DOI:
10.1039/D2GC00684G
(Paper)
Green Chem., 2022,
24, 3697-3703
Electrooxidative tricyclic 6–7–6 fused-system domino assembly to allocolchicines by a removable radical strategy†
Received
19th February 2022
, Accepted 9th March 2022
First published on 13th April 2022
Abstract
Natural allocolchicine and analogues derived thereof a tricyclic 6–7–6-system have been found as key scaffold of various biologically relevant molecules. However, the direct preparation of the allocolchicine motif remains difficult to date. Herein, we report on an electrooxidative radical cyclization of biarylynones with various carbon- and heteroatom-centered radical precursors via a sequential radical addition/7-endo-trig/radical cyclization domino reaction. This approach provides a step-economical and strategically novel disconnection for the facile assembly of a wide range of carbocyclic 6–7–6 fused ring systems. Remarkably, the sulfonyl group on the products could be easily removed by photocatalysis at room temperature with high yields.
Introduction
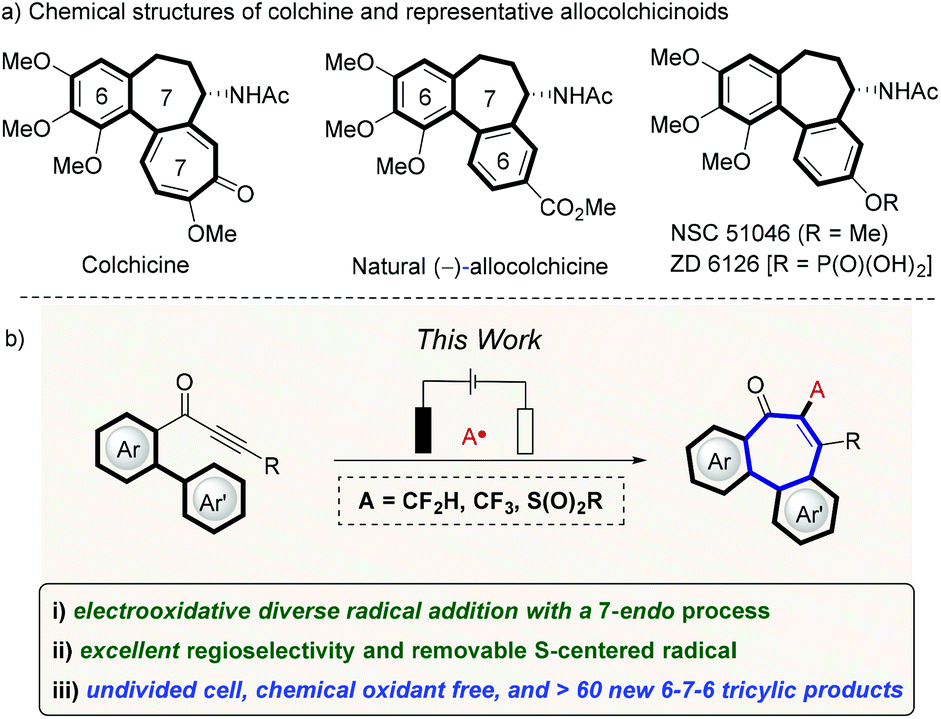
Seven-membered carbocycles are privileged structural motifs found in natural products and pharmaceutical compounds with important biological properties.1 Among these, the 6–7–6 benzo-fused rings has been found as a classic scaffold, which is present in natural allocolchicine and its analogues, such as ZD6126 and N-acetylcolchicinol methyl ether (NSC 51046) (Fig. 1a, top).2,3 Fortunately, in these synthetic colchicine derivatives, the 6–7–6 carbocyclic framework have promising anticancer bioactivities, but with reduced toxicity as compared to the 6–7–7 tricyclic system present in colchicine,4 which has translated into limitations in the treatment of human neoplasm and proved ineffective for therapeutic studies.5 Therefore, the development of modular approaches that provide a direct access to a variety of allocolchicine analogues continues to be in high demand. During the past decades, major momentum has been gained in the construction of such tricyclic frameworks, including enyne ring-closing metathesis/Diels–Alder approaches,6,7 palladium-catalyzed direct C–H