
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From A3/KA2 to AYA/KYA multicomponent coupling reactions with terminal ynamides as alkyne surrogates – a direct, green route to γ-amino-ynamides†
Fabian
Schlimpen
 ,
Tun
Ast
,
Valérie
Bénéteau
,
Patrick
Pale
* and
Stefan
Chassaing
*
,
Tun
Ast
,
Valérie
Bénéteau
,
Patrick
Pale
* and
Stefan
Chassaing
*
Laboratoire de Synthèse, Réactivité Organiques et Catalyse (LASYROC), Institut de Chimie, CNRS-UMR7177, Université de Strasbourg, 4 rue Blaise Pascal, 67070 Strasbourg, France. E-mail: ppale@unistra.fr; chassaing@unistra.fr
First published on 6th August 2022
Abstract
A copper-catalysed three-component coupling reaction between a carbonyl derivative, a terminal ynamide and an amine has been developed for the one-pot green construction of γ-amino-ynamides from simple starting materials and with a broad functional group tolerance.The copper-zeolite-catalysed three-component coupling reaction between an aldehyde or a ketone, a terminal ynamide and an amine is reported. The so-elaborated methods, dubbed AYA (i.e. Aldehyde-Ynamide-Amine) and KYA (i.e. Ketone-Ynamide-Amine), enable the straightforward one-pot synthesis of γ-amino-ynamides from simple starting materials and with a broad functional group tolerance. Moreover, AYA/KYA processes are performed at room temperature under solvent-free conditions or in ethyl acetate as green solvent in the presence of a copper-based easy-to-recover/-remove catalyst, at low loading (3 mol%), making these synthetic methods fully green.
Introduction
Multicomponent reactions (MCRs) are among the most important and powerful synthetic tools for implementing Green Chemistry to organic & pharmaceutical chemistry.1,2 Allowing the convergent one-pot assembly of at least three readily available starting materials, MCRs are indeed in line with most of the standards set by Green Chemistry,3 such as excellent atom and step economy, no (or very limited) waste generation, resources/assets saving and minimal energy consumption. As a consequence, synthetic organic chemists pay an ever-increasing attention to this field, resulting in the continuous discovery of novel MCRs which allow access to an exceptional level of molecular complexity and diversity.Although serendipity played a role in the MCR discovery, the emergence of design strategies allowed to explore the chemical space in a more rational way.4 Among them, the most intuitive one is the so-called Single Reactant Replacement (SRR) strategy,5 that is the replacement of one reactant with a structurally different reactant which features the same essential reactivity mode for the related MCR. As the structure of the surrogate usually differs by the presence of additional/distinct functional group(s), the synthesis of more complex, and occasionally unprecedented, molecular scaffolds could be accomplished. The popular UGI reaction, allowing access to α-acylamino-amides,6 actually is a SRR-guided MCR of the equally popular, 100-year-old PASSERINI reaction,7 with the use of an imine as surrogate of the native carbonyl component.
Here, we describe an SRR-guided extension of the A3/KA2 coupling reactions, pioneered by C.-J. Li and coworkers,8 which combine aldehyde/ketone, terminal alkyne and amine into propargylamine in a one-pot transition metal-catalysed coupling (Scheme 1, top).9,10 Replacing the alkyne component by a terminal ynamide11,12 could lead to a one-pot synthesis of γ-amino-ynamides (Scheme 1, bottom). This approach was driven by the following reasoning: (i) despite its interest,13 accesses to the latter scaffold are so far limited and based on multi-step sequences,14 a simpler way would thus be beneficial and probably open up new perspectives; (ii) since ynamides are more nucleophilic than alkynes (see N parameters),15,16 they should be more reactive in A3/KA2-type reactions; (iii) although green by essence (see above),8 A3/KA2 reactions could be made greener by avoiding solvent and/or by using recyclable catalyst. For the latter aspect, we already developed the 1st heterogenous A3 coupling based on CuI-zeolites as catalysts.17 Other heterogenous versions have since been proposed.9
 | ||
| Scheme 1 From propargylamines synthesis via popular A3/KA2 3-CRs (previous works, top) to γ-amino-ynamides synthesis via unprecedented AYA/KYA 3-CRs (this work, bottom) (EWG = Electron-Withdrawing Group). | ||
Since we have proven the compatibility of ynamides with zeolitic materials,18 we embarked on the development of CuI-catalysed MCRs combining aldehyde or ketone, ynamide and amine (respectively AYA or KYA) and we report here the first one-step method for preparing γ-amino-ynamides, without solvent or in ethyl acetate19 as an environmentally benign solvent, catalysed by copper-doped zeolite CuI-USY,20 as well as a comparison with their parent A3/KA2 reactions. It is worth noticing that this approach is quite challenging due to the known high reactivity of the ynamide moiety.11
Results and discussion
Conditions set up
To assess the feasibility of the AYA/KYA approach towards γ-amino-ynamides, initial investigations were conducted under conditions close to our previously reported A3 and KA2 reaction conditions,17,21 with benzaldehyde 1, N-ethynylsulfonamide 2 and pyrrolidine 3 as model coupling partners and a CuI-zeolite as catalyst (Table 1). Due to its large cage-shaped porous system, the commercial and cheap USY zeolite‡ has often given the best results in catalysing various transformations,20 the easy-to-prepare CuI-doped USY was thus used here as catalyst.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | T (°C) | Time (h) | 4a | 4a′ |
| a Reactions run in a sealed tube with benzaldehyde 1 (1.4 equiv.), ynamide 2 (1.0 equiv. with a 1.3 M concentration), pyrrolidine 3 (1.1 equiv.) and CuI-USY (10 mol%), unless otherwise stated. b Isolated yield (%) after purification by column chromatography. c Reactions performed under neat conditions. d Yield (%) estimated by 1H NMR of the crude using 1,3,5-trimethoxybenzene as internal standard. e Not detected in the crude mixture. | |||||
| 1 | PhMe/MeCN (1:1) |
80 | 3 | 35b | 59b |
| 2 | Nonec | 90 | 1 | < 5 | 43b |
| 3 | Nonec | 40 | 2 | 83d | —e |
| 4 | 2-MeTHF | rt then 40 | 2 then 1 | 61d | —e |
| 5 | AcOEt | 30 | 0.5 | 82d | —e |
Mixing these three reagents in toluene/acetonitrile (1:1) and heating the resulting mixture to 80 °C for three hours led to the formation of the expected γ-amino-ynamide 4a in a promising isolated yield (entry 1), together with sulfonamide 4a′ as the major product (respectively 35% and 59% yield).§ Performing the reaction under neat conditions at 90 °C also led to complete consumption of 2 but with the major formation of 4a′, although in moderate yield after column chromatography, and with only trace amounts of 4a (entry 2). Lowering the reaction temperature proved beneficial. Indeed, the coupling efficiency and selectivity could be dramatically improved at 40 °C under neat conditions (entry 3). For comparison, and as solubility and stirring problems could occur depending on the components, reaction in green solvents was also evaluated. Stirring for two hours at room temperature in green 2-methylTHF only led to slow but clean reaction, but after one hour at 40 °C, an encouraging increase in yield and selectivity was achieved (entry 4 vs. 1). Interestingly, ethyl acetate as green solvent allowed at 30 °C to produce the expected product 4a in yield similar to those achieved under neat conditions (entry 5 vs. 3). It is worth noticing that in this solvent, the reaction proved to be much faster (30 min vs. 2 h).
These results highlight the key influence of the reaction temperature on the coupling selectivity and suggest that the reaction could be efficiently performed with or without solvent.
Following these preliminary results, the solvent-free AYA reaction between 1, 2 and 3 was used as a bench reaction to perform further optimisation studies via the so-called Design of Experiments (DoE) method.22 We chose to perform a simple experimental design to identify the most crucial variables affecting the yield of γ-amino-ynamides. Six variables, namely temperature, catalyst loading, aldehyde equivalents, stirring rate, stirring bar type and reaction time, were studied in a 26-3 fractional factorial design (FFD) giving a total number of eight experiments, which were run in parallel (see ESI for details†). The results of this short DoE study revealed that temperature, followed by reaction time, are the two critical variables providing γ-amino-ynamide 4a in high yield, while catalyst loading (in a range of 3 to 10 mol%) or the type of stirring bar are not pertinent. Although statistically not significant in this short study, the two last variables, that are the number of aldehyde equivalents and the stirring rate, positively correlated with the yield of 4a. In light of these DoE results, we turned to the use of equimolar amounts of each coupling partner and set reaction temperature to 30 °C, catalyst loading to the lowest evaluated (i.e. 3 mol%) and stirring rate to the highest value investigated (i.e. 600 rpm).
Because A3/KA2 reactions can be promoted with various metal cations,9 we next evaluated the catalytic potential of USY zeolites doped with CuII, ZnII and MnII (Table 2). Under the conditions set above in ethyl acetate, CuI-USY clearly appeared as the best catalytic system, with high conversion and 4a yield after 3 hours (entry 1). Its oxidized CuII-form led to a slow and messy reaction, which resulted in dramatic drop in yield despite nearly full conversion after 19 hours (entry 2). Similar poor yields were also obtained with MnII-USY and ZnII-USY (entries 3 and 4 vs. 2) but in sharp contrast to CuII-USY, the lower yields are not due here to selectivity issues but mainly to significantly lower conversions compared to CuI-USY (entries 3 and 4 vs. 1). With CuCl alone as CuI source, the expected product 4a was again formed in poor yield despite full conversion (entry 5 vs. 1 and 2). The same issue was obtained with CuBr and CuI, two commercial copper sources commonly used in A3/KA2 coupling reactions (entries 6, 7). Further control experiments confirmed that the coupling reaction was not promoted neither in the presence of the native H-USY (entry 8) nor without catalyst (entry 9). Overall, these data revealed the key, dual role played by the CuI ions and the USY-zeolite for efficiently catalysing this coupling reaction. Accordingly, CuI-USY, at low Cu loading of only 3 mol%, was further employed to investigate the scope and limitations of this three-component reaction.
a
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Yieldb (%) | Conversionb (%) |
| a Reactions run in a sealed tube with benzaldehyde 1 (1.0 equiv. with a 0.85 M concentration), ynamide 2 (1.0 equiv.) and pyrrolidine 3 (1.0 equiv.). b Yield and conversion estimated by 1H NMR of the crude using 1,3,5-trimethoxybenzene as internal standard. c Formation of several unidentified byproducts. d Selective but slow conversion. e No traces of 4a detected and no conversion observed after 19 h. | ||||
| 1 | CuI-USY | 3 | 84 | 88 |
| 2 | CuII-USY | 19 | 21c | 95 |
| 3 | ZnII-USY | 3 | 31d | 46 |
| 4 | MnII-USY | 3 | 16d | 23 |
| 5 | CuCl | 3 | 32c | 100 |
| 6 | CuBr | 3 | 13c | 100 |
| 7 | CuI | 3 | 32c | 100 |
| 8 | H-USY | 19 | —e | —e |
| 9 | None | 19 | —e | —e |

Reusability and catalyst stability
Stability and reusability are key features for heterogeneous catalyst. We have thus evaluated the reusability of the CuI-USY catalyst using the same model reaction involving partners 1, 2 and 3 in ethyl acetate under the conditions described in Table 1, entry 5. After each reaction run, the reaction mixture was centrifuged and the supernatant was decanted; the recovered zeolite was washed with the same solvent and centrifuged twice. The recovered catalyst was directly engaged in the next run, without cleaning it by heating as usual. The catalyst could be used four times with an excellent selectivity towards 4a and without much difference in efficiency (68% in average). However, a substantial decrease in product yield was observed on the fifth run (41%) (see ESI for details†). Despite this decrease, it is worth noting that even after the fifth run, the recovered zeolitic materials remain more efficient and selective than typical CuX salts (Table 2, entries 5–7). Such decrease could be due to the slight loss of solids during the recycling/recovery process. Nevertheless, a control experiment (Sheldon test) showed that the reaction could keep running after removal of the solid catalyst by filtration, although at a lower rate. Copper ions could have been extracted by the amine, as one would expect, or tiny amount of copper zeolite could go through and keep being active as catalyst. Such behaviour is common with coordinating substrates and was already observed in the CuI-USY catalysed synthesis of ynamides.18As in other CuI-USY catalysed reactions, XPS analysis revealed that copper ions within the zeolite do not change their oxidation state upon reaction and remain at +I state.
Scope and limitations
With optimised conditions in hand, we investigated the possibility offered by the AYA/KYA reactions. We first examined the AYA potential with ynesulfonamide 2, pyrrolidine 3 and various aldehydes as coupling partners (Scheme 2).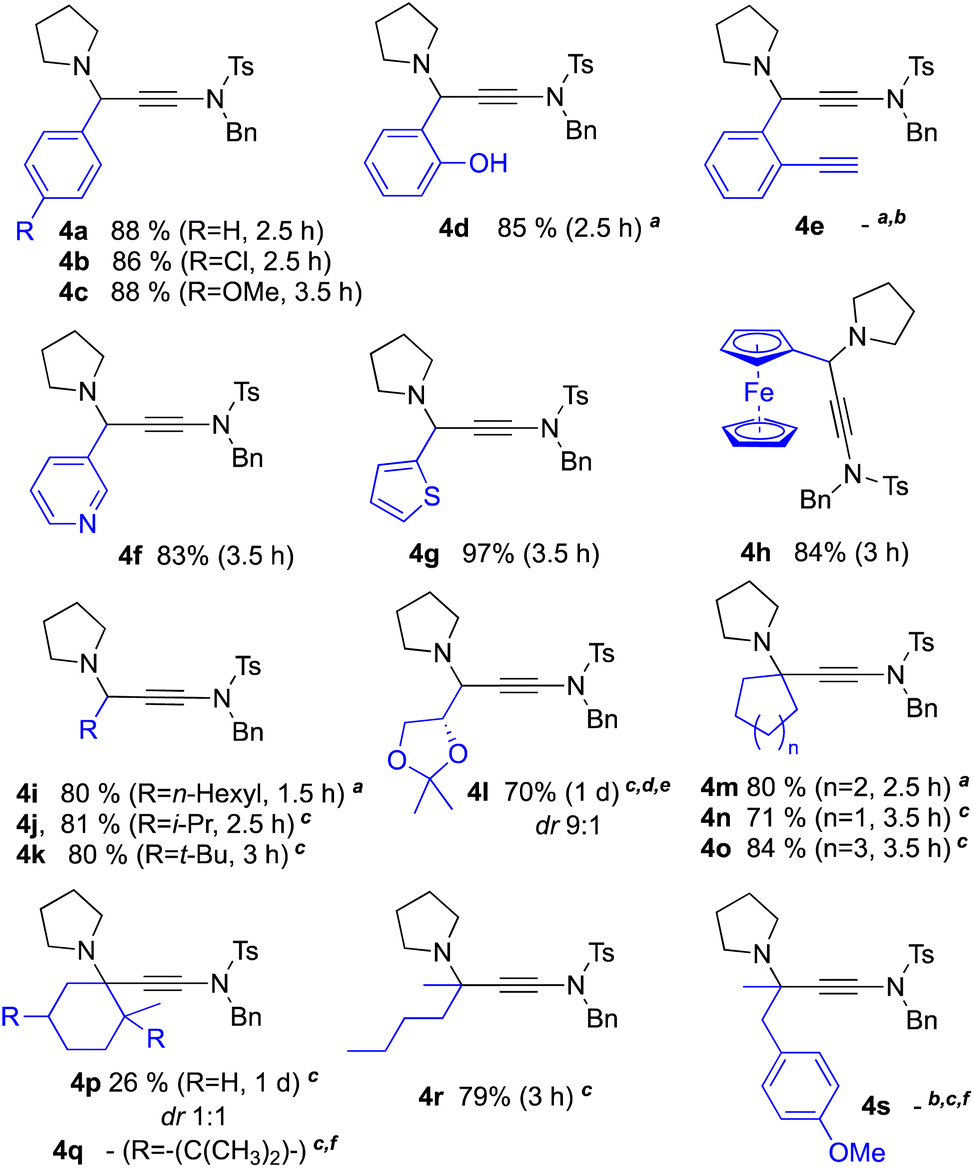 | ||
| Scheme 2 Carbonyl scope of the CuI-USY-catalysed AYA/KYA coupling reaction. General conditions: reactions run neat at 30 °C in a sealed tube with a carbonyl compound (1.0 equiv.), ynamide 2 (1.0 equiv.), pyrrolidine 3 (1.0 equiv.) and CuI-USY (3 mol%), unless otherwise stated. aReaction run in EtOAc (0.85 M). bComplex mixture of unidentified difficult-to-purify products. cReaction run in EtOAc (0.85 M) in the presence of molecular sieves (4 Å, 300 mg mmol−1). dReaction set up in EtOAc (0.85 M) at 0 °C and then warmed to room temperature overnight. eLower yield in 4l obtained at room temperature. fNo conversion observed for the corresponding ketone. | ||
4-Chloro- and 4-methoxybenzaldehydes reacted similarly to benzaldehyde, thus furnishing the expected γ-amino-ynamides 4b,c without much electronic effects. Salicylaldehyde reacted as well and selectively yielded the AYA product 4d in high yield. However, 2-ethynylbenzaldehyde led to a complex reaction mixture without traces of the desired product 4e. Heteroaryl aldehydes, such as 3-pyridinecarboxaldehyde and 2-thiophenecarboxaldehyde, as well as organometallic aldehydes such as ferrocenecarboxaldehyde, also proved to be suitable coupling partners, providing 4f–h in high to excellent yields within similar reaction times.
Shifting to aliphatic aldehydes had no impact on coupling efficiency. γ-Amino-ynamides 4i–k could indeed be prepared in similar high yields from either linear n-heptanal (cf.4i) or branched 2-methylpropanal and 2,2-dimethylpropanal (cf.4j,k). Interestingly enough, the use of chiral 2,3-O-isopropylideneglyceraldehyde led to the substrate-controlled asymmetric synthesis of γ-amino-ynamide 4l in good yield and diastereoselectivity.
The behaviour of ketones was then explored. Cyclohexanone was selected as model partner due to the well-established, beneficial strain energy release during nucleophilic additions involving this cyclic ketone and its iminium form.23 Satisfyingly, cyclohexanone reacted in the same way as aromatic and aliphatic aldehydes without the need to adjust the optimised reaction conditions. The expected product 4m was indeed obtained with comparable yield and reaction time. The more challenging cyclopentanone- and cycloheptanone-derived ynamides 4n and 4o were obtained with similar efficiency in the presence of molecular sieves (4 Å). Nevertheless, increasing the steric hindrance of the ketones had a deleterious effect on the efficiency of the KYA coupling reaction. Shifting from cyclohexanone to 2-methylcyclohexanone led to a significant drop in yield (cf. 80% for 4mvs. 26% for 4p), whereas the more hindered (+)-camphor did not react. Noteworthy is that 4p was obtained as a 1:1 diastereoisomer mixture, while a diastereoselectivity was observed in related KA2 reactions.9d Acyclic aliphatic ketones also reacted similarly, as shown by the coupling of hexan-2-one. The resulting γ-amino-ynamide 4r was indeed obtained in mostly the same yield as its cyclic counterpart (79% for 4rvs. 80% for 4m). In sharp contrast, 4-methoxyphenylacetone as model α-arylated ketone did not provide the desired KYA product 4s even at 55 °C, probably due to the high stability of the resulting conjugated enamine which prevents the coupling from taking place.
In a second series of experiments, we varied the amine component, while keeping benzaldehyde 1 and ynesulfonamide 2 (Scheme 3). In ethyl acetate as solvent using additional molecular sieves, cyclic secondary amines, such as pyrrolidine, piperidine, morpholine and tetrahydroisoquinoline, afforded the corresponding AYA products 4a and 5a–c in high to excellent yields. Chiral amines were then scrutinised to evaluate the potential of amine-controlled stereoselective AYA reactions. Under standard conditions and without further optimisation, (R)-prolinol led to the efficient formation of the expected γ-amino-ynamide 5d with a high level of diastereoselectivity. Crystallization from a CH2Cl2/Et2O mixture by slow evaporation furnished 5d as fine pale-yellow needles, whose X-ray analysis24,25 unambiguously confirmed the structure and absolute configuration of this chiral γ-amino-ynamide. This diastereoselective induction is similar to the one reported in related A3 reactions using (R)-prolinol.26 In sharp contrast, the AYA coupling proved ineffective with α-methyl-L-proline as chiral amine, probably for solubility and/or reactivity issues. Shifting from cyclic to acyclic secondary amines led to more or less efficient coupling reactions. For example, N-allyl-4-methoxybenzylamine allowed the formation of the AYA product 5f together with an unidentified by-product in a yield slightly lower than those achieved so far. However, N,N-diethylamine reacted cleanly but much more slowly than its cyclic pyrrolidine surrogate (cf. 53% for 5g after 69 hours vs. 88% for 4a after 2.5 hours), and the less nucleophilic N,N-diphenylamine did not even react.
 | ||
| Scheme 3 Amine scope of the CuI-USY-catalysed AYA coupling reaction. General conditions: reactions run neat at 30 °C in a sealed tube with benzaldehyde 1 (1.0 equiv.), ynamide 2 (1.0 equiv.), an amine (1.0 equiv.) and CuI-USY (3 mol%), unless otherwise stated. aReaction run in EtOAc (0.85 M) in the presence of molecular sieves (4 Å, 300 mg mmol−1). bSee ref. 24 for CCDC† deposition number. cNo conversion of the amine observed. dIncomplete conversion. eReaction run in EtOAc (0.85 M). fImine detected. gTraces of the expected product detected. hComplex mixture of unidentified difficult-to-purify products. | ||
Primary amines were found even trickier to work with. Aniline, as well as 4-fluoroaniline¶, proved to be reactive but both reactions stalled at the imine intermediates. Aliphatic primary amines, including benzylamine and n-propylamine, also proved to react but unfortunately led to complex mixtures in which trace amounts of the expected products 5k,l could be detected together with ynamide-derived oligomers probably resulting from the initial formal hydroamination of the amine onto ynamide 2 (Scheme 4). Preforming the imines and trapping them by the ynamide partner was envisaged as a two-step approach to circumvent the suggested hydroamination pathway. The feasibility of this alternative approach was demonstrated with the formation of the γ-amino-ynamide 5k from preformed N-benzylaldimine and ynamide 2 in the presence of titanium tetraethoxide as additional Lewis acid (Scheme 4). Applying similar conditions to N-phenylbenzaldimine unfortunately failed to form the γ-amino-ynamide 5i.
