
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFully biomass-derived vitrimeric material with water-mediated recyclability and monomer recovery†
Zhuang Mao
Png‡
 *ab,
Jie
Zheng‡
ab,
Sirin
Kamarulzaman‡
a,
Sheng
Wang
ab,
Zibiao
Li
*abc and
Shermin S.
Goh
*a
*ab,
Jie
Zheng‡
ab,
Sirin
Kamarulzaman‡
a,
Sheng
Wang
ab,
Zibiao
Li
*abc and
Shermin S.
Goh
*a
aInstitute of Materials Research and Engineering (IMRE), A*STAR (Agency for Science, Technology and Research), 2 Fusionopolis Way, Innovis #08-03, 138634, Singapore. E-mail: lizb@imre.a-star.edu.sg; gohsms@imre.a-star.edu.sg
bInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), A*STAR, 2 Fusionopolis Way, Innovis #08-03, 138634, Singapore. E-mail: png_zhuang_mao@isce2.a-star.edu.sg
cDepartment of Materials Science and Engineering, National University of Singapore, 9 Engineering Drive 1, 117576, Singapore
First published on 18th July 2022
Abstract
Covalent adaptable networks (CANs) are polymers which demonstrate both high mechanical strength and self-healing/recyclability, which are important for extending material service lifespan and meeting sustainability demands. However, these materials are often derived from non-renewable petrochemical feedstocks, or by multi-step modification of bio-based feedstocks. Here, we disclose a fully bio-based vitrimeric poly(acetal) prepared from high volume sugar derivatives in a single step. The polymer was hard and rigid in the glassy state, exhibiting a tensile strength of 13 MPa and a storage modulus of 3300 MPa at room temperature, transitioning to the rubbery state at ca. 120 °C. Stress relaxation studies revealed an Arrhenius fit with an apparent activation energy of 110 kJ mol−1. Small molecule model studies indicate that the dynamic exchanges occur via an initial dissociation of the acetal into the aldehyde upon heating followed by reformation of new acetal bonds, rapidly reaching steady state. Thus, the polymer network was mechanically reprocessable simply by hot-pressing at 140 °C; with water-mediated recycling capable of restoring the material strength to its pristine state. Furthermore, the constituent monomers were recoverable simply by heating in green solvents (i.e. water or ethanol), meeting the closed-loop requirement of circular materials and fulfilling the principles of green chemistry.
Introduction
Sustainability has surged to the forefront of research areas, driven by pressing environmental issues such as global warming and plastic pollution.1–4 Concerns surrounding plastics such as its carbon footprint (as it is derived from petroleum feedstock), as well as its environmental persistence, have resulted in calls for development of more sustainable alternatives.5,6 The ideal plastic would possess beneficial properties such as durability, low cost, and malleability, while being derived from sustainable feedstock, be infinitely recyclable, as well as bio-degradable so as to meet the needs of a sustainable circular economy.7 Covalent adaptable networks (CANs) are materials which come close to meeting these requirements, combining the ease of processibility and recyclability of traditional thermoplastics with the strength and chemical stability of thermosets.8–10 Broadly speaking, two types of CANs are discernable, namely those which operate via a dissociative mechanism, whereby the cross-links are first disconnected before reformation, and those which operate via an associative mechanism, whereby the cross-links first undergo nucleophilic attack.11,12 The latter type of CANs were first discovered by Leibler's group in 2011 who coined the term vitrimers, alluding to how their viscosity–temperature relationship resembles that of vitreous silica.13 Although the constant cross-link density of associative CANs can impart greater solvent resistance and strength, the distinction between dissociative and associative CANs is not always obvious from a physical point of view, with many dissociative CANs also exhibiting vitrimer-like behavior.14 A multitude of dynamic covalent bond exchanges have been investigated, including transesterification,13,15,16 disulfide exchange,17,18 imine exchange,19–21 boronic ester exchange,22,23 and transalkylation.24,25 These new generation materials have been explored in many applications, including soft actuators,26,27 3D printing,28,29 recyclable adhesives,30,31 and healable electronics.32,33Significant efforts have been devoted to improving the sustainability of CANs and vitrimers by following the principles of green chemistry.34,35 For instance, many reports of bio-derived CANs have surfaced in recent years in an effort to utilise renewable feedstock.36 In these examples, there have been efforts to demonstrate the material's recyclability by mechanical reprocessing or chemical degradation;37,38 some have even examined polymer biodegradability.39 However, to date, very limited works have demonstrated the full recovery and isolation of monomers from CANs.40,41 This is perhaps unsurprising as most polymers are unable to truly return to their monomeric forms, notably in the commonly used acid-epoxy networks.42 Even in cases where this is possible, monomer separation may be non-trivial, requiring either multiple separation steps41 or cross-linker resynthesis.44
Acetal bonds are well known in small molecule chemistry as a useful protecting group due to its acid lability and base stability.45 It has also been used in the making of polyacetals such as polyoxymethylene, an engineering thermoplastic.46 Recently, Ma and coworkers demonstrated the asymmetric acetal motif as a dynamic covalent cross-linker, synthesizing vitrimers with mechanical and chemical recyclability.44,47 Impressively, desirable properties, including catalyst-free reprocessing, multiple recyclability, thermal stability, malleability, and rapid stress relaxation at elevated temperatures, were observed. Nevertheless, the cyclic divinyl ether cross-linker could not be directly obtained after chemical degradation, requiring an additional synthetic step before polymer reformation. In addition, subsequent work using bio-based feedstocks were still at least partially derived from fossil fuel-based starting materials.31,48 These works inspired us to design fully recoverable acetal CANs from biomass sources, which is desirable for the development of sustainable and circular economy.