arylations,8 intramolecular Nicholas reaction,9 and oxidative couplings,10 among others.11,12 In the meantime, the design of novel radical cascade cyclizations has emerged as an increasingly-powerful strategy to construct complex molecular scaffolds,13 as this approach generally features mild reaction conditions, high functional group tolerance and diverse viable radical precursors. Despite these indisputable advances, almost all radical sources will unfortunately leave behind an undesired chemical footprints, which jeopardizes the resource-economy towards the desired skeleton of the target products. Consequently, we wondered whether we could devise a removable radical cascade strategy by assembling the 6–7–6 tricyclic motif in a more straightforward manner. While we have recently reported on the construction of 6–7–6-system by a removable P-centered radical, the need for silver catalysts and stoichiometric amounts of chemical oxidants significantly limited this approach.14
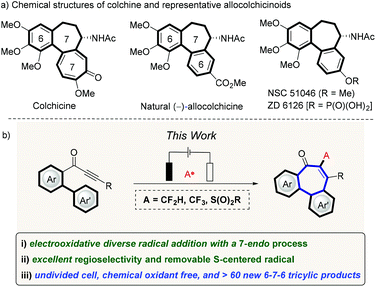 |
| | Fig. 1 Tricyclic 6–7–6-system construction (a) selected examples of bioactive 6–7–6 tricyclic compounds, (b) reaction design. | |
During the past few years, organic electrochemistry has been identified as an uniquely-effective and robust tool for the generation of reactive intermediates, such as radicals and radical ions, due to its inherent tunability.15,16 Based on our continued interest in electrochemical syntheses17 and radical formation using electricity as the sacrificial oxidant,18 we questioned whether the efficiency noted in a electrooxidative removable radical formation could be translated into a practical strategy towards 6–7–6 scaffolds. However, there are two key challenges. First, the regioselectivity of vinyl radical cyclization needs to be well controlled by adjusting the influence of substituents on the arene moiety. Compared to five- or six-membered ring formation, the radical cyclization of the seven-endo-trig approach is indeed rare.19 Second, the chemical footprints derived from various radical sources should be easily removable during the late-stage derivatization. We, herein, report on