
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDisproportionation of nitrogen induced by DC plasma-driven electrolysis in a nitrogen atmosphere†
C.
Pattyn
 a,
N.
Maira
a,
M.
Buddhadasa
a,
E.
Vervloessem
bc,
S.
Iseni
d,
N. C.
Roy
a,
A.
Remy
a,
M.-P.
Delplancke
e,
N.
De Geyter
b and
F.
Reniers
*a
a,
N.
Maira
a,
M.
Buddhadasa
a,
E.
Vervloessem
bc,
S.
Iseni
d,
N. C.
Roy
a,
A.
Remy
a,
M.-P.
Delplancke
e,
N.
De Geyter
b and
F.
Reniers
*a
aUniversité Libre de Bruxelles, Faculty of Sciences, Chemistry of Surfaces Interfaces and Nanomaterials (ChemSIN), Avenue F. D. Roosevelt 50, B-1050 Brussels, Belgium. E-mail: francois.reniers@ulb.be
bResearch Unit Plasma Technology (RUPT), Department of Physics, Ghent University, 9000 Ghent, Belgium
cResearch group PLASMANT, Department of Chemistry, University of Antwerp, Universiteitsplein 1, 2610 Antwerp, Belgium
dGREMI – Groupe de Recherches sur l'Energétique des Milieux Ionisés, UMR 7344 CNRS/Université d'Orléans, 14 rue d'Issoudun, BP6744, 45067 ORLEANS Cedex 2, France
e4MAT, Université Libre de Bruxelles, 50 Av. F.D. Roosevelt, 1050 Brussels, Belgium
First published on 30th August 2022
Abstract
Nitrogen disproportionation i.e. its simultaneous conversion to compounds of higher (NOx) and lower (NH3) oxidation states in a N2 DC plasma-driven electrolysis process with a plasma cathode is investigated. This type of plasma–liquid interaction exhibits a growing interest for many applications, in particular nitrogen fixation where it represents a green alternative to the Haber–Bosch process. Optical emission spectroscopy, FTIR and electrochemical sensing systems are used to characterize the gas phase physico–chemistry while the liquid phase is analyzed via ionic chromatography and colorimetric assays. Experiments suggest that lowering the discharge current enhances nitrogen reduction and facilitates the transfer of nitrogen compounds to the liquid phase. Large amounts of water vapor appear to impact the gas discharge physico–chemistry and to favor the vibrational excitation of N2, a key parameter for an energy-efficient nitrogen fixation.
Introduction
Nitrogen is one of the most abundant elements in the Earth's atmosphere. It is commonly found in the form of N2 molecules which represent about 78% of the earth's atmosphere. Being an essential component of biomolecules such as proteins or nucleic acids, a supply of nitrogen is crucial for any form of life on earth. Nonetheless, the triple bond connecting two nitrogen atoms is one of the strongest chemical bonds (9.79 eV), making molecular nitrogen gas kinetically inert and inaccessible to most organisms. Nitrogen fixation (NF) is a natural process in which atmospheric nitrogen is fixed into the soil or aquatic systems (via microorganisms and lightning) in the form of ammonia and NOx, which are accessible compounds to living organisms. Thereafter, following a series of transformations, nitrogen is released back into the atmosphere in the form of N2 molecules, constituting the so-called nitrogen cycle, essential for life. At the beginning of the 20th century, the development of new technologies for artificial NF enabled the rapid expansion of human activity and greatly impacted the global nitrogen cycle, using the Birkeland–Eyde process and later the notable Haber–Bosch (HB) process, which converts nitrogen (N2) and hydrogen (H2) into ammonia (NH3).1–5 The latter is a feedstock for the production of nitrogen fertilizers (such as ammonium nitrate or urea), essential for modern agriculture which followed the green revolution, but is also used as a precursor for the synthesis of most compounds containing nitrogen. As an example, ammonia is routinely converted into nitric acid (HNO3) using the Ostwald process, which is a key precursor for the synthesis of organic nitro compounds (Nitration).Despite being extensively optimized, the HB process alone accounts for 1–2% of the global energy consumption and more than 1% of the global carbon emission (300 million metric tons per year).6–8 Additionally, it requires H2 as a precursor, which is commonly produced by steam reforming of hydrocarbons, typically methane. This makes the HB process one of the biggest greenhouse gas emitters while being dependent on a hydrocarbon precursor – mainly from fossil origin – which is not part of a sustainable development approach.9 The carbon cycle is resilient and thousands of years are required to naturally absorb the excess of CO2 released in the atmosphere from human activity.10,11 This excess of CO2 has already triggered an uncontrollable climate drift and put an end to the climate stability of the Holocene. The devastating consequences, such as glacier melting, sea level rise, extreme weather events or biodiversity collapse represent an existential threat for human civilizations.12–18 Therefore, there is an urgent need to find a “greener” alternative for artificial NF, one which would allow supporting the growing population needs while reaching net-zero carbon emission and not being dependent on rapidly depleting reserves of fossil fuels.
Non-thermal plasma based processes have been one of the most prominent alternatives studied over the past few years for artificial NF.6,19–24 Their unique non-equilibrium properties give them the potential to be theoretically less energy intensive than the HB process.9 This is mainly related to the capability of plasmas to transfer energy from hot electrons specifically to other gas components through inelastic collisional interactions. Thereby, for molecular gases, rotational, vibrational and electronic excitations are typically transferred to molecules, along with dissociation and ionization reactions, instead of reactions based mainly on thermal energy. For nitrogen-based plasmas, the vibrational excitation of N2 is a crucial parameter on the molecular dissociation. In particular, vibrational–vibrational relaxation between vibrationally excited nitrogen molecules allows for the population of high vibrational levels through the so-called vibrational ladder climbing, which reduces the activation energy for N2 dissociation.25–27 According to Cherkasov et al.9 High degrees of vibrational excitation of N2 is a key condition for reaching a competitive energy cost for NF by plasma. For practical reasons, igniting gas discharges at atmospheric pressure and (near) ambient temperature is also more advantageous than the HB process which is typically carried out at high pressures (about 200 atm) and temperatures (typically 450 °C) in large scale facilities. Plasmas can also be easily operated with renewable electricity as a power source, which is a way to “store” an intermittent renewable energy supply in the form of chemical energy.19,28
Before developing and optimizing plasma-based systems for NF which can be used in the chemical industry, fundamental knowledge still needs to be revealed to enable efficient and selective synthesis of valuable nitrogen compounds such as nitrates or ammonia. There is a great diversity of non-equilibrium plasma sources (DBDs, gliding arcs, microwave plasmas, DC, AC or RF discharges…) and possible gas mixtures (using N2, O2, H2, H2O…) currently being studied for NF.26,29–37 Amid various different designs, plasmas interacting with water38–41 exhibit a particular interest and can supply hydrogen from water molecules for the reduction of N2 (ammonia synthesis).42 It was recently demonstrated37,43–45 that N2 plasmas ignited in the presence of water vapor or on/in liquids such as water or ethanol allow the formation of ammonia gas or ammonium ions dissolved into the liquid. These results were achieved using either catalyzed or non-catalyzed systems, with increasing energy efficiencies over the years. Additionally, plasmas interacting with liquids are also known to generate NOx, which in some cases result in gas discharges that can simultaneously oxidize and reduce nitrogen. The simultaneous synthesis of compounds containing nitrogen in states (compared to N2) called nitrogen disproportionation, is of high interest for the green synthesis of nitrogen fertilizers. It is indeed widely accepted that a co-supply of different nitrogen sources such as ammonium and nitrates is critical to maximize plant growth.46–49 The synergism between these compounds varies for different plant types. Thus, developing novel systems that allow adjusting the ratio of different oxidized/reduced nitrogen species is essential for an optimized synthesis of nitrogen fertilizers, in particular considering the significant energy cost associated with its production.
A plasma-driven electrolysis, hereafter referred to as “plasma-electrolysis” is presented in this work where pure N2 is used as the working gas. It should be noted that due to water evaporation related to plasma–liquid interaction, “humid N2” i.e. a mixture of N2 and H2O describes the gas composition better. Plasma-driven electrochemistry has attracted growing attention as a method to electrify the chemical industry thereby providing novel green chemical transformation processes, including NF.50,51 In this interesting approach, hydrogen (H2) and oxygen (O2) are supplied through the hydrogen evolutions reaction (HER) and oxygen evolution reaction (OER) driven by water electrolysis, while the nitrogen plasma allows excitation and dissociation of N2 molecules and their subsequent reaction with other species in the gas discharge, in particular H2O molecules. As this system appears suitable for nitrogen disproportionation, the mechanisms for nitrogen oxidation and reduction need to be clearly understood in order to control the (energy-efficient) synthesis of specific compounds. Gas phase diagnostics are performed using optical emission spectroscopy (OES) and industrial electrochemical sensors (Testo Inc.) while the solution is analyzed following plasma treatment using ionic chromatography and colorimetric assays (via UV-visible spectrophotometry). The influence of the discharge/electrolysis current on nitrogen disproportionation is investigated and a calculation of the energy cost is presented. Different (initial) amounts of water vapor are used and experiments are divided into two groups: with “normal experimental conditions” and with “H2O admixtures” which corresponds to a condition with a higher amount of water vapor. This allows studying the influence of water vapor on the properties the discharge and its physico–chemical reactions for which different mechanisms are discussed.
Experimental section
The plasma-electrolysis process is operated using a platinum wire (Pt, Sigma-Aldrich CAS 7440-06-4) immersed in a 50 ml aqueous solution of NaCl (1 mM, VWR Chemical, CAS 54-21-7) acting as anode and a pointy stainless steel capillary needle (outside diameter 1.5 mm) acting as a “plasma cathode electrode” (see Fig. 1). A DC plasma is ignited between the surface of the solution and the capillary needle: for this purpose, a negative high voltage ranging from 900 V to 2.3 kV is supplied to the needle by a Technix DC generator (maximum voltage of 5 kV), which ignites a discharge with a current ranging from 1 to 10 mA, using a ballast resistor of 150 kΩ. For all experimental conditions, the plasma-electrolysis is run for 30 min. The plasma is operated at atmospheric pressure in a closed Plexiglas vessel filled with N2 (Air Liquide, Alphagaz1) with a constant gas glow of 1 standard liter per minute (slm). The gap between the needle and the water surface is fixed at 0.7 ± 0.1 mm during operation (including the dip induced by the discharge on the water surface). | ||
| Fig. 1 Schematic of the plasma-electrolysis experimental set-up. The gas lines are heated in some experiments using a heating cable maintaining a constant temperature of 67 °C to the heated parts. | ||
Before being introduced to the working vessel, the nitrogen gas is bubbled through 250 ml of Milli-Q water in order to increase the humidity of the feed gas. The outlet gas stream is also bubbled through Milli-Q water which allows to collect soluble compounds that are formed in the gas phase during the plasma-electrolysis process. The exhaust bubbler is rinsed and refilled with 50 ml of fresh water prior to each plasma ignition. This bubbler, obviously, was not used when gas phase diagnostics were required on the outflow. A heating cable made of a tungsten wire insulated with glass wool and connected to a voltage generator is used to heat the gas lines, the vessel and the humidifier at a constant temperature of 67 °C (see Fig. 1).
Experiments are divided into two groups for which two different humidity levels are used in the vessel to run plasma-electrolysis. In the first group, referred to as “normal conditions”, the heating cable is off and the nitrogen gas flows directly to the chamber, keeping the humidifier bubbler gas line isolated. In these conditions, the evaporation of the working solution at 1 atm and 20 °C under a nitrogen gas flow of 1 slm leads to an initial humidity of approximately 0.8 mol%. In the second group, referred to as “H2O admixtures”, the heating cable is turned on and the nitrogen gas inflow is driven through the humidifier bubbler, closing the valve of the parallel gas line. This results in an initial humidity ranging from 6 to 8 mol% (molar proportion). In this group the working solution is introduced into the vessel a few seconds prior to plasma ignition.
Gas phase diagnostics
In this work, the analysis of plasma at different experimental conditions is carried out by means of OES. The spectrograph used is an absolutely calibrated Andor Technology SR-500i-D2-R spectrometer and an Andor DU420 CCD camera with an opened electrode. The light is collected using an optical fiber through a LiF window installed on the vessel and centered on the discharge. Each optical emission spectrum is acquired using a 2400 grooves per mm grating providing a resolution of 0.0176 nm. Each step of the scan accumulates 4 acquisitions of 3 seconds each (full vertical binning). To perform the measurements, the width of the entrance slit is set to 160 μm.OES is used in this work to get a better insight on the energetic properties of the plasma. This is performed by recording the molecular spectra of the second positive system of nitrogen, N2 (C3Πu → B3Πg) in the UV. At atmospheric pressure, the condition of high collision frequency between atoms and molecules is well assumed. This means that in the case of N2 molecules, each rotational population distribution is in thermodynamic equilibrium. The population energy distribution of the vibrational states can be described by the Boltzmann distribution, too. However, the rotational and the vibrational states are very likely not in equilibrium all together, meaning that Trot does not equal Tvib. With the assumption to consider the translational temperature in equilibrium with the excitation of N2 (C3Πu), a detailed study of the N2 (C3Πu → B3Πg) spectra will provide an estimation of the rotational and vibrational temperatures. While Trot will give a realistic measurement of the neutral gas temperature,52Tvib is often related to the dissociation efficiency of the plasma. As suggested by Andre et al.53 in the context of DC discharge with a liquid water electrode, only vibrational transition bands with Δv = −2 will be considered in this work. The analyses of the recorded spectra is carried out with the help of the simulation of the N2 (C3Πu → B3Πg) together with an optimization curve fitting routine (Fig. 2).54 The relative intensity of each ro-vibrational transition  is calculated according to,
is calculated according to,

 | ||
| Fig. 2 (a) Optical emission spectroscopy spectra of the N2 plasma electrode for different currents and (b) fitting of the N2 (C3Πu → B3Πg) rovibrational bands for the calculation of Trot and Tvib. | ||
The composition of the gas exiting the vessel is analyzed in real time using a Testo350 device, equipped with electrochemical sensors calibrated for the measurement of the density of NO, NO2 and H2 in ppmv. A Vertex 70/70 V Fourier-transform infrared spectrometer (FTIR) system is also used for the same purpose. However, due to high amount of water vapor and the substantial condensation in the system, the use of FTIR was restricted and only provided qualitative information.
The relative humidity and temperature inside the vessel are monitored in real time using a DHT22 sensor, which is equipped with a capacitive humidity sensor and a thermistor. The partial pressure of H2O is determined by multiplying the measured relative humidity with the saturated vapor pressure Psat which is estimated using the Buck equation:

Thereafter, the partial pressure of water is divided by the total pressure in the chamber (101![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 325 Pa) to obtain the molar proportion (in mol%) of H2O in the gas.
325 Pa) to obtain the molar proportion (in mol%) of H2O in the gas.
Liquid phase diagnostics
Following the end of the plasma-electrolysis, the solution is immediately analyzed by ionic chromatography using a 930 compact IC flex from Thermo Scientific with a DionexIonPac AS9-HC Analytical column for anions (NO2− and NO3−) and a Metrosep C6 – 250/4.0 Analytical column for cations (NH4+). Based on the NO2− concentration measured by ionic chromatography and the solution pH, the proportion of NO2− and HNO2 is determined using the pKa of the reaction: HNO2 + H2O → NO2− + H3O+.The measurement of the concentration of NH3(aq)/NH4+(aq) in the working solution is performed with the Nessler Reagent (K2HgI4, Sigma Aldrich)58,59 using the following procedure: after the plasma-electrolysis process, 5 ml of the solution is sampled and mixed with 0.25 ml of Nessler Reagent, leading to a very alkaline solution in which all NH4+(aq) is turned into NH3(aq). Thereafter, ammonia leads to the formation of a yellow-orange complex with a maximum absorption at 394 nm, following the reaction:
| NH3(aq) + 2 HgI42−(aq) + 3OH−(aq) → OHg2NH2I(aq) + 7I−(aq) + 2H2O(aq) |
The concentration of NH3(aq) is subsequently estimated using UV-VIS spectrophotometry (UV-3100PC, VWR equipped with a tungsten and a deuterium lamp), for which a calibration was carried out based on a standard solution of NH4Cl (Merck, certified Certipur, 1000 mg l−1). The proportion of NH3(aq)/NH4+(aq) is eventually evaluated based on the pH of the analyzed solution and the concentration of NH3(aq) measured by the Nessler Reagent.
Ionic chromatography measurements were used to validate the reliability of the Nessler Reagent method and the results showed a relative difference of less than 10% between the two methods.
Energy cost calculation
The energy cost is presented in Joule per mole of valuable components generated, both in the gas phase (NO and H2) and the liquid phase (NO2−, NO3−, NH4+). The energy consumption is calculated based on the product of the discharge current and either the voltage supplied by the generator (for the energy provided by the generator) or the voltage between the needle and the water (for the energy supplied to the discharge) over half an hour, i.e. E(J) = I(A) × U(V) × 1800(s). This comparison is made because a significant fraction of the energy supplied is lost due to Joule heating in the ballast resistor. The amounts (in moles) of NO2−(aq), NO3−(aq) and NH4+(aq) are obtained from the measured concentration (in ppm) in the liquid phase via ionic chromatography using the following calculation: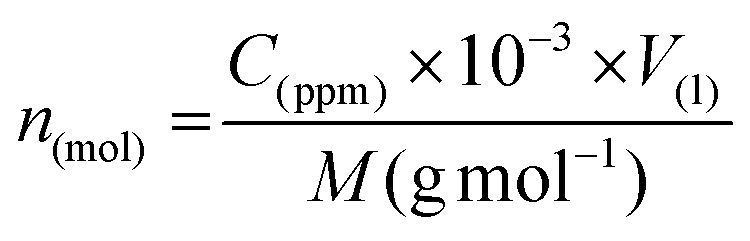
For NO(g) and H2(g), whose concentrations (in ppmv) at the gas outlet were measured with the Testo350, the amounts were calculated every second of the measurement and thereafter integrated over the 30 min of plasma operation (i.e. 1800 seconds) as following:
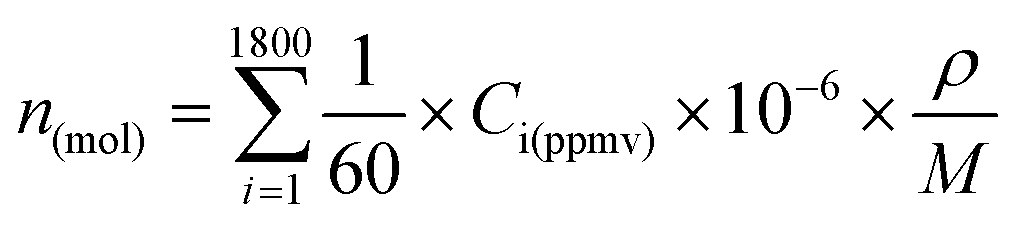
Here ρ is the density of the gas (in g l−1), M is the molar mass (in g mol−1) of the molecule studied and Ci(ppmv) its concentration at each step i (in ppmv, or μl l−1). Because the Testo350 device conducts one concentration measurement per second and considering a gas flow rate of 1 slm, the approximation that 1/60 l of gas is analyzed every second is made. By multiplying the concentration by 1/60 (l) and by a factor 10−6, the volume of the measured gas (in l) is obtained. Eventually, the amount (in mol) is obtained by multiplying with the gas density and dividing by its molar mass. The following standard gas densities were used for the calculation: 1.3402 g l−1 for NO and 0.0852 g l−1 for H2.
Results and discussion
(1) Simultaneous oxidation and reduction of nitrogen
In the DC discharge ignited between the needle and the water surface, the needle acts as the plasma cathode to which the negative high voltage is applied and the platinum wire immersed in the conductive solution acts as the anode. Thus, hydrogen gas is formed at the plasma–liquid interface by reduction of hydrogen cations by solvated electrons (HER, reaction (R1))60–64 while oxygen is formed on the platinum wire by the OER (R2). Besides HER and OER, the electron impact dissociation of H2O molecules in the plasma leads to the formation of H atoms and OH radicals (R3). The subsequent recombination of H atoms in the discharge also leads to the formation of H2(R4) in addition to its formation by the HER. OH radicals are highly oxidizing in nature and previous studies51 have demonstrated their importance in NOx formation (in particular NO) during N2 plasma electrolysis ((R5) and (R6), Extended Zeldovich mechanism).| 2H+(aq) + 2e−(aq) → H2(g) | (R1) |
| 2H2O(aq) → O2(g) + 4H+(aq) + 4e−(aq) | (R2) |
 | (R3) |
 | (R4) |
 | (R5) |
 | (R6) |
Therefore, for plasma-electrolysis performed in N2, the supply of oxidative species (O2, OH) and reductive species (H2, H) for nitrogen disproportionation strongly depends on the amount of water vapor in the gas phase, which increases following plasma ignition. To illustrate this, the molar percentage of H2O(g) inside the vessel is calculated as a function of time during plasma electrolysis under different currents ranging from 1 to 10 mA (Fig. 3(c), see Experimental section for calculation details). Within 30 min, a substantial increase in the water vapor content inside the vessel is observed, especially when using a 10 mA current where the molar proportion of H2O increases from less than 1 mol% up to approximately 6 mol%. This is mostly due to two reasons: first, with higher currents i.e. higher powers, the average gas temperature inside the vessel increases over the treatment time (+20 °C in 30 min at 10 mA, +2 °C at 1 mA), which results in a higher saturation vapor pressure, thereby increasing water vapor transfer to the gas phase. Secondly, in addition to the increase in the gas and plasma temperatures (see section 2.b.), the solution temperature also rises at higher discharge currents (+13 °C after 30 min for 10 mA, no change at 1 mA), which results in a stronger evaporation of water, especially at the plasma–liquid interface where local heating is significant.
 | ||
| Fig. 3 Temporal evolution of the (volumetric) concentration of NO (a) and H2 (b) in the gas outlet of the vessel following plasma ignition and (c) the corresponding molar fraction of H2O in the gas phase, for different plasma-electrolysis currents. The discharge is ignited at t = 0 and stopped at t = 30 min in N2 (1 slm) at atmospheric pressure using a water volume of 50 ml and a needle-water distance of 0.7 mm. | ||
The evolution of the water vapor content in the gas phase impacts the discharge properties, as illustrated in Fig. 3(a) and (b), which presents the temporal evolution of the concentration of NO and H2 at the outlet of the reactor vessel. Following the initial sharp increase in the species concentration after plasma ignition, a slower increase is observed over longer periods of time and this increase is more pronounced for higher currents. A clear correlation is observed between the rise of the gas temperature, the solution temperature, the water vapor content in the gas phase and the production of NO and H2 in the plasma-electrolysis system. It is expected that the increase in the solution temperature impacts liquid phase reactions such as HER and OER and causes a higher production of H2. However, the formation of NO occurs in the gas phase and is most likely affected by different plasma properties caused by the increase in the amount of water vapor in the gas phase on one hand, and the possibly greater production of highly oxidizing species such as OH˙ radicals on the other. It is reasonable to assume that water evaporation provides more H2O molecules which can undergo electron impact dissociation (R3) and provides H atoms for the formation of H2(R4) and OH˙ radicals. However, water vapor influences the discharge properties complexly in many aspects, influencing for instance the inelastic collisional mechanisms65,66 or the electron density and electron energy distribution function, given the different ionization potential between nitrogen (15.58 eV)67 and water (12.62 eV).68
Nitrogen oxidation
The plasma reaction between N atoms and OH˙ radicals is the main route for the initial formation of NO through the extended Zeldovich mechanism (R6). The presence of NO in the plasma, previously measured using the Testo electrochemical sensing system, is confirmed by OES with the presence of NO (A2Σ+ → X2Π) bands in the region 200–280 nm. While the Testo device is equipped with NO2 sensors, the latter molecule could not be detected in any experimental condition. NO2 is typically formed by the reaction between NO and O˙ (R7), or in the presence of ozone. However, the gas temperature (≥1000 K, see section 2.b.) is too high to form ozone in this system and the absence of O emission lines in the optical emission spectra suggests that O atoms are also not formed in significant amount in the discharge. Consequently, NO cannot be further oxidized to NO2 in our system. Traces of N2O are however measured in the gas outlet of the vessel by FTIR (see Fig. 4), but no other NOx (e.g. N2O4 and N2O5) could be identified by FTIR. With the formation of O2 at the anode (i.e. the platinum wire) which is only 2 cm away from the plasma cathode, it is expected that small fractions of O2 are present in the gas discharge which can influence its physico–chemistry. It is possible that NO2 is an intermediary species in the plasma for the formation of N2O through reactions with N atoms (R8). | (R7) |
 | (R8) |
 | (R9) |
 | (R10) |
 | ||
| Fig. 4 FTIR spectra of the gas flow exiting the vessel during an N2 DC plasma electrolysis process performed at atmospheric pressure with a 50 ml solution, in a water anode configuration and with a needle-water distance of 0.7 mm. | ||
In the liquid phase, nitrites (NO2−) and nitrates (NO3−) are measured by ionic chromatography following plasma treatment, and NO2− ions are in this study clearly much more abundant than NO3− (see further on in Fig. 5). Their formation results from the synergistic effect between gas phase and liquid phase chemistry in the plasma electrolysis cell, in which gaseous H2O2 and NOx are transported into the solution where they further react, even after the plasma is switched off.69 Because NO and NO2 have a low solubility in water according to Henry's law,70 their further oxidation by OH radicals (similarly to what is described in ref. 51) to the highly soluble HNO2 and HNO3 species (reactions (R9) and (R10)) should be considered as one of the main mechanism for the formation of nitrites and nitrates in the solution. The formation of significant amounts of HNO3 appears unlikely in this study (also confirmed by the results shown in Fig. 5) considering that no NO2 is measured using the Testo system in any experimental condition, which might explain – at least partly – the low amount of NO3− anions in the solution.
 | ||
| Fig. 5 Molar fraction of stable nitrogen species of interest produced during the N2 plasma-electrolysis process for different discharge currents, under (a) normal experimental conditions (b) and with H2O admixtures. | ||
Nitrogen reduction
The reduction of nitrogen in the used plasma electrolysis cell is observed simultaneously with its oxidation through the formation of NH4+ ions in the solution (detected using the Nessler Reagent and ionic chromatography). Traces of NH3 are also detected in the gas outlet by FTIR (see Fig. 4). The formation of these compounds can occur through several different pathways which are still under debate in literature. Hawtof et al.64 presented an N2 DC plasma–water electrolytic system for the production of ammonia for which a model suggests that (activated) N2 molecules are first dissolved into the liquid where they further react in the solution with H˙ radicals to form NH3, with N2H as an intermediary species. In a similar set-up, Haruyama et al.71 assumed that NH˙ was primarily formed at the plasma–water interface from reactions between N atoms and H2O molecules, before being further reduced in the liquid phase to NH2 and NH3. This result is supported by the findings of Tsuchida et al. which demonstrated that atomic nitrogen transported from a gas discharge to a water surface has a substantial impact on the selective synthesis of ammonia (in the form of NH4+ ions in the liquid phase).72 In the plasma–liquid interface, which is known to be a crucial region with a highly complex (and not yet fully understood) physico–chemistry, Sakakura et al.73 highlighted that ultraviolet (UV) irradiation could enhance the formation of H˙ radicals and thereby impact ammonia production. In addition, it was suggested in72,74 that, in spite of their lower reactivity, N2* and N2+ could react with dissociated water molecules at the plasma–liquid interface and form ammonia gas.Here, as a first observation, NH˙ radicals are identified in the discharge by OES (≈336 nm). There are several possible mechanisms for their formation ((R11)–(R13)).75,76 It is likely that the further reaction of NH˙ with H˙ atoms in the discharge leads to the formation of NH2˙ and eventually NH3(R14). Despite the solubility of ammonia, it is expected that a significant amount of this gas is carried away after its formation to the gas outlet, which is confirmed by FTIR (Fig. 4), with the identification of the specific absorption peaks of ammonia in the region 850–1150 cm−1. Therefore, the exhaust bubbler is used to collect ammonia assuming that it is completely dissolved into the solution as NH3(aq) and NH4+(aq)(R15). Following the 30 min plasma-electrolysis, Nessler Reagent is added to the exhaust bubbler water in order to quantify the amount of NH3 in this solution, and the total amount of NH3(aq) + NH4+(aq) is assumed to be equal to the amount of ammonia that did flow through it during the plasma-electrolysis process.
As a result, the estimated amount of ammonia going through the gas outlet of the reactor accounts for 23 to 60% of the total ammonia/ammonium formed during the plasma-electrolysis process (see Fig. 5). It suggests that the main route for the production of ammonia occurs in the gas phase, most likely through the mechanism discussed previously. It is not clear in which region of the plasma ammonia is preferably formed, but it is possible that the plasma–liquid interface, a region of high interest and complexity, plays a significant role in the formation of ammonia. Furthermore, ammonia decomposition occurs at elevated temperatures,21 and may be a limiting factor for its formation within the discharge volume.
 | (R11) |
 | (R12) |
 | (R13) |
 | (R14) |
| NH3(aq) + H2O(aq) ⇌ NH4+(aq) + OH−(aq) | (R15) |
(2) Plasma electrolysis at low currents
In the next experiment, the influence of the discharge/electrolysis current (ranging from 1 to 10 mA) on nitrogen oxidation/reduction is evaluated. A reduction in the discharge current naturally decreases the electron density, but nonetheless here with a limited impact on the electron temperature. Indeed, the decrease in the applied high voltage is counterbalanced by a reduction in the voltage drop across the ballast resistor, resulting in a rather limited variation in the inter-electrode potential difference (up to 18% variation between 1 and 10 mA, with a tendency to increase with decreasing applied voltage/discharge current). It should be noted that the voltage drop through the positive column (which represents a large majority of the discharge volume) is obtained after subtraction of the normal cathode fall (estimated to be approximately 215 V).77 Next to its influence on the discharge behavior, the use of different currents is known to also impact the HER in the liquid phase close to the plasma–water interface.To study the influence of water vapor on plasma-electrolysis, focus is put on two hydrogen production routes: HER in the liquid phase, and water dissociation in the gas phase. For this purpose, two experimental conditions are defined and experiments are divided into two groups: in the first group named “normal conditions”, plasma-electrolysis is carried out in a dry N2 flow of 1 slm. The plasma is ignited at ambient temperature, with an initial water content, before plasma ignition, of around 0.8 mol% (Fig. 3(c)) which then rises up to a few percentages, reaching a maximum of 7 mol% after 30 min of plasma electrolysis at 10 mA. In the second group named “H2O admixtures”, all experimental conditions remain the same as in the previous group except for the N2 flow which is bubbled through Milli-Q water (maintained at 67 °C) prior to being introduced in the vessel. In this group, the gas lines and the vessel are also kept at 67 °C (see Fig. 1) to prevent condensation and to maintain the ambient gas temperature at a constant value at which the saturated vapor pressure is higher. The resulting water content in this group ranges from 6 to 8 mol% prior to plasma ignition, and above 13 mol% at least after 30 min of plasma electrolysis in all conditions studied. A precise number could not be obtained because the relative humidity and gas temperature reached values above the sensor boundaries. All experiments are carried out using a treatment time of 30 min.