 | ||
| Scheme 4 Attempts towards γ-amino-ynamides derived from primary amines – the one-pot AYA approach vs. a two-step alternative with imine preformation as initial step. aImine preformed under conventional conditions. | ||
In a last series of experiments, the scope of the three-component AYA/KYA coupling reactions was investigated by varying the ynamide partner (Scheme 5). The influence of the electron-withdrawing group substituting the nitrogen atom was first explored. Regarding sulfonyl groups, the tosyl, nosyl and mesyl groups were tested and found amenable to AYA/KYA reactions to access the corresponding γ-amino-ynamides 4a,m and 6a–d in moderate to high yields. Nevertheless, the sulfonyl group nature proved to exhibit a significant impact on the reaction efficiencies and kinetics. While tosylated products 4a and 4m were obtained in only 2.5 hours, their nosyl derivatives 6a and 6c necessitated the addition of molecular sieves to be formed with similar rates and yields.|| For mesyl derivatives 6b and 6d, several days were required to reach full conversions. γ-Amino-ynamides bearing a lactam (cf.6e) or an oxazolidinone (cf.6f) moiety were also obtained, in low and high yields respectively. Unfortunately, the vinylogue indole-based product 6g could only be detected as trace amounts in a complex mixture of unidentified products. Diverse substitutions on the amino group of N-tosyl-ynamides were also scrutinised. In addition to the benzyl group (cf.4a and 4m), phenyl and allyl groups were perfectly tolerated (cf.6h–k). However, and not so surprisingly, the propargyl group was found to be incompatible with our catalytic conditions with no traces of products 6l,m detected. Other functional groups, such as ester and indole, were also compatible in the AYA reaction; the corresponding γ-amino-ynamides 6n and 6o were indeed obtained in good to excellent yields.
 | ||
| Scheme 5 Ynamide scope of the CuI-USY-catalysed AYA/KYA coupling reactions. General conditions: reactions run neat at 30 °C in a sealed tube with benzaldehyde or cyclohexanone (1.0 equiv.), an ynamide (1.0 equiv.), pyrrolidine 3 (1.0 equiv.) and CuI-USY (3 mol%), unless otherwise stated. aReaction run in EtOAc (0.85 M) in the presence of molecular sieves (4 Å, 300 mg mmol−1). bReaction run in EtOAc (0.85 M). cTraces of the expected AYA product. dComplex mixture of unidentified difficult-to-purify products. | ||
Comparison and mechanistic aspects
These results allow to start comparing the present reactions to their parent A3/KA2 versions. From a practical point of view, the former can only be performed using CuI-USY as catalyst (see Table 2), while the more classical A3/KA2 reactions can be performed under homogeneous as well as heterogeneous conditions. When strictly comparing the present AYA/KYA reactions to their parent A3/KA2 versions,17,21 both catalysed by CuI-USY (Table 3), some discrepancies can be highlighted, although both are closely related in term of efficiency and scope. The major one has to do with primary amines and anilines. Although effective in KA2 reaction, primary amines failed as coupling partners for the AYA/KYA processes. In a similar way, anilines were found ineffective in the AYA/KYA coupling, while enabling the A3/KA2 coupling depending on their substitution pattern. On the contrary, hexan-2-one as model aliphatic acyclic ketone proved as effective as its cyclic surrogates under KYA conditions (cf.4rvs.4m in Scheme 2), while appearing less effective and thus more challenging under KA2 conditions.9d,21**
| AYA/KYAa | A3/KA2b | |
|---|---|---|
| a This work. b Previous works. For details, see ref. 17 and 21. c Observed in most cases. d In the case of KA2 reactions. | ||
| Challenging substrates | Primary amines | Acyclic ketones |
| Anilines | ||
| Reaction time | <5 hc | >15 h |
| Optimal temperature | 30 °C | 80 °C |
| Competing pathway | Hydroamination with primary amines | Hydroamination with secondary aminesd |
The comparison of reaction times and optimal temperatures clearly shows that shifting from a terminal alkyne to a terminal ynamide induces a huge rate acceleration of such MCRs. Obviously, both CuI-USY-catalysed A3/KA2 and AYA/KYA reaction mechanisms are similar, with the in situ formation of copper acetylide which adds to in situ formed imine or iminium as the key steps (Scheme 6). Due to the polarisation of their triple bond, ynamides are nevertheless more nucleophilic than their alkyne derivatives (see N parameters),15,16 and so may be the corresponding copper acetylides. However, the formation of the latter requires a C–H activation step – most probably a deprotonation – which should be disfavoured due to increasing electron density throughout this bond.
 | ||
| Scheme 6 Mechanistic rationale for the AYA/KYA reactions. | ||
Because much faster reactions were achieved with ynamides than with alkynes (2–5 vs. 15–18 h), as well as milder conditions (30 vs. 80 °C), it seems that the zeolitic CuI-USY catalyst overcomes these opposite trends, suggesting confinement effect. It is worth noticing that related enhanced reactivity has been assigned to differences in the respective rates of acetylide formation in CuAAC reactions.27 It is thus probable here that the enhanced π-nucleophilicity of the ynamide favours the formation of the π-ynamide complex I1 and thus accelerates the formation of the copper acetylide intermediate I2, possibly in a rate-determining step. In this scenario, the observed reaction kinetics should be in line with the donation ability of the N lone pair to the π system of the alkyne moiety: the most donating the N lone pair, the faster the formation of the π-ynamide complex I1. This trend is well confirmed by the observed differences in rate when comparing reactions from ynesulfonamides and ynecarbamates with those from ynelactams or yneindoles (respectively 2–4 h and more than 2 d; see Scheme 5).
Hydroamination of the π-complex I1 proved to take place in both cases as a competing pathway, with contrasting results depending on the class of the amine partner (Scheme 6). While π-alkynyl complexes require secondary amines to undergo the hydroamination event (as previously discussed in ref. 21), π-ynamide complexes proved here enough reactive to undergo this unwished event with less nucleophilic primary amines.
When compared to homogeneous conditions, the present zeolite-based conditions clearly offer an efficient and fruitful alternative. Indeed, decomposition-polymerisation occurred with simple copper(I) halides as catalysts (see Table 2), while cleaner and efficient reactions were achieved with CuI-USY as catalyst. The latter seems to act as nanoreactor, confining the various partners within zeolite pores and avoiding/limiting such degradation.
Conclusions
In summary, we presented herein the first one-pot synthesis of γ-amino-ynamides. The developed approach relies on the copper-zeolite-catalysed reaction between a carbonyl compound, a terminal ynamide and an amine as readily available coupling partners. These AYA/KYA three-component coupling reactions were shown to proceed under simple, mild conditions (i.e. room temperature or 30 °C, neat or ethyl acetate as benign solvent, easy-to-remove/-recover catalyst), with low copper loading (3 mol%) and generally short reaction times. It is worth highlighting here that the present CuI-USY-catalysed reactions are the only one able to readily produce γ-amino-ynamides, since homogeneous conditions with classical copper halides as catalysts induce decomposition.Moreover, the present conditions offer a wide scope and tolerate a variety of functional groups (i.e. halide, alkene, hydroxy, ether, acetal, ester, sulfonyl, lactam, oxazolidinone, etc.). Water being moreover the sole by-product formed, AYA/KYA processes emerge as powerful atom-, energy- and step-economical methods for preparing γ-amino-ynamides, in full agreement with Green Chemistry standards.
Further works are under way in our group to explore the synthetic potential of γ-amino-ynamides.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Centre National de la Recherche Scientifique (CNRS), University of Strasbourg, and the French Ministry of Research for financial support. F. S. thanks the French Ministry of Research for a Ph.D. fellowship. The authors also thank Dr Benoit Louis (ICPEES, Strasbourg, France) for X-ray photoelectron spectroscopy (XPS) analyses.Notes and references
- For selected books, see: (a) J. Zhu and H. Bienaymé, Multicomponent Reactions, Wiley-VCH, Weinheim, 2005 CrossRef; (b) J. Zhu, Q. Wang and M.-X. Wang, Multicomponent Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2015 Search PubMed; (c) R. P. Herrera and E. Marqués-Lopez, Multicomponent Reactions: Concepts and Applications for Design and Synthesis, John Wiley & Sons, Hoboken, 2015 Search PubMed; (d) M. M. Heravi and V. Zadsirjan, Recent Advances in Applications of Name Reactions in Multicomponent Reactions, Elsevier, Amsterdam, 2020 Search PubMed.
- For selected reviews, see: (a) A. Domling, W. Wang and K. Wang, Chemistry and biology of multicomponent reactions, Chem. Rev., 2012, 112, 3083–3135 CrossRef CAS PubMed; (b) P. Slobbe, E. Ruijter and R. V. A. Orru, Recent applications of multicomponent reactions in medicinal chemistry, MedChemComm, 2012, 3, 1189–1218 RSC; (c) T. Zarganes-Tzitzikas and A. Doemling, Modern multicomponent reactions for better drug syntheses, Org. Chem. Front., 2014, 1, 834–837 RSC; (d) L. Reguera, Y. Mendez, A. R. Humpierre, O. Valdes and D. G. Rivera, Multicomponent reactions in ligation and bioconjugation chemistry, Acc. Chem. Res., 2018, 51, 1475–1486 CrossRef CAS; (e) O. Ghashghaei, F. Seghetti and R. Lavilla, Selectivity in multicomponent reactions: types and synthetic applications, Beilstein J. Org. Chem., 2019, 15, 521–534 CrossRef CAS PubMed; (f) S. E. John, S. Gulati and N. Shankaraiah, Recent advances in multicomponent reactions and their mechanistic insights: a triennium review, Org. Chem. Front., 2021, 8, 4237–4287 RSC.
- (a) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed; (b) R. C. Cioc, E. Ruijter and R. V. A. Orru, Multicomponent reactions: advanced tools for sustainable organic synthesis, Green Chem., 2014, 16, 2958–2975 RSC.
- (a) R. Scheffelaar, E. Ruijter and R. V. A. Orru, Multicomponent Reaction design strategies: towards scaffold and stereochemical diversity, in Synthesis of heterocycles via multicomponent reactions II. Topics in heterocyclic chemistry, ed. R. Orru and E. Ruijter, Springer, Berlin, 2010, vol. 25, pp. 95–126 Search PubMed; (b) E. Ruijter, R. Scheffelaar and R. V. A. Orru, Multicomponent reaction design in the quest for molecular complexity and diversity, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS.
- B. Ganem, Strategies for innovation in multicomponent reaction design, Acc. Chem. Res., 2009, 42, 463–472 CrossRef CAS.
- (a) I. Ugi, R. Meyr, U. Fetzer and C. Steinbrückner, Versuche mit Isonitrilen, Angew. Chem., 1959, 71, 386–388 Search PubMed; (b) P. Lemmen, E. Fontain, J. Bauer and K. Ivar, Ugi (1930–2005): Multi-component reactions, computer and phosphorus chemistry, Angew. Chem., Int. Ed., 2006, 45, 193 CrossRef CAS.
- (a) M. Passerini, Gazz. Chim. Ital., 1921, 51, 126–129 CAS; (b) L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, The 100 facets of the Passerini reaction, Chem. Sci., 2021, 12, 15445–15472 RSC.
- (a) C.-J. Li and C. Wei, Highly efficient Grignard-type imine additions via C-H activation in water and under solvent-free conditions, Chem. Commun., 2002, 268–269 RSC; (b) C. Wei and C.-J. Li, A highly efficient three-component coupling of aldehyde, alkyne, and amines via C-H activation catalyzed by gold in water, J. Am. Chem. Soc., 2003, 125, 9584–9585 CrossRef CAS.
- For selected reviews on A3/KA2 coupling reactions, see: (a) W.-J. Yoo, L. Zhao and C.-J. Li, The A3-coupling (Aldehyde-Alkyne-Amine) reaction: a versatile method for the preparation of propargylamines, Aldrichimica Acta, 2011, 44, 43–51 CAS; (b) V. A. Peshov, O. P. Pereshivko and E. V. van der Eycken, A walk around the A3-coupling, Chem. Soc. Rev., 2012, 41, 3792–3807 Search PubMed; (c) I. Jesin and G. C. Nandi, Recent advances in the A3 coupling reactions and their applications, Eur. J. Org. Chem., 2019, 2704–2720 CrossRef CAS; (d) L. P. Zorba and G. C. Vougioukalakis, The Ketone-Amine-Alkyne (KA2) coupling reaction: transition metal-catalyzed synthesis of quaternary propargylamines, Coord. Chem. Rev., 2021, 429, 213603 CrossRef CAS.
- For selected reviews on propargylamine chemistry, see: (a) K. Lauder, A. Toscani, N. Scalacci and D. Castagnolo, Synthesis and reactivity of propargylamines in organic chemistry, Chem. Rev., 2017, 117, 14091–14200 CrossRef CAS; (b) X. Sheng, K. Chen, C. Shi and D. Huang, Recent advances in reactions of propargylamines, Synthesis, 2020, 1–20 Search PubMed.
- For selected reviews on ynamide chemistry, see: (a) C. A. Zificsak, J. A. Mulder, R. P. Hsung, C. Rameshkumar and L.-L. Wei, Recent advances in the chemistry of ynamines and ynamides, Tetrahedron, 2001, 57, 7575–7606 CrossRef CAS; (b) G. Evano, A. Coste and K. Jouvin, Ynamides: versatile tools in organic synthesis, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed; (c) K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Ynamides: a modern functional group for the millenium, Chem. Rev., 2010, 110, 5064–5106 CrossRef CAS PubMed; (d) A. M. Cook and C. Wolf, Terminal ynamides: synthesis, coupling reactions, additions to common electrophiles, Tetrahedron Lett., 2015, 56, 2377–2392 CrossRef CAS PubMed.
- For selected examples of MCRs involving terminal ynamides, see: (a) Y. Shen, B. Huang, L. Zeng and S. Cui, Single reactant approach of Passerini reaction: one-pot synthesis of β-acyloxyamides and phthalides, Org. Lett., 2017, 19, 4616–4619 CrossRef CAS PubMed; (b) B. Huang and S. Cui, Homologation of Ugi and Passerini reactions using ynamides, Drug Discovery Today, 2018, 29, 43–49 CrossRef; (c) R. Chen, Y. Liu and S. Cui, 1,4-Conjugate addition/esterification of ortho-quinone methides in a multicomponent reaction, Chem. Commun., 2018, 54, 11753–11756 RSC; (d) X. Li, H. Zeng, L. Lin and X. Feng, Catalytic asymmetric hydroacyloxylation/ring-opening reaction of ynamides, acids and aziridines, Org. Lett., 2021, 23, 2954–2958 CrossRef CAS PubMed.
- For selected synthetic applications of γ-amino-ynamides, see: (a) K. C. M. Kurtz, R. P. Hsung and Y. Zhang, A ring-closing yne-carbonyl metathesis of ynamides, Org. Lett., 2006, 8, 231–234 CrossRef CAS; (b) K. A. DeKorver, R. P. Hsung, W.-Z. Song, X.-N. Wang and M. C. Walton, An intramolecular [2 + 2] cycloaddition of ketenimines via palladium-catalyzed rearrangements of N-allyl-ynamides, Org. Lett., 2012, 14, 3214–3217 CrossRef CAS PubMed; (c) T. Nishimura, Y. Takiguchi, Y. Maeda and T. Hayashi, Rhodium-catalyzed asymmetric cycloisomerization of 1,6-ene-ynamides, Adv. Synth. Catal., 2013, 355, 1374–1382 CrossRef CAS; (d) X. Zhou, I. Zafar and G. Dong, Catalytic intramolecular decarbonylative coupling of 3-aminocyclobutenones and alkenes: a unique approach to [3.1.0] bicycles, Tetrahedron, 2015, 71, 4478–4483 CrossRef CAS PubMed.
- For multi-step methods allowing the preparation of γ-amino-ynamides, see: (a) X. Zhang, Y. Zhang, J. Huang, R. P. Hsung, K. C. M. Kurtz, J. Oppenheimer, M. E. Petersen, I. K. Sagamanova, L. Shen and M. R. Tracey, Copper(II)-catalyzed amidations of alkynyl bromides as a general synthesis of ynamides and Z-enamides. An intramolecular amidation for the synthesis of macrocyclic ynamides, J. Org. Chem., 2006, 71, 4170–4177 CrossRef CAS PubMed; (b) R. Qi, X.-N. Wang, K. A. DeKorver, Y. Tang, C.-C. Wang, Q. Li, H. Li, M.-C. Lv, Q. Yu and R. P. Hsung, A convenient synthesis of γ-amino-ynamides via additions of lithiated ynamides to aryl imines; observation of an Aza-Meyer-Schuster rearrangement, Synthesis, 2013, 1749–1758 CAS; (c) S. J. Mansfield, C. D. Campbell, M. W. Jones and E. A. Anderson, A robust and modular synthesis of ynamides, Chem. Commun., 2015, 51, 3316–3319 RSC.
- H. A. Laub, G. Evano and H. Mayr, Hydrocarbation of C≡C bonds: quantification of the nucleophilic reactivity of ynamides, Angew. Chem., Int. Ed., 2014, 53, 4968–4971 CrossRef CAS PubMed.
- For a data compilation, see the Mayr's database of reactivity parameters: https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank/.
- For our CuI-USY-catalyzed version of A3 coupling reactions, see: M. K. Patil, M. Keller, B. M. Reddy, P. Pale and J. Sommer, Copper zeolites as green catalysts for multicomponent reactions of aldehydes, terminal alkynes and amines: an efficient and green synthesis of propargylamines, Eur. J. Org. Chem., 2008, 4440–4445 CrossRef CAS.
- H. Harkat, S. Borghèse, M. de Nigris, S. Kiselev, V. Bénéteau and P. Pale, Zeo-Click synthesis: copper-zeolite-catalyzed synthesis of ynamides, Adv. Synth. Catal., 2014, 356, 3842–3848 CrossRef CAS.
- For selected reviews, see: (a) R. A. Sheldon, Green solvents for sustainable organic synthesis: state of the art, Green Chem., 2005, 7, 267–278 RSC; (b) F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. J. Sherwood, Tools and techniques for solvent selection: green solvent selection guides, Sustainable Chem. Processes, 2016, 4, 7 CrossRef; (c) C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Green and sustainable solvents in chemical processes, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- For reviews dealing with CuI-USY as a catalytic system for organic synthesis, see: (a) S. Chassaing, A. Alix, T. Boringari, A. Sani Souna Sido, M. Keller, P. Kuhn, B. Louis, P. Pale and J. Sommer, Copper(I)-zeolites as new heterogeneous and green catalysts for organic synthesis, Synthesis, 2010, 1557–1567 CAS; (b) S. Chassaing, V. Bénéteau, B. Louis and P. Pale, Zeolites as green catalysts for organic synthesis: the cases of H-, Cu- & Sc-zeolites, Curr. Org. Chem., 2017, 21, 779–793 CrossRef CAS; (c) S. Chassaing, V. Bénéteau and P. Pale, Green catalysts based on zeolites for heterocycle synthesis, Curr. Opin. Green Sustainable Chem., 2018, 10, 35–39 CrossRef.
- For our CuI-USY-catalyzed version of KA2 coupling reactions, see: F. Schlimpen, C. Plaçais, E. Starck, V. Bénéteau, P. Pale and S. Chassaing, α-Tertiary propargylamines synthesis via KA2-type coupling reactions under solvent-free CuI–zeolite catalysis, J. Org. Chem., 2021, 86, 16593–16613 CrossRef CAS PubMed.
- For a review dealing with Design of Experiments (DoE), see: S. A. Weissman and N. G. Anderson, Design of Experiments (DoE) and process optimization. A review of recent publications, Org. Process Res. Dev., 2015, 19, 1605–1633 CrossRef CAS.
- For relevant works dealing with such torsional strain aspects, see: (a) H. C. Brown and K. Ichikawa, Chemical Effects of Steric Strains - XIV: The effect of ring size on the rate of reaction of the cyclanones with sodium borohydride, Tetrahedron, 1957, 1, 221–230 CrossRef CAS; (b) H. C. Brown and J. Muzzio, Rates of reaction of sodium borohydride with bicyclic ketones. Steric approach control and steric departure control in the reactions of rigid bicyclic systems, J. Am. Chem. Soc., 1966, 88, 2811–2822 CrossRef CAS; (c) P. Müller, P. AA and J.-C. Perlberger, Steric effects in the reduction of ketones with sodium borohydride, Helv. Chim. Acta, 1976, 59, 1880–1885 CrossRef.
- CCDC2155844† for compound 5a contains the supplementary data for this paper.
- Selected data for compound 5a: C28H30N2O3S, M = 474.60, orthorhombic, space group P212121, a = 10.6469(3) Å, b = 15.8266(4) Å, c = 28.9384(7) Å, β = 90°, V = 4876.2(2) Å3, Z = 8, crystal size 0.10 × 0.20 × 0.20 mm3, 52110 reflections collected (8605 independent, Rint = 0.0496), 617 parameters, R1[I > 2σ(I)] = 0.0327, wR2 [all data] = 0.0907, largest diff. peak and hole: 0.298 and −0.306 eÅ−3.
- O. Cortezano-Arellano, M. A. Hernandez-Gasca, D. Angeles-Beltran, G. E. Negron-Silva and R. Santilan, Diastereoselective synthesis of propargylamines catalyzed by Cu-MCM-41, Tetrahedron Lett., 2018, 59, 2403–2406 CrossRef CAS.
- (a) V. O. Rodionov, V. V. Fokin and M. G. Finn, Mechanism of the ligand-free CuI-catalyzed azide-alkyne cycloaddition reaction, Angew. Chem., Int. Ed., 2005, 44, 2210–2215 CrossRef CAS; (b) G.-C. Kuang, P. M. Guha, W. S. Brotherton, J. T. Simmons, L. A. Stankee, B. T. Nguyen, R. J. Clark and L. Zhu, Experimental investigation on the mechanism of chelation-assisted, copper(II) acetate-accelerated azide-alkyne cycloaddition, J. Am. Chem. Soc., 2011, 133, 13984–14001 CrossRef CAS; (c) X. Zhang, P. Liu and L. Zhu, Structural determinants of alkyne reactivity in copper-catalyzed azide-alkyne cycloadditions, Molecules, 2016, 21, 1697 CrossRef PubMed; (d) M. Z. C. Hatit, J. C. Sadler, L. A. McLean, B. C. Whitehurst, C. P. Seath, L. D. Humphreys, R. J. Young, A. J. B. Watson and G. A. Burley, Chemoselective sequential click ligations directed by enhanced reactivity of an aromatic ynamine, Org. Lett., 2016, 18, 1694–1697 CrossRef CAS PubMed; (e) C. P. Seath, G. A. Burley and A. J. B. Watson, Determining the origin of rate-independent chemoselectivity in CuAAC reactions: an alkyne-specific shift in rate-determining step, Angew. Chem., Int. Ed., 2017, 56, 3314–3318 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2155844. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2gc00966h |
| ‡ For information, the cost of the commercial NH4-USY used as zeolitic precursor of CuI-USY is 0.6€ per gram, thus cheap. CuI-USY is readily produced by heating under N2 flux a mixture of H-USY and CuCl (see ESI for details†). |
| § As sulfonamide 4a′ was not detected in the 1H NMR spectrum of the crude mixture, this unanticipated compound is thought to result from a by-product which is unstable in the presence of silica and thus prone to decompose during column chromatography. |
| ¶ When 4-fluoroaniline was used, the hydrated form of terminal ynamide 2a was detected among other unidentified reaction products. |
| || In the absence of molecular sieves, the model reactions provided 6a and 6c in lower yields (i.e. 48% for 6a and 62% for 6c), together with nonnegligible amounts of the hydrated ynamide as side-product. |
| ** Under our KA2 coupling conditions, hexan-2-one proved much less effective than its cyclic surrogate (cf. 54% vs. 92%). For more details, see ref. 21. |
| This journal is © The Royal Society of Chemistry 2022 |