an unprecedented cascade cyclization of biaylynones under operationally-simple electrochemical conditions, which can be performed with commercially available, inexpensive radical precursors under water-tolerant radical reaction conditions (Fig. 1b, bottom). Notable features of our strategy include (a) the first Ts˙, CF2H˙ and CF3˙ addition to internal alkynes resulting in a 7-endo-trig process, (b) the thus-obtained S-motif can be removed traceless under operationally-simple conditions, (c) the absence of catalyst and chemical oxidants, (d) simple reaction conditions, and (e) ample substrate scope.
Results and discussion
Optimization of reaction condition
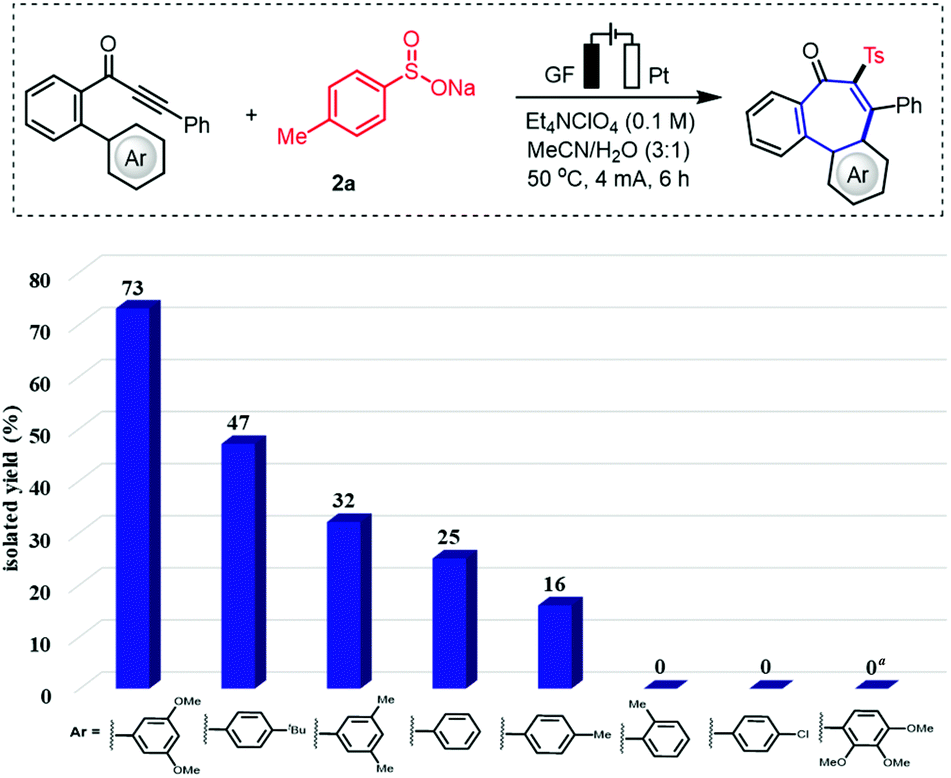
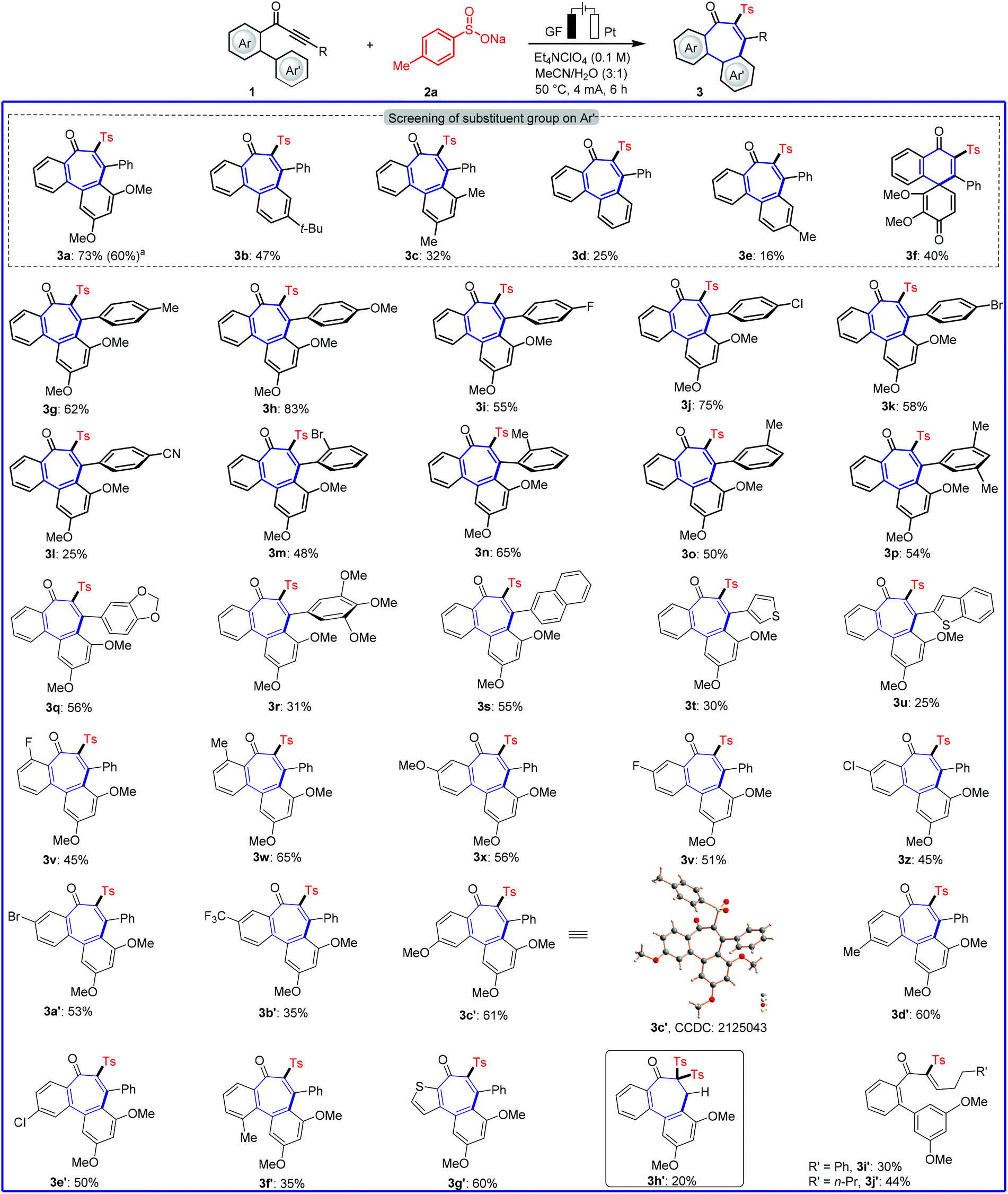
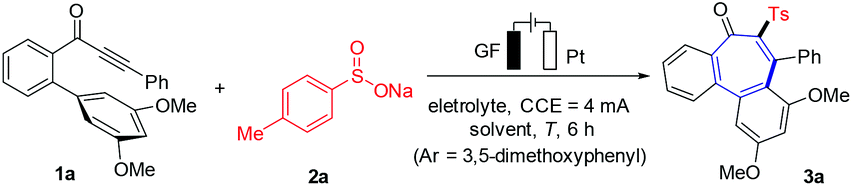
We initiated our studies by probing various reaction conditions for the envisioned domino cyclization of 1-(3′,5′-dimethoxy-[1,1′-biphenyl]-2-yl)-3-phenylprop-2-yn-1-one (1a) as the model substrate (Table 1 and Table S-2 in the ESI†), using the inexpensive p-toluenesulfinate (2a) as the sulfonyl radical source. After considerable preliminary experimentation, we observed that the desired 6–7–6 tricyclic fused product 3a was isolated in 73% yield with a mixed solvent system consisting of MeCN/H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and Et4NClO4 as the electrolyte (entry 1). Different solvents and a series of supporting electrolytes were tested, but showed not to be beneficial (entries 2–6, see also Table S-2 in the ESI†). Either decreasing or increasing the reaction temperature and the current failed to improve the yield of 3a (entries 7–9). During the optimization of the electrode material, it was found that the use of a nickel cathode, as well as a platinum anode led to a decrease in the yield or did not afford any product (entries 10 and 11). Control experiments confirmed the essential role of the electricity for the electrooxidative cyclization (entry 12). Not surprisingly, we observed a strong influence of the arene Ar′. Therefore, the substitution effect was examined under the optimized electrooxidative conditions. Thus, the type and