
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
AqSO biorefinery: a green and parameter-controlled process for the production of lignin–carbohydrate hybrid materials†
Dmitry
Tarasov
 a,
Philipp
Schlee
a,
Andrey
Pranovich
b,
Adrian
Moreno
c,
Luyao
Wang
b,
Davide
Rigo
a,
Mika H.
Sipponen
c,
Chunlin
Xu
b and
Mikhail
Balakshin
*a
a,
Philipp
Schlee
a,
Andrey
Pranovich
b,
Adrian
Moreno
c,
Luyao
Wang
b,
Davide
Rigo
a,
Mika H.
Sipponen
c,
Chunlin
Xu
b and
Mikhail
Balakshin
*a
aDepartment of Bioproducts and Biosystems, Aalto University, Vuorimiehentie 1, Espoo, 02150, Finland. E-mail: mikhail.balakshin@aalto.fi
bLaboratory of Natural Materials Technology, Åbo Akademi University, Henrikinkatu 2, Turku, 20500, Finland
cDepartment of Materials and Environmental Chemistry, Stockholm University, Svante Arrhenius väg 16C, Stockholm, 10691, Sweden
First published on 30th July 2022
Abstract
The current biorefineries are focused on the comprehensive fractionation of biomass components into separate lignin and carbohydrate fractions for the production of materials, platform chemicals and biofuel. However, it has become obvious that the combination of lignin and carbohydrates can have significant technical, environmental, and economic benefits as opposed to their separate use. Herein, we developed a green, simple, and flexible biorefinery concept for the integrated utilization of all major biomass components for high-value applications with the focus on functional lignin–carbohydrate hybrids (LCHs). The established process consisted of a modified hydrothermal treatment (HTT) of birch wood followed by solvent extraction of the resulting solids and is therefore named AquaSolv Omni (AqSO) biorefinery. The AqSO biorefinery produces three major streams: hydrolysate (hemicellulose-derived products), solvent-extracted lignin–carbohydrate complexes (LCCs) and cellulose-rich fibers. Specific process conditions were found to facilitate the production of LCCs of different types in high yields as a new valuable and industrially realistic process stream. The effect of the process severity and liquid to solid (L/S) ratio on the yields and compositions of the produced fractions as well as on the structure and properties of the extracted LCCs was investigated using state of the art NMR spectroscopy and molar mass distribution analysis among other methods. The high flexibility of the process allows for engineering of the resulting products in a wide range of chemical compositions, structures and physicochemical properties and therefore gives a good opportunity to optimize the products for specific high-value applications. The process can be easily combined with other biorefinery operations (e.g., enzymatic hydrolysis, pulping, bleaching) to be incorporated into existing value chains or create new ones and thus is suitable for different biorefinery scenarios. First examples of high-value applications of AqSO biorefinery LCHs are reported. LCC nanoparticles (LCCNPs) were produced for the first time directly from the solvent extract and their properties were investigated. LCCNPs could efficiently stabilize Pickering emulsions of tetrahydrofurfuryl methacrylate and allowed their free radical polymerization. In addition, AqSO LCHs showed promising results as wood adhesives. Overall, our results provide detailed information on the complex lignocellulosic fractions and bridge the gap from process engineering to sustainable product development.
1. Introduction
Plant biorefinery is a valuable alternative to the current petroleum-based industry and thus it is of key importance to address UN Sustainability Goals.1 Plant biomass is one of a few potential sources of renewable energy, but not the only one; there are other alternatives, such as solar, wind and hydroelectric power of energy, green hydrogen fuel, etc. However, plant biomass is the most abundant and readily available source of renewable chemicals. Therefore, the utilization of biomass components for high-value chemicals/materials can be more economically profitable than biofuel (e.g., bioethanol) production, which can thus be considered a co-product rather than the main target in the new scenario. The main goal of the new biorefinery approach is to maximize technical and therefore economic outcomes, considering high-value products from all biomass components, i.e., creation of an advanced integrated biorefinery.Current industrial sugar-based biorefineries are focused on ethanol (biofuel) among other monosaccharide-derived platform chemicals leaving very little attention to lignin and potential polymeric carbohydrate streams. However, lignin-containing streams, current low-value by-products, could be exploited to replace chemicals with significantly higher revenue than biofuel alone.2,3 They can be the game-changer for sugar-based biorefineries rather than just an added value. However, the ability to optimize the properties of lignins for targeting characteristics (i.e., lignin engineering) via biomass processing is vital to meet the requirements of precursors in high-value applications. This is not the case in the current biorefinery where lignin is obtained as a by-product after process optimization for bioethanol production.
In addition to lignin, the valorization of polymeric and oligomeric hemicelluloses and cellulose (including cellulose nanocrystals/cellulose nanofibers (CNCs/CNFs)) for high-value products, such as environmentally compatible and human-friendly materials for various applications targeting health, food, cosmetics, polymeric composites for 3D-printing, and the paper/textile industry,4–7 is desirable for the best benefits from integrated biorefineries as a sound alternative to the current bioethanol or monomeric sugar production. This also allows one to take advantage of the unique polymeric features of the plant polysaccharides rather than losing these properties when breaking the polysaccharides into the respective monomers.
The predominant industrial wood biorefinery process, Kraft pulping, is focused on cellulosic pulp production, i.e. utilizing only about 30–60% of biomass (including minor side streams, such as Kraft lignin, turpentine, tall oil, etc.) while the rest is incinerated to generate the energy required for the recovery of the Kraft process chemicals. Isolation of Kraft lignin (KL) is technically limited to a small portion of the extracted lignin2,3,8 while dissolved carbohydrates cannot be recovered due to their further transformation to a complex mixture of products. A good example of an integrated biorefinery is the current sulfite process, producing pulp, lignosulphonates (LS), bioethanol and other valuable products, such as vanillin,9 from all components of wood. However, the process has serious drawbacks limiting its share in the industry to a few percent.10 All current pulping processes are optimized for specific pulp characteristics, thus dramatically limiting the range for lignin optimization. In addition, both sulfite and Kraft processes use sulfur-containing chemicals and therefore are not very environmentally friendly. Furthermore, the presence of sulfur in KLs and LSs is often undesirable in different high-value applications.
Organosolv pulping processes can overcome the limitations of the current industrial pulping and can potentially serve (if all industrial issues are addressed) as an integrated biorefinery11–15 utilizing all process streams. Nevertheless, the production costs are significantly higher than the traditional pulping and sugar-based biorefineries.3,8 More advanced “lignin-first” approaches suggest process optimization for specific lignin characteristics.12–16 However, the suggested methods also possess certain limitations. The current “protective” strategies imply much more complex processes using expensive and sometimes toxic chemicals (such as formaldehyde, dioxane, etc.). Sophisticated valorization routes are suggested for targeted lignin degradation products as an alternative to the inefficient drop-in strategy.15 Thus, very high production costs would be expected limiting the current “lignin-first” approach only to the production of chemicals of very high value (e.g., pharmaceuticals), which, however, have a very limited market size.3,8,16 The most economically attractive lignin applications of high market size, but of lower (as compared to pharmaceuticals) value, such as thermoplastics, polyurethanes (PU), and phenolic resins, will be likely more cost-challenging for these sophisticated processes.
Importantly, the traditional biorefinery approaches, both industrial and emerging, imply maximal separation of the biomass components targeting high-purity products; there is also an assumption on direct correlation between the purity of a product and its value. However, harnessing the lignin–carbohydrate synergism of biorefinery streams, demonstrated recently,17–22 is a very promising direction for advanced biorefineries showing better performance (at lower costs) of lignocellulosic hybrids (LCHs). A few examples are the so-called cellolignins (lignins with minor amounts of cellulose) and lignocelluloses (celluloses with minor amounts of lignin) which perform better in specific applications than pure lignin or cellulose. This approach may be considered as novel because it shows a conceptional difference compared to the exhaustive biomass fractionation.
Lignin–carbohydrate complexes (LCCs) represent another good example for the synergism between lignin and carbohydrates and therefore can be very valuable biorefinery products. LCCs combine chemically linked aromatic and carbohydrate moieties exhibiting amphiphilic properties that create unique possibilities for their compatibility with different bioproducts.23–26 For example, polysaccharides increase the emulsification performance of lignin and lignin nanoparticles (LNPs).27,28 On the other hand, a positive effect of lignin moieties on the stabilization of galactoglucomannan (GGM)-based emulsions was recently claimed.29,30 It was explained by the natural amphiphilicity of LCCs and its capacity to anchor into the oil–water interface and contribute to interfacial stabilization. LCCs demonstrated better anti-HIV properties than tannins and flavonoids,31 as well as good biological compatibility allowing their application in different areas of medicine.31–34 It was claimed that lignin moieties in LCCs were more important for anti-HIV activity than carbohydrate units. At the same time, application of pure lignin did not show any anti-HIV activity, indicating the synergism between lignin and polysaccharide domains in LCCs.31 In another study, carbohydrate-rich LCCs obstructed the development of breast tumor cells, whereas lignin-rich LCCs effectively activated macrophage cells that are considered as the first line of defense in the immune system.35 These reports underline that the ability to engineer the LCC structure should be critical in LCC valorization.
The presence of LCCs in spent liquors from neutral sulfite semichemical pulping36 and hydrothermal treatment (HTT) hydrolysates of soft-, hardwood and non-wood biomasses25,37–39 has been reported. Isolation of LCCs from these solutions usually implied the use of XAD resins and methanol.25,39 Maple and bagasse LCCs isolated via this route are composed of about 34.5% and 24% lignin, respectively, and about 48% carbohydrates;39 about 10% of lignin monomeric units were involved in LCC linkages. Giummarella and Lawoko38 reported much higher amounts of LCC linkages in HTT hydrolysates (without LCC fractionation) composed of ca. 10% lignin; up to 50% of lignin monomeric units were claimed to be involved in LCC linkages. The main limitation in the LCC valorization is the lack of industrially relevant methods for LCC isolation as the use of polymeric resins is neither facile nor cost-efficient for industry. Moreover, the yields of LCCs isolated via this route were rather low, only 0.4–0.8% of the original biomass.25
Thus, biorefinery approaches providing different high-value streams from all biomass components are of top importance. In particular, engineering of LCHs in the new biorefinery concept and their further exploitation are of high interest. To develop an advanced integrated and industrially feasible biorefinery approach, HTT is a simple and efficient industrially relevant technology currently used for hemicellulose extraction from biomass10,40,41 in processes targeting dissolving grade pulps or bioethanol. HTT was also optimized for the valorization of polymeric and oligomeric hemicelluloses.5 Nevertheless, few attempts have been made related to lignin valorization in this process. Recently, we have suggested a lignin-centered HTT process42 that showed a proof-of-concept for the extraction of pure lignins in high yields from HTT solids produced under specific HTT conditions. Direct utilization of the lignin extracts in lignin micro- and nanoparticle (LMNP) manufacture allowed for low production costs. However, the engineering of lignin and other valuable process streams has not been addressed yet.
In this study, we propose a green, simple and flexible biorefinery process for integrated utilization of all biomass components for high-value applications with the focus on functional lignin–carbohydrate hybrids. The suggested process consists of HTT followed by a solvent extraction of the resulting solids (under ambient conditions) and was therefore named AquaSolv Omni (AqSO) Biorefinery. In particular, process conditions resulting in a new LCC stream of high yields that can be easily implemented by the industry were discovered. The main objectives of this work were (1) to study the effect of process conditions on the yield, structure and physicochemical properties of the AqSO products to evaluate the ability to engineer precursors with desirable characteristics; (2) to produce nano-particles from the LCC streams and elucidate their properties and application potential and (3) to evaluate different biorefinery scenarios for the AqSO process.
2. Experimental
2.1. Materials and chemicals
Acetone (C3H6O, 95 vol%), sulfuric acid (H2SO4, 98 wt%), and deuterated dimethyl sulfoxide (DMSO-d6), (all analytical grade) were purchased from Sigma-Aldrich.Sawdust of silver birch (Betula pendula) was screened to collect 0.55–0.125 mm particle size and air-dried. To eliminate the effect of lipophilic extractives, they were removed prior to HTT using acetone extraction of the sawdust in a Soxhlet apparatus (Table 1).
2.2. AqSO process (Fig. 1)
HTT of the extractive-free sawdust was carried out in a swing reactor (inner volume = 120 cm3; Fig. SI1†), equipped with temperature control in both the heating block and inside the reactor, at 195 °C and at the liquid to solid (L/S) ratio range of 1–8. The temperature profile is shown in Fig. SI2.† As the heating period had a significant effect on the course of the process, the severity of HTT was expressed as P-factor as follows (assuming activation energy as 125.6 kJ mol−1):10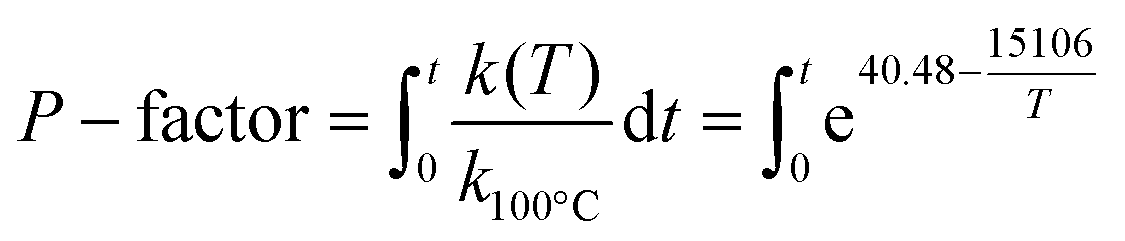 | (1) |
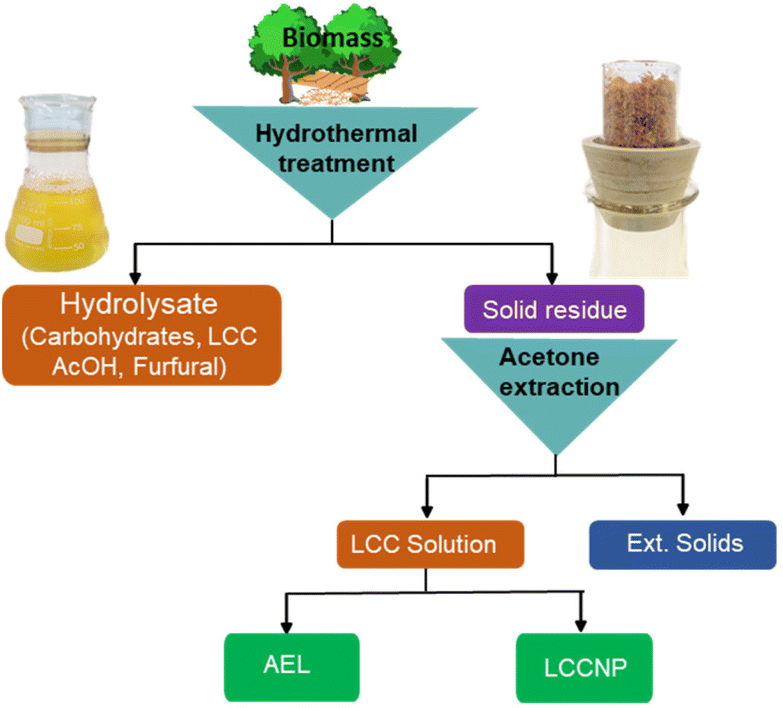 | ||
| Fig. 1 Experimental design. | ||
One example of a typical experimental procedure is given (conditions: P-factor = 500, L/S = 1). Birch wood sawdust (4.21 g with 4.90% moisture content corresponding to 4.00 g o.d. wood) were placed inside the swing reactor together with the required amount of water (3.79 g). Further details are given in the ESI.† The reactor was closed and then set to swing until the required P-factor was reached. Once the reaction was complete, the reactor was cooled down fast (from 195 °C to T < 100 °C within 2 min). The resulting HTT solids and hydrolysate were separated by filtration using a glass crucible (pore size 3 μm) and the solids were exhaustively washed with deionized water. The washed HTT solids were exhaustively extracted with aqueous acetone solutions of various concentration to extract the maximal amount of acetone-extractable LCC (AEL). A comprehensive sequence was tested starting with the acetone concentration of 90% (v/v) followed by consequent extractions with 75 and then with 50% acetone. However, separate experiments showed that the yield and the properties of the AELs extracted directly with 75% acetone were the same as those of AEL obtained from the sequential extraction; therefore, direct extraction with 75% acetone was used on a routine basis. The acetone solution was rotary evaporated (T = 40 °C, p = 20 mbar) to produce AEL, which was vacuum-dried to a constant mass at 40 °C over P2O5. The corresponding solids (ext. solids) were dried and weighed to determine the yield. The AELs were coded as follows: the ones produced at different L/S ratios (at constant P-factor of 500) were coded as AEL-1, AEL-2 … AEL-8. Those produced at different P-factors (at constant L/S = 1) were coded as AEL-400, AEL-500 … AEL-2500.
2.3. Compositional analysis of HTT solids
The compositional analysis of ext. solids included the quantification of acid insoluble lignin (AIL), acid soluble lignin (ASL) and carbohydrate composition using standard protocols.43 A dry sample (ca. 0.3 g) was hydrolyzed with 3 mL of 72% H2SO4 at 30 °C for 1 h. The acid solution was diluted to 4% concentration of H2SO4, and autoclaved at 121 °C for 1 h. The amount of AIL was determined gravimetrically upon filtration, while ASL was evaluated by UV spectrophotometry at 205 nm. The carbohydrate monomers released by the acid hydrolysis were analyzed by ion chromatography using a Dionex ICS 3000A machine equipped with a CarboPac PA1 column.2.4. Carbohydrate composition in AELs
To characterize the chemical composition of non-cellulosic carbohydrate in the AELs, acid methanolysis and trimethylsilylation were carried out following the procedures described by Sundberg et al.44 In short, approximately 10 mg (±0.01 mg) of freeze-dried samples were methanolysed into monosaccharides using 2 mL of 2 M HCl in anhydrous methanol. The mixture was kept at 105 °C for 4 hours in a 10 mL pear-shaped flask equipped with a hermetically sealing Teflon-coated screw cap. A 1 mL carbohydrate calibration solution containing an equal amount of monosaccharide (0.1 mg mL−1 in methanol/water 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v)) and uronic acid (except 4-O-MeGlcA) was also subjected to the same acid methanolysis procedure. The mixture was cooled to ambient temperature and neutralized with 150 μL of pyridine. Subsequently, 1 mL of 0.1 mg mL−1 resorcinol in methanol/water 95:5 (v/v) was added as the internal standard and the mixture was then evaporated to dryness by nitrogen (N2) flow in a 50 °C water bath. The completely dried samples were trimethylsilylated using 150 μL of pyridine, 150 μL of hexamethyldisilazane (HMDS), and 70 μL of trimethylchlorosilane (TMCS) at ambient temperature for at least 12 hours. The trimethylsilylated derivatives of sugar and uronic acid units were analyzed with a gas chromatograph (GC, PerkinElmer AutoSystemXL) equipped with a flame ionization detector (FID) on Agilent capillary columns coated with crosslinked methyl polysiloxane (HP-1, 25 m × 0.20 mm i.d., 0.11 μm film thickness; HP-5, 25 m × 0.20 mm i.d., 0.11 μm film thickness). The analytes were quantified against resorcinol using correction factors determined by calibration. Then, the carbohydrate content in the AEL samples was calculated from the corresponding monosaccharide amount using an anhydrosugar coefficient of 0.88 for pentoses, 0.90 for hexoses, and 0.91 for uronic acid.
:1 (v/v)) and uronic acid (except 4-O-MeGlcA) was also subjected to the same acid methanolysis procedure. The mixture was cooled to ambient temperature and neutralized with 150 μL of pyridine. Subsequently, 1 mL of 0.1 mg mL−1 resorcinol in methanol/water 95:5 (v/v) was added as the internal standard and the mixture was then evaporated to dryness by nitrogen (N2) flow in a 50 °C water bath. The completely dried samples were trimethylsilylated using 150 μL of pyridine, 150 μL of hexamethyldisilazane (HMDS), and 70 μL of trimethylchlorosilane (TMCS) at ambient temperature for at least 12 hours. The trimethylsilylated derivatives of sugar and uronic acid units were analyzed with a gas chromatograph (GC, PerkinElmer AutoSystemXL) equipped with a flame ionization detector (FID) on Agilent capillary columns coated with crosslinked methyl polysiloxane (HP-1, 25 m × 0.20 mm i.d., 0.11 μm film thickness; HP-5, 25 m × 0.20 mm i.d., 0.11 μm film thickness). The analytes were quantified against resorcinol using correction factors determined by calibration. Then, the carbohydrate content in the AEL samples was calculated from the corresponding monosaccharide amount using an anhydrosugar coefficient of 0.88 for pentoses, 0.90 for hexoses, and 0.91 for uronic acid.
2.5. Hydrolysate analysis
:1) was added into a sample vial containing 2.0 mg (±0.01 mg) of the freeze-dried sample. The solvent was evaporated under a N2 flow in a 50 °C water bath and the sample was completely dried in a vacuum desiccator at 40 °C. The dried sample was dissolved in 150 μL of pyridine and then trimethylsilylated with 150 μL of HMDS and 70 μL of TMCS overnight. The trimethylsilylated derivatives of sugars (mono- and disaccharides) were determined with the same GC-FID instrument and columns used for the carbohydrate analysis of AELs. The correction coefficient were determined by calibration.45
The analysis of silylated oligosaccharides containing 3 to 5 monosaccharide units was performed with a GC-FID (PerkinElmer, Clarus 500) using an Agilent J&W HP-1/SIMDIST short capillary column (6 m × 0.535 mm i.d., 0.15 μm film thickness). The chromatogram quantitation was performed by the peak area normalization method.46
2.6. Nuclear magnetic resonance (NMR) spectroscopy
| G + S = G2 + S2,6/2 = 100Ar. |
This assumption implies that the condensation (substitution) at the positions of G2 and S2,6 of lignin is insignificant, which would be verified in more comprehensive further NMR studies. However, it is still valuable for relative comparison with the literature data as this normalization is used when only HSQC spectra of lignins are available.49,52,53
2.7. Glass transition temperature (Tg)
The glass transition temperature was determined by using a Q2000 (TA Instruments, USA) differential scanning calorimeter (DSC). Each specimen (ca. 7 mg) was heated under a nitrogen flow (50 ml min−1) at a rate of 20 °C min−1 from 25 °C to 150 °C and held for five minutes to remove any remaining moisture and cooled to 25 °C. Subsequently, the sample was heated at the same rate to 200 °C and 220 °C. The last trace was used to determine the Tg with the TA Universal Analysis software. The DSC curves are shown in the ESI (Fig. SI3†).2.8. Molar mass distribution
Determination of molar mass characteristics was performed on an HPSEC (Agilent 1100/1200/1260 Series, USA) equipped with an RI detector (Wyatt Technology Optilab High Concentration Differential Refractometer with a Laser Option 785 nm, Wyatt Technology, USA) and a MALS detector (Wyatt Technology DAWN 8 ambient with a Laser Option 785 nm, Wyatt Technology, USA) following the recently described protocol.54 This method allows for the detection of the absolute molar mass in contrast to the commonly used calibration with specific standards.where M is the molar mass, I is the light scattering intensity, dn/dc is the refractive index increment, and c is the mass concentration.
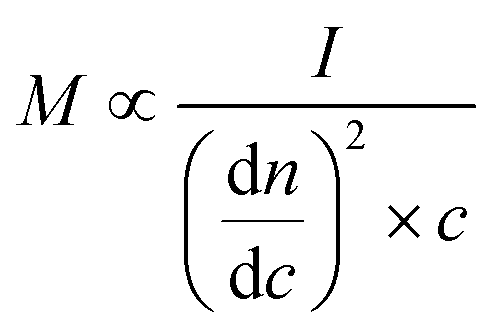
Dry AELs were dissolved in 0.05 M LiBr in DMSO (10 mg mL−1), filtered and analyzed with a series of connected columns (Jordi Gel Glucose Mixed-bed guard column (50 × 10 mm i.d.) and a Jordi Gel DVB Mixed-bed column (250 mm × 10 mm i.d.)) using 0.05 M LiBr in DMSO as the eluent. The dn/dc value of 0.15 mL g−1 was used for calculations.
2.9. Surface tension analysis
The surface tension of the sample solution (0.1 mg mL−1, pH 12.5) against air was measured with a force tensiometer-K100 (Krüss, Germany) using a Wilhelmy plate at 25 °C.2.10. Preparation and characterization of colloidal LCC nanoparticles (LCCNPs)
The preparation of LCCNPs from the extracted AELs was performed via the solvent-exchange methodology adapted from a previous work.55 The particles were produced by rapid pouring of water into a vigorously stirred LCC solution in acetone–water (3:1, w/w). The final aqueous dispersion of LCCNPs (0.4 wt%, 4 g L−1) was obtained after filtration with a mass yield of 87%. A Zetasizer Nano ZS (Malvern, UK) equipment was used to characterize LCCNPs for the hydrodynamic particle diameter by dynamic light scattering (DLS). Zeta potential measurements were made using a dip cell probe in the same instrument. Scanning electron microscopy (SEM) images were recorded on a JEOL JSM-7401F (JEOL Ltd, Japan) operating at 5 kV. The colloidal dispersions of LCCNPs were deposited onto a silicon wafer substrate and evaporated at room temperature for the SEM investigation. For particle size measurements and electron microscopy imaging colloidal dispersions of LCCNPs were diluted in deionized water by a factor of 1:40.
2.11. Pickering emulsion polymerization of tetrahydrofurfuryl methacrylate
The Pickering emulsions were formed by gradually adding tetrahydrofurfuryl methacrylate (THFMA) and azobisisobutyronitrile (AIBN, 2 wt% relative to THFMA) to a water dispersion of LCCNPs (9 g L−1) under vigorous stirring (final o/w ratio 10/90 v/v), following a previously described methodology for the preparation of styrene-based Pickering emulsions using hybrid LNPs.56 Prior to polymerization, Pickering emulsions were degassed by bubbling with nitrogen gas for 10 minutes. Polymerizations were conducted by heating the degassed Pickering emulsions stabilized with LCCNPs at 65 °C in a thermostatic bath for 12 h. After polymerization, the resulting LCCNP-coated polymeric microparticles were purified by three centrifugation/re-dispersion cycles replacing each decanted supernatant with deionized water.Optical microscopy analysis of Pickering emulsions and latex dispersions was carried out in a Nikon (Alphaphot2) optical microscope. The emulsion and latex dispersions were previously diluted by adding one drop of the sample in 1 mL of deionized water. ImageJ software was used to process the recorded images and calculate the mean droplet size based on 50 measurements for each sample.
2.12. Automated bond evaluation system (ABES) method
Veneer strips (20 × 117 mm2) were cut from conditioned veneer sheets (silver birch, Betula pendula Roth). An industrial liquid phenol formaldehyde (PF) resin (Prefere 14J021, Dynea Chemicals Oy, Hamina, Finland) was modified by substituting 30% of the solid content with HTT solids. This means that the PF load was 30% less than in the control and HTT solids were added instead of it in the equivalent mass amount. The resins were applied to an area of 5 × 20 mm2 at one end of the veneer strips to give a resin spread rate of ∼200 g m−2. After adhesive application, the veneer-resin assembly was placed in the ABES equipment (Adhesive Evaluation Systems, Incorporated, Corvallis, OR, USA) and hot pressing was started almost immediately. The assembly time for specimens was approx. 10 s. The environmental conditions during all tests were fixed at 20 (±1) °C and 65 (±2)%. The shear strength was measured after various pressing times ranging from 25 to 150 s. The platen temperature was set to 130 °C and the press pressure was 2.0 MPa. At least seven replicated measurements were made for each datapoint.3. Results and discussion
The overall goal of the present study was to elucidate the distribution of the main wood components in the process streams and changes in the AqSO Biorefinery products, such as their yields, compositions, chemical structures, and other key characteristics, through the course of the reaction in order to evaluate the applicability of the process for different biorefinery scenarios. Therefore, the process was carried out at different HTT severity and L/S ratios but at a fixed reaction temperature (195 °C).3.1. Yields and composition of the main products
The previous study42 implied that the L/S ratio significantly affected the yields of the extracted lignins (AELs). However, recently we found out that the Parr reactor used in the previous work was not suitable to carry out HTT at L/S ≤ 4 due to inhomogeneous heat distribution through the reactor, and the quantitative data reported there for the experiments at L/S = 4 should be reconsidered. The use of the new reactor type solved the problem; the temperature of the heating block (set at 195 °C to avoid overheating) and the temperature inside the reactor matched closely after the heating up period (see Fig. SI2†). After confirming this, the role of L/S ratio was revisited in a wider range of L/S ratios using the reliable reactor type.The effect of the L/S ratio was investigated at a fixed P-factor of 500. No significant differences were found between the experiments at the L/S ratio in the range of 3–8 (Fig. 2). However, a further decrease in the L/S ratio (L/S = 2) resulted in a substantial increase in the AEL yield and the corresponding decrease in the amount of residual lignin (RL) in the extracted solids. Surprisingly, AEL with a significant carbohydrate content, implying the presence of LCC linkages, was produced at L/S = 1 (Fig. 2c and d). LCC linkages were confirmed by 2D NMR (see below). A higher concentration of LCC at L/S = 1 vs. that at L/S = 2 can be a reason for retaining both lignin and xylan (Xyl) in the HTT solids (Fig. 2b) and decreasing the AEL yield (Fig. 2a).
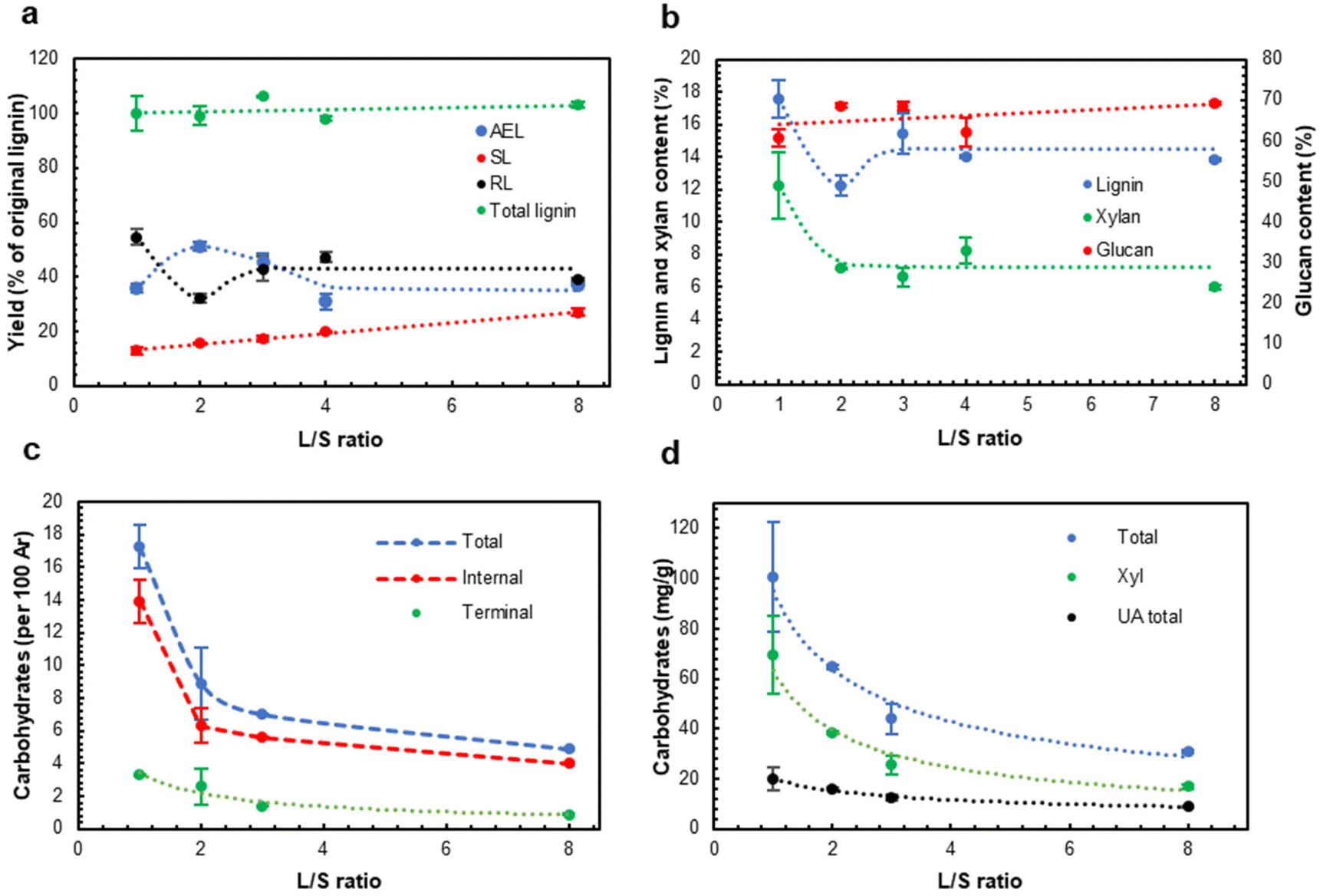 | ||
| Fig. 2 The effect of the L/S ratio (P-factor = 500) on the yield of different lignin fractions (a), extracted solid composition (b), carbohydrate contents in AELs by the HSQC NMR (c) and methanolysis methods (d). AEL – acetone extracted LCC, SL – water soluble lignin in the hydrolysate, RL – residual lignin in the extracted HTT solids. | ||
Although the highest AEL yield was observed at L/S = 2 (Fig. 2a), the highest LCC content was found in AEL produced at L/S = 1 (Fig. 2c and d). Due to high interest in LCC production as a valuable stream, L/S = 1 was selected for the next set of experiments at different process severities.
As expected, an increase in the process severity (P-factor) resulted in progressive wood dissolution, mainly due to xylan hydrolysis. Of note is that the total lignin closure (Fig. 3a) increased from about 100% (per original lignin) in the low severity range (P < 1000) to about 140% at P = 2500 indicating the formation of pseudo-lignin at P > 1000. Pseudo-lignin is defined as carbohydrate transformation products (such as polyfurans (aka humins) formed from furfural, a xylan degradation product), contributing to the analysis of lignin by the Klason method.43
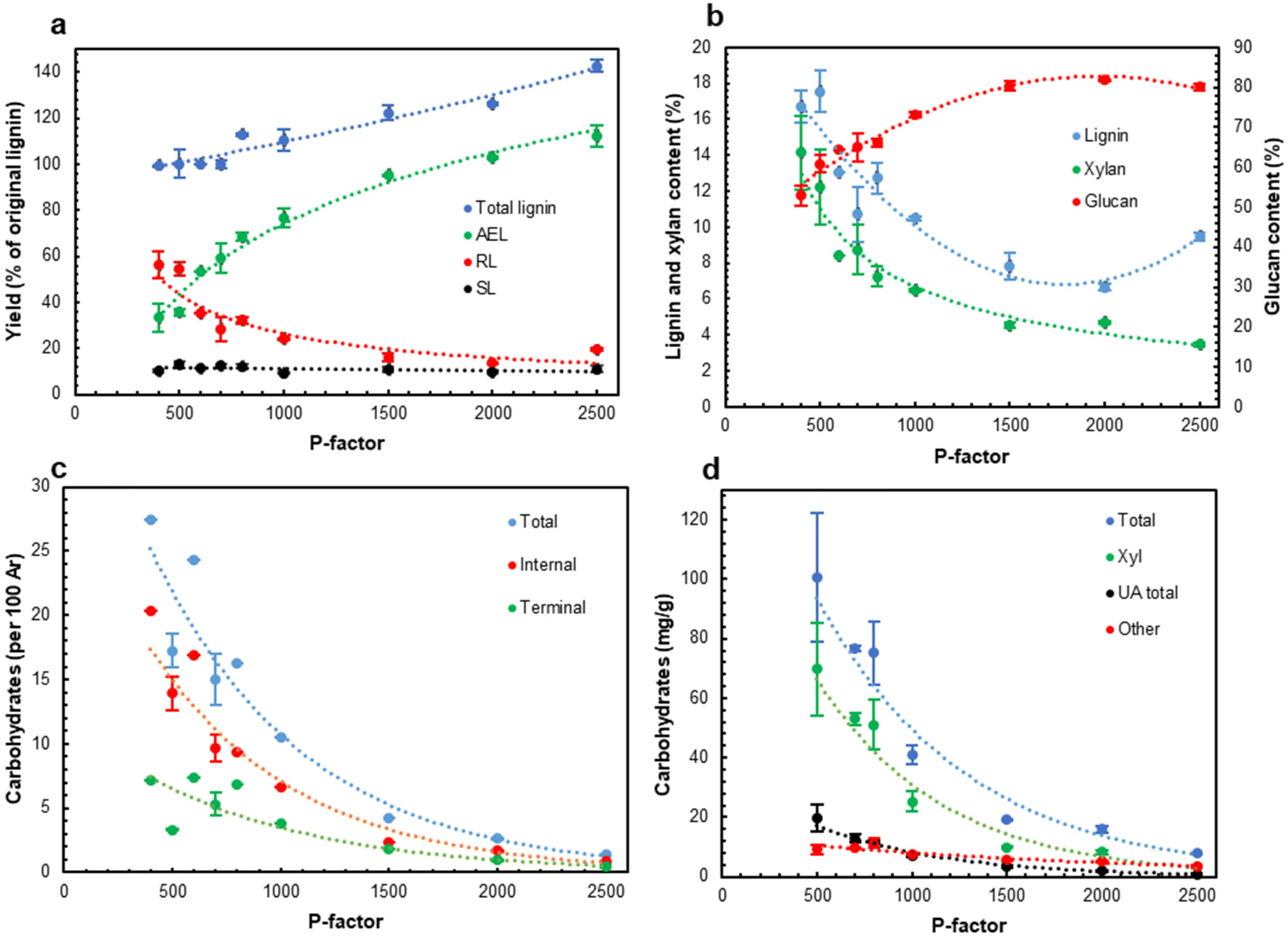 | ||
| Fig. 3 The effect of P-factor (L/S = 1) on yields of different lignin (including pseudo-lignin) streams (a), extracted solid composition (b), carbohydrate amounts in AELs by the HSQC (c) and methanolysis (d) methods. | ||
The yield of AEL also increased in the whole severity range and reached a very high value of ca. 110% at a P-factor of 2500 (Fig. 3a). Correspondingly, the amount of residual lignin (RL) in the extracted solids decreased reaching the minimum at P = 2000. The amount of dissolved lignin in the hydrolysate (SL) was rather low, about 10% of the original biomass lignin, and was independent of the process severity.
As expected, glucan (as cellulose) was very stable; its retention in the solids was above 90% of the original glucan content over the entire severity range. The glucan content in the extracted solids increased from 60 to 80% due to xylan and lignin removal (Fig. 3b). The lowest lignin content in the extracted solids was 6.7% at P = 2000. These solids also contained a low amount of xylan (ca. 4%) and have a high cellulose content (above 85%). A further increase in the severity resulted in an increase in the residual lignin content (Fig. 3b), probably due to re-polymerization of lignin and/or the contribution of pseudo-lignin.
3.2. Structure of acetone-extractable LCCs
A low HTT severity (P ∼ 400–600) resulted in AELs with the highest carbohydrate content, which was significantly reduced as the severity increased (Fig. 3c and d). A part of the extractable LCC apparently originated from partially cleaved native LCC that enables its extraction with the solvent. Another part of LCC was hypothesized to be formed during HTT. This was in agreement with the highest LCC content at the lowest L/S ratio (AEL-1); the low L/S ratio increased the concentrations of lignin and carbohydrates in the reaction mixture and thus facilitated the formation of new LCC linkages. An increase in the L/S ratio resulted in the degradation of LCC linkages (Fig. 2c and d), likely due to a higher amount of water in the reaction system shifting the competition between the hydrolysis and formation of LCC to the former. Eventually, the amount of carbohydrates in AELs decreased with increasing severity (Fig. 3c and d) due to the degradation of LCC linkages as well as carbohydrate (mainly Xyl) moieties in LCCs themselves.The main carbohydrate component in the LCCs was xylan (Fig. SI4 and Table SI1†). However, the share of Xyl and uronic acids (UA) in the total carbohydrate content decreased (from 69 to 42% and from 20 to 12%, correspondingly) while the share of the minor carbohydrates (called “others”) correspondingly increased from ca. 11 to 46%. This again illustrates the cleavage of xylan chains (with associated UA) in LCCs with increasing process severity. The average carbohydrate chain length can be calculated from the ratio of the terminal carbohydrates to the total carbohydrates. The results showed (Table SI2†) that the extracted LCCs were of a rather special type having a short (2.3–5 units) average carbohydrate chain attached to lignin. We expect that the amounts and composition of carbohydrates and the carbohydrate chain length are among significant factors determining LCC properties.
Assuming that each carbohydrate chain was attached to lignin by only one linkage,57 the total amount of LCC linkages could be evaluated by the amount of terminal carbohydrates (reducing end units) plus phenyl glycoside (PhGly) moieties. The amount of γ-ester LCC linkages was usually determined by the corresponding γ-CH2 resonance at ca. 62–65/4–4.5 ppm.51 However, we realized that when the amount of these moieties is not great, this signal was overestimated due to partial overlap with a large resonance of other lignin γ-CH2 signals (Fig. 4 and Table SI2†). Therefore, in this study, the amounts of γ-ester LCC linkages were quantified from the CH-1 signal of the esterified uronic acid (GlcUE) at ca. 101.1/4.65 ppm.50 Of note is that there was a reasonable correlation between the amount of uronozyl moieties (sum of esterified and free COOH) quantified by the methanolysis and HSQC methods. Benzyl ether (BE) LCC moieties resonate at ca. 80–81/4.5–4.8 ppm (primary carbohydrate OH, BE1)48,50 and ca. 80–81/4.9–5.1 ppm (secondary carbohydrate OH, BE2).51 The amount of the latter in AELs was negligible, whereas the signals of BE1 were rather significant (Fig. 4). However, lignin–lignin benzyl ether Alk-O-Alk moieties also contribute to this region58 and therefore, the resonance in this area should be assigned to the total BE moieties (both LCC and lignin ones). The amount of the total BE was rather stable (ca. 4/100 Ar) through the whole severity range (Table SI2†) and did not show any correlation with the amount of carbohydrates in AELs. This implied that the contribution of lignin–lignin ethers in BE resonance was significant, and it was not reliable to evaluate the amount of BE LCC linkages separately from the HSQC spectra. However, the maximal possible amounts of these linkages (BE1max) can be evaluated by the amount of minor carbohydrate units that may form BE1 LCC linkages, i.e., arabinose, mannose, galactose and glucose units quantified by the methanolysis method (Table SI1†).
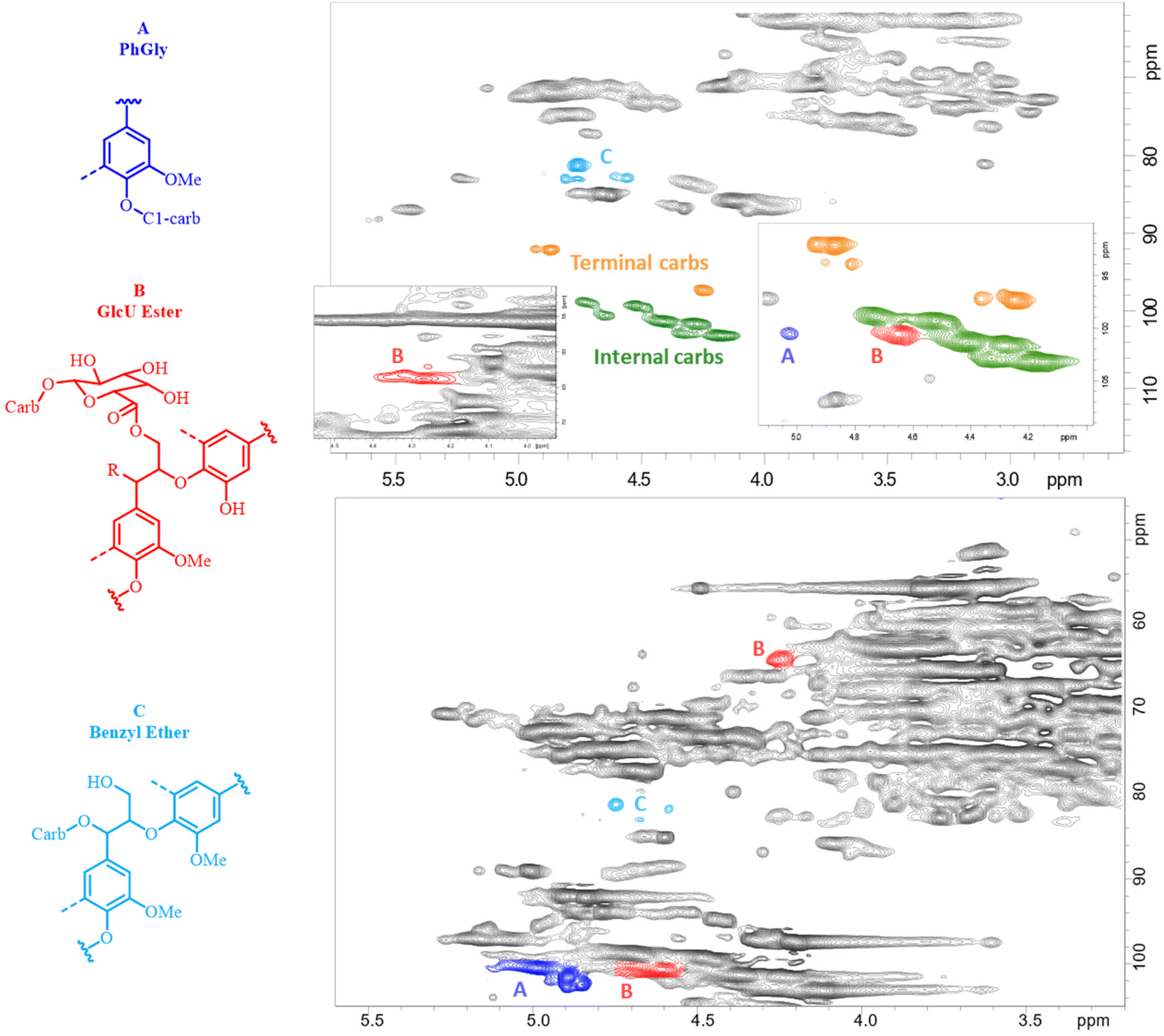 | ||
| Fig. 4 2D HSQC NMR spectra of AEL-500 (top) and H500 (bottom). The colored signals indicate the LCC moieties of the main interest quantified by the resonances of these signals. | ||
Of note is that the sum of ether (even as a maximum, BE1max) and ester LCC linkages was significantly less than the total LCC linkages (excluded PhGly) quantified as the amount of terminal carbohydrates units (Fig. 3). This implied the presence of LCC linkages of unidentified types; e.g., a possibility of semi-acetal and acetal LCC linkages between carbonyl groups of lignin and OH groups of carbohydrates could not be excluded. In addition, the amounts of carbohydrates determined by the methanolysis were always somewhat lower than that by the NMR method (Tables SI1 and SI2†) indicating that certain linkages between monomeric units are stable under acidic methanolysis conditions. The GC chromatograms of the methanolysis products showed significant amounts of non-identified components at retention times higher than those for carbohydrate monomers (Fig. SI5†). These suggested a possibility of the presence of non-hydrolysable (methanolysis-resisted) LCC moieties, e.g., those of C–C linkage types. However, further studies are needed to elucidate new potential types of LCC linkages.
The amounts of LCC linkages of all types decreased with increases in P-factor and L/S ratio (Fig. 2 and 3). A similar tendency was observed for acetyl (Ac) groups, likely from acetylated xylan units. The average carbohydrate chain length (DP) decreased from 4–5 at lower HTT severity to ca. 2.5 at higher severity (P-factor >1500) (Table SI2†).
3.3. Properties of acetone-extractable LCCs (AEL)
The molar mass data showed (Fig. 5 and Table SI3†) that the L/S ratio did not have a significant effect on the weight-average molar mass (Mw) except the experiment at the L/S ratio of 2 when Mw of AEL was about 2.5 times higher (Fig. 5a). Mw dramatically increased, more than 15 times, with the increase in the P-factor from 500 to 2500 following an exponential trendline (Fig. 5b). The molar mass dispersity (ĐM) of AELs also significantly increased with increasing P-factor as well as with increasing L/S ratio from 1 to 2 (at P-factor = 500) (Fig. SI6 and Table SI3†). Furthermore, positive correlations between Mw and the AEL yields were found (Fig. SI7†). This tendency was very interesting and indicated that higher AEL yields caused by better solubility of AEL were due to the chemical structure despite the significantly higher molar mass of AEL. In particular, the higher solubility of AEL in the aqueous organic solvents can be due to a partial cleavage of LCC linkages resulting in further extraction of AEL with lower carbohydrate contents as the solubility of lignin in acetone is much higher than that of carbohydrates.
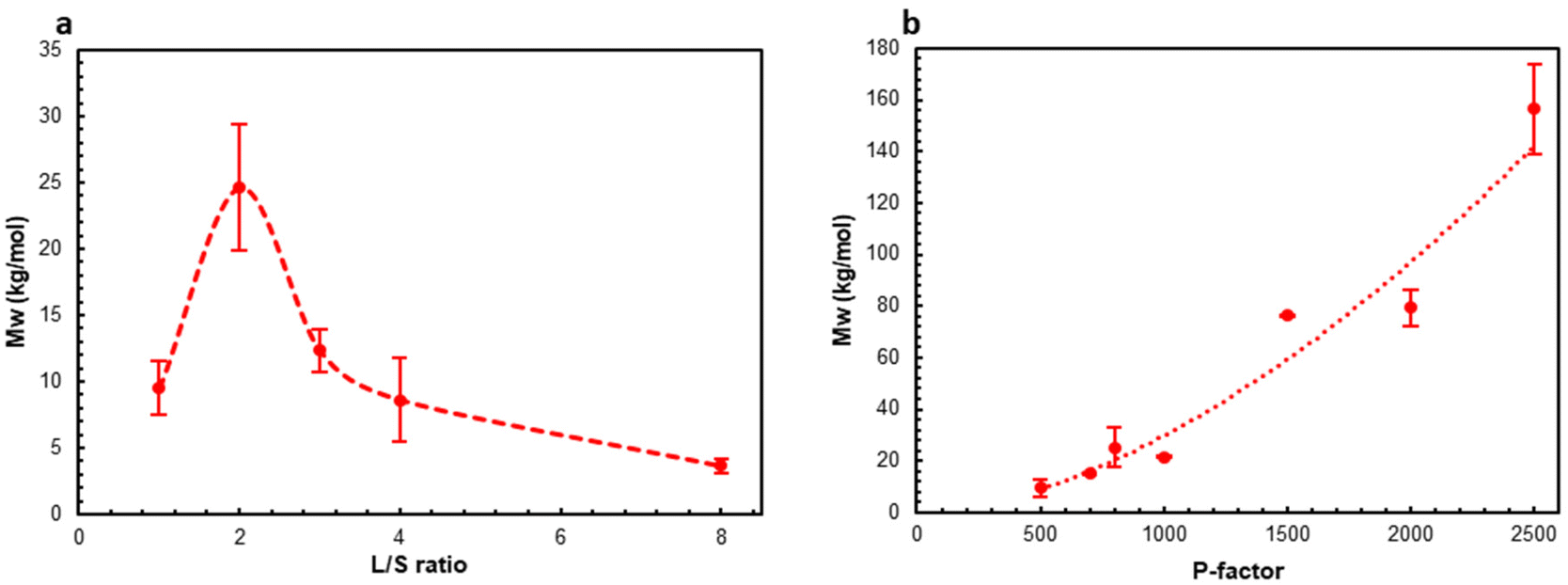 | ||
| Fig. 5 The effect of L/S ratio (a) and P-factor (b) on the molar mass (Mw) of AELs. | ||
Even though low molar mass lignins with low dispersity are often targeted for various material applications, such as thermoplastic or thermosetting polymer blends,59,60 this narrows the window of opportunity for lignin valorization, which is highly biased by the nature of synthetic polymers produced in reactors under highly controlled conditions rather than in very variable environments (biosynthesis of biopolymers in plants). The scope of application needs to be widened to a more flexible lignin property space to enable cost-effective and hence industrially scalable lignin valorization. There are examples where lignins with variable characteristics are successfully used in plastics, such as hydroxyalkylated lignin derivatives with a wide range of molar masses (Mw = 2000–50000 g mol−1) and dispersities (2.5–25).61 The AELs produced by the facile AqSO process exhibit a similar range of Mws and dispersity without further modification reactions. Of special interest is the fact that the low molar mass LCCs produced at low P-factors incorporate a substantial amount of “hydroxyalkylated” groups due to carbohydrates. Thus, these highly functional LCC substrates can open the bottleneck to environmentally benign bio-based plastics potentially without the need for further modification reactions. Other fields of application, in which the variable molar mass and ĐM have been appreciated, are micro-/nanoparticles for dispersants, emulsions, colloids, coatings, paints and adhesives.62
The Tg of AELs changed in a rather wide range of about 110–150 °C, depending on the reaction conditions (Fig. 6 and Table SI4†). This was in the range of two different technical lignin standards, indulin (Tg = 143 °C) and Alcell (Tg = 91 °C), indicating the possibility to produce a target value with the same process by simply changing the process parameters.
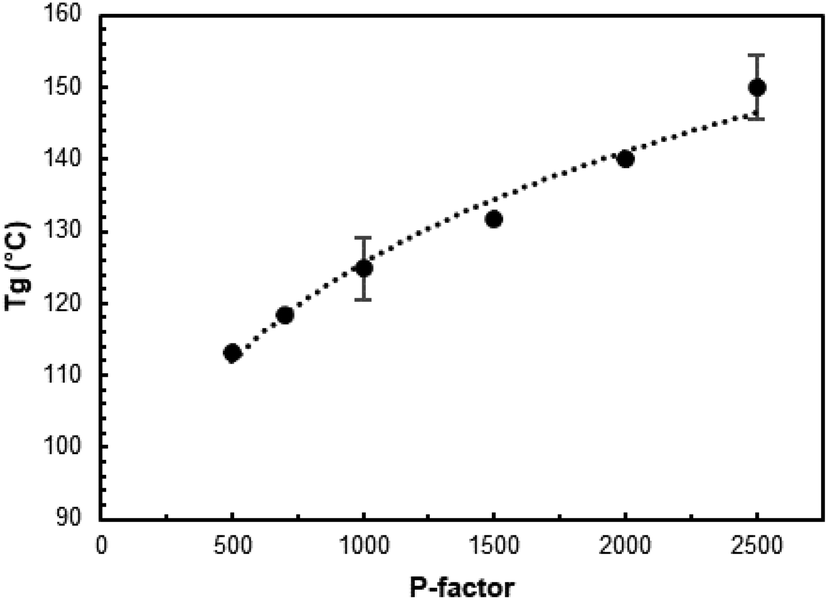 | ||
| Fig. 6 The effect of the P-factor on the Tg of AELs. | ||
| Sample | Surface tension (mN m−1) |
|---|---|
| AEL-500 (L/S = 1) | 62.6 ± 1.17 |
| AEL-8 (P = 500) | 60.9 ± 0.25 |
| AEL-2000 | 67.3 ± 0.05 |
| BCEL | 59.7 ± 0.08 |
| PLCC | 69.9 ± 0.08 |
| Alcell | 60.3 ± 0.05 |
| Indulin AT | 65.0 ± 0.03 |
3.4. Hydrolysate composition
The focus of this research was on the solvent extracted LCCs (AELs) as a new process stream. The composition of HTT hydrolysate was addressed in previous publications and therefore little attention was paid to it in the current research. However, general information on the main hydrolysate products would be useful to see the whole picture of the overall biorefinery approach. In addition, our process was carried out at much lower L/S ratio than most of the previous studies and comparison with literature data would be useful to indicate potential differences.The results showed that the hydrolysate produced at moderate severity (H500) was suitable for oligo-xylan production; it also contained some xylose (monomers) while the amounts of the degradation products were rather minor (Fig. 7). In particular, only 17% of the original acetyl groups were released under these conditions and therefore the majority of xylan units still possessed their acetylation pattern. In contrast, xylan was almost completely (ca. 90%) degraded to furfural and products of its further transformation (e.g., polyfurans) at high process severity (P = 2500) and the hydrolysate (H2500) was more appropriate for furfural and acetic acid production. The amounts of glucan and its degradation products in the hydrolysates were very minor, as about 90% of glucan remained in the solids. Of note is that the amount of non-identified products was rather negligeable in H500 (Fig. 7a and b), but significant in H2500 representing ca. 40% of dissolved biomass (Fig. 7a) or above 50% of the original xylan (Fig. 7c). They likely belong to various polyfuran compounds, being in good agreement with the amount of pseudo-lignin reported above. Even though pseudo-lignin (also called “humins”) formation was often considered as a negative phenomenon, some literature reported the suitability of humins for potential high-value applications, such as activated carbon or adhesives.3,66,67
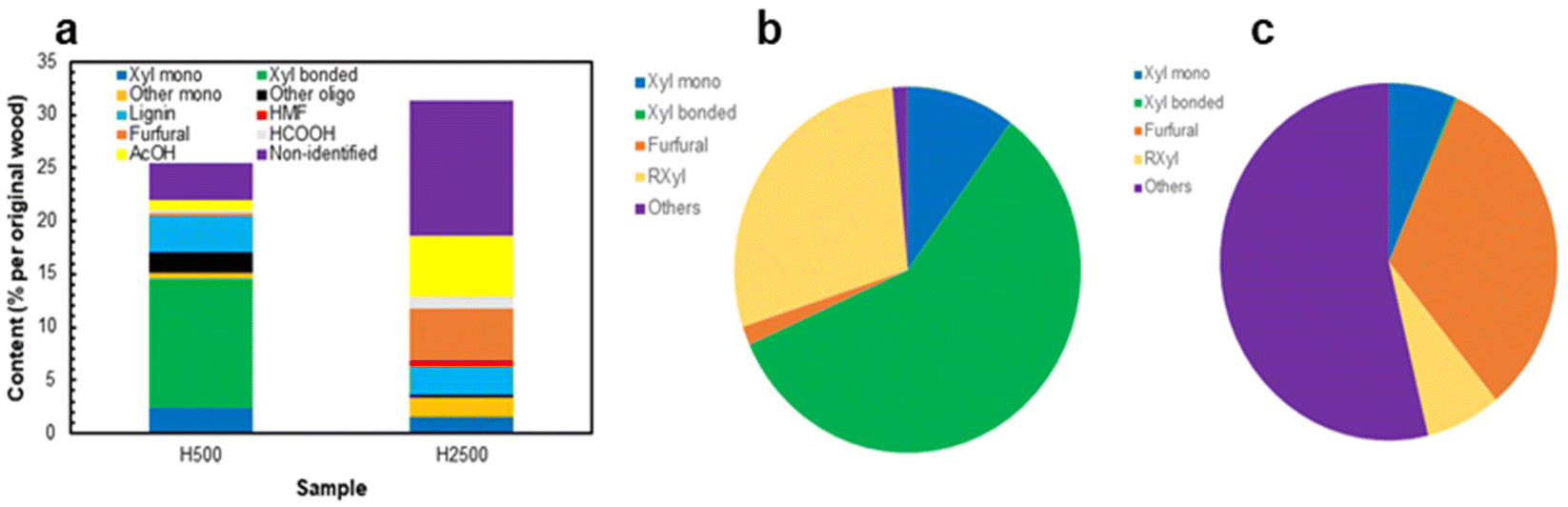 | ||
| Fig. 7 Composition of hydrolysates (a); overall xylan balance (mol%) in HTT experiments at P = 500 (b) and P = 2500 (c). | ||
Carbohydrates in the hydrolysate were accessed by two methods: direct analysis of monosaccharides in the hydrolysate as is and the analysis of monosaccharides released after mild acid hydrolysis of the samples. The latter gave the sum of free mono- and originally “bonded” carbohydrates, such as free oligosaccharides and carbohydrates involved in LCC (via hydrolysable linkages). Importantly, the amount of “bonded” carbohydrates in H500 was an order of magnitude higher than the values obtained by a direct analysis of free oligosaccharides (with DP = 2–5) by GC using a short column (Tables SI6–SI8†). This implied that a high portion (above 70% by mass) of carbohydrates in H500 was linked to lignin. On the other hand, a very high portion of lignin monomeric units linked to carbohydrates was determined by the HSQC method (Fig. 4, bottom). The amounts of γ-esters (GlcE), BE and PhGly LCC linkages were about 18, 11 and 47 per 100 Ar, correspondingly, i.e. 76/100 Ar in total. The evaluated amount of LCC (by mass % and the frequency of LCC linkages) was much higher than that reported in the literature for HTT hydrolysates produced under different conditions (higher L/S ratio, lower temperature)25,38,68 and once again confirmed our suggestion on the excessive formation of LCC linkages in different process steams under AqSO process conditions. At the same time, further validation of the HSQC analysis of LCC in hydrolysates (which composition and origin is very different from more traditional LCC preparations we have studied with the quantitative HSQC method23,51 by now) is needed to confirm these values, that should be considered as relative so far.
The amount of water-soluble lignin (including lignin domain in LCC) in the hydrolysates was rather stable (ca. 10% of the original lignin or about 2.7% per biomass) at different process severities at L/S = 1 (Fig. 3a) and significantly increased (to about 5.8% per biomass) when the L/S ratio increased (Fig. 2a). Of note is that the amount of lignin in the hydrolysate in our experiments at L/S = 1 was about 3.7 times less than that reported earlier.69 This was beneficial for the corresponding increase in the yield of the target AELs. More importantly, the significantly lower amounts of lignin in our hydrolysates should mitigate a problem related to lignin precipitation on equipment, which is a very serious obstacle in hydrolysate industrial processing.70,71
Comparison of our results with literature data indicated that the yield of xylan (especially oligo-) in H500 was about 1.5–1.8 times higher than that under similar severity, but at a higher L/S ratio and lower temperature,69,72 which is an additional advantage of the AqSO process.
3.5. Production of colloidal nanoparticles from the AELs
Lignin nanoparticles (LNPs) have emerged as a rapidly growing field in lignin research due to their easy preparation and uniform spherical shape, which offer benefits over structurally complex technical lignins.62,73,74 Such particles are negatively charged in water dispersions, owing to the presence of hydrophilic groups on their surface and have been found to be relevant building blocks in many applications ranging from biomedicine to composites.75–78 However, to our knowledge there are no previous attempts related to the production of colloidal nanoparticles from LCCs. Therefore, we evaluated the possibility to produce such colloidal nanoparticles and elucidate their basic properties and potential applications.The preparation of LNPs from the AELs, (hereafter called LCCNPs) was performed via the solvent-exchange methodology adapted from a previous work.55 The particles were produced by rapid pouring of water into the LCC solution in aqueous acetone. In all cases, colloidally stable LCCNPs could be obtained from all AELs regardless of the HTT process conditions (Table 3 and Fig. SI9†). In contrast, these particles cannot be produced from the hydrolysate LCCs (e.g., H500) naturally due to their solubility in water and therefore this route is possible for AELs only, but not for earlier studied LCC samples.26,27,30,37,40
| Samples | Diameter (nm) | PDI | Zeta potential (mV) | Native pH |
|---|---|---|---|---|
| a At least three measurements were completed for each parameter. Error ranges correspond to one standard deviation. | ||||
| AEL500 | 180 ± 6.7 | 0.127 ± 0.010 | −34.5 ± 0.9 | 5.5 |
| AEL600 | 128 ± 1.3 | 0.137 ± 0.021 | −26.4 ± 0.8 | 5.5 |
| AEL700 | 109 ± 1.5 | 0.075 ± 0.010 | −25.6 ± 0.9 | 5.7 |
| AEL1000 | 108 ± 1.5 | 0.129 ± 0.085 | −33.4 ± 0.6 | 5.9 |
| AEL2000 | 94 ± 1.2 | 0.128 ± 0.025 | −37.8 ± 1.2 | 5.5 |
| BCEL | 89 ± 2.7 | 0.153 ± 0.092 | −38.9 ± 1.8 | 6.1 |
| PLCC | 155 ± 3.2 | 0.101 ± 0.091 | −27.4 ± 1.3 | 5.6 |
Dynamic light scattering (DLS) results revealed a clear trend, where the particle size decreased with increasing P-factor, which is most probably attributed to the amount of carbohydrates in LCCs, frequency of lignin–carbohydrate linkages and differences in the molecular weights of the carbohydrate moieties that change the hydrophilic/hydrophobic balance of the different samples (Table 3). Native LCC preparations (BCEL and PLCC) also fit this tendency. However, no simple correlation between LCC characteristics and zeta potential was found.
Scanning electron microscopy (SEM) micrographs also confirmed the formation of spherical LCCNPs from different AELs and native LCCs (Fig. 8a and b). Interestingly, two main populations of spherical particles (200 nm and 90 nm) could be appreciated in AEL-500, while only one main population of spherical particles (90 nm) was observed in AEL-1000. This was also in agreement with DLS measurements (Table 3). For the native LCCs (PLCC and BCEL), spherical particles could also be observed (Fig. 8c and d). However, most of these particles were agglomerated, and only a few individual particles could be identified (highlighted in red sections). This high tendency to agglomerate upon drying may be related to a high concentration of carbohydrate chains in PLCC, which would act as a bridging point and promote the interaction between different particles.
 | ||
| Fig. 8 SEM micrographs of LCCNPs from (a) AEL-500, (b) AEL-1000, (c) PLCC and (d) BCEL. Red marked sections highlight the presence of individual particles in samples with high tendency to form particle agglomeration. | ||
Among different potential applications of LNPs, their use as stabilizers for oil–water Pickering emulsions is very attractive due to not only the generation of durably stable emulsions,28,79 but also the possibility to be used as a vessel to polymerize hydrophobic monomers and generate latex dispersions that allow the preparation of composites with improved properties and favorable carbon footprint.27,56 In this context, the behavior of LCCNPs for the stabilization and radical polymerization of tetrahydrofurfuryl methacrylate (THFMA)-based Pickering emulsions was investigated (Fig. 9a). THFMA was selected as the monomer due to its bio-sourced origin, hydrophobic nature and potential to replace petroleum-sourced monomers such as butyl methacrylate.80 LCCNPs from AEL-500 and PLCC were selected as model particles due to their different tendency for interfacial agglomeration (Fig. 9, compare a and c).