Herein, we disclose a fully biomass-derived vitrimeric material (Table 1) synthesised from furan-2,5-dicarboxylaldehyde (a derivative of 5-(hydroxymethyl)furfural, which is a commodity chemical derived from dehydration of fructose),49 and sorbitol (a sugar alcohol which is obtained from reduction of glucose) with high atom economy and water as the only by-product. The polymer was prepared via a green process using DMSO, a benign solvent,43 as well as catalytic acid (0.5 mol%). The acid-labile cyclic acetal bonds present was identified as a key factor influencing material rigidity as well as reprocessibility.44,47,48,50,51 The obtained polymer demonstrated both mechanical and chemical reprocessability, with closed-loop monomer-to-polymer-to-monomer recycling by facile aqueous hydrolysis and simple monomer separation by extraction with green solvents. From cradle to cradle, this circular material fulfils 11 out of the 12 principles of green chemistry,34,35 as presented in Table 1. Such a fully bio-based vitrimeric material which can undergo facile recycling without sacrificing its performance can offer inspiration for the design of next-generation circular materials to contribute towards sustainability.
|
|
|
|---|---|
| Principles of green chemistry34,35 | Justification of principles |
| (1) Prevention of waste | Mechanical and water-mediated recycling of polymer network; and closed-loop recycling to recover both monomers are demonstrated. |
| (2) Atom economy | >90% atom economy during polymer synthesis; water is the only by-product. |
| (3) Less hazardous chemical syntheses | Hazardous/toxic reagents and solvents are avoided; water is the only by-product. |
| (4) Designing safer chemicals | Sorbitol and furan-2,5-dicarbaldehyde monomers are non-toxic. |
| (5) Safer solvents and auxiliaries | Only green/benign solvents43 – water, DMSO, ethanol and ethyl acetate – are used. |
| (6) Design for energy efficiency | Simple and green monomer recovery process reduces the energy cost of chemical recycling. |
| (7) Use of renewable feedstocks | Sorbitol and furan-2,5-dicarbaldehyde are bio-based chemicals derived from sugars. |
| (8) Reduce derivatives | One-step polymer synthesis from high-volume chemicals with the desired functionalities. |
| (9) Catalysis | Catalytic (0.5 mol%) p-toluenesulfonic acid is used for polymer synthesis and reprocessing. |
| (10) Design for degradation | Water-mediated hydrolysis of polymer to monomer enables closed-loop recycling. |
| (11) Inherently safer chemistry for accident prevention | Simple protocol performed in ambient atmosphere; no hazardous/air-sensitive reagents or pressurised environment required. |

Results and discussion
Small molecule model studies of acetal exchange
We began our investigation by modelling the dynamic exchange with furfural–ethylene acetal and sorbitol, monitoring the reaction by NMR (Fig. 1a). When heated to 120 °C in the absence of catalyst, no reaction was observed for the first 400 min (Fig. 1b). From 400 min, furfural peaks (ca. 9.6 ppm) began to appear, indicating the decomposition of the acetal into its constituent aldehyde and ethylene glycol. This is likely due to autocatalysis by oxidation of minute quantities of furfural to furan–carboxylic acid species, which was observable by gas chromatography-mass spectroscopy (Fig. S2†). Over time, increasing amount of furfural–sorbitol acetal peaks were observed until a steady state was reached after about 850 min. On the other hand, when TsOH, a common catalyst used for acetylation,52 was added (0.5 mol%), the reaction was extremely rapid. Almost all the starting furfural–ethylene acetal converting into other forms within 15 min, and steady state was achieved in about 70 min (Fig. 1c). A similar experiment conducted with furfural–propylene acetal to simulate the six-membered cyclic acetal (Fig. S1†) yielded similar observations. Furfural–propylene acetal converted to other forms within about 20 min, and steady state was achieved at about 200 min. We also examined the relationship between reaction kinetics and temperature (Fig. 1d). Even at 50 °C, the exchange reaction was still possible, with formation of furfural–sorbitol acetal being observed, though at a much slower rate. Previous studies have shown that the initial rate of acid-catalysed acetal hydrolysis/transacetalisation can be described by pseudo-first-order kinetics, with protonation of the acetal the rate-limiting step.53 Pleasingly, an Arrhenius relationship was observed, with an activation energy (Ea) of 76.4 kJ mol−1 (Fig. 1e), which is in the range of other reported acetal-based networks.44,47,48,50,51 Various mechanisms have been identified for acetal exchanges, ranging from transacetalisation/metathesis of spiroacetals,50 to dissociative mechanisms via a vinyl ether44,47 or carbocation intermediates.48 Here, we observe the presence of furfural, indicating that the dynamic exchanges for furfural–acetal with sorbitol proceeds via an initial dissociation into the aldehyde furfural, followed by sorbitol attack to rapidly reach an equilibrium mixture of acetal and furfural. This gives us confidence that an acetal network comprising furan-2,5-dicarbaldehyde and sorbitol would also be able to undergo productive dynamic acetal exchanges via such a dissociative mechanism, while still exhibiting vitrimeric properties since a steady state with both acetal and aldehyde is rapidly established and maintained thereafter.54 | ||