position of the substituents on the arene moiety determines the efficacy of the 7-endo-trig cyclization process, while five- or six-membered products were not observed (Fig. 2 and Scheme 1). In contrast to other substituted arenes, a substrate bearing the 2,3,4-trimethoxy phenyl moiety featured high reactivity, but in this case the reaction followed a 6-exo-trig process yielding the non-aromatic product 3f (Scheme 1).
:1) and Et4NClO4 as the electrolyte (entry 1). Different solvents and a series of supporting electrolytes were tested, but showed not to be beneficial (entries 2–6, see also Table S-2 in the ESI†). Either decreasing or increasing the reaction temperature and the current failed to improve the yield of 3a (entries 7–9). During the optimization of the electrode material, it was found that the use of a nickel cathode, as well as a platinum anode led to a decrease in the yield or did not afford any product (entries 10 and 11). Control experiments confirmed the essential role of the electricity for the electrooxidative cyclization (entry 12). Not surprisingly, we observed a strong influence of the arene Ar′. Therefore, the substitution effect was examined under the optimized electrooxidative conditions. Thus, the type and position of the substituents on the arene moiety determines the efficacy of the 7-endo-trig cyclization process, while five- or six-membered products were not observed (Fig. 2 and Scheme 1). In contrast to other substituted arenes, a substrate bearing the 2,3,4-trimethoxy phenyl moiety featured high reactivity, but in this case the reaction followed a 6-exo-trig process yielding the non-aromatic product 3f (Scheme 1).
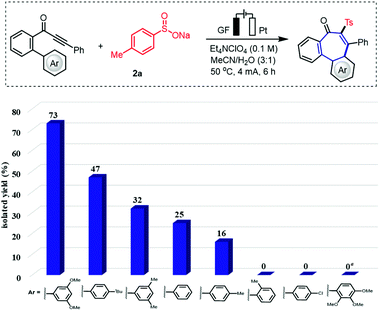 |
| | Fig. 2 Substituent impact on electrooxidative 7-endo radical cyclization process. Standard conditions: undivided cell, GF anode, Pt cathode, constant current (CCE) = 4 mA, 1 (0.3 mmol), 2a (0.6 mmol), Et4NClO4 (0.1 M, 0.4 mmol), MeCN/H2O (3:1, 4 mL), 50 °C, under air, 6 h. Yield of the isolated products.a6-exo product 3f formed in 40% yield. | |
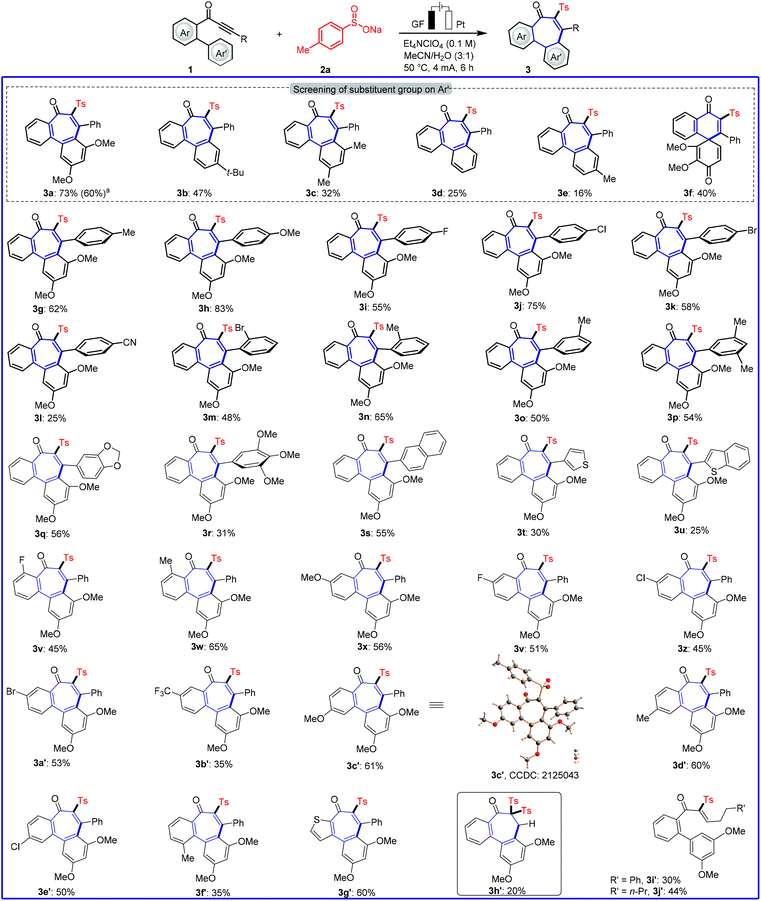 |