Selectivity of nitrogen compounds
The nitrogen compounds of interest i.e. NO2−, NO3−, HNO2 and NH4+ in the activated water and NO and NH3 in the gas outlet are quantified directly after the 30 min plasma electrolysis process. The molar fraction of each species with regards to the total amount formed is presented on Fig. 5(a) for normal conditions and on Fig. 5(b) for conditions with H2O admixtures. Although the decrease of the discharge current lowers the electron density and thus the amount of nitrogen fixed, as confirmed by the experimental results, a clear difference is observed in the relative proportion of each synthesized compound. Firstly, the liquid/gas phase ratio of nitrogen compounds drastically decreases as the discharge current increases (Fig. 5). This means, at lower currents the loss of nitrogen compounds to the gas phase is reduced, hence more of the fixed nitrogen is trapped in the liquid. This illustrates that a reduction in the “losses” of nitrogen compounds to the gas outlet is obtained when using lower currents, with a better transfer of nitrogen species from the gas phase to the liquid phase. The higher amounts of NO in the “H2O admixture” group can be explained by the (probably) higher amount of OH radicals in the gas discharge, resulting from the high amount of water vapor. This increase in the production of NO directly causes a globally higher conversion of nitrogen for this group, with a conversion ranging from 8.12 × 10−6 to 5.69 × 10−5 (increasing along with the discharge current). In comparison, the conversion rate ranges from 4.72 × 10−6 to 2.43 × 10−5 for normal conditions.Overall, the decrease in the discharge current increases the proportion (in moles) of reduced nitrogen compounds in the fixated nitrogen (Fig. 6(a)), with a molar proportion increasing from 15 mol% at 10 mA to approximately 35 mol% at 1 mA. Similar trends, regarding reduction vs. oxidation selectivity are obtained for both normal conditions and with H2O admixtures, indicating that this tendency is close to independent for the initial humidity in the gas phase and depends mainly on the discharge current. Nitrogen reduction is here possibly favored by a proportionally higher supply of N atoms to the plasma–liquid interface with a reduced presence of oxidative species.72 Hence, NH˙ may be subsequently forming in this region before being further reduced in the liquid phase to NH3/NH4+.
 | ||
| Fig. 6 (a) Molar proportion of reduced nitrogen species in the fixated nitrogen (b) molar ratio of ammonium (measured in the plasma electrolysis beaker) to ammonia gas, for different discharge currents and experimental conditions. | ||
Yet, though only significant at currents above 2.8 mA, the molar ratio of ammonium ions to ammonia gas is slightly higher when the plasma-electrolysis process is performed at high water vapor contents (H2O admixtures, Fig. 6(b)). The reason for this tendency is unclear, but it may be related to a higher supply of reactive reducing species (in particular H atoms) at the plasma–liquid interface.
Discharge properties
To understand how the variation of the discharge current and the water content affects the plasma properties, OES is used to acquire and fit the N2 (C3Πu → B3Πg) emission bands and to estimate the rotational (Trot) and vibrational (Tvib) temperatures of N2. In the present N2/H2O plasma-electrolysis system, it is reasonable to assume a Boltzmann distribution for the rotational states of N2(C), a necessary condition for the approximation Trot = Tgas thus allowing the estimation of the gas temperature.54 In this study, for both experimental conditions, Trot is estimated to be approximately 1500 K at 10 mA, and decreases with the decrease in current. A drop of 500 K and 300 K down to 1 mA in average for normal conditions and for H2O admixtures respectively, owing to a lower electron density and thermalization of the discharge.78In normal experimental conditions, while Tvib exhibits a similar behavior as Trot, the ratio Tvib/Trot increases slightly with the discharge current, from 2.03 to 2.52, which may indicate higher non-equilibrium properties for the plasma at high discharge current, which is assumed to be more favorable for an efficient nitrogen fixation. A much stronger difference is however observed when experiments are performed with H2O admixtures. While Trot in both conditions remain in a very similar range, the estimation of Tvib shows much higher values for H2O admixtures, with an increase of ∼1000 K compared to that under normal conditions. Tvib appears to slightly increase at lower currents, providing two maximums at the extremes of the range of currents studied: 1 and 10 mA. However, it should be noted that the raio Tvib/Trot linearly increases here with decreasing current, ranging from 2.52 at 10 mA to 3.05 at 1 mA. While Tvib is only representative of electronically excited species and thus only partially define the plasma properties, the increase in the ratio Tvib/Trot suggests favorable experimental conditions for NF since vibrational excitation of N2 is essential for its efficient dissociation whereas gas heating, which depopulates the vibrational states through vibrational-translational relaxation, is counterproductive and should be reduced. This is particularly important to promote vibrational–vibrational relaxation (vibrational ladder climbing) to populate high energy vibrational levels.25
Energy cost
Fig. 8 presents the calculation of the energy cost in MJ mol−1 for the production of the nitrogen compounds of interest described in the previous section -for different discharge currents and water vapor contents. At first if one considers the complete system, it appears evident that the energy losses due to Joule heating in the ballast resistor, a phenomenon proportional to the square of the electrical current, can be reduced by lowering the latter. While 74% of the energy is converted to heat in the ballast resistor at 10 mA, it represents only 17% at 1 mA. If the energy deposited to the discharge is solely considered (by considering the voltage between the needle and the water, see Experimental section), the reduction in the discharge current also leads to an improvement in the energy efficiency (down to about 100 MJ mol−1 in normal conditions, see Fig. 8(a)). This correlates with the increase in the Tvib/Trot ratio and thus of the non-equilibrium properties of the discharge with the increase in discharge current under normal conditions as previously discussed. It might be assumed that the proportionally higher vibrational excitation of N2 favors its dissociation and allows for an improved system energy efficiency, as already pointed out in the literature.9,25,26,42,79 However, as simultaneous increase of atomic hydrogen and OH occur also at high humidity in the discharge, further research should be carried out in order to separate the different variables.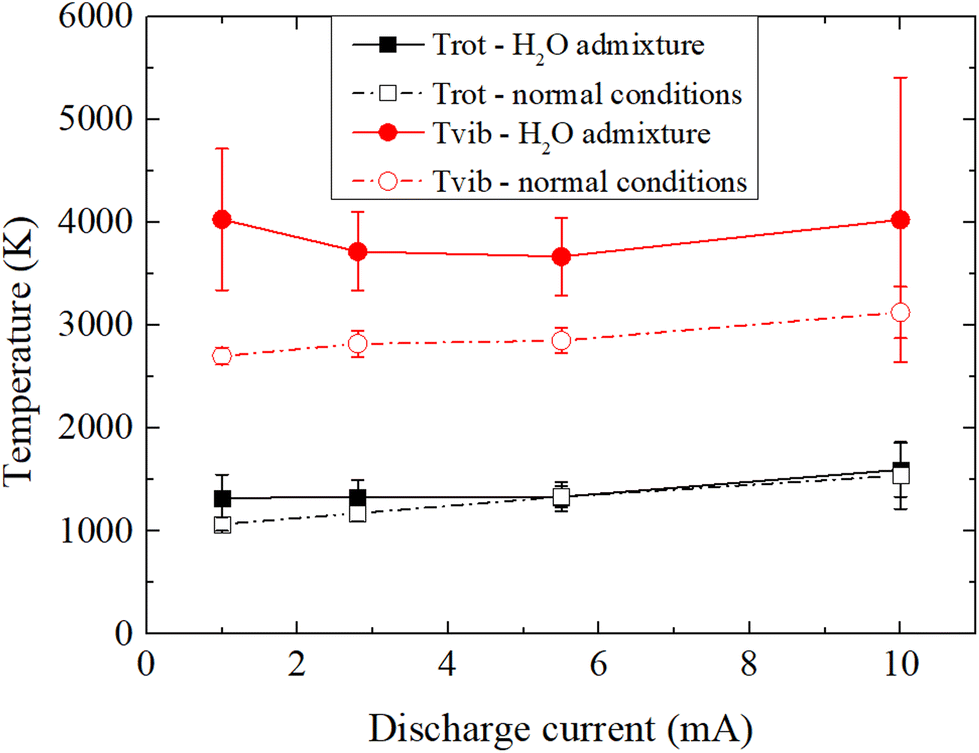 | ||
| Fig. 7 Rotational and vibrational temperatures of N2 in the discharge at different plasma-electrolysis currents and for different experimental conditions. | ||
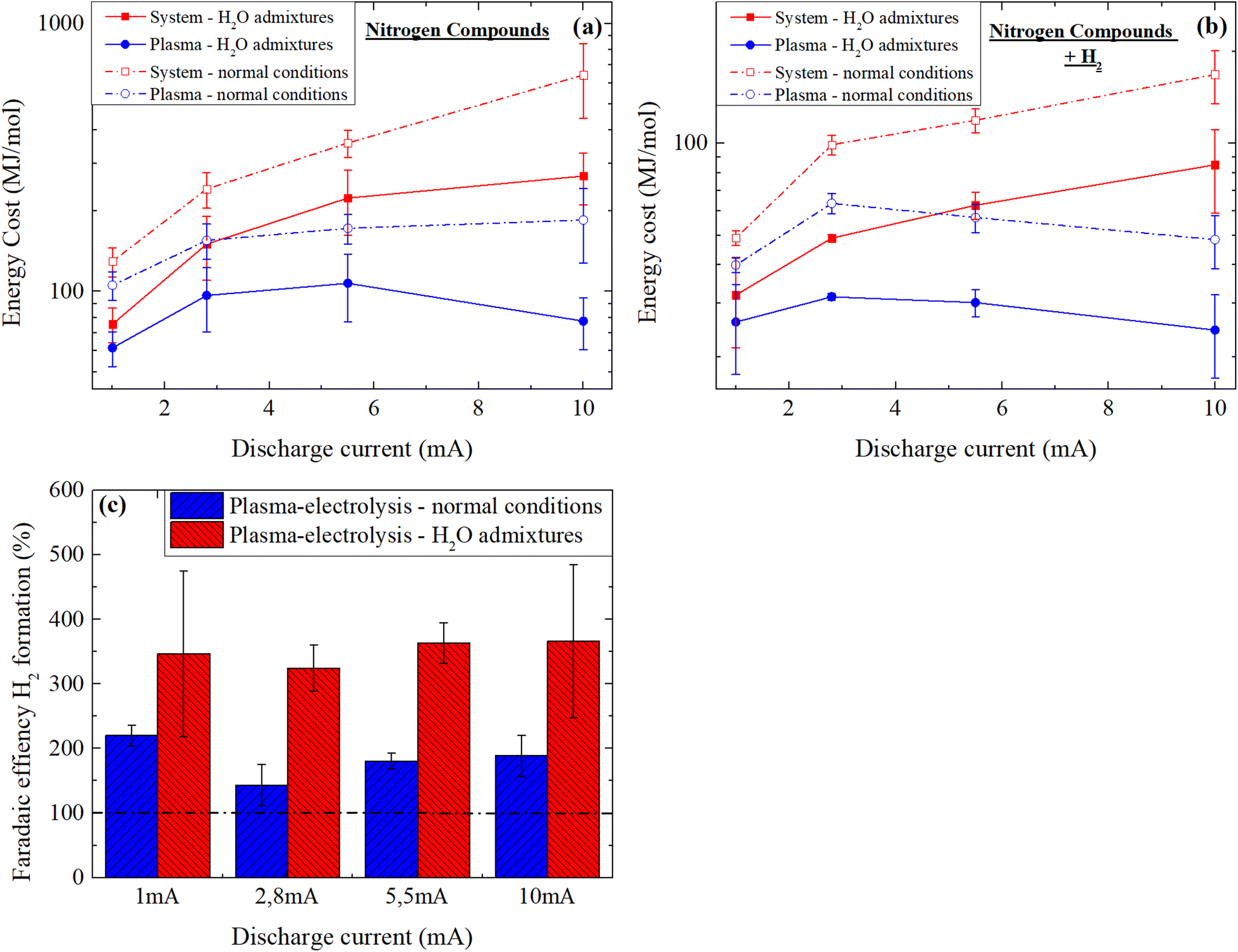 | ||
| Fig. 8 Energy cost for the production of nitrogen compounds (a) and nitrogen compounds + H2 gas (b) and (c) faradaic efficiency for the production of H2 gas in the N2 plasma-electrolysis system at different discharge currents. | ||
Interestingly, when experiments are started with a high water vapor content (H2O admixtures), the relationship between the energy efficiency and the discharge current is completely different, if one considers only the energy deposited to the discharge. As shown on Fig. 8(a), the energy efficiency reaches a minimum on both extremes of the current range i.e. at 1 and 10 mA, and is higher in between. Although this tendency is not fully understood, the lowest energy cost is here correlated to Tvib, which is highest for 1 and 10 mA (Fig. 7). It should be noted that the energy efficiency is globally higher (i.e. the energy cost is lower) for the conditions with H2O admixtures. This is also correlated to Tvib, which is globally much higher compared to normal experimental conditions. The best energy efficiency that could be obtained, with regards to nitrogen compounds, is 61.6 MJ mol−1, which is reasonable for an experimental reactor designed for the purpose of fundamental studies, even though it is still about 100 times higher than the Haber Bosch process.
If H2 gas, a valuable side product of the process, is also considered (Fig. 3(b)) in the calculation of the energy cost, a similar tendency is obtained, with the exception that in normal experimental conditions, the lowest energy cost is also obtained at both 1 and 10 mA, if the energy deposited to the plasma is solely considered. This is most likely related to the high water evaporation and thus higher hydrogen production at 10 mA. The lowest energy cost calculated for fixated nitrogen + hydrogen is 26 MJ mol−1, which is significantly better but not yet competitive. However, the cogeneration of nitrogen compounds and hydrogen gas becomes more attractive when the faradaic efficiency for H2 formation is estimated. For this purpose, the theoretical amount of H2(g) which is expected to be formed in an ideal electrolysis is calculated using the relation:
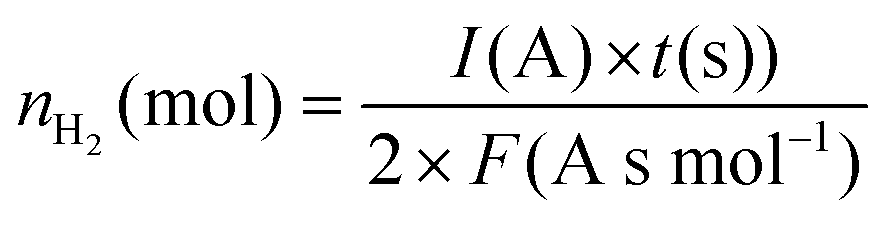
I (in A) refers to the plasma-electrolysis current, t (in seconds) to the duration of the process and F (in A s mol−1) to the faradaic constant. For a plasma-electrolysis process of 30 min, the amount of hydrogen that should ideally be formed ranges from 9.33 × 10−6 to 9.33 × 10−5 mol for currents ranging from 1 to 10 mA. When the amount of hydrogen (in mol) measured using the testo350 device is divided by these values to estimate the faradaic efficiency in each experimental condition and in each group, values ranging from 143 to 366% are obtained (Fig. 8(c)). These unexpected values are probably due to the high production of hydrogen in the plasma following electron impact dissociation of H2O, in parallel to the HER. The generally higher faradaic efficiencies obtained for the group “H2O admixtures” confirms this point. This result illustrates that plasma-electrolysis systems could have a great potential for the cogeneration of nitrogen compounds and hydrogen, and industrially optimized reactors could become competitive compared to the Haber–Bosch process with regards to the hydrogen supply.
Conclusions
In this study, a N2 plasma-electrolysis system was used to study the disproportionation of nitrogen, opening the possibility of a carbon-free synthesis of ammonium nitrate with nitrogen gas and water as precursors. Experiments have highlighted that ammonia is formed mainly in the gas phase through multiple pathways which benefit from the hydrogen evolution reaction at the plasma–liquid interface on one hand, and from electron impact dissociation of H2O molecules on the other, with faradaic efficiencies for hydrogen formation above 100%. The reduction of the plasma-electrolysis current from 10 mA to 1 mA enhanced the reduction of nitrogen as opposed to its oxidation, and seemed to aid the transport of more nitrogen compounds to the liquid phase (in the form of stable secondary species such as NO2−, NO3− and NH4+).The amount of water vapour in the gas phase, which increases following plasma ignition, had a significant impact on the discharge properties. In particular, the energy cost for nitrogen fixation was lower with higher humidity (61.6 MJ mol−1). Temperature calculation from OES suggests that increasing the amount of water vapor in the gas phase enhances the vibrational excitation of N2 molecules. However, it also raises the question of the interpretation of data solely from electronically excited species, which only partially describes the discharge. Future work on nitrogen fixation by plasma will require use state of the art plasma diagnostics in combination with calculations to precisely understand the vibrational kinetics of nitrogen. For plasma interacting with liquid, one of the biggest challenges that is yet to overcome concerns the highly complex plasma–liquid interface, which is of great interest.
Author contributions
CP has conducted the majority of experiments and written the manuscript. NM, MB, NCR and AR have contributed to the design of experiments and the interpretation of results. SI has conducted spectroscopy analysis. EV has conducted gas phase electrochemical analysis. MPD and NDG have provided expertise on gas and liquid phase diagnostics. FR has supervised the research. All authors have agreed to the publication of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Excellence of Science FWO-FNRS project (FWO grant ID GoF9618n, EOS ID 30505023).Notes and references
- V. Smil, Sci. Am., 1997, 277, 76–81 CrossRef CAS.
- J. N. Galloway, J. D. Aber, J. A. N. W. Erisman, P. Sybil, R. W. Howarth, E. B. Cowling and B. J. Cosby, Bioscience, 2003, 53, 341–356 CrossRef.
- J. N. Galloway and E. B. Cowling, Ambio, 2002, 31, 64–71 CrossRef PubMed.
- J. N. Galloway, A. R. Townsend, J. W. Erisman, M. Bekunda, Z. Cai, J. R. Freney, L. A. Martinelli, S. P. Seitzinger and M. A. Sutton, Science, 2008, 320, 889–893 CrossRef CAS PubMed.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- A. Anastasopoulou, S. Butala, J. Lang, V. Hessel and Q. Wang, Ind. Eng. Chem. Res., 2016, 55, 8141–8153 CrossRef CAS.
- Y. Tanabe and Y. Nishibayashi, Coord. Chem. Rev., 2013, 257, 2551–2564 CrossRef CAS.
- C. P. Chanway, R. Anand and H. Yang, IntechOpen, 2014, 3–22 CAS.
- N. Cherkasov, A. O. Ibhadon and P. Fitzpatrick, Chem. Eng. Process.: Process Intensif., 2015, 90, 24–33 CrossRef CAS.
- I. Ganesh, Renewable Sustainable Energy Rev., 2016, 59, 1269–1297 CrossRef CAS.
- S. Hernandez, M. A. Farkhondehfal, F. Sastre, M. Makkee, G. Saracco and N. Russo, Green Chem., 2017, 19, 2326–2346 RSC.
- C. Bellard, C. Bertelsmeier, P. Leadley, W. Thuiller and F. Courchamp, Ecol. Lett., 2012, 15, 365–377 CrossRef PubMed.
- A. J. Challinor, J. Watson, D. B. Lobell, S. M. Howden, D. R. Smith and N. Chhetri, Nat. Clim. Change Lett., 2014, 4, 287–291 CrossRef.
- A. Hunt and P. Watkiss, Clim. Change, 2011, 104, 13–49 CrossRef.
- S. Nunez, E. Arets, R. Alkemade, C. Verwer and R. Leemans, Climatic, 2019, 154, 351–365 CrossRef.
- R. K. Mall, R. Singh, A. Gupta, G. Srinivasan and L. S. Rathore, Clim. Change, 2006, 78, 445–478 CrossRef.
- F. N. Tonmoy, A. El-zein and J. Hinkel, WIREs Clim. Change, 2014, 5, 775–792 CrossRef.
- A. Raza, A. Razzaq, S. S. Mehmood, X. Zou, X. Zhang, Y. Lv and J. Xu, Plants, 2019, 8, 1–29 CrossRef PubMed.
- A. Bogaerts and E. C. Neyts, ACS Energy Lett., 2018, 3, 1013–1027 CrossRef CAS.
- A. Anastasopoulou, R. Keijzer, S. Butala, J. Lang, G. Van Rooij and V. Hessel, J. Phys. D: Appl. Phys., 2020, 53, 234001 CrossRef CAS.
- P. Peng, P. Chen, C. Schiappacasse, N. Zhou, E. Anderson, D. Chen, J. Liu, Y. Cheng, R. Hatzenbeller, M. Addy, Y. Zhang, Y. Liu and R. Ruan, J. Cleaner Prod., 2018, 177, 597–609 CrossRef CAS.
- S. Li, J. A. Medrano, V. Hessel and F. Gallucci, Processes, 2018, 6, 248 CrossRef CAS.
- A. Anastasopoulou, Q. Wang, V. Hessel and J. Lang, Processes, 2014, 2, 694–710 CrossRef.
- B. S. Patil, Q. Wang, V. Hessel and J. Lang, Catal. Today, 2015, 256, 49–66 CrossRef CAS.
- S. Van Alphen, V. Vermeiren, T. Butterworth, D. C. M. Van den Bekerom, G. J. Van Rooij and A. Bogaerts, J. Phys. Chem. C, 2020, 124, 1765–1779 CrossRef CAS.
- E. Vervloessem, M. Aghaei, F. Jardali, N. Hafezkhiabani and A. Bogaerts, ACS Sustainable Chem. Eng., 2020, 8, 9711–9720 CrossRef CAS.
- M. Capitelli, G. Colonna, G. D. Ammando, V. Laporta and A. Laricchiuta, Chem. Phys., 2014, 438, 31–36 CrossRef CAS.
- Y. He, Z. Chen, Z. Li, G. Niu and J. Tang, Front. Optoelectron., 2018, 11, 92–96 CrossRef.
- A. W. Wang, B. S. Patil, S. Heijkers and A. Bogaerts, ChemSusChem, 2017, 10, 2145–2157 CrossRef PubMed.
- B. S. Patil, V. Hessel and Q. Wang, Plasma Chem. Plasma Process., 2016, 36, 241–257 CrossRef CAS.
- V. Hessel, A. Anastasopoulou, Q. Wang, G. Kolb and J. Lang, Catal. Today, 2013, 211, 9–28 CrossRef CAS.
- M. J. Pavlovich, D. S. Clark and D. B. Graves, J. Phys. D: Appl. Phys., 2014, 47, 505202 CrossRef.
- Y. Gorbanev, E. Vervloessem, A. Nikiforov and A. Bogaerts, ACS Sustainable Chem. Eng., 2020, 8, 2996–3004 CrossRef CAS.
- S. Van Alphen, V. Vermeiren, T. Butterworth, D. C. M. Van den Bekerom, G. J. Van Rooij and A. Bogarts, J. Phys. Chem. C, 2019, 124, 1765–1779 CrossRef.
- Q. Sun, A. Zhu, X. Yang, J. Niu, Y. Xu and L. Beams, Chem. Commun., 2003, 5, 1418–1419 RSC.
- H. Patel, R. K. Sharma, V. Kyriakou, A. Pandiyan, S. Welzel, M. C. M. Van de Sanden and M. N. Tsampas, ACS Energy Lett., 2019, 4, 2091–2095 CrossRef CAS.
- Y. Kubota, K. Koga, M. Ohno and T. Hara, Plasma Fusion Res. Lett., 2010, 5, 042 CrossRef.
- P. Bruggeman and C. Leys, J. Phys. D: Appl. Phys., 2009, 42, 053001 CrossRef.
- P. J. Bruggeman, M. J. Kushner, B. R. Locke, J. G. E. Gardeniers, W. G. Graham and D. Fernandez Rivas, Plasma Sources Sci. Technol., 2016, 25, 053002 CrossRef.
- Y. Mizukoshi, R. Katagiri, H. Horibe, S. Hatanaka, M. Asano and Y. Nishimura, Chem. Lett., 2015, 44, 495–496 CrossRef CAS.
- F. Girard, M. Peret, N. Dumont, V. Badets, S. Blanc, K. Gazeli, C. Nöel, T. Belmonte, L. Marlin, J.-P. Cambus, G. Simon, N. Sojic, B. Held, S. Arbault and F. Clément, Phys. Chem. Chem. Phys., 2018, 20, 9198–9210 RSC.
- Y. Gorbanev, E. Vervloessem, A. Nikiforov and A. Bogaerts, ACS Sustainable Chem. Eng., 2020, 8, 2996–3004 CrossRef CAS.
- P. Peng, C. Schiappacasse, N. Zhou, M. Addy, Y. Cheng, Y. Zhang, E. Anderson, D. Chen, Y. Wang, Y. Liu, P. Chen and R. Ruan, J. Phys. D: Appl. Phys., 2019, 52, 494001 CrossRef CAS.
- G. Akay, Catalysts, 2020, 10, 1–65 CrossRef.
- S. Zen, T. Abe and Y. Teramoto, Plasma Chem. Plasma Process., 2018, 38, 347–354 CrossRef CAS.
- T. Hachiya and H. Sakakibara, J. Exp. Bot. Adv. Access, 2016, 1–12 Search PubMed.
- H. J. Kronzucker, M. Y. Siddiqi, A. D. M. Glass and G. J. D. Kirk, Plant Physiol., 1999, 119, 1041–1045 CrossRef CAS PubMed.
- S. J. Tabatabaei, L. S. Fatemi and E. Fallahi, J. Plant Nutr., 2006, 29, 1273–1285 CrossRef CAS.
- F. Wiesler, Pfanzenernähr. Bodenk., 1997, 160, 227–238 CrossRef CAS.
- P. J. Bruggeman, G. C. Schatz, R. R. Frontiera, S. Exarhos, M. J. Kushner, S. Linic, H. Andaraarachchi, L. O. Jones, C. M. Mueller, C. C. Rich, C. Xu, Y. Yue and Y. Zhang, J. Appl. Phys., 2021, 129, 200902 CrossRef CAS.
- C. Pattyn, N. Maira, A. Remy, N. C. Roy, S. Iseni and D. Petitjean, Phys. Chem. Chem. Phys., 2020, 22, 24801–24812 RSC.
- P. J. Bruggeman, N. Sadeghi, D. C. Schram and V. Linss, Plasma Sources Sci. Technol., 2014, 23, 023001 CrossRef CAS.
- P. Andre, Y. A. Barinov, G. Faure and S. M. Shkol, J. Phys. D: Appl. Phys., 2011, 44, 375203 CrossRef.
- S. Iseni, R. Michaud, P. Lefaucheux, G. B. Sretenovi, V. Schulz von der Gathen and R. Dussart, Plasma Sources Sci. Technol., 2019, 28, 065003 CrossRef CAS.
- I. Kovàcs, Rotational structure in the spectra of diatomic molecules, 1969 Search PubMed.
- F. Roux and F. Michaud, J. Mol. Spectrosc., 1993, 158, 270–277 CrossRef CAS.
- G. Faure, Plasma Sources Sci. Technol., 2012, 14, 192–200 CrossRef CAS.
- Y. Zhao, R. Shi, X. Bian, C. Zhou, Y. Zhao, S. Zhang, F. Wu, G. I. N. Waterhouse, L. Wu, C. Tung and T. Zhang, Adv. Sci., 2019, 6, 1802109 CrossRef PubMed.
- L. Zhou and C. E. Boyd, Aquaculture, 2016, 450, 187–193 CrossRef CAS.
- C. Richmonds, M. Witzke, B. Bartling, S. W. Lee, J. Wainright, C. Liu and R. M. Sankaran, J. Am. Chem. Soc., 2011, 133, 17582–17585 CrossRef CAS PubMed.
- M. Witzke, P. Rumbach, D. B. Go and R. M. Sankaran, J. Phys. D: Appl. Phys., 2012, 46, 442001 CrossRef.
- P. Rumbach, D. M. Bartels, R. M. Sankaran and D. B. Go, J. Phys. D: Appl. Phys., 2015, 48, 424001 CrossRef.
- R. Akolkar and R. M. Sankaran, J. Vac. Sci. Technol. A, 2013, 31, 050811 CrossRef.
- R. Hawtof, S. Ghosh, E. Guarr, C. Xu, R. M. Sankaran and J. N. Renner, Sci. Adv., 2019, 5, 1–9 Search PubMed.
- S. Collette, T. Dufour and F. Reniers, Plasma Sources Sci. Technol., 2016, 25, 025014 CrossRef.
- F. Fresnet, G. Baravian, L. Magne, S. Pasquiers, C. Postel and V. Puech, Plasma Sources Sci. Technol., 2002, 11, 152–160 CrossRef CAS.
- T. Chen, W. D. Smith and J. Simons, Chem. Phys. Lett., 1974, 26, 296–300 CrossRef CAS.
- Y. V. Kovtun, At. Mol. Phys., 2015, 60, 1110–1118 CAS.
- P. Lukes, E. Dolezalova, I. Sisrova and M. Clupek, Plasma Sources Sci. Technol., 2014, 23, 015019 CrossRef CAS.
- R. Sander, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef CAS.
- T. Haruyama, T. Namise, N. Shimoshimizu, S. Uemura, Y. Takatsuji, M. Hino, R. Yamasaki, T. Kamachi and M. Kohno, Green Chem., 2016, 18, 4536–4541 RSC.
- Y. Tsuchida, N. Murakami, T. Sakakura, Y. Takatsuji and T. Haruyama, ACS Omega, 2021, 6, 29759–29764 CrossRef CAS PubMed.
- T. Sakakura, S. Uemura, M. Hino, S. Kiyomatsu, Y. Takatsuji, R. Yamasaki, M. Miromoto and T. Haruyama, Green Chem., 2018, 20, 627–633 RSC.
- T. Sakakura, N. Murakami, Y. Takatsuji, M. Morimoto and T. Haruyama, ChemPhysChem, 2019, 20, 1467–1474 CAS.
- J. T. Herron and D. S. Green, Plasma Chem. Plasma Process., 2001, 21, 459–481 CrossRef CAS.
- D. L. Baulch, C. J. Cobos, R. A. Cox, P. Frank, G. Hayman, T. Just, J. A. Kerr, T. Murrells, M. J. Pilling, J. Troe, R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data, 1994, 23, 847–1033 CrossRef CAS.
- Y. P. Raizer, Gas Discharge Physics, 1991 Search PubMed.
- X. L. Deng, A. Y. Nikiforov, P. Vanraes and C. Leys, Appl. Phys. Lett., 2013, 113, 023305 Search PubMed.
- A. Bogaerts and G. Centi, Front. Energy Res., 2020, 8, 1–23 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc01013e |
| This journal is © The Royal Society of Chemistry 2022 |