
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Racemization-free and scalable amidation of L-proline in organic media using ammonia and a biocatalyst only†
Julia
Pitzer
a,
Kerstin
Steiner
a,
Christian
Schmid
a,
Viktor K.
Schein
b,
Christoph
Prause
b,
Claudia
Kniely
c,
Michaela
Reif
c,
Martina
Geier
a,
Elena
Pietrich
a,
Tamara
Reiter
ab,
Philipp
Selig
d,
Clemens
Stückler
d,
Peter
Pöchlauer
d,
Georg
Steinkellner
ae,
Karl
Gruber
e,
Helmut
Schwab
c,
Anton
Glieder
*c and
Wolfgang
Kroutil
 *b
*b
aACIB GmbH, Petersgasse 14, 8010 Graz, Austria
bDepartment of Chemistry, Organic & Bioorganic Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: wolfgang.kroutil@uni-graz.at; Tel: +43-316-380-5350
cInstitute of Molecular Biotechnology, Graz University of Technology, Petersgasse 14, 8010 Graz, Austria. E-mail: a.glieder@tugraz.at; Tel: +43-316-873-4074
dThermo Fisher Scientific Linz, St-Peter-Straße 25, 4020 Linz, Austria
eInstitute of Molecular Biosciences, University of Graz, Humboldtstrasse 50, 8010 Graz, Austria
First published on 25th May 2022
Abstract
Efficient amide formation is of high importance for the chemical and pharmaceutical industry. The direct biocatalytic one-pot transformation of acids into amides without substrate activation is a highly desirable but highly challenging reaction; this is why in general the acid is activated using additional reagents before amide formation occurs. In particular, amidation of α-amino acids is challenging and in general requires protection strategies for the amino functionality. A further challenge is the low solubility of the unprotected amino acids in organic solvents. Furthermore, the amidation process is prone to racemisation as observed for the acyl chloride derivative. These three challenges may be addressed using biocatalysis. Here the enzyme catalyzed, racemization-free amidation of unprotected L-proline with ammonia in an organic solvent is described. Comprehensive reaction, solvent and enzyme engineering allowed obtaining high L-prolinamide concentrations. For instance at 145 mM substrate concentration, 80% conversion was achieved employing an immobilized CalB variant and ammonia in 2-methyl-2-butanol at 70 °C. A two-fold increase in L-prolinamide formation was achieved employing the immobilized and engineered enzyme variant CalBopt-24 T245S compared to wild type CalB. In contrast to chemical processes, racemization, halogenated solvents and waste are avoided/minimized and atom efficiency is significantly improved from 45.5% to 86.4%. The excellent optical purity of the obtained product (ee >99%) and the stability of immobilized CalB pave the way for an innovative industrial process to produce L-prolinamide, a key intermediate in drug synthesis.
Introduction
The amide moiety constitutes an essential functional group of numerous natural as well as synthetic structures including proteins, pharmaceuticals and polymers; consequently, it represents an important target for the development of chemical as well as biocatalytic synthetic methods. Numerous strategies for amide synthesis have been described.1–10 Despite impressive success in selected cases, the scalability of biocatalytic amide synthesis for small molecules for industrial applications is still limited.10 The importance and the requirement for improvements of amide formation processes were underlined in a voting of the ACS GCI Pharmaceutical Roundtable: ‘Amide formation avoiding poor atom economy reagents’ was selected as the reaction with the highest interest of pharmaceutical companies for better and greener reagents.11Previous biocatalytic amidation efforts mostly employed lipases as catalyst transforming activated carboxylic acid derivatives, e.g. esters12–15 and anhydrides,16 or lipophilic substrates, such as long chain fatty acids, with amines.17–21 For instance the amidation of octanoic acid was performed via the ester intermediate, since direct ammonolysis led to the formation of salts instead of the desired octanamide.13 As an alternative, selective enzymatic kinetic resolution of various primary amines was reported for long chain carboxylic esters and acids with Candida antarctica lipase B (CalB) in hexane.22 Solvent free systems for biocatalytic amidation of aliphatic acids with rac-2-ethylhexyl amine23 and continuous flow systems for amidation of esters15 and oleic acid24 have been developed. The most straightforward way – the reaction of carboxylic acids with ammonia – has been rarely achieved.24,25
Most recently, amide formation was accomplished in an aqueous environment using acyltransferases7,26–28 or carboxylic acid reductases (CARs).8,29,30 In an alternative approach an engineered peptide amidase has been applied for the amidation of the C-terminal end of peptides in organic (co-)solvents.31 The enantioselective amidation of mandelic acid with different immobilized lipases and aqueous ammonium bicarbonate was reported in biphasic systems.32
Cbz-protected amino acids and peptides have been successfully converted to the amide with Alcalase CLEA and CalB using ammonium salts or ammonia.33 Considering the principles of green chemistry, thus, avoiding derivatization steps, like introducing protecting groups, is highly desirable.34 Yet, amidation of underivatized amino acids has not been accomplished yet, probably due to the low solubility of unprotected amino acids in organic solvents; consequently their direct amidation was not expected to be feasible.35 For instance, L-prolinamide is an important raw material and intermediate for pharmaceuticals, e.g. for antidiabetic drugs such as vildagliptin,36,37 and agrochemicals and is also used as an organocatalyst.38,39
Currently amidation of L-proline is performed chemically with thionyl chloride,36,40 which leads to the formation of hazardous waste (e.g. SO2, HCl) and results in partial racemization of the α-chiral centre. Consequently, a racemisation free and a minimum-reagent method is needed.
Results and discussion
In an ideal case the biocatalytic transformation of amino acid L-proline to the corresponding primary amide would require just ammonia, producing only water as a by-product and may be performed in one step (Scheme 1). To circumvent and avoid hydrolysis of the formed amide, the reaction is expected to be performed best in an organic solvent with the minimum water content. Thus, one of the challenges is the low solubility of the highly polar substrate proline in lipophilic solvents.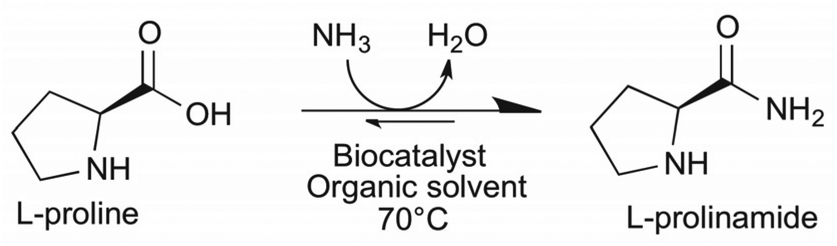 | ||
| Scheme 1 Enzymatic amidation of L-proline in the organic solvent with ammonia forming L-prolinamide and water. | ||
Selection of the enzyme
Various hydrolases, including lipases, esterases, proteases and amidases, might be expected to catalyse the target reaction, as they are in general able to perform the reverse reaction, the hydrolysis. Reports in the literature describe successful amidation of carboxylic esters13 and acids32,41 with lipases, e.g. from Candida rugosa, Pseudomonas sp.,32Rhizopus arrhizus13 and Rhizomucor miehei19,41 and proteases, e.g. subtilisin A.33 For initial experiments, a panel of enzymes obtainable from commercial suppliers was selected (e.g. Hydrolase Enzyme Screening Kit from Almac) or prepared by expression in E. coli or Pichia pastoris (Komagataella phaffii). Testing these preparations for the target reaction in 2-methyl-2-butanol (2M2B) with ammonia (0.5 M) led in most cases only to traces of product formation as observed e.g. with Lipolase 100T, Lipozyme TL IM and a lipase from Alcaligenes sp. (in this case 1,4-dioxane was used as the solvent) (ESI†). Out of the broad spectrum of hydrolases tested, immobilized CalB seemed to be promising and was therefore chosen for further tests.Selection of the solvent
The choice of the solvent is crucial influencing the solubility of the substrate, the product, and the stability of the enzyme preparation. The solubility of L-proline was found to be the highest in water (162 w% at 25 °C), followed by methanol (17.5 w% at 21 °C) and short chain alcohols (ESI†). However, as water enables the reverse hydrolysis reaction and competes with ammonia as a nucleophile, it was omitted. Amidation activity in methanol was very low, which seemed to be due to deactivating effects of methanol on CalB by partial unfolding or inhibition of lipase.42,43The substrate solubility of L-proline in primary alcohols such as n-butanol (3.1 w% at 21 °C) was significantly higher than in iso-butanol (0.4 w% at 21 °C), a secondary alcohol, or the tertiary alcohol 2-methyl-2-butanol (2M2B, 0.02 w% at 21 °C) (ESI†). It has to be noted, that the solubility increased upon the addition of NH3 or the reaction product, L-prolinamide. Although this might suggest primary alcohols as suitable solvents, they were omitted due to observed ester formation (∼10%) as undesired side products. Previous experiments with N-Cbz-L-proline suggested the use of toluene or t-BuOH/DMF.33 Consequently selected organic solvents not prone to ester formation were tested as solvents for the transformation of L-proline into L-prolinamide (Fig. 1). Most solvents tested, including 1,4-dioxane, tetrahydrofuran (THF), 2-methyl-THF, toluene and t-BuOH, resulted in the conversions of around 10–30% at 33 mM substrate concentration.