| | Scheme 1 Robustness of electrooxidative 7-endo-trig cyclization. Reaction conditions of Table 1, entry 1, alkynone (1, 0.30 mmol), 6 h. a1 mmol scale (5 mL solvent). | |
Table 1 Optimization of the cascade cyclization/sulfonylationa
|
|
| Entry |
Deviation from standard conditions |
Yield/% |
|
Standard conditions: undivided cell, graphite felt (GF) anode, Pt cathode, constant current = 4 mA, 1a (0.30 mmol), 2a (0.60 mmol, 2.0 equiv.), Et4NClO4 (0.1 M), MeCN/H2O (3:1, 4.0 mL), under air, 6 h, 3.0 F mol−1. Yield of the isolated product.
|
| 1 |
no change |
73 |
| 2 |
EtOH/H2O (3:1) |
0 |
| 3 |
DMF/H2O (3:1) |
0 |
| 4 |
1,4-Dioxane /H2O (1:1) |
39 |
| 5 |
DCE/MeCN/H2O (5:5:1) |
37 |
| 6 |
No electrolyte |
36 |
| 7 |
Reaction at 25 °C |
0 |
| 8 |
CCE = 3 mA |
32 |
| 9 |
CCE = 6 mA |
60 |
| 10 |
GF(+)|Ni(−) instead of GF(+)|Pt(−) |
54 |
| 11 |
Pt(+)|Pt(−) instead of GF(+)|Pt(−) |
0 |
| 12 |
No electricity |
0 |
Robustness
With the optimized reaction conditions in hand, we became interested in investigating the substrate scope of this electrooxidative radical cyclization, and we tested a diverse range of biarylyone substrates 1 with different substitution patterns (Scheme 1). The reaction tolerated a variety of substituents with diverse electronic properties in all positions of the arene R (3g–3l). We were pleased to find that ortho- and meta-substituted arenes underwent this transformation efficiently, despite a possible steric repulsion (3m–3r), even for polysubstituted substrates. Furthermore, heterocyclic substrates bearing thiophene and benzothiophene as well as naphthyl substituents were also tolerated in this transformation (3s–3u). Having demonstrated the broad applicability with respect to the arene, substitutions on the phenone scaffold were examined. Substrates decorated with both, electron-withdrawing and electron-donating groups on aryl ring had a significant effect on the yield of the reaction. Namely, electron-withdrawing groups somewhat blocked the reaction (3l and 3z–3b′). Furthermore, the reaction of substrate 1f′ was found with lower chemical yield (3f′), maybe due to a steric hindrance effect. A heterocyclic substrate proved also applicable in the electrