers were further evaluated by measuring their total transmittance (Fig. 5c) and haze (Fig. 5d). As the thickness of the nanopaper plays an important role in light propagation,46 it should be mentioned that the thicknesses of the D-CNF and K-CNF nanopapers were 42 and 32 μm, respectively. The total transmittance for the D-CNF nanopaper was 63%, with a high haze of 78%. The K-CNF nanopaper demonstrated a higher total transmittance of 86% and a lower haze of 43%, indicating much lower light scattering. The higher total transmittance was mainly attributed to two factors. First, the porosity of the K-CNF nanopaper was lower (5.5%) than that (12.9%) of the D-CNF nanopaper (Table S2†). In addition, the thin width of the K-CNFs (3.7 nm) likely ensured better nanoparticle packing efficiency during vacuum filtration, as demonstrated also in the smaller thickness of the nanopaper. An interesting observation is that the total transmittance of the K-CNF nanopaper (86%) was in fact comparable to that of a thinner nanopaper (20 μm) prepared from TEMPO-oxidised CNFs, which showed a total transmittance of 90%.47
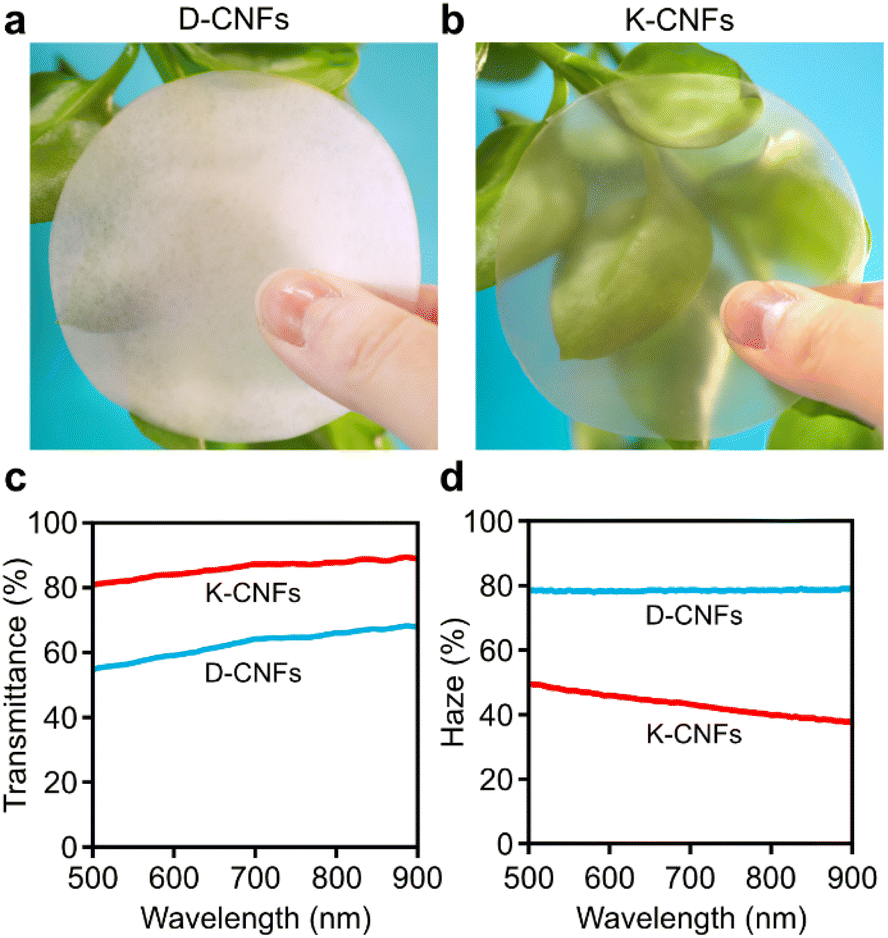 | ||
| Fig. 5 Photographs of nanopapers prepared from (a) D-CNFs and (b) K-CNFs. (c) Total transmittance and (d) haze of the nanopapers. | ||
The mechanical properties of the nanopapers were measured by tensile testing. Fig. 6 presents the stress–strain curves under uniaxial tension, and the physical and mechanical properties of the nanopapers are summarised in Table S2.† The mechanical performance of the K-CNF nanopaper was much higher than that of the D-CNF nanopaper. The average tensile strength of the K-CNF nanopaper was 260 MPa, 88% higher than that of the D-CNF nanopaper (138 MPa). The toughness (work-to-fracture) of the K-CNF nanopaper was 10.6 MJ m−3, twice as much as that (5.1 MJ m−3) for the D-CNF nanopaper owing to the increases in the modulus, tensile strength and strain-to-failure at the same time, a result of the higher carboxyl content and enhanced nanofibrillation of the kraft pulp fibres.
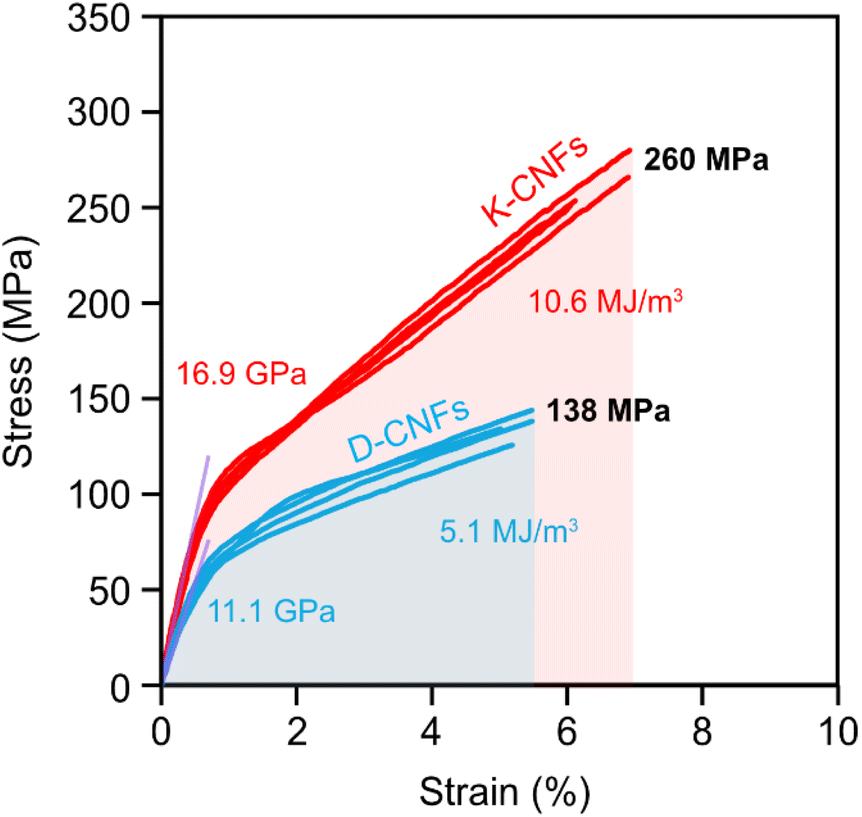 | ||
| Fig. 6 Stress–strain curves of nanopapers prepared from D-CNFs and K-CNFs. | ||
Gelation behaviour of the LPMO-oxidised CNFs
Previous studies have shown that by taking advantage of the carboxyl group, well-dispersed TEMPO-oxidised CNF suspensions with low nanocellulose content at pH 8 can be converted into strong hydrogels by decreasing the pH with dilute acid.48 Nanofibrils that do not contain surface charges, obtained by e.g. endoglucanases, require approximately an order of magnitude higher solid content in water suspension to become physically entangled into an equally strong hydrogel.6 Therefore, evaluating the potential of LPMO-oxidised K-CNFs to form hydrogels is of great interest. Due to the lack of colloidal stability of D-CNFs in water, their rheological properties were not studied.Rheological measurements showed that G′ and G′′ were not much affected by the applied angular frequency across all the investigated CNF concentrations at both pH 8 (Fig. 7a) and pH 2 (Fig. 7b). In addition, G′ was an order of magnitude higher than G′′ (Fig. S5†). These results revealed that the dispersions behaved as elastic gels. However, lower nanofibril concentrations of 0.125–0.25% (w/v) demonstrated low G′ values, less than 10 Pa at pH 8 (Fig. 7a). At these concentrations, the G′ values were slightly lower than CNF dispersions with a similar solid content obtained by endoglucanases.6 This was attributed to the much thinner nanofibril width of 3.7 nm obtained by TtLPMO9E-oxidation compared to that of the endoglucanase-derived CNFs (10–20 nm). When the solid content was 0.5% and higher, the gel network was approximately five times stronger than that of endoglucanase-derived CNFs which could be attributed to enhanced interfibrillar interactions of entangled nanofibrils with a high surface area. After the pH of the CNF dispersion was adjusted with dilute acid, the strength of the gels was further enhanced (Fig. 7b), similar to TEMPO-oxidised CNFs.48
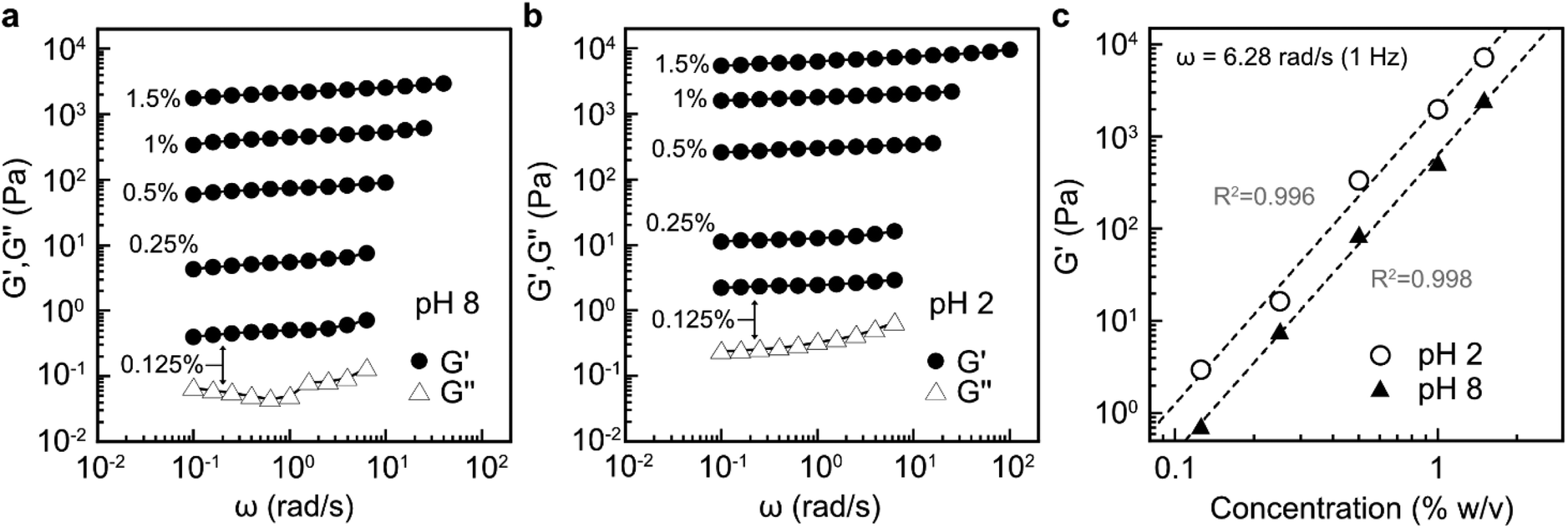 | ||
| Fig. 7 Rheological behaviour of 0.125, 0.25, 0.5, 1.0, and 1.5% (w/v) water dispersions of K-CNFs (a) at pH 8 and (b) at pH 2, where the storage modulus (G′) and the loss modulus (G′′) are plotted as a function of frequency. The measurements were conducted at 25 °C. (c) The storage moduli of K-CNFs at an angular frequency of 1 Hz against the CNF concentration. | ||
The obtained elastic moduli are shown as a function of CNF content for both pH values in Fig. 7c. The gelation was strongly dependent on the CNFs concentration, as the G′ values increased linearly upon increasing the CNF content. In addition, approximately five times increase in the G′ values was observed upon the addition of acid over the entire studied CNF concentration range. For instance, the 1.5% (w/v) nanofibril suspension at pH 8 had a G′ value of ca. 2 × 103 Pa, while the acid-treated suspension at the same concentration had a G′ value of approximately 104 Pa. This value is comparable to that for a 3% endoglucanase-derived CNF dispersion,6 indicating an enhanced ability of the LPMO-oxidised nanofibrils to form strong network structures in a dilute suspension.
Discussion
In the wood cell wall, hemicellulose exists in close association with cellulose microfibrils, where the two structural polysaccharides interact mainly through hydrogen bonding.49,50 Hemicellulose is known to significantly influence the cell wall morphology owing to its ability to prevent cellulose microfibril coalescence.51,52 Hemicellulose acts as a spacer between the basic structural units of the cell wall, i.e. cellulose microfibril bundles, and effectively inhibits the aggregation of cellulose.53,54 Indeed, it has been shown by small-angle X-ray scattering analysis that the removal of hemicellulose from the wood cell wall results in an increased cellulose microfibril bundle diameter, a phenomenon which arises from the aggregation of cellulose.55 The dense hydrogen bonding formed between the cellulose microfibril bundles makes the cell wall poorly accessible to liquid water, limiting the access of enzymes.28,56 Thus, the accessibility of TtLPMO9E to the dissolving pulp that comprises >92% cellulose (4% hemicellulose and 3% lignin) is limited due to the presence of excessive cellulose aggregation. Although dissolving pulp could also be oxidised by TtLPMO9E to a lesser extent as revealed by the release of soluble sugars, the cellulose chain cleavages could be expected to be limited on the outer surfaces of the fibre cells.Hemicellulose was not found to cause steric hindrance on the cellulose microfibril surfaces and block the access of the LPMO in the kraft pulp. This resulted from the preferential location of hemicellulose on the hydrophilic faces of the cellulose crystallites,49,57 instead of the hydrophobic ones that may serve as binding sites for TtLPMO9E.58 Therefore, the presence of hemicellulose in the kraft pulp was found to effectively enhance the accessibility for TtLPMO9E by inducing swelling between the cellulose microfibril bundles, thus keeping the microfibril surfaces well-exposed and water-accessible. Our results are consistent with a recent study that evaluated two different delignification processes on wheat straw. The harsher of the two studied processes led to the concomitant removal of hemicelluloses from the cell wall, increasing interfibrillar interactions of cellulose which consequently decreased enzyme accessibility.59 Reversely, bacterial cellulose produced in the presence of hemicelluloses has been shown to form a composite material with enhanced accessibility to hydrolytic cellulases.60
In addition to enhancing the substrate accessibility for TtLPMO9E, hemicelluloses also affected the properties of the produced nanopapers. Interestingly, the tensile strength (260 MPa) and Young's modulus (16.9 GPa) of the K-CNF nanopaper were higher than those for nanopapers of TEMPO-oxidised CNFs from kraft pulp, which showed a typical tensile strength of ca. 230 MPa and a modulus of ca. 6.9 GPa.47,61 Although the width of the obtained nanofibrils by LPMO-catalysed oxidation and TEMPO-mediated oxidation are similar, it is possible that the intact hemicellulose fraction in the LPMO-oxidised CNFs contributes to the higher tensile strength and modulus of the resulting nanopaper. Hemicelluloses act as a glue-like matrix in the interphase between the load bearing nanofibrils, enhancing the interfacial interactions.62 TEMPO-oxidation is known to cause partial degradation of hemicelluloses,63 which makes the ability of LPMOs to selectively target cellulose while leaving hemicellulose intact particularly attractive. Indeed, the obtained results indicate that LPMO-oxidation might offer significant advantages in terms of the mechanical performance of nanocellulose-based materials.
Although cellulose degradation by the LPMOs can be boosted by the addition of H2O2 as a co-substrate,20,21 in this work, H2O2 was not introduced in the reaction with commercial pulp fibres due to a potential risk of overdosing and enzyme inactivation as previously reported.18,64 The LPMO reactions were performed with continuous mixing for 72 h to ensure homogeneous oxidation of the pulp fibres under standard aerobic conditions. However, it would be desirable to increase the reaction speed for larger scale industrial CNF production. Therefore, further optimisation of the reaction conditions including controlled feeding of H2O2 is necessary in the future.
Conclusions
In summary, the cellulose-active and C1-oxidising TtLPMO9E from T. terrestris was found to introduce almost a three-fold higher carboxyl content on kraft pulp fibres (0.22 mmol g−1) than on dissolving pulp fibres (0.08 mmol g−1). This led to enhanced nanofibrillation of the kraft pulp fibres and production of more individualised CNFs with a thinner width (3.7 nm) than those from dissolving pulp fibres. The enhanced nanofibrillation also resulted in better optical and mechanical properties of the subsequently prepared nanopapers. More pronounced loss in DP, and enhanced release of soluble sugars confirmed that the kraft pulp fibre cell wall was more accessible to the LPMO than was the dissolving pulp. These results suggested that instead of being a steric hindrance on the cellulose microfibril surfaces, hemicellulose is critical for the accessibility of LPMOs owing to the enhanced swelling of the wood fibre cell wall. Water suspensions of the CNFs from the kraft pulp also exhibited acid-triggered gelation behaviour deriving from the enzymatically introduced carboxylic acid groups. This gelation behaviour can be further exploited in an environmentally friendly preparation of hydrogels with a highwater content. Our work demonstrated that native hemicelluloses in the wood pulp fibres are crucial for the LPMO-assisted CNF production that is environmentally friendly, contributing to the adaptation of LPMOs in industrial nanofibrillation processes.Materials and methods
Materials
Never-dried softwood kraft pulp was kindly provided by Nordic Paper, Karlstad, Sweden. Never-dried softwood dissolving pulp was kindly provided by Domsjö Fabriker AB, Sweden. Both pulp fibres were used as received.LPMO selection and sequence analysis
The selected LPMO (TtLPMO9E) has been previously characterised as cellulose-active and C1 regioselectivite.36 The amino acid sequence of TtLPMO9E was obtained from UniProt (protein-ID: XP_003657366.1). The native signal peptide of TtLPMO9E was predicted using SignaIP software 4.1. The theoretical molecular mass of the mature recombinant LPMO was calculated from the amino acid sequence without the signal peptide using the Expasy ProtParam tool. The calculated theoretical molecular mass of TtLPMO9E is 23 kDa.Gene synthesis and cloning
The gene encoding TtLPMO9E was synthesised, and codon optimised by GenScript, USA. The synthetic gene encoded a native secretion signal at the N-terminus and (His)6 tag at the C-terminus of the corresponding protein. The gene was inserted into the EcoRI-NotI position of the P. pastoris expression vector pPICZB, in which the c-myc epitope and polyhistidine tag were omitted. The construct was transformed into E. coli TOP10 cells (Thermo Fisher Scientific) by electroporation, and the transformants were cultivated in a low salt Luria–Bertani (LB) broth containing 25 μg mL−1 zeocin (Invitrogen). Plasmid DNA was extracted using a GeneJet Plasmid Miniprep kit (Thermo Scientific). The construct was confirmed by restriction pattern analysis (EcoRI and NotI – Thermo Scientific), and the positive clones were sequenced by Eurofins Genomics, Germany. A construct with a verified sequence was linearised using PmeI and transformed into P. pastoris X-33 cells by electroporation. The transformants were selected on YPDS agar containing 100 μg mL−1 zeocin. Integration of the LPMO-encoding gene into P. pastoris genomic DNA was confirmed by colony PCR utilising AOX1 primers and a standard protocol.Production and purification of TtLPMO9E
The yeast was initially cultivated in 50 mL of glycerol complex medium (BMGY). For the protein production phase, the overnight cultivation was used to inoculate Fernbach flasks containing 300 mL of buffered methanol complex medium (BMMY). First, the cells were collected by centrifugation (850g, 10 min, RT) and then suspended in BMMY for a final cell density (OD600) of 1. The cultivations were performed at 30 °C and 250 rpm for 4 days. To induce recombinant protein production, methanol was added daily to the cultivations for a final concentration of 2%. BMGY and BMMY contained 1% yeast extract, 2% peptone, 1.34% yeast nitrogen base, 4 × 10−5% biotin, and 100 mM potassium phosphate buffer pH 6.0. BMGY also contained 1% glycerol, and BMMY contained 0.005% antifoam 204 (Sigma Aldrich). The cultivations were ended by separating the cells from the growth liquid by centrifugation (4300g, 30 min, 4 °C). A protease inhibitor (Pierce Protease Inhibitor Tablets, EDTA-free, Sigma-Aldrich) was added to the supernatant containing the extracellular LPMO, and the supernatant was further purified by filtering through a 0.45 μm PES membrane (Filtropur BT50, Sarstedt).For biochemical characterisations, the His-tagged TtLPMO9E was purified by immobilised metal-ion affinity chromatography (IMAC) using a fast protein liquid chromatography system (FPLC Äkta, GE Healthcare, USA) equipped with IMAC FF HiTrap column (Bio-Rad). The binding buffer was 50 mM NaCl–20 mM sodium phosphate, pH 7.4. Gradient elution was performed using 500 mM imidazole and 500 mM NaCl. The LPMO was buffer exchanged on a Biogel P-6 (Bio-Rad) column to 20 mM sodium acetate pH 5.0. For large reactions on pulp fibres, the filtered supernatant containing TtLPMO9E was buffer-exchanged to 20 mM sodium acetate pH 5.0 in an ultrafiltration unit (Amicon Stirred Cell, Merch Millipore) equipped with a 10 kDa cut-off PES membrane. In both protocols, the obtained enzyme was copper saturated by the addition of CuSO4 to a final concentration of 500 μM. Excess copper was removed by extensive washing in 20 mM sodium acetate buffer, and the enzyme was further concentrated in 5 kDa cut-off Vivaspin 20 centrifugal concentrators (GE Healthcare). The obtained LPMO was analysed by SDS-PAGE and Western-blot according to previously described protocols.23 The protein concentrations were determined using a Pierce BCA protein assay kit (Thermo Scientific).
LPMO activity assays
The peroxidase activity of TtLPMO9E was determined spectrophotometrically (Cary 50 UV-VIS) against the soluble synthetic substrate 2,6-dimethoxyphenol (2,6-DMP; Sigma Aldrich). The formation of the reaction product (coerulignone) was monitored at a wavelength of 469 nm. The assay contained an appropriate amount of the enzyme, 1 mM 2,6-DMP, 100 μM H2O2 and 50 mM sodium phosphate buffer pH 6.5 in a final volume of 1 mL, as described by Breslmayr et al.35 The reaction temperature was 25 °C. Three technical replicates were used for all assays. To determine the temperature stability, TtLPMO9E was incubated for 0, 3, 6, 12, 24, 36, 48, and 72 at 30–90 °C, after which the residual activity was assayed at 25 °C as described above.The optimum pH was determined based on the amount of released total soluble sugars (cello-oligosaccharides) from a natural insoluble cellulosic substrate, microcrystalline cellulose (MCC). The reactions were performed at 40 °C in a series of 50 mM sodium malonate buffer with pH in the range of 4.0–8.0. The reactions contained 0.4 wt% MCC (Avicel PH-101) and 2 mM gallic acid as the electron donor for the enzyme in a total volume of 500 μL. The reactions were performed under magnetic stirring for 20 h. All reactions were performed in triplicate, and reactions without LPMOs were conducted as controls. The reactions were stopped by separating the enzyme from the remaining substrate by filtering the reaction liquid through 0.2 μm nylon syringe filters (Thermo Scientific). The released soluble cello-oligosaccharides were quantified as glucose-equivalents with a phenol–sulphuric acid assay according to a previously reported method.65
LPMO oxidation of the pulp and preparation of CNFs
Dissolving pulp and kraft pulp fibres (2.5 g of each) were dispersed in 20 mM sodium acetate buffer, pH 5 and soaked overnight. For the LPMO reactions, the fibres were diluted to a final concentration of 0.4% (w/v). The LPMO reaction contained 90 mg g−1TtLPMO9E at an approximate molar ratio of 4 μmol g−1 of enzyme to cellulose, and 2 mM gallic acid as the electron donor for the enzyme. The LPMO reactions were performed at 40 °C under magnetic stirring for 72 h. The reactions were stopped by separating the fibres from the suspension. To remove the enzyme, the fibres were washed with 10 μM NaOH pH 9. NaOH was removed by extensive washing with ultrapure water in a vacuum filtration unit equipped with a Durapore® 0.65 μm DVPP membrane (Merck). The LPMO-oxidised fibres were adjusted to 1% (w/v) in ultrapure water and passed once through an M-110EH microfluidizer (Microfluidics Ind., USA), equipped with 200 and 100 μm chambers. The microfluidizer was operated at a pressure of 1600 bar and at room temperature.Preparation of nanopapers
To prepare the nanopapers, 200 mg of the nanofibrils were suspended in ultrapure water at a final concentration of 0.1% (w/v). Water was removed from the suspensions in a vacuum-filtration setup equipped with a filter funnel (Φ = 70 mm) and a filter membrane (0.22 μm, DVPP). The wet cakes obtained from filtration were carefully peeled off and placed between two stainless steel meshes. The films were dried under pressure at 50 °C for 24 h.Characterisation
An inverted light microscope (Nikon Eclipse Ti-S, USA) running imaging software Nis-Elements version 4.60 was used for the optical microscopy analyses. The solid content of the samples was adjusted to 0.1% (w/v) before observation. Fourier transform infrared spectroscopy (FTIR) was performed using a PerkinElmer Spectrum 2000 instrument equipped with an MKII Golden Gate Single Reflection ATR system from Specac Ltd, UK. The ATR crystal was an MKII heated diamond 45° ATR top plate. The measurement was performed in a spectral range of 600–4000 cm−1 with a resolution of 4 cm−1. Carboxyl contents were determined by conductometric titration. Briefly, suspensions containing 100 mg of cellulose were diluted in ultrapure water and the pH and conductivity of the suspensions were adjusted with 0.1 M HCl and 0.01 M NaCl to 2.5 and ∼1200 μS cm−1, respectively. The suspensions were stirred and titrated with 0.04 M NaOH by adding 0.1 mL aliquots at 60 s intervals, and the conductivity was monitored with a conductometric station (SevenCompact, Mettler-Toledo, USA). The zeta (ζ) potentials were measured at 25 °C using a Zetasizer nano ZS instrument (Malvern, UK) and following the Smoluchowski method. Before measurements, the solid contents were adjusted to 0.05% (w/v) and NaCl was added to a final concentration of 1 mM. Atomic force microscopy (AFM) analyses were performed on a MultiMode 8 atomic force microscope (Digital Instruments, USA) operated in the ScanAsyst-air mode. Widths of the nanofibrils were measured from the obtained images using Gwyddion software. Powder X-ray diffraction (XRD) patterns were recorded using an X'Pert Pro X-ray diffractometer (PW 3040/60, Philips, Netherlands) in the reflection mode (5–35° 2θ angular range, steps of 0.05°). Cu Kα radiation (λ = 1.5418 Å) was generated at 45 kV and 40 mA and monochromatised using a 20 μm Ni-filter. Diffractograms were recorded from rotating specimens using a position sensitive detector. The crystallinity index was calculated from the ratio between the intensity of the (200) peak (I200) and the intensity of the minimum (IAM) between the (200) and (110) peaks. The degree of polymerisation (DP) was calculated from intrinsic viscosity number measured according to the ISO 5351:2000 standard. Briefly, 30 mg of the samples was dissolved in 50 mL of 0.5 M copper ethylenediamine for 30 min before measurements. DP was converted from intrinsic viscosity number using the equations: η = 0.42 × DP for DP < 950 and η = 2.28 × DP0.76 for DP > 950. The optical transmittance of nanopapers was measured with a spectrophotometer having a supercontinuum white-light source (SC-5, YSL Photonics, China) as the incident beam. Transmitted light was captured in an integrating sphere. The transmittance values are reported as an average from the measurement range of 500 to 900 nm. Haze was measured based on the ASTM D1003 standard and a previously published method.66 The mechanical properties of nanopapers were tested under uniaxial tension at 25 °C with an universal mechanical tester (Instron 5944, USA) equipped with a 500 N load cell and a video extensometer. The specimens were cut into strips of 3 mm in width and 50 mm in length and conditioned at 25 °C and 50% relative humidity for 48 h before testing. At least five specimens were tested from each sample and the results are reported as an average of at least four specimens. The tests were performed using a strain rate of 2 mm min−1 and a gauge length of 20 mm. The modulus was determined from the slope of the initial linear deformation range (0–0.1%) of the stress–strain curve. Toughness, defined as work to fracture, was calculated as the area under the stress–strain curve. Microstructures of the nanopaper surfaces and freeze-fractured cross-sections were imaged with a field-emission scanning electron microscope (FE-SEM, S-4800 Hitachi, Japan). For observations, the samples were attached with carbon tape onto a specimen holder and coated with a 5 nm layer of platinum/palladium using a sputter coater (208HR, Cressington Scientific Instruments, UK). Rheology analyses were performed using a Discovery HR-2 rheometer (TA Instruments, USA) equipped with a 20 mm parallel plate geometry. The gap size was 1 mm and the temperature was set to 25 °C. Briefly, the frequency sweeps of 0.1–100 rad s−1 were measured in the linear viscoelastic regime with a controlled strain rate. Strain rates (%) were 5, 1, 0.5, 0.5 and 0.2 for the 0.125, 0.25, 0.5, 1 and 1.5% (w/v) samples, respectively. Acid crosslinking of the samples was performed by surrounding the sample with 0.05 M HCl, which was allowed to stand for 2 h before the measurement. To avoid evaporation, the samples were sealed with oil.Author contributions
S. K. and Q. Z. conceptualized the work. S. K. performed cloning, recombinant production and purification of the enzyme, biochemical characterizations, produced the CNF samples, performed rheological experiments, FTIR, XRD, conductometric titration, ζ-potential measurements, optical microscopy, imaging of the samples, sedimentation test, and phenol–sulphuric acid assay, interpreted the results and wrote the manuscript. S. K. and L. Z. performed microfluidization and nanopaper preparation. L. Z. performed the tensile test, transmittance measurements and SEM. M. Y. supervised the transmittance measurements. S. W. performed AFM, histogram analysis, DP measurements, and sugar analysis. Q. Z. supervised the whole work and revised the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Wallenberg Wood Science Center (WWSC) and the Swedish Research Council – Vetenskapsrådet (2015-05030) for the financial support.References
- K. Heise, E. Kontturi, Y. Allahverdiyeva, T. Tammelin, M. B. Linder, Nonappa and O. Ikkala, Adv. Mater., 2021, 33, 2004349 CrossRef CAS PubMed.
- E. Kontturi, P. Laaksonen, M. B. Linder, Nonappa, A. H. Gröschel, O. J. Rojas and O. Ikkala, Adv. Mater., 2018, 30, 1703779 CrossRef PubMed.
- O. Nechyporchuk, M. N. Belgacem and J. Bras, Ind. Crops Prod., 2016, 93, 2–25 CrossRef CAS.
- Q. Li, S. McGinnis, C. Sydnor, A. Wong and S. Renneckar, ACS Sustainable Chem. Eng., 2013, 1, 919–928 CrossRef CAS.
- J. R. Cherry and A. L. Fidantsef, Curr. Opin. Biotechnol., 2003, 14, 438–443 CrossRef CAS PubMed.
- M. Pääkkö, M. Ankerfors, H. Kosonen, A. Nykänen, S. Ahola, M. Österberg, J. Ruokolainen, J. Laine, P. T. Larsson, O. Ikkala and T. Lindström, Biomacromolecules, 2007, 8, 1934–1941 CrossRef PubMed.
- M. Henriksson, G. Henriksson, L. A. Berglund and T. Lindström, Eur. Polym. J., 2007, 43, 3434–3441 CrossRef CAS.
- G. L. Berto, B. D. Mattos, O. J. Rojas and V. Arantes, ACS Sustainable Chem. Eng., 2021, 9, 2260–2270 CrossRef CAS.
- K. M. Kleman-Leyer, M. Siika-Aho, T. T. Teeri and T. K. Kirk, Appl. Environ. Microbiol., 1996, 62, 2883–2887 CrossRef CAS PubMed.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sørlie and V. G. Eijsink, Science, 2010, 330, 219–222 CrossRef CAS PubMed.
- M. Eibinger, T. Ganner, P. Bubner, S. Rosker, D. Kracher, D. Haltrich, R. Ludwig, H. Plank and B. Nidetzky, J. Biol. Chem., 2014, 289, 35929–35938 CrossRef CAS PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- F. L. Aachmann, M. Sørlie, G. Skjåk-Bræk, V. G. Eijsink and G. Vaaje-Kolstad, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18779–18784 CrossRef CAS PubMed.
- W. T. Beeson, C. M. Phillips, J. H. Cate and M. A. Marletta, J. Am. Chem. Soc., 2012, 134, 890–892 CrossRef CAS PubMed.
- S. J. Horn, G. Vaaje-Kolstad, B. Westereng and V. G. Eijsink, Biotechnol. Biofuels, 2012, 5, 45 CrossRef CAS PubMed.
- B. Wang, Z. Wang, G. J. Davies, P. H. Walton and C. Rovira, ACS Catal., 2020, 10, 12760–12769 CrossRef CAS.
- S. Kim, J. Ståhlberg, M. Sandgren, R. S. Paton and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 149–154 CrossRef CAS PubMed.
- B. Bissaro, A. K. Røhr, G. Müller, P. Chylenski, M. Skaugen, Z. Forsberg, S. J. Horn, G. Vaaje-Kolstad and V. G. H. Eijsink, Nat. Chem. Biol., 2017, 13, 1123–1128 CrossRef CAS PubMed.
- R. Kont, B. Bissaro, V. G. H. Eijsink and P. Väljamäe, Nat. Commun., 2020, 11, 5786 CrossRef CAS PubMed.
- S. Kuusk, B. Bissaro, P. Kuusk, Z. Forsberg, V. G. H. Eijsink, M. Sørlie and P. Väljamäe, J. Biol. Chem., 2018, 293, 523–531 CrossRef CAS PubMed.
- G. Müller, P. Chylenski, B. Bissaro, V. G. H. Eijsink and S. J. Horn, Biotechnol. Biofuels, 2018, 11, 209 CrossRef PubMed.
- A. Villares, C. Moreau, C. Bennati-Granier, S. Garajova, L. Foucat, X. Falourd, B. Saake, J. G. Berrin and B. Cathala, Sci. Rep., 2017, 7, 40262 CrossRef CAS PubMed.
- S. Koskela, S. Wang, D. Xu, X. Yang, K. Li, L. A. Berglund, L. S. McKee, V. Bulone and Q. Zhou, Green Chem., 2019, 21, 5924–5933 RSC.
- J. Hu, D. Tian, S. Renneckar and J. N. Saddler, Sci. Rep., 2018, 8, 3195 CrossRef PubMed.
- C. Moreau, S. Tapin-Lingua, S. Grisel, I. Gimbert, S. Le Gall, V. Meyer, M. Petit-Conil, J. G. Berrin, B. Cathala and A. Villares, Biotechnol. Biofuels, 2019, 12, 156 CrossRef PubMed.
- C. Chen, C. Duan, J. Li, Y. Liu, X. Ma, L. Zheng, J. Stavik and Y. Ni, BioResources, 2016, 11, 5553–5564 Search PubMed.
- L. Östberg, N. Kvarnlöf and U. Germgård, Nord. Pulp Pap. Res. J., 2013, 28, 377–380 CrossRef.
- T. Oksanen, J. Buchert and L. Viikari, Holzforschung, 1997, 51, 355–360 CrossRef CAS.
- H. Nilsson, S. Galland, P. T. Larsson, E. K. Gamstedt and T. Iversen, Cellulose, 2012, 19, 751–760 CrossRef CAS.
- P. A. Moss and J. Pere, Nord. Pulp Pap. Res. J., 2006, 21, 8–12 CrossRef CAS.
- Y. Okita, T. Saito and A. Isogai, Biomacromolecules, 2010, 11, 1696–1700 CrossRef CAS PubMed.
- J. Zhang, M. Tang and L. Viikari, Bioresour. Technol., 2012, 121, 8–12 CrossRef CAS PubMed.
- R. Kumar and C. E. Wyman, Biotechnol. Bioeng., 2014, 111, 1341–1353 CrossRef CAS PubMed.
- J. Li, L. Solhi, E. D. Goddard-Borger, Y. Mathieu, W. W. Wakarchuk, S. G. Withers and H. Brumer, Biotechnol. Biofuels, 2021, 14, 29 CrossRef CAS PubMed.
- E. Breslmayr, M. Hanžek, A. Hanrahan, C. Leitner, R. Kittl, B. Šantek, C. Oostenbrink and R. Ludwig, Biotechnol. Biofuels, 2018, 11, 79 CrossRef PubMed.
- B. Westereng, D. Cannella, J. Wittrup Agger, H. Jørgensen, M. Larsen Andersen, V. G. Eijsink and C. Felby, Sci. Rep., 2015, 5, 18561 CrossRef CAS PubMed.
- J. Velasco, B. Oliva, A. L. Goncalves, A. S. Lima, G. Ferreira, B. A. Franca, E. J. Mulinari, T. A. Goncalves, F. M. Squina, M. A. S. Kadowaki, A. Maiorano, I. Polikarpov, L. C. Oliveira and F. Segato, Appl. Microbiol. Biotechnol., 2020, 104, 8309–8326 CrossRef CAS PubMed.
- A. Zerva, A. Limnaios, A. S. Kritikou, N. S. Thomaidis, P. Taoukis and E. Topakas, New Biotechnol., 2021, 63, 45–53 CrossRef CAS PubMed.
- M. B. Keller, C. Felby, C. A. Labate, V. O. A. Pellegrini, P. Higasi, R. K. Singh, I. Polikarpov and B. M. Blossom, Biotechnol. Lett., 2020, 42, 93–102 CrossRef CAS PubMed.
- B. Westereng, J. W. Agger, S. J. Horn, G. Vaaje-Kolstad, F. L. Aachmann, Y. H. Stenstrøm and V. G. Eijsink, J. Chromatogr. A, 2013, 1271, 144–152 CrossRef CAS PubMed.
- J. Wei, Y. Chen, H. Liu, C. Du, H. Yu, J. Ru and Z. Zhou, Ind. Crops Prod., 2016, 92, 227–235 CrossRef CAS.
- I. Usov, G. Nyström, J. Adamcik, S. Handschin, C. Schütz, A. Fall, L. Bergström and R. Mezzenga, Nat. Commun., 2015, 6, 7564 CrossRef CAS PubMed.
- A. B. Fall, S. B. Lindström, O. Sundman, L. Ödberg and L. Wågberg, Langmuir, 2011, 27, 11332–11338 CrossRef CAS PubMed.
- A. Pei, N. Butchosa, L. A. Berglund and Q. Zhou, Soft Matter, 2013, 9, 2047–2055 RSC.
- H. Zhu, S. Zhu, Z. Jia, S. Parvinian, Y. Li, O. Vaaland, L. Hu and T. Li, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8971–8976 CrossRef CAS PubMed.
- G. Jacucci, L. Schertel, Y. Zhang, H. Yang and S. Vignolini, Adv. Mater., 2021, 33, 2001215 CrossRef CAS PubMed.
- H. Fukuzumi, T. Saito, T. Iwata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2009, 10, 162–165 CrossRef CAS PubMed.
- T. Saito, T. Uematsu, S. Kimura, T. Enomae and A. Isogai, Soft Matter, 2011, 7, 8804–8809 RSC.
- N. J. Grantham, J. Wurman-Rodrich, O. M. Terrett, J. J. Lyczakowski, K. Stott, D. Iuga, T. J. Simmons, M. Durand-Tardif, S. P. Brown, R. Dupree, M. Busse-Wicher and P. Dupree, Nat. Plants, 2017, 3, 859–865 CrossRef CAS PubMed.
- D. J. Cosgrove and M. C. Jarvis, Front. Plant Sci., 2012, 3, 204 Search PubMed.
- S. Väisänen, R. Pönni, A. Hämäläinen and T. Vuorinen, Cellulose, 2018, 25, 6923–6934 CrossRef.
- R. Pönni, T. Vuorinen and E. Kontturi, BioResources, 2012, 7, 6077–6108 CrossRef.
- Y. Zhang, J. Yu, X. Wang, D. M. Durachko, S. Zhang and D. J. Cosgrove, Science, 2021, 372, 706–711 CrossRef CAS PubMed.
- L. Donaldson, Wood Sci. Technol., 2007, 41, 443–460 CrossRef CAS.
- E. Brännvall, P. T. Larsson and J. S. Stevanic, Cellulose, 2021, 28, 3951–3965 CrossRef.
- R. Pönni, L. Galvis and T. Vuorinen, Carbohydr. Polym., 2014, 101, 792–797 CrossRef PubMed.
- T. J. Simmons, J. C. Mortimer, O. D. Bernardinelli, A. C. Pöppler, S. P. Brown, E. R. deAzevedo, R. Dupree and P. Dupree, Nat. Commun., 2016, 7, 13902 CrossRef CAS PubMed.
- M. Eibinger, J. Sattelkow, T. Ganner, H. Plank and B. Nidetzky, Nat. Commun., 2017, 8, 894 CrossRef PubMed.
- V. Pihlajaniemi, M. H. Sipponen, H. Liimatainen, J. A. Sirviö, A. Nyyssölä and S. Laakso, Green Chem., 2016, 18, 1295–1305 RSC.
- P. A. Penttilä, T. Imai, J. Hemming, S. Willfor and J. Sugiyama, Carbohydr. Polym., 2018, 190, 95–102 CrossRef PubMed.
- H. Fukuzumi, S. Fujisawa, T. Saito and A. Isogai, Biomacromolecules, 2013, 14, 1705–1709 CrossRef CAS PubMed.
- X. Yang, E. Jungstedt, M. S. Reid and L. A. Berglund, Macromolecules, 2021, 54, 4443–4452 CrossRef CAS.
- G. Rodionova, T. Saito, M. Lenes, Ø. Eriksen, Ø. Gregersen, R. Kuramae and A. Isogai, J. Polym. Environ., 2012, 21, 207–214 CrossRef.
- F. Calderaro, M. Keser, M. Akeroyd, L. E. Bevers, V. G. H. Eijsink, A. Várnai and M. A. van den Berg, Biotechnol. Biofuels, 2020, 13, 195 CrossRef CAS PubMed.
- S. Koskela, S. Wang, P. M. P. Fowler, F. Tan and Q. Zhou, ACS Sustainable Chem. Eng., 2021, 9, 11331–11341 CrossRef CAS.
- E. Vasileva, H. Chen, Y. Li, I. Sychugov, M. Yan, L. A. Berglund and S. Popov, Adv. Opt. Mater., 2018, 6, 1800999 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02237k |
| This journal is © The Royal Society of Chemistry 2022 |