| Fig. 1 NMR kinetics study of the exchange reaction between furfural–ethylene acetal (0.2 mmol) and sorbitol (0.2 mmol) with 1,3,5-trimethoxybenzene (0.06 mmol) as an internal standard in DMSO-d6 (0.55 mL). For the catalysed reaction, 5 mol% TsOH was added. (a) Initial and final NMR spectra of the catalysed reaction at 120 °C. Reaction profile of the (b) uncatalysed and (c) catalysed reaction at 120 °C. The time taken to reach steady state was 850 min for the uncatalysed reaction and 70 min for the catalysed reaction; (d) kinetics of the catalysed reaction at different temperatures; (e) Arrhenius plot to obtain Ea of the small molecule dynamic acetal exchange. | ||
Synthesis and optimization
Having evidenced the dynamic acetal exchange, we proceeded to synthesise a polymer network with furan-2,5-dicarbaldehyde and sorbitol. In our optimization of polymerization conditions, we began with catalyst-free and melt-polymerization type conditions, under steady stream of nitrogen (Table 2, entries 1–3).55,56 At temperatures higher than 130 °C, some reaction occurred, with dimer and trimer species observed by mass spectroscopy. However, under these conditions, furan-2,5-dicarbaldehyde was observed to sublime (typically at temperatures higher than 100 °C), resulting in loss of material. As such, we introduced DMSO as a solvent (entry 4). However, the results were similar to that without the solvent, as the stream of nitrogen gradually removed the solvent over time. When the reaction was instead performed in a sealed tube under N2, little reaction was observed, and the starting materials were recovered (entry 5). We then added 0.5 mol% of p-toluenesulfonic acid (TsOH) (entry 6).52 Pleasingly, a gel-like solid was observed at the end of the reaction. Even at temperatures as low as 100 °C, the reaction was still productive, and a gel-like solid is observed (entry 7). Unfortunately, attempts to remove the solvent from the reaction resulted in the reaction mixture rapidly turning black and the formation of a hard black solid (entry 8).|
|
||||||
|---|---|---|---|---|---|---|
| No. | DMSO | T/°C | TsOHa, mol% | FDC, eq. | Pre-curing observation | Tensile strength post hot-pressd |
| Reaction conditions: (1) sorbitol (1 mmol) and indicated amount of furan-2,5-dicarbaldehyde (FDC) and catalyst were dissolved in DMSO (0.5 mL, where indicated) and stirred for 16 h under a constant stream of N2; (2) vacuum oven, 24 h, 120 °C; (3) hot-press, 140 °C, 5 bar, 30 min.a TsOH was added as catalyst.b In a sealed tube instead of under N2 stream.c N.R. = no reaction.d Only the gel-like polymers in entries 7, 9 and 10 were hot-pressed.e N.T. = cannot be tested as samples easily broke upon handling. | ||||||
| 1 | N | 120 | 0 | 0.8 | N.R.c | — |
| 2 | N | 130 | 0 | 0.8 | Oligomers | — |
| 3 | N | 140 | 0 | 0.8 | Oligomers | — |
| 4 | Y | 130 | 0 | 0.8 | Oligomers | — |
| 5b | Y | 130 | 0 | 0.8 | N.R.c | — |
| 6 | Y | 130 | 0.5 | 0.8 | Gel-like polymer | — |
| 7 | Y | 100 | 0.5 | 0.8 | Gel-like polymer | N.T.e |
| 8 | N | 100 | 0.5 | 0.8 | Decomposed | — |
| 9 | Y | 100 | 0.5 | 1.0 | Gel-like polymer | N.T.e |
| 10 | Y | 100 | 0.5 | 0.6 | Gel-like polymer P1 | 13 MPa |
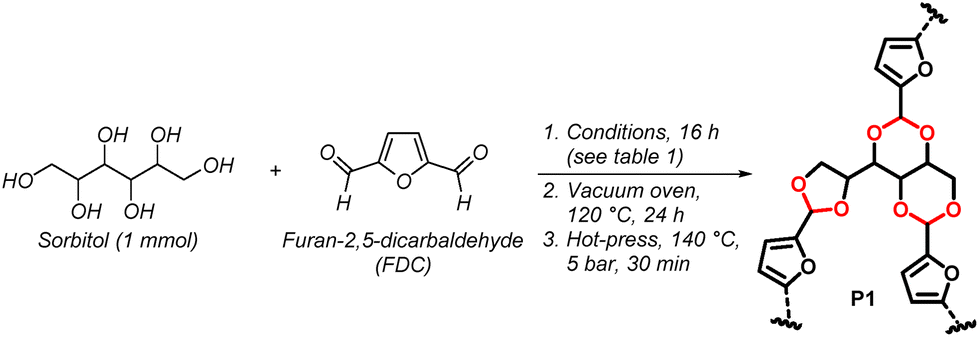
We thus proceeded with the reaction conducted in DMSO, with 0.5 mol% of TsOH, under stream of nitrogen. The reaction was scalable to 50 mmol (∼10 g) through careful adjustment of the stream of nitrogen to ensure that most of the DMSO is removed at the end of the reaction. The gel-like solid was then fully dried in a vacuum oven at 120 °C for 24 hours to obtain a hard brown solid. The obtained polymer could then be directly hot-pressed at 140 °C, then cut into strips, or blended into fine powder before being hot-pressed in a mold. However, after hot-pressing, the polymer was found to be brittle and easily broke upon handling. Thus, the ratio of sorbitol and furan-2,5-dicarbaldehyde was adjusted. Increasing the amount of furan-2,5-dicarbaldehyde to 1 equivalent (eq.) still resulted in a very brittle material (entry 9). However, when the amount of furan-2,5-dicarbaldehyde was decreased to 0.6 eq., the polymer (P1) was observed to be significantly less brittle (entry 10). Polymer P1 was found to be hard and rigid at room temperature, but flexible and malleable at elevated temperatures. This was attributed to the high density of rings (furans and acetals) in the network, which has been shown to improve mechanical strength and rigidity due to inhibition of rotation of the polymer chain.57,58
NMR analysis of the pre-cured sample (Fig. S3†) revealed that only traces (∼1%) of the starting furan-2,5-dicarbaldehyde was present, with most of the dialdehyde material converted to either the singly or doubly coupled moiety. Distinctive acetal peaks were also observed in the 5.5–7.0 ppm region, confirming the formation of acetal bonds.59 FTIR analysis of the sample showed that the strong OH peak (∼3400 cm−1) of sorbitol and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak (1682 cm−1) of furan-2,5-dicarbaldehyde were present in P1 both before and after curing (Fig. S4†). A noticeable decrease in intensity of the CO peak was observed from the monomer to the pre-cured polymer to the post-cured polymer, indicating an increasing extent of acetal formation. However, extended curing time in the vacuum oven did not result in complete disappearance of the CO peak, suggesting that residual free aldehyde groups are present even in the fully cured sample, likely due to steric hindrance in the highly cross-linked network with multiple cyclic moieties.
O peak (1682 cm−1) of furan-2,5-dicarbaldehyde were present in P1 both before and after curing (Fig. S4†). A noticeable decrease in intensity of the CO peak was observed from the monomer to the pre-cured polymer to the post-cured polymer, indicating an increasing extent of acetal formation. However, extended curing time in the vacuum oven did not result in complete disappearance of the CO peak, suggesting that residual free aldehyde groups are present even in the fully cured sample, likely due to steric hindrance in the highly cross-linked network with multiple cyclic moieties.
The gel fraction and swelling ratio of P1 were measured in toluene, chloroform, and water (Fig. S5†). A high gel fraction was observed in toluene (97%) and chloroform (89%), while that in water is lower (74%). This is attributed to the high polarity of the starting materials, which would likely make even oligomers soluble in water. It is also possible that some degree of hydrolysis occurs over time in water due to the presence of TsOH catalyst, resulting in degradation to oligomers. Solvent polarity also directed the compatibility and thus swelling of the polymers: while the swelling ratio was relatively low in toluene (104%), that in water and chloroform was considerably higher (194 and 196% respectively). These results indicate a relatively high degree of organic solvent resistance at room temperature, though the resistance was modest for water.
Thermomechanical properties
We went on to investigate the thermomechanical behaviors of the acetal network P1via TGA, DSC, and DMA. P1 displayed a Td5% (temperature corresponding to a 5% weight loss) of 216 °C (Fig. S6†), demonstrating the good thermal stability of the resultant polymer network. The storage modulus (E′) curve revealed a temperature-dependent viscoelastic behavior with a high E′ of 4000 MPa in the glass state, and a low E′ of 11.82 MPa in the rubbery state (Fig. 2a). The Tg of P1 was determined to be 120 °C based on the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ trace (peak value). This value is consistent with the DSC analysis, where Tg was located at 134 °C (Fig. 2b, red line), with the slight difference commonly observed between measurement methods.60 Notably, in the DSC curve, another transition was observed at around 55 °C (Tα). However, when the TsOH was removed by soaking the sample in KOH, this transition completely disappeared (Fig. 2b, black line), even though FTIR shows that the chemical composition of the polymer network remains intact (Fig. S4†). As such, we attribute transition Tα to the small-scale local network rearrangement enabled by acid-catalysed endothermic dissociation and reassociation of some acetal bonds.61 This also corresponds to a small peak observed in the tanδ trace at about 60 °C. The cross-linking density (ρ) of the polymer network was calculated to be 1095 mol m−3 based on the E′ value in the rubbery state according to the following equation:48,62
δ trace (peak value). This value is consistent with the DSC analysis, where Tg was located at 134 °C (Fig. 2b, red line), with the slight difference commonly observed between measurement methods.60 Notably, in the DSC curve, another transition was observed at around 55 °C (Tα). However, when the TsOH was removed by soaking the sample in KOH, this transition completely disappeared (Fig. 2b, black line), even though FTIR shows that the chemical composition of the polymer network remains intact (Fig. S4†). As such, we attribute transition Tα to the small-scale local network rearrangement enabled by acid-catalysed endothermic dissociation and reassociation of some acetal bonds.61 This also corresponds to a small peak observed in the tanδ trace at about 60 °C. The cross-linking density (ρ) of the polymer network was calculated to be 1095 mol m−3 based on the E′ value in the rubbery state according to the following equation:48,62where E′ refers to the storage modulus of P1 network at (Tg + 40 °C), R and T represent the gas constant and absolute temperature, respectively. The high cross-linking density arises from the large number of cross-linking sites in the polymer network and contributes to the rigidity of the network.