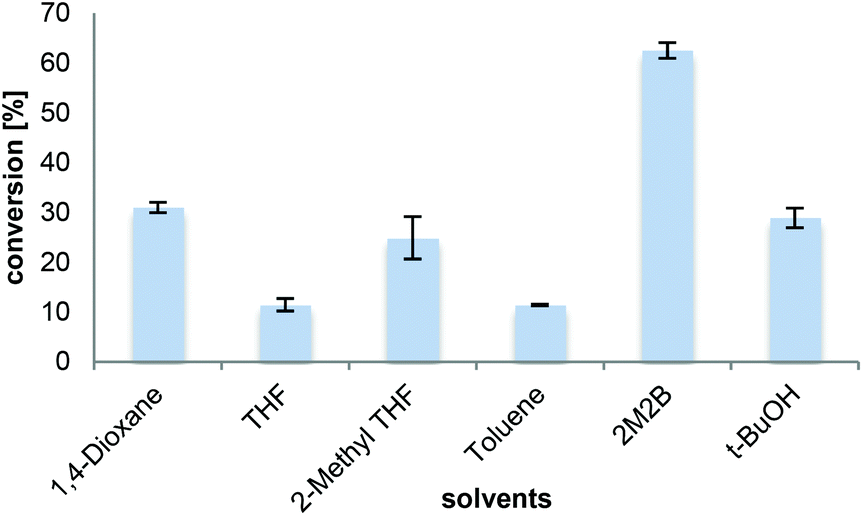 | ||
| Fig. 1 Transformation of L-proline (33 mM) in different solvents with 0.5 M NH3 in general (0.5 mL total volume). In 2-methyl-THF ∼0.2 M NH3 was dissolved. The reactions were performed with Novozym 435 (8.3 mg) for 16 h at 700 rpm and 70 °C and analyzed by HPLC-MS. | ||
Despite the low solubility of L-proline in 2M2B, this solvent led to 60% conversion without any observable ester formation. 2M2B has previously been described for amidation reactions with CalB for fatty acids with N-methyl-glucamine,19 hydroxy fatty acids20,21 and for the synthesis of oleamide in continuous flow.24 In contrast to the well-soluble substrate oleic acid, the solubility of L-proline in 2M2B was very low (1.2 mM at 21 °C, 3.5 mM at 70 °C, see the ESI†). However, it can be fairly assumed that the solid substrate L-proline (33 mM apparent concentration) was gradually dissolved during the course of the reaction allowing high product concentrations. Alcohols are ranked as one of the ‘greenest’ solvent classes in solvent selection guides, clearly superior to hydrocarbons, ethers or halogenated solvents.44–46 Therefore, 2M2B was preferred based on the high conversions obtained as well as due to the environmental aspects concerning its sustainability.
Effect of the water content
As even trace quantities of water in the organic solvent can cause hydrolysis of the acyl donor in lipase-catalysed aminolysis,47 the effect of the water concentration (addition of 0.5–2.5 v/v% H2O) in 2M2B was investigated (Fig. 2). The water content in the solvent before reaction was <0.1% v/v as determined by the Karl–Fischer titration. The experiment showed that low water concentrations (max 0.6% v/v) were essential for efficient amidation. A decrease in amide formation was seen when the water concentration was increased further. These results are relevant for long-term production on a larger scale, e.g. indicating how much water needs to be removed from the media in case of solvent recycling.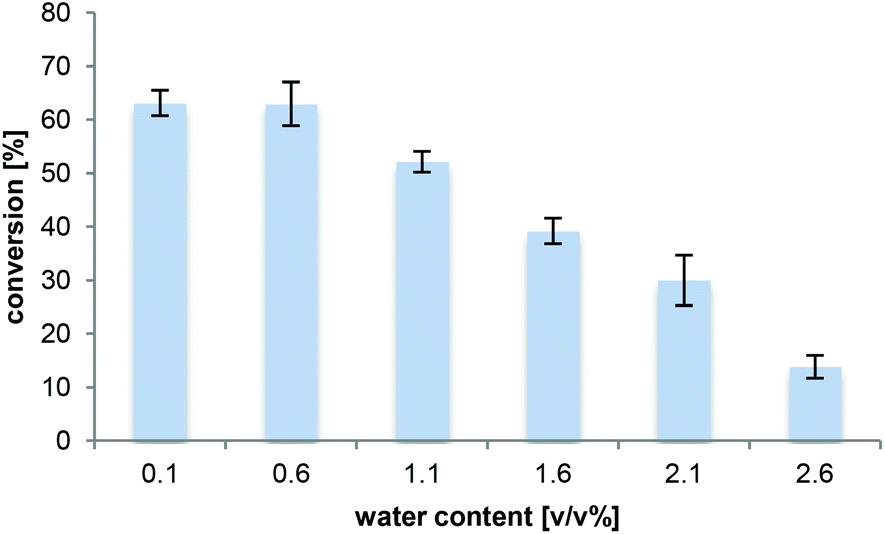 | ||
| Fig. 2 Effect of the water content on the amidation of L-proline. L-Proline (33 mM) with 0.5 M NH3 (Vtot = 500 μL) in 2M2B. The reactions were performed at varied water contents (0.1 to 2.6 v/v%) with Novozym 435 (8.3 mg) for 16 h at 700 rpm and 70 °C and analysed by HPLC-MS. | ||
Effect of temperature
The reaction temperature influences (i) the substrate solubility, (ii) the reaction rate as well as (iii) the enzyme stability and thus represents a key parameter. Increasing the temperature from 50 °C to 70 °C when using Novozym 435 improved conversions substantially (Fig. 3). Raising the temperature further was not beneficial. This was not surprising considering that it is usually accompanied by a decrease in enzyme stability,48,49 especially for long-term reactions. Additionally, loss of NH3 from the solution is expected at elevated temperatures.50 Consequently, 70 °C was selected as the preferred temperature, although in this reaction set-up also a reduction in the ammonia concentration was observed in 1,4-dioxane by 80% after 20 h at 70 °C. Gratifyingly, neither racemization nor dimerization of L-proline into diketopiperazine occurred at this temperature.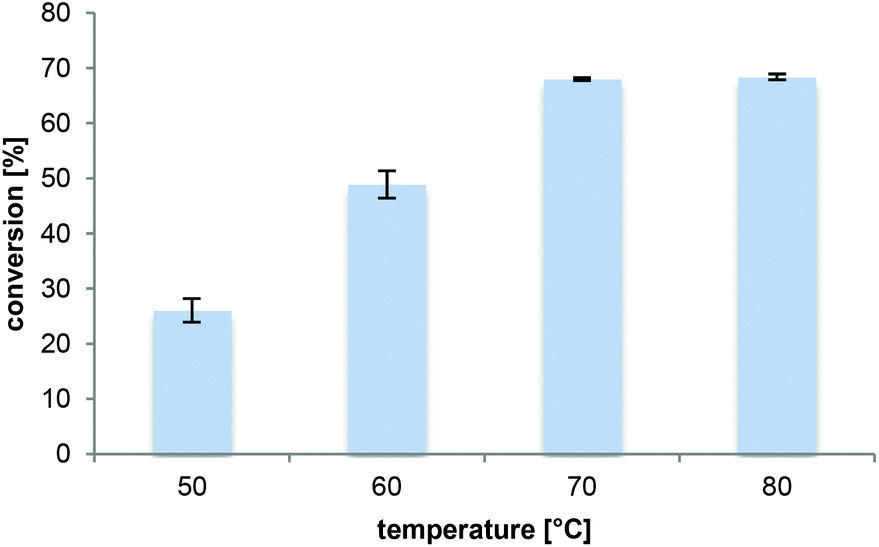 | ||
| Fig. 3 Effect of temperature on the amidation of L-proline. Transformation of L-proline (33 mM) in 0.5 M NH3 in 2M2B (Vtot = 500 μL) and Novozym 435 (8.3 mg) for 16 h at 700 rpm and 50–80 °C; analyzed by HPLC-MS. | ||
Enzyme preparation
For efficient amide formation in an organic solvent, the enzyme had to be stabilized by immobilization. The commercial wild type CalB preparation Novozym 435 was included in all experiments as a reference. That preparation was reported to be made by immobilization via adsorption. In this work, two complementary methods of immobilization were applied. Immobilization on resin ECR8806 was achieved via adsorption, while immobilization on resin ECR8285 (both obtained from Purolite) was achieved via covalent binding. The self-made preparations of wild type CalB (CalB WT) were obtained by employing the enzyme produced by secretion employing the host P. pastoris and immobilized on the two resins. Higher product formation was obtained with resin ECR8806 compared to ECR8285 (see the ESI†), suggesting the use of adsorption resin for scale-up.To evaluate the recyclability of the immobilized enzymes, the same enzyme preparation was reused for 20 days for several amidation reactions at 70 °C in 2M2B and NH3. The immobilized beads were washed between each use with 2M2B. After 20 days in 2M2B/70 °C, the reused Novozym 435 enzyme preparation led to slightly reduced conversion in comparison to a fresh Novozym 435 preparation. In contrast, the variant CalBopt-24 (see below, immobilized on resin 8806) was still performing better than fresh Novozym 435 (see Fig. S6 in the ESI†).
In order to evaluate the mechanical stability of the beads,51 the beads were examined by transmitted-light microscopy (see the ESI†) after the amidation reaction in 2M2B, whereby no mechanical destruction was detected, which is in accordance with previous reports.24
Furthermore, different drying strategies were compared for the removal of water from the beads after enzyme immobilisation. The enzyme preparations used in this work were dried by freezing the beads for 1 h at −80 °C followed by lyophilization for 24 h.
Enzyme engineering
To check for even better suited variants, promising amino acid residues with general beneficial effects were selected from the literature (see the ESI†). In these studies various objectives were pursued, which ranged from improving the solvent-52–54 or thermostability55–57 to altering substrate access by modifying the water tunnel towards the active site.58By modelling L-proline into the active site of CalB considering the tetrahedral intermediate of the acyl-enzyme intermediate with Ser105 and ammonia, residues possibly involved in substrate binding (T40, D134, T138, Q157, I189, V190) were selected and mutated (Fig. 4). One idea was to reduce the rather spacy active site to possibly tighter binding of the substrate. The intermediate structure is stabilized via neighbouring residues His224 and Asp187.
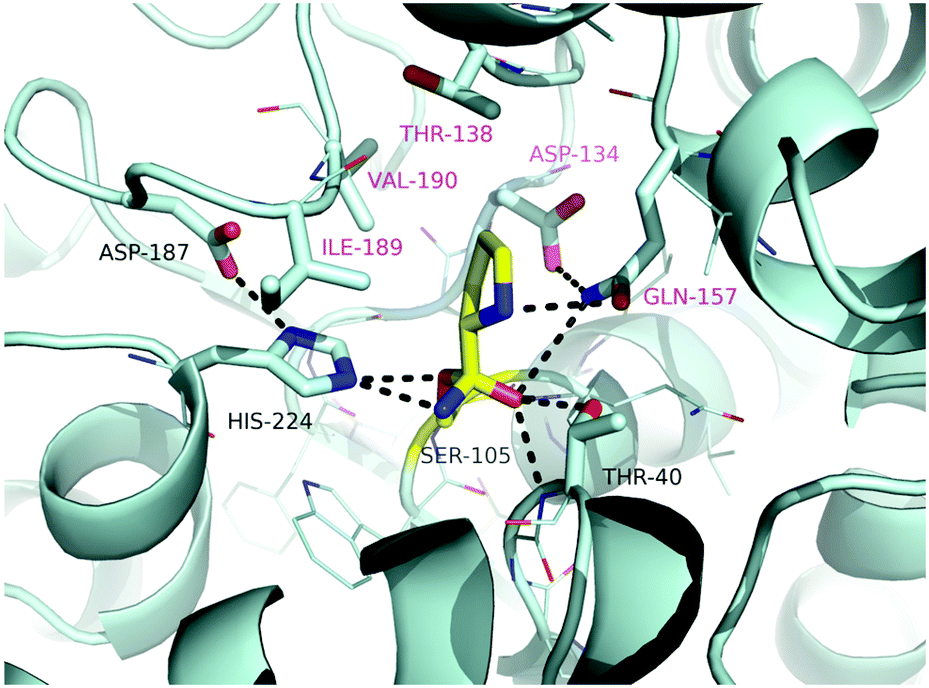 | ||
| Fig. 4 Model of complex of L-proline as a covalent tetrahedral acyl enzyme intermediate (energy minimization with a YASARA Structure utilizing the AMBER03 Force Field and the standard YASARA minimization protocol). | ||
All the variants (see ESI†) were prepared by individual site directed single-point mutagenesis, expressed in P. pastoris, concentrated by filtration, immobilized and then tested for the synthesis of L-prolinamide. Amino acid exchanges derived from the docking study in the active site (V190F, V190P, V190H, V190A, T40V, Q157L, I189P, I189F, T138L, T318H, T138F, etc.) significantly reduced amide formation (Fig. 5). However, increased L-prolinamide formation was observed e.g. for solvent- and thermotolerant variants55–57 leading overall to the identification of several amino acid exchanges (N264Q, A251E, T245S, N97Q, A8T, Q46A/S47L, K308C, T57A/A89T/G226R/R168K, etc.) with beneficial effects. The reduced conversion observed with Novozym 435 in Fig. 5 and 6 compared to Fig. 1–3 was due to the reduced amount of enzyme preparation used in these reactions (2 mg compared to 8.3 mg).
