
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of a green chemoenzymatic cascade for scalable synthesis of bio-based styrene alternatives†
Philipp
Petermeier
 a,
Jan Philipp
Bittner
b,
Simon
Müller
b,
Emil
Byström
c and
Selin
Kara
*ad
a,
Jan Philipp
Bittner
b,
Simon
Müller
b,
Emil
Byström
c and
Selin
Kara
*ad
aBiocatalysis and Bioprocessing Group, Department of Biological and Chemical Engineering, Aarhus University, 8000 Aarhus C, Denmark. E-mail: selin.kara@bce.au.dk
bInstitute of Thermal Separation Processes, Hamburg University of Technology, 21073 Hamburg, Germany
cSpinChem AB, Tvistevägen 48C, 90736 Umeå, Sweden
dInstitute of Technical Chemistry, Leibniz University Hannover, 30167 Hannover, Germany
First published on 8th August 2022
Abstract
As renewable lignin building blocks, hydroxystyrenes are particularly appealing as either a replacement or addition to styrene-based polymer chemistry. These monomers are obtained by decarboxylation of phenolic acids and often subjected to chemical modifications of their phenolic hydroxy groups to improve polymerization behaviour. Despite efforts, a simple, scalable, and purely (chemo)catalytic synthesis of acetylated hydroxystyrenes remains elusive. We thus propose a custom-made chemoenzymatic route that utilizes a phenolic acid decarboxylase (PAD). Our process development strategy encompasses a computational solvent assessment informing about solubilities and viable reactor operation modes, experimental solvent screening, cascade engineering, heterogenization of biocatalyst, tailoring of acetylation conditions, and reaction upscale in a rotating bed reactor. By this means, we established a clean one-pot two-step process that uses the renewable solvent CPME, bio-based phenolic acid educts and reusable immobilised PAD. The overall chemoenzymatic reaction cascade was demonstrated on a 1 L scale to yield 18.3 g 4-acetoxy-3-methoxystyrene in 96% isolated yield.
Introduction
Following the call for sustainable production, increasing research focus is put on the utilization of renewable resources and on efficient environmentally benign manufacturing methods. Such efforts are of particular importance for industrial sectors of high mass throughputs, such as the plastics industry with a global annual production of 368 million tons (2019).1 The recently soaring number of publications about polymerization and material characteristics of bio-based polymers is emblematic of said potential. Among these new strategies many build on the use of hydroxystyrene monomers.2–13 Their great appeal resides in their aromatic hydroxy groups, as they offer scope for modifications to tune mechanical properties and to design self-healing materials.3,4 In addition, hydroxystyrenes are common building blocks in lignocellulosic biomass, meaning that they can be derived from the most abundant renewable feedstock there is.14 Typical precursors of hydroxystyrenes are lignin-based phenolic acids such as ferulic acid, p-coumaric acid, caffeic acid, and sinapinic acid. These can be obtained from, e.g., rice bran,15 corn stover,16 or sugarcane bagasse17 and need to undergo decarboxylation to obtain the monomers of interest. Additionally, van Schijndel et al.10 reported a protocol in which 4-hydroxybenzaldehydes are converted into mentioned phenolic acids by means of a green Knoevenagel reaction with malonic acid followed by decarboxylation. This allows to funnel an even broader spectrum of bio-based resources towards hydroxystyrenes. These, however, are reactive and are thought to undergo uncontrolled oligomerization complicating their isolation, storage and polymerization.8–10 Although controlled polymerization of unprotected hydroxystyrenes has been demonstrated, it requires demanding conditions (−40 °C, N2 atmosphere)6 or yields drastically lower conversion than styrene.2 This is due to their protic phenol groups, which are detrimental to many conventional polymerization techniques. Therefore, chemical modifications are used to make these monomers available for a wider range of applications.8 Both acetylation and silylation have been found to produce monomers that enable efficient polymerization and result in well controlled molecular weight distributions and material properties.8–13 For cases in which these protection groups are of transient utility, selective deprotection protocols have been established.7,8,10–13 Analogous to the well-known duo of polyvinyl alcohol and polyvinyl acetate, both protected and deprotected polyhydroxystyrenes are of interest.Given the range and potential of functionalized polystyrenes, the question arises to what extent the respective monomers are accessible via both scalable and sustainable synthesis. For a detailed assessment of a wider range of published routes to lignin-derived monomers, we refer to a recent review by Fadlallah et al.18 For the specific case of protected hydroxystyrenes we want to highlight the work by Takeshima et al.12 and van Schijndel et al.10 In 2018, Takeshima12 presented a straightforward one-pot two-step process characterized by cheap reagents and good overall yields on multi-gram scale (Fig. 1A). However, in this route the tertiary amine base is used in excess, and it is both flammable and harmful. Also, the solvent DMF is classified as ‘undesirable’ due to its toxicity according to Pfizer's solvent selection guide.19 In 2020, van Schijndel10 followed up with a two-step process in which the first step is run in a green solvent and the second one under neat conditions (Fig. 1B). Also, no excess amounts of amine are used and the inorganic catalyst in the second reaction step is environmentally benign and harmless. Yet, the use of inert gas atmosphere and intermediate work-up steps add practical constraints to this proposed synthetic route.
 | ||
| Fig. 1 Comparison of synthetic routes to acetyl-protected hydroxystyrenes. The approaches are classified according to their use of chemical (CHEMO) and biocatalytic (BIO) steps. The use of unspecific R and R′ residues illustrate that above synthetic strategies apply to a range of phenolic acid substrates such as ferulic acid, p-coumaric acid, caffeic acid, and sinapinic acid. The given atom efficiencies were calculated for the reference substrate ferulic acid, the reported reagent equivalents and under disregard for purely catalytic species such as HMTA, BsPAD and NaOAc. Abbreviations: DMF, N,N-dimethylformamide; HMTA, hexamethylenetetramine; PC, propylene carbonate; BsPAD, phenolic acid decarboxylase from Bacillus subtilis; CPME, cyclopentyl methyl ether. | ||
We hypothesized that these limitations could be circumvented by a tailored combination of a more active decarboxylation catalyst, milder reaction conditions and a compatible reaction medium. As a renewable, non-toxic, and highly active catalyst, phenolic acid decarboxylase from Bacillus subtilis (BsPAD) was the biocatalyst of our choice to embark on this process development. Herein, we describe our systematic efforts to design an alternative, scalable, chemo-enzymatic route to access bio-based styrene alternatives in an environmentally friendly and efficient manner.