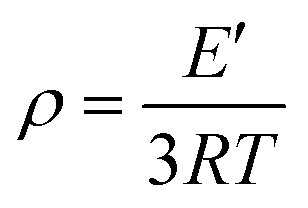
 | ||
| Fig. 2 Thermomechanical properties of P1. (a) DMA curves of P1 showing variation of storage modulus and tanδ with temperature. (b) DSC curves of P1 with acid catalyst (red line) and without catalyst (black line). (c) Stress relaxation curves of P1 at temperatures from 120 °C to 150 °C. Dotted line shows 1/e of initial stress. (d) Arrhenius analysis of relaxation time against 1000/T, showing an Ea of 110 kJ mol−1. | ||
We next studied the dynamic properties of the P1 network based on stress relaxation analysis in the temperature range 120–150 °C (at the initial stages of the rubbery region, to avoid thermal decomposition) with an applied strain of 1% via DMA (Fig. 2c). An exponential decrease in stress over time was observed, with a decrease in time required at higher temperature. For instance, the relaxation time (τ*) required to reach 1/e of the initial stress was only 8 s at 150 °C, but this increased to 26 s at 130 °C and 115 s at 120 °C, respectively. A plot of ln(τ*) against 1000/T demonstrated an Arrhenius relationship63,64 with Ea of 110 kJ mol−1 (Fig. 2d), similar to that reported by Ma and coworkers for linear acetals (126–136 kJ mol−1).44Ea is an important indicator not only of the bond exchange reactivity, but also network viscosity change upon heating. The high Ea of the P1 network with acetal linkage suggests a high energy barrier of bond exchange, leading to the good dimensional stability of P1. Furthermore, the high Ea also results in high sensitivity of viscosity upon temperature change, enabling fast processing on demand. The topology freezing transition temperature (Tv) is another important parameter for dynamic network, at which point the bond exchange rate is sufficiently fast to endow the material transition from a viscoelastic solid to a viscoelastic liquid. This transition is conventionally chosen at the point where a viscosity of 1012 Pa s is reached.63,64 The Tv of P1 was thus calculated to be 45 °C based on the extrapolation of the Arrhenius fitted line when τ* is 2.5 × 106 s, which corresponds to the minor transition Tα observed in the DSC and DMA tanδ traces. At this temperature, even though the rate of acetal bond exchange is rapid, the polymer network is unable to achieve macroscopic flow since the dynamic linkages are trapped in a glassy matrix (Tg ∼ 120 °C), with only some localized network rearrangements possible. When P1 is further heated to above Tg, the long-range polymer chain segmental motion is accessible, imparting the network with viscoelastic-liquid-like properties.
Closed-loop recycling
Taking advantage of the versatile dynamic acetal bonds, the P1 network permits both mechanical and chemical recycling (Fig. 3a). To investigate mechanical reprocessability, the P1 specimen was cut into small pieces, then hot-pressed at 140 °C and 5 bars. P1 could reform complete films (Fig. 3a, left), with FTIR analysis confirming that no significant change occurred during the reprocessing of the sample (Fig. S4†). Fig. 3b depicts the tensile stress–strain curves of the original and mechanically reprocessed P1 network after the first and second reprocessing cycles. The pristine polymer exhibited a tensile strength of about 13 MPa at a strain of 0.35% before breaking. A slight decrease in tensile strength to about 9 and 7 MPa was observed for the first and second reprocessed specimen, respectively, demonstrating mechanical recoverability and reprocessibility. The reduction of mechanical strength of the recycled samples could be due to high humidity in the environment (average relative humidity ∼ 83.9%), which could hydrolyse the acetal bonds under elevated temperatures, such as in the hot press, over an extended time. | ||
| Fig. 3 (a) Mechanical and chemical recycling of P1. (b) Tensile–strain curves, and (c) tensile strength value of pristine, mechanically reprocessed, and water-mediated recycling samples. (d) NMR study of degradation of P1 in water at 80 °C into furan and sorbitol at time 0 and 500 min; inset: reaction profile of P1 degradation. | ||
In addition to mechanical recycling, we also examined the chemical recyclability of the polymer. Acetal bonds are known to be labile under acidic conditions but stable under basic conditions.65 As such, when the polymer was refluxed in KOH, no obvious degradation was observed and FTIR showed the structure remained the same (Fig. S4†). However, when heated in water, methanol, or ethanol, rapid dissolution was observed (Fig. 3a, right). This is likely mediated by the TsOH used in the preparation of the polymer. We went on to study its degradation in D2O by NMR with DMSO as an internal standard. Furan and sorbitol peaks rapidly appeared within the first 100 min (Fig. 3d, inset). After 500 min, about 86% of the original sample has been broken down into its constituent furan and sorbitol small molecules, which can be clearly identified in the NMR spectrum (Fig. 3d). This indicated that water-mediated recycling of the polymer was possible. Thus, we dissolved the twice reprocessed polymer by heating in water, filtered off the residue (∼5%), and removed the water in vacuo. The polymer was then reformed by our optimised procedure (Table 2, entry 10), and strips were reformed via hot press. Pleasingly, these reformed polymers exhibited tensile strength comparable to that of the pristine polymer (Fig. 3b and c, blue line/bar), circumventing the loss in tensile strength observed after direct mechanical reprocessing. FTIR analysis also confirmed the recovery of the water-recycled polymer (Fig. S4†), demonstrating the reformation of the acetal network. Moreover, instead of reforming the polymer, the monomers could be isolated and recovered. The small molecules were separable by extraction, with furan-2,5-dicarbaldehyde partitioning in organic solvents such as ethyl acetate (86% recovery), and sorbitol partitioning in water (90% recovery). While many other polymers have been reported to be chemically degradable, to our knowledge, monomer recovery via water as medium and separation is considerably less common.41 We believe these closed-loop recyclable polymers which are synthesized and recycled via green processes would grow in importance in tandem with the development of a truly circular economy.
Conclusion
We have demonstrated the green synthesis and recycling of a fully biomass-based vitrimeric poly(acetal). The polymer P1 was synthesised from bio-derived chemicals furan-2,5-dicarbaldehyde and sorbitol with >90% atom economy. Although P1 was hard and rigid at room temperature, with a storage modulus of 3300 MPa and tensile strength of 13 MPa, like other previously reported acetal based vitrimeric materials,44,47,48,50 it could be recycled mechanically at temperatures around its Tg of 120 °C. Small molecule studies established that the dynamic acetal exchanges proceeded via a dissociative mechanism through an aldehyde, with the rapid equilibration to a steady state of acetal and aldehyde allowing P1 to exhibit vitrimeric properties. In the polymeric form, its vitrimeric properties was demonstrated by temperature dependent stress relaxation time analysis which revealed an Arrhenius fit with an apparent activation energy of 110 kJ mol−1 and a Tv of 55 °C. A calculated crosslinking density of 1095 mol m−3 as well as a gel fraction of 89% in chloroform also supports our proposed crosslinked network structure. Although a slight reduction in tensile strength was observed after mechanical reprocessing, P1 could be fully restored to pristine strength by water-mediated recycling. Most impressively, this material could be degraded in water and separated with green solvents to recover its constituent monomers with high yield. Such bio-derived recyclable materials, which are reusable multiple times, and ultimately degradable into its starting material in a closed-loop process following green principles, will play an increasingly important role in the development of circular materials, contributing towards a more sustainable Earth.Materials and methods
General experimental procedure
Furan-2,5-dicarbaldehyde, sorbitol, TsOH and all other reagents were used as supplied from Sigma Aldrich, Alfa Aesar, or Combi-blocks without further purification.Nuclear magnetic resonance (NMR) spectra were recorded on a JEOL ECA-II 500 MHz spectrometer using DMSO-d6 as the solvent. Data are reported as follows: chemical shift (integration, splitting (s = singlet, d = doublet, t = triplet, q = quartet, qt = quintet, sxt = sextet, spt = septet, m = multiplet), coupling constant, assignment).
Fourier-transform infrared spectra (FTIR) were recorded on a PerkinElmer Paragon 1000 FTIR spectrometer and using samples pelletized with KBr. Absorption maxima (λmax) are quoted in wavenumbers (cm−1).
Samples were prepared by using Collin hot press (140 °C, 5 bar, 10–30 min) and cut into strips or blended into a fine powder and pressed into a dog bone shape using a mold. Mechanical re-processibility was conducted by cutting up processed strips, followed by hot pressing the sample under the same conditions, and cutting into strips for tensile strength analysis.
Thermogravimetric analysis (TGA) was determined via TA Instruments Q500. About 10 mg of samples was placed into an alumina pan and heated from 30 to 900 °C with a ramp rate of 20 °C min−1 in nitrogen at a flow rate of 60 mL min−1.
Differential scanning calorimetry (DSC) was performed on a TA Instruments Q100 calorimeter. About 10 mg of sample was placed into an aluminum hermetic pan. The first heating scan was from 25 to 120 °C to erase any thermal history, followed by isothermal for 2 min, cooling to −20 °C, and subsequent heating (second heating scan) to 160 °C. The heating and cooling rate for the scans was 10 °C min−1 under a nitrogen flow of 50 mL min−1.
Dynamic Mechanical Analysis (DMA) was performed on a TA Instruments Q800. The samples (100 × 5 × 1 mm3) were measured using a single cantilever mode from −10 to 160 °C at a frequency of 1 Hz, amplitude of 20 μm, and a temperature ramp of 3 °C min−1. The storage modulus (E′) and tan delta (δ) were recorded as a function of temperature. Stress relaxation experiments were also performed on the TA Instruments Q800 in “stress relaxation mode”, where a 1% relaxation strain was applied at temperature of 120, 130, 140, 145, and 150 °C, respectively.
Tensile strength measurements were evaluated by the Instron 5569 Table Universal testing machine in accordance with ISO 527. Five samples were stretched at a crosshead speed of 1 mm min−1 at ambient temperature.
Swelling and gel fraction measurements were conducted by immersing samples of P1 (mass m0 ∼ 50 mg) in the respective solvents for 72 hours, drying them by paper towel, and measuring the mass (m1). The sample was then dried in a vacuum oven (60 °C), and the final mass measured (m2).


Chemical reprocessibility was performed by heating P1 (∼100 mg) in the relevant solvent (∼2 mL) at 80 °C for 16 hours. Dissolution was observed when methanol, ethanol, or water was used as the solvent. For the sample using ethanol as the solvent, the mixture was then cooled to room temperature, filtered, and 15 mL of water was added to it. Extraction was carried out by ethyl acetate (20 mL × 10), and the combined organic extract was dried with magnesium sulfate and the solvent removed in vacuo to obtain furan-2,5-dicarbaldehyde (86% recovery). The aqueous portion was also dried in vacuo to obtain sorbitol (90% recovery).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Agency for Science, Technology and Research (A*STAR), Science and Engineering Research Council for this work under its AME Young Individual Research Grant (YIRG) Grant (Grant No. A2084c0166) and UGT HTPO seeds fund (Grant No. C211718004).References
- Ó. Ögmundarson, M. J. Herrgård, J. Forster, M. Z. Hauschild and P. Fantke, Nat. Sustain., 2020, 3, 167–174 CrossRef.
- G. Caniglia, C. Luederitz, T. von Wirth, I. Fazey, B. Martín-López, K. Hondrila, A. König, H. von Wehrden, N. A. Schäpke, M. D. Laubichler and D. J. Lang, Nat. Sustain., 2021, 4, 93–100 CrossRef.
- P. Shani, S. Chau and O. Swei, Resour., Conserv. Recycl., 2021, 173, 105701 CrossRef.
- E. Rossi, A. C. Bertassini, C. d. S. Ferreira, W. A. N. do Amaral and A. R. Ometto, J. Cleaner Prod., 2020, 247, 119137 CrossRef.
- S. Walker and R. Rothman, J. Cleaner Prod., 2020, 261, 121158 CrossRef CAS.
- Z. Wen, Y. Xie, M. Chen and C. D. Dinga, Nat. Commun., 2021, 12, 425 CrossRef CAS.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- B. Krishnakumar, R. V. S. P. Sanka, W. H. Binder, V. Parthasarthy, S. Rana and N. Karak, Chem. Eng. J., 2020, 385, 123820 CrossRef CAS.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- J. Zheng, Z. M. Png, S. H. Ng, G. X. Tham, E. Ye, S. S. Goh, X. J. Loh and Z. Li, Mater. Today, 2021, 51, 586–625 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- B. R. Elling and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 1488–1496 CrossRef CAS.
- M. Delahaye, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2019, 141, 15277–15287 CrossRef CAS.
- S. Debnath, S. Kaushal and U. Ojha, ACS Appl. Polym. Mater., 2020, 2, 1006–1013 CrossRef CAS.
- F. Fan, S. Ji, C. Sun, C. Liu, Y. Yu, Y. Fu and H. Xu, Angew. Chem., Int. Ed., 2018, 57, 16426–16430 CrossRef CAS PubMed.
- S. Nevejans, N. Ballard, J. I. Miranda, B. Reck and J. M. Asua, Phys. Chem. Chem. Phys., 2016, 18, 27577–27583 RSC.
- P. Taynton, K. Yu, R. K. Shoemaker, Y. Jin, H. J. Qi and W. Zhang, Adv. Mater., 2014, 26, 3938–3942 CrossRef CAS.
- P. Taynton, H. Ni, C. Zhu, K. Yu, S. Loob, Y. Jin, H. J. Qi and W. Zhang, Adv. Mater., 2016, 28, 2904–2909 CrossRef CAS PubMed.
- S. Dhers, G. Vantomme and L. Avérous, Green Chem., 2019, 21, 1596–1601 RSC.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- M. Röttger, T. Domenech, R. Van Der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2017, 6, 930–934 CrossRef CAS PubMed.
- D. W. Hanzon, N. A. Traugutt, M. K. McBride, C. N. Bowman, C. M. Yakacki and K. Yu, Soft Matter, 2018, 14, 951–960 RSC.
- Y. Yang, H. Wang, S. Zhang, Y. Wei, X. He, J. Wang, Y. Zhang and Y. Ji, Matter, 2021, 4, 3354–3365 CrossRef CAS.
- Q. Shi, K. Yu, X. Kuang, X. Mu, C. K. Dunn, M. L. Dunn, T. Wang and H. Jerry Qi, Mater. Horiz., 2017, 4, 598–607 RSC.
- E. Rossegger, R. Höller, D. Reisinger, J. Strasser, M. Fleisch, T. Griesser and S. Schlögl, Polym. Chem., 2021, 12, 639–644 RSC.
- S. Zhang, T. Liu, C. Hao, L. Wang, J. Han, H. Liu and J. Zhang, Green Chem., 2018, 20, 2995–3000 RSC.
- A. Moreno, M. Morsali and M. H. Sipponen, ACS Appl. Mater. Interfaces, 2021, 13, 57952–57961 CrossRef CAS PubMed.