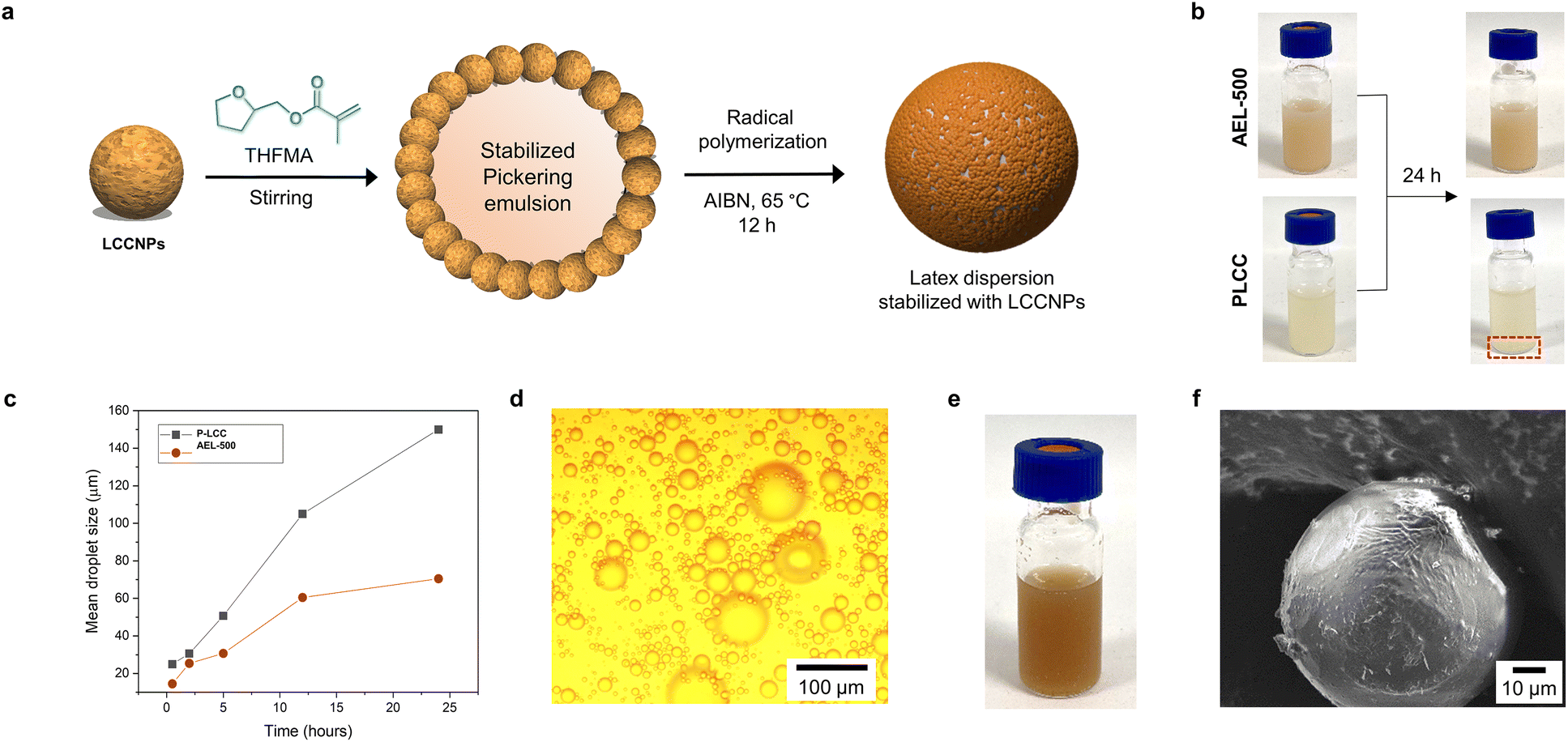 | ||
| Fig. 9 (a) Schematic diagram of the use of LCCNPs as surfactants in the Pickering emulsion polymerization process. (b) Digital images of THFMA-Pickering emulsions stabilized with LCCNPs from AEL-500 and PLCC at different times. (c) Time evolution of mean droplet size of THFMA-Pickering emulsions stabilized with PLCC NPs and AEL-500 NPs. (d) Optical microscopy and (e) digital images of latex dispersion after polymerization process. (f) A SEM image of the P-LCC NPs-coated poly(THFMA) microbeads after free radical polymerization at 65 °C. | ||
The Pickering emulsions were formed by gradually adding THFMA to a water suspension of LCCNPs (9 g L−1) under vigorous stirring (final o/w ratio 10/90 v/v), following a previously described methodology for the preparation of styrene-based Pickering emulsions using hybrid LNPs.57 After immediate preparation, THFMA-Pickering emulsions were stable regardless of the LCCNPs employed (Fig. 9b). However, time-dependent visual inspection and mean droplet size measurements by optical microscopy revealed partial THFMA migration from the emulsion phase (Fig. 9b) and a significant increase in emulsion droplet size at longer times (Fig. 9c and Fig. SI10†). This coalescence process occurs due to the inefficient coverage of the oil–water interface with the particles. Here, it is important to note that THFMA-Pickering emulsions stabilized with LCCNPs from AEL-500 were found to be significantly more stable in contrast to those obtained from PLCC (smaller droplet size and no visual phase separation, compare Fig. 9b and c). This fact could be associated with the high tendency for PLCC to form aggregates as mentioned above, which most probably promoted a destabilization of the emulsion, and consequently a lower concentration of particles was available to cover the emulsion droplet.
Therefore, free radical polymerization (FRP) of THFMA-based Pickering emulsions using LCCNPs from AEL-500 and AIBN as a thermal initiator at 65 °C was studied (Fig. 9a). Stable latex dispersions were obtained from the polymerization (Fig. 9e). SEM and optical light microscopy methods revealed uniform spherical polymeric microbeads with a similar size to that of the initial monomer droplets (Fig. 9d and f). The microscopy images clearly showed that the poly(THFMA) microspheres contained a thin amorphous layer of LCCNPs (Fig. 9f), which indicate that LCCNPs can enable the polymerization process in aqueous media. Thus, AqSO LCCNPs have the potential to be used as building blocks to produce bio-based composites, similarly to recently reported studies on LNPs coated with chitosan and glucose oxidase.56 Importantly, the best AEL preparation was found and its properties were more suitable than those of reference native LCCs.
3.6. Application of lignocellulosic hybrids in wood adhesives
Replacement of phenol-formaldehyde (PF) resins, petrochemical-based products derived from unsustainable fossil resources using toxic formaldehyde and phenol in their formulations, with sustainable and green lignocellulosic precursors is of great importance. Recent studies17,81,82 showed the good potential of lignocellulosic hybrids (LCH) produced by a biorefinery process based on supercritical water hydrolysis to replace PF adhesives. It was speculated that other biorefinery LCH can perform similarly. Therefore, it was of interest to evaluate the performance of AqSO LCH in this application. For this purpose, HTT solids were tested for replacement of a PF resin used for wood adhesives.In this initial trial, 30% of PF was replaced by the non-extracted HTT solids produced at P = 2000. The results showed that the bonding strength of the LCH-PF blend was very close to that of the control PF resin (Fig. 10) indicating good potential of using AqSO LCH in replacing PF wood adhesive. Importantly, not-optimized AqSO LCH showed significantly better performance as compared to traditional technical lignins, such as Indulin (Fig. 10) and others reported earlier.3 Following studies are expected to further optimize the substrate for this application.
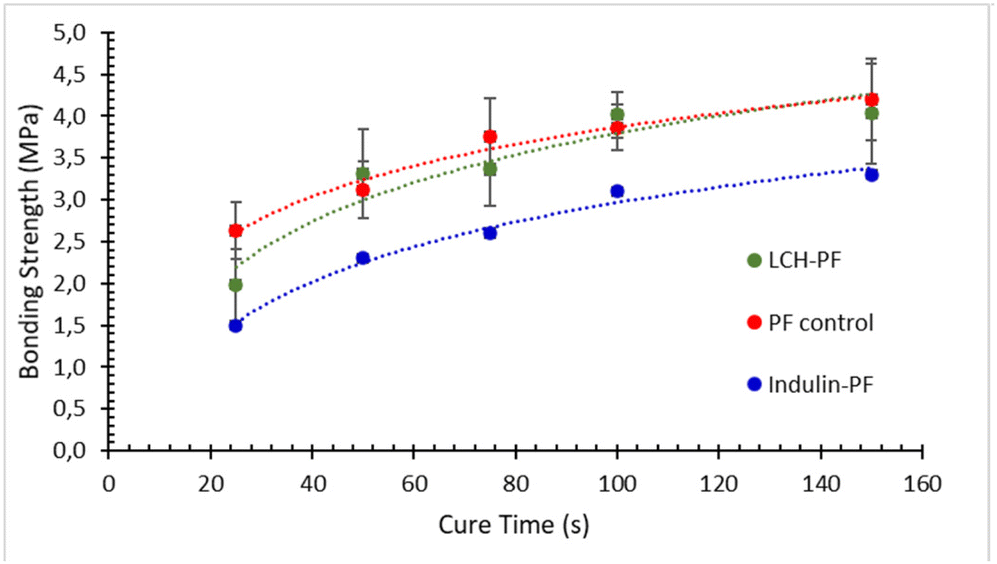 | ||
| Fig. 10 Adhesive performance of an LCH-PF blend (3:7, w/w) vs. the control PF resin and indulin-PF blend by ABES test; non-extracted HTT solids produced at P = 2000 were used as LCH. | ||
Of note is that the LCH used in the ABES test contains a significant amount (ca. 40%) of pseudo-lignin (as polyfurans), the presence of which is often considered negative. However, our results confirm the earlier suggestion3 that polyfurans incorporated into lignin may play a positive role in certain applications. It can be speculated that the incorporated polyfurans possessing potential crosslinking centers, such as aldehyde groups and/or double bonds, facilitate resin curing during hot pressing.
Importantly, 30% of the total PF resin replacement corresponds, by the quantity of lignin used, to a situation when ca. 70% phenol is replaced with lignin during resin formulation (as the phenol share in the PF resin is about 40% by dry weight).3 In addition, the logistics of the direct PF resin replacement is much simpler compared to inclusion of lignin in the resin formulation. Furthermore, the price of PF resin is somewhat higher than that of phenol.8 Pilot experiments showed that, in addition of PF resin, LCHs successfully replace (100%) a set of four fillers used in plywood manufacture.82 Thus, this way of LCH valorization is very attractive from a practical point of view.
4. Outlook
Herein we present a general overview of the AqSO process in light of an advanced integrated biorefinery and discussed various scenarios for best utilization of the entire biomass as well as pros and cons of this biorefinery approach (Fig. 11).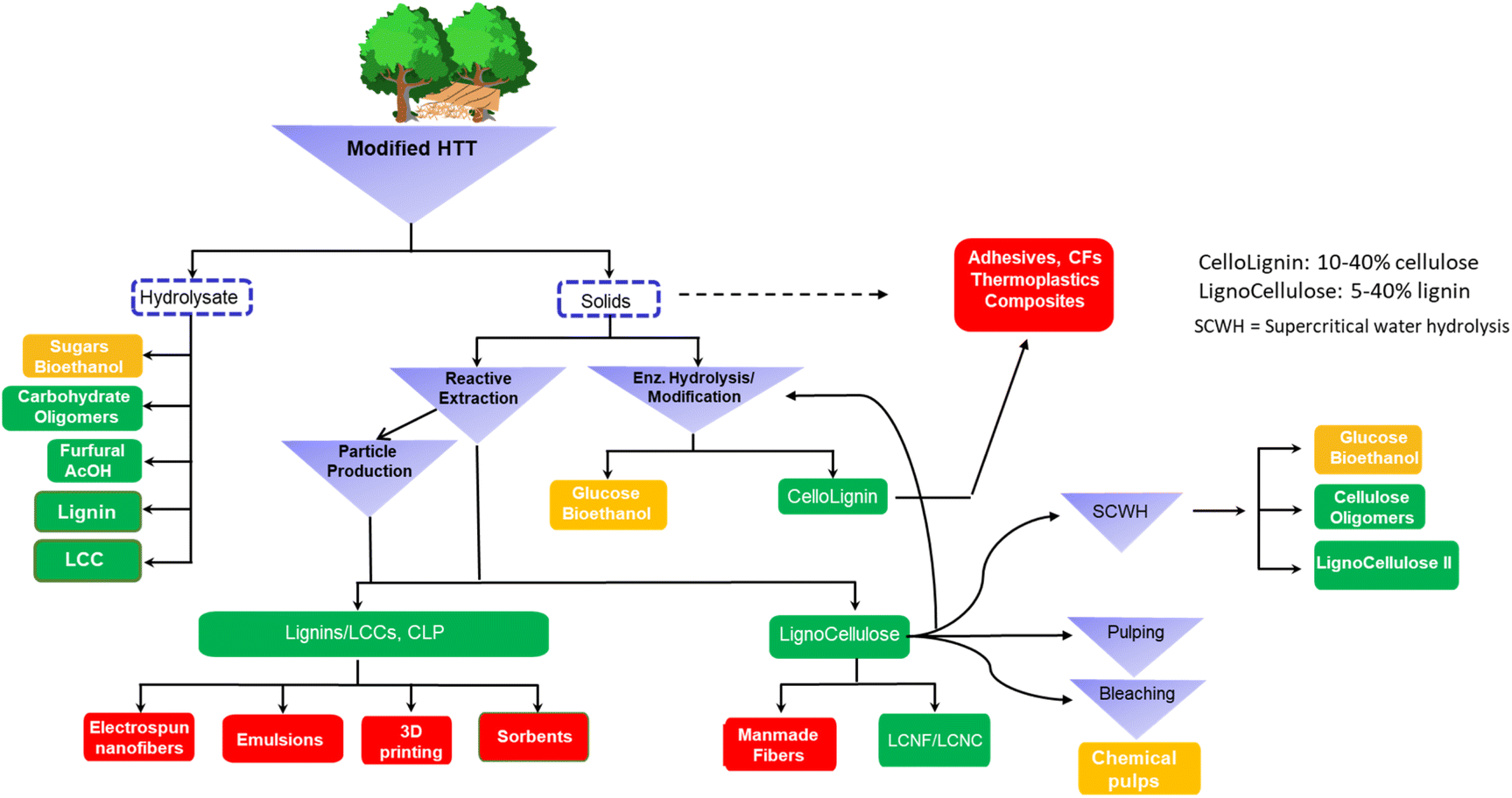 | ||
| Fig. 11 Examples of different scenarios for the AqSO process as an advanced integrated biorefinery. Orange boxes show the traditional biorefinery products while green ones indicate additional valuable AqSO biorefinery streams and their potential applications (red). | ||
The AqSO biorefinery provides three major streams, namely hydrolysate (mainly hemicellulose-derived products), solvent-extracted lignin/LCC (AEL) and cellulose-rich fiber (lignocellulose). Additional streams are possible under more advanced scenarios, e.g., adding enzymatic hydrolysis/modification to the overall process. Although the process can provide rather efficient fractionation of wood components producing high purity products, it is of higher interest to explore the use of ligno–carbohydrate hybrids (LCH), such as cellolignins and lignocelluloses (Fig. 11) in high-value applications where they have better performance than pure lignin and carbohydrates.3 For example, a proof of concept for the use of AqSO LCH in replacing PF wood adhesives was demonstrated in this work. The unnecessity of deep fractionation of the wood components would also simplify the process and reduce its production costs. Furthermore, this option gives us a greater degree of freedom in product engineering.
In addition to cellolignins and lignocelluloses, our studies showed the possibility to produce LCC-type precursors as a new valuable process stream. This finding is of great importance as, despite the great interest in potential LCC applications, there is almost no process to manufacture industrially relevant LCC precursors at high yields yet; our discovery creates this opportunity. The produced LCCs possess special characteristics (e.g., higher share of lignin content, short carbohydrate chains) that should allow this LCC type to find an optimal niche in the application roadmap. One of them is LCCNP production, for the first time, directly from the process stream and their application in specialty products, such as Pickering emulsion stabilizers that enable free radical polymerization reactions.
The hydrolysate can be traditionally used for sugars/bioethanol or/and preferably further fractionated to isolate specific useful products, such as oligosaccharides,4,5 lignin and LCC or used as is for high-value applications, e.g. surfactants.83 These possibilities have been already described in the literature and therefore were not studied for the AqSO process yet.
Lignocellulose (e.g., extracted solids with a low lignin content) has the potential to be a suitable precursor for Ioncell textile fiber production3 based on earlier studies with unbleached Kraft pulps.84,85 Further optimization of the properties of the solids to the application requirements is likely possible by manipulating the process variables. On the other hand, solids with a higher lignin content should be suitable for Ioncell CF production.3,86 Importantly, a high concentration of LCC linkages expected in the substrate can be critical (i). to avoid phase separation between cellulose and lignin and (ii). to prevent lignin leaching in the coagulation bath and thus facilitate the recovery of ionic liquid used in the Ioncell process. In addition, lignocelluloses can be used for the production of lignin-containing LCNF/LCNC3,19,22 or/and processed through supercritical water hydrolysis biorefinery, pulping and/or bleaching.
In addition to new value chains, the AqSO biorefinery can be easily incorporated into the current biorefinery scenarios as an added-value stream. Analysis of literature data allows specifying the following optimal severity ranges in different HTT current scenarios
1. Prehydrolysis for paper grade pulp: P < 500 (ref. 87)
2. Prehydrolysis for dissolving grade pulps: P = 500–1000 (ref. 10)
3. Pretreatment for the enzymatic hydrolysis of cellulose to glucose: P > 1000 (P 2500 optimal).88,89
Based on the literature reports and our results, two apparent optimal ranges can be suggested for the AqSO biorefinery (Fig. 11):
• A low severity process (P-factor of 400–800), at a low L/S ratio would be optimal for LCC engineering in the AEL stream and, likely, for the extraction of oligomeric hemicelluloses (hydrolysate stream).90 The extracted solids could be further used for dissolved grade pulp production and even for paper grade pulps (at a low HTT severity), providing these traditional pulping processes with additional revenue stream(s) without any significant modifications required. Alternatively, the HTT solids (extracted or not) can be used for Ioncell CF production.3
• A high severity process (P-factor of around 2000) seems optimal for the production of high yield pure lignins and cellulosic fibers with low lignin and xylan contents. The hydrolysate can be used for furfural and acetic acid production and for humin valorization3,66,67 (along with dissolved lignin). As the lignin content in the residual extracted solids is very low, the solids can be directly used for Ioncell textile fibers3 or further delignified (e.g., with the current oxygen process) and/or bleached to fit the specifications for current cellulosic products. Importantly, the expensive pulping process can be thus omitted.
Enzymatic hydrolysis or/and different types of enzymatic modifications of the solids can also be used to optimize the produced LCHs (e.g., their lignin:cellulose ratio, lignin and cellulose characteristics optimal for CFs, PF resins etc.) and to produce glucose as a co-product. It is important to highlight that, in contrast to the traditional biorefinery, the application of enzymes will be focused on the LCH engineering rather than on the glucose yield. Thus, a high variety of process scenarios by flexible combinations of different process elements (not limited to the examples above) allows for considering the suggested approach as rather universal (i.e., Omni).
As our goal is to use the AqSO biorefinery streams as precursors for high-value applications, the possibility of their optimization for specific applications is very important. These studies showed that the AqSO biorefinery allows for a wide range of structure and property characteristics giving a large amount of room for product engineering per required characteristics. Therefore, it can provide LCH precursors for a wide variety of applications: from lignin-based carbons or carbon fibers to lignin containing thermoplastic blends, colloids and surfactants by simply tuning the process conditions without any additional input of chemicals or additives. Such properties as Tg and MWD can be further tuned by post-processing, e.g., solvent fractionation.3,91 Moreover, the fractionation can be coupled with reactive extraction of different types to increase the lignin performance by introduction of favorable functionalities, such as alkyl or acyl groups.92 This high flexibility of the AqSO process is a big advantage over the current industrial processes (e.g., Kraft pulping) that have very limited room for process variation and therefore for product engineering.
The AqSO biorefinery has some specific process-related advantages. The process is free from toxic chemicals and therefore is green by definition. A variety of different solvent mixtures to extract lignin/LCC can be used. For example, our preliminary experiments showed that ethanol could be used for lignin/LCC extraction; the yield of the LCC extracted with 70% ethanol from the solids produced at P = 500 was only slightly lower (ca. 20%, relative) than that of AEL and the LCC content was about the same. Thus, from a practical point of view, there would be no need for an external solvent as ethanol is a typical biorefinery product. Alternatively, diluted (e.g., 1%) NaOH can be used for LCC/lignin extraction; this can be easily incorporated into the existing Kraft mill process.
The extraction of AELs from HTT solids under ambient conditions is very easy in strict contrast to other biorefinery lignins where solid/liquid separation (e.g., by filtration) is very challenging. The easy processibility should also minimize the amount of solvent needed, e.g., via countercurrent extraction in a column. Alternatively, as low lignin/LCC concentration is needed for LCCNP production42,62 and the extraction solution is used directly, a larger amount of solvent would not be a problem and might be helpful in AEL extraction. The low L/S ratio in the HTT stage results in much lower energy requirements for HTT heating, distillation to recover hydrolysate products, etc. than the traditional HTT processes.
It is important to mention that, in spite of the use of an organic solvent, the AqSO process is very different from the common organosolv pulping, both from fundamental and engineering points of view. In the AqSO biorefinery all chemical reactions occur during HTT, i.e., in the absence of organic solvents, which are used for lignin dissolution after the process without any chemical modification. From the practical point of view, the absence of solvent in the reactor (at high temperature and pressure) mitigates many problems of organosolv delignification associated with the use of high amounts of solvent required for successful pulping,11 high pressure, flammability, etc.
AqSO biorefinery should have a rather different niche vs. various organosolv and new advanced “lignin-first” biorefinery processes.14,15,52 The latter is designed to provide lignins suitable for aromatic monomer production via lignin decomposition and has a clear advantage in this area. However, the AqSO biorefinery is much simpler (and therefore cheaper) than the OS and “lignin-first” processes. Therefore, the application of its products in replacement of commodity petrochemicals of high market sizes would be more economically feasible, while OS and “lignin-first” biorefinery products should be used in specialty applications. Thus, these biorefinery approaches complement each other in expanding the area of sustainable biomass-derived products rather than competing for the same market.
5. Conclusions
A green, simple, and flexible biorefinery process for integrated utilization of all biomass components in high-value applications has been suggested. It consists of a modified HTT treatment of wood biomass followed by solvent extraction of LCC from the resulting solids under ambient conditions and therefore named AquaSolve Omni (AqSO) Biorefinery. Although the process allows for the production of high purity lignins in high yields, ligno–carbohydrate hybrids (LCH) are of specific interest. In particular, the AqSO biorefinery produces, under specific conditions (moderate severity, low L/S ratio), LCCs of different types in high yields as a very valuable process stream. Thus, the AqSO biorefinery allows for the fractionation of biomass into 3 process streams: hemicellulose-derived products in the hydrolysate, solvent extracted LCCs and cellulose-rich solids.First examples of high-value applications of AqSO products were reported. LCC nanoparticles were produced for the first time directly from the solvent extract. The LCCNPs had unique properties and showed good application potential, e.g., in stabilization of THFMA-Pickering emulsions for the corresponding polymer production. In addition, AqSO LCHs showed promising results in the direct replacement of PF resins used in wood adhesives. The cellulose-rich solids with low lignin content (<7%) can be a good precursor for Ioncell textile fiber production.
The high flexibility of the process enables engineering of the resulting products in a wide range of chemical compositions, structures and properties and therefore allows a large space to optimize the products for specific high-value applications.
Conflicts of interest
No conflict of interest is declared.Acknowledgements
D. T. and D. R. gratefully acknowledge the support from the Academy of Finland through the FinnCERES BioEconomy flagship. M. H. S. acknowledges financing from The Swedish Research Council (grant number 2020-03752). L. Wang would like to acknowledge the China Scholarship Council (Student ID 201804910639) as well as the Business Finland Project (43674/31/2020) for their funds to her doctoral study at Åbo Akademi University. Both the Aalto University and Åbo Akademi University research teams would like to acknowledge the Academy of Finland (grant 341596) for funding support.Notes and references
- United Nations, The Sustainable Development Goals.
- M. Balakshin and E. Capanema, 18th ISWFPC, Porto Seguro, 2017, pp. 178–182.
- M. Y. Balakshin, E. A. Capanema, I. Sulaeva, P. Schlee, Z. Huang, M. Feng, M. Borghei, O. J. Rojas, A. Potthast and T. Rosenau, ChemSusChem, 2021, 14, 1016–1036 CrossRef CAS PubMed.
- A. Ebringerová, Z. Hromádková and T. Heinze, in Polysaccharides, ed. T. Heinze, Springer-Verlag Berlin Heidelberg, Berlin, 2005, pp. 1–67 Search PubMed.
- S. Willför, K. Sundberg, M. Tenkanen and B. Holmbom, Carbohydr. Polym., 2008, 72, 197–210 CrossRef.
- W. Xu, A. Pranovich, P. Uppstu, X. Wang, D. Kronlund, J. Hemming, H. Öblom, N. Moritz, M. Preis, N. Sandler, S. Willför and C. Xu, Carbohydr. Polym., 2018, 187, 51–58 CrossRef CAS PubMed.
- K. S. Mikkonen, S. Kirjoranta, C. Xu, J. Hemming, A. Pranovich, M. Bhattarai, L. Peltonen, P. Kilpeläinen, N. Maina, M. Tenkanen, M. Lehtonen and S. Willför, Ind. Crops Prod., 2019, 133, 212–220 CrossRef CAS.
- L. Dessbesell, M. Paleologou, M. Leitch, R. Pulkki and C. (Charles) Xu, Renewable Sustainable Energy Rev., 2020, 123, 109768 CrossRef CAS.
- M. Lersch, Creating value from wood – The Borregaard biorefinery sulfite pulping, 2009 Search PubMed.
- H. Sixta, Handbook of Paper and Board, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006 Search PubMed.
- C. Arato, E. K. Pye and G. Gjennestad, Appl. Biochem. Biotechnol., Part A, 2005, 123, 871–882 CrossRef.
- M. Galkin, Curr. Opin. Green Sustainable Chem., 2021, 28, 100438 CrossRef CAS.
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS.
- S. Bertella and J. S. Luterbacher, Trends Chem., 2020, 2, 440–453 CrossRef CAS.
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS.
- A. W. Bartling, M. L. Stone, R. J. Hanes, A. Bhatt, Y. Zhang, M. J. Biddy, R. Davis, J. S. Kruger, N. E. Thornburg, J. S. Luterbacher, R. Rinaldi, J. S. M. Samec, B. F. Sels, Y. Román-Leshkov and G. T. Beckham, Energy Environ. Sci., 2021, 14, 4147–4168 RSC.
- E. A. Capanema and M. Y. Balakshin, Adhesive compositions comprising type-II cellulose, Patent WO 2016/049569Al, 2016.
- M. Y. Balakshin and E. A. Capanema, in 14th European Workshop on Lignocellulosics and Pulp, 2016, pp. 63–66 Search PubMed.
- E. A. Capanema, M. Y. Balakshin, P. D. Fritzgibbon, M. Kosa, T. M. Mclarty and C. S. Sanderson, Cellulose-containing compositions and methods of making the same, Patent WO 2016/049567Al, 2016.
- L. Dehne, C. V. Babarro, B. Saake and K. U. Schwarz, Ind. Crops Prod., 2016, 86, 320–328 CrossRef CAS.
- A. Stücker, F. Schütt, B. Saake and R. Lehnen, Ind. Crops Prod., 2016, 85, 300–308 CrossRef.
- G. N. Rivière, F. Pion, M. Farooq, M. H. Sipponen, H. Koivula, T. Jayabalan, P. Pandard, G. Marlair, X. Liao, S. Baumberger and M. Österberg, Sustainable Mater. Technol., 2021, 28, e00269 CrossRef.
- M. Y. Balakshin, E. A. Capanema and A. Berlin, in Studies in natural products chemistry, ed. Atta-ur-Rahman, Elsevier, 2014, pp. 83–115 Search PubMed.
- D. M. De Carvalho, M. H. Lahtinen, M. Lawoko and K. S. Mikkonen, ACS Sustainable Chem. Eng., 2020, 8, 11795–11804 CrossRef CAS.
- I. Sumerskiy, A. Pranovich, B. Holmbom and S. Willför, J. Wood Chem. Technol., 2015, 35, 387–397 CrossRef CAS.
- D. M. D. Carvalho, M. H. Lahtinen, M. Bhattarai, M. Lawoko and K. S. Mikkonen, Green Chem., 2021, 23, 9084–9098 RSC.
- A. Moreno and M. H. Sipponen, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- T. Zou, M. H. Sipponen and M. Österberg, Front. Chem., 2019, 7, 1–12 CrossRef PubMed.
- M. Lehtonen, M. Merinen, P. O. Kilpeläinen, C. Xu, S. M. Willför and K. S. Mikkonen, J. Colloid Interface Sci., 2018, 512, 536–547 CrossRef CAS PubMed.
- M. H. Lahtinen, F. Valoppi, V. Juntti, S. Heikkinen, P. O. Kilpeläinen, N. H. Maina and K. S. Mikkonen, Front. Chem., 2019, 7, 1–18 CrossRef PubMed.
- H. Sakagami, T. Kushida, T. Oizumi, H. Nakashima and T. Makino, Pharmacol. Ther., 2010, 128, 91–105 CrossRef CAS PubMed.
- H. Sakagami, H. Sheng, N. Okudaira, T. Yasui, H. Wakabayashi, J. Jia, T. Natori, M. Suguro-Kitajima, H. Oizumi and T. Oizumi, In Vivo, 2016, 30, 331–339 CAS.
- H. Zhao, Q. Feng, Y. Xie, J. Li and X. Chen, BioResources, 2017, 12, 8490–8504 CAS.
- H. Zhao, J. Li, P. Wang, S. Zeng and Y. Xie, Int. J. Polym. Sci., 2017, 4915185 Search PubMed.
- C. Huang, S. Tang, W. Zhang, Y. Tao, C. Lai, X. Li and Q. Yong, ACS Sustainable Chem. Eng., 2018, 6, 12522–12531 CrossRef CAS.
- P. Fatehi, W. Gao, Y. Sun and M. Dashtban, Bioresour. Technol., 2016, 218, 518–525 CrossRef CAS PubMed.
- D. Tarasov, M. Leitch and P. Fatehi, Cellulose, 2018, 25, 1377–1393 CrossRef CAS.
- N. Giummarella and M. Lawoko, ACS Sustainable Chem. Eng., 2017, 5, 5156–5165 CrossRef CAS.
- R. H. Narron, H. M. Chang, H. Jameel and S. Park, ACS Sustainable Chem. Eng., 2017, 5, 10763–10771 CrossRef CAS.
- F. Carvalheiro, L. C. Duarte and F. M. Gírio, J. Sci. Ind. Res., 2008, 67, 849–864 CAS.
- H. Sixta, M. Iakovlev, L. Testova, A. Roselli, M. Hummel, M. Borrega, A. van Heiningen, C. Froschauer and H. Schottenberger, Cellulose, 2013, 20, 1547–1561 CrossRef CAS.
- T. V. Lourencon, L. G. Greca, D. Tarasov, M. Borrega, T. Tamminen, O. J. Rojas and M. Y. Balakshin, ACS Sustainable Chem. Eng., 2020, 8, 1230–1239 CrossRef CAS.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of structural carbohydrates and lignin in biomass, 2012 Search PubMed.
- A. Sundberg, K. Sundberg, C. Lillandt and B. Holmbom, Nord. Pulp Pap. Res. J., 1996, 11(04), 216–219 CrossRef CAS.
- B. Holmbom and F. Örså, in Proceedings of the 7th International Symposium on Wood Pulping Chemistry, Beijing, 1993, pp. 810–817.
- C. Xu, A. Pranovich, J. Hemming, B. Holmbom, S. Albrecht, H. A. Schols and S. Willför, Holzforschung, 2009, 63, 61–68 CAS.
- J. Ralph and L. L. Landucci, in Lignin and Lignans, ed. C. Heitner, D. Dimmel and J. Schmidt, CRC Press, Taylor & Francis, Boca Raton, 2010, pp. 137–243 Search PubMed.
- M. Y. Balakshin, E. A. Capanema, C. L. Chen and H. S. Gracz, J. Agric. Food Chem., 2003, 51, 6116–6127 CrossRef CAS PubMed.
- C. S. Lancefield, H. J. Wienk, R. Boelens, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Sci., 2018, 9, 6348–6360 RSC.
- M. Y. Balakshin, E. A. Capanema and H. M. Chang, Holzforschung, 2007, 61, 1–7 CAS.
- M. Balakshin, E. Capanema, H. Gracz, H.-m. Chang and H. Jameel, Planta, 2011, 233, 1097–1110 CrossRef CAS PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- E. A. Capanema, M. Yu. Balakshin, H.-m. Chang and H. Jameel, Quantitative analysis of technical lignins by a combination of 1H-13C HMQC and 13C NMR methods (oral presentation), Proceedings of International Conference on Pulping, Papermaking and Biotechnology. Nov. 4–6, Nanjing, China, 2008, pp. 647–651 Search PubMed.
- G. Zinovyev, I. Sulaeva, S. Podzimek, D. Rössner, I. Kilpeläinen, I. Sumerskii, T. Rosenau and A. Potthast, ChemSusChem, 2018, 11, 3259–3268 CrossRef CAS PubMed.
- G. N. Rivière, A. Korpi, M. H. Sipponen, T. Zou, M. A. Kostiainen and M. Österberg, ACS Sustainable Chem. Eng., 2020, 8, 4167–4177 CrossRef PubMed.
- A. Moreno, M. Morsali, J. Liu and M. H. Sipponen, Green Chem., 2021, 23, 3001–3014 RSC.
- J. Obst, Tappi, 1982, 65, 109–112 CAS.
- M. Y. Balakshin, E. A. Capanema, X. Zhu, I. Sulaeva, A. Potthast, T. Rosenau and O. J. Rojas, Green Chem., 2020, 22, 3985–4001 RSC.
- K. Hofmann and W. G. Glasser, J. Wood Chem. Technol., 1993, 13, 73–95 CrossRef CAS.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- W. G. Glasser, C. A. Barnett, T. G. Rials and V. P. Saraf, J. Appl. Polym. Sci., 1984, 29, 1815–1830 CrossRef CAS.
- M. Österberg, M. H. Sipponen, B. D. Mattos and O. J. Rojas, Green Chem., 2020, 22, 2712–2733 RSC.
- P. Schlee, S. Herou, R. Jervis, P. R. Shearing, D. J. L. Brett, D. Baker, O. Hosseinaei, P. Tomani, M. M. Murshed, Y. Li, M. J. Mostazo-López, D. Cazorla-Amorós, A. B. Sobrido and M. M. Titirici, Chem. Sci., 2019, 10, 2980–2988 RSC.
- O. Hosseinaei, D. P. Harper, J. J. Bozell and T. G. Rials, ACS Sustainable Chem. Eng., 2016, 4, 5785–5798 CrossRef CAS.
- W. G. Glasser, Front. Chem., 2019, 7, 1–17 CrossRef PubMed.
- S. Kang, S. Jiang, Z. Peng, Y. Lu, J. Guo, J. Li, W. Zeng and X. Lin, Biomass Convers. Biorefin., 2018, 8, 889–897 CrossRef CAS.
- T. M. C. Hoang, L. Lefferts and K. Seshan, ChemSusChem, 2013, 6, 1651–1658 CrossRef CAS PubMed.
- N. Giummarella and M. Lawoko, ACS Sustainable Chem. Eng., 2016, 4, 5319–5326 CrossRef CAS.
- L. Testova, K. Vilonen, H. Pynnönen, M. Tenkanen and H. Sixta, Lenzinger Ber., 2009, 87, 58–65 CAS.
- J. S. Gütsch and H. Sixta, Ind. Eng. Chem. Res., 2012, 51, 8624–8630 CrossRef.
- J. S. Gütsch and H. Sixta, Lenzinger Ber., 2011, 142–151 Search PubMed.
- J. S. Gütsch, T. Nousiainen and H. Sixta, Bioresour. Technol., 2012, 109, 77–85 CrossRef PubMed.
- M. H. Sipponen, H. Lange, C. Crestini, A. Henn and M. Österberg, ChemSusChem, 2019, 12, 2039–2054 CrossRef CAS PubMed.
- A. Moreno and M. H. Sipponen, Mater. Horiz., 2020, 7, 2237–2257 RSC.
- N. Chen, L. A. Dempere and Z. Tong, ACS Sustainable Chem. Eng., 2016, 4, 5204–5211 CrossRef CAS.
- M. H. Sipponen, H. Lange, M. Ago and C. Crestini, ACS Sustainable Chem. Eng., 2018, 6, 9342–9351 CrossRef CAS PubMed.
- D. Tian, J. Hu, J. Bao, R. P. Chandra, J. N. Saddler and C. Lu, Biotechnol. Biofuels, 2017, 10, 1–11 CrossRef PubMed.
- E. Lizundia, M. H. Sipponen, L. G. Greca, M. Balakshin, B. L. Tardy, O. J. Rojas and D. Puglia, Green Chem., 2021, 23, 6698–6760 RSC.
- M. H. Sipponen, M. Smyth, T. Leskinen, L. S. Johansson and M. Österberg, Green Chem., 2017, 19, 5831–5840 RSC.
- A. Moreno, N. Bensabeh, J. Parve, J. C. Ronda, V. Cádiz, M. Galià, L. Vares, G. Lligadas and V. Percec, Biomacromolecules, 2019, 20, 1816–1827 CrossRef CAS PubMed.
- M. Y. Balakshin and E. A. Capanema, in 14th European Workshop on Lignocellulosics and Pulp, Grenoble, France, 2016 Search PubMed.
- E. Capanema and M. Balakshin, 2017, 18th ISWFPC, Porto Seguro, 2017, 65–69.
- K. S. Mikkonen, Green Chem., 2020, 22, 1019–1037 RSC.
- L. K. J. Hauru, Y. Ma, M. Hummel, M. Alekhina, A. W. T. King, I. Kilpeläinen, P. A. Penttilä, R. Serimaa and H. Sixta, RSC Adv., 2013, 3, 16365–16373 RSC.
- Y. Ma, J. Stubb, I. Kontro, K. Nieminen, M. Hummel and H. Sixta, Carbohydr. Polym., 2018, 179, 145–151 CrossRef CAS PubMed.
- M. Trogen, N. D. Le, D. Sawada, C. Guizani, T. V. Lourençon, L. Pitkänen, H. Sixta, R. Shah, H. O'Neill, M. Balakshin, N. Byrne and M. Hummel, Carbohydr. Polym., 2021, 252, 117133 CrossRef CAS PubMed.
- M. Borrega, P. T. Larsson, P. Ahvenainen, S. Ceccherini, T. Maloney, L. Rautkari and H. Sixta, Carbohydr. Polym., 2018, 190, 212–221 CrossRef CAS PubMed.
- A. Romaní, G. Garrote, J. L. Alonso and J. C. Parajó, Bioresour. Technol., 2010, 101, 8706–8712 CrossRef PubMed.
- A. Romaní, G. Garrote and J. C. Parajó, Fuel, 2012, 94, 305–312 CrossRef.
- A. Pranovich, B. Holmbom and S. Willför, J. Wood Chem. Technol., 2016, 36, 140–156 CrossRef CAS.
- H. Sadeghifar and A. Ragauskas, ACS Sustainable Chem. Eng., 2020, 8, 8086–8101 CrossRef CAS.
- M. Y. Balakshin, E. A. Capanema, M. Colakyan and F. Lipiecki, Upgrading lignin from lignin - containing residues through reactive extraction, Patent US 10240006 B2, 2019.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02171d |
| This journal is © The Royal Society of Chemistry 2022 |