Results and discussion
Computational solvent assessment
In the spirit of time and resource-efficiency, we started our process development with computational means to estimate fundamental constraints such as substrate solubilities in a range of common and renewable solvents. Here, we used the highly predictive capabilities of COSMOtherm 2020 (ref. 35–38) to screen thermodynamic properties of ferulic acid, p-coumaric acid, caffeic acid, and sinapinic acid. Since these structurally related compounds gave comparable trends, only results for the model compound ferulic acid are shown here, whereas the others are compiled in the ESI (Fig. S3 and 4†). The calculated solubilities of ferulic acid are illustrated in Fig. 2 for both dry and water saturated (from now on called ‘wet’) solvents. Humidification was considered, as enzyme-catalysed reactions in non-conventional media often depend on the availability of residual water. The results indicate that also substrate solubility is significantly depending on co-solubilised water. Moreover, we found that (i) all substrates have drastically increased solubility in 2MeTHF, PC, 1-octanol, MTBE, and CPME compared to water (∼1 g L−1), (ii) all substrates are more soluble in polar organic solvents than in apolar ones, (iii) hydroxy groups of substrates reduce their solubility in screened solvents, and (iv) methoxy groups of substrates increase their solubility in screened solvents (ESI, Fig. S3 and 4†). | ||
| Fig. 2 Calculated solubilities of ferulic acid in selected dry and water saturated (wet) organic solvents at +30 °C using COSMOtherm 2020. 2MeTHF: 2-methyltetrahydrofurane; MTBE: methyl tert-butyl ether. | ||
To ensure that COSMOtherm 2020 gave reliable trends, they were successfully validated against experimental data (ESI, Fig. S5†). As detailed in the Experimental section, calculations of substrate solubilities in organic media were based on reference solubilities in water. This data was not accessible for labile hydroxystyrenes, which is why we resorted to an alternative method: We estimated enthalpies of fusion by the Joback group-contribution method,20 and used reported melting points10,21 to derive their respective solubilities. For details we refer to the computational methodology and ESI.† Although this approach is based on sensitive approximations and is thus less reliable, it still indicates manyfold greater solubilities of decarboxylation products. Hence, solubilities of hydroxystyrenes are not a limiting factor for the design of the first catalytic step.
In addition, partition coefficients of phenolic acids and hydroxystyrenes were calculated for the same series of immiscible organic solvent–water pairs (Fig. 3). They show that hydroxystyrenes are more preferably extracted into organic phases than phenolic acids, which is in line with their predicted higher solubilities. Given our objective of an efficient synthesis at high product titers, these findings indicate the use of either a fed-batch process or a two-liquid phase system with in situ product removal to alleviate solubility limitations. In the first case enzyme activity should not suffer from product inhibition, in the second case the enzyme should not suffer from severe interfacial toxicity, and in both the enzyme must be productive in the presence of the given solvent. As such, the next step was to systematically investigate the solvent tolerance of BsPAD.
 | ||
Fig. 3 Calculated partition coefficients at infinite dilution (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Korg/aq) of ferulic acid and its decarboxylated derivative 4-vinylguaiacol in equilibrated biphasic organic solvent-water mixtures at +30 °C using COSMOtherm 2020. Korg/aq) of ferulic acid and its decarboxylated derivative 4-vinylguaiacol in equilibrated biphasic organic solvent-water mixtures at +30 °C using COSMOtherm 2020. | ||
Experimental solvent screening
First, all four substrates were subjected to a kinetic assay in potassium phosphate buffer (50 mM KPi, pH 6.0, 5 vol% DMSO). In agreement with literature on purified wild-type BsPAD,22 our lyophilised whole-cell preparations displayed the highest activity with ferulic acid, followed by p-coumaric acid and caffeic acid (ESI, Fig. S6†). Only sinapinic acid was not converted. Still, as Morley et al.23 showed, the BsPAD I85A variant – with an individual substitution in the active site – accepts sinapinic acid. Knowing that all four phenolic acids can be decarboxylated in a biocatalytic manner by BsPAD, ferulic acid (FA) was selected as model substrate for all further investigations. Next, a standardised assay (100 mM FA, 100 mg L−1 whole-cells, 16 h, +30 °C, 1000 rpm, triplicates) was used to analyse a range of water-miscible and water-immiscible solvents (ethylene glycol, Cyrene™, PC, DMF, THF, 2MeTHF, MTBE, CPME) and binary eutectic mixtures such as choline chloride:ethylene glycol (ChCl:EG (1:2)), choline chloride:urea (ChCl:U (1:2)), choline chloride:glycerol (ChCl:Gly (1:2)). None of which worked when used directly. Accordingly, water-immiscible solvents were equilibrated over water at +25 °C and others were charged with a comparably low amount of 5 vol% water before the assay was repeated. Whilst again no conversion was observed for most solvents, conversions improved for wet ethers such as THF (<1%), 2MeTHF (13 ± 5%), MTBE (99.8 ± 0.1%), and CPME (≥99.9%). Interestingly, we found an inverse correlation between the dipole moments of varyingly bulky ethers and the conversion of ferulic acid by BsPAD. Based on this, we hypothesize that less polar ethers are less capable of competing with structural water of the enzyme and at the same time they (R–O–R) do not introduce functional groups that are structurally too foreign to the natural aqueous (H–O–H) environment.
At this point it shall be noted that reference experiments in KPi buffer at the same substrate load did not exceed conversions of 44 ± 5%. This is rationalized as follows: the buffer capacity was raised to 500 mM and the buffer pH prior substrate addition to 7.3 to dissolve 100 mM FA. By this means, the initial pH was within the operational range of the enzyme. However, as the decarboxylation of the acidic substrate proceeded, the pH increased, stalling conversion. It is important to note that aqueous reaction systems allow, in principle, to solubilise and convert the same amount of substrate, but either require active pH control24 or high buffer concentrations to do so effectively. As these results show, such resource demanding measures can be avoided by proper solvent choice. Additionally, it shall be mentioned that the product 4-vinylguaiacol (4VG) precipitated in the aqueous reaction media but was well solubilised in the other solvents.
Finally, comparing the two best performing organic solvents CPME and MTBE, we selected CPME as starting point for further cascade engineering due to its lower heat of vaporization (69.2 kcal kg−1vs. 81.7 kcal kg−1), comparable low peroxide formation tendency, lower water uptake potential, its promising prospect as eco-friendly solvent,25 and its wider operational window, i.e., liquidity range (ϑb: +105 °C vs. +55 °C). The last point is of special importance as it allows for subsequent reactions at elevated temperatures without the need for a solvent switch or pressurisation.
Cascade engineering
In a first effort at this stage, we aimed to verify computational results that point towards increased substrate solubility in wet CPME. As shown in Fig. 4, co-solubilisation of water in CPME indeed increases ferulic acid solubility.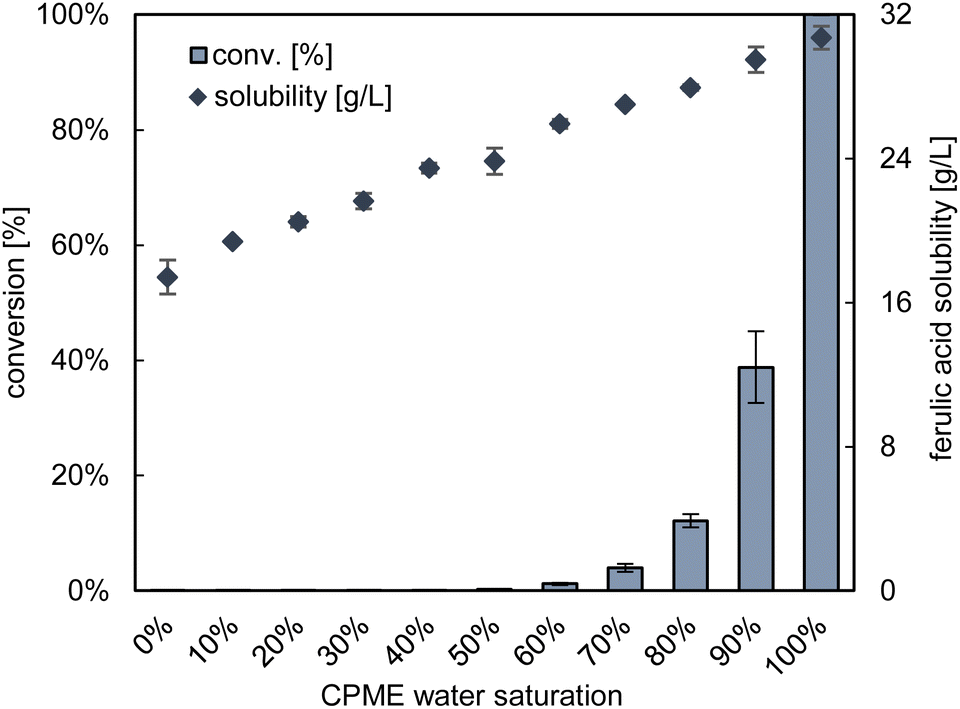 | ||
| Fig. 4 Influence of dissolved water in CPME on the solubility of ferulic acid and the activity of BsPAD on its decarboxylation to 4-vinylguaiacol at +30 °C. A saturation of 100% refers to CPME that was equilibrated over water for 24 h at room temperature. 0% refers to CPME dried over molecular sieves under the same conditions. Blends gave intermediate levels of 10–90%. Reaction conditions: 100 mM FA, 100 mg L−1 whole-cells, wet CPME, 1000 rpm, +30 °C, 5.5 h, experimental duplicates. | ||
Also, the influence of water on the catalytic performance of BsPAD was investigated and the results in Fig. 4 identify water saturation as a highly sensitive influencing parameter.
For water saturations below 60%, hardly any product formation was observed even after 24 h. More detailed progress curves are given in Fig. S7.† For saturation levels of 60% and 70%, we not only observed drastically reduced initial enzymatic activity but also pronounced degressive reaction trends. This indicates ongoing enzyme deactivation over the course of the reaction. With an exponential decay fit, we estimated the respective half-life times (t1/2) with 11.8 h and 17.0 h. Thus, increased CPME water saturation benefits substrate solubility as well as enzyme activity and stability.
We carefully controlled that the observed activity trend (Fig. 4) is not caused by contamination of CPME after contact with desiccant. These efforts confirmed water content as the determining factor while discarding the desiccant as influencing factor (ESI, Fig. S8†).
After identification of wet CPME (100% water saturation, Fig. 4) as suitable solvent for the first step of the reaction cascade, we focused on its extension to the second step. Here, the direct continuation of the overall cascade in the same reaction medium would be most straightforward. Building on the green approach by van Schijndel et al.,10 we decided for NaOAc as inorganic base catalyst. We wondered to which degree co-solubilised water competes with hydroxystyrenes for the carboxylic anhydride and thereby affects overall conversion. To answer this, a 100 mM solution of 4VG in wet CPME was prepared in situ using whole-cells. The solution was dried over desiccant, re-wetted with varying amounts of water, and subjected to chemical acetylation. The results are illustrated in Fig. 5. Interestingly, they indicate that a minimum of 40% water saturation is even beneficial for the overall reaction performance. We conjecture that these small amounts of water promote solubilisation of the inorganic base which fosters its activity. Moreover, the fact that we did not see any significantly reduced conversion even at full water saturation might be ascribed to the low solubility of water in CPME (∼0.7 wt% at room temperature). In any case, this clearly demonstrates that hydroxystyrenes can be directly acetylated in wet CPME, eliminating the need for an intermediate drying step or solvent switch.
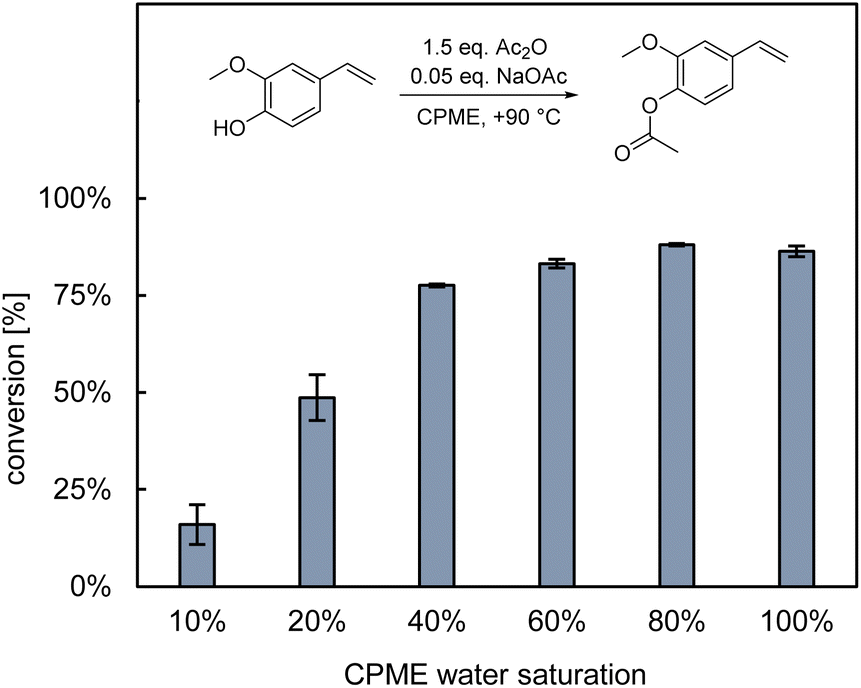 | ||
| Fig. 5 Influence of dissolved water in CPME on the base-catalysed chemical acetylation of 4-vinylguaiacol. Reaction conditions: 100 mM 4VG, wet CPME, 150 mM Ac2O, 5 mM NaOAc, 1000 rpm, +90 °C, 1 h, experimental duplicates. | ||
Since both Fig. 4 and 5 only refer to substrate conversion, it is worth noting that the entire reaction cascade proceeded without noteworthy formation of by-products.
Next, we considered heterogenization of the biocatalyst as it would come with two main benefits: first, removing the protein load prior to acetylation prevents unspecific consumption of acetylating agent by unwanted side reactions. Second, a heterogenized biocatalyst allows compartmentalization, recovery, and reuse, all potentially enhancing its practical applicability and productivity.
Enzyme immobilization
Considering our objective of a simple, yet still robust and scalable process, we screened widely available methacrylate Lifetech™ carries. Their broad selection of diverse carriers with well-defined specifications allows finding a commercial enzyme support with high process compatibility.All immobilisations were conducted with crude cell-free extract (CFE) and assessed with regards to immobilization yield, specific activity, and activity yield. To obtain initial rates and specific activities, standardized activity assays were set up as reported for both the CFE and the immobilized enzyme. The assay conditions were defined in line with substrate solubility, enzyme pH tolerance, and zero-order kinetics according to Michaelis–Menten (cS ≫ KM). Immobilization conditions and results are summarized in Table 1. In general, we report very high immobilization yields but widely varying biocatalytic activities. To this end, amino-functionalized carriers outperformed their epoxy-functionalized counterparts in all cases. Also, activities are indirectly correlated with bead size and directly correlated with pore diameter. This signifies mass transfer limitations, a conclusion that was further substantiated by even greater specific activities and activity yields with ground carrier beads. Yet, the use of increasingly smaller beads does not pose a particularly practical solution as it runs into other problems associated with packing densities and back pressure in flow systems or containment measures in compartmentalized applications. We thus favoured the smallest commercially available beads with the biggest pores. Lastly, we evaluated the resilience to enzyme leaching in aqueous media. No evidence of leached active enzyme was found for any of the covalently immobilised preparations (ESI, Fig. S10†) indicating strong covalent linkages. However, both ionic immobilized preparations of 8315F and 8415F showed enzyme leaching. Here the results indicate more severe leaching with the shorter C2 linker on 8315F. Noteworthy, by switching to wet CPME as reaction medium, leaching was effectively suppressed (ESI, Fig. S11†). From these findings, we conclude that covalent immobilization of BsPAD on small-sized amino carrier beads with large pore diameters (8315F and 8415F) give the most viable heterogenized biocatalyst preparations. It shall also be noted that at +4 °C these immobilized enzyme preparations have a remarkable, months-long storage stability.
| Carrier specifications | Experimental specifications | |||||||
|---|---|---|---|---|---|---|---|---|
| ECR | Type | Linker | Pore Φ [Å] | Bead size [μm] | Immobilization | Immobilization yield [%] | Specific activity [U g−1] | Activity yield [%] |
| 8285 | Epoxy | C2 | 400–600 | 250–1000 | Covalent | 97 | 15 ± 1 | 0.9 ± 0.1 |
| 8209F | Epoxy | C2 | 600–1200 | 150–300 | Covalent | 95 | 99 ± 9 | 6.1 ± 0.5 |
| 8215F | Epoxy | C2 | 1200–1800 | 150–300 | Covalent | 96 | 81 ± 3 | 4.9 ± 0.2 |
| 8309F | Amino | C2 | 600–1200 | 150–300 | Covalent | 93 | 166 ± 9 | 11.8 ± 0.6 |
| 8315F | Amino | C2 | 1200–1800 | 150–300 | Covalent | 96 | 213 ± 9 | 14.8 ± 0.6 |
| 8315F | Amino | C2 | 1200–1800 | 150–300 | Ionic | 92 | 186 ± 20 | 14.1 ± 1.5 |
| 8315F | Amino | C2 | 1200–1800 | Ground | Ionic | 92 | 244 ± 4 | 19.9 ± 0.3 |
| 8409F | Amino | C6 | 600–1200 | 150–300 | Covalent | 93 | 181 ± 0 | 12.7 ± 0.0 |
| 8415F | Amino | C6 | 1200–1800 | 150–300 | Covalent | 95 | 213 ± 3 | 15.1 ± 0.2 |
| 8415F | Amino | C6 | 1200–1800 | 150–300 | Ionic | 91 | 209 ± 4 | 15.9 ± 0.3 |
| 8415F | Amino | C6 | 1200–1800 | Ground | Ionic | 93 | 469 ± 5 | 38.3 ± 0.4 |
Finally, we evaluated the reusability of such immobilized BsPAD in a repetitive batch experiment. This was done in duplicates, using wet CPME, a substrate load of 100 mM FA, and 4 g L−1BsPAD-8415F. We saw exceptionally high catalytic activity in the first batch (>1000 U g−1) followed by reduced but constant performance in three subsequent batches (ESI, Fig. S9†). In all consecutive batches, full conversion was achieved in less than 90 min.
Knowing about the catalytic activity and reusability of immobilized BsPAD in wet CPME, we turned again to the second cascade step. Our next objective was to optimize chemical acetylation of hydroxystyrenes in water saturated CPME in absence of residual biocatalyst.
Optimization of acetylation step
To efficiently tailor reaction parameters to fast conversions, we opted for a design of experiment (DoE) approach in which the concentration of base (NaOAc), reagent (Ac2O), and temperature (T) were considered as influencing factors. The analysed ranges of these factors were based on literature10 and initial experiments:– NaOAc [0.02–0.08 eq.],
– Ac2O [1.0–3.0 eq.],
– Reaction temperature [70–100 °C].
Other parameters such as substrate concentration (100 mM 4VG), solvent (wet CPME), volume, mixing, and reaction time (10 min) were kept constant. The software MODDE®13 (Sartorius Stedim Analytics AB, Umeå, Sweden) was used for experimental design. After performing the experiments, the data were subjected to multiple linear regression (MLR) modelling to calculate the coefficients of each single factor (NaOAc, Ac2O, T) and their interaction terms. During model refinement two interaction terms (NaOAc2 and NaOAc·Ac2O) were dropped due to insignificance, yielding a model with no lack-of-fit and given summary of fit: R2 = 0.978, Q2 = 0.948, validity = 0.994. The relative contributions of model parameters on reaction conversion can best be seen from the model coefficients (Fig. 6 bottom). Inspection of those identifies temperature and Ac2O as most impactful parameters. In contrast, NaOAc is of comparable low priority (cf. Fig. S12†). Thus, Fig. 6 illustrates the impact of temperature and Ac2O on the conversion of 4VG to 4-acetoxy-3-methoxystyrene (AMS) for a reaction time of 10 min and 0.08 eq. NaOAc. Both the curvature of the surface area plot as well as the model coefficients give another valuable insight: although the linear Ac2O term is as determining as the linear temperature term, its quadratic term bears a negative coefficient. This suggests that its initially strong positive influence gradually declines at higher Ac2O equivalents. This was further substantiated by a second model fit for a reaction time of 20 min as illustrated in Fig. S13.†
 | ||
| Fig. 6 3D response plot of refined MLR model illustrating influence of temperature and Ac2O equivalents on conversion of 4VG to AMS. Data for a reaction time of 10 min and 0.08 eq. NaOAc. Scaled and centred coefficients of model terms are given. software: MODDE®13.0.1, Umetrics, SARTORIUS. | ||
Following these findings, we again subjected 4VG to acetylation in water saturated CPME but this time in the absence of NaOAc. Thereby we aimed to ascertain whether the use of NaOAc is required at all. The experiment yielded more than 4-fold reduced conversion, corroborating the use of minimum amounts of NaOAc.
With these insights and in line with our objective for resource efficiency, we decided to (i) minimize use of NaOAc, (ii) limit the use of Ac2O equivalents, and (iii) maximize temperature within operational limits.
Preparative scale implementation
Lastly, we demonstrated the overall bio-chemo cascade on a synthetically tangible 1 L scale. To facilitate easy biocatalyst recovery after the first reaction step, we decided for a rotating bed reactor in which the heterogeneous biocatalyst beads are enclosed in a cage-like containment that also serves as agitator. Just as on small scale, water saturated CPME was charged with 100 mM ferulic acid and the use of 5 g L−1BsPAD-8415F beads at 30 °C resulted in full conversion (>99%) in 3 h. The exchange of the stirring module with a propeller stirrer removed the biocatalyst and prepared the reactor setup for the second reaction step. Following the guidelines of the previous chapter, the reactor content was heated to 90 °C by use of the water jacket and acetylation of 4VG to AMS successfully achieved by use of 2.0 eq. Ac2O and 0.03 eq. NaOAc. The desired product was obtained in high yield (18.3 g, 96%) and high HPLC purity (99%) after only two washing steps, demonstrating the practical utility and scalability of the presented synthetic strategy.Conclusions
In this work, we describe the systematic development of a tailor-made chemoenzymatic cascade to produce bio-based styrene alternatives in a solvent that can likewise be derived from biogenic resources.25 Besides the deliberate choice of renewable starting materials, our catalytic route is characterized by negligible by-product formation, no intermediate workup steps as well as by simple product isolation. In particular, our mild reaction conditions and solvent system proved beneficial as they stabilize reactive hydroxystyrene intermediates and thereby drastically simplify the overall route. This provides heightened intrinsic process robustness, obviates the need for protective atmospheres, and minimizes equipment requirements for both reaction and workup.Even if the synthetic strategy was easily scaled up by a factor of 1000 from test tubes to a 1 L benchtop reactor and the product obtained in high yield, the volumetric productivity (1.6 g L−1 h−1) and the product titer (19 g L−1) still have potential for further improvement. Therefore, we see measures to refine the reaction setup and reaction control on preparative scale as necessary next steps. Above all, these will need to establish the fed-batch strategy highlighted by our solubility calculations, as well as, e.g., optimized biocatalyst loading, stirring rate or rotating bed basket type. These efforts shall be compiled and discussed in an upcoming technical work.
Experimental
General information
1H and 13C NMR spectra were recorded in CDCl3 at +25 °C on a 600 MHz Bruker NMR. Chemical shifts are given in parts per million (ppm) relative to the residual CHCl3 peak (1H: δ = 7.24 ppm, 13C: δ = 77.2 ppm). Thin layer chromatography was carried out on silica gel 60 F254 plates and compounds were visualised by alkaline permanganate. The phenolic acid decarboxylase used in this study was heterologously expressed in E. coli cells as described in the ESI.†Materials
All chemicals, materials and solvents were obtained from commercial suppliers (Acros Organics, Alfa Aesar, Sigma-Aldrich, VWR International, Carl-Roth GmbH, Merck KGaA, Thermo Fisher Scientific, TCI Europe, Biowest) and used as received: trans-ferulic acid (FA, ≥99% grade), p-coumaric acid (pCA, ≥98%), caffeic acid (CA, ≥98%), K2HPO4 (≥99%), KH2PO4 (≥99%), NaOH (≥98%, p.a.), Na2HPO4·2 H2O (≥99.5%), citric acid monohydrate (≥99%), NaOAc (≥99%), NaOAc·3H2O (99.8%), H3PO4 (85%), Na2SO4 (anhydrous, 99%), aqueous HCl (37%), acetic anhydride (≥99%), formic acid (98–100%), molecular sieves 3A (4 to 8 mesh), cyclohexane (CH, ≥99.5%), n-heptane (≥99.0%), toluene (≥99.9%), cyclopentyl methyl ether (CPME, ≥99.0%, stabilized), Cyrene™ (≥98.5%), propylene carbonate (PC, for synthesis), N,N-dimethylformamide (DMF, ≥99.5%), 2-methyltetrahydrofuran (2MeTHF, ≥99%), dimethyl sulfoxide (DMSO, ≥99.5%), methyl tert-butyl ether (MTBE, ≥99%), tetrahydrofuran (THF, ≥99%, BHT stabilized), choline chloride (ChCl, 99%), glycerol (Gly, 99.5%), ethylene glycol (EG, ≥98.0%), urea (U, ≥99.5%), ethyl acetate (EtOAc, ≥99.5%) Karl-Fischer solvent CM (Art. No. 85461.290), Karl-Fischer reagent TitrANT 5 (Art. No. 85468.320), HYDRANAL™ Standard 5.0 (Art. No. 34813), Bradford assay solution (Art. No. B5702), bovine serum albumin (BSA, lyophilized). Acetonitrile (≥99.95%, UHPLC grade) used for HPLC was purchased from VWR International. Lifetech™ carriers for enzyme immobilization were generously provided by Purolite Ltd (UK): ECR8285, ECR8209F, ECR8215F, ECR8309F, ECR8315F, ECR8409F, ECR8415F. Glutaraldehyde (50% solution in water) used for activation of amino carriers was obtained from Sigma-Aldrich.Computational methodology
All calculations of organic solvent-water partition coefficients at infinite dilution (logKorg–aq), solubilities (xi,S) of substrate and intermediates in mixed solvent systems were performed using BIOVIA COSMOtherm 2020.35–38 The conformer sets were generated with COSMOconf v3.0 and the COSMO calculations were performed using Turbomole v6.6 using the TZVPD-FINE parameter set. Infinite dilution activity coefficients γ∞i of substrates and intermediates were calculated in different organic–aqueous biphasic systems. Using the definition of the partition coefficient Korg−aq = xorgi/xaqi and the equilibrium condition xorgi·γorgi = xaqi·γaqi allows calculation of infinite dilution partition coefficient by: | (1) |
For calculating the solid–liquid equilibrium (SLE) following equation was employed according to Tosun:26
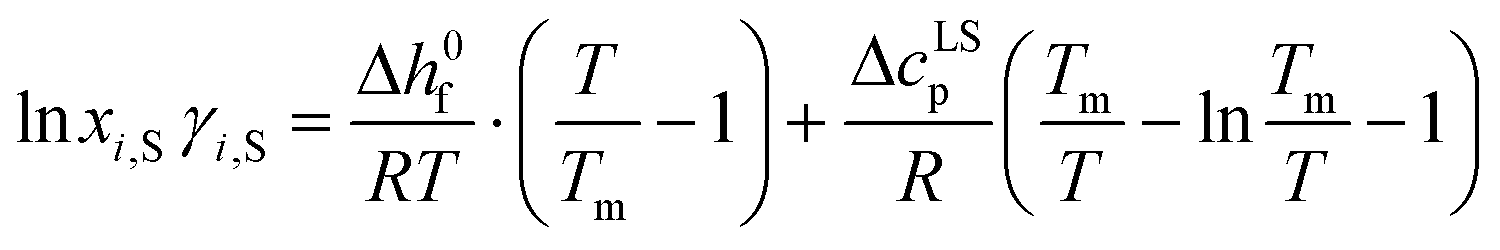 | (2) |
| lnxLi,S·γLi,S = lnxrefi,S·γrefi,S | (3) |
As all considered substrates are thermally labile compounds and disintegrate upon melting, available literature values for enthalpies of fusion were not deemed particularly reliable. Thus, the latter strategy using reference solubilities of phenolic acids in pure water at +30 °C was used to estimate substrate solubilities in various dry and wet organic solvents. Thereby COSMOtherm accounts for the transfer from the aqueous reference system to the organic solvent mixture. For input data see Table 2.
Literature data on thermodynamic liquid–liquid equilibrium mixtures were used for calculations of systems with water-saturated organic solvents.30–34 For validation (see Fig. S5†), solvents were equilibrated over water at room temperature (24 h), their moisture content analysed, the water-saturated solvents used for substrate solubility tests, and the measured water contents used for calculations.
Substrate solubility evaluation
500–1500 μL of the liquid phase for which substrate solubility was to be analysed were transferred to a 4 mL glass vial, stirred with a magnetic stirring bar at 450 rpm, and tempered to +30 °C. Substrate was added in excess and suspensions were stirred for 2–5 h. The mixtures were transferred to 1.5 mL microcentrifuge tubes, centrifuged (13400 rpm, 2 min), supernatants sampled and diluted in a series of dilutions (10×, 100×, 500×, 1000×) using water/acetonitrile (1/1). These dilutions were subjected to HPLC-UV analytics. From peaks within calibration range, solubilities were derived in [mM]. Considering the straightforward experimental setup, we consider our results as practical solubilities relevant for process design, which may differ slightly from precise thermodynamic solubilities after more extended equilibration periods.
Validation of computational efforts required experimental solubilities in [gFA/kgsolvent] and thus densities of ferulic acid saturated solvents at +30 °C. For this, 300 μL samples of tempered saturated ferulic acid solutions were transferred to 1.5 mL glass vials and weighed. From weight and volume experimental densities were derived. All solutions were prepared in duplicates and measured in triplicates.
Karl-Fischer titration
Quantitative determination of water content in organic media was done using a volumetric titrator of the TitraLab KF1000 Series (Hach Lange GmbH, Germany). Prior measurement of any samples, the titrator setup was validated using the HYDRANAL™ Standard 5.0. For both standard and samples, the KF titration solvent was titrated with the KF reagent to remove excess moisture. Samples and standards were drawn in 2–10 mL syringes, weighed, added to the dry titration solution and the syringes weighed again. From the mass difference before and after sample injection the mass fractions of water were derived from the titration results in [wt%].Activity assay in aqueous media
This standard assay was performed in KPi buffer (50 mM, pH 6.0) using the model substrate ferulic acid at an initial concentration of 10 mM ensuring zero-order kinetics. The assay was used for both free and immobilized enzyme. For the former, 900 μL KPi buffer were mixed with 50 μL FA stock (200 mM in DMSO) and tempered to +30 °C on a Thermoshaker. To start the reaction, 50 μL resuspended enzyme preparation (1 mg mL−1 whole-cells or CFE in KPi buffer) were added and the mixture subjected to +30 °C and 1000 rpm. For immobilized enzyme, roughly 5 mg enzyme preparation were resuspended in 1425 μL KPi buffer and tempered to +30 °C. To start the reaction, 75 μL FA stock (200 mM in DMSO) were added and the mixture subjected to +30 °C and 1000 rpm. Samples of 100 μL were quenched and diluted by addition to 900 μL water/acetonitrile (1/1), vortexed, centrifuged (13200 rpm, 2 min), and subjected to HPLC analysis. Standard sampling times for whole-cell and CFE preparations were 120, 160, 200, 240, and 285 s, whereas for immobilized enzyme preparations they were 2, 5, 10, 15, 20, 30, 45, 60, and 90 min. All derived kinetic data is based on substrate depletion. For immobilized enzyme, initial rates were obtained by progress curve analysis as described below.
Activity assay in CPME
This standard assay was performed in wet CPME (equilibrated over water at room temperature) using the model substrate ferulic acid at an initial concentration of 100 mM. The assay was used for both free and immobilized enzyme. For the former, 19 mg FA were dissolved in 900 μL of wet CPME and tempered to +30 °C on a Thermoshaker. To start the reaction, 100 μL of resuspended enzyme preparation (1 mg mL−1 in wet CPME) were added and the mixture subjected to +30 °C and 1000 rpm. For immobilized enzyme, roughly 5 mg enzyme preparation were charged with 1.5 mL of FA solution (100 mM in wet CPME, tempered to +30 °C) and the mixture subjected to +30 °C and 1000 rpm. Samples of 10 μL were quenched and diluted by addition to 990 μL water/acetonitrile (1/1), vortexed, centrifuged (13200 rpm, 2 min), and subjected to HPLC analysis. Standard sampling times for whole-cell and CFE preparations were within 2–80 min, whereas for immobilized enzyme preparations they were 2, 5, 10, 15, 20, 30, 45, 60, and 90 min. All derived kinetic data is based on substrate depletion. For immobilized enzyme, initial rates were obtained by progress curve analysis as described below.
Protein assay
A 500 mg L−1 BSA solution was prepared in a 15 mL Falcon tube by dissolving 4–6 mg BSA in aqueous NaCl solution (0.15 M). Further standards (400, 300, 200, and 100 mg L−1) were prepared by diluting the 500 mg L−1 stock with 0.15 M NaCl solution. The NaCl solution was also used as a blank (0 mg L−1 BSA) and – if necessary – to dilute samples. Samples and standards were measured analogously by given procedure: 20 μL of sample were transferred to a cuvette and mixed with 980 μL of ready-to-use Bradford assay solution. The mixture was incubated at room temperature (5 min) and absorbance measured at 595 nm using a U-1900 HITACHI spectrophotometer (Hitachi, Japan). The calibration was done new for every measurement series with an R2 > 0.995.Covalent enzyme immobilization on epoxy carriers
Prior to enzyme immobilization, carrier beads were washed three times with KPi buffer (50 mM, pH 6.0) using a resin/buffer ratio of 1/1 (w/v). A solution of 25 mg mL−1 crude CFE (protein content ∼50%) was prepared in immobilization buffer (KPi, 1 M, pH 6.0). Washed carrier beads were mixed with the crude enzyme solution in a resin/buffer ratio of 1/4 (w/v). The slurry was gently mixed for 19 h at room temperature on a self-made end-over-end mixer before it was left without mixing at +4 °C for another 24 h. Lastly, the mixtures were spun down (13200 rpm, 1 min) the supernatant removed, the beads washed twice with KPi buffer (50 mM, pH 6.0) at a resin/buffer ratio of 1/2 (w/v) each, and all supernatants (original and after both washing steps) combined. The protein content in the collected liquid phase was determined using the protein assay reported. Wet immobilized enzyme preparations were stored in closed vessels at +4 °C.
Covalent enzyme immobilization on amino carriers
Prior to enzyme immobilization, carrier beads were washed three times with KPi buffer (50 mM, pH 6.0) using a resin/buffer ratio of 1/1 (w/v). Next, the beads were incubated with glutaraldehyde buffer (2 vol% in KPi buffer, 50 mM, pH 6.0) in a resin/buffer ratio of 1/4 (w/v). The slurry was mixed for 1 h at room temperature on a self-made end-over-end mixer before the supernatant was removed and the beads washed 4 times with KPi buffer (50 mM, pH 6.0) at a resin/buffer ratio of 1/1 (w/v). A solution of 25 mg mL−1 crude CFE (protein content ∼50%) was prepared in immobilization buffer (KPi, 50 mM, pH 6.0). Washed carrier beads were mixed with the crude enzyme solution in a resin/buffer ratio of 1/4 (w/v). The slurry was gently mixed for 18 h at room temperature on a self-made end-over-end mixer. Lastly, the mixtures were spun down (13200 rpm, 1 min) the supernatant removed, the beads washed twice with KPi buffer (50 mM, pH 6.0) at a resin/buffer ratio of 1/2 (w/v) each, and all supernatants (original and after both washing steps) combined. The protein content in the collected liquid phase was determined using the protein assay reported. Wet immobilized enzyme preparations were stored in closed vessels at +4 °C.
Ionic enzyme immobilization on amino carriers
Prior to enzyme immobilization, carrier beads were suspended in deionized water and the mixture titrated with diluted HCl until a stable pH of 5.8–6.0 was obtained. The resin was filtered and rinsed with deionized water. A solution of 25 mg mL−1 crude CFE (protein content ∼50%) was prepared in immobilization buffer (KPi, 50 mM, pH 6.0). Washed carrier beads were mixed with the crude enzyme solution in a resin/buffer ratio of 1/4 (w/v). The slurry was gently mixed for 24 h at room temperature on a self-made end-over-end mixer. Lastly, the mixtures were spun down (13200 rpm, 1 min) the supernatant removed, the beads washed twice with KPi buffer (50 mM, pH 6.0) at a resin/buffer ratio of 1/2 (w/v) each, and all supernatants (original and after both washing steps) combined. The protein content in the collected liquid phase was determined using the protein assay reported. Wet immobilized enzyme preparations were stored in closed vessels at +4 °C.
Immobilization yield
The immobilization yield was derived from the offered amount of protein in the immobilization buffer and the remaining protein concentration in the collected supernatants after immobilization.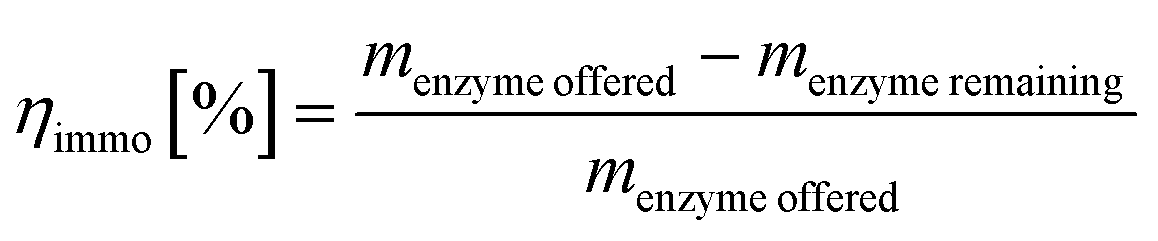 | (4) |
Progress curve analysis
All kinetic data is based on substrate depletion [S]t. For flat and highly linear progress curves, initial rates were obtained by linear regression of data points at <10% conversion. Progress curves with steep initial reaction progress and subsequent slowdown were fitted with first order kinetics assuming no significant inhibition or deactivation phenomena.| [S]t = [S]0·e−kE·t | (5) |
The fits of experimental datasets gave coefficients of determination of R2 = 0.994 ± 0.002, indicating excellent descriptive power of employed model and justifying above assumptions. From these, initial rates ri were calculated:
| ri = kE·[S]0 | (6) |
Specific activity and activity yield
Specific activities were derived from initial rates ri in [μmol L−1 min−1] according to following correlation: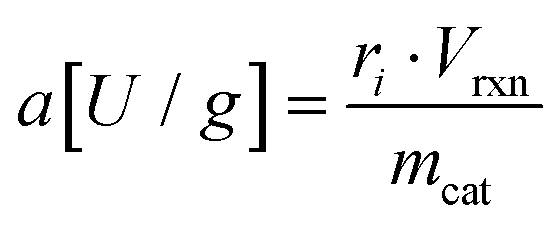 | (7) |
This equation was used for free and immobilized enzyme. From their respective specific activities under otherwise same reaction conditions activity yields were calculated.
 | (8) |
High-performance liquid chromatography with mass spectrometry
Both qualitative and quantitative analysis were done with an Agilent Technologies 1260 Infinity II high-performance liquid chromatography system (Germany) consisting of a G7111B quaternary pump, a G7129A vial sampler, a G7116A multi-column thermostat, and a G7117C DAD detector interfaced with a G6125C LC-ESI-MS detector. The column used was a Kinetex® 5 μm C18 100 Å LC column (250 × 4.6 mm, Phenomenex). The sample injection volume was 10 μL, the column oven temperature 35 ± 0.5 °C, and the total flow rate 0.7 mL min−1. A solvent mixture of water/acetonitrile (45/55) with 0.1 vol% formic acid was used for isocratic separation of analytes. The 300 nm channel of the diode array detector (DAD) was used for calibration and quantification of ferulic acid, p-coumaric acid and caffeic acid. For sinapinic acid the 310 nm channel was used. A ferulic acid standard curve and an exemplary chromatogram showing separation of FA, 4VG and AMS are provided in the ESI.†Synthesis of 4-acetoxy-3-methoxystyrene
A SpinChem V3 double wall glass reactor was charged with 1.0 L CPME, the solvent tempered to 30 °C, stirred at 500 rpm with a propeller mixer, and saturated with water by addition of 6.3 mL MilliQ water. Next, 19.4 g ferulic acid (0.1 mol) were added and the mixture was stirred at 700 rpm and 30 °C for 2.5 h. 5 g wet BsPAD-8415F beads were mixed with 10 g untreated ECR8415F beads and filled into a SpinChem RBR S2 rotating bed basket. After above substrate solubilisation period, the basket replaced the propeller mixer, was set to 300 rpm, and lowered into the reaction mixture. The formation of gas bubbles indicated ongoing decarboxylation and reaction progress was tracked by HPLC analysis. After 4 h, full conversion as achieved, the rotating bed basket was washed in 100 mL CPME, and the wash solution was added to the reactor content. The mixture was stirred at 500 rpm using the propeller mixer and tempered to 90 °C. The acetylation was started by addition of 163 mg anhydr. NaOAc (2 mmol, 0.02 eq.) and 14.25 mL Ac2O (15.3 g, 0.15 mol, 1.5 eq.) and was again tracked by HPLC-UV. After 2.5 h, conversion was 81% and reaction progress slowed down, which was why another 4.75 mL Ac2O (0.05 mol, 0.5 eq.) and 100 mg NaOAc (1.2 mmol, 0.01 eq.) were added. At a conversion of 99% the reaction was stopped by addition of 100 mL aqueous Na2CO3 solution (0.5 mol L−1). After vigorous mixing, the phases were separated, the organic phase was washed with 100 mL saturated aqueous NaCl solution, the phases separated, and the organic phase dried over anhydrous Na2SO4. The desiccant was filtered off, and the solvent was evaporated under reduced pressure to give 18.3 g of the product AMS (96% yield, 99% HPLC purity) as a slightly greenish clear liquid. TLC (silica gel 60, cyclohexane/EtOAc = 4:1): Rf = 0.53. 1H-NMR (600 MHz, CDCl3): δ [ppm] = 2.29 (3H, s, –CO–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3), 3.83 (3H, s, –O–C3), 5.23 (1H, d, J = 10.9 Hz, cis-CH
3), 3.83 (3H, s, –O–C3), 5.23 (1H, d, J = 10.9 Hz, cis-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2), 5.68 (1H, d, J = 17.5 Hz, trans-CHC2), 6.66 (1H, dd, J1 = 17.6 Hz, J2 = 10.9 Hz, –C=CH2), 6.96–6.99 (3H, m, ar-H); 13C-NMR (600 MHz, CDCl3): δ [ppm] 20.9, 56.0, 110.0, 114.3, 119.1, 122.9, 136.5, 136.8, 139.6, 151.2, 169.2; HPLC-MS (70 eV): m/z (%) = 407 (2M + Na+, 14), 252 (24), 231 (12), 215 (M + Na+, 19), 193 (M + H+, 100), 151 (12), 119 (12), 101 (27), 83 (62).
C2), 5.68 (1H, d, J = 17.5 Hz, trans-CHC2), 6.66 (1H, dd, J1 = 17.6 Hz, J2 = 10.9 Hz, –C=CH2), 6.96–6.99 (3H, m, ar-H); 13C-NMR (600 MHz, CDCl3): δ [ppm] 20.9, 56.0, 110.0, 114.3, 119.1, 122.9, 136.5, 136.8, 139.6, 151.2, 169.2; HPLC-MS (70 eV): m/z (%) = 407 (2M + Na+, 14), 252 (24), 231 (12), 215 (M + Na+, 19), 193 (M + H+, 100), 151 (12), 119 (12), 101 (27), 83 (62).
Author contributions
P. P.: methodology, investigation, validation, formal analysis visualization, writing – original draft & editing. J. P. B.: methodology, formal analysis. S. M.: methodology, resources. E. B.: conceptualization, resources. S. K.: conceptualization, resources, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 860414 and from the Deutsche Forschungsgemeinschaft (DFG) under grant number KA 4399/3-1 and JA 2500/5-1. The authors thank Prof. Robert Kourist from TU Graz (AT) for the kind provision of the BsPAD gene and Michelle Leganger Juul Sørensen from Aarhus University (DK) for her kind technical support in enzyme production.For access to NMR spectrometry, we thank Dr Tobias Sparrman from Umeå University (SE) and for the opportunity to run HPLC analysis we gladly acknowledge Dr Tobias Jonsson and Dr Ngoc Phuoc Dinh from Diduco AB (SE).
We also thank Dr Alessandra Basso and Dr Simona Serban from Purolite Ltd (UK) for the provision of Lifetech™ immobilization carriers.
Notes and references
- PlasticsEurope, Plastics – the Facts 2020, [Online Resource accessed 20.02.2022], 2020 Search PubMed.
- K. Degawa and A. Matsumoto, Chem. Lett., 2019, 48, 928–931 CrossRef CAS.
- Y. Na and C. Chen, Angew. Chem., Int. Ed. Engl., 2020, 59, 7953–7959 CrossRef CAS PubMed.
- B. K. Ahn, D. W. Lee, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2014, 13, 867–872 CrossRef CAS PubMed.
- H. Watanabe, M. Takahashi, H. Kihara and M. Yoshida, Langmuir, 2019, 35, 4534–4539 CrossRef CAS PubMed.
- H. Takeshima, K. Satoh and M. Kamigaito, Polym. Chem., 2019, 10, 1192–1201 RSC.
- T. Hirai, J. Kawada, M. Narita, T. Ikawa, H. Takeshima, K. Satoh and M. Kamigaito, Polymer, 2019, 181, 121667 CrossRef CAS.
- M. Messerschmidt, M. Millaruelo, H. Komber, L. Häussler, B. Voit, T. Krause, M. Yin and W.-D. Habicher, Macromolecules, 2008, 41, 2821–2831 CrossRef CAS.
- Z. Qian, Y. Lou, Q. Li, L. Wang, F. Fu and X. Liu, ACS Sustainable Chem. Eng., 2021, 9, 10929–10938 CrossRef CAS.
- J. van Schijndel, D. Molendijk, K. van Beurden, L. A. Canalle, T. Noël and J. Meuldijk, Eur. Polym. J., 2020, 125, 109534 CrossRef.
- H. Takeshima, K. Satoh and M. Kamigaito, Macromolecules, 2017, 50, 4206–4216 CrossRef CAS.
- H. Takeshima, K. Satoh and M. Kamigaito, ACS Sustainable Chem. Eng., 2018, 6, 13681–13686 CrossRef CAS.
- H. Takeshima, K. Satoh and M. Kamigaito, J. Polym. Sci., 2019, 58, 91–100 CrossRef.
- J. S. Mahajan, R. M. O'Dea, J. B. Norris, L. T. J. Korley and T. H. Epps, ACS Sustainable Chem. Eng., 2020, 8, 15072–15096 CrossRef CAS.
- H. T. Truong, M. Do Van, L. Duc Huynh, L. Thi Nguyen, A. Do Tuan, T. Le Xuan Thanh, H. Duong Phuoc, N. Takenaka, K. Imamura and Y. Maeda, Appl. Sci., 2017, 7, 796 CrossRef.
- V. I. Timokhin, M. Regner, A. H. Motagamwala, C. Sener, S. D. Karlen, J. A. Dumesic and J. Ralph, ACS Sustainable Chem. Eng., 2020, 8, 17427–17438 CrossRef CAS.
- P. B. Dhamole, S. Chavan, R. G. Patil, H. Feng, M. Bule and P. Kinninge, Korean J. Chem. Eng., 2016, 33, 1860–1864 CrossRef CAS.
- S. Fadlallah, P. Sinha Roy, G. Garnier, K. Saito and F. Allais, Green Chem., 2021, 23, 1495–1535 RSC.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- CheCalc, Chemical Engineering Calculations, https://www.checalc.com/, (accessed 25/03/2021) Search PubMed.
- E. Nomura, A. Hosoda, H. Mori and H. Taniguchi, Green Chem., 2005, 7, 863–866 RSC.
- A. K. Schweiger, N. Ríos-Lombardía, C. K. Winkler, S. Schmidt, F. Morís, W. Kroutil, J. González-Sabín and R. Kourist, ACS Sustainable Chem. Eng., 2019, 7, 16364–16370 CrossRef CAS.
- K. L. Morley, S. Grosse, H. Leisch and P. C. K. Lau, Green Chem., 2013, 15, 3312–3317 RSC.
- L. Pesci, M. Baydar, S. Glueck, K. Faber, A. Liese and S. Kara, Org. Process Res. Dev., 2017, 21, 85–93 CrossRef CAS.
- G. de Gonzalo, A. R. Alcántara and P. Domínguez de María, ChemSusChem, 2019, 12, 2083–2097 CrossRef CAS PubMed.
- İ. l. Tosun, in The Thermodynamics of Phase and Reaction Equilibria, 2021, pp. 561–599, DOI:10.1016/b978-0-12-820530-3.00022-2.
- J. A. P. Coutinho, S. I. Andersen and E. H. Stenby, Fluid Phase Equilib., 1995, 103, 23–29 CrossRef CAS.
- F. L. Mota, A. J. Queimada, S. P. Pinho and E. A. Macedo, Ing. Eng. Chem. Res., 2008, 47, 5182–5189 CrossRef CAS.
- F. Shakeel, N. Haq, M. M. Salem-Bekhit and M. Raish, J. Mol. Liq., 2017, 225, 833–839 CrossRef CAS.
- B. Wang, W. Ran, Y. Li, J. Xiong and S. Li, J. Chem. Eng. Data, 2012, 57, 3309–3314 CrossRef CAS.
- M. Männistö, J.-P. Pokki, L. Fournis and V. Alopaeus, J. Chem. Thermodyn., 2017, 110, 127–136 CrossRef.
- J. A. Alkandary, A. S. Alijmaz, M. S. Fandary and M. A. Fahim, Fluid Phase Equilib., 2001, 187-188, 131–138 CrossRef CAS.
- A. Dallos and J. Liszi, J. Chem. Thermodyn., 1995, 27, 447–448 CrossRef CAS.
- S. Shekarsaraee, J. Chem. Thermodyn., 2017, 104, 16–23 CrossRef CAS.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- A. Klamt, V. Jonas, T. Bürger and J. C. W. Lohrenz, J. Phys. Chem. A, 1998, 102, 5074–5085 CrossRef CAS.
- F. Eckert and A. Klamt, AIChE J., 2002, 48, 369–385 CrossRef CAS.
- BIOVIA COSMOtherm, Release 2020; Dassault Systèmes, http://www.3ds.com.
Footnote |
| † Electronic supplementary information (ESI) available: Enzyme expression, HPLC-UV analytics, COSMOtherm results, substrate and water dependent activities, details on immobilized enzyme activity, reusability, and leaching, additional DoE model results, NMR spectra of isolated product. See DOI: https://doi.org/10.1039/d2gc01629j |
| This journal is © The Royal Society of Chemistry 2022 |