- J. Deng, X. Kuang, R. Liu, W. Ding, A. C. Wang, Y. C. Lai, K. Dong, Z. Wen, Y. Wang, L. Wang, H. J. Qi, T. Zhang and Z. L. Wang, Adv. Mater., 2018, 30, 1705918 CrossRef PubMed.
- J. Zhang, Z. Lei, S. Luo, Y. Jin, L. Qiu and W. Zhang, ACS Appl. Nano Mater., 2020, 3, 4845–4850 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green chemistry: theory and practice, Oxford University Press, Oxford [England], New York, 1998 Search PubMed.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS.
- T. Vidil and A. Llevot, Macromol. Chem. Phys., 2022, 2100494 CrossRef CAS.
- Q. Yu, X. Peng, Y. Wang, H. Geng, A. Xu, X. Zhang, W. Xu and D. Ye, Eur. Polym. J., 2019, 117, 55–63 CrossRef CAS.
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Macromolecules, 2018, 51, 389–397 CrossRef CAS.
- A. Zych, J. Tellers, L. Bertolacci, L. Ceseracciu, L. Marini, G. Mancini and A. Athanassiou, ACS Appl. Polym. Mater., 2021, 3, 1135–1144 CrossRef CAS.
- W. A. Ogden and Z. Guan, J. Am. Chem. Soc., 2018, 140, 6217–6220 CrossRef CAS PubMed.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS.
- L. Zhong, Y. Hao, J. Zhang, F. Wei, T. Li, M. Miao and D. Zhang, Macromolecules, 2022, 55, 595–607 CrossRef CAS.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- Q. Li, S. Ma, S. Wang, W. Yuan, X. Xu, B. Wang, K. Huang and J. Zhu, J. Mater. Chem. A, 2019, 7, 18039–18049 RSC.
- M. Schelhaas and H. Waldmann, Angew. Chem., Int. Ed. Engl., 1996, 35, 2056–2083 CrossRef CAS.
- A. Hufendiek, S. Lingier and F. E. Du Prez, Polym. Chem., 2019, 10, 9–33 RSC.
- Q. Li, S. Ma, S. Wang, Y. Liu, M. A. Taher, B. Wang, K. Huang, X. Xu, Y. Han and J. Zhu, Macromolecules, 2020, 53, 1474–1485 CrossRef CAS.
- Q. Li, S. Ma, P. Li, B. Wang, H. Feng, N. Lu, S. Wang, Y. Liu, X. Xu and J. Zhu, Macromolecules, 2021, 54, 1742–1753 CrossRef CAS.
- A. Shrotri, H. Kobayashi and A. Fukuoka, in Advances in Catalysis, ed. C. Song, Academic Press, 2017, vol. 60, pp. 59–123 Search PubMed.
- S. Yu, S. Wu, C. Zhang, Z. Tang, Y. Luo, B. Guo and L. Zhang, ACS Macro Lett., 2020, 9, 1143–1148 CrossRef CAS PubMed.
- D. Boucher, J. Madsen, L. Yu, Q. Huang, N. Caussé, N. Pébère, V. Ladmiral and C. Negrell, Macromolecules, 2021, 54, 6772–6779 CrossRef CAS.
- G. Z. Raskildina, Y. G. Borisova, S. S. Dzhumaev and S. S. Zlotsky, Russ. J. Gen. Chem., 2019, 89, 2341–2344 CrossRef CAS.
- E. H. Cordes and H. G. Bull, Chem. Rev., 2002, 74, 581–603 CrossRef.
- P. Chakma, C. N. Morley, J. L. Sparks and D. Konkolewicz, Macromolecules, 2020, 53, 1233–1244 CrossRef CAS.
- S. Pasupuleti and G. Madras, J. Appl. Polym. Sci., 2011, 121, 2861–2869 CrossRef CAS.
- B. L. Wegenhart, S. Liu, M. Thom, D. Stanley and M. M. Abu-Omar, ACS Catal., 2012, 2, 2524–2530 CrossRef CAS.
- X. Yang, L. Guo, X. Xu, S. Shang and H. Liu, Mater. Des., 2020, 186, 108248 CrossRef CAS.
- J. Wu, X. Yu, H. Zhang, J. Guo, J. Hu and M.-H. Li, ACS Sustainable Chem. Eng., 2020, 8, 6479–6487 CrossRef CAS.
- Y.-J. Kim, N.-K. Kim, W.-T. Park, C. Liu, Y.-Y. Noh and D.-Y. Kim, Adv. Funct. Mater., 2019, 29, 1807786 CrossRef.
- Y. Li, T. Liu, S. Zhang, L. Shao, M. Fei, H. Yu and J. Zhang, Green Chem., 2020, 22, 870–881 RSC.
- M. Rahaman, N. S. Graça, C. S. M. Pereira and A. E. Rodrigues, Chem. Eng. J., 2015, 264, 258–267 CrossRef CAS.
- C. di Mauro, T.-N. Tran, A. Graillot and A. Mija, ACS Sustainable Chem. Eng., 2020, 8, 7690–7700 CrossRef CAS.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS PubMed.
- J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, London, 2nd edn, 2012 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional information on small molecule and polymer characterisation. See DOI: https://doi.org/10.1039/d2gc01556k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |