
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formate as a key intermediate in CO2 utilization
Eric
Schuler
a,
Michele
Morana
 a,
Pavel A.
Ermolich
a,
Kristian
Lüschen
a,
Adam J.
Greer
b,
S. F. Rebecca
Taylor
b,
Christopher
Hardacre
b,
N. Raveendran
Shiju
a and
Gert-Jan M.
Gruter
*ac
a,
Pavel A.
Ermolich
a,
Kristian
Lüschen
a,
Adam J.
Greer
b,
S. F. Rebecca
Taylor
b,
Christopher
Hardacre
b,
N. Raveendran
Shiju
a and
Gert-Jan M.
Gruter
*ac
aVan‘t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1090 GD Amsterdam, The Netherlands. E-mail: g.j.m.gruter@uva.nl
bDepartment of Chemical Engineering & Analytical Science, University of Manchester, Oxford Road, Manchester M13 9PL, UK
cAvantium Chemicals BV, Zekeringstraat 29, 1014 BV Amsterdam, The Netherlands
First published on 5th August 2022
Abstract
Replacing fossil feedstocks for chemicals and polymers in the chemical industry is a key step towards a future circular society. Making use of CO2 as a starting material in Carbon Capture and Utilization (CCU) or Carbon Capture and Storage (CCS) processes presents a great opportunity. Unfortunately, converting CO2 is not easy – due to its stability and inherently low reactivity either high energy inputs or nifty catalytic systems are required for its conversion. An electrochemical cell using a gas-diffusion electrode to convert CO2 into formate is such a promising system. But making formate alone does not allow us to substitute many fossil carbon-fed processes. Oxalic acid on the other hand is a potential new platform chemical for material production as useful monomers such as glycolic acid can be derived from it. Fortunately, formate can be converted into oxalate (and subsequently oxalic acid) by coupling two formates in a formate to oxalate coupling reaction (FOCR). The FOCR is a reaction that has been studied for more than 175 years and has seen widespread industrial use in the past. In this work, we critically discuss the history of the FOCR, present the most recent advances and draw a perspective for its future. We provide an overview of all (side)products obtained in FOCR and examine the various reaction parameters and their ability to influence the reaction. To understand the reaction better and improve it in the future, we critically discuss the many mechanisms proposed for the various catalytic systems in the FOCR. At last, we explore the potential to introduce new catalytic and solvent systems or co-reactants to the FOCR to improve reaction performance and broaden the range of products from CO2 derived formate.
1 Introduction
When John Louis Jullion first patented the coupling reaction of two formate molecules to oxalate in 1846, the industrial revolution was in full swing.1 Today, 175 years later, we have seen the industrial use and even the formation of a world-spanning cartel around this reaction to produce oxalic acid. Yet, the advent of petrochemistry brought about alternative routes or products and made the formate coupling process obsolete. In those 175 years, we have developed into a society emitting ever-more greenhouse gases into our atmosphere causing severe environmental problems.2–4 Society is looking at the chemical industry to play its part in a transition to a circular economy by reducing its CO2 footprint by utilizing it rather than emitting it.5–7 CO2 can become an attractive building block for producing organic chemicals and materials, as it is an economical, abundant, and nontoxic carbon source that can be incorporated in downstream products with high ‘atom efficiency’.8–14 The capturing of CO2 and its subsequent conversion can be integrated in one process to utilize energy and heat efficiently.15,16 However, activation of CO2 is still costly due to its thermodynamic stability and kinetic inertness.17Electrochemistry is a promising way to utilize CO2 as a resource for chemicals.18,19 It provides ways for sustainable electricity to be stored in chemical bonds and can contribute to solving the storage and intermittency problem of renewable energy. Compared to other pathways, hydrogen is produced rather than consumed. Out of all options for electrochemical CO2 conversion, the production of alkali formate and CO are the most advanced and most promising because of high volume potential.20,21 The value of CO (syngas) in conversions such as Fischer–Tropsch processes allows the production of a broad variety of chemicals and is well known and established.11,22
Formate is today mainly used as an anti-freezing or cooling liquid or for producing formic acid.23 To unlock an interesting C2 product tree, via the implementation of formate from CO2 at a large scale, we require a formate to oxalate coupling process. The 175-year-old formate to oxalate coupling route fits right in here and we are currently developing such a route from CO2 to polymers in the European Horizon 2020 “OCEAN” project (Scheme 1).24
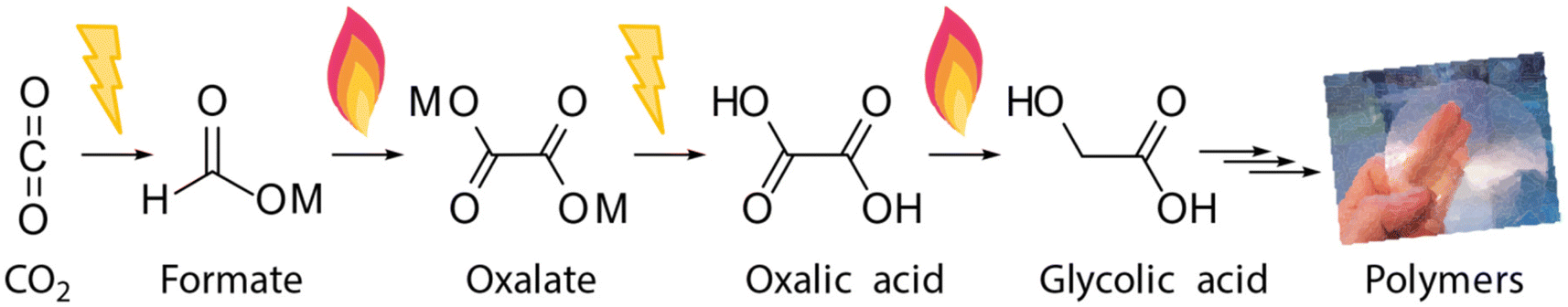 | ||
| Scheme 1 “OCEAN” process for CO2 utilization via (i) electrochemical reduction to formate, (ii) thermal formate coupling to oxalate, (iii) electrochemical oxalate acidification, (iv) thermocatalytic reduction of oxalic acid to glycolic acid, and (v) polymer production from oxalic acid and its derivatives. | ||
In the first step, CO2 undergoes electrochemical conversion to formate salt. Our research focuses on the electrochemical reduction of CO2 to formate and to CO, which both do not require hydrogen and elevated temperatures.25,26 As formate and CO can both be obtained via a 2 electron electrochemical reduction of CO2, the electrochemical production of formate from CO2 aligns well with the ambition to use CO2 as a renewable C1 feedstock.27,28 In a next step, formate must be removed from water for the subsequent reactions. This poses a great separation challenge due to the high solubility. Separation cost and energy consumption may represent a big share of the overall process cost and thus this step must not be overlooked in the overall process design. The formate can be effectively recovered from the solution by a combination of solvent extraction, evaporation and cooling crystallization. The combination of these three techniques allows to reduce the overall energy consumption compared to evaporation alone.29 In the second step, these formates are catalytically coupled to oxalate in the formate coupling reaction (FOCR), which is the subject of this paper. Formate is then acidified to oxalic acid in the third step. The fourth step of the technology targets the derivatization of oxalic acid to esters or its conversion to produce monomers such as glycolic acid. In the fifth (and final) step, we investigate new high-performing polymers from these CO2-based monomers.30–33 Polymers can be especially interesting as they allow for long term storage of sequestered CO2 in materials.34,35 CO2-Based chemicals such as oxalic acid will become new platform chemicals for a wide range of downstream products such as MEG, glycolic, and glyoxylic acid that all can be obtained from oxalic acid in various sustainable routes.30,36
The formate to oxalate coupling reaction has been discussed in the scientific literature with major contributions from Freidlin, Górski, and most recently Lakkaraju and our group.37–56 Yet, the story concerning both crucial (industrial) reaction parameters and (scientific) mechanisms is neither clear nor complete. Many potential reaction parameters and their effect on the conversion and selectivity of the reaction have been reported in scientific publications and patents over the years but many are contradicting each other as details are tightly related to the reactor designs or reaction systems that were used to generate the data. Many mechanisms were proposed and to date, carbonite ([CO2]2−), first proposed as an intermediate by Freidlin, is accepted as the main intermediate. The activation pathways to obtain said carbonite from formate – which appear to be diverse and interconnected – are not fully understood yet. Such understanding is required to explain the many observations made by scientists and alkali oxalate producers and then optimize for the reaction further.
Given the potential importance of this reaction in electrochemical CO2 utilization in the future, we aim to first offer a complete overview of reported observations and their relevance followed by a critical discussion of all existing and potential mechanisms. Finally, we will extrapolate improvement potentials and new opportunities even beyond the production of alkali oxalate.
2 History and factors influencing the FOCR
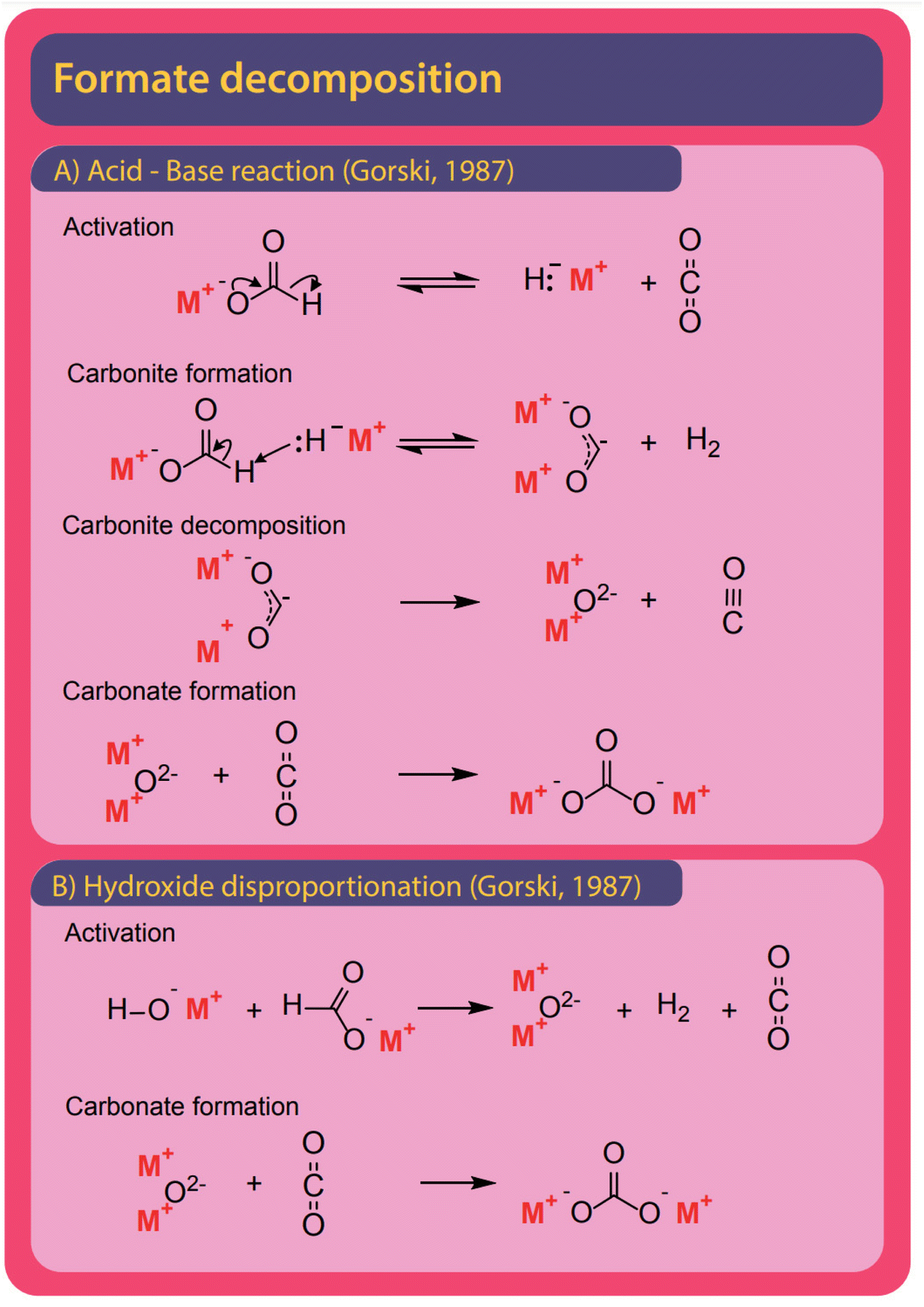
To understand the formate to oxalate coupling reaction (FOCR) we will first look at the research and patent history of the reaction. A brief timeline of the history is shown in Fig. 1. Investigations started already in the 19th century shortly after the discovery of oxalic acid. However, the route starting from formate was discovered by John Louis Jullion during his attempts to improve the paper-making process and he patented the process in 1852.1 Merz and Weith were the first to describe the required process conditions in more detail in 1882.57,58 Goldschmidt in 1900 was the first to use a catalyst, in this case, carbonate base (CO32−), which also is a product in the reaction. It was suggested that the carbonate and formate would decompose upon heating to CO and hydrogen. Then the carbon monoxide will react with the carbonate to oxalate.59 The reaction was performed in an iron vessel above 400 °C under the exclusion of air and the reaction required 45 minutes. Wiens et al. continued the optimization of oxalate production in 1902, which led to the suggestion to use oxalate as a (base) catalyst for the reaction, which avoided the separation of the carbonate after the reaction.60 In 1912, Strauss patented a reactor design that relies on adding pure alkali formate to a pre-heated reactor. He claimed that no catalyst is required and that air is sufficient as atmosphere. However, he did not provide any specific examples and there are no reports of successful application of his reactor in the historic archives we visited.61 Mewburn suggested a reactor where the reaction mixture consisting of only alkali formate is pre-heated to 270 °C and then introduced to the main reactor which is preheated to 440 °C. According to their invention, this procedure reduced the reaction time to 5 minutes and they obtained an oxalate yield of 90%.62 Paulus et al. were the first to invent a continuous process for oxalate production from formate. This involved pre-heating of formate followed by rapid heating through spraying the molten reactant on a pre-heated rotating drum. They claimed that this resulted in a much more rapid reaction and an instantaneous conversion within 2–3 seconds without the use of any catalyst and the absence of side-product formation.63 Wallace adopted the pre-heating strategy for the batch process shortly after this using a different reactor design but he didn't report reaction times.64 In the invention of Bredt et al. in 1927, for the first time, the use of alkali earth metal formates was reported. They converted alkali earth metals to sodium formate using sodium hydroxide. The remaining sodium hydroxide was discovered to work as an efficient FOCR catalyst for the first time.65 Enderli and Hene for the first time directly used potassium formate as a reactant in 1935 and recognized that they were required to use active removal of gases during the reaction and hydroxide as a catalyst to obtain similar yields as in the sodium formate reaction.66–68 To actively remove gaseous reaction products, they exchanged the gas in the reactor by purging it with nitrogen. They optimized the reaction by varying the required flow rates of gas-removal, reaction temperatures and catalyst loadings. To our knowledge, their process was in commercial use for several decades at one of the main oxalate producers in Europe at the time.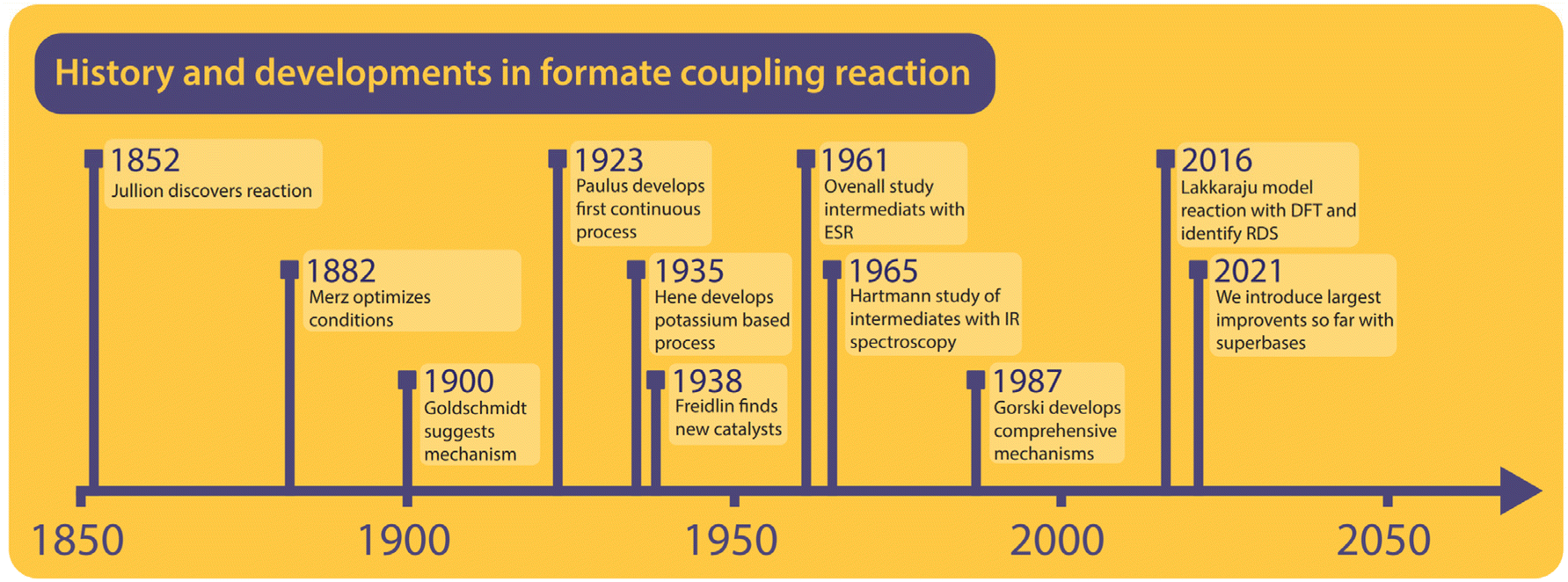 | ||
| Fig. 1 Brief history with some of the most important milestones in the FOCR. | ||
Freidlin et al. have studied the formate coupling reaction intensely between 1937 and 1940 and published their findings in 14 scientific papers.43,45,46,69–79 They looked at various aspects from the reaction conditions to the role of reactor materials and different (gas) atmospheres both from a scientific perspective but always with process development and application in mind. Most notably they investigated the suitability of a broad range of basic catalysts including the use of alkali metals, their amalgams, and superbases. Ovenall et al. studied the reaction of sodium formate incorporated in a crystal matrix and activated with high-energy γ-radiation.80 Unlike previous studies, they claimed that formate radicals rather than ionic intermediates were formed by homolytic splitting of the C–H bond. Subsequently, oxygen radicals were obtained from the subsequent reaction of the formate radical into CO and oxygen radical. They used electron spin resonance (ESR) to analyse the structure and the electronic state of these intermediates. The only carbonate was formed as a product from the reaction of the oxygen radical with the formate radical.80 Canning et al. turned their interest towards the thermal decomposition of alkaline earth formates but did not observe any oxalate formation. They were the first to establish that the nature of the cation influences the decomposition of the formates and argued that in the primary stage of the decomposition the transfer of electrons from the acid radical to the alkaline earth metal ion is required.53
Hartmann et al. studied the decomposition of a wide range of metal formates into salt matrices by pyrolysis in 1965 and 1966 and analysed the products using IR spectroscopy.47,48 No oxalate formation was suggested in their studies and it was claimed that two formate molecules recombine into a transition complex which then decomposes to carbonate and a formaldehyde moiety. Meisel et al. studied the thermal decomposition of several alkali metal formates by a complex dynamic thermo-analytical method.54 During this study, it was found that the FOCR with different alkali metal formates results in different oxalate yields and the atmosphere influences the ratio of the formate decomposition products. They achieved the highest yields with potassium formate followed by reactions with sodium and rubidium formate. With lithium and caesium formate, no oxalate was formed.54 In 1976, Shishido et al. found that the decomposition temperature ranges for Li, Na, K, Rb, and Cs formates differ and that transition metal formates decompose at even lower temperatures but don't lead to oxalate formation.52,55 Based on the findings of Ovenall et al., Shishido et al. proposed the formation of oxalate from two CO2 radicals and hydrogen stems from the recombination of two hydrogen radicals.80 They also established that the formation of oxalate depends on the stability of the formed oxalate which follows the sequence of K > Na > Rb > Cs. In 1979, Baraldi et al. investigated the thermal behaviour of metal formates with IR spectroscopy.81 In this paper, they found that the decomposition pathways of formates could be divided into two groups concerning the metals that were used. The first group, Na, K, Ca, and barium formate, mainly decomposed into carbonate at 500 °C. Yet they reported the formation of a stable intermediate from dehydrated formate, indicating that oxalate was formed in the process. All metal formates from other main groups decomposed to metal oxides or metals and gaseous carbon compounds.
Górski et al. investigated the formate coupling reaction in 1987 from four different angles including the role of the atmosphere, solid reactants, intermediates, decomposition of oxalate, and the formation of organic gaseous products.37,38,40,82 They were the first to suggest a full mechanism with carbonite, the di-anion of CO2, as a reactive intermediate in the FOCR as shown in Fig. 6.37 The underlying mechanism suggests that the pathways depend on the nature of the metal cation–hydride bond during the formate activation. A covalent bond leads to the formation of free hydrogen and carbonite whilst an ionic bond leads to formaldehyde and metal–oxides.40 In the first step, independent of the metal cation, formate decomposes to hydride (H−) and CO2. The subsequent reaction pathway in the second step depends on the metal cations. If they form mainly strongly polarized ionic bonds such as Li, Na, or K, then the hydride abstracts a proton from formate leading to the formation of hydrogen and the active carbonite intermediate which subsequently reacts with another formate to form oxalate. If metal cations form weakly polarized bonds with a largely covalent character, then the hydride and formate form a tetrahedral [HCHO]− intermediate which decomposes to formaldehyde and an oxygen dianion (O2−) to form a metal oxide. The formation of organic gaseous products was related to a heterogeneous reaction of the intermediate formaldehyde with metal oxides in the reaction medium. As metal oxides only form with certain formats, the formation of organic gases can be avoided by the choice of metal.82 For the FOCR they showed that the oxalate yield from the alkali metal formate is the highest when the same alkali metal hydroxide was used. Atmospheres also influence the reaction and whilst nitrogen, carbon monoxide, and hydrogen are inert towards the FOCR, carbon dioxide, water vapour and oxygen retard the reaction.38,83 Masuda studied the thermal phase transformations of lithium, sodium, and potassium formate. For these three formates, small endothermic peaks without any weight changes were observed at 230, 237, and 135 °C, respectively.84 Li et al. patented various designs of continuous processes for the production of sodium oxalate.85–89
In their earliest process, they aimed to improve the oxalate yield by improving the formate dehydrogenation by quickly heating the sodium formate from 300 °C to 420 °C with hot nitrogen at high flow velocities.85 In their second design, they replaced nitrogen with superheated steam for rapid heating.87 A fluidized bed reactor was patented by Cao et al. in 2009 for which they claimed to improve the oxalate yield by avoiding the decomposition of oxalate to carbonate.90 Microwave-assisted dehydrogenation of formate was patented by Ep et al. in 2015. They only achieved 75% oxalate yield but increased the energy efficiency for the heating.91 In 2016, Lakkaraju et al. performed a mechanistic study of the coupling reaction and confirmed carbonite as the active intermediate with DFT calculations and Raman spectroscopy in a reaction mixture of sodium formate with sodium hydride catalyst.41 The formation of carbonite was claimed as the rate-determining step for the reaction. They could reduce the reaction time with hydride, which is a stronger base relative to NaOH but could not reduce the reaction temperature. This work was patented in 2017.92 Most recently, we confirmed the presence of carbonite in the FOCR catalysed when using hydrides as superbases.42 We were also able to reduce the reaction times from minutes to seconds and temperature by 200 °C (now 170–200 °C) for the first time.
2.1 Products obtained during attempted formate coupling
The coupling of formate to oxalate ideally only produces oxalate and hydrogen as products. However, several side reactions occur due to non-optimal conditions. The side-products can be divided into solid and gaseous and a complete overview of them is shown in Fig. 2.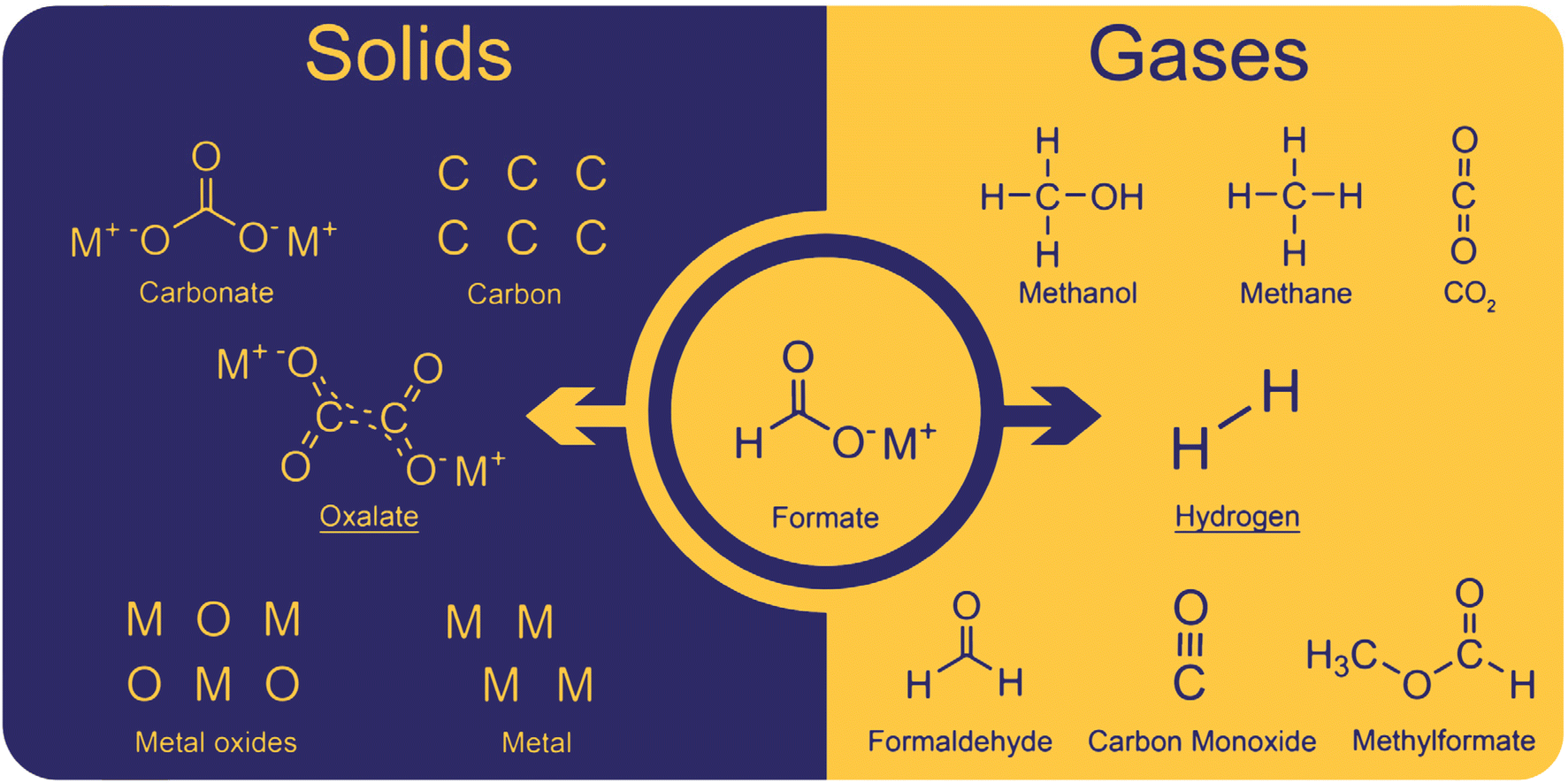 | ||
| Fig. 2 Solids and gases obtained as products in the formate coupling reaction. The desired products are oxalate and hydrogen. Solid side products include carbonate and as well as carbon, metal oxides, and metals. Gaseous side products were found to be methanol, methane, CO2, formaldehyde, carbon monoxide, and methyl formate.82 | ||
Solid oxalate is the desired product; however, carbonate was often observed as a side product. With hydroxide bases, carbonate was obtained at relatively low temperatures at which the formation of oxalate does not yet occur. However, in our experiments, we never observed carbonate formation at low temperatures apparently due to the dry conditions we maintained. Additionally, carbonate can be formed by oxalate decomposition at high temperatures which is helped by the presence of hydroxide. Another decomposition product is elemental carbon which is produced from oxalate, formate, or carbonate at high temperatures. Górski et al. reported a pronounced carbon formation when lithium formate was used in the FOCR but only traces of carbon were formed with sodium- and potassium formate. They explained this with the preferred formation of carbonate and CO instead of oxalate for lithium formates.93 The CO formed is then disproportionate to elemental carbon and CO2. If borohydride is used as a catalyst in the FOCR with sodium and potassium formate, the formation of meta-borate can be observed. The formation of metals and metal oxides has been reported when strong bases were used in the reaction. Górski et al. suggested the disproportionation of two carbonite intermediates to oxalate and metals as a potential pathway. However, we believe that, due to the strong repulsion of the two molecules and low overall concentration, this is unlikely to occur. We never observed metal or metal oxides in our reactions with hydrides as catalysts.
In the formate coupling reaction not only oxalate is produced but also one equivalent of the valuable gas hydrogen. Additionally, a wide range of other gases has been reported including formaldehyde, methanol, methyl formate, methane, carbon monoxide, and CO2. Whilst these gases are products formed from formates, also water contained in the highly hygroscopic reaction mixture leaves the reactor at increased temperatures. The formation of CO and CO2 as intermediates in the reaction pathway from formate to oxalate was suggested by Górski et al. The loss of these gases from the reaction was proposed as the reason for the low oxalate yields. The formation of the gaseous organic compounds such as methanol, methane, formaldehyde, and methyl formate depends on the formation of formaldehyde as an intermediate. Potential pathways for these reactions were discussed in detail by Górski et al.40 They found that the formation of formaldehyde as an intermediate was mainly dependent on the metal counter ion. From formaldehyde, methanol and methyl formate can be formed in Cannizzaro or Tishchenko reactions as shown in Scheme 2.94,95
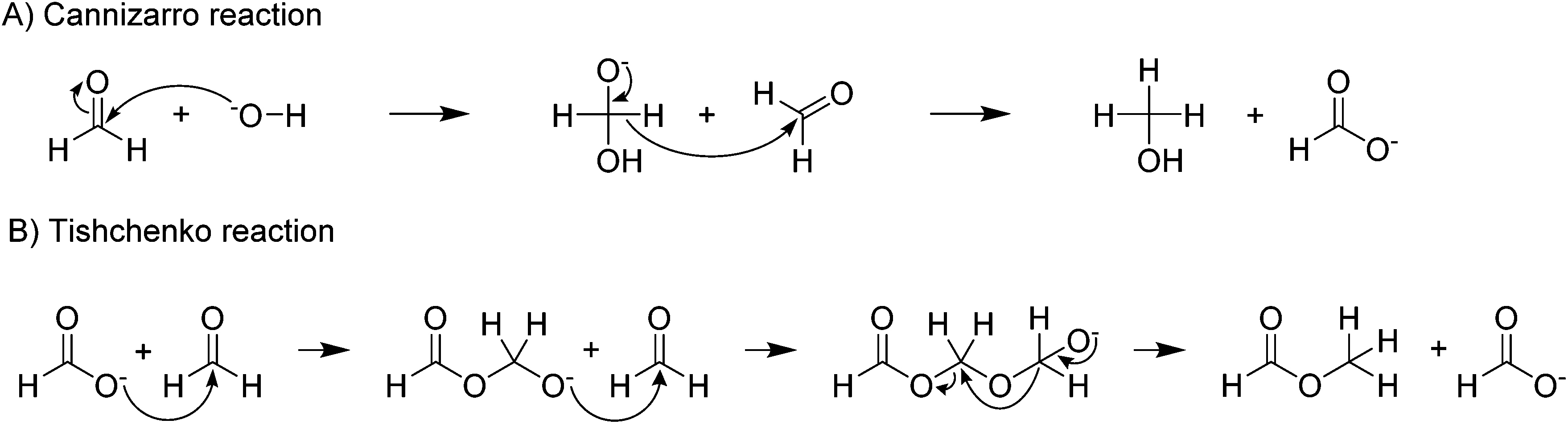 | ||
| Scheme 2 Formation of (A) methanol via Cannizzaro reaction,94 and (B) methylformate via Tishchenko reaction.95 | ||
To gain more insight into gas evolution, we studied the time-resolved formation of gases during the reaction with different catalysts including hydroxides, superbases, or titanium hydrides in non-isothermal reactions with heating rates of 10 °C per minute (Fig. 3). We employed both GC and mass spectrometry, which were directly connected to the outlets of our reactor. Independent of the catalysts used in the reaction system, hydrogen was observed as the main gaseous product in the temperature regime where oxalate was produced. CO2 and CO were formed in the potassium hydroxide catalysed reactions when the reaction mixture was not dried sufficiently. We observed the formation of CO in trace amounts at the beginning of the reaction during the evolution of residual water when hydroxides were used as catalysts. Once the hydrogen formation increased at higher temperatures, CO was not detected anymore. In the absence of a catalyst, CO2 and CO were formed at temperatures below 380 °C during carbonate formation. If the reaction temperature is set too high, the formation of CO2 was also observed during oxalate decomposition. In the presence of super bases such as hydrides or amides also CO was produced during oxalate decomposition at high temperatures.
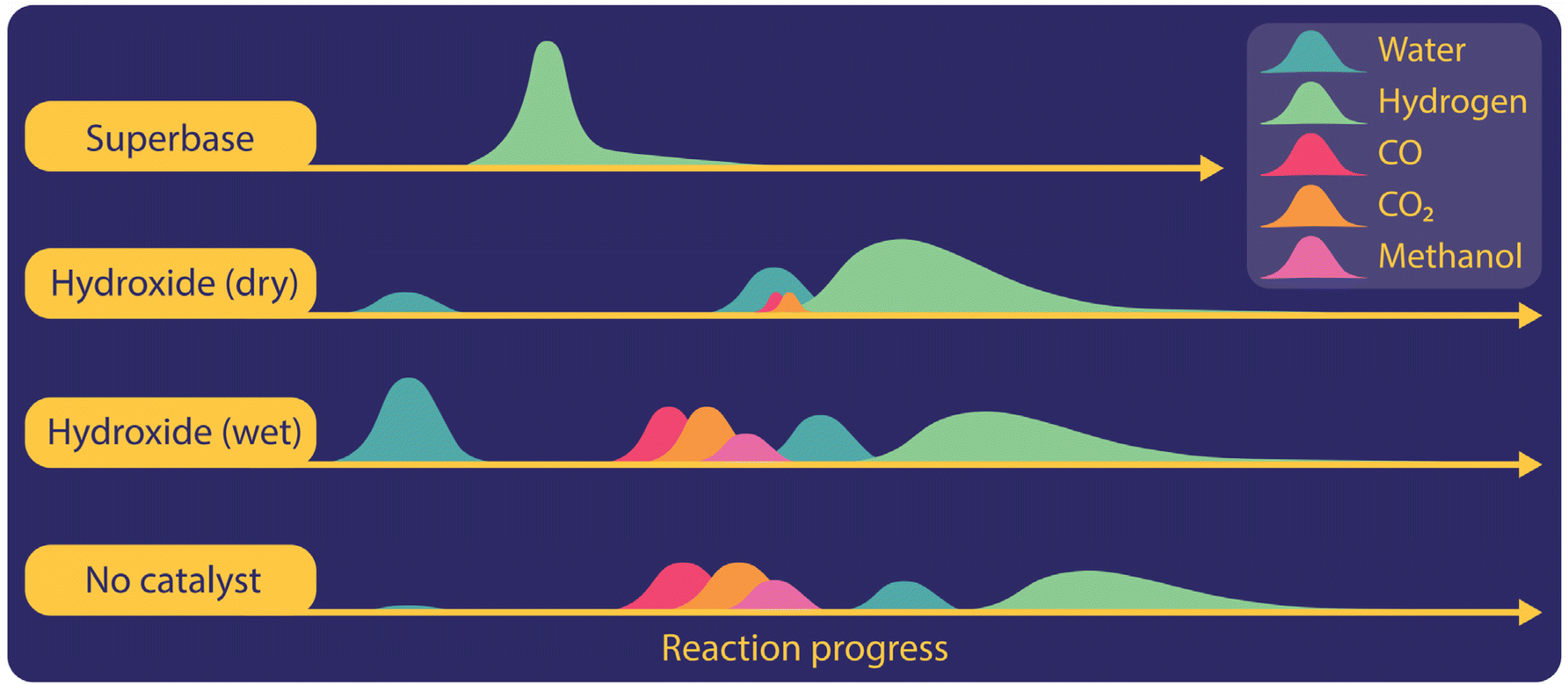 | ||
| Fig. 3 Gases produced during heating of formate with various catalysts. | ||
We also observed various organic gaseous compounds in uncatalyzed reactions or with hydroxide or titanium hydride as a catalyst. Methanol formation was only observed shortly after the appearance of carbon monoxide and carbon dioxide from pure formate or insufficiently dried hydroxide catalysed formate reactions. Methane was detected in the presence of titanium hydride as a catalyst and coincided here with the release of hydrogen stored in the titanium hydride. We did not, however, observe any formaldehyde or methyl-formate.
The evolution of water could be observed in hydroxide catalysed reactions, even if the reaction mixture was dried in a vacuum oven. The magnitude of water release increases with higher loadings of hydroxide catalyst and occurred in two stages at different temperature ranges, which became even more visible at lower heating rates. We believe that this indicates that the released water is of different origins. The first one is crystal water strongly attached to hygroscopic hydroxide. The second source is water produced to initiate the FOCR itself. For potassium hydroxide catalysed potassium formate reactions, water was released first, followed by the subsequent production of CO, CO2, and methane. Finally, hydrogen was produced and none of the other gases appeared any longer in the effluent gas.
2.2 Parameters used in past
The production of oxalate and hydrogen, as well as the side products, is influenced by various process variables as shown in Fig. 4. We can divide the variables into six categories including the formate metal cation, heating of the reaction, the atmosphere, use and nature of catalysts, potential poisons, and the reaction time. For each category several continuous and categorial factors are tuneable, yet not all of them lead to the desired selective formation of oxalate nor have they necessarily turned out to be of influence at all. Catalysts have a special role in this list as they can drastically change the required reaction time and temperature of the FOCR.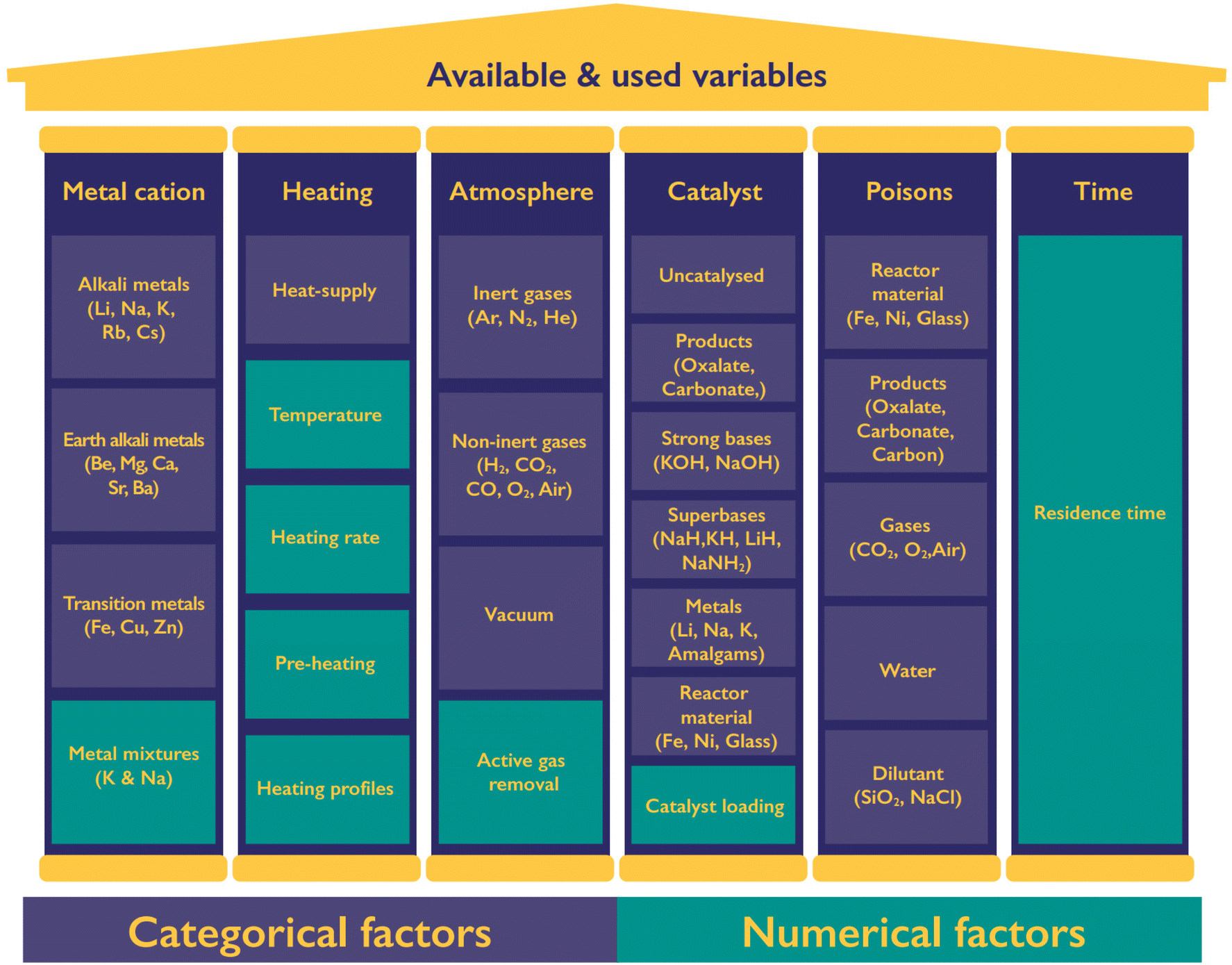 | ||
| Fig. 4 The reaction parameter choices made in the past can be categorized into 6 principal pillars of formate metal ion, heating method, atmosphere, catalyst choice, presence of poisons, and reaction time. For each of these pillars, several subcategories are available which contain either categorial or numerical variables. | ||
Only alkaline metal formates produce oxalate in the decomposition process. Whilst potassium and sodium metal formates reliably allow oxalate production, this is not the case for rubidium, caesium, and lithium formates. The oxalate yield that can be obtained from formate decomposition follows the sequence of K > Na > Rb > Cs > Li.53–55 Shishido et al. reasoned that the stability of the produced oxalate is responsible for this observation.55 The production of oxalates from lithium formate appears the most challenging as not all authors report oxalate formation but rather decomposition to elemental carbon and carbon monoxide.38 Because of this absence or very limited oxalate production from Li, Cs, Rb, and transition metal formates we focus on the decomposition behaviour of potassium and sodium formate in our work. We were the first to look into mixtures of Na/K formates as these have the potential to fine-tune the reaction conditions allowing us a broader array of potential reactor designs. For the remaining parameters, we restricted our literature research to parameters used in sodium and potassium formate reaction systems.99 If the FOCR is part of a CO2 to chemicals process starting with the electrochemical reduction of CO2 to formate, then potassium is the most desirable metal anion as it promises the highest CO2 conversion efficiencies and yields.33,100
The weak base alkali carbonate was the first suggested catalyst for FOCR by Goldschmidt in 1900.59 Later Górski et al. found that adding equimolar amounts of a base harms the reaction.37 We investigated various carbonates as catalysts encouraged by the activity of caesium carbonate, a stronger base that was active in similar reactions.102 Unfortunately, none of the tested carbonates had any visible effect on the reaction in our tests.42 Also, oxalate, the main product of the reaction, was used as a catalyst in the process developed by Wiens, yet we could not observe any catalytic activity when adding oxalates to the reaction mixture.60 This suggests that a stronger base is required to drive the reaction.
Alkali hydroxides have been most popular and still are the only ones used in commercial processes. With hydroxide, the reaction start-temperature, and optimal reaction temperature at which the reaction proceeds at its highest rate could be lowered on average by 40 °C. More importantly, the reaction completes in a matter of minutes rather than hours. Unfortunately, hydroxides do not only function as a catalyst but also as stoichiometric reactants with both formates and oxalates leading to the formation of carbonate and CO2. Hence an optimal amount of catalyst loading exists which strikes the balance between accelerating the FOCR without causing too much carbonate formation. The optimal amount of hydroxide was reported within a range of 1–10 wt%. Hydroxide has the advantage of being relatively cheap and it's recoverable from the products after the reaction.
Sodium borohydride is an even stronger base that is stable in the presence of water. It was used in the formate coupling reaction and reduced the reaction temperature by up to 100 °C. Although high oxalate yields can be achieved, 5 wt% catalyst loading is still required. Also, metaborate is formed leading to difficulties to recover the catalyst and the higher initial cost prevented commercial application.40
Given that the reaction appears to improve with increasingly basic catalysts superbases are interesting. Already Freidlin et al. had made use of them and tested hydrides, amides as well as alkali metals as catalysts.103–105 They showed that amides could lower the reaction temperature by 150 °C and alkali metals were performing even better. Alkali metals, however, are difficult to handle and their recovery poses a major challenge. Freidlin solved this by using amalgams of alkali metals which were high-density non-miscible liquids at the applied reaction conditions.105 Due to their toxicity, this is not a sustainable option in large-scale operations today. Hydrides have been the most investigated superbases and were used by Freidlin, Górski, and Lakkaraju. While they all reported strong improvements in reaction rates, only a small decrease in reaction temperatures was observed compared with hydroxides.41,43 We, however, showed most recently that these superbases can facilitate the reaction at much lower temperatures and that reactions are limited by the availability of formate once the reaction mixture melts.42 The reaction rates are greatly increased and no side products are produced under optimized conditions. This however requires an absolute absence of moisture. The recovery of the more expensive superbases catalysts is still very challenging, but only 0.5–1% of the catalyst is required which outweighs the cost of their loss as our calculations show.42 Whilst reactor materials including glass, iron, and nickel were also reported as potential catalysts, we recently showed that they influence the reaction as poisons and not as catalysts.83
The ideal reaction temperature differs depending on the formate metal ion, catalyst used, the atmosphere, water content in the reaction mixture, reaction time, and reactor design. Hence, the reported ideal reaction temperature differs across the literature.
In Fig. 5 we illustrate the different processes happening when potassium format is used as a reactant. We found that formate does not react in its solid form. In the presence of a superbase catalyst, oxalate is formed rapidly once the formate melts (at 170 °C and 248 °C for K- and Na-formate, respectively).42 In the absence of superbases, catalyst-free decomposition of formate to carbonate dominate from melting up to about 360 °C. We, however, found that this decomposition does not occur in the absence of moisture.83 With hydroxide as a catalyst, oxalate formation can already be achieved at 320 °C and the ideal reaction temperature window is lowered to 410–430 °C.42 Without a catalyst, oxalate formation starts at 360 °C and the reaction rate towards oxalate increases up to 440 °C. Above 440 °C the decomposition of oxalate towards carbonates, volatiles and elemental carbon occurs primarily. The initial decomposition temperature of oxalate depends mainly on the atmosphere, metal ions and can be lowered when catalysts are used. Especially the use of CO2 in the atmosphere can lower the oxalate decomposition temperature.38 Hence the ideal temperature window for oxalate production historically was between 360 and 440 °C. We found that without catalyst, the required reaction temperature was higher and optimally between 420–440 °C.42
 | ||
| Fig. 5 Reactions occurring during formate to oxalate coupling at different temperatures. | ||
Rapid heating of the formate was proposed in order to avoid the side reaction to carbonate in the low-temperature region.90 In our experiments, however, we found no noticeable impact of slow heating rates on the oxalate yields in our system. Conversely, fast heating causes strong foaming of the reactant caused by very rapid gas formation. This harms heat transfer and increases the reaction temperatures or the reaction times to achieve oxalate yields like reactions with slower heating rates. We furthermore observed uncomplete reactions with strong foaming. To overcome these problems, Hene proposed two-stage heating and showed that a longer first reaction stage at a lower temperature followed by a short second reaction stage could improve oxalate yields significantly.67 While this was carried out in one reaction vessel, other two-stage heating processes rely on pre-heating the reaction mixture in a separate vessel to decrease the required heating towards the desired reaction temperature. Mewburn showed earlier that pre-heating formate to 270 °C in a separate heater before rising it quickly to the desired reaction temperature can reduce the reaction times significantly and improve the yield.62 This concept was adapted in many continuous process designs as they require the reaction mixture in a liquid state.
The second most common atmosphere used in FOCR is nitrogen which showed a higher conversion towards oxalate compared to air.38,41,42,54,55,67,68,81,85,92 Nitrogen has been used with all available catalysts and the reaction performs similarly in the inert atmospheres of argon or helium which indicates that nitrogen is inert, too.93
If the FOCR involves equilibrium reactions, the produced gases should affect the achievable yields or reaction times. In the FOCR mainly hydrogen is formed, and water is released from the hygroscopic reaction mixture. Additionally, CO and CO2 can be produced in undesired side reactions. A potential influence of these gases on the course of the reaction was investigated by Górski et al. who performed the uncatalysed FOCR in CO and CO2 atmospheres.37 They did not observe any detrimental effect of CO and, therefore, concluded it to be an inert gas. In our investigations with hydroxide catalysed reactions, however, we found that carbon monoxide reduces the conversion but has no effect on selectivity towards oxalate.83 The story for CO2 is, however, different and Górski et al. found that carbon dioxide did not affect required reaction temperatures but observed that high CO2 concentrations reduced the conversion significantly. They argued that the concentration of CO2 during the uncatalysed reaction needs to be well balanced. In their proposed mechanism, CO2 is a reaction intermediate that is first formed in the decomposition of formate to form active hydride species and later reacts with the active carbonite intermediate as shown in Fig. 6. They assumed that too much CO2 prevents the activation which is an equilibrium reaction. CO2 also prevented the decomposition reaction of the formed oxalate to carbonate and CO2 as this reaction is an equilibrium reaction and the presence of CO2 shifts the equilibrium to the oxalate side.37 In a hydroxide catalysed FOCR, we, however, found that CO2 inhibits the reaction and blocks the formation of oxalate in favour of carbonate formation.83 We could recently show that CO2 reacts with both hydride and carbonite, the two highly reactive species in the reaction system.117 Hydride and CO2 react towards formate. Carbonite and CO2 react towards oxalate. In both reactions, no new reactive species are formed. Therefore, the presence of CO2 removes the active species and inhibits the reaction.
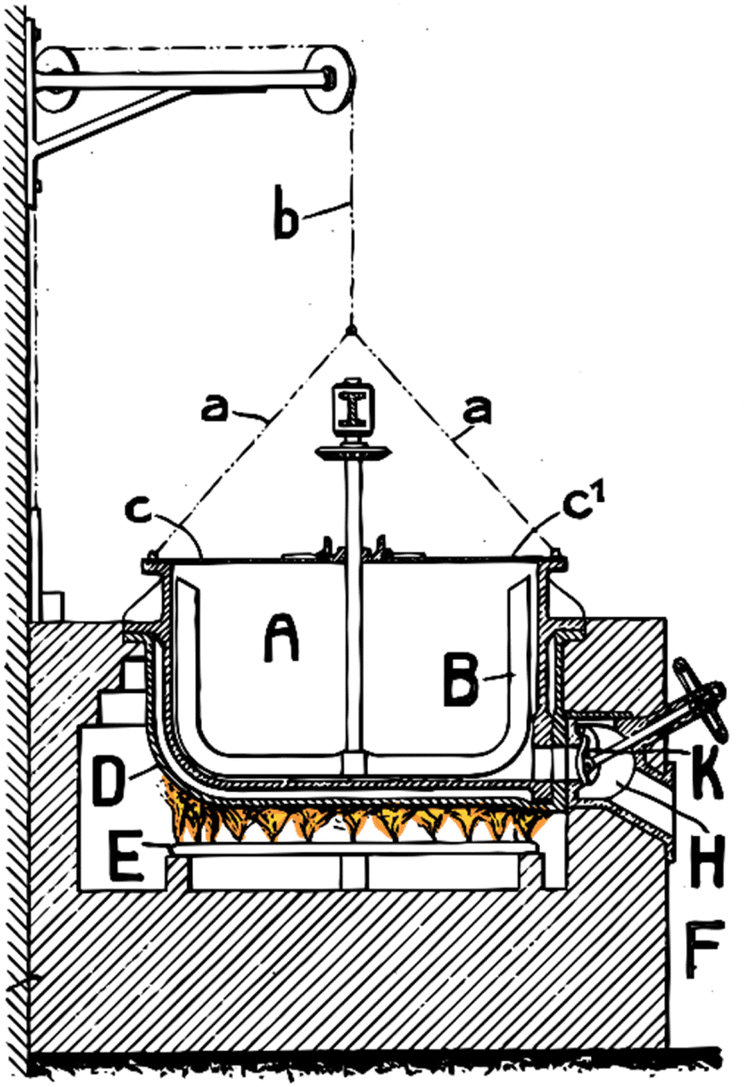 | ||
| Fig. 6 Reactor apparatus used for formate coupling reaction 100 years ago. A device with two pivoted lids c, c1 which are connected by chains a, a, b with a weight C tending to hold the lids in their position when opened. Pan is surrounded by a receptacle D confining an air bath between itself and pan A and disposed above a series of gas burners E. The said receptacle D and the gas burners E is contained in the interior of a masonry mass F provided with the necessary flue for the combustion gases. Pan A is provided with a tube E for the evacuation of the steam and gases resulting from the reaction in pan A. The socket H on the bottom of pan A and normally closed by a valve K allows removing from pan A the oxalate produced at the end of the operation. The formate is fed via the lid c and stirred via the stirring device B. Ideally 150–200 kg of formate are heated per m2 of heating area. The ideal temperature for the burner is between 500 to 570 °C, the temperature in the molten formate is above 400 °C.61 | ||
Hydrogen is the main gaseous product of the FOCR and a valuable feedstock with ever-increasing interest. As it is most favourable to capture it undiluted in commercial applications, hydrogen is the most interesting atmosphere. Fortunately, Górski et al. have shown that nitrogen behaves as an inert gas in the sodium formate coupling reaction.40 During the development of a commercial hydroxide catalysed potassium formate coupling process, however, Enderli et al. observed that hydrogen has an inhibiting effect above a certain partial pressure.68 Our results, however, do not show inhibition by hydrogen on the hydroxide catalysed FOCR. Conversely, hydrogen appears to be the most suitable atmosphere.83 We suggest that not the removal of hydrogen as suggested by Enderli et al., but rather the removal of water which is released from the reaction mixture improves the oxalate yields. In particular, we could observe the negative effect of water when steam was used as an atmosphere. The active removal of water is therefore critical in potassium-based systems but also beneficial in sodium-based systems. The removal can be achieved by either flowing an inert gas, ideally hydrogen, over or through the reaction mixture or by applying a vacuum.67,115
Reactors for the FOCR have traditionally been made from metals, most commonly steel. This, however, may lead to the formation of iron oxalates which are difficult to remove and cause a reduction in oxalate yields.71 Especially in a process where the FOCR is paired with electrochemical cells, the introduction of iron ions is undesirable. In these processes, the electrolytes used for downstream acidification of oxalate to oxalic acid and the upstream CO2-to-formate reaction are mixed. Iron is a known poison to the upstream electrochemical CO2-to-formate reaction.36 The use of nickel or glass as reactor materials has shown to be inert towards the reaction and therefore provide alternatives to iron.71 Oxalate and carbonate are both produced in the FOCR and have been suggested as potential catalysts in the reaction.60,118 Other reports however suggest that especially carbonate, if present in larger quantities, reduces the achievable oxalate yield by up to 23%.40 We found that only carbonate if added in equimolar amounts harms the oxalate formation. Gases such as CO2, oxygen, and steam can also negatively affect the FOCR, as discussed above. Dilutants are technically not necessarily poisoning the formate coupling reaction itself. However, as the reaction is relying on mass and heat transfer in the molten salt, the introduction of dilutants such as silica or carbon powders can reduce the obtainable oxalate yield. We have seen this especially with low-density materials such as silica aerogels.83
Whilst most patents suggest reaction times from 15 minutes to 1.5 hours, it can require up to 8 hours to reach completion.59–62,64,66–68,106–109,119–123 The slowest reaction times were reported for large-scale drum reactors which were operated in batch mode with hydroxide as catalysts (Fig. 6). The latter limits the reaction rate and convectional heating in a high volume-to-surface ratio reduces the heating rate, increasing the overall residence time of the reactants in the reactor. With increasing reaction times, oxalate can decompose to carbonate or elemental carbon. Hence, Paulus proposed a different reactor design that should allow for more rapid heating and claims an instant conversion in his patent, yet he does not back this claim with any evidence.107 On the other end of the scale of the required reaction, times are superbase catalysed systems in which the reaction reaches completion as quickly as 30 seconds. The reaction rate with superbases is increased by several orders of magnitude and the lower reaction temperature reduces the time required to heat the reaction mixture.
3 Mechanistic pathways
The history of the FOCR literature is rich in proposed mechanisms.37–42,47–50,52,53,55,56,81,118,124–130 Interestingly, these pathways differ not only depending on the study but also depending on the catalyst used in the FOCR. In many cases such as in the use of the commercial hydroxide “catalyst,” the so-called catalyst is a catalyst precursor. The active catalyst during the reaction is produced in situ. The first suggestion known to us is by Goldschmidt, involving decomposition of carbonate and formate upon heating to carbon monoxide and hydrogen which then react with the carbonate to oxalate.59 Takagi and Freidlin et al. were the first to systematically study the FOCR to unveil its mechanism.46,126 Hartmann et al. studied the decomposition of metal formate into salt matrices by pyrolysis in 1965–1966.47,48 In the 1960s radical formations were proposed and studied by Brivati, Ovenall, Atkins, Whiffen, and Bellis.124,129–131 Based on the work of Ovenall et al., Shishido and Masuda proposed the formation of oxalate from two CO2-radicals and the recombination of two hydrogen radicals to hydrogen.52,80 Yet, Górski et al. suggested a multi-step reaction cycle involving carbonite as a reactive intermediate. Górski et al. themselves were not able to directly prove the presence of carbonite but Lakkaraju later showed spectroscopic evidence.41 We added further proof recently with D2O quenching studies.42 Most recently, we showed the role of CO2 as a reaction partner or poison of the reaction, depending on the presence and quantity of base catalysts.117 In another publication, we address the role of the metal counterion and gas-removal in hydroxide catalysed reactions.99Overall, the FOCR proceeds through various stages illustrated in Fig. 7. All reactions start with the activation of formate by proton abstraction, followed by oxalate formation via carbon–carbon coupling (carbanion attack on electrophilic carbonyl of another formate) and – although undesired – decomposition of oxalate. In parallel, the decomposition of formate to other organic compounds presents further undesired side reactions.
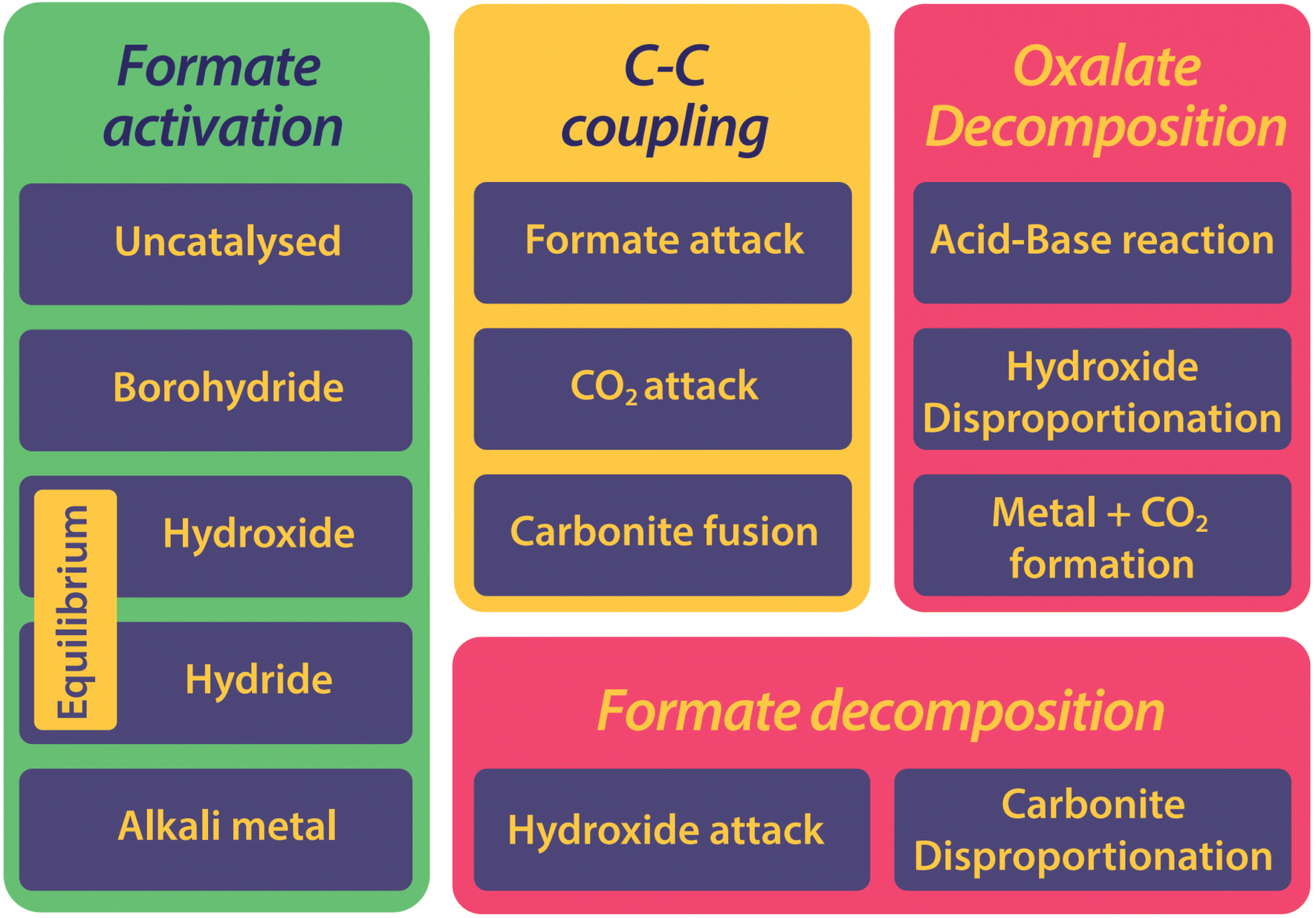 | ||
| Fig. 7 The FOCR can be divided into three stages including formate activation, carbon–carbon coupling, and at last decomposition reactions. Various mechanisms were proposed for each of the stages which differ for catalysts or reaction principles. | ||
3.1 Carbon–carbon coupling
Although only the second step in the reaction, we find it helpful to first discuss potential pathways to couple two carbons starting from formate. All proposed mechanisms, presented in Fig. 8, are depending on the presence of a curious reactive species called carbonite. Carbonites are very short-lived due to their high reactivity. A comprehensive review of carbonites was recently published by Paparo et al.128 Freidlin et al. were the first to suggest the presence of carbonite in the FOCR but it was Górski et al. who based all of their pathways on the presence of carbonite.38 They suggested the reaction of carbonite with CO2 as the main pathway as shown in Fig. 8B. The electrons are located on the carbonite's carbon which hence acts as a carbanion nucleophile to attack CO2 molecule. Alternatively, Górski et al. proposed that two carbonite molecules could disproportionate and fuse to form oxalate and alkali metal following Fig. 8C. Due to the low apparent concentration of carbonite, this reaction appears rather unlikely. If borohydride is used as a catalyst, the carbonite is stabilized as borocarbonite. In the reaction with formate, the formate proton is exchanged for the carboxyl group (Fig. 8D). Górski et al. do not provide any detailed reaction mechanisms or computational proof for their suggested mechanisms but only rely on observations during thermogravimetric experiments and product analysis. Although they argue that superbases such as hydrides or amides should favour oxalate formation a dedicated carbon–carbon coupling mechanism was not discussed.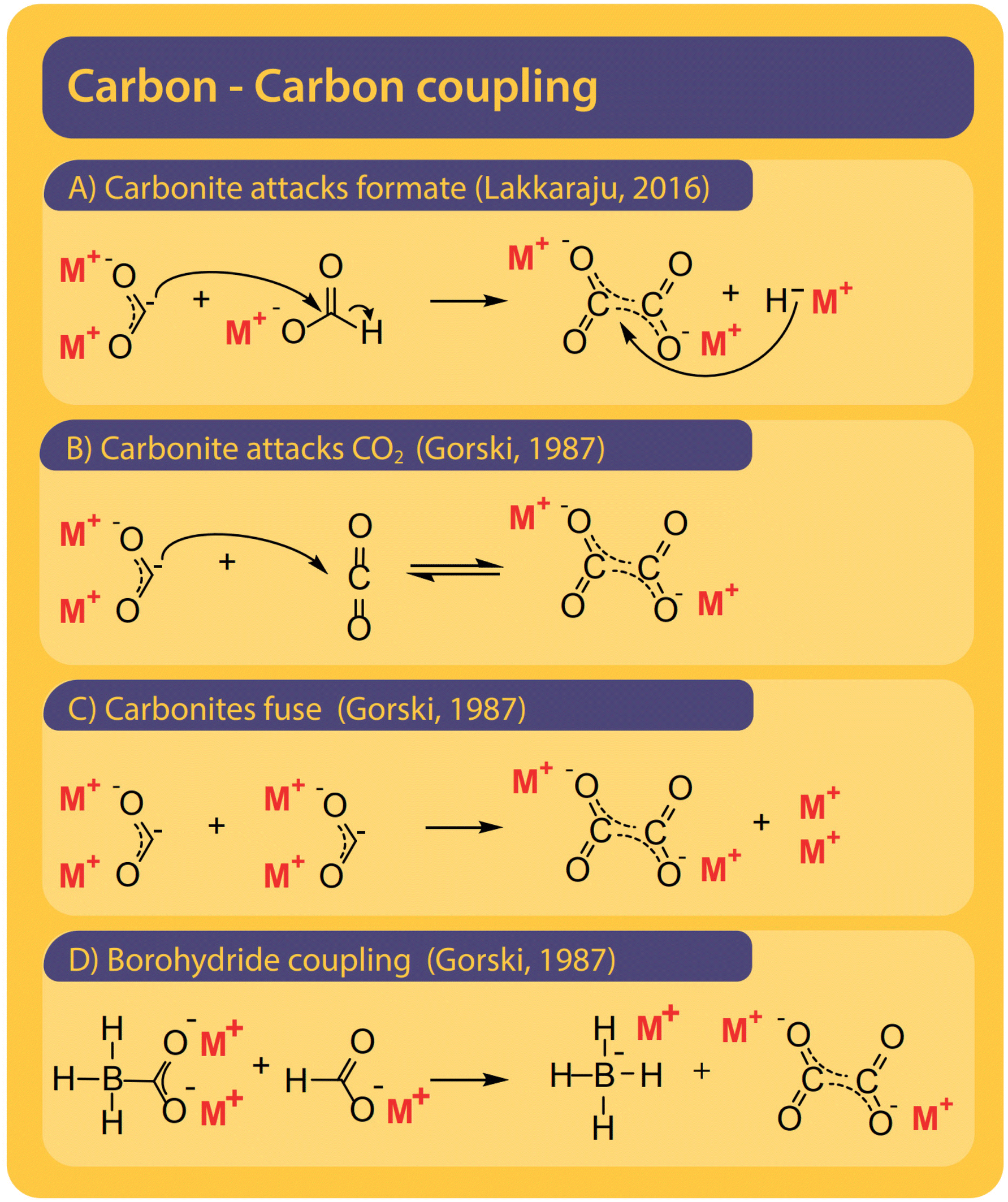 | ||
| Fig. 8 Four pathways were suggested for the carbon–carbon coupling reaction in the FOCR in literature.41 (A) The attack of the active carbonite intermediate on formate to form oxalate and hydride. (B) Carbonite attacking a CO2 molecule to form oxalate by no hydride.38 (C) The disproportionation of two carbonite intermediates to fuse towards oxalate and metal.38 (D) Attack of borohydride stabilized carbonite on formate with the formation of metal borohydride. The coupling of borocarbonite is restricted to the use of borohydrides as a catalyst.38 | ||
Lakkaraju et al. for the first time suggested a detailed mechanism involving intermediate states and the DFT calculations for both potassium and sodium systems. They suggest a direct attack of the active carbonite on surrounding formate followed by the release of a new hydride which can function as a catalyst again (Fig. 8A). It is, however, not the C–C coupling step in their mechanism which was found to be rate determining but the generation of the reactive carbonite species in the first place. In conclusion, it is imperative to produce carbonite from formate to produce carbonate. The differences in required reaction conditions, rates, and reaction orders suggest that several different mechanisms exist for different catalysts or catalyst precursors which are all converted to the active catalyst in situ. We showed that both coupling options of carbonite with either formate or CO2 are possible and the strong poisoning behaviour of CO2 suggests that the reaction of carbonite with CO2 might be preferred over the reaction of carbonite with formate.117 DFT calculations of Lakkaraju et al. indicate a strong exothermicity for this reaction.41 The strong poisonous character of CO2 we have seen in the FOCR when a base is added in catalytic amounts underlines this.83
3.2 Uncatalyzed formate activation
The first step is the activation of formate by breakage of the carbon–hydrogen bond to form carbonite, for which several options exist as shown in Fig. 9.132 The reaction mechanisms might differ depending on the presence and type of a catalyst, which we discuss individually.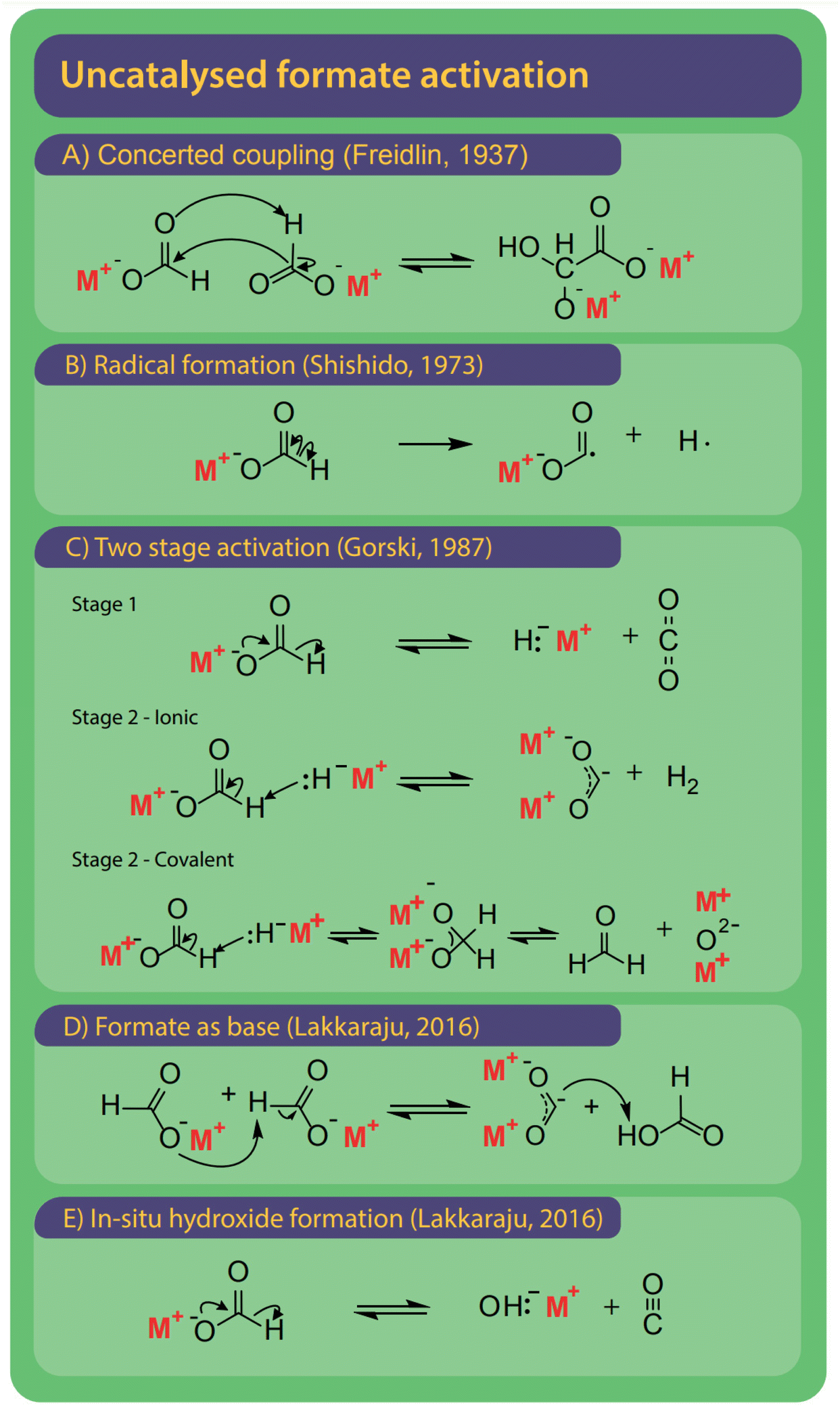 | ||
| Fig. 9 The FOCR can be performed without the presence of an additional catalyst. Overall, five different mechanisms for the activation of formate were suggested historically. (A) Oxalate is formed in a concerted coupling of formate, strictly this combines activation and coupling in one step.78 (B) Homolytic cleavage of the C–H bond to form formate and hydrogen radical.78 (C) Two-step activation starting with the decomposition of formate to hydride and CO2. Step 2 is depending on the metal–hydrogen (M–H) bond of hydride formed in step 1. If the M–H bond has an ionic character, then carbonite and hydrogen are formed. If the M–H bond has a covalent character, then formaldehyde and metal oxide are formed.37 (D) Formate acts as a strong base itself to form formic acid and carbonite.41 (E) Formate decomposes to hydroxide and CO to in situ form hydroxide catalyst precursor. | ||
A first mechanism for the uncatalyzed reaction, shown in Fig. 9A, was proposed by Freidlin et al. and involves the concerted coupling of two formate molecules to form a glyoxylate intermediate. Subsequently, oxalate is formed via the release of hydrogen.78 The driver for the reaction suggested by Freidlin is unclear and we deem such a multistep concerted reaction unlikely. Later Brivati, Ovenall, Atkins, Whiffen and Hartman studied the decomposition of formates in the presence of γ-radiation. They however aimed to study the decomposition behaviour of organic mediator substances in nuclear reactors rather than the production of oxalate.124,129–134 Ovenall et al. established that a CO2 radical anion is formed by homolytic cleavage of the C–H bond in formate at elevated temperatures after irradiation.135 They studied sodium formate incorporated in crystal matrix with high-energy γ-radiation and used electron spin resonance (ESR) to determine the electronic state of the CO2 free-radical.
If two calcium formate molecules were used, they recombined into a transition complex and then decomposed to carbonate and formaldehyde. Shishido et al. translated the radical mechanism to the formation of oxalate from two CO2 radicals and the recombination of two hydrogen radicals to hydrogen as shown in Fig. 9B.52 Yet, Górski et al. showed by ESR studies that these radicals were only observed if the samples were irradiated with γ-radiation before the measurement and not during thermal decomposition alone.37,124,129 As an alternative they introduced carbonite, a di-valent carbon, as the reactive intermediate.
Already Freidlin et al. had considered a second route involving the formation of carbonite when using metals as a catalyst but rejected this intermediate as they thought the formation of a two-valent enol-like species was highly unlikely.78 Górski et al. used thermogravimetric analysis to study the FOCR and showed that synthesis gas was not the precursor to organic species formed during the formate decomposition.93 In their two-step activation, shown in Fig. 9C, Górski et al. hence took the influence of the metal into account. In the first step, formate decomposes to hydride and CO2, which is the reverse reaction of formate formation from CO2 and hydrides. They observed CO2 amongst the products for all uncatalyzed reactions independent of the formate metal cation used. Therefore, they concluded this reaction to be independent of the formate metal cation and non-reversible as the hydride rapidly reacts with another formate anion. Hence, this step is also not influenced by the presence of CO2. Whilst this first step was proposed to be independent, the second step depended on the metal cation. Already Meisel et al. studied the influence of the metal ion on the FOCR with thermogravimetric analysis and concluded that the increasingly covalent character of the metal–carboxyl bond with increasing radii of the metal ion hinders oxalate formation.54 They suggested various decomposition paths, but without any detail on the actual mechanism. Górski et al. incorporated this in their proposition for the second step reaction step. Here the degree of covalence of the metal–hydride bond determines the further reaction path. Metal cations with low electron affinity such as sodium, potassium, or rubidium form strongly polarized bonds with the hydride anion resulting in an ionic character. The hydride acts as a strong Lewis base and abstracts a proton to form molecular hydrogen and reactive carbonite. This reaction was thought to be favourable due to the increased bond strength of the newly formed H–H bond compared to the original C–H bond. For metal cations that form weaker polarized bonds, the hydride–metal interaction is largely covalent. Consequently, the hydride acted as a nucleophile leading to the formation of the tetrahedral methanebisolate which decomposed subsequently to formaldehyde and metal oxides. Gaseous organic products observed in the decomposition of most metal formates stemmed from the formation of formaldehyde and its subsequent decomposition helped by the metal oxides. Methanol was proposed to form in a Cannizzaro reaction on metal oxides with strong basicity, whilst weakly basic metal oxides favour the Tishchenko reaction towards methyl formate.136,137 All of Górski et al.'s suggestions were based on thermogravimetric studies with analysis of the obtained gases but no spectroscopic or computational evidence was provided. Hence, they were not able to directly prove the presence of the reactive carbonite. Lakkaraju et al. showed spectroscopic evidence recently, however only for hydride catalysed reactions.41 We added further experimental proof to the presence of carbonite recently with D2O quenching studies.42 Lakkaraju et al. however disagree with the in situ formation of hydride and CO2 from a formate, as their calculations show a very high energy barrier for this reaction.41 They propose the in situ formation of hydroxide and carbon monoxide as shown in Fig. 9E instead. After this initial in situ formation of hydroxide, the reaction proceeds like hydroxide catalysed reactions as introduced below.
Finally, it has been proposed to include a new potential pathway in which the formate itself acts as a proton abstracting base. This would lead to the formation of formic acid and carbonite as intermediates. We include this pathway for mainly two reasons: it is in line with the carbonite formation via base abstraction mechanisms suggested for other catalysts and secondly, we did observe this transition during our molecular dynamic calculations, although as a rare event with a very high activation barrier. The rarity and high activation barrier make it difficult to simulate this reaction as it requires computing the behaviour of the system over a relatively long time. Additionally, the high energies caused by the high reaction temperature make other side reactions difficult to control. Lakkaraju et al. argue that the proton abstraction by formate itself has a too high energy barrier and deem the in situ formation of hydroxide via formate decomposition to hydroxide and CO is more likely to occur.41 Interestingly, other than in catalysed reactions we don't see any indication for autocatalytic behaviour with an acceleration of the reaction rate once an active catalyst is formed. In a purely carbonite-based mechanism, this should be induced as once carbonite is formed, the reactive hydride is liberated in the C–C coupling process and subsequently available as a potent catalyst. Given the high temperatures required for the uncatalyzed reaction, it may also be possible that several pathways occur.
To date, it is not yet clear which mechanism the uncatalyzed FOCR follows. Experimental evidence of intermediates or dedicated computational models is not yet available. We know from ESR studies, that radicals as intermediates can be ruled out, yet the formation of carbonites could not be proven as their lifetime and especially concentration in uncatalyzed systems is expected to be very low. In our recent publication, we could show that the activation energies are much higher for uncatalyzed systems compared to catalysed ones and may rely upon multiple activation pathways.42 Due to the low chance of success for spectroscopic evidence, we suggest exploring the reaction mechanism with dynamic computational models in a form we have used for the hydroxide catalysed reaction recently.99
3.3 Base catalysed formate activation
Bases are ideal catalysts for the FOCR and their effectiveness increases with basicity.42 Varying mechanisms specific to hydroxide, borohydride, and superbases have been suggested as shown in Fig. 10. Formate activation with superbases is of the most simple nature as the superbases which include metal hydrides, amides or methoxides act as a proton abstractor to form hydrogen, ammonia, or methanol, respectively and the active carbonite species.38,41,42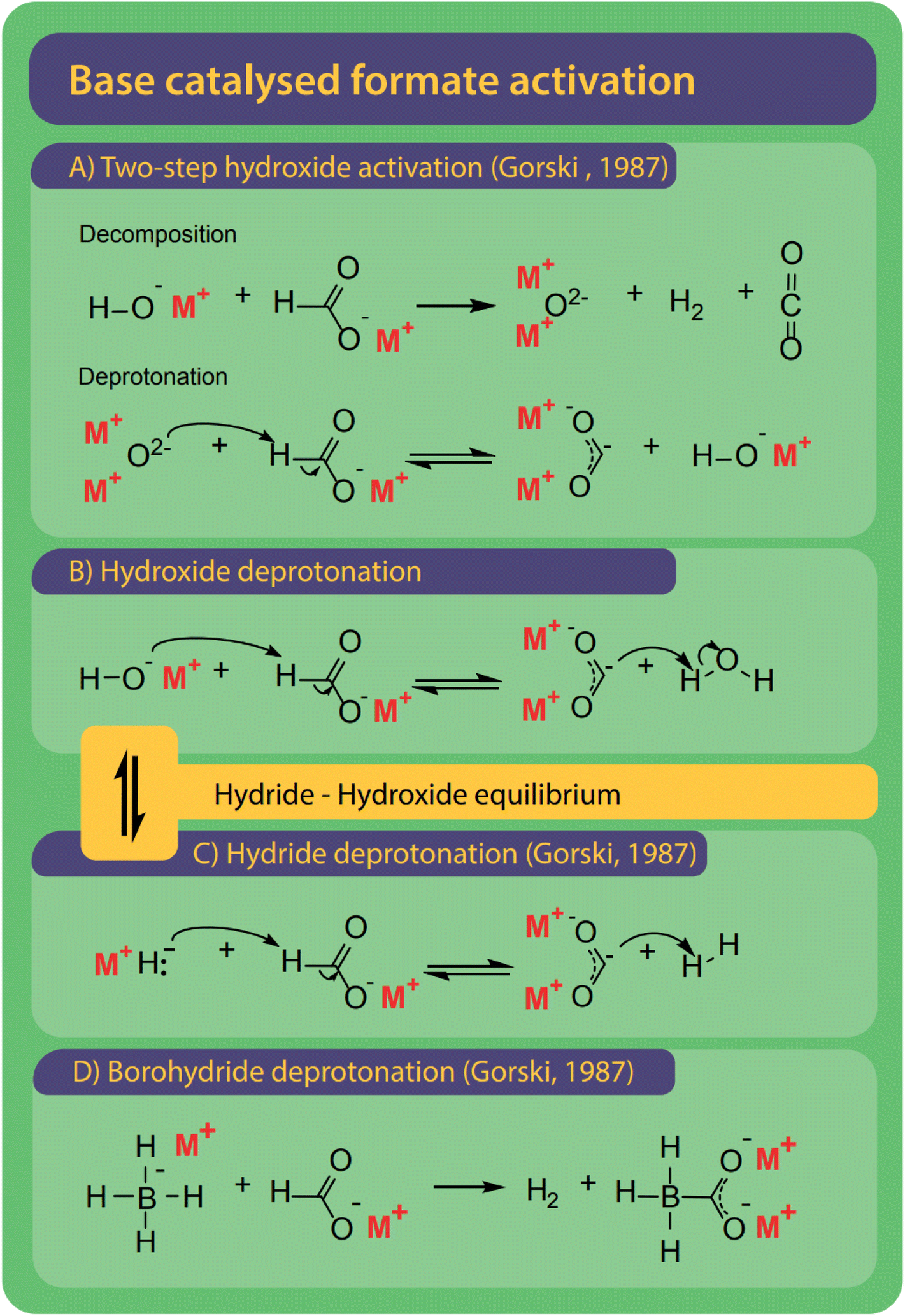 | ||
| Fig. 10 Bases are the most common catalysts for the FOCR and four different mechanisms were proposed. (A) With hydroxide catalysts Górski et al. suggested a two-step activation involving the formation of a metal–oxide which ultimately leads to the formation of carbonite with another formate.38 (B) Lakkaraju et al. suggest direct deprotonation of formate by hydroxide. We include the potential presence of equilibrium in this reaction.41,99 (C) Hydrides and superbases are the most active catalysts and activate the formate by direct deprotonation to form the active carbonite.37,41,42,99 (D) Borohydride activates formate similarly but benefits from the formation of a stabilized boro-carbonite species.38 | ||
The reaction of hydrides or other superbases with formate was first described by Freidlin et al. yet no mechanism was proposed.103 Górski et al. were the first to include hydrides in their reaction mechanism as described above and shown in Fig. 9C.37 Lakkaraju et al. were the first to use a combination of experimental and computational methods in the form of DFT calculations to develop and prove a reaction mechanism for base catalysed formate coupling.41 They calculated the respective energies of possible intermediates at different temperatures and proposed the formation of the active carbonite intermediate by deprotonation of formate with a base as the rate-determining step (RDS) with a 41 kcal mol−1 energy gap.41 The importance of the proton abstraction as the elementary step suggests that the basicity of the catalyst plays a major role in the reaction. The stronger the base the higher its capability to abstract the proton from the formate molecule. Lakkaraju et al. also proved the presence of carbonite by Raman spectroscopy when using sodium formate with hydride catalysts in the presence of high catalyst loadings. Despite many attempts, we were not able to reproduce this spectroscopic work with either sodium or potassium formate and their respective hydrides as catalysts. Hence, we looked for other potential proof and recently could show this with the use of D2O quenching experiments in which we produce deuterated formate [DCOO]− from carbonite after reaction of potassium formate with stoichiometric amounts of sodium- or potassium hydride.42
Borohydride is a well-known hydride donor and is frequently used in organic synthesis or as a reducing agent.138,139 It is easy to handle compared to other hydrides which make it an interesting catalyst for the FOCR. Górski et al. showed in their thermogravimetric studies that the reaction between sodium formate and sodium borohydride occurs at a lower temperature of 278 °C compared to 380 °C with weaker bases. For the activation of formate with borohydride, they suggested the formation of a carbonite dianion facilitated by the proton abstraction from formate by the hydride as shown in Fig. 10D. The carbonite can be stabilized by forming an adduct with the boron cation. Our group also investigated the reaction and observed similar behaviour when using borohydride as a catalyst for formate coupling with potassium formate.42 Our kinetic studies showed a strong improvement in reaction rates compared to reactions with weaker bases.
Hydroxides are the most used and only commercial catalyst for the FOCR, yet the reaction mechanisms are still unclear. Górski et al. proposed a mechanism that differs from the hydride and uncatalyzed reaction. Fig. 10A shows their suggested reaction which starts with a non-reversible decomposition of formate and hydroxide to form hydrogen, CO2, and metal–oxide. In a second step, the metal–oxide reacts with another formate molecule to form the reactive carbonite and hydroxide. This second reaction was suggested to be reversible due to its similarity with the oxalate decomposition reaction, but no further explanation was given. They concluded that the reaction's equilibrium position depends on the binding strength of the O–H bond in the hydroxide. For the FOCR with lithium hydroxide, this dependency on the O–H bond strength explains the formation of carbonate rather than oxalate. With lithium hydroxide, the equilibrium is on the left causing an increased apparent concentration of metal oxide in the system which facilitates the carbonate formation. Whether this reaction mechanism is possible is still not clear.
In our recent publication, we investigated the hydroxide catalysed reaction with a focus on the roles of metal ions and purging in the reaction.99 The strong influence of the metal ions on the reaction in the molten salt motivated us to develop a computational model. We investigated the course of the reaction and the likeliness of intermediates formation in a realistic molecular dynamic system. All molecules and neighbouring atoms were freely available for reactions with all species in the system and were allowed to form intermediates without predetermination.99 We accompanied this computational work with high-resolution kinetics and operando spectroscopy. In our own path-independent molecular dynamic calculations we did not observe the formation of metal oxide species or decomposition of formate to CO2 as suggested by Górski et al. During our Raman studies we have not observed the formation of metal–oxides either.
In our experience potassium formate and sodium formate in FOCR catalysed by their respective hydroxides showed very different reaction rates and conversion efficiencies. For potassium formate FOCR with potassium hydroxide, we noticed that reaction rates were two degrees of magnitude lower than for sodium-based systems. We found that the low oxalate yields obtained without purging were caused by low formate conversion rather than the production of carbonate. Interestingly, we could improve the reaction rates by an order of magnitude by active gas removal from the reaction. We could show for sodium-based systems, that rates closer to the hydride systems are possible in moisture-free conditions even without active removal of gases. When we analysed the off-gases, we could show that water is removed initially before the reaction rate increases (resulting from formate proton abstraction by hydroxide). The water content in the purging stream then decreased and only hydrogen was produced (after one catalytic cycle, the base is hydride). Additionally, we investigated the influence of using mixtures of sodium and potassium formates and hydroxides in various ratios.99 We could see that already adding a small amount of 1–10% sodium was sufficient to negate the effect of gas removal. Purging also had a positive effect on the sodium-based systems and allowed them to reach higher yields, however, the effect was much less pronounced. Already Hene et al. found that with potassium formate active purging of the reaction mixture is imperative to achieve high yields and reduce required reaction times.67 Surprisingly, this was never reported or mentioned in any scientific literature. Górski et al. also did not address the effect of active gas removal in their mechanism.
We studied this reaction and recently proposed a new mechanism for the hydroxide catalysed FOCR, a combination of Fig. 10B and C connected by an equilibrium reaction.99 We found that the direct proton abstraction from formate by the hydroxide to form the active carbonite species has a much high energy barrier of 40 kcal mol−1. Interestingly, our calculations show that the in situ formation of hydride and water in an equilibrium reaction with hydroxide and hydrogen is much more likely in molten formate melt and only require 30 kcal mol−1. The formed hydride species can then abstract a proton from formate to form the reactive carbonite species identical to the formate activation with superbases described above. The active removal of water can shift this equilibrium towards the hydride side as it prevents the back-reaction of newly formed hydrides to less-active hydroxide. As this reaction requires hydrogen, the catalytic cycle still needs to be induced by the less likely proton abstraction by hydroxide. Yet, this event is followed by an exponential increase in the hydride, limited only by the available hydroxide concentration, via the equilibrium reaction, which we observe in our reactions as the reaction rate increases exponentially after an induction period and even reaction rates comparable to hydride catalysed systems could be achieved. In the presence of higher amounts of water, we do not observe this acceleration as the hydride coming off the carbon–carbon coupling is converted back to hydroxide. The in situ hydride formation also explains the difference between potassium and sodium-based systems in the FOCR. Smaller metal anions such as sodium can stabilize the formed hydride better compared to their bulky potassium counterparts. This helps in the initial hydride formation and makes a quick back-reaction towards hydroxide and hydrogen less likely.
3.4 Metal catalysed formate activation
Freidlin et al. were the first to employ and test (alkali) metals as catalysts and even suggested alternative systems to solve the issue of catalyst recovery by using amalgams of the active metals.43 Freidlin et al. suggested two different mechanisms shown in Fig. 12A and B.46 The first mechanism (Fig. 11A) involves the formation of a metal complex by adding two metal atoms to the formate. The carbon atom becomes a much stronger nucleophile and can attack another formate after which the metals and hydrogen are released. The second mechanism (Fig. 11B) functions via the transfer of electrons from the metal to the formate and the formation of a di-valent carbon intermediate, later known as carbonite. This second mechanism was rejected by Freidlin et al. as they perceived the formation of two valent enol-like species as highly unlikely.78 Górski, however, have suggested various mechanisms involving carbonite, suggested this mechanism once again.38 However, it is yet unclear via which pathway the hydrogen is released.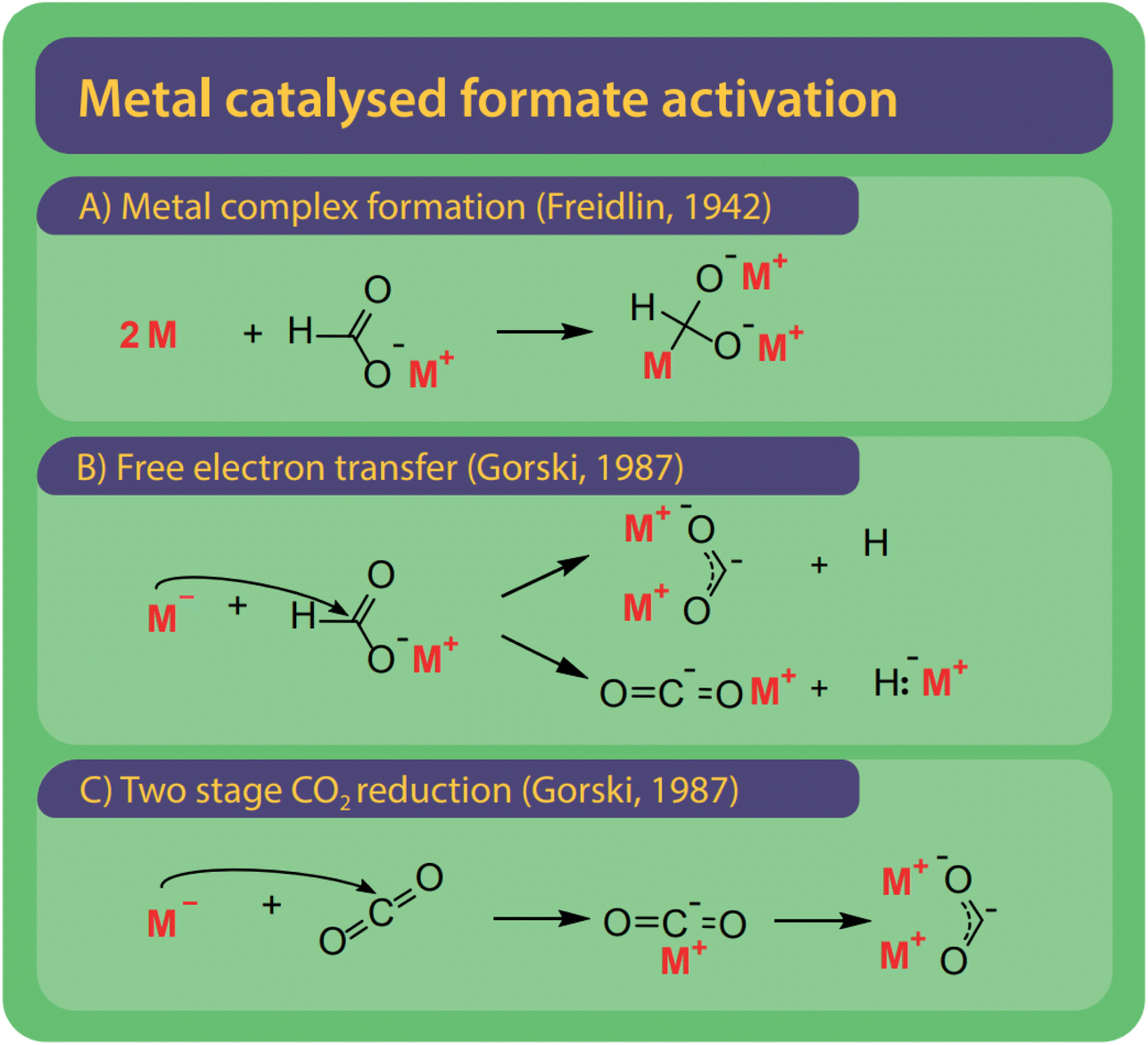 | ||
| Fig. 11 Three reaction activation pathways were suggested for alkali metals as catalysts which are very active catalysts in the FOCR and three reaction pathways have been suggested. (A) Freidlin et al. suggested activation via the formation of a highly active metal complex.46 (B) Although initially rejected by Freidlin et al., Górski et al. argued that the reductive potential via the free metal electrons drives reaction and can lead to the formation of carbonite or a carbon dioxide anion and active hydride. (C) As a second option, Górski et al. proposed the two-stage reduction of CO2 obtained from formate decomposition to carbonate was suggested. | ||
 | ||
| Fig. 12 Formate can decompose mainly to carbonate and carbonous gases CO and CO2. (A) In the absence of catalyst Górski et al. propose the reaction to follow an acid–base reaction with a total of four reaction steps. In the first step formate spontaneously decomposes to hydride and CO2. The formed hydride abstracts a proton form formate in the absence of CO2 to form hydrogen and carbonite. In a third step, the unstable carbonite decomposes to metal oxide and carbon monoxide. At last, carbonate is formed in the reaction of metal oxide and CO2.38 (B) In the presence of hydroxide, formate is first decomposed in a concerted reaction to form a metal oxide, hydrogen, and CO2. The metal–oxide and CO2 form carbonate in a second step. | ||
Alternatively, Górski et al. proposed that the hydride from formate can be released as a metal hydride with the formation of a single-valent carbon dioxide anion. The formed hydride is then available to also activate further formate molecules. The stability and further reactions of the single-valent carbon dioxide anion remain a mystery though. A third option considered by Górski et al. was the direct reduction of CO2 by the metals as shown in Fig. 11C. This was inspired by their earlier suggestion of the decomposition of formate to hydride and CO2 as the first step in their uncatalyzed two-step activation. Whatever the true mechanism for this reaction is, the presence of alkali metals, metallic sodium and potassium would increase the concentration of carbonite or hydride anions.
3.5 Decomposition reactions
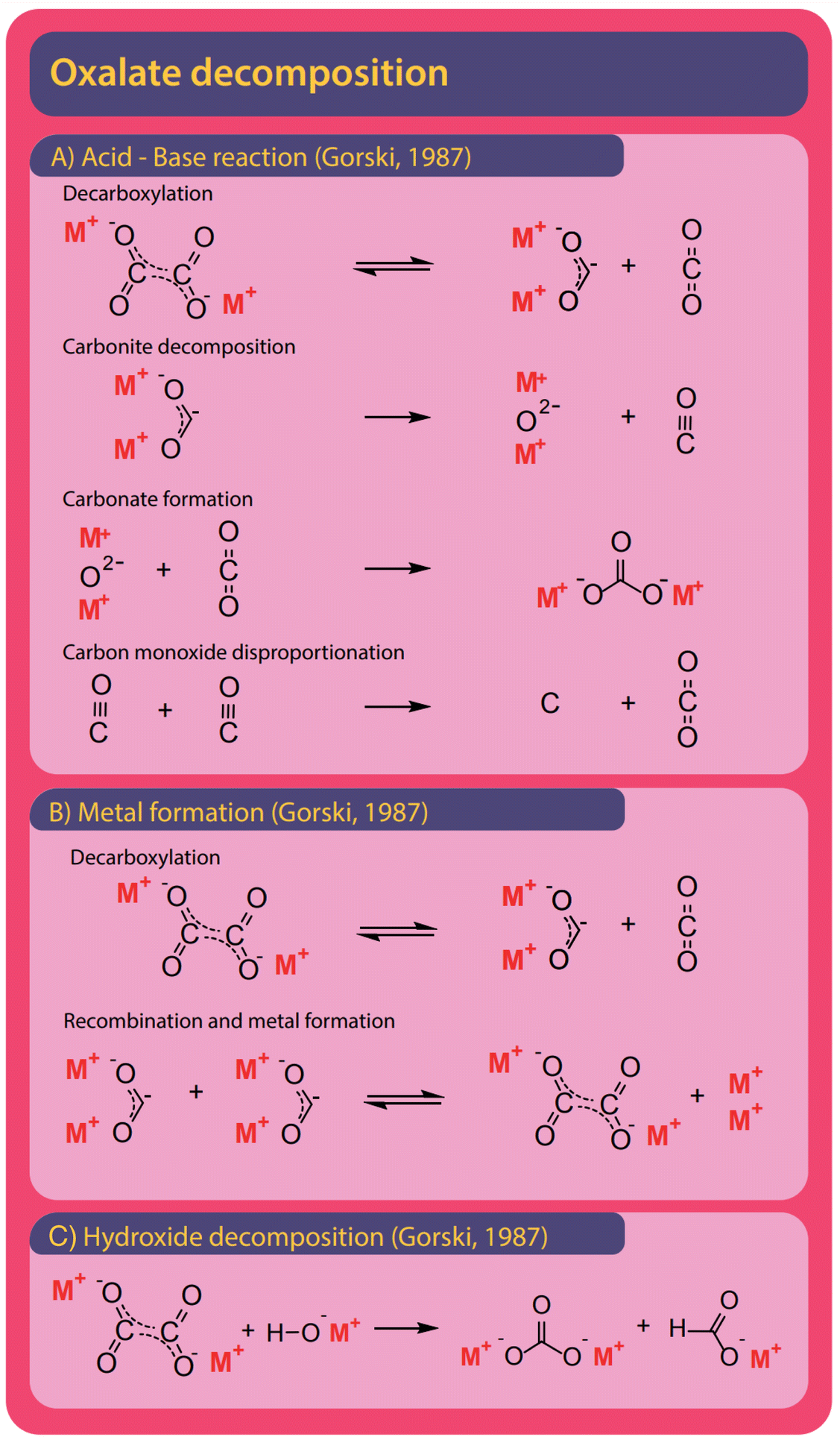
Both formate and oxalate decompose at elevated temperatures and form elemental carbon, CO2, CO, and carbonate and various reaction pathways were proposed. Hydroxide was found to facilitate decomposition reactions.The formation of carbonate from formate is the most widely observed side reaction and appears in the absence of catalysts or when hydroxide is used as a catalyst. Górski et al. proposed an acid–base reaction mechanism for the uncatalyzed carbonate formation which is a variation of the oxalate formation pathway and shown in Fig. 12A. It starts with the activation of formate where hydride and CO2 are formed. Subsequently, carbonite and hydrogen are formed when the hydride reacts with another formate.38 To facilitate the formation of carbonate, the carbonite was suggested to intermediately decompose to a metal oxide and carbon monoxide in the absence of CO2. To form carbonate, however, the metal oxide reacts with a CO2 molecule. Hence this proposed mechanism requires both the absence and presence of CO2. The disappearance and reappearance of CO2 lead us to question this mechanism. The proposed mechanism for carbonate formation from formate in the presence of hydroxide, shown in Fig. 12B, avoids such contradictions. Górski et al. suggested that it proceeds via the direct concerted decomposition of formate and hydroxide to hydrogen, metal oxide, and CO2. This in effect is the same reaction that takes place in the hydroxide activation proposed by Górski et al. to form oxalate. Instead of the metal oxide reacting with another formate, carbonate is then formed in a second step due to the recombination of metal oxide and CO2. In our experiments, at temperatures lower than required for the FOCR, we did not observe any carbonate formation. We observed the formation of carbonate only at high temperatures after oxalate formation. Oxalate, unfortunately, also decomposes further to form mainly carbonate and CO2 but also elemental carbon, metals, and carbon monoxide was observed. Also, the decomposition of oxalate to formate and CO2 has been reported recently by our group.
As shown in Fig. 13A, Górski et al. proposed an acid–base reaction cascade of a total of four reactions starting with the decarboxylation of oxalate to form CO2 and the active carbonite.37 This reaction is the reverse reaction of their proposed pathway for initial oxalate formation in the FOCR. They proposed that, after decarboxylation, the carbonite readily decomposes to form a metal oxide and carbon monoxide. This step must occur in the absence of CO2 to prevent the reverse reaction, oxalate formation. Górski showed that the decomposition temperature increases by 50 °C in CO2 atmospheres for sodium (466 to 503 °C), barium (396 to 463 °C), calcium (369 to 423 °C), and lithium (450 to 495 °C) oxalates. In a third step, carbonate is formed in the reaction of CO2 with the metal oxide. At last, the carbon monoxide, if present in sufficient amounts, can disproportionate to form CO2 and elemental carbon in the Boudouard reaction. Like Górski et al. proposed for the formate decomposition, the alternating absence and presence of CO2 lead us to question this mechanism. Alternatively, Górski et al. suggested that oxalate can decompose towards metal and CO2 as shown in Fig. 13B. Again, the oxalate formation is reversed, and carbonite and CO2 are formed. Two carbonites can now recombine to form oxalate and metallic species. The newly formed oxalate can then decompose again to build up more metal. Notably, the absence or removal of CO2 is consistent in this mechanism.
 | ||
| Fig. 13 The decomposition of oxalate can proceed via three routes, all suggested by Górski et al. (A) In a four step acid-base reaction suggested by Górski, oxalate first decarboxylates to form carbonite an CO2. In a second step, carbonite decomposes to metal-oxide and CO. The Metal-oxide and CO2 (formed in step 1) react in the third step to form metal carbonate. The CO formed in the third step disproportionates to elemental carbon and CO2 in the fourth step. (B) (Alkali)metal-formation also starts with the decarboxylation of oxalate to carbonite and CO2. In a subsequent step, two carbonites recombine to form oxalate and (alkali)metal. (C) The presence of hydroxide can stimulate the decomposition of oxalate to form carbonate and formate. | ||
Oxalate can be split to form carbonate and formate in the presence of hydroxide. A detailed reaction mechanism was not proposed for this reaction. In Fig. 13C we propose that it most likely proceeds via the attack of the negatively charged oxygen of the hydroxide on one of the two oxalate carbons and the subsequent splitting of the carbon–carbon bond to form carbonite and carbonate. The carbonite could abstract the proton, once belonging to the hydroxide, and form formate. In our recent work, we found that the formation of carbonate in potassium-based systems is independent of hydroxide content and therefore the decomposition of oxalate without the involvement of hydroxide appears to be preferred.83 In the decomposition of formate, however, hydroxide is consumed as a reactant and not recovered as when oxalate is formed. Hence, the potential for decomposition towards carbonate is limited by the added amount of catalyst. At the same time, the catalyst is consumed. In conclusion, it is desirable to choose reaction conditions such that the oxalate formation is favoured. Temperature, reaction times, and CO2 availability are crucial here. If too much CO2 is present, the catalyst is consumed as a reactant in the carbonate forming reaction. We recently found that in absence of CO2 and by active removal of gaseous reaction products, the formation of carbonate is independent of the amount of hydroxide added. Only longer residence times at higher temperatures increased carbonate formation.83 Yet we could not distinguish whether the formed carbonate originated from formate or oxalate decomposition. Carbonate yields increase at high temperatures and reaction times whilst oxalate yields are higher at lower temperatures and shorter reaction times. This indicates an increased likeliness of oxalate decomposition as the origin of carbonate formation.
In the presence of hydride, we never observed the formation of carbonate or other decomposition products from potassium formate due to the relatively low reaction temperatures.42 Hartmann and Hisatsune studied the decomposition of metal formate into salt matrices by pyrolysis in 1965–1966.47,48 They claimed that two calcium formate molecules recombine into a transition complex and then decomposes to carbonate and formaldehyde. The absence of formaldehyde formation for alkali metal formates indicates the absence of this mechanism for these formates.
For none of the decomposition reactions of both formate and oxalate are any computational models nor spectroscopic evidence available. In the interest of understanding the full reaction, we encourage the study of these processes to fully understand the reactions and to be able to avoid carbonate formation.
4 Future of formate coupling
Although formate has been coupled to oxalate industrially for over a century now, there are still new opportunities lying ahead. We aim to provide an insight into potential gains from performing the reaction in solution and attempts to harness the reactivity of the active carbonite intermediate to couple formate with alternative molecules.4.1 The FOCR in solution
We have shown that the FOCR starts with the formation of carbonite. If superbases are used for this reaction, the reaction start is determined by the melting point of the used formate, i.e., 168 °C for potassium formate and 251 °C for sodium formate. The fast reaction and independence from the basicity of superbases indicate that this reaction temperature could be lowered even further. We propose that the formation of a solution or salt melt is required to dissolve hydride and formate and make the two reaction partners available for a reaction. Therefore, performing the reaction in solution allows lower reaction temperature and introduction of alternative reaction partners. Additionally, the FOCR in solution would allow to perform more in situ kinetic studies with superbase catalysts. We first tested various solvents for this reaction. The combination of requiring a polar solvent to dissolve the hydride and formate, whilst being aprotic and stable towards contact with superbases is a great challenge. To our knowledge, we are the first to report the FOCR in solvents. Overall, we chose to test nine different solvents with high thermal stability which are shown in Table 1. Unfortunately, we did not succeed in making oxalate in these systems as they either suffered from low solvation of formates or a visible reaction of the solvent with the superbase.| Solvent | Chemical structure |
|---|---|
| Glycerol |
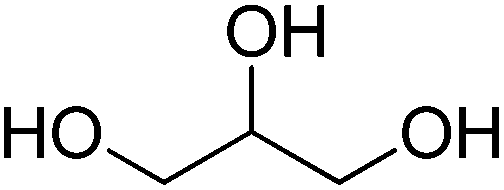
|
| Hexadecane |

|
| 1-Decene |

|
| N-Methyl-2-pyrrolidone |
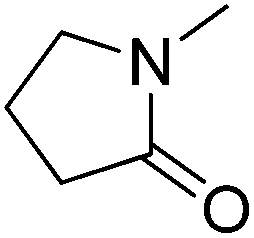
|
| Di(propylene glycol) methyl ether |
|
| Phenoxyethanol |

|
| Benzaldehyde |
|
| Cinnamaldehyde |
|
| Sulfolane |
|
In search of a suitable solvent for the FOCR, we then turned to ionic liquids (ILs). They consist of large anions and cations which are sterically hindered and exhibit a delocalized charge. These attributes prevent efficient packing and lead to those salts being liquid even at temperatures below 100 °C but can have high thermal stability with their structure determining their hydrophilicity. Overall, those molten salts are not dissimilar to the formate melt which facilitates the formate to oxalate reaction. Conveniently some ILs have been used with superbases before and were stable.140–145 In Fig. 14 we show eight ILs we purchased or synthesised as combinations with three different cations and six different anions. We focused on combinations that promised to be inert towards superbases at higher temperatures due to their absence of acidic protons. Initially, we tested the solubility of formates in the ILs. In a glovebox, we transferred 750 mg of each IL into a 2 ml glass vial equipped with a stirring bar. To the ILs we added 25 mg of potassium formate and 20 mg of sodium hydride. Formate and hydride only dissolved in [P6,6,6,14][DCA], [C4mim][DCA], and [C2mim][NTf2] in a mass ratio of 375![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 IL to formate. The high molar mass ILs appears to be limiting the solvation due to the relatively low ion-to-mass ratio compared with formate. To observe the suitability of the systems for the FOCR we subsequently heated the mixtures to 175 °C and analysed them using 1H-NMR, 13C-NMR, and 31P-NMR. We observe that the ILs stayed intact even upon contact with hydride at elevated temperatures. We visibly observed a reaction in all systems which caused the formation of gas and a colour change. Unfortunately, also formate appears to have stayed intact and the formation of oxalate could not be observed even after extended reaction times of up to 3 hours. We could not identify which reaction occurred chemically. We conclude that whilst the search for a suitable solvent system promises further reductions in reaction temperature in the FOCR and allow for more insights into the reaction mechanism, this proves a difficult task and requires more work in the future.
:1 IL to formate. The high molar mass ILs appears to be limiting the solvation due to the relatively low ion-to-mass ratio compared with formate. To observe the suitability of the systems for the FOCR we subsequently heated the mixtures to 175 °C and analysed them using 1H-NMR, 13C-NMR, and 31P-NMR. We observe that the ILs stayed intact even upon contact with hydride at elevated temperatures. We visibly observed a reaction in all systems which caused the formation of gas and a colour change. Unfortunately, also formate appears to have stayed intact and the formation of oxalate could not be observed even after extended reaction times of up to 3 hours. We could not identify which reaction occurred chemically. We conclude that whilst the search for a suitable solvent system promises further reductions in reaction temperature in the FOCR and allow for more insights into the reaction mechanism, this proves a difficult task and requires more work in the future.
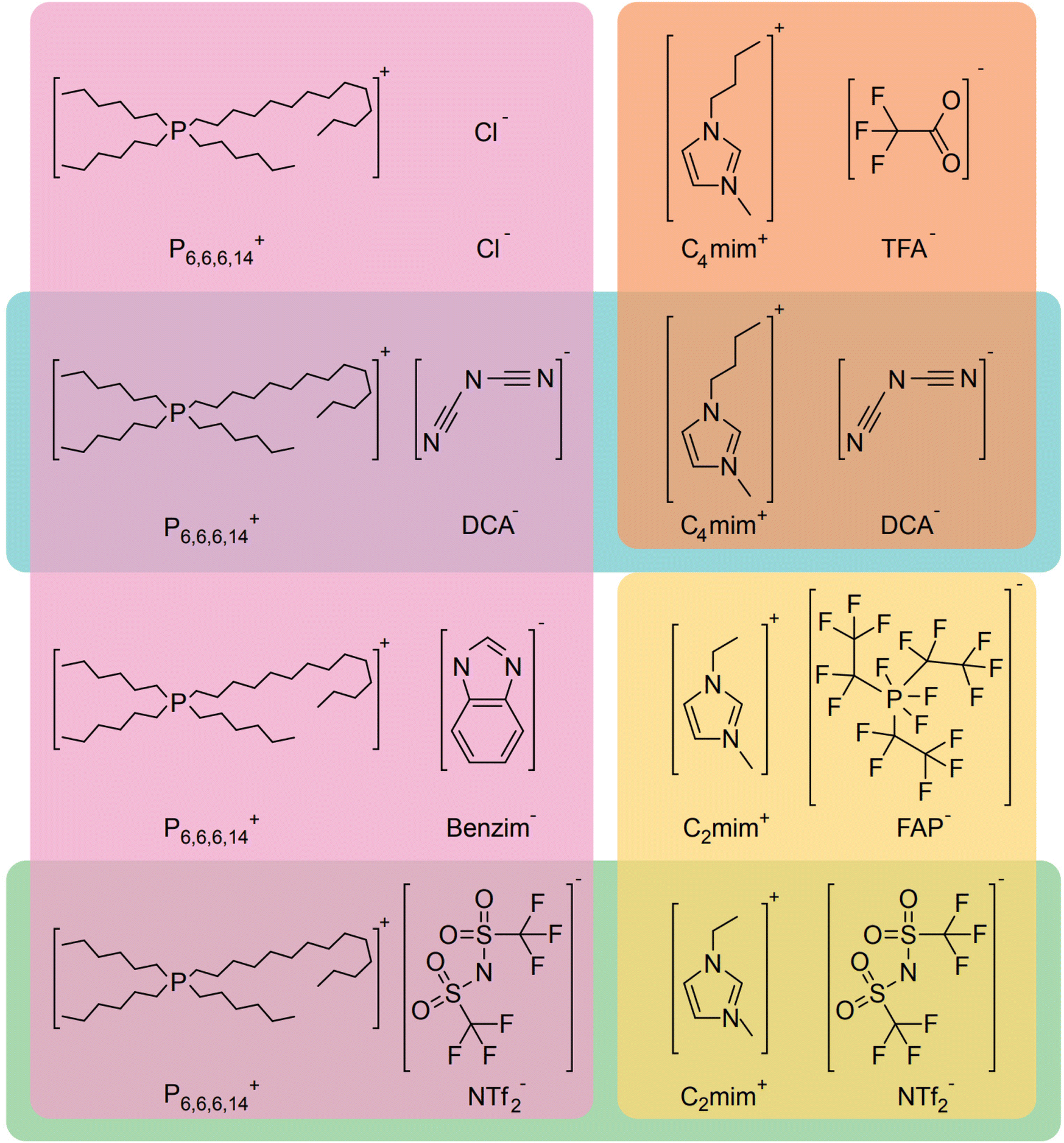 | ||
| Fig. 14 Structures of the eight ionic liquids used to facilitate the formate to oxalate coupling reaction. | ||
4.2 Opportunities of the active carbonite intermediate in formate coupling
Formate made from CO2 is arguably an expensive reactant as it requires the investment of two electrons. To make oxalate in the FOCR, two formates and thus the investment of four electrons are required. Hence, we think it is interesting to see if it is possible to use the carbonite intermediate to create other valuable products by coupling it with alternative reaction partners. As the carbonite intermediate is very reactive due to its high negative charge density, it can react as a Lewis base towards a nucleophilic attack of an electrophile. The reactive intermediate of the FOCR, carbonite, is a reactive carbanion nucleophile not dissimilar to well-known organometallic carbanions such as organolithium or organomagnesium reagents.Like the carbonite dianion, these organometallic compounds exhibit the presence of a free electron pair on a carbon atom. In consequence, the carbon-atom becomes a nucleophile and turns those reagents into extremely reactive compounds and even have been used to capture CO2.146 Other than carbonites, however, organomagnesium or organolithium compounds can be stored in solutions (such as apolar solvents). This makes them attractive as reagents in organic synthesis. They are broadly used in nucleophilic addition reactions to form new carbon–carbon bonds. The most common examples are the alkylation of aldehydes and ketones in the Grignard reaction, worth a Nobel Prize in 1912, the alkylation of metals and metalloids or the coupling with organic halides.147 Due to the similarity between carbonite and those organometallic compounds, we expect carbonite to also react with a broad variety of molecules.128 The carbonite shares the two surplus electrons to form a new covalent bond between its carbon atom and the targeted species. In Fig. 15 we summarize the potential alternative reactant groups with electrophilic carbonyl groups we base on proven reactants for organomagnesium and organolithium compounds which include aldehydes, ketones, esters, and alkenes as well as molecules with good leaving groups.
 | ||
| Fig. 15 The top shows the proposed general mechanism for a potential reaction of carbonite an electrophile. At the bottom, the various potential routes of alternative reactants and the resulting products are shown. | ||
Due to its instability, the carbonite must be provided in situ by the abstraction of a proton from formate with a superbase. The greatest challenge is providing the active carbonite whilst avoiding side reactions between the alternative reactant and the superbase. Due to the current unavailability of a suitable solvent, it is necessary to perform the reaction in a formate melt and therefore a temperature of at least 170 °C is required. A potential reaction partner must not decompose in these conditions. If the use of stoichiometric amounts of superbase wants to be avoided, the reaction partner should release a hydride or other superbases after the coupling to allow the formation of another carbonite in a catalytic cycle.
Potential side reactions of the alternative reactants with the superbase include the attack of the superbase as a reducing agent on the carbonyl carbon by nucleophilic addition.148 Some carbonyl compounds also undergo enolization if treated with strong bases or with molecules with higher proton acidity than formate. If aldehydes are used, the enolate can further react at the α-carbon with the carbonyl of another aldehyde molecule to obtain β-hydroxy aldehyde. Hence molecules with lower proton acidity than formate (exclusion of e.g. –OH; –COOH, –NH4) and not prone to form enols for α-hydrogen abstraction are most likely more suitable to avoid condensation reactions with reagents and products. Physical properties, especially the phase of the reactant, influence the reactant introduction and product separation and determine the required reactor design. Using a liquid is preferable as it eases its introduction, and it could act as a solvent/dispersant for formate and hydride. Gaseous reaction partners need to be provided in large excess in a pressurized reactor or bubbled through the molten formate. We evaluated a variety of potential compounds shown in Table 2 and chose CO2, paraformaldehyde, benzaldehyde and cinnamaldehyde for proof-of-principle experiments.
| Reactant | Reactivity with alkali hydridea | Phase at formate activation | Melting point (°C) | Boiling point (°C) | |
|---|---|---|---|---|---|
| a In the evaluation process the reactivity of the reactants with alkali hydride driven proton abstraction was ranked in the categories low, medium, high. We did not consider reduction of aldehydes or esters. Low reactivity includes molecules with protons with lower acidity then formate and not prone to form enols for α-hydrogen abstraction. Medium reactivity includes molecules with protons with similar acidity then formate and/or prone to form enol for α-hydrogen abstraction. High reactivity includes molecules with protons with higher acidity then formate (e.g. –OH; –COOH, –NH4) and/or highly prone to form enol for α-hydrogen abstraction. The hydride ion could attack the carbonyl carbon by nucleophilic addition, acting as a reducing agent. This might happen according to the basic strength of the metal hydride and the reactivity of the carbonyl compound.148 | |||||
| Aldehydes | Paraformaldehyde | Low | Gas | 120–170 | |
| Acetaldehyde | High | Gas | 21 | ||
| Propionaldehyde | High | Gas | −80 | 49 | |
| Butyraldehyde | High | Gas | 76 | ||
| Acrolein | Low | Gas | 53 | ||
| Crotonaldehyde | Low | Gas | 104 | ||
| Formylcyclohexane | Medium | Gas/liquid | 161 | ||
| Benzaldehyde | Low | Liquid | −26 | 178 | |
| Glucose | High | Liquid | 150 | ||
| Furfural | Low | Gas/liquid | −37 | 162 | |
| Methylbenzaldehyde | Low | Liquid | 200 | ||
| Cinnamaldehyde | Low | Liquid | 248 | ||
| Glutaraldehyde | High | Liquid | −14 | 187 | |
| Phthalaldehyde | Low | Liquid | 56 | 266 | |
| Esters | Dimethyl terephthalate | Low | Gas/liquid | 142 | 288 |
| Methyl butyrate | High | Gas | −85 | 102 | |
| Ethyl butyrate | High | Gas | −101 | 121 | |
| Pentyl acetate | High | Gas/liquid | −71 | 148 | |
| Isopentyl acetate | High | Gas/liquid | −79 | 142 | |
| Benzyl acetate | High | Liquid | −51 | 215 | |
| Pentyl butyrate | High | Liquid | −73 | 185 | |
| Octyl acetate | High | Liquid | −39 | 210 | |
| Methyl benzoate | Low | Liquid | −12.5 | 199 | |
| Methyl cinnamate | Low | Liquid | 35 | 261 | |
| Ketones | Acetone | High | Gas | −95 | 56 |
| Ethyl methyl ketone | High | Gas | −86 | 80 | |
| Diethyl ketone | High | Gas | −40 | 102 | |
| Cyclohexanone | High | Gas/liquid | −31 | 155 | |
| Methyl phenyl ketone | High | Liquid | 20 | 202 | |
| Acetic acid anhydride | High | Liquid | −73 | 140 | |
| Alkenes | 5-Decene | Medium | Gas | −73/−112 | 170 |
| 2-Heptene | Medium | Gas | — | 99 | |
| Toluene | Medium | Gas | −95 | 110 | |
| Xylene | Medium | Gas/liquid | −10 | 140 | |
| Leaving group | Chlorobenzene | Low | Gas | −45 | 132 |
| Chloronaphthalene | Low | Liquid | — | 256 | |
| Bromobenzene | Low | Gas | −30 | 156 | |
| Bromonaphthalene | Low | Gas | 2 | 145 | |
| 1-Chlorohexane | Medium | Gas | −94 | 135 | |
| 1-Bromohexane | Low | Gas | −84 | 155 | |
| CO | Carbon monoxide | Low | Gas | −205 | −191 |
| CO2 | Carbon dioxide | Low | Gas | −78 | −78 |
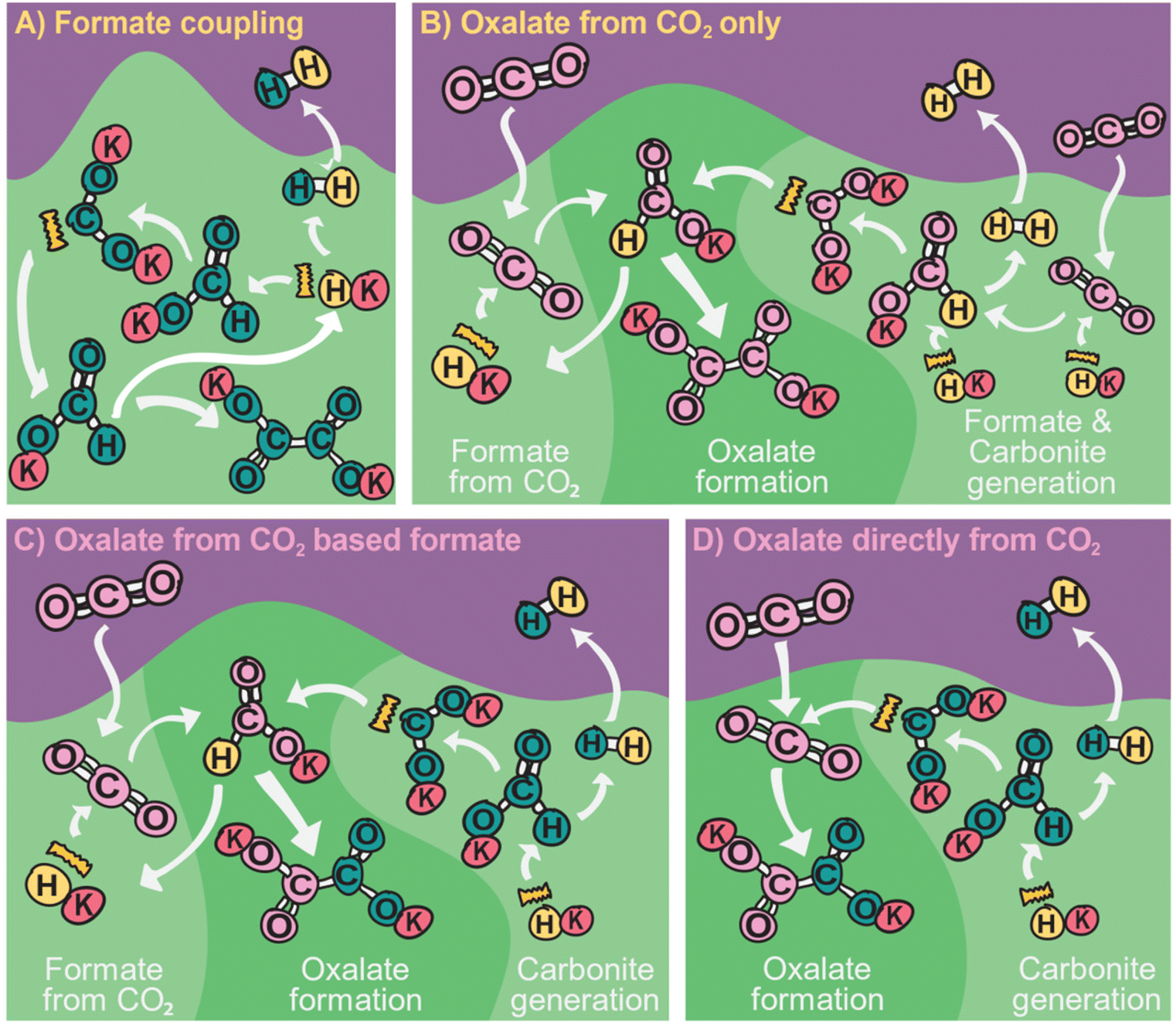 | ||
| Fig. 16 Three fundamental pathways for oxalate production are consisting of either (A) classic FOCR without the involvement of CO2, or (B–D) with the involvement of CO2. (A) Oxalate and hydrogen can be formed by traditional formate to oxalate coupling (FOCR) from two formates catalysed by hydride via the carbonite intermediate in the formate melt. CO2 can dissolve in the formate melt and react towards oxalate in two ways. (B) Two formates can be formed from CO2 in a reaction between CO2 and hydride. One of these two formates is activated to carbonite and react with the other formate to oxalate in a FOCR reaction as in (A). (C) It can react with hydride to form formate which can subsequently react towards oxalate in a FOCR reaction. (D) Alternatively, CO2 can be attacked by the reactive carbonite intermediate formed from the reaction of formate with hydride to form oxalate directly in a stoichiometric reaction. | ||
Whilst the stability of formaldehyde in the required reaction conditions is unlikely, cinnamaldehyde and benzaldehyde promise to be stable in the reaction conditions. They have low enolization potential and exhibit a high thermal stability, and boiling point. Formaldehyde can be introduced to the reaction in solid para-benzaldehyde form. Benzaldehyde and cinnamaldehyde are even easier to introduce as they are liquids and lead to desirable products which could be used e.g., in polymer applications.
Other than in the alternative reaction with CO2, for the aldehyde reactions, no pressurised reactor was required. We used inert conditions with a constant nitrogen flow in a Schlenk line to prevent the intrusion of water. This setup allowed us to introduce the reactants in different orders and at different times. We analysed the reaction products using IR, NMR and GC-MS techniques to detect the formation of the desired reaction products or known potential side-products such as benzyl benzoate. During this work, we faced several hurdles and could not obtain any coupling of formate and aldehydes. Therefore, we did not observe the anticipated coupling products. Initially, we mixed all reaction partners and heated them together but did not show any formation of the desired product. In the reaction of paraformaldehyde with formate, we observed the complete evaporation or decomposition of formaldehyde during heating. When heating benzaldehyde and cinnamaldehyde together with formate we surprisingly saw the formation of a thick suspension and no reaction. Even the addition of equimolar amounts of hydride did not lead to any reaction in the suspension. In the end, a liquid mixture could only be obtained when benzaldehyde or cinnamaldehyde was first heated under reflux, and formate was added in small amounts up to 5 mol%. Even with the addition of hydride, no reaction occurred. Subsequently, we introduced sulfolane as a solvent or diluent but could not increase the amount of formate that could be added before the formation of a suspension. Interestingly, the addition of hydride to the mix of formate and benzaldehyde in sulfolane did cause a reaction, yet not the formation of desired product but benzyl benzoate. A similar reaction of benzaldehyde and hydride in toluene was reported before.157
In conclusion, the attempt to couple formate with aldehydes led to the formation of a thick unreactive suspension when the reactants were mixed in equimolar amounts. The addition of hydride to benzaldehyde dissolved in sulfolane lead to the formation of benzyl benzoate and not the desired phenyl glyoxylic acid. Cinnamaldehyde dissolved in sulfolane did not appear to react with hydride. None of these experiments yielded the desired coupling of K-formate and benzaldehyde or cinnamaldehyde.
5 Conclusions and outlook
Many parameters are available to reduce the residence times of the reaction mixture in the reactor, increasing yield and reactor cost. It becomes apparent that the plethora of different factors is likely to be related to or affect each other. Through the publication history on the FOCR contradicting reports are present. The best example is water, which is considered the most common and potent poison, and yet reactor designs in which the reaction is performed using an aqueous solution have been patented. Overall, this makes it difficult to optimize the reaction and avoid faults in reactor designs beforehand. We provide a complete overview and systematically assess the effect of all reported reaction parameters which we divided into six sub-categories. This provides a starting point to uncover intercorrelations and the relative impact of each parameter. Recently, we used this overview to evaluate the influence and importance of reaction parameters in hydroxide catalysed FOCR systems given their high relevance when coupled with electrochemical CO2 reduction to formates.20,83,100 Within this realm, the absence of water and choice of temperature were found most important. The former can be achieved through drying of the reactant prior and its active removal once released as a gas. We found, that the required temperature is dependent on catalyst choice but dictates the required residence time and formation of side products. With new superbase catalysts, the reaction temperature and time could be lowered drastically for the first time.42Independent of the catalyst, we show, that carbonite today is established as the reactive intermediate in the FOCR. Its presence is proven spectroscopically as well as by D2O quenching.41,42 We recently contributed new insights into the role of counter-ions and the presence of equilibrium reactions in the hydroxide-based system.99 Whilst we shine a light on the reaction towards oxalate in hydroxide catalysed or uncatalyzed systems, the decomposition reactions towards carbonate and gaseous or elemental carbon compounds are still not fully understood. Further work is needed to understand the decomposition reactions especially the formation of carbonate in hydroxide-catalysed systems, as the avoidance of this side reaction allows for major improvements of the process.
The use of carbonite as a reactant to form interesting products with the introduction of alternative reaction partners opens new possibilities. We assessed over 30 potential alternative coupling partners for coupling with active carbonite intermediate and especially their reactivity of the hydride catalyst poses a major challenge. We performed proof-of-principle reactions with CO2, formaldehyde, benzaldehyde and cinnamaldehyde but failed in the coupling of carbonites with aldehydes due to competing reactions. For CO2 we could show the successful coupling with carbonite using isotope labelling. Unfortunately, also the production of carbonate was increased in high hydride concentrations and the presence of excess amounts of supercritical CO2. As equimolar amounts of hydride are required for the coupling of CO2 and formate, and the carbonate formation occurs it is questionable if this presents a viable alternative to the FOCR.117
The development of commercial FOCR processes in conjunction with electrochemical CO2 reduction to formate is required to close the gap between CO2 reduction and the production of valuable chemicals starting from oxalic acid. Many reactor designs were proposed in the past but the knowledge on how to build and operate them has been lost over time. The next step to bring FOCR back to life, require the assessment and testing of various reactor types which conform with the knowledge about the influence of reaction parameters on the FOCR. For the newly discovered superbase catalysed FOCR, suitable large-scale reactors must be developed. A further reduction in reaction temperature or harnessing the reactivity of the carbonite intermediate in alternative reactions is an interesting scientific challenge that requires more work in the future but is at this point far from industrial relevance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 767798.References
- C. Jullion and R. MacDougall, Ueber die Fabrication von Kleesäure, Polytech. J., 1852, 124, 175–181 Search PubMed
https://dingler.culture.hu-berlin.de/article/pj124/ar124042, (accessed October 24, 2019).
- P. Friedlingstein, M. O'Sullivan, M. W. Jones, R. M. Andrew, J. Hauck, A. Olsen, G. P. Peters, W. Peters, J. Pongratz, S. Sitch, C. Le Quéré, J. G. Canadell, P. Ciais, R. B. Jackson, S. Alin, L. E. O. C. Aragão, A. Arneth, V. Arora, N. R. Bates, M. Becker, A. Benoit-Cattin, H. C. Bittig, L. Bopp, S. Bultan, N. Chandra, F. Chevallier, L. P. Chini, W. Evans, L. Florentie, P. M. Forster, T. Gasser, M. Gehlen, D. Gilfillan, T. Gkritzalis, L. Gregor, N. Gruber, I. Harris, K. Hartung, V. Haverd, R. A. Houghton, T. Ilyina, A. K. Jain, E. Joetzjer, K. Kadono, E. Kato, V. Kitidis, J. I. Korsbakken, P. Landschützer, N. Lefèvre, A. Lenton, S. Lienert, Z. Liu, D. Lombardozzi, G. Marland, N. Metzl, D. R. Munro, J. E. M. S. Nabel, S. I. Nakaoka, Y. Niwa, K. O'Brien, T. Ono, P. I. Palmer, D. Pierrot, B. Poulter, L. Resplandy, E. Robertson, C. Rödenbeck, J. Schwinger, R. Séférian, I. Skjelvan, A. J. P. Smith, A. J. Sutton, T. Tanhua, P. P. Tans, H. Tian, B. Tilbrook, G. Van Der Werf, N. Vuichard, A. P. Walker, R. Wanninkhof, A. J. Watson, D. Willis, A. J. Wiltshire, W. Yuan, X. Yue and S. Zaehle, Global Carbon Budget 2020, Earth Syst. Sci. Data, 2020, 12, 3269–3340, DOI:10.5194/essd-12-3269-2020
- W. Steffen, K. Richardson, J. Rockström, S. E. Cornell, I. Fetzer, E. M. Bennett, R. Biggs, S. R. Carpenter, W. de Vries, C. A. de Wit, C. Folke, D. Gerten, J. Heinke, G. M. Mace, L. M. Persson, V. Ramanathan, B. Reyers and S. Sörlin, Planetary boundaries: Guiding human development on a changing planet, Science, 2015, 347, 1259855, DOI:10.1126/science.1259855
- Global warming of 1.5 °C. An IPCC Special Report on the impacts of global warming of 1.5 °C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, 2018.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, The teraton challenge. A review of fixation and transformation of carbon dioxide, Energy Environ. Sci., 2010, 3, 43–81, 10.1039/b912904a
- A. J. Hunt, E. H. K. Sin, R. Marriott and J. H. Clark, Generation, Capture, and Utilization of Industrial Carbon Dioxide, ChemSusChem, 2010, 3, 306–322, DOI:10.1002/cssc.200900169
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Closing the loop: Captured CO2 as a feedstock in the chemical industry, Energy Environ. Sci., 2015, 8, 3283–3297, 10.1039/c5ee02591e
- C. Song, Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing, Catal. Today, 2006, 115, 2–32, DOI:10.1016/j.cattod.2006.02.029
- M. Aresta and A. Dibenedetto, Utilisation of CO2 as a chemical feedstock: opportunities and challenges, Dalton Trans., 2007, 2975, 10.1039/b700658f
- C. Ampelli, S. Perathoner and G. Centi, CO2 utilization: an enabling element to move to a resource- and energy-efficient chemical and fuel production, Philos. Trans. R. Soc., A, 2015, 373, 20140177, DOI:10.1098/rsta.2014.0177
- G. Centi, E. A. Quadrelli and S. Perathoner, Catalysis for CO2 conversion: a key technology for rapid introduction of renewable energy in the value chain of chemical industries, Energy Environ. Sci., 2013, 6, 1711, 10.1039/c3ee00056g
- T. Sakakura, J. C. Choi and H. Yasuda, Transformation of carbon dioxide, Chem. Rev., 2007, 107, 2365–2387, DOI:10.1021/cr068357u
- C. Maeda, Y. Miyazaki and T. Ema, Recent progress in catalytic conversions of carbon dioxide, Catal. Sci. Technol., 2014, 4, 1482–1497, 10.1039/c3cy00993a
- M. N. Anwar, A. Fayyaz, N. F. Sohail, M. F. Khokhar, M. Baqar, A. Yasar, K. Rasool, A. Nazir, M. U. F. Raja, M. Rehan, M. Aghbashlo, M. Tabatabaei and A. S. Nizami, CO2 utilization: Turning greenhouse gas into fuels and valuable products, J. Environ. Manage., 2020, 260, 110059, DOI:10.1016/j.jenvman.2019.110059
- S. Kar, A. Goeppert and G. K. S. Prakash, Integrated CO2 Capture and Conversion to Formate and Methanol: Connecting Two Threads, Acc. Chem. Res., 2019, 52, 2892–2903, DOI:10.1021/ACS.ACCOUNTS.9B00324/ASSET/IMAGES/LARGE/AR9B00324_0011.JPEG
- Z.-Z. Yang, L.-N. He, Y.-N. Zhao, B. Li and B. Yu, CO2 capture and activation by superbase/polyethylene glycol and its subsequent conversion, Energy Environ. Sci., 2011, 4, 3971, 10.1039/c1ee02156g
-
P. Styring, E. A. Quadrelli and K. Armstrong, Carbon Dioxide Utilisation: Closing the Carbon Cycle: First Edition, Elsevier Inc., 2014. DOI:10.1016/C2012-0-02814-1
-
M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley, 2010. DOI:10.1002/9783527629916
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364, eaav3506, DOI:10.1126/science.aav3506
- M. F. Philips, G. J. M. Gruter, M. T. M. Koper and K. J. P. Schouten, Optimizing the Electrochemical Reduction of CO2 to Formate: A State-of-the-Art Analysis, ACS Sustainable Chem. Eng., 2020, 8, 15430–15444, DOI:10.1021/acssuschemeng.0c05215
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes, Chem. Rev., 2017, 117, 9804–9838, DOI:10.1021/acs.chemrev.6b00816
- M. Ronda-Lloret, G. Rothenberg and N. R. Shiju, A Critical Look at Direct Catalytic Hydrogenation of Carbon Dioxide to Olefins, ChemSusChem, 2019, 12, 3896–3914, DOI:10.1002/cssc.201900915
-
J. Hietala, A. Vuori, P. Johnsson, I. I. Pollari, W. Reutemann and H. Kieczka, Formic acid, Ullmann's Encycl. Ind. Chem, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2016, pp. 335–340. DOI:10.1002/14356007.a12
- Oxalic acid from CO2 using Eletrochemistry At demonstratioN scale|OCEAN Project|H2020|CORDIS|European Commission, 2020, https://cordis.europa.eu/project/id/767798, (accessed February 4, 2020).
- F. Jin, Y. Gao, Y. Jin, Y. Zhang, J. Cao, Z. Wei and R. L. Smith, High-yield reduction of carbon dioxide into formic acid by zero-valent metal/metal oxide redox cycles, Energy Environ. Sci., 2011, 4, 881–884, 10.1039/c0ee00661k
- H. Zhong, L. Wang, Y. Yang, R. He, Z. Jing and F. Jin, Ni and Zn/ZnO Synergistically Catalyzed Reduction of Bicarbonate into Formate with Water Splitting, ACS Appl. Mater. Interfaces, 2019, 11, 42149–42155, DOI:10.1021/acsami.9b14039
- M. Jouny, W. Luc and F. Jiao, General Techno-Economic Analysis of CO2 Electrolysis Systems, Ind. Eng. Chem. Res., 2018, 57, 2165–2177, DOI:10.1021/acs.iecr.7b03514
- R. Aldaco, I. Butnar, M. Margallo, J. Laso, M. Rumayor, A. Dominguez-Ramos, A. Irabien and P. E. Dodds, Bringing value to the chemical industry from capture, storage and use of CO2: A dynamic LCA of formic acid production, Sci. Total Environ., 2019, 663, 738–753, DOI:10.1016/j.scitotenv.2019.01.395
- A. T. Laitinen, V. M. Parsana, O. Jauhiainen, M. Huotari, L. J. P. Van Den Broeke, W. De Jong, T. J. H. Vlugt and M. Ramdin, Liquid-Liquid Extraction of Formic Acid with 2-Methyltetrahydrofuran: Experiments, Process Modeling, and Economics, Ind. Eng. Chem. Res., 2021, 60, 5588–5599, DOI:10.1021/ACS.IECR.1C00159/ASSET/IMAGES/LARGE/IE1C00159_0010.JPEG
- M. A. Murcia Valderrama, R.-J. van Putten and G.-J. M. Gruter, The potential of oxalic – and glycolic acid based polyesters (review). Towards CO2 as a feedstock (Carbon Capture and Utilization – CCU), Eur. Polym. J., 2019, 119, 445–468, DOI:10.1016/j.eurpolymj.2019.07.036
- M. A. Murcia Valderrama, R.-J. van Putten and G.-J. M. Gruter, PLGA Barrier Materials from CO2. The influence of Lactide Co-monomer on Glycolic Acid Polyesters, ACS Appl. Polym. Mater., 2020, 2, 2706–2718, DOI:10.1021/acsapm.0c00315
-
B. Wang and G. J. M. Gruter, Polyester copolymer, WO2018211132A1, 2017
-
B. Wang and G. J. M. Gruter, Polyester copolymer, WO2018211133A1, 2017
- F. D. Meylan, V. Moreau and S. Erkman, CO2 utilization in the perspective of industrial ecology, an overview, J. CO2 Util., 2015, 12, 101–108, DOI:10.1016/j.jcou.2015.05.003
- R. M. Cuéllar-Franca and A. Azapagic, Carbon capture, storage and utilisation technologies: A critical analysis and comparison of their life cycle environmental impacts, J. CO2 Util., 2015, 9, 82–102, DOI:10.1016/j.jcou.2014.12.001
- E. Schuler, M. Demetriou, N. R. Shiju and G.-J. M. Gruter, Towards Sustainable Oxalic Acid from CO2 and Biomass, ChemSusChem, 2021, 14, 3636–3664, DOI:10.1002/cssc.202101272
- A. Górski and A. D. Kraśnicka, The importance of the CO22− anion in the mechanism of thermal decomposition of oxalates, J. Therm. Anal., 1987, 32, 1229–1241, DOI:10.1007/BF01905177
- A. Górski and A. D. Kraśnicka, Formation of oxalates and carbonates in the thermal decompositions of alkali metal formates, J. Therm. Anal., 1987, 32, 1895–1904, DOI:10.1007/BF01913982
- A. Górski and A. D. Kraśnicka, The importance of the CO22− anion in the mechanism of thermal decomposition of oxalates, J. Therm. Anal., 1987, 32, 1229–1241, DOI:10.1007/BF01905177
- A. Górski and A. D. Kraśnicka, Influence of the cation on the formation of free hydrogen and formaldehyde in the thermal decomposition of formates, J. Therm. Anal., 1987, 32, 1345–1354, DOI:10.1007/BF01913334
- P. S. Lakkaraju, M. Askerka, H. Beyer, C. T. Ryan, T. Dobbins, C. Bennett, J. J. Kaczur and V. S. Batista, Formate to Oxalate: A Crucial Step for the Conversion of Carbon Dioxide into Multi-carbon Compounds, ChemCatChem, 2016, 8, 3453–3457, DOI:10.1002/cctc.201600765
- E. Schuler, P. A. Ermolich, N. R. Shiju and G.-J. M. Gruter, Monomers from CO2: Superbases as Catalysts for Formate–to–Oxalate Coupling, ChemSusChem, 2021, 14, 1517–1523, DOI:10.1002/cssc.202002725
- L. K. Freidlin, Kinetics of potassium formate decomposition in presence of alkali metals, Zh. Obshch. Khim., 1937, 7, 1675–1683 CAS
- L. K. Freidlin, Thermal transformation of potassium and sodium formates in presence of alkali hydroxides, Zh. Prikl. Khim. (S.-Peterburg, Russ. Fed.), 1937, 10, 1086–1094 CAS
- L. K. Freidlin, Kinetics of sodium formate thermal decomposition, Trans. All-Union Acad. Food Ind. Named after Stalin, 1939, 145–157 CAS
- L. K. Freidlin, A. A. Balandin and A. I. Lebedeva, On the consecutive stages of the thermal conversion of formates into oxalates, Russ. Chem. Bull. (Izvestiya Akad. Nauk SSSR), 1941, 2, 275–288 Search PubMed
- K. O. Hartman and I. C. Hisatsune, The Kinetics of Calcium Formate Pyrolysis in Potassium Bromide Matrix, J. Phys. Chem., 1965, 69, 583, DOI:10.1021/j100886a037
- K. Hartman and I. C. Hisatsune, The Kinetics of Formate Ion Pyrolysis in Alkali Halide Matrices, J. Phys. Chem., 1966, 583, 1281–1287, DOI:10.1021/j100876a051
- G. D. Buttress and M. A. Hughes, The Thermal Decomposition of Uranium(IV) and of Uranyl(VI) Formates, J. Chem. Soc. A, 1968, 1272–1277, 10.1039/J19680001272
- G. D. Buttress and M. A. Hughes, The Thermal Decomposition of Uranyl(VI) Oxalate, J. Chem. Soc. A, 1968, 1985–1989, 10.1039/J19680001985
- G. D. Buttress and M. A. Hughes, The thermal decomposition of uranyl(VI) oxalate, J. Chem. Soc. A, 1968, 1985–1989, 10.1039/J19680001985
- S. Shishido and Y. Masuda, The Gaseous Products formed in the Thermal Decompositions of Formates, Nippon Kagaku Kaishi, 1973, 185–188, DOI:10.1246/nikkashi.1973.185
- R. Canning and M. A. Hughes, The thermal decomposition of alkaline earth formates, Thermochim. Acta, 1973, 6, 399–409, DOI:10.1016/0040-6031(73)87006-6
- T. Meisel, Z. Halmos, K. Seybold and E. Pungor, The thermal decomposition of alkali metal formates, J. Therm. Anal., 1975, 7, 73–80, DOI:10.1007/BF01911627
- S. Shishido and Y. Masuda, Thermal Decomposition of Alkali Metal Formates, Nippon Kagaku Kaishi, 1976, 5, 325–1675, DOI:10.1246/nikkashi.1976.66
- Y. Masuda, A. Yahata and H. Ogawa, Thermal Phase Transition of Cesium Formate, Inorg. Chem., 1995, 34, 3130–3133, DOI:10.1021/ic00116a002
- V. Merz and W. Weith, Ueber synthetische Oxalsäure, Ber. Dtsch. Chem. Ges., 1882, 15, 1507–1513, DOI:10.1002/cber.18820150215
- V. Merz, Herstellung von Oxalsäure, Polytech. J., 1882, 245, 312 Search PubMed
-
M. Goldschmidt, Process of making oxalates, US659733A, 1900
-
A. Wiens, Process of making oxalates, US714347A, 1902
-
D. Strauss, Manufacture of oxalates, US1038985A, 1912
-
E. Mewburn, Manufacture of Oxalates and Oxalic acid, GB160747, 1922
-
H. W. Paulus, Method of Converting Formate into Oxalates, US1445163A, 1923
-
W. Wallace, Producing oxalates from formates, US1506872A, 1924
-
P. C. Bredt Otto, Treatment of formates of metals of the alkali-earth group, US1622991A, 1927
-
M. Enderli, Process for the production of caustic potash and oxalic acid, US2002342A, 1935
-
E. Hene, Process for making potassium oxalate, US2004867A, 1935
-
M. Enderli and A. Schrodt, Process for the preparation of potassium oxalate from potassium formate, US2033097A, 1936
- L. K. Freidlin, Volumetric Determination of Alkali Metal Carbonates and Oxalates in Mixture with Formates, Sci. Rep. Moscow Univ., 1936, 152–156 Search PubMed
- L. K. Freidlin, A. A. Balandin and A. I. Lebedeva, Thermal decomposition of tallium formate, Bull. Acad. Sci. USSR, Cl. Sci. Math. Nat., Ser. Chim., 1940, 6, 955–962 Search PubMed
- L. K. Freidlin, On the Production of Oxalic Acid in the USSR, Prom-st. Org. Khim., 1937, 3, 681–686 CAS
- L. K. Freidlin, Selectivie effect of different caltalysts on converting fusible formates to oxalates, Zh. Prikl. Khim. (S.-Peterburg, Russ. Fed.), 1938, 11, 975–980 CAS
- A. A. Balandin, L. K. Freidlin and D. N. Vaskevich, Kinetics of thermal decomposition of potassium formate, Sci. Rep. Moscow State Univ., 1936, 6, 321–345 Search PubMed
- L. K. Freidlin, A. A. Balandin and A. I. Lebedeva, Thermal decomposition of lithium formate, Russ. Chem. Bull. (Izvestiya Akad. Nauk SSSR), 1941, 2, 261–267 Search PubMed
- L. K. Freidlin and A. I. Lebedeva, Thermal transformation of potassium and sodium formates in presence of alkali hydroxides, Zh. Prikl. Khim. (S.-Peterburg, Russ. Fed.), 1937, 10, 1086–1094 CAS
- L. K. Freidlin, A. A. Balandin and A. I. Lebedeva, Thermal decomposition of caesium formate, Russ. Chem. Bull. (Izvestiya Akad. Nauk SSSR), 1941, 2, 255–262 Search PubMed
- A. A. Balandin and L. K. Freidlin, Reaction of sodium formate with sodium hydroxide, Zh. Obshch. Khim., 1935, 6, 868–872 Search PubMed
- L. K. Freidlin, A. A. Balandin and A. I. Lebedeva, On the capacity of metal formates to be transformed into oxalates, Russ. Chem. Bull. (Izvestiya Akad. Nauk SSSR), 1941, 2, 268–274 Search PubMed
- L. K. Freidlin, A. A. Balandin and A. I. Lebedeva, Thermal decomposition of rubidium formate, Russ. Chem. Bull. (Izvestiya Akad. Nauk SSSR), 1941, 2, 247–256 Search PubMed
- D. W. Ovenall and D. H. Whiffen, Electron spin resonance and structure of the co-2 radical ion, Mol. Phys., 1961, 4, 135–144, DOI:10.1080/00268976100100181
- P. Baraldi, Thermal behaviour of metal carboxylates: Metal formates, Spectrochim. Acta, Part A, 1979, 35, 1003–1007, DOI:10.1016/0584-8539(79)80027-6
- A. D. Górski and A. Kraśnicka, Origin of Organic Gaseous Products Formed in the Thermal Decomposition of Formates, J. Therm. Anal., 1987, 32, 1243–1251, DOI:10.1007/BF01905178
- E. Schuler, M. Stoop, N. R. Shiju and G.-J. M. Gruter, Stepping Stones in CO2 Utilization: Optimizing the Formate to Oxalate Coupling Reaction Using Response Surface Modeling, ACS Sustainable Chem. Eng., 2021, 9, 14777–14788, DOI:10.1021/acssuschemeng.1c04539
- Y. Masuda, K. Hashimoto and Y. Ito, The thermal phase transformations of lithium, sodium and potassium formates, Thermochim. Acta, 1990, 163, 271–278, DOI:10.1016/0040-6031(90)80407-P
-
L. Li, Three-phase fluidized bed double cycle continuous dehydrogenation production of sodium oxalate process and equipment, CN1166482A, 1997
-
A. Li and Y. Li, Process and equipment for manufacturing sodium oxalate by jet-mixing of overheated steam, rapid heating, and continuous dehydrogenation, CN1927805A, 2007
-
A. Li and Y. Li, Method and equipment for producing sodium oxalate by spray dehydrogenation of sodium formate, CN1948260A, 2007
-
A. Li and Y. Li, Technology and equipment for production of sodium oxalate by continuous dehydrogenation, CN101823950A, 2010
- Y. Yu, B. Chen, W. Qi, X. Li, Y. Shin, C. Lei and J. Liu, Enzymatic conversion of CO2 to bicarbonate in functionalized mesoporous silica, Microporous Mesoporous Mater., 2012, 153, 166–170, DOI:10.1016/j.micromeso.2011.12.005
-
Z. Cao and Y. Cao, Production of oxalates, oxalic acid and dihydrogen phosphate salt by dehydrogenation of formate salts, CN101462943A, 2009
-
X. Kuojun, A method for producing sodium oxalate by microwave dehydrogenation, CN1727322A, 2018
-
J. J. Kaczur, P. P. Lakkaraju and R. R. Parajuli, Process for producing oxalic acid, WO2017121887A1, 2017
- A. Górski and A. Kraśnicka, Origin of organic gaseous products formed in the thermal decomposition of formates, J. Therm. Anal., 1987, 32, 1243–1251, DOI:10.1007/BF01905178
- S. Cannizzaro, Ueber den der Benzoësäure entsprechenden Alkohol, Justus Liebigs Ann. Chem., 1853, 88, 129–130, DOI:10.1002/JLAC.18530880114
- V. E. Tishchenko, J. Russ. Phys. – Chem. Soc., 1906, 38, 355 Search PubMed
- F. Weigel, N. Ter Meer and H. E. mult Wiberg, Der thermische Abbau von Americium(III)-oxalat, -formiat und -carbonat Thermal Decomposition of Americium(III)-oxalate, -formiate, and -carbonate, Z. Naturforsch., 1971, 26, 504–512 CrossRef CAS
- E. L. Head and C. E. Holley, The thermal decomposition of scandium formate and Oxalate, Nucl. Chem., 1964, 26, 525–530 CrossRef CAS
- D. Shyamala, Thermal decomposition of europium formate and oxalate, Thermochim. Acta, 1982, 56, 135–146 CrossRef
- E. Schuler, A. Perez de Alba Ortiz, B. Ensing, N. R. Shiju and G.-J. M. Gruter, Understanding a key step in CO2 to polymers: The role of alkali hydroxides in formate coupling reactions, J. Am. Chem. Soc., 2022 Search PubMed
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution, Nat. Catal., 2021, 4, 654–662, DOI:10.1038/s41929-021-00655-5
- H. Oehme, Verfahren zur Gewinnung von Oxalsäure, DE414376, 1925, https://translationportal.epo.org/emtp/translate?ACTION=description-retrieval&COUNTRY=DE&ENGINE=google&FORMAT=docdb&KIND=C&LOCALE=en_EP&NUMBER=414376&OPS=ops.epo.org%2F3.2&SRCLANG=de&TRGLANG=en&apikey=TSMqTfrVAvNtryGI8Qlfbozj8DnAGlqJ&PDF=vGcgfViHwGj2iP3zpT, (accessed April 18, 2018).
- A. Banerjee and M. W. Kanan, Carbonate-Promoted Hydrogenation of Carbon Dioxide to Multicarbon Carboxylates, ACS Cent. Sci., 2018, 4, 606–613, DOI:10.1021/acscentsci.8b00108
- L. K. Freidlin and A. I. Lebedeva, Interaction of sodium amide with formates, Dokl. Akad. Nauk SSSR, 1938, 19, 701–705 CAS
- L. K. Freidlin, Selective action of the catalysts on the transformation of fusible formates to oxalates, Zh. Prikl. Khim. (S.-Peterburg, Russ. Fed.), 1938, 11, 975–980 CAS
- L. K. Freidlin, The kinetics of the thermal decomposition of potassium formate in the presence of alkali metals, Zh. Obshch. Khim., 1937, 7, 1675–1682 CAS
-
H. W. Paulus, Method of converting formates into oxalates, US1445163A, 1923
-
H. W. Paulus, Apparatus for converting formates into oxalates, US1445162A, 1923
-
O. P. C. Bredt, Treatments of formates of metals of the alkali-earth group, US1622991A, 1927
- J. Wimmer, Verfahren zur Darstellung von Oxalsäure, DE588159, 1933, https://translationportal.epo.org/emtp/translate?ACTION=description-retrieval&COUNTRY=DE&ENGINE=google&FORMAT=docdb&KIND=C&LOCALE=en_EP&NUMBER=561180&OPS=ops.epo.org%2F3.2&SRCLANG=de&TRGLANG=en&apikey=TSMqTfrVAvNtryGI8Qlfbozj8DnAGlqJ&PDF=vGcgfViHwGj2iP3zpT, (accessed April 18, 2018).
-
Z. Cao and Y. Cao, Apparatus for continuous dehydrogenation of formate for combined production of oxalate, oxalic acid and dihydrogen phosphate, CN201343509Y, 2009
-
Z. Xu, X. Cheng and W. Xia, Method and apparatus for production of sodium oxalate by double-fluid spraying and continuous dehydrogenation, CN102391099A, 2012
-
H. Jiang, D. Li, Z. Jiang, D. Wang, Z. Ban and B. Zhang, Chemical industrial reaction system for producing oxalic acid diester and oxalic acid as main products from sodium formate, CN102659556A, 2012
-
A. Li, C. Liu and Q. Li, Preparation of sodium oxalate by continuous dehydrogenation of sodium formate using dehydrogenation surplus heat, CN101077855A, 2007
-
X. Yu, Preparation of sodium oxalate by liquid-spraying sodium formate dehydrogenation and equipment applied thereof, CN1502599A, 2004
- R. Sabbah, J. Haladjian and P. Bianco, Oxalate des formiates de sodium et de potassium, Bull. Soc. Chim. Fr., 1969, 7, 2304 Search PubMed
- M. I. Diaz-Guemes, A. S. Bhatti and D. Dollimore, The thermal decomposition of oxalates, Thermochim. Acta, 1987, 111, 275–282, DOI:10.1016/0040-6031(87)88054-1
- E. Schuler, M. Morana, N. R. Shiju and G. M. Gruter, A New way to make Oxalate from CO2, Sustain. Chem. Clim. Action, 2022, 1, 100001 CrossRef
-
M. Goldschmidt, Process of making oxalates, US659733A, 1900 Search PubMed
-
H. Hoff, Caustic potash and oxalic acid, FR759216, 1934
- L. W. Andrews, Process for manufacturing oxalates, US1281117A, p. 1918.
- A. Hempel, Process of making oxalates, US1070806A, p. 1913.
- J. Weise, F. Rieche and A. Barth, Process of making formates, US820159A, p. 1906.
- H. W. Paulus, Manufacture of Oxalate, US1420213A, p. 1922.
- D. W. Ovenall and D. H. Whiffen, Electron spin resonance and structure of the CO2 radical ion, Mol. Phys., 1961, 4, 135–144, DOI:10.1080/00268976100100181
- J. S. Jestilä, J. K. Denton, E. H. Perez, T. Khuu, E. Aprà, S. S. Xantheas, M. A. Johnson and E. Uggerud, Characterization of the alkali metal oxalates (MC2O4−) and their formation by CO2 reduction via the alkali metal carbonites (MCO2−), Phys. Chem. Chem. Phys., 2020, 22, 7460–7473, 10.1039/D0CP00547A
- S. Takagi, Study on the formation of Sodium Oxalate and Carbonate by the Pyrolysis of Sodium Formate and its Mechanism. I, Nippon Kagaku Kaishi, 1939, 60, 625–631, DOI:10.1246/nikkashi1921.60.625
- B. Andresen, Synthesis of Sodium Formate 13C Oxalic Acid-13C2, J. Org. Chem., 1977, 42, 2790 CrossRef CAS
- A. Paparo and J. Okuda, Carbonite, the dianion of carbon dioxide and its metal complexes, J. Organomet. Chem., 2018, 869, 270–274, DOI:10.1016/j.jorganchem.2017.10.005
- R. E. Bellis and S. Clough, An electron spin resonance study of a free radical reaction in crystals of sodium formate, Mol. Phys., 1965, 10, 33–39, DOI:10.1080/00268976600100051
- G. W. Chantry and D. H. Whiffen, Electronic absorption spectra of CO2-trapped in γ-irradiated crystalline sodium formate, Mol. Phys., 1962, 5, 189–194, DOI:10.1080/00268976200100191
- P. W. Atkins, N. Keen and M. C. R. Symons, 561. Oxides and oxyions of the non-metals. Part II. CO2? and NO2, J. Chem. Soc., 1962, 2873, 10.1039/jr9620002873
- E. S. Lewis and M. C. R. Symons, Mechanisms for carbon–hydrogen bond breakage, Q. Rev., Chem. Soc., 1958, 12, 230–249, 10.1039/QR9581200230
- M. C. R. Symons, 48. Unstable intermediates. Part III. Proton interaction in aliphatic free radicals, J. Chem. Soc., 1959, 277, 10.1039/jr9590000277
- J. A. Brivati, N. Keen and M. C. R. Symons, 44. Unstable intermediates. Part XIV. The formyl radical, J. Chem. Soc., 1962, 237, 10.1039/jr9620000237
- D. W. Ovenall and D. H. Whiffen, Electron spin resonance and structure of the CO−2 radical ion, Mol. Phys., 1961, 4, 135–144, DOI:10.1080/00268976100100181
- G. Darzens and M. Meyer, Methnaol de Cannizzaro, C. R. Hebd. Seances Acad. Sci., 1953, 1496 CAS
- I. Lin and A. R. Day, A Study of the Mixed Tischtschenko Reaction, J. Am. Chem. Soc., 1952, 74, 5133–5135, DOI:10.1021/ja01140a041
- M. Paskevicius, L. H. Jepsen, P. Schouwink, R. Černý, D. B. Ravnsbæk, Y. Filinchuk, M. Dornheim, F. Besenbacher and T. R. Jensen, Metal borohydrides and derivatives-synthesis, structure and properties, Chem. Soc. Rev., 2017, 46, 1565–1634, 10.1039/c6cs00705h
- H. C. Brown, E. J. Mead and B. C. S. Rao, A Study of Solvents for Sodium Borohydride and the Effect of Solvent and the Metal Ion on Borohydride Reductions, J. Am. Chem. Soc., 1955, 77, 6209–6213, DOI:10.1021/ja01628a044
- M. Paskevicius, M. B. Ley, D. A. Sheppard, T. R. Jensen and C. E. Buckley, Eutectic melting in metal borohydrides, Phys. Chem. Chem. Phys., 2013, 15, 19774–19789, 10.1039/c3cp53920b
- L. Lombardo, H. Yang and A. Züttel, Study of borohydride ionic liquids as hydrogen storage materials, J. Energy Chem., 2019, 33, 17–21, DOI:10.1016/j.jechem.2018.08.011
- C. W. Duan, L. X. Hu and J. L. Ma, Ionic liquids as an efficient medium for the mechanochemical synthesis of α-AlH3 nano-composites, J. Mater. Chem. A, 2018, 6, 6309–6318, 10.1039/C8TA00533H
- T. Biedroń and P. Kubisa, Imidazolium ionic liquids with short polyoxyethylene chains, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6961–6968, DOI:10.1002/pola.23005
- E. Amigues, C. Hardacre, G. Keane, M. Migaud and M. O'Neill, Ionic liquids – Media for unique phosphorus chemistry, Chem. Commun., 2006, 72–74, 10.1039/b509248e
- H. Fu, Y. Wu, J. Chen, X. Wang, J. Zheng and X. Li, Promoted hydrogen release from alkali metal borohydrides in ionic liquids, Inorg. Chem. Front., 2016, 3, 1137–1145, 10.1039/c6qi00167j
- G. R. M. Dowson, I. Dimitriou, R. E. Owen, D. G. Reed, R. W. K. Allen and P. Styring, Kinetic and economic analysis of reactive capture of dilute carbon dioxide with Grignard reagents, Faraday Discuss., 2015, 183, 47–65, 10.1039/C5FD00049A
-
V. Grignard, Sur les combinaisons organomagnésiennes mixtes, PhD thesis, Université de Lyon, 1901 Search PubMed
- A. S. Kiselyov and L. Strekowski, The trifluoromethyl group in organic synthesis. A review, Org. Prep. Proced. Int., 1996, 283, 289–318, DOI:10.1080/00304949609356536
- A. M. Bahmanpour, A. Hoadley, S. H. Mushrif and A. Tanksale, Hydrogenation of Carbon Monoxide into Formaldehyde in Liquid Media, ACS Sustainable Chem. Eng., 2016, 4, 3970–3977, DOI:10.1021/acssuschemeng.6b00837
- G. J. Millar and M. Collins, Industrial Production of Formaldehyde Using Polycrystalline Silver Catalyst, Ind. Eng. Chem. Res., 2017, 56, 9247–9265, DOI:10.1021/acs.iecr.7b02388
- A. Saravanan, P. Senthil kumar, D.-V. N. Vo, S. Jeevanantham, V. Bhuvaneswari, V. Anantha Narayanan, P. R. Yaashikaa, S. Swetha and B. Reshma, A comprehensive review on different approaches for CO2 utilization and conversion pathways, Chem. Eng. Sci., 2021, 236, 116515, DOI:10.1016/j.ces.2021.116515
- A. J. L. Cooper, J. Z. Ginos and A. Meister, Synthesis and Properties of the α-Keto Acids, Chem. Rev., 1983, 83, 321–358, DOI:10.1021/cr00055a004
- Y. Song, J. Li, H. d. Shin, L. Liu, G. Du and J. Chen, Biotechnological production of alpha-keto acids: Current status and perspectives, Bioresour. Technol., 2016, 219, 716–724, DOI:10.1016/j.biortech.2016.08.015
- S. Kobayashi, T. Yokoyama, K. Kawabe and T. Saegusa, Deoxy-polymerization of phenylglyoxylic acid using a cyclic phosphite as deoxygenating agent a new synthesis of poly(α-ester), Polym. Bull., 1980, 3, 585–591, DOI:10.1007/BF01135327
- M. T. Xiao, Y. Y. Huang, X. A. Shi and Y. H. Guo, Bioreduction of phenylglyoxylic acid to R-(−)-mandelic acid by Saccharomyces cerevisiae FD11b, Enzyme Microb. Technol., 2005, 37, 589–596, DOI:10.1016/j.enzmictec.2005.02.018
- M. Shiri, M. M. Heravi and B. Soleymanifard, Arylidene pyruvic acids (APAs) in the synthesis of organic compounds, Tetrahedron, 2012, 68, 6593–6650, DOI:10.1016/j.tet.2012.05.009
- T. Werner and J. Koch, Sodium hydride catalyzed Tishchenko reaction, Eur. J. Org. Chem., 2010, 6904–6907, DOI:10.1002/ejoc.201001294
| This journal is © The Royal Society of Chemistry 2022 |