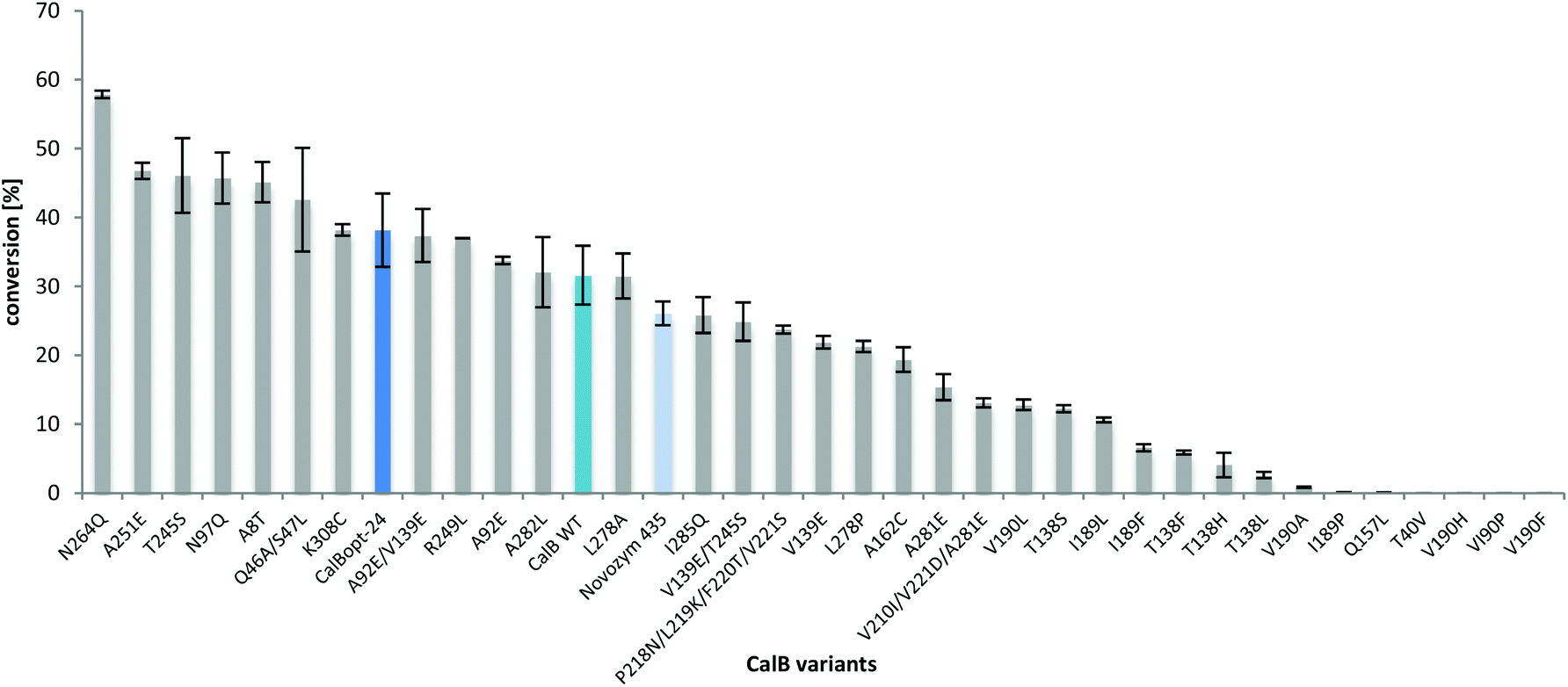 | ||
| Fig. 5 Evaluation of rationally designed CalB variants and variants described in the literature. Transformations of L-proline (33 mM) were performed in 2M2B with 0.5 M NH3 (500 μL) for 16 h at 700 rpm and 70 °C. The reactions were catalyzed by different CalB variants (all produced by P. pastoris with similar enzyme titers as the wild type) immobilized by covalent binding on resin ECR8285 from Purolite and lyophilized (2 mg). Original Novozym 435, immobilized by adsorption on a different carrier by Novozymes was used for comparison. Analysis was performed by HPLC-MS and the results were normalized to the amount of protein bound on the beads. | ||
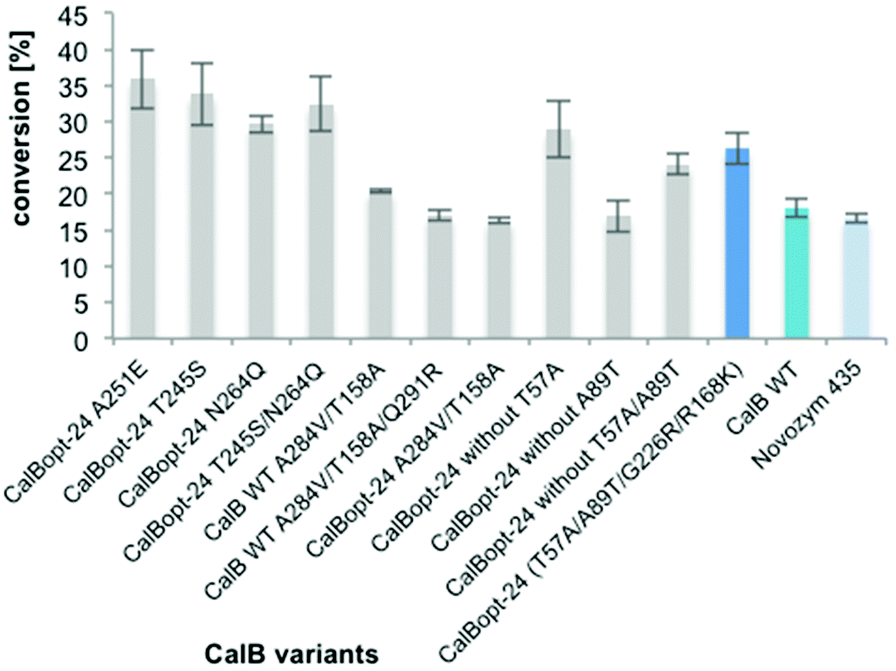 | ||
| Fig. 6 Evaluation of combined amino acid exchanges. Transformation of L-proline (33 mM) in 2M2B with 0.5 M NH3 (500 μL). The reactions were performed with different variants of CalB (all resulting in similar enzyme titers as the wild type) immobilized on resin ECR8806 by adsorption and lyophilized (2 mg) for 16 h at 700 rpm and 70 °C; addition of MeOH and HPLC-MS analysis of the reactions. The results were normalized to the protein loading on the beads. | ||
In an initial screening with 1,4-dioxane as the solvent, the thermostabilized variant T57A/A89T/G226R/R168K,57 which was named CalBopt-24, performed the best and was selected for further investigations. In contrast to the CalB wild type protein sequence (NCBI accession number: P41365) from the strain used in this work (CBS 214.83 Candida antarctica, also named Pseudozyma antarctica, ATCC 34888),59 another CalB homologue from ATCC 32657 Pseudozyma aphidis already contains the two amino acid exchanges T57A and A89T. The latter one was, e.g., used as a template for mutagenesis in circular permutation studies.60 Therefore, depending on the wild type sequence used as a reference, CalBopt-24 represents either a double or quadruple variant. In 2M2B, some variants led to conversions similar to or even higher than that of CalBopt-24, e.g. the solvent stabilized variants T245S54 and N264Q53 and the thermostabilized variant A251E.54,61 Furthermore, variant Q46A/S47L,58 having a modified water tunnel leading to the active site, enabled high conversion. A detailed list of the selected amino acid exchanges, the intention behind it, the references and the obtained conversions in the amidation reaction is provided in the ESI.†
Random mutagenesis
To investigate the potential to improve the stability and activity of CalBopt-24 further, random mutagenesis by error-prone PCR, combined with screening for improved solvent- and thermostability was performed. A shortened protocol based on an ARS (autonomous replicating sequence) plasmid and homologous recombination by P. pastoris was applied without sub-cloning into E. coli.62 The remaining hydrolytic activity after incubation at 60 °C in 1,4-dioxane (which was seen as the best solvent at the time when these experiments were performed) was determined using p-nitrophenyl butyrate as a surrogate substrate. In most cases, increased hydrolytic activity in the screening correlated with increased or at least retained amidation activity in the target reaction compared to the starting protein (Table 1).| Exchange | Conv. [%], in 1,4-dioxanea | Conv. [%], in 2M2Ba |
|---|---|---|
| a Transformation of L-proline (33 mM) in 2M2B and 1,4-dioxane with 0.5 M NH3 (500 μL). The reactions were performed with different CalB variants immobilized by adsorption on resin ECR8806 and lyophilized (3 mg) for 16 h at 700 rpm and 70 °C; conversions were determined by HPLC-MS. For all variants, the results were normalized to the amount of protein bound on the beads. | ||
| CalBopt-24 A284V | 26.2 | 33.9 |
| CalBopt-24 A284T | 28.5 | 33.6 |
| CalBopt-24 I285T | 28.9 | 29.3 |
| CalBopt-24 V154A | 10.1 | 19.2 |
| CalBopt-24 L278P | 21.9 | 28.2 |
| CalBopt-24 T158A/Q291R | 18.6 | 37.3 |
| CalBopt-24 | 20.2 | 30.2 |
| Novozym 435 | 9.0 | 22.9 |
Interestingly, several of the subsequently identified amino acid residues have already been described as hot spots for engineering efforts (L278P,63 Leu278,64–67 Ile28568,69 and Thr15861). Nevertheless, in addition to the already reported amino acid exchanges, several new ones were found, namely A284V/T, V154A and Q291R. Best results concerning the targeted L-proline amidation in 2M2B were obtained with CalBopt-24 T158A/Q219R and CalBopt-24 A284V.
Combining amino acid exchanges
In the next step the amino acid exchanges of interesting variants identified by random and rational engineering were combined. The introduction of A251E for improved thermostability54,61 or T245S for enhanced solvent stability54 into CalBopt-24 turned out to be beneficial. Furthermore, the contribution of T57A and A89T to the beneficial effect of CalBopt-24 (T57A/A89T/G226R/R168K) was investigated. The results indicated that T57A was not contributing to the positive effect of CalBopt-24. When CalBopt-24 was combined with the amino acid exchanges Q46A/S47L, Q46A/S47L/T245S and Q46A/S47L/A251E no CalB activity was measured in the supernatant after 96-well DWP cultivation. Therefore, combinations with Q46A/S47L were not pursued further.In the transformation with 33 mM proline (Fig. 6) CalBopt-24 T245S led to 20% higher amide formation than CalBopt-24 and was superior to other variants at 200 mM substrate loading (see Fig. S1†). Furthermore, CalBopt-24 T245S led to two-fold higher L-prolinamide formation compared to immobilized CalB WT, thus representing the best CalB variant found in this work for the amidation of L-proline.
Preparative scale
While enzyme engineering to identify improved enzyme variants was still ongoing, scale up experiments were performed to evaluate the feasibility of preparative scale amide formation. Performing the reaction of suspended L-proline (250 mg, apparent conc.: 145 mM) in 2M2B containing NH3 (0.5 M) with immobilized enzymes at 15 mL scale led to a product concentration of up to 117 mM using CalBopt-24 immobilized on ECR8806 (Table 2). This corresponds to a conversion of 80%. For product isolation, the reaction mixture was filtered and L-prolinamide was isolated by fractionated crystallization from 2M2B/n-heptane (128 mg isolated from the reaction with CalB WT immobilized on resin ECR8806 in Table 2; see 1H-NMR in the ESI†).| L-Prolinamidea,b [mM] | |
|---|---|
| a The reactions were performed with Novozym 435 as well as CalB WT and CalBopt-24 immobilized on resin ECR8806 (250 mg) at 95 rpm shaking speed and 70 °C in a shaking incubator for 4.5 days (112 h). A substrate loading of 250 mg L-proline (corresponding to a max. concentration of 145 mM, partly present in solid form) in 2M2B (15 mL) with 0.5 M NH3 was used. b Concentrations in soluble phase determined by HPLC-MS analysis after filtration of the reaction solution. | |
| Novozym 435 | 101.5 |
| CalBopt-24 | 117.2 |
| CalB WT | 107.2 |
Assessment of chemical and biocatalytic methods
By following the principles of green chemistry it is possible to achieve two goals at the same time, namely, to develop economically profitable as well as environmentally compatible processes.70 A comparison of established chemical strategies for L-prolinamide synthesis with the present study showed that when using thionyl chloride (either via the methyl ester or the chloride)36,40 and the amidation on a cationic ion exchange resin,71 two steps are required (Table 3). The process via proline chloride requires dichloromethane as the solvent, which is ranked as hazardous,72 and generates SO2 and HCl as waste. The biocatalytic process with a recyclable alcohol as the solvent does not produce waste apart from water. After the removal of formed water, e.g. by distillation, the solvent can be recycled. Due to the impressive long-term stability of immobilized CalB, the biocatalyst can be reused as well.| Reference | Catalyst | Reagents | Reaction solvents | Solvents work-up | Wastea | Optical purity | Steps | Atom efficiency [%] |
|---|---|---|---|---|---|---|---|---|
| a H2O not considered. b Assuming that MeOH can be recycled. c Represents the maximum. | ||||||||
| CN102180823B, via methylester | — | NH3, SOCl2, KOH | MeOH | CH2Cl2, EtOAc | SO2, KCl | Racemization | 2 | 37.2b |
| CN102491928A, via chloride | — | NH3, SOCl2 | CH2Cl2 | CH2Cl2 | SO2, HCl | Racemization | 2 | 45.5 |
| US6271394B1 | Amberlyst | NH3, MeOH | MeOH | Toluene, n-heptane | Up to 4% side product | Not given | 2 | 86.4a,c |
| This study | Biocatalyst | NH3 | 2M2B | 2M2B, n-heptane | — | >99% | 1 | 86.4c |
The chemical method via activation with thionyl chloride leads to an atom economy of 45.5% and considerable amounts of hazardous waste and requires two process steps. The enzymatic reaction, on the other hand, achieves an atom economy of 86.4% and requires only one process step. No racemization of the substrate and product was observed under the reaction conditions, thus enantiomerically pure L-prolinamide (ee >99%) was obtained.
Conclusions
A scalable, efficient biocatalytic one-pot reaction for the amidation of L-proline with ammonia was developed. By employing a tert-alcohol (2-methyl-2-butanol) as the solvent and ammonia as the only stoichiometric reagent, the amount of required chemicals and waste produced can be kept to a minimum. Water, which is here formed as a by-product, can be removed by distillation. Consequently, the dry solvent can be reused and the process costs and environmental impact are reduced. Strongly corrosive chemicals, toxic off-gases like SO2 and HCl and expensive reagents are avoided, which are common side products in amide formation when using thionyl chloride as the activating reagent. Increased conversions were achieved with optimized variants of CalB. This enzyme is easily well expressible in P. pastoris, and immobilization by adsorption provides a very stable enzyme preparation. The reaction system represents an innovative new approach for L-prolinamide production in optically pure form on a large scale. Preparative scale conversions so far were performed either with immobilized CalB WT or CalBopt-24 reaching up to 117 mM product concentration. The evaluation of further identified improved enzyme variants after enzyme engineering experiments in analytical biotransformations on the mg scale indicated the feasibility of further process improvements for example by using variants such as CalBopt-24 T245S.Experimental
General materials and methods
Reagents and organic solvents were obtained from chemical suppliers in reagent grade quality and applied without further purification. L-Proline and L-prolinamide were obtained from Thermo Fisher Scientific (Linz, Austria). Novozym 435 was obtained from Novozymes (Bagsvaerd, Denmark).NH3 quantification
The NH3 concentration in organic solvents, e.g. 2M2B, was determined after bubbling NH3 through the solvent in an open round bottom flask while stirring with a magnetic stir bar for a few hours. The Ammonia Assay Kit from Sigma-Aldrich (MAK310) was used according to the manufacturer's instructions. The samples were diluted with dH2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :700) to obtain concentrations in the linear range of the kit of 0.012–1 mM. After the addition of the reaction mix, the plate was incubated in the dark at 21 °C for 15 min and the fluorescence intensity was measured (λex = 360 nm, λem = 450 nm).
:700) to obtain concentrations in the linear range of the kit of 0.012–1 mM. After the addition of the reaction mix, the plate was incubated in the dark at 21 °C for 15 min and the fluorescence intensity was measured (λex = 360 nm, λem = 450 nm).
Amidation of L-proline
L-Proline (1.9 mg, 33 mM) and immobilized CalB (8.3 mg), Novozym 435 or self-immobilized CalB variants, were used (unless otherwise stated). The reactions were performed in 0.5 M NH3 in an organic solvent (2M2B, 500 μL) in small glass vials sealed with a cap and a septum and shaken for 16 h at 700 rpm and 70 °C in an Eppendorf Thermoshaker Comfort.After the reaction, MeOH (500 μL) was added to dissolve all substrates and products. The samples were diluted to 1 mM substrate concentration (1:33) with MeOH. As an internal standard D,L-norvaline (0.5 mM in MeOH) was added and the samples were vortexed, centrifuged (20000g, 2 min), filtered through cotton wool and analyzed by HPLC-MS.
Reactions at the preparative scale
The reactions were performed by shaking in a thermo incubator with 0.5 M NH3 in 2M2B (15 mL) using L-proline (250 mg, 2.18 mmol, 145 mM) as the substrate and immobilized CalB (250 mg). Novozym 435, as well as CalB WT and CalBopt-24, immobilized on resin ECR8806 were investigated at 95 rpm shaking speed and 70 °C for 4.5 days (112 h). After filtration of the reaction solution, L-prolinamide was isolated by fractionated crystallization from 2M2B/n-heptane and analysed by NMR.1H-NMR (300 MHz, MeOD): δ [ppm] = 1.76 (m, 2H, C4H2), 2.12 (m 2H, C3H2), 2.15 (s, 1H, NH), 2.88 (m, 1H, C5H), 2.99 (m, 1H, C5H), 3.31 (MeOD), 3.61 (m, 1H, C2H), 4.82 (br s 2H, NH2).
13C-NMR (75 MHz, MeOD): δ [ppm] = 26.0 (C4), 31.3 (C3), 48.05 (C5), 60.4 (C2), 179.0 (CO). The NMR results were in accordance with the data from the literature.73
HPLC-MS analysis
For HPLC-MS analysis an Agilent 1200 Infinity system was equipped with a Zorbax 300-SCX 4.6 × 250 mm 5 Micron column from Agilent. Analysis was performed by isocratic elution using 90% H2O + 0.1% formic acid and 10% MeOH + 0.1% formic acid for ∼20 min. The flow rate was set to 1 mL min−1 and the maximal pressure limit to 400 bar. Analysis was performed in positive SCAN (114.5–300 m/z) and SIM modes (115 m/z for L-prolinamide, 116 m/z for L-proline, and 118 m/z for D,L-norvaline as the internal standard). A sample volume of 2 μL was injected.L-Proline and L-prolinamide calibration curves were obtained with reference solutions from 0.03–4 mM in MeOH containing DL-norvaline (0.5 mM) as internal standards.
Analysis of optical purity
For analysis derivatization was required using chloroformate and DMAP.74 The solvent was removed by air, then the residue was dissolved in MeOH (500 μL, containing 5% DMAP) and ethyl chloroformate (100 μL) was added. The derivatization reaction was performed for 1 h at 50 °C and 700 rpm. MeOH was removed by air and 2% HCl (500 μL) was added before extraction with EtOAc (2 times with 500 μL). The combined organic phases were dried over Na2SO4, and after centrifugation and filtration 5 μL of the sample solution were injected for GC analysis. A CP Chiralsil Dex CB column was used for analysis with a flow rate of 1 mL min−1, injection temperature of 250 °C and detection temperature of 250 °C. A temperature gradient starting at 60 °C (5 min hold) and increasing at 3 °C min−1 to 180 °C (5 min hold) was used.Cloning, enzyme expression and engineering
Cloning, primer sequences, random and rational mutagenesis and enzyme expression in P. pastoris are described in detail in the ESI.† For the production of the integrative Zeocin selection vector pBSY3S1Z (from Bisy, Austria) with an optimized Dα signal sequence was used, based on the pPpT4_S plasmid described previously.75 Cloning was performed by Gibson assembly76 or overlap extension PCR.77 Enzymes, chemicals, cloning kits and cultivation of E. coli were used and performed as detailed before.78 Transformation of P. pastoris BSY BG11 (from Bisy, Austria) for genomic integration was performed with SwaI linearized DNA (1 μg) according to the protocol.79 Cultivation of transformants in 96-deep-well plates were performed by growth on glycerol containing media (buffered minimal medium with 1% w/v glycerol, BMG1%) followed by methanol induction, based on a published protocol.80The hydrolytic activity of CalB and its variants was determined using p-nitrophenyl butyrate as the substrate according to the literature.81 Transformants with the highest activity were selected, streaked for single colonies and their uniform expression level was verified by rescreening, before they were cultivated in 2.5 L Ultra Yield™ flasks (from Thomson Instrument Company, CA, USA). After inoculation of BMG1% (450 mL), the flasks were incubated at 28 °C and 105 rpm for 60 h. Methanol was added daily to induce and maintain the expression of CalB. First BMM5% (50 mL) was added followed by addition of pure MeOH (5 mL) every 12 h. After 72 h of protein expression, the cultures were harvested by centrifugation at 6000g and 4 °C for 20 minutes. The supernatant was filtered through a membrane with a minimum pore size diameter of 0.45 μm by vacuum filtration and concentrated by centrifugation using Vivaspin 20, 10000 MW CO centrifugation tubes from Sartorius at full speed and 8 °C.
Immobilization of CalB
The protein concentration in the samples was determined using the Bradford Assay (Bio-Rad).
Immobilization on ECR8285 by covalent binding. The beads (100 mg, wet weight) were filled into a 1.5 mL tube and washed four times with the washing buffer (100 μL, 10 mM Na-phosphate buffer, pH 7.5). Protein solution (5 mg protein in 600 μL immobilization buffer 0.5 M Na-phosphate buffer, pH 7.5) was added to the washed beads and incubated for 18 hours at 50 rpm on a spinning wheel and for 20 h on the bench at 21 °C. After washing the beads two times with washing buffer (100 μL, 10 mM Na-phosphate buffer, pH 7.5) and once with washing buffer containing 0.5 M NaCl (100 μL) for desorption of non-covalently bound proteins, the enzyme beads were frozen at −80 °C for 1 h. The tubes were opened and placed in a lyophilisator for 24 h at −40 °C with 50 μbar vacuum. Subsequently, they were stored at 4 °C and tested for the target amidation reaction with L-proline.
Immobilization on ECR8806 by adsorption. The beads (100 mg, wet weight) were filled into a 1.5 mL tube and washed twice with the washing buffer (120 μL, 10 mM Na-phosphate buffer, pH 7.5). Protein solution (5 mg protein in 600 μL immobilization buffer, 20 mM Na-phosphate buffer, pH 7.5) was added to the washed beads and incubated for 24 hours at 50 rpm at 21 °C on a spinning wheel. After washing the beads once with washing buffer (150 μL, 10 mM Na-phosphate buffer, pH 7.5), the enzyme beads were frozen at −80 °C for 1 h. The tubes were opened and placed in a lyophilisator for 24 h at −40 °C with 50 μbar vacuum. Subsequently, they were stored in the fridge at 4 °C and tested for the target amidation reaction with L-proline.
The immobilization supernatant and the wash fractions were collected and the remaining protein concentration was measured, to determine the immobilization efficiency.
Structural modelling
The structure of CalB with the PDB ID 5A71, chain A in the ‘open’ form (which is identical to the CalB WT sequence P41365 used in this study) was used for the modelling. CalB has a homodimer structure and contains a glycosylation site and disulfide bridges. Chain A represents the ‘open’ form and Chain B the closed form of this particular CalB. Alternates were removed using PyMOL. CASoX analysis of the active site pocket was performed. The tetrahedral intermediate was modelled into the open form. Both chains (chain A and chain B) are very similar regarding the active site arrangement and space. The modelled acetamide complex was prepared for energy minimization using YASARA structure using AMBER03 Force Field and the standard YASARA minimization protocol. Based on this tetrahedral intermediate the L-proline substrate was modelled into a structure and minimized using the above protocol. Pictures of the final complex were obtained using PyMOL 1.7, Pymol BNI-Tools 0.31.Conflicts of interest
J. P., P. P., P. S., C. Z., K. S., A. G. and W. K. are authors of the patent WO2020/126484A1.Acknowledgements
The COMET Center: acib: Next Generation Bioproduction is funded by BMK, BMDW, SFG, Standortagentur Tirol, Government of Lower Austria und Vienna Business Agency in the framework of COMET – Competence Centers for Excellent Technologies. The COMET-Funding Program is managed by the Austrian Research Promotion Agency FFG. The University of Graz and the Field of Excellence BioHealth are acknowledged for financial support. The funding agencies had no influence on the conduct of this research.References
- H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714–2742 RSC.
- J. Pitzer and K. Steiner, J. Biotechnol., 2016, 235, 32–46 CrossRef CAS PubMed.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- M. R. Petchey and G. Grogan, Adv. Synth. Catal., 2019, 361, 3895–3914 CrossRef CAS.
- D. Gahloth, G. A. Aleku and D. Leys, J. Biotechnol., 2020, 307, 107–113 CrossRef CAS PubMed.
- E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org. Chem., 2020, 4641–4651 CrossRef CAS.
- M. L. Contente, D. Roura Padrosa, F. Molinari and F. Paradisi, Nat. Catal., 2020, 3, 1020–1026 CrossRef CAS.
- M. Winn, M. Rowlinson, F. Wang, L. Bering, D. Francis, C. Levy and J. Micklefield, Nature, 2021, 593, 391–398 CrossRef CAS PubMed.
- M. Zhu, L. Wang and J. He, Angew. Chem., Int. Ed., 2021, 60, 2030–2035 CrossRef CAS PubMed.
- B. M. Dorr and D. E. Fuerst, Curr. Opin. Chem. Biol., 2018, 43, 127–133 CrossRef CAS PubMed.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. a. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- M. C. de Zoete, A. C. Kock-van Dalen, F. van Rantwijk and R. A. Sheldon, J. Chem. Soc., Chem. Commun., 1993, 1831–1832, 10.1039/c39930001831.
- M. C. de Zoete, A. C. Kock-Van Dalen, F. van Rantwijk and R. A. Sheldon, Biocatalysis, 2009, 10, 307–316 CrossRef.
- M. C. de Zoete, A. C. Kock-van Dalen, F. van Rantwijk and R. A. Sheldon, J. Mol. Catal. B: Enzym., 1996, 2, 19–25 CrossRef CAS.
- L. H. Andrade, B. A. Sousa and T. F. Jamison, J. Flow Chem., 2016, 6, 67–72 CrossRef CAS.
- K. J. Liu, K. M. Liu and H. M. Chang, Food Chem., 2007, 102, 1020–1026 CrossRef CAS.
- N. P. Awasthi and R. P. Singh, J. Oleo Sci., 2007, 56, 507–509 CrossRef CAS PubMed.
- M. Fernández-Pérez and C. Otero, Enzyme Microb. Technol., 2001, 28, 527–536 CrossRef.
- T. Maugard, M. Remaud-Simeon, D. Petre and P. Monsan, J. Mol. Catal. B: Enzym., 1998, 5, 13–17 CrossRef CAS.
- W. E. Levinson, T. M. Kuo and C. P. Kurtzman, Enzyme Microb. Technol., 2005, 37, 126–130 CrossRef CAS.
- S. K. Khare, A. Kumar and T. M. Kuo, Bioresour. Technol., 2009, 100, 1482–1485 CrossRef CAS PubMed.
- M. Nechab, N. Azzi, N. Vanthuyne, M. Bertrand, S. Gastaldi and G. Gil, J. Org. Chem., 2007, 72, 6918–6923 CrossRef CAS PubMed.
- A. K. Prasad, M. Husain, B. K. Singh, R. K. Gupta, V. K. Manchanda, C. E. Olsen and V. S. Parmar, Tetrahedron Lett., 2005, 46, 4511–4514 CrossRef CAS.
- W. F. Slotema, G. Sandoval, D. Guieysse, A. J. J. Straathof and A. Marty, Biotechnol. Bioeng., 2003, 82, 664–669 CrossRef CAS PubMed.
- R. Madeira Lau, F. Van Rantwijk, K. R. Seddon and R. A. Sheldon, Org. Lett., 2000, 2, 4189–4191 CrossRef CAS PubMed.
- A. Zadlo-Dobrowolska, N. G. Schmidt and W. Kroutil, Chem. Commun., 2018, 54, 3387–3390 RSC.
- M. L. Contente, A. Pinto, F. Molinari and F. Paradisi, Adv. Synth. Catal., 2018, 360, 4814–4819 CrossRef CAS.
- H. Muller, S. P. Godehard, G. J. Palm, L. Berndt, C. P. S. Badenhorst, A. K. Becker, M. Lammers and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 2013–2017 CrossRef PubMed.
- A. J. L. Wood, N. J. Weise, J. D. Frampton, M. S. Dunstan, M. A. Hollas, S. R. Derrington, R. C. Lloyd, D. Quaglia, F. Parmeggiani, D. Leys, N. J. Turner and S. L. Flitsch, Angew. Chem., Int. Ed., 2017, 56, 14498–14501 CrossRef CAS PubMed.
- M. Lubberink, C. Schnepel, J. Citoler, S. R. Derrington, W. Finnigan, M. A. Hayes, N. J. Turner and S. L. Flitsch, ACS Catal., 2020, 10, 10005–10009 CrossRef CAS.
- B. Wu, H. J. Wijma, L. Song, H. J. Rozeboom, C. Poloni, Y. Tian, M. I. Arif, T. Nuijens, P. J. L. M. Quaedflieg, W. Szymanski, B. L. Feringa and D. B. Janssen, ACS Catal., 2016, 6, 5405–5414 CrossRef CAS.
- D. Yildirim and S. S. Tükel, Biocatal. Biotransform., 2014, 32, 251–258 CrossRef CAS.
- T. Nuijens, E. Piva, J. A. W. Kruijtzer, D. T. S. Rijkers, R. M. J. Liskamp and P. J. L. M. Quaedflieg, Tetrahedron Lett., 2012, 53, 3777–3779 CrossRef CAS.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- M. J. J. Litjens, A. J. J. Straathof, J. a. Jongejan and J. J. Heijnen, Tetrahedron, 1999, 55, 12411–12418 CrossRef CAS.
- K. Xiang and M. Xu, CN102180823A, 2011.
- M. Castaldi, M. Baratella, I. G. Menegotto, G. Castaldi and G. B. Giovenzana, Tetrahedron Lett., 2017, 58, 3426–3428 CrossRef CAS.
- C. Yue, Y. Yamashita and S. Kobayashi, Green Chem., 2021, 23, 1989–1994 RSC.
- G. Tang, X. Hu and H. J. Altenbach, Tetrahedron Lett., 2011, 52, 7034–7037 CrossRef CAS.
- H. Zhengliang, W. Wanchun and W. Bo, CN102491928A, 2011.
- T. Maugard, M. Remaud-Simeon, D. Petre and P. Monsan, Tetrahedron, 1997, 53, 7587–7594 CrossRef CAS.
- T. Kulschewski, F. Sasso, F. Secundo, M. Lotti and J. Pleiss, J. Biotechnol., 2013, 168, 462–469 CrossRef CAS PubMed.
- M. Lotti, J. Pleiss, F. Valero and P. Ferrer, Biotechnol. J., 2015, 10, 22–30 CrossRef CAS PubMed.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7–7 CrossRef.
- F. van Rantwijk, M. A. P. J. Hacking and R. A. Sheldon, Monatsh. Chem., 2000, 131, 549–569 CrossRef CAS.
- V. G. H. Eijsink, S. GÅseidnes, T. V. Borchert and B. Van Den Burg, Biomol. Eng., 2005, 22, 21–30 CrossRef CAS PubMed.
- P. V. Iyer and L. Ananthanarayan, Process Biochem., 2008, 43, 1019–1032 CrossRef CAS.
- A. Seidell and W. F. Linke, Solubilities of Inorganic and Organic Compounds: A Compilation of Solubility Data from the Periodical Literature. Supplement to ed. 3, Van Nostrand, New York, 1952 Search PubMed.
- T. Siódmiak, D. Mangelings, Y. Vander Heyden, M. Ziegler-Borowska and M. P. Marszałł, Appl. Biochem. Biotechnol., 2015, 175, 2769–2785 CrossRef PubMed.
- C. F. J. Yagonia, H. J. Park, S. Y. Hong and Y. J. Yoo, Biotechnol. Bioprocess Eng., 2015, 20, 218–224 CrossRef CAS.
- H. J. Park, J. C. Joo, K. Park and Y. J. Yoo, Biotechnol. Bioprocess Eng., 2012, 17, 722–728 CrossRef CAS.
- H. J. Park, J. C. Joo, K. Park, Y. H. Kim and Y. J. Yoo, J. Biotechnol., 2013, 163, 346–352 CrossRef CAS PubMed.
- H. S. Kim, Q. A. T. Le and Y. H. Kim, Enzyme Microb. Technol., 2010, 47, 1–5 CrossRef CAS.
- Q. A. T. Le, J. C. Joo, Y. J. Yoo and Y. H. Kim, Biotechnol. Bioeng., 2012, 109, 867–876 CrossRef CAS PubMed.
- X.-Q. Peng, Appl. Biochem. Biotechnol., 2013, 169, 351–358 CrossRef CAS PubMed.
- M. Wittrup Larsen, D. F. Zielinska, M. Martinelle, A. Hidalgo, L. Juhl Jensen, U. T. Bornscheuer and K. Hult, ChemBioChem, 2010, 11, 796–801 CrossRef CAS PubMed.
- T. B. Nielsen, M. Ishii and O. Kirk, Lipases A and B from the yeast Candida antarctica, in Biotechnological Applications of Cold-Adapted Organisms, ed. R. Margesin and F. Schinner, Springer-Verlag, Berlin Heidelberg, 1999, pp. 49–61. DOI:10.1007/978-3-642-58607-1_4.
- Z. Qian and S. Lutz, J. Am. Chem. Soc., 2005, 127, 13466–13467 CrossRef CAS PubMed.
- H. J. Park, K. Park, Y. H. Kim and Y. J. Yoo, J. Biotechnol., 2014, 192, 66–70 CrossRef CAS PubMed.
- T. Vogl, A. Glieder and R. Wasmayer, WO2017055436A1, 2015.
- S. Y. Kim, J. H. Sohn, Y. R. Pyun, I. S. Yang, K. H. Kim and E. S. Choi, J. Microbiol. Biotechnol., 2007, 17, 1308–1315 CAS.
- Z. Marton, V. Léonard-Nevers, P. O. Syrén, C. Bauer, S. Lamare, K. Hult, V. Tranc and M. Graber, J. Mol. Catal. B: Enzym., 2010, 65, 11–17 CrossRef CAS.
- M. Takwa, M. W. Larsen, K. Hult and M. Martinelle, Chem. Commun., 2011, 47, 7392–7394 RSC.
- Y. Xie, J. An, G. Yang, G. Wu, Y. Zhang, L. Cui and Y. Feng, J. Biol. Chem., 2014, 289, 7994–8006 CrossRef CAS PubMed.
- D. Liu, P. Trodler, S. Eiben, K. Koschorreck, M. Müller, J. Pleiss, S. C. Maurer, C. Branneby, R. D. Schmid and B. Hauer, ChemBioChem, 2010, 11, 789–795 CrossRef CAS PubMed.
- M. R. Hediger, L. De Vico, J. B. Rannes, C. Jäckel, W. Besenmatter, A. Svendsen and J. H. Jensen, PeerJ, 2013, 1, e145–e145 CrossRef PubMed.
- P.-O. Syrén, E. Lindgren, H. W. Hoeffken, C. Branneby, S. Maurer, B. Hauer and K. Hult, J. Mol. Catal. B: Enzym., 2010, 65, 3–10 CrossRef.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- R. Shekhar and M. J. Girgis, U.S. Pat, US6271394B1, 2001 Search PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- F. Herold, M. Dawidowski, I. Wolska, A. Chodkowski, J. Kleps, J. Turło and A. Zimniak, Tetrahedron: Asymmetry, 2007, 18, 2091–2098 CrossRef CAS.
- G. Gourinchas, E. Busto, M. Killinger, N. Richter, B. Wiltschi and W. Kroutil, Chem. Commun., 2015, 51, 2828–2831 RSC.
- L. Näätsaari, B. Mistlberger, C. Ruth, T. Hajek, F. S. Hartner and A. Glieder, PLoS One, 2012, 7, e39720–e39720 CrossRef PubMed.
- D. G. Gibson, L. Young, R.-y. Chuang, J. C. Venter, C. A. Hutchison and H. O. Smith, Nat. Methods, 2009, 6, 343–345 CrossRef CAS PubMed.
- Z. Liu, B. Pscheidt, M. Avi, R. Gaisberger, F. S. Hartner, C. Schuster, W. Skranc, K. Gruber and A. Glieder, ChemBioChem, 2008, 9, 58–61 CrossRef CAS PubMed.
- T. Vogl, C. Ruth, J. Pitzer, T. Kickenweiz and A. Glieder, ACS Synth. Biol., 2014, 3, 188–191 CrossRef CAS PubMed.
- J. Lin-Cereghino, W. W. Wong, S. Xiong, W. Giang, L. T. Luong, J. Vu, S. D. Johnson and G. P. Lin-Cereghino, BioTechniques, 2005, 38, 44–48 CrossRef CAS PubMed.
- R. Weis, R. Luiten, W. Skranc, H. Schwab, M. Wubbolts and A. Glieder, FEMS Yeast Res., 2004, 5, 179–189 CrossRef CAS PubMed.
- F. W. Krainer, C. Dietzsch, T. Hajek, C. Herwig, O. Spadiut and A. Glieder, Microb. Cell Fact., 2012, 11, 22–22 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc00783e |
| This journal is © The Royal Society of Chemistry 2022 |