
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interfacial catalysis and lignin nanoparticles for strong fire- and water-resistant composite adhesives†
K. Alexander
Henn
 a,
Susanna
Forssell
b,
Antti
Pietiläinen
a,
Nina
Forsman
a,
Ira
Smal
a,
Paula
Nousiainen
a,
Rahul Prasad
Bangalore Ashok
b,
Pekka
Oinas
b and
Monika
Österberg
*a
a,
Susanna
Forssell
b,
Antti
Pietiläinen
a,
Nina
Forsman
a,
Ira
Smal
a,
Paula
Nousiainen
a,
Rahul Prasad
Bangalore Ashok
b,
Pekka
Oinas
b and
Monika
Österberg
*a
aDepartment of Bioproducts and Biosystems, Aalto University, Espoo, Uusimaa, Finland. E-mail: monika.osterberg@aalto.fi.
bDepartment of Chemical and Metallurgical Engineering, Aalto University, Espoo, Uusimaa, Finland
First published on 1st August 2022
Abstract
Wood is increasingly replacing concrete to reduce CO2 emissions in buildings, but fossil-based adhesives are still being used in wood panels. Epoxidized lignin adhesives could be a potential replacement, but their preparation has so far required low-molecular weight lignin and long reaction times. Here we show a new efficient method to produce epoxidized kraft lignin (EKL) from regular kraft lignin by using interfacial catalysis. We demonstrate that EKL combined with biocolloids in the form of lignin nanoparticles (LNPs) produces a strong adhesive comparable to commercially available ones when cross-linked at 130–160 °C for only 3–5 minutes. The adhesive was free of phenol or formaldehyde, had a lignin content of over 80% and still showed impressive wet strength and incredible thermal stability. The process was shown to be scalable and environmentally more sustainable than resins from fossil-based feedstock or currently available ones from renewable resources.
Introduction
Climate change and rising fossil raw-material prices are pushing industries toward technologies with a lower carbon footprint. The construction industry can lower its carbon footprint significantly by replacing concrete with wood.1–3 The plywood and woodboard adhesive industry largely rely on formaldehyde as a cross-linker for phenol, urea, and melamine. Formaldehyde is an efficient cross-linker, but is also toxic,4,5 and although formaldehyde can be obtained from natural sources, it is mostly produced from fossil-based raw materials.6 However, most adhesives from biomaterials (e.g., proteins or polysaccharides) are either too expensive or too hydrophilic, the former hampering scale-up and the latter resulting in poor wet bonding strength.7 Nevertheless, plywood manufacturers have used lignin as a filler in adhesives to decrease raw-material costs since before the 1940's.8,9 Lignin is produced as a side product in pulp and biorefinery industries in huge amounts annually but is astoundingly underutilized despite its low price and abundance. Only 2–5% of the produced lignin is used while the rest is incinerated for energy recovery in pulp mills.10 Burning lignin results in significant CO2 emissions, so using noncombustible energy sources in the pulp mills and valorizing lignin as a material would both decrease carbon dioxide emissions and lead to more sustainable material flows.Applying significant amounts of lignin in adhesives is challenging because it often reduces the adhesive's performance.11–13 Modern lignin-containing adhesives use phenolated lignin to increase its reactivity with formaldehyde.14,15 This strategy allows the lignin content to be increased but the formaldehyde content still cannot be decreased by more than a few percent.15,16 Being able to also decrease formaldehyde would indeed reduce the use of fossil-based chemicals, but it would also be good for the health of people working in fields like resin manufacture or firefighting who have a high risk of exposure to significant levels of formaldehyde daily.4,17–20 Some studies even suggest that the general public frequently get exposed to significant levels of formaldehyde from wood-panels.18,21 Because concrete is increasingly being replaced by wood as a building material to reduce CO2 emissions,22–24 it is expected that the demand for adhered wood-boards like plywood and particleboard will increase, as these products are widely used in walls, roofs and flooring.23 The drawbacks of modern commercial wood-panel adhesives should be solved to make the transition to wood-based construction safe and sustainable.
Epoxies could be an alternative to formaldehyde as a cross-linker in resins.25 Lignin epoxidation strategies have been developed for years and can be used to make lignin-based cross-linkers. Lignin epoxidation is generally done by reacting lignin with epichlorohydrin under highly alkaline conditions (Fig. 1a).26–29 Lignin is first dissolved in an aqueous solution of sodium hydroxide or an organic solvent with a high boiling point,26,30,31 to which epichlorohydrin is added, or vice versa. There are nevertheless unsolved challenges in the current lignin epoxidation methods, like the need for pre-processed low molecular weight lignin as raw material as well as the chemical- and energy-intensive reactions. Reported reaction times for lignin epoxidation processes have been around 1–14 hours at 60 °C or above. The long reaction times gives room for homopolymerization, which causes a high yield of insoluble epoxidized lignin, which is why low molecular weight lignin is needed.26,31,32 This approach results in an rather expensive process and thereby removes one of the main benefits of using lignin as raw material. While there are many studies on lignin-epoxidation, only a few demonstrate its use in applications,28,33 probably due to its poor solubility. It is nevertheless likely that lignin epoxidation could be a promising way to turn lignin into a valuable functional raw material if the above-mentioned challenges are solved. In addition, recent studies have shown that epichlorohydrin can feasibly be made from bio-based glycerol.34,35 Epoxidation via epichlorohydrin could thus enable the production of fully bio-based composite materials when glycerol-based epichlorohydrin becomes more accessible.
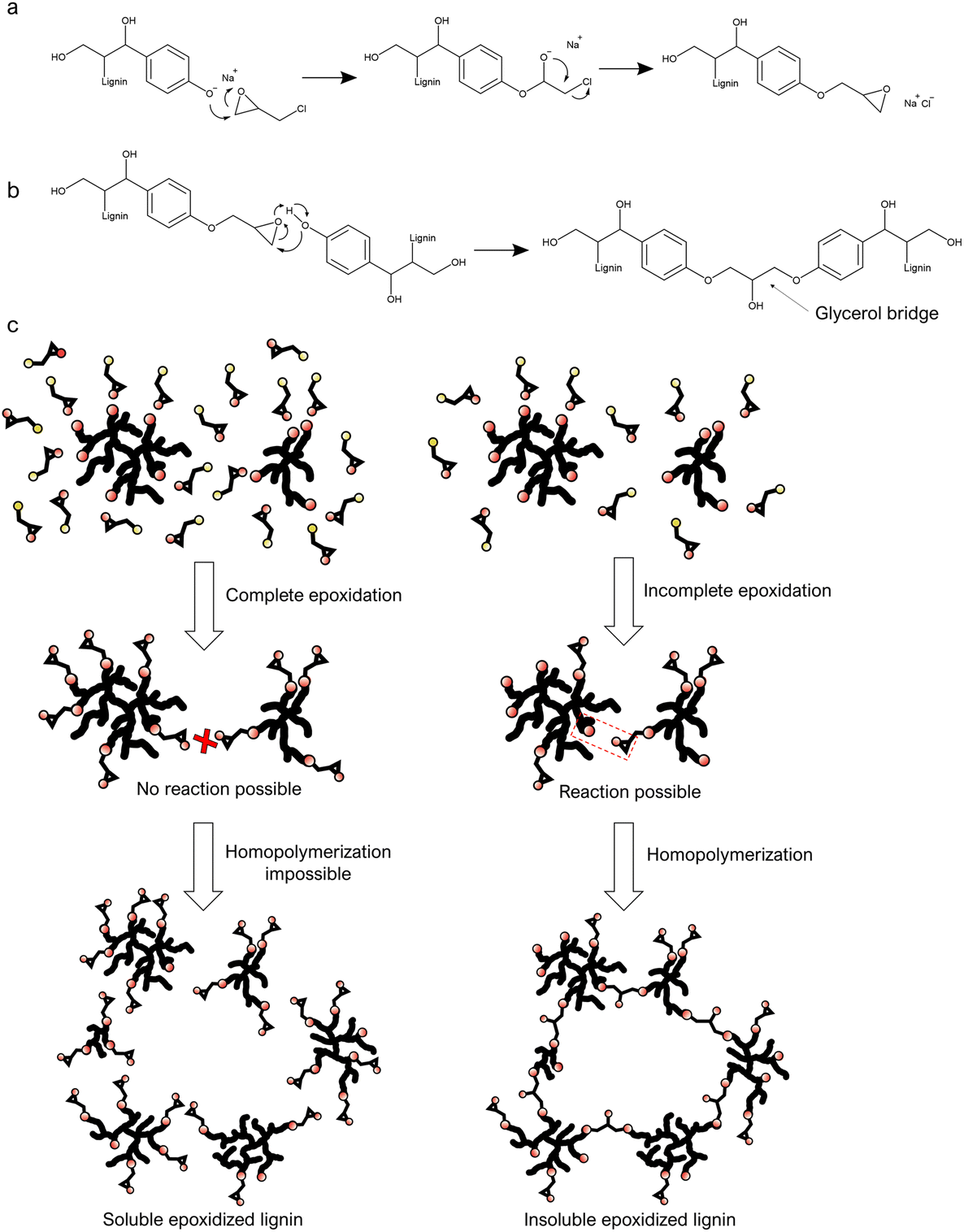 | ||
| Fig. 1 Lignin epoxidation and homopolymerization mechanisms. (a) Lignin epoxidation via epichlorohydrin in alkaline conditions. (b) Homopolymerization between lignin and epoxidized lignin. (c) Complete versus incomplete epoxidation respectively leading to soluble and insoluble epoxidized lignin, here due to insufficient amounts of epichlorohydrin. The black lines represent the lignin backbone, the red dots represent oxygen groups, and the yellow dots represent chlorine. | ||
A lignin-based resin needs to be reactive and preferably water soluble. Here we achieve both by conducting the reaction in an emulsion-like medium. This enables the epoxidation reaction to be catalyzed at the water–oil interface which minimizes the reaction time and simultaneously reduces homopolymerization. This resin is then cured with water-dispersible colloidal lignin particles (LNPs). The resulting adhesive uses no volatile organic solvents, has a strength comparable to phenol-formaldehyde resins, and contains over 80% of lignin. The resin can be made 100% bio-based when using epichlorohydrin from glycerol and is therefore a great step in the direction toward sustainable and formaldehyde-free resins and composites. The scalability and sustainability of the process is evaluated by a pre-feasibility techno-economic analysis (TEA) and life cycle assessment (LCA).
Results
Design of epoxidized lignin synthesis
Lignin epoxidation reactions often take ca. 1–8 hours and is usually done in mostly organic solvents,26–28,30,31,36,37 because epichlorohydrin dissolves poorly in water. However, the intermediate product in the lignin-epoxidation reaction is highly polar (Fig. 1), and the presence of water could perhaps stabilize the intermediate product and thus increase the reaction rate. Certain reactions even experience a catalytic effect in the presence of non-miscible compounds (like water and epichlorohydrin) because of interfacial catalysis,38–40 and it was examined whether this phenomenon could be used in lignin epoxidation. It was observed that lignin-stabilized epichlorohydrin droplets could be formed in aqueous alkaline lignin solutions by stirring (Fig. 2 & Fig. S2†). The epoxidation reaction is highly exothermic, and changes in temperature were therefore used to monitor the progress of the reaction. An exponential increase in temperature was observed at ∼43 °C, and this was used as initiation temperature. The temperature was kept below 56 °C by gradually adding the lignin solution (Fig. S3†), thus avoiding homopolymerization.26,29 The temperature started to drop after 11 minutes, indicating completion of the reaction. Prolonging the reaction time beyond this did not seem to increase the epoxidation significantly since the intensity of the epoxy groups’ Fourier-transform infrared (FTIR) absorption signal did not increase (Fig. S5 & S6†). HSQC analysis also showed that the epoxy/methoxy ratio increased logarithmically with reaction time but started to level out after 6 minutes (Fig. S10 & S12†). The amount of insoluble aggregates nevertheless increased, indicating homopolymerization upon prolonged reaction times. This finding is in agreement with previous work where the reaction time was found to have an almost insignificant effect on epoxy content (EC) while increase in temperature had a negative effect.29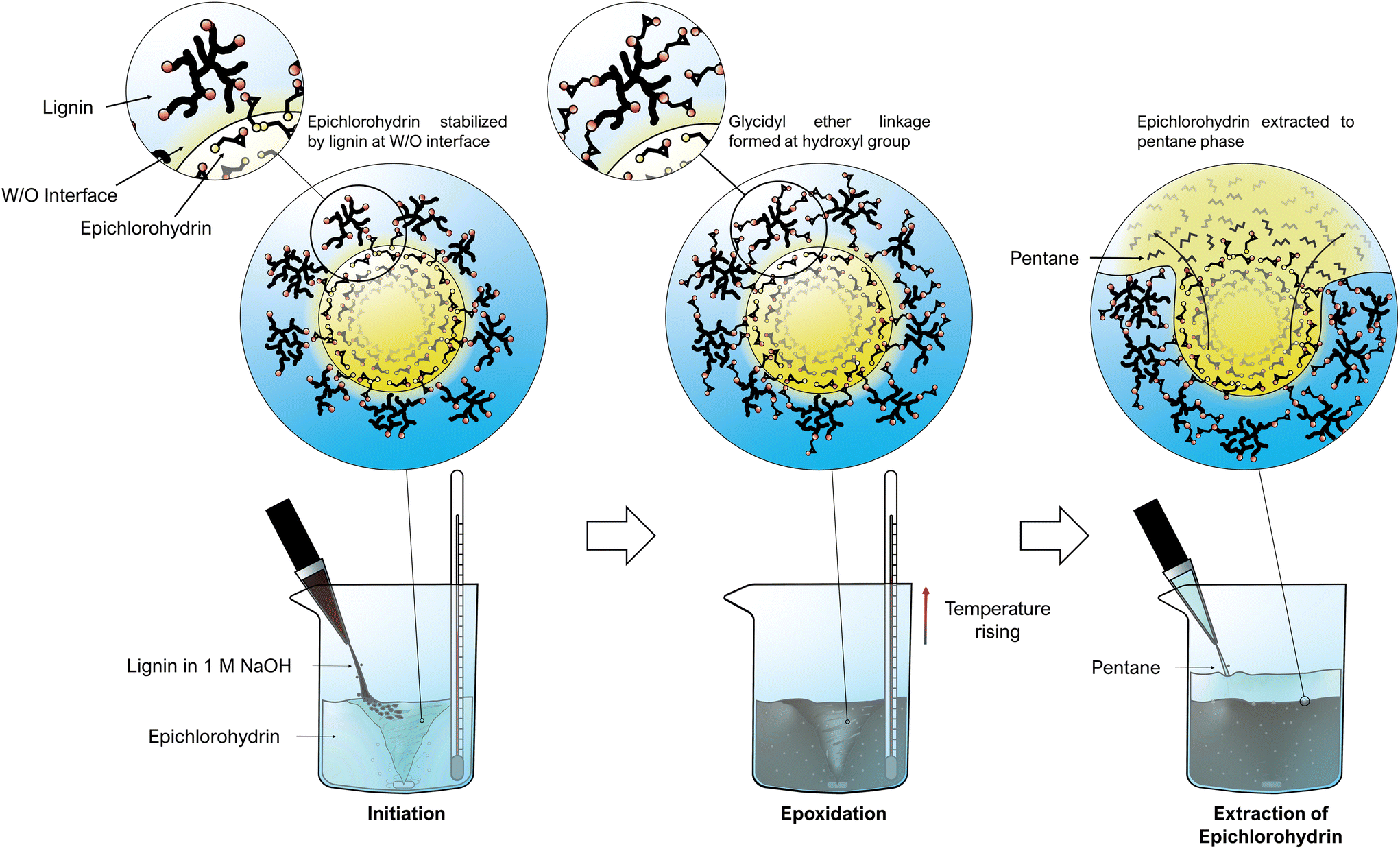 | ||
| Fig. 2 Water-based lignin epoxidation via interfacial catalysis at epichlorohydrin droplets (reaction mechanism in Fig. 1a), followed by the simple recovery of unreacted epichlorohydrin via pentane-extraction. | ||
The epoxidized lignin obtained from 11-minute reactions was water-soluble at pH above 8, in contrast to unmodified lignin, which is poorly soluble in aqueous solutions below pH 10. The improved solubility may be due to reduced intermolecular hydrogen bonding by phenolic end groups.41 As expected, FTIR and NMR analyses showed reduced numbers of phenolic hydroxyl groups, increased numbers of methylene groups, and the presence of epoxide groups. Fig. S4 and Table S1† presents the FTIR spectra and chemical structures that correspond to the obtained FTIR bands, while 1H-NMR and 31P-NMR spectra are presented in Fig. S8, S9 and Table S2.† According to EKL's epoxy/methoxy ratio by HSQC analysis (Fig. S10†), the epoxy index (EI) was 2.3 mmol g−1, meaning an epoxy equivalent weight (EEW) of 430 g mol−1. 1H-NMR analysis gave slightly differing results with an EI and EEW of 1.8 mmol g−1 and 556 g mol−1 (Fig. S8†). The relatively large molecular size could make the quantification in especially 1H-NMR less precise. This problem also reduces signal intensity in HSQC, but the ratios of structural features is unchanged which still enables comparison. Previous studies have obtained comparable EI values of 1–3 mmol g−1 from the epoxidation of non-fractionated or depolymerized lignin with reaction times of 3–7 h at 50–90 °C, but with the formation of an insoluble fraction.26,37 Some studies have also presented the epoxidation of solvent fractionated low molecular-weight lignin using a two-step reaction system, taking ca. 10–12 h in total at 50–80 °C. The resulting epoxidized lignin had EEW values of ca. 330–415 g mol−1 (EI ca. 2–3 mmol g−1), depending on the lignin source and the fractionation method.27,28,36 The most significant difference compared to previous studies on lignin epoxidation is the fast reaction rate observed here. We speculate that the presence of water indeed helps to stabilize the polar intermediate product in the reaction between lignin and epichlorohydrin. Water could also make chlorine a better leaving group by increasing the solubility of the formed sodium chloride salt, which can further drive the reaction. This could favor the grafting of epichlorohydrin onto lignin (Fig. 1a & c) rather than ring-opening leading to homopolymerization (Fig. 1b & c). In fact, water has been used to increase the reaction rate in the production of commercial epoxies (e.g. bisphenol-A diglycidyl ether) for decades,42,43 although lignin epoxidation reactions have been performed in organic solvents with little or no water.27,28,30,31,36
The two-phased nature of the reaction mixture here made it easier to recover the excess epichlorohydrin after the reaction through extraction with pentane. The epichlorohydrin recovery was examined using a separation funnel. Using three consecutive pentane extractions, ca. 98% of the unreacted epichlorohydrin could be recovered. The number of extractions could likely be reduced with some optimization, as the first extraction recovered 91% of the epichlorohydrin. In an industrial process, mixer-settlers and extraction columns could be used to increase the recovery yield further. Methods to optimize the extraction process in industrial scale to maximize the epichlorohydrin recovery will be discussed further in upcoming work focusing on process optimization and scale-up. In laboratory scale, precipitating and washing EKL in acetone could be used to remove all of the epichlorohydrin in one step. The advantage of using solvent extraction instead of precipitation via acidification, the latter being more commonly used, was that all of the product remained in one phase, instead of two (Fig. S7†).26,37 At an industrial scale, solvent exchange allows for easy and energy-efficient recovery of epichlorohydrin (Fig. S7†). Although the use of pentane creates a risk due to its flammability and highly volatile nature,44 it requires relatively little energy to distill from the epichlorohydrin after the extraction. The pentane's volatility should nevertheless be considered to guarantee safety both in large and small scale, but since epichlorohydrin is hazardous,44 its efficient recovery is very important to minimize waste.
The effect of the lignin concentration on the formed EKLs solubility and molecular size was also examined. Higher concentrations reduce the need for the removal of water and reduces the total filtration volume in an up-scaled process. The concentration was therefore increased from 10 wt% to 20 and 30 wt% (Fig. 3). Light microscopy showed larger and denser aggregates for EKL produced with higher lignin concentrations. For all samples, dynamic light scattering (DLS) showed a higher intensity-average than number average particle size, and gel permeation chromatography (GPC) showed a much smaller number-average (Mn) than weight-average (Mw) molecular size (Table 1). GPC showed a 48% higher Mw and a 32% higher Mn for the EKL made using 20 wt% lignin solutions compared to the EKL made with 10 wt% solutions. GPC could however not be efficiently used on EKL from 30 wt% solutions because the particle size was way above the filter's pore size (0.2 μm), and most suspended particles were therefore filtered away before the measurements. DLS measurements nevertheless showed larger particle size for EKL from 30 wt% solutions compared to the other samples, indicating a larger molecular weight. Both GPC and DLS results suggest that high initial concentrations of lignin increase the probability of homopolymerization and therefore lead to the formation of large insoluble aggregates. The increased homopolymerization in concentrated solutions can be explained by the increased probability of two lignin molecules colliding and cross-linking. Large aggregates may decrease the adhesives’ ability to penetrate into the wood, and the EKL from 10 wt% solutions was therefore chosen for adhesive tests. A discussion of a large-scaled implementation of the process is presented in a later section.
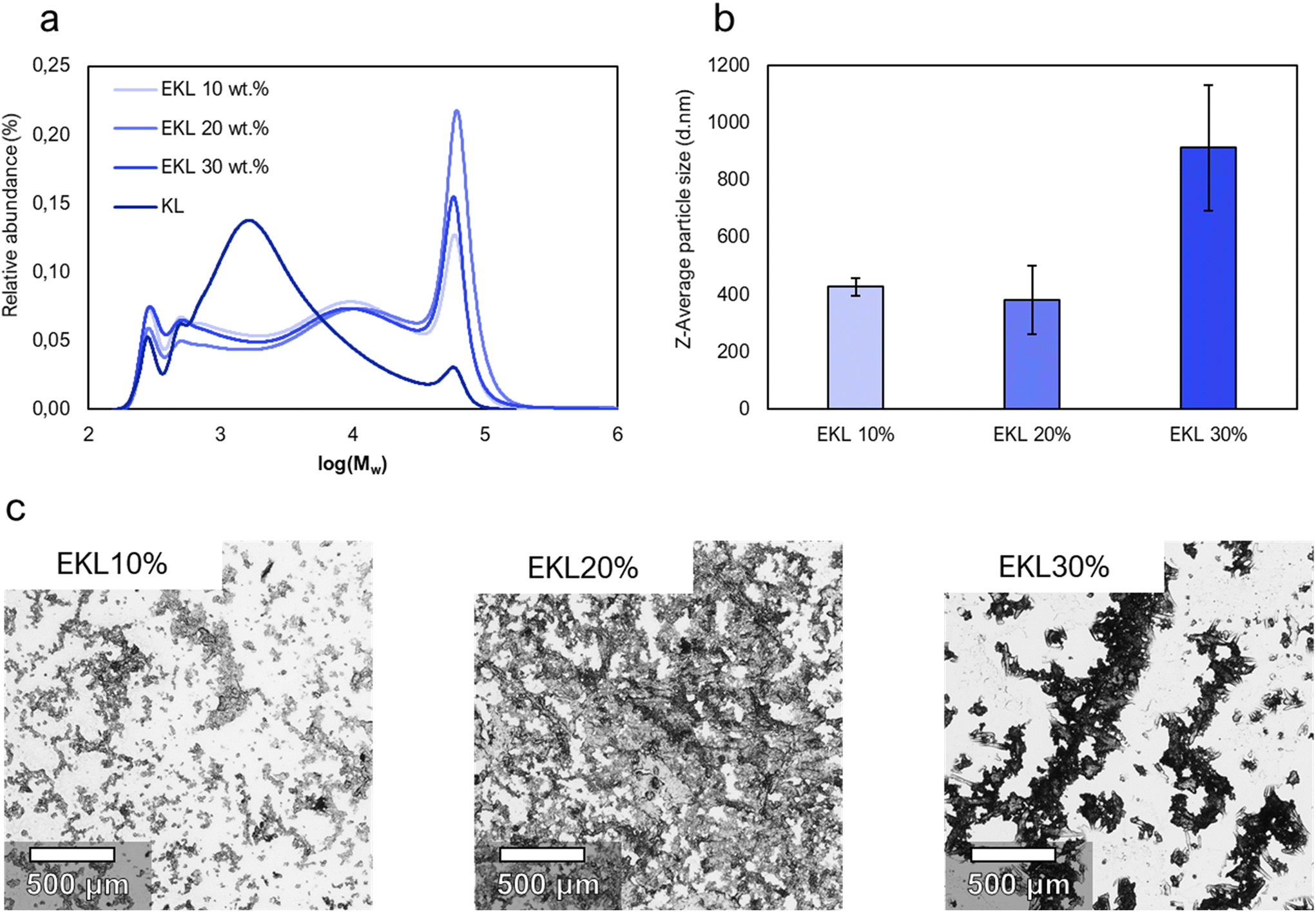 | ||
| Fig. 3 The appearance and molar weight distributions of unmodified kraft lignin and different epoxidized kraft lignin (EKL) samples. (a) The molar mass distributions of unmodified kraft lignin and EKL samples according to high-performance liquid chromatography measurements. (b) Z-Average particle diameters of EKL samples measured by dynamic light scattering. (c) Light-microscope images of dried of EKL samples. Photographs of solutions of EKL10–30% are presented in Fig. S1.† | ||
| Sample | M w × 103 (g mol−1) | M n × 103 (g mol−1) | PDI |
|---|---|---|---|
| KL | 6.68 | 1.30 | 5.1 |
| EKL10% | 20.1 | 1.55 | 13.0 |
| EKL20% | 29.9 | 2.04 | 14.7 |
| EKL30% | 20.5 | 1.53 | 13.4 |
Curing and adhesive testing
Epoxy-based adhesives require both an epoxy compound and a hardener. Hydroxyl groups, especially phenolic hydroxyls, are susceptible to epoxy-polymerization reactions, making LNPs interesting hardeners due to their hydroxyl-rich surface.45,46 The effect of the EKL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :LNP ratio, the particle size, the curing heat, the curing time, and the adhesive spread was examined to find optimal conditions for adhesive performance. The EKL:LNP ratio was optimized first. In traditional epoxy chemistry where small molecules are used as starting material, the molar ratio of epoxy groups and hardener should be close to 1:1. However, because LNPs in this case may also function as a particle-reinforcer, and because the reactivity and hydrogen bonding capacity of different hydroxyl and oxirane groups can differ, the optimal ratio was determined based on adhesive strength (Fig. 4a). EKL adhered better to the wood and was therefore spread first. The LNPs were then added in the middle as a reactive “bridge”. It was found that the tensile shear strength of EKL:LNP with a mass ratio of 1:1 (hereafter referred to as EKL:LNP if the components have been cured, and EKL + LNP if components are combined but not cured) was similar to that of (PF) resins and stronger than commercial epoxy resins, reaching a strength of over 12 MPa (Fig. 4a). EKL without LNPs achieved a surprisingly satisfactory adhesive strength. The LNPs size's effect on the lap shear strength was also examined, but the overall differences were small (Fig. 4d) and particles of 400–500 nm in diameter were therefore used for all the subsequent tests. More information about the LNPs is found in ESI Note 6, Table S4 and Fig. S23.†
:LNP ratio, the particle size, the curing heat, the curing time, and the adhesive spread was examined to find optimal conditions for adhesive performance. The EKL:LNP ratio was optimized first. In traditional epoxy chemistry where small molecules are used as starting material, the molar ratio of epoxy groups and hardener should be close to 1:1. However, because LNPs in this case may also function as a particle-reinforcer, and because the reactivity and hydrogen bonding capacity of different hydroxyl and oxirane groups can differ, the optimal ratio was determined based on adhesive strength (Fig. 4a). EKL adhered better to the wood and was therefore spread first. The LNPs were then added in the middle as a reactive “bridge”. It was found that the tensile shear strength of EKL:LNP with a mass ratio of 1:1 (hereafter referred to as EKL:LNP if the components have been cured, and EKL + LNP if components are combined but not cured) was similar to that of (PF) resins and stronger than commercial epoxy resins, reaching a strength of over 12 MPa (Fig. 4a). EKL without LNPs achieved a surprisingly satisfactory adhesive strength. The LNPs size's effect on the lap shear strength was also examined, but the overall differences were small (Fig. 4d) and particles of 400–500 nm in diameter were therefore used for all the subsequent tests. More information about the LNPs is found in ESI Note 6, Table S4 and Fig. S23.†
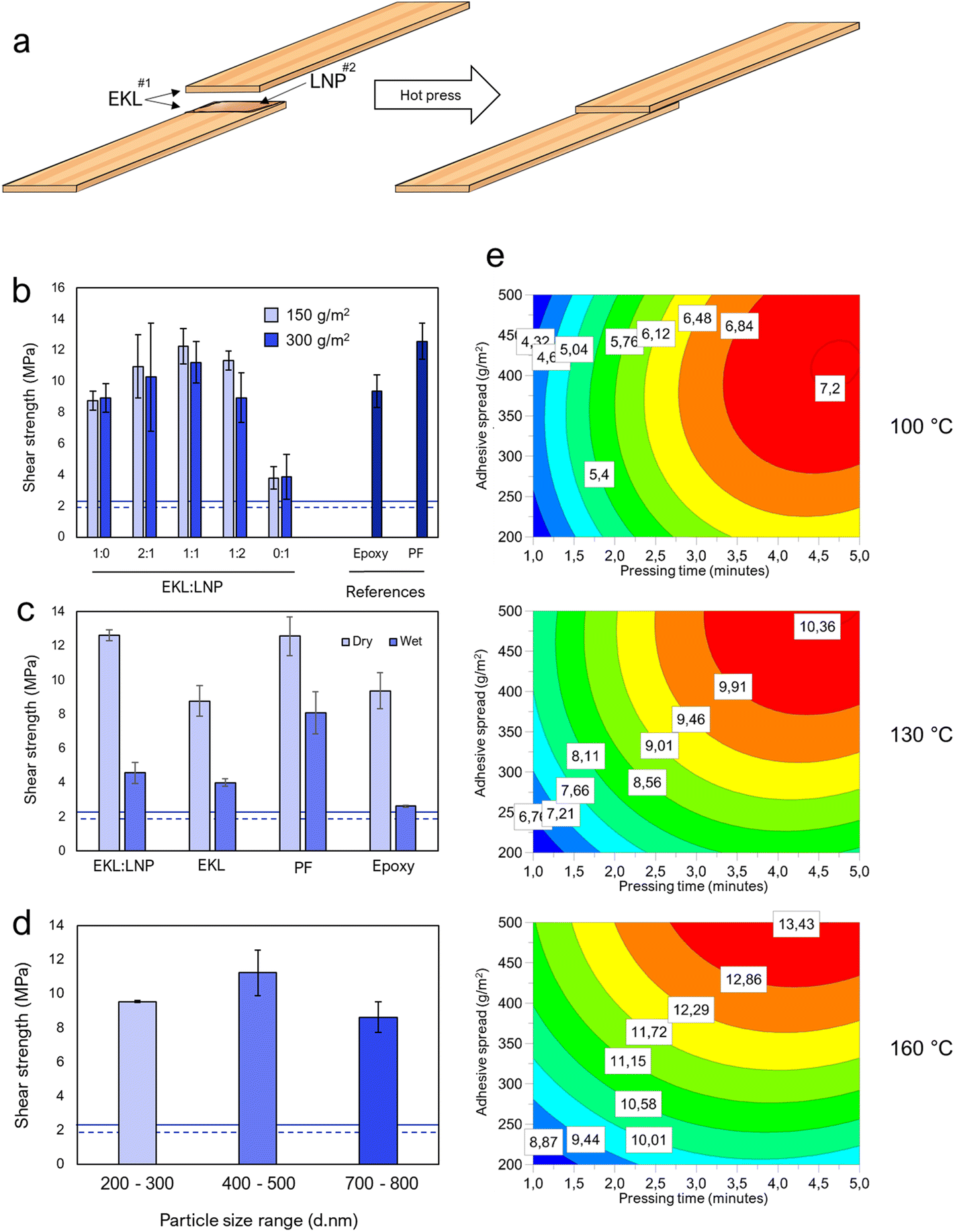 | ||
| Fig. 4 The adhesive strength of epoxidized lignin hardened with LNPs. (a) Illustration of adhesive sample preparation. (b) The effect of different EKL:LNP mass ratios on adhesive strength and a commercial epoxy adhesive. The epoxy and PF references were made with a spread of 300 g m−2. (c) The dry and wet strength of the EKL:LNP and EKL adhesive with a spread of 150 g m−2. (d) The effect of LNP's particle size on dry adhesive strength. All samples in (b–d) were pressed at 145 °C for 5 minutes. The solid and dotted lines in (b and c) represent the respective dry and wet strength required by urea-formaldehyde type adhesives according to the ASTM-D4690 standard. (e) Surface response model prediction of adhesive strengths (in MPa) with different hot press temperatures and times, and adhesive spreads. The EKL:LNP ratio was 1:1 except in (b). | ||
Achieving competitive wet-strength is one of the major challenges in research on bio-based formaldehyde-free adhesives.47,48 Although the EKL:LNP adhesive's wet strength was 44% lower than PF adhesives, the achieved wet strength of 4.5 MPa is remarkable for a formaldehyde-free lignin-based adhesive and clearly passes the requirements, and can very likely be increased with further research. Wood deforms and swells in the presence of excessive amounts of water, which can disrupt a resin's structure and bonding to the wood.49–51 In addition, because of cellulose's hydrophilicity, hydrogen bonding with water is likely to compete with those of the resin in wet conditions. Still, the wet strength of the joints was surprisingly high considering the lignin-content and the absence of formaldehyde. Lignin-containing plywood adhesives often contain less phenol than commercial PF adhesives, but very rarely contain less formaldehyde.11–14,52 One benefit of formaldehyde as a cross-linker is that it is able to directly cross-link with lignin and phenol-containing extractives, and to some degree cellulose and hemicellulose,53,54 making it practically undetachable from wooden surfaces when cured. Epoxies are not known to react with cellulose, but reactions with phenolic groups in lignin and extractives are likely. Because we did not observe significant differences in wet strength between adhesives of only EKL and EKL:LNP, it is possible that the wet strength is mainly achieved by cross-links between EKL and phenolic components, such as lignin, within the wood. The resin's viscosity is another important factor, since it affects the ability to penetrate into the wood's structure. Higher penetration allows for stronger joints. The resin viscosity was not optimized in this study, but could have a significant effect on both wet and dry strength. This understanding is important to be able to improve the wet strength further.
The effect of the adhesive spread and the hot-press time and temperature were then further examined using surface response models (Fig. 4e and Fig. S13–S15†). The optimal hot-pressing temperature was shown to be between 145–160 °C, depending on the curing time and the spread. The results showed that the energy input needed for cross-linking could be achieved by increasing the curing time or curing temperature, and higher amounts of adhesive required higher amounts of energy. Excessively high temperatures were shown to cause some degradation at prolonged curing times, but good performance with short curing times, while thin adhesive spreads required lower temperatures and shorter curing times. Very thin spreads, between 100–200 g m−2, could induce wood failures (maximal adhesive performance) even when cured at 130 °C. Achieving shear strengths of over 10 MPa, even the samples with thin spreads hot-pressed at 130 °C for 5 minutes could be used to lift more than 10 kg (Fig. S18†). More information about the overall adhesive performance, differences between thin and thick adhesive layers, and the mode of failure is provided in ESI Note 4.†
Thermal stability
Thermal stability is an indication of structural rigidity, compactness, and bond strength,55,56 and it is therefore relevant also in applications where thermal stability is not required. Compared to kraft lignin, both EKL and EKL:LNP showed significantly increased thermal stability (Table 2) especially at temperatures above 400 °C, which is unseen in studies where an epoxy with longer chain length is used.27,57 One reason may be the high density of thermally stable aromatic structures that the short cross-link bridges enable. The low mass loss of EKL:LNP composites upon heating suggested that the material may possess flame resistance, which was hence tested with a Bunsen burner at close range to wooden surface coated with EKL:LNP. The coated side was not ignitable by the flame at close range, further verifying this conclusion. The sample only burned from its uncoated wooden backside, although the flame was directed towards the glued side (Fig. 5d and video ESI†). The EKL:LNP formulation thus has potential as a fire-proof coating as well. We suggest two reasons for the good thermal stability and flame resistance, namely the high lignin content and short spacer length of EKL. The aromatic groups of lignin are thermally stable and the short cross-linking spacer length of EKL minimizes voids within the structure, which is beneficial for thermal resistance.58,59 In future studies the benefit of flame resistance of the EKL:LNP composites in coatings should be more thoroughly evaluated with standard testing. Differential scanning calorimetry (DSC) was used to determine glass transition temperatures (Tg), indicating molecular mobility,55,56 and also to examine whether curing could be observed between a mixture of LNPs and EKL (EKL + LNP) (Fig. 5c). Lignin (especially in the form of LNPs) quickly adsorb moisture from the air, which is why the second heating cycle, was analyzed (Fig. 5c). In contrast to the uncured EKL + LNP mixtures, the cured EKL:LNP sample had no clear Tg. The thermogram of uncured EKL + LNP was more endothermic than the two other samples which could be due to energy absorbed by curing. The broad curing signal is expected considering lignin's heterogeneous structure, although complete curing is very unlikely due to the restricted molecular mobility in the dry samples. More detailed information about the DSC results can be found in the ESI (Note 5 and Fig. S22†).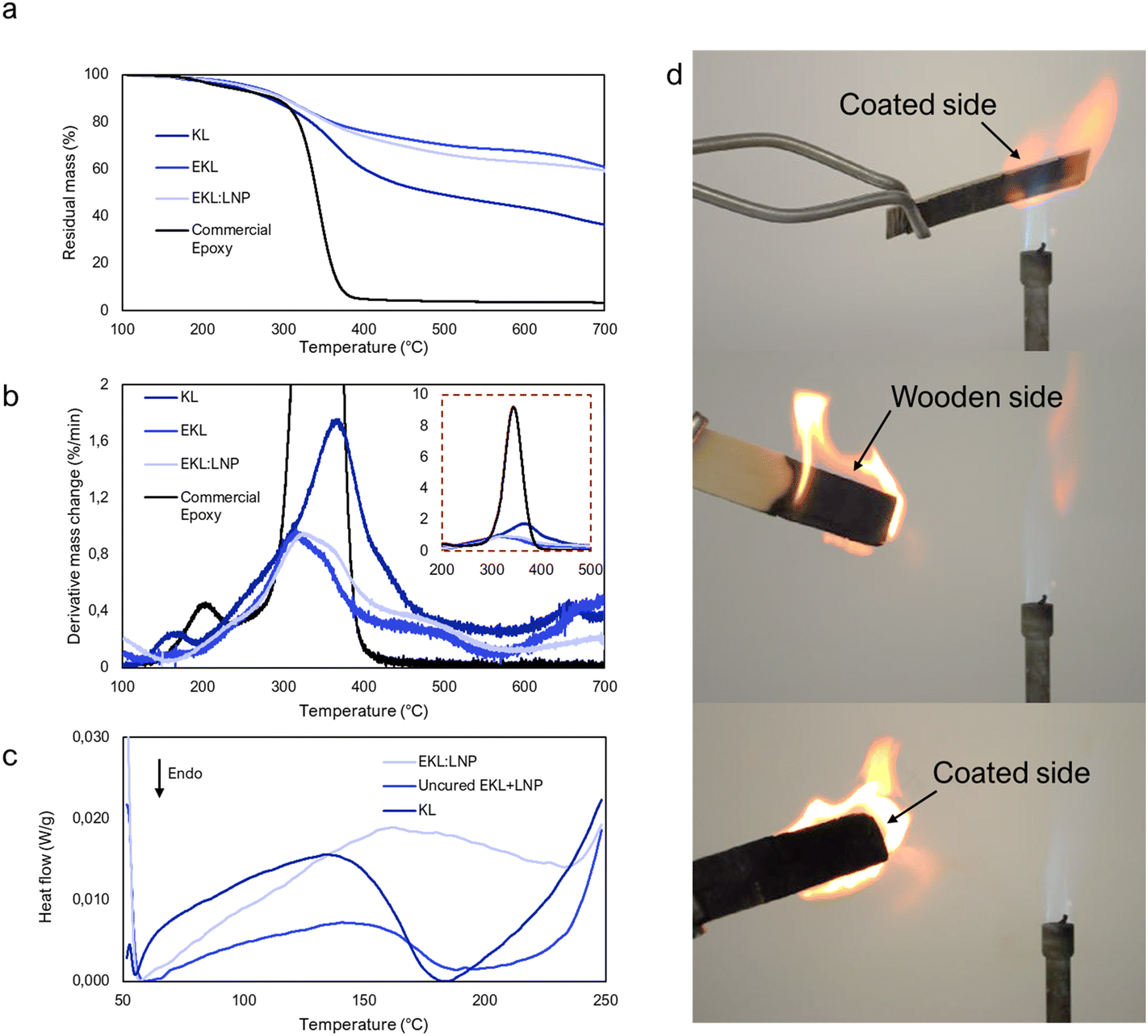 | ||
| Fig. 5 Thermal stability of cured and uncured EKLs compared to lignin and a commercial epoxy. (a) TGA and (b) DTG curves of cured EKL:LNP composite, uncured EKL, and unmodified kraft lignin (KL). (c) Second heating cycle DSC thermogram of cured EKL:LNP and uncured EKL + LNP mixtures and KL. (d) Fire resistance of EKL:LNP structure, showing inability of the coated EKL:LNP to burn in contrast to the wooden substrate. | ||
| Sample |
T
d5 (°C) |
T
d10 (°C) |
T
d30 (°C) |
T
dmax (°C) |
T HRI (°C) | Total loss (%) |
|---|---|---|---|---|---|---|
| KL | 237 | 282 | 366 | 359 | 154 | 64 |
| EKL | 260 | 301 | 506 | 315 | 200 | 40 |
| EKL:LNP | 253 | 300 | 446 | 311 | 181 | 41 |
| Commercial epoxy | 224 | 289 | 329 | 348 | 141 | 97 |
Industrial process design and scalability
For the large-scale production of epoxidized lignin-based adhesives, the EKL process must be developed for facilitating industrial scalability. The commercial scale process to be designed, could preferably have a capacity of approximately 70 kt per annum, and be integrated with an existing pulp mill or biorefinery. The main process steps and the system boundary of the LCA analysis are shown in Fig. 6 as a block diagram. It is worth noting that based on experimental results, the EKL process can be combined with the LNP production process to manufacture adhesives with exceptional properties. The possibility of scale-up of the production of LNPs has been examined in our previous research.60–62 Furthermore, Ashok et al.60 carried out a techno-economic assessment for the large-scale production of LNPs and found the process to be fully scalable with the potential to be viable and profitable, resulting in a LNP manufacturing cost of less than 1 € per kg.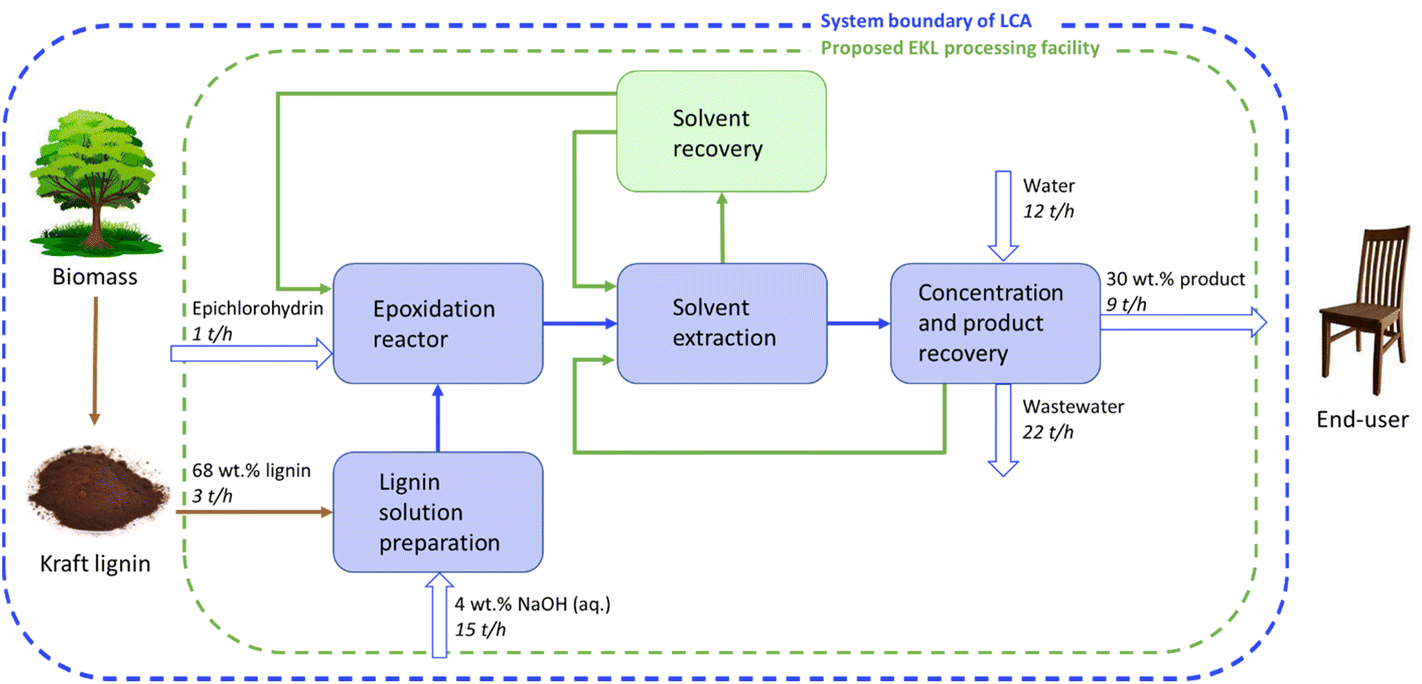 | ||
| Fig. 6 A block flow diagram for large-scale manufacturing of EKL and system boundary of the life cycle assessment. | ||
In the industrial scale EKL production process lignin is dispersed in a mixture of water and sodium hydroxide to create a 10 wt% lignin solution. The lignin has been assumed to be purified and dried at the pulp mill or refinery, whereas ash is removed by filtration before use of the dissolved lignin. The lignin solution is mixed with epichlorohydrin in a jacketed stirred tank reactor to carry out lignin epoxidation. Excess epichlorohydrin from the epoxidation reactor is recovered by solvent extraction with pentane. The organic phase, i.e., pentane and epichlorohydrin, is sent to the solvent recovery unit for separation of the components. In the solvent recovery unit, the organic phase is distilled to recover epichlorohydrin and pentane to be reused in the epoxidation reactor and extraction, respectively. The EKL-containing aqueous layer from the epoxidation reactor will be sent to the product recovery unit. This aqueous phase includes some residual epichlorohydrin and NaOH, as well as the NaCl that was formed in the reaction.
The concentrated aqueous product (EKL) is sent to an ultrafiltration unit and diluted with water to reduce the concentration of NaCl and NaOH in the final product to approximately 1 wt%. The final product is 30 wt% EKL in an aqueous solution. The permeate from the concentration device (ultrafilter) is sent for decantation, where the upper organic phase containing the solvents is recycled and the lower, aqueous phase is sent to wastewater treatment.
This new, sustainable process utilizes lignin, a biobased raw material, which is typically used in low-value applications, such as energy-input in a pulp mill.62 The developed process is energy efficient requiring low heating duty. The energy consumption of the plant was estimated with an Aspen Plus simulation model. The total heating and cooling duty of the plant was estimated to be 15–20 MW. The distillation unit, separating epichlorohydrin and pentane for recycling, corresponds to more than 85% of the energy. However, the separation and recovery efficiencies are high, thus reducing the need for additional solvent make-up and consequently, the operating costs. The process requires only conventional process equipment, and hence the industrial scalability and technical and economic feasibility is favorable.
The operating costs of the EKL production plant were estimated to be approximately 0.6 € per kg in a prefeasibility study of the process. Further information about the operating costs can be found in the ESI (Note 7†). With an estimated price of 0.9–1.4 € per kg for phenol formaldehyde adhesives, an 1:1 EKL:LNP adhesive has the potential to be competitive to phenol formaldehyde resins.63 A full techno-economic assessment of the large-scale production of EKL will be elucidated in a future publication.
Environmental impact of the process
In order to approximate the environmental sustainability of the EKL process, a LCA calculation was carried out according to the ISO 14044 standard.64 A brief overview will be presented here, and the full assessment process along with complete results will be described in a forthcoming article. More information about the methods is found in the ESI Note 8.† The system boundary for the calculations was a cradle to gate model in which calculations begin from harvesting of the wood material and finish at the final product, of which 1 ton was determined as the functional unit. The impacts were calculated using GaBi software and the Ecoinvent LCI database along with some literature values for kraft lignin production65 and competing renewable resins based on kraft lignin, soy and tannin.66 The impact assessment methodology used was ReCiPe 2016 Midpoint (H). The impact categories were global warming potential and fossil depletion.The calculations were made for two cases: a standalone plant and a plant integrated to a kraft pulp mill. In the case of the standalone mill, all energy is produced by Finnish power grid mix and in the integrated mill all heat comes from waste low-pressure (LP) steam and electricity is 90% from surplus electricity from the pulp mill and 10% from Finnish grid mix. The calculations were made on the grounds of laboratory experiments and process modelling, so these results should not be treated as exact references of the industrial process but rather as an implication of how the environmental impacts are formed from the laboratory to industrial production. The results were compared to three resins from fossil-based feedstocks (melamine-formaldehyde (MF), urea-formaldehyde (UF) and phenol-formaldehyde (PF) resins) as well as three adhesives from renewable feedstocks.
The global warming potential for the EKL process was 3580 kg(CO2-eq.) per t(product) for the standalone plant and approximately 1550 kg(CO2-eq.) per t(product) for the integrated plant. As can be seen from Fig. 7a, most of the effect for the standalone plant comes from heat production, hence integrating to a pulp mill reduces the impact to a level lower than any fossil or renewable based product. This is due to the carbon-heavy energy production mix of Finland. The fossil depletion for the EKL process was 1323 kg(oil-eq.) per t(product) for the standalone plant and 583 kg(oil-eq.) per t(product) for the integrated plant. Fig. 7b shows that, again, most of the impact is from heat production. Similarly, the impact for the integrated plant is markedly lower than any of the competing products. It can be seen that the EKL process is more environmentally feasible than competing processes, especially when it has access to a renewable energy source such as a kraft pulp mill. This holds true for both fossil-based products as well as resins from renewable materials.
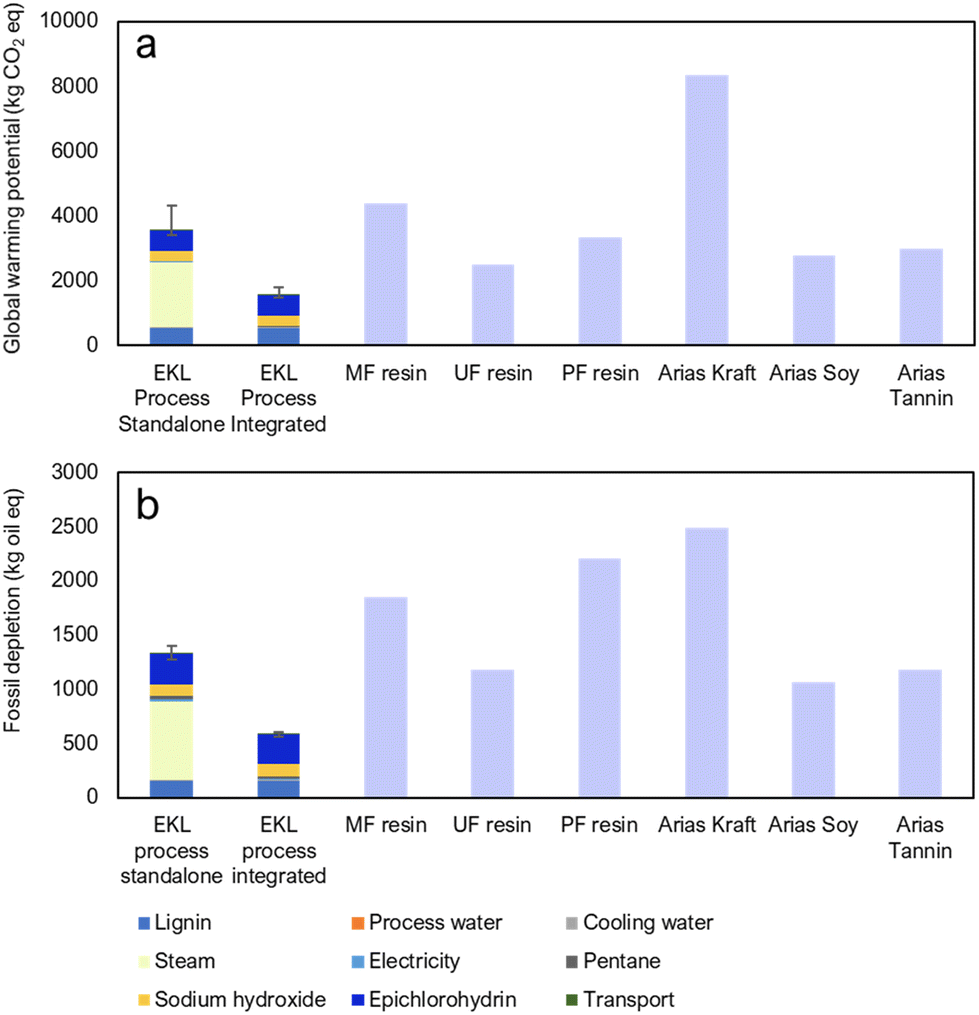 | ||
| Fig. 7 (a) Global warming potential and (b) fossil depletion of epoxidized kraft lignin production compared to melamine formaldehyde (MF), urea formaldehyde (UF), phenol formaldehyde (PF), and three renewable resins. | ||
In the actual manufacture of wood-panels (like plywood), differences in the compositing step (veneer stacking and hot-pressing) between EKL:LNP and traditional PF resins must be taken into consideration. Although the hot-press time is similar, the EKL:LNP adhesive is hot-pressed at a 15–20 °C higher temperature compared to PF resins (145–150 °C compared to ∼130 °C) to reach its maximum strength, which would increase energy consumption in the compositing step by ca. 10–15%. The energy consumption of the board forming step according to relevant LCAs is approximately 15–18% of the energy consumption of the overall production process,67–69 which would translate to a 1.5–3% increase in electricity overall for EKL:LNP adhesives compared to PF resins. The veneer manufacturing, including debarking, compositing (including the production of the resin), and drying, is responsible for the majority of energy depletion.69 However, compositing releases approximately 40% of the overall pollutants, mainly formaldehyde when using phenol-formaldehyde resins.68,69 LCAs of plywood and hardwood production list phenol and formaldehyde as the major cause of human toxicity potential, freshwater ecotoxicity potential, terrestrial ecotoxicity potential, photochemical oxidants potential, and significant causes of marine aquatic ecotoxicity potential and abiotic depletion potential.69,70 Epichlorohydrin is nevertheless also a hazardous chemical with toxic and mutagenic health effects. However, the release of toxic volatile compounds, including epichlorohydrin, is avoided in the EKL:LNP adhesive. After the concentration and product recovery step there are only trace amounts of epichlorohydrin left, and no volatile solvents are needed to dissolve the EKL. The recyclability of wood-panels could be also considered. Plywood can be recycled after use, and is usually then shredded into chips that can be used in particle boards or incinerated for energy. By utilizing lignin in the adhesive, the length of its lifetime is improved immensely compared to the current practice, where lignin is incinerated immediately after being isolated from lignocellulosic biomass. In contrast to PF adhesives, the EKL:LNP adhesive is unlikely to form covalent bonds with the cellulose in the wood plies. Therefore, it could even be possible to use recycled plywood chips as raw materials in pulp mills if they are made with the EKL:LNP adhesive.
Discussion
Wood panels are becoming increasingly important in the construction industry now that concrete is starting to be replaced with wood, but adhesives for wood panels are still fossil-based and sometimes even toxic. In this paper, we present a new water-based lignin-epoxidation strategy and a method for using a mixture of epoxidized lignin and colloidal lignin particles (LNPs) as composite adhesive for wood panels. The synthesis of epoxidized kraft lignin (EKL) relied on interfacial catalysis at the oil water interface,38–40 which reduced the reaction time and homopolymerization significantly. Kraft lignin could be used as such without need for depolymerization or fractionation, and the overall process was comparatively simple.26–29,31 Moreover, we demonstrate that high degrees of epoxidation can be achieved in only a few minutes at the interface between aqueous media and organic solvent without need for any additional phase-transfer catalysts, such as tetramethylammonium chloride. In that, these findings open completely new possibilities in the field of epoxidation strategies and epoxy chemistry in general.The water solubility of the epoxidized kraft lignin enabled its use in adhesives with aqueous dispersions of LNPs. The adhesive mass, hot-press time, and hot-press temperature were optimized for high shear strength, which was above 13 MPa in the best conditions. The wet strength was very good compared to that of existing lignin-based resins despite containing no formaldehyde. We furthermore noticed that cured EKL:LNP showed excellent thermal stability and was even flame resistant. Now, the EKL:LNP-composite's aging by environmental factors, such as UV-light, air, changes in temperature and humidity, and moisture, should be evaluated to guarantee its long-term performance when applied. Especially moisture is challenging for adhesives, as it causes wood to swell and deform.50,51 For exterior use, the adhesive needs to retain its adhesive performance in changing moisture-levels for prolonged periods of time. Because the product does not contain any harmful volatile compounds, the material is a promising replacement for phenol-formaldehyde resins. The EKL solutions’ viscosity and concentration are easily adjusted to resembling existing phenol-formaldehyde resins, making them applicable without large investments for changes in production lines. A pre-feasibility study showed that the production of EKL in large scale seems feasible and economical, with an estimated manufacturing cost of ca. 600 € per t. A life cycle assessment on the process showed low impact values in all relevant categories. Given the adhesive's sustainable production and lignin-content over 80%, the adhesive could be a big step towards finally achieving high performance sustainable bio-based adhesives.
Methods
Materials
BioPiva 100 kraft LignoBoost® lignin, purchased from UPM (Finland) was used in this study. Epichlorohydrin (≥99%), dimethylformamide (99.8%), pyridine (99.8%), N-hydroxy-5-norbornene-2,3-dicarboxylic acid imine (97.0%), chromium(III) acetylacetonate (≥98.0%), 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (95%), chloroform-D (99.8%), phenol (≥99.5), and formaldehyde (36.5–38.0%) was purchased from Sigma-Aldrich. Pentane (≥99%) was obtained from Riedel-de Haën. Analytical grade acetone, tetrahydrofuran (AnalaR NormaPur, ≥99.5%) and sodium hydroxide granules (≥99.1%) was purchased from VWR chemicals. p-Nitrobenzaldehyde (≥97.0%) by Fluka Analytical was purchased from Sigma-Aldrich. All chemicals were used as received.Preparation of epoxidized lignin (EL)
A 10 wt% lignin solution was prepared by dissolving 1.0 gram of lignin in 9 g of 1 M NaOH. Then, 7 ml epichlorohydrin, was heated to 43 °C in a glass beaker while being stirred. The reaction beaker was insulated using two layers of aluminum foil around the beaker and on top, with a small opening for the thermometer and addition of lignin solution. The lignin solution was added to the epichlorohydrin at a rate of 1 ml per minute, until all had been added. Note that proper ventilation should be used when working with epichlorohydrin, as well as protective glasses and gloves. The temperature of the lignin solution was 25 °C. The reaction was stopped 11 minutes after the first addition of lignin-solution. The temperature change in the solution during the reaction was carefully monitored. Some variations in temperature were observed in different batches, but the reaction was considered successful only if the temperature increased to a maximum of 52–54 °C after 3–4 minutes and then stabilized at 48–50 °C after 10 minutes. Each batch was analyzed with FTIR spectroscopy to ensure structural similarity between batches, and deviant batches were excluded from any further experiments. After the reaction, the reaction beaker was cooled to room temperature in an ice bath, after which 7 ml pentane was added and a transparent organic phase was formed on top of the dark aqueous phase. The contents of the beaker were calmly stirred for approximately 2 minutes, and the aqueous phase was isolated. The epoxidized kraft lignin (EKL) was precipitated by the addition of acetone (≥3 times the volume of the EKL solution). The liquid was removed, and the precipitate was washed with acetone once more to ensure removal of unreacted epichlorohydrin. The precipitated EKL was re-dissolved in 2 ml deionized water. The EKL was then carefully rotary evaporated at max 40 °C and 40 mbar for 5–10 minutes to remove the acetone. The epichlorohydrin could also be extracted without using acetone with three consecutive pentane-extractions. In this method, 12 ml pentane is added into a separation funnel, followed by the EKL solution. Then, the funnel was closed and shaken thoroughly for ca. 10 seconds, to combine the organic and the aqueous phases. The phases were allowed to separate for four minutes, after which the organic phase was isolated and rotary evaporated to recover the epichlorohydrin. The pH of EKL solutions was ∼12.5. The EKL was cooled to 0 °C and used within 6 hours. The concentration of EKL solutions in adhesives were 18–22 wt%. The EKL used for NMR analysis was neutralized and dried after the acetone washing and ground into a fine powder to prevent homopolymerization. The epoxy index (EI) and the epoxy equivalent weight (EEW) was calculated via the number of epoxide groups in moles per gram of sample (NE) obtained through 1H-NMR and HSQC, using eqn (1) and (2).71 The epoxy content (EC) is defined as the mass percent of epoxy groups in an epoxy resin, and is calculated according to eqn (3).| EI = NE × 1000 g | (1) |
| EEW = (NE)−1 | (2) |
| EC = NE × 43.04 g mol−1 × 100% | (3) |
Preparation of lignin particles
First, a solution of 4.7 wt% BioPiva 100 kraft lignin in a mixture of 30.7 wt% ethanol, 34.6 wt% analytical grade THF and 30.0 wt% deionized water, was prepared and stirred for 3 hours in a closed flask. The solution was decanted to remove insoluble aggregates. To make particles sized 400–500 nm in diameter, the solution was swiftly added to 1.72 times its mass of deionized water that was stirred into a vigorous vortex using a magnet stirrer, forming a dispersion of colloidal lignin particles (LNPs). To form particles sized 600–700 nm in diameter, the same method was used but with slightly calmer stirring. To form particles of 200–300 nm in diameter, the solution was swiftly added to 2 times its mass of deionized water, and stirred aggressively using an overhead stirrer. The dispersions were stirred for at least 15 minutes after the addition of water. The solvent-concentration was reduced to below 15 wt% by dilution with deionized water. Then, the dispersions could be concentrated by ultrafiltration, using an OptiFilter CR250 (Valmet Technologies Inc.), and the remaining organic solvents were removed by rotary evaporation at 50 °C and 50 mbar. The LNP dispersion, free of organic solvents, could be further concentrated by centrifugation at 4000 rpm for one hour. The supernatant was removed, and the particles were re-dispersed by mixing and vortexing. The concentration of dispersions used in adhesive samples were adjusted to ca. 30 wt%Dynamic light scattering (DLS)
The particle size of LNPs and EKL was measured using a Zetasizer Nano-ZS90 (Malvern, U.K.) instrument. A zeta dip cell was used to measure the surface charge, and the ζ-potential values were calculated from the obtained electrophoretic mobility data using the Smoluchowski model. DLS particle size distribution data and ζ-potentials of LNPs can be found in Fig. S23 and Table S4.†Fourier-transform infrared (FTIR) spectroscopy
FTIR spectroscopy with attenuated total reflectance (ATR) module was used to characterize the EKLs and unreacted kraft lignin. The measurements were done right after sample preparation using a Spectrum Two™ FT-IR (PerkinElmer, Massachusetts, USA) and the background extraction was done in air. Only dry samples were used for measurement. The resolution was 1 cm−1, and 10 scans were collected for each measurement between 4000–600 cm−1. Min–max normalization was used for all samples. The normalizations were done between 4000–600 cm−1 (full spectra), 1700–600 cm−1 (fingerprint region) and 1024–890 cm−1 (epoxy region).31P-NMR
Dried EKL was used for all analyses within 3 hours from its synthesis. Samples were solvent exchanged with acetone immediately after synthesis, air-dried until seemingly fully dry, and then vacuum dried at 0.03 mbar for at least 1 hour before re-dissolution. 30 mg of each sample was accurately weighed into separate glass vials. First, 150 μl dimethylformamide and 100 μl pyridine were added to the samples. Then 200 μl of the internal standard solution containing 10.2 μmol N-hydroxy-5-norbornene-2,3-dicarboxylic acid imine dissolved in 1.6 parts pyridine and 1 part CDCl3 (v/v) was added, followed by the addition of 50 μl of a solution containing 1.6 μmol chromium(III) acetylacetonate, also dissolved in pyridine and CDCl3 (1.6:1, v/v). Right before the measurement was conducted, 150 μl 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, as phosphitylation agent, was added dropwise with approximately 2 seconds between each drop, while the sample was stirred using a magnet. Last, 300 μl chloroform-d was added and the vials were stirred for approximately 10 minutes.
The 31P-NMR spectra were measured using a Bruker Avance 400 MHz spectrometer (Massachusetts, USA) equipped with 5 mm broadband (BB) probe. A total of 128 scans were performed using the pulse sequence zgig with a pulse angle of 90°, an acquisition time of 1 second, and a pulse relaxation delay of 5 seconds. The spectral width was 185 ppm. The chemical shifts were referenced to the phosphitylated water signal at 132.2 ppm in CDCl3. Different regions were integrated according to literature and compared to internal standard.72
1H-NMR and HSQC NMR
EL samples were prepared as described previously, and were dried by rotary evaporation after being precipitated and washed with acetone. Then, 30 mg of the preapred samples were dissolved in 400 μl D2O and 600 μl DMSO-d6. para-Nitrobenzaldehyde (PNB) (∼5 mg) was used as an internal standard.73 The solution was warmed to ∼40 °C and stirred in a water bath for a 3–5 minutes to dissolve the PNB. The residual solvent peak (δC 39.52/δH 2.50) was used as an internal reference. In the case of EKL, samples were prepared the same day as analyzed. The measurement was performed using a Bruker Avance 400 MHz spectrometer equipped with 5 mm BB probe with the standard pulse sequence zgig with 16 scans, a pulse angle of 90°, an acquisition time of 1 second and a pulse delay of 5 seconds. Spectral width of 16 ppm was used.The HSQC spectra were acquired from the same samples with a Bruker standard pulse sequence (hsqcetgpsisp.2) with the following conditions: 11 ppm spectral width in F2 (1H) dimension with 1024 data points and 215 ppm spectral width in F1 (13C) dimension with 256 increments of 32 scans and using 2 seconds pulse delay and J(C,H) 145 Hz (D24 0.89 ms).
Gel permeation chromatography
The molecular weight distribution was analyzed using gel permeation chromatography. The measurements were performed by using an Agilent 1100 (Agilent Technologies, USA) high-performance liquid chromatography HPLC-system with GPC-Add-on. The samples were prepared by first dissolving 2 mg of lignin or epoxidized lignin in 2 ml 0.1 M NaOH solution. The samples were stirred until dissolved and stored at room temperature overnight. The samples were then filtered with 0.2 μm syringe filters before measurement. A 0.1 M NaOH solution was used as eluent with 0.8 ml min−1 elution rate. Three different PSS MCX columns (Polymer Standards Service, Germany), all with particle sizes of 5 μm and with porosities of 1000, 500, and 100 Å columns were used in that order, and both UV (at 280 nm) and RI detectors were used for monitoring. A series of polystyrene standards with molar masses 1000, 3420, 6430, 15800, 20500, 65400, and 208000 g mol−1 as well as ascorbic acid and NaCl, all with a concentration of 2 mg ml−1 in 0.1 M NaOH, were used for calibration and were prepared and measured the same way as the other samples. A blank 0.1 M NaOH sample was also used. A third order polynomial fit was used for the calibration curve. From the data, Mn (number-average molecular weight), Mw (weight-average molecular weight) and polydispersity PDI (Mw/Mn) values were obtained.
Mechanical tests and sample preparation
A reference phenol-formaldehyde resin was prepared. First, 1.24 g phenol was weighed and dissolved in 2.0 g formaldehyde solution (37 wt%, 0.75 g pure formaldehyde). Then, 0.25 g of an aqueous solution 50 wt% NaOH was added. The solution was heated to 95 °C and maintained at this temperature while stirring for about 20 minutes to initiate polymerization, visible by an increased viscosity. The solution was cooled to 80 °C, and 0.25 g of a 50 wt% aqueous NaOH solution was added. The stirring was continued for 10 minutes, whereafter another 0.25 g of the same NaOH solution was added. The solution was allowed to stir for an additional 10 minutes at 60 °C, achieving a honey-like viscosity, and was then frozen until used. The samples adhered with the phenol-formaldehyde resin were prepared similarly to those prepared with EKL, using a spread of 300 g m−2 pressed at 145 °C with a pressure of 1.1 MPa for 5 minutes. A commercial two-component bisphenol-A-based epoxy resin was applied following the manufacturer's instructions using a spread of 300 g m−2. The epoxy resin was pressed without heat at 1.1 MPa for 20 minutes, and then allowed to cure for an additional day with no pressure applied before tensile testing.
| Parameter | Low | Center | High | Additional |
|---|---|---|---|---|
| a The curing time was 5 minutes for all samples. | ||||
| Model 1 | ||||
| Time (minutes) | 1 | 3 | 5 | — |
| Temperature (°C) | 100 | 130 | 160 | — |
| Dry spread (g m−2) | 200 | 350 | 500 | — |
| Model 2 | ||||
| Time (minutes) | 3 | 5 | 7 | — |
| Temperature (°C) | 130 | 160 | 190 | — |
| Dry spread (g m−2) | 350 | 500 | 650 | — |
| Model 3 | ||||
| Temperature (°C) | 130 | 145 | 160 | — |
| Dry spread (g m−2) | 50 | 100 | 150 | 200 |
Thermal stability
Glass transition temperatures (Tg) and curing temperatures were examined using a DSC 6000 (PerkinElmer, Massachusetts, USA) differential scanning calorimeter. Pierced aluminum sample crucibles were used for the analysis. The samples were first air dried until they were seemingly fully dry, and then vacuum dried at 0.03 mbar at least one hour prior to the measurement. The samples were analyzed between 50 °C to 250 °C at a heating rate of 5 °C per minute. Every measurement began with a 3-minute isothermal step at 50 °C. Heat-degradation was examined using a Q500 thermogravimetric analyzer (TA Instruments, Delaware, USA) with a heating rate of 10 °C per minute in nitrogen from 30–700 °C. Thermal heat resistance indexes (THRI) were calculated according to eqn (4).| THRI = 0.49 × (Td5% + 0.6 × (Td30% − Td5%)) | (4) |
where Td5% and Td30% are the temperatures at which 5% and 30% of the initial mass of the sample has been lost, respectively.
Microscopy
The cured EKL:LNP and EKL adhesives were characterized using a Phenom Pure 5G tabletop scanning electron microscope (SEM) (Thermo Scientific, Massachusetts, U.S.A.) using a standard sample holder. Before SEM analysis, the samples were coated with a gold–palladium mixture (Au80Pd20) with a Q150R S plus rotary-pumped coater (Quorum Technologies, U.K.). The used sputter current, sputter time, and tooling factor was 20 mA, 20 s, and 1.0, respectively. The EKL aggregate size was analyzed using an Axio Vert A1 inverted light microscope (Zeiss, Germany). The epoxidation reaction liquid content was analyzed using an Olympus (Japan) SZX10 light microscope.Author contributions
KAH planned all experiments and performed most of them, as well as analyzed data and prepared the text and figures. SF designed the large-scale production process, developed the simulation model, calculated the operating costs of the process and prepared the related text and figures with scientific guidance from RPBA and PO. IS helped in preparing adhesive samples and performed adhesive strength testing together with KAH. IS also performed some of the FTIR, DLS, DSC and TGA measurements. PN was responsible for NMR procedures. AP was responsible for the LCA. NF provided guidance for experimental plans and reviewed and revised manuscript and provided scientific guidance. MÖ reviewed and revised manuscript as well as provided scientific guidance.Conflicts of interest
The authors declare the following competing financial interest(s): authors Alexander Henn, Susanna Forssell, Rahul Bangalore Ashok, Nina Forsman, Pekka Oinas, and Monika Österberg declare that they have a financial interest in the development and commercialization of the research presented in this article. Aalto University has filed a provisional patent application (FI20217173).Acknowledgements
This work was a part of the Academy of Finland's Flagship Programme under projects no. 318890 and 318891 (Competence Center for Materials Bioeconomy, FinnCERES). KAH thanks FinnCERES for their financial and organizational support. KAH, SF, NF and RPBA thank Business Finland for financing via the LignoSphere project (project number: 2117744). PN acknowledges receiving funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 869993. The authors thank Dr Heidi Henrickson for her generous help with proofreading and linguistic guidance. KAH also thanks Dr Tao Zou for sharing his knowledge and expertise in the characterization of lignin-epoxy materials. Lastly, KAH and SF are grateful for Dr Esko Tirronen's excellent and generous guidance in plant and process design.References
- J. F. Bastin, Y. Finegold, C. Garcia, D. Mollicone, M. Rezende, D. Routh, C. M. Zohner and T. W. Crowther, Science, 2019, 364, 76–79 CrossRef PubMed.
- D. Peñaloza, M. Erlandsson and A. Falk, Constr. Build. Mater., 2016, 125, 219–226 CrossRef.
- M. Asif, T. Muneer and R. Kelley, Build. Environ., 2007, 42, 1391–1394 CrossRef.
- K. H. Kim, S. A. Jahan and J. T. Lee, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2011, 29, 277–299 CrossRef CAS PubMed.
- A. Duong, C. Steinmaus, C. M. McHale, C. P. Vaughan and L. Zhang, Mutat. Res., Rev. Mutat. Res., 2011, 728, 118–138 CrossRef CAS PubMed.
- A. M. Bahmanpour, A. Hoadley and A. Tanksale, Rev. Chem. Eng., 2014, 30, 583–604 CAS.
- L. A. Heinrich, Green Chem., 2019, 21, 1866–1888 RSC.
- C. A. Hochwalt and M. Plunguian, US2168160A, 1936.
- E. E. Novotny and C. J. Romieux, US1886353A, 1922.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
- C. H. Ludwig and A. W. Stout, US3658638A, 1970.
- S. Pietarinen, S. Valkonen and O. Ringena, JP2019108554A, 2013.
- G. A. Doering, US5202403A, 1992.
- S. Kalami, M. Arefmanesh, E. Master and M. Nejad, J. Appl. Polym. Sci., 2017, 134, 45124 CrossRef.
- S. Pietarinen, S. Valkonen and O. Ringena, JP6588433B2, 2013.
- T. M. McVay, G. F. Baxter and F. C. Dupre Jr., US5866642A, 1992.
- J. A. Jurvelin, R. D. Edwards, M. Vartiainen, P. Pasanen and M. J. Jantunen, J. Air Waste Manage. Assoc., 2003, 53, 560–573 CrossRef CAS PubMed.
- M. G. D. Baumann, L. F. Lorenz, S. A. Batterman and G. Z. Zhang, For. Prod. J., 2000, 50, 75–82 CrossRef CAS PubMed.
- G. McGwin, J. Lienert and J. I. Kennedy, Environ. Health Perspect., 2010, 118, 313–317 CrossRef CAS PubMed.
- P. Kurttio, H. Norppa, H. Jarventaus, M. Sorsa and P. Kalliokoski, Scand. J. Work, Environ. Health, 1993, 19, 132–134 CrossRef CAS PubMed.
- K. H. Kim, S. A. Jahan and J. T. Lee, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2011, 29, 277–299 CrossRef CAS PubMed.
- S. Elbein, Natl. Geogr. Mag., 2020.
- J. Salazar and J. Meil, J. Cleaner Prod., 2009, 17, 1563–1571 CrossRef.
- J. Savolainen and M. Oinas, YIT Corp. News, 2021.
- R. J. Li, J. Gutierrez, Y. L. Chung, C. W. Frank, S. L. Billington and E. S. Sattely, Green Chem., 2018, 20, 1459–1466 RSC.
- T. Malutan, R. Nicu and V. I. Popa, BioResources, 2008, 3, 1371–1376 Search PubMed.
- C. Asada, S. Basnet, M. Otsuka, C. Sasaki and Y. Nakamura, Int. J. Biol. Macromol., 2015, 74, 413–419 CrossRef CAS PubMed.
- C. Sasaki, M. Wanaka, H. Takagi, S. Tamura, C. Asada and Y. Nakamura, Ind. Crops Prod., 2013, 43, 757–761 CrossRef CAS.
- F. Ferdosian, Z. Yuan, M. Anderson and C. Xu, RSC Adv., 2014, 4, 31745–31753 RSC.
- A. Zafar and D. Areskogh, WO2015044893 (A1), 2016.
- A. Jablonskis, A. Arshanitsa, A. Arnautov, G. Telysheva and D. Evtuguin, Ind. Crops Prod., 2018, 112, 225–235 CrossRef CAS.
- F. Ferdosian, Z. Yuan, M. Anderson and C. C. Xu, J-FOR, 2012, 2, 11–15 Search PubMed.
- J. R. Gouveia, G. E. S. Garcia, L. D. Antonino, L. B. Tavares and D. J. Dos Santos, Molecules, 2020, 25, 2513 CrossRef CAS PubMed.
- E. Santacesaria, R. Tesser, M. Di Serio, L. Casale and D. Verde, Industrial and Engineering Chemistry Research, 2010, vol. 49, pp. 964–970 Search PubMed.
- A. Almena and M. Martín, Ind. Eng. Chem. Res., 2016, 55, 3226–3238 CrossRef CAS.
- C. Asada, M. Fujii, A. Suzuki and Y. Nakamura, Biomass Convers. Biorefin., 2021, 1–10 Search PubMed.
- N. E. El Mansouri, Q. Yuan and F. Huang, BioResources, 2011, 6, 2492–2503 CAS.
- T. Kitanosono and S. Kobayashi, Chem. – Eur. J., 2020, 26, 9408–9429 CrossRef CAS PubMed.
- M. F. Ruiz-Lopez, J. S. Francisco, M. T. C. Martins-Costa and J. M. Anglada, Nat. Rev. Chem., 2020, 4, 459–475 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
- E. I. Evstigneev, Russ. J. Appl. Chem., 2010, 83, 509–513 CrossRef CAS.
- S. R. Sandler, W. Karo, J.-A. Bonesteel and E. M. Pearce, Polymer Synthesis and Characterization, A laboratory manual, 2018, vol. 1 Search PubMed.
- E. Bryan, Chemistry and Technology of Epoxy Resins, 1993 Search PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- M. Farooq, Z. Tao, J. J. Valle-Delgado, M. H. Sipponen, M. Morits and M. Österberg, Langmuir, 2020, 36, 15592–15602 CrossRef CAS PubMed.
- I. V. Pylypchuk, P. A. Lindén, M. E. Lindström and O. Sevastyanova, ACS Sustainable Chem. Eng., 2020, 8, 13805–13812 CrossRef CAS.
- J. Luo, J. Luo, C. Yuan, W. Zhang, J. Li, Q. Gao and H. Chen, RSC Adv., 2015, 5, 100849–100855 RSC.
- S. Chen, Y. Chen, Z. Wang, H. Chen and D. Fan, RSC Adv., 2021, 11, 11724–11731 RSC.
- D. Yang, J. L. Diflavio, E. Gustafsson and R. Pelton, Nord. Pulp Pap. Res. J., 2018, 33, 632–646 CrossRef CAS.
- A. Ranta-Maunus, Wood Sci. Technol., 1975, 9, 189–205 CrossRef.
- M. Wolcott, F. Kamke and D. Dillard, Wood Fiber Sci., 1994, 26, 496–511 CAS.
- W. Zhang, Y. Ma, Y. Xu, C. Wang and F. Chu, Int. J. Adhes. Adhes., 2013, 40, 11–18 CrossRef CAS.
- X. Wang, Y. Deng, Y. Li, K. Kjoller, A. Roy and S. Wang, RSC Adv., 2016, 6, 76318–76324 RSC.
- X. Wang, X. Chen, X. Xie, Z. Yuan, S. Cai and Y. Li, Nanomaterials, 2019, 9, 1409 CrossRef CAS PubMed.
- M. Hasegawa, Y. Hoshino, N. Katsura and J. Ishii, Polymer, 2017, 111, 91–102 CrossRef CAS.
- P. Sheth, S. Mestry, D. Dave and S. Mhaske, J. Coat. Technol. Res., 2020, 17, 231–241 CrossRef CAS.
- K. A. Henn, N. Forsman, T. Zou and M. Österberg, ACS Appl. Mater. Interfaces, 2021, 13, 34793–34806 CrossRef CAS PubMed.
- M. Patel, S. Mestry, S. P. Khuntia and S. Mhaske, J. Coat. Technol. Res., 2020, 17, 293–303 CrossRef CAS.
- Y. Wang, S. Wang, C. Bian, Y. Zhong and X. Jing, Polym. Degrad. Stab., 2015, 111, 239–246 CrossRef CAS.
- R. P. Bangalore Ashok, P. Oinas, K. Lintinen, G. Sarwar, M. A. Kostiainen and M. Österberg, Green Chem., 2018, 20, 4911–4919 RSC.
- T. Leskinen, M. Smyth, Y. Xiao, K. Lintinen, M. L. Mattinen, M. A. Kostiainen, P. Oinas and M. Österberg, Nord. Pulp Pap. Res. J., 2017, 32, 586–596 CrossRef CAS.
- R. P. Bangalore Ashok, Y. Xiao, K. Lintinen, P. Oinas, M. A. Kostiainen and M. Österberg, Colloids Surf., A, 2020, 587, 124228 CrossRef CAS.
- Price quotation of phenol-formaldehyde adhesive, https://www.alibaba.com/showroom/phenol+formaldehyde+adhesive.html?fsb=y&IndexArea=product_en&CatId=&SearchText=phenol+formaldehyde+adhesive&isGalleryList=G.
- ISO (International Standardisation Organisation), Environmental management—Life cycle assessment—Requirements and guidelines, 2006, vol. 14044 Search PubMed.
- E. Bernier, C. Lavigne and P. Y. Robidoux, Int. J. Life Cycle Assess., 2013, 18, 520–528 CrossRef CAS.
- A. Arias, S. González-García, S. González-Rodríguez, G. Feijoo and M. T. Moreira, Sci. Total Environ., 2020, 738, 140357 CrossRef CAS PubMed.
- D. Kaestner, Master's thesis, University of Tennessee, 2015.
- M. Puettman, D. Kaestner and A. Taylor, CORRIM Report: Life Cycle Assessment for the Production of PNW Softwood Plywood, 2016 Search PubMed.
- L. Jia, J. Chu, L. Ma, X. Qi and A. Kumar, Int. J. Environ. Res. Public Health, 2019, 16, 2037 CrossRef CAS PubMed.
- S. González-García, G. Feijoo, P. Widsten, A. Kandelbauer, E. Zikulnig-Rusch and M. T. Moreira, Int. J. Life Cycle Assess., 2009, 14, 456–466 CrossRef.
- European Committee for Standardization, 1999.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- C. Gioia, G. Lo Re, M. Lawoko and L. Berglund, J. Am. Chem. Soc., 2018, 140, 4054–4061 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional NMR FTIR, and DSC data of EKL and LNPs, dynamic light scattering (DLS) results of LNPs, and additional results from mechanical tests and surface response models (with statistical data) including a discussion of the mode of adhesive failure and images of representative samples. Video of close-ranged ignition test of EKL:LNP-coated adhesive joint and lifting 10 kg with an adhesive sample glued with a spread of 100 g m−2 and respective curing time and temperature of 5 min and 130 °C. See DOI: https://doi.org/10.1039/d2gc01637k |
| This journal is © The Royal Society of Chemistry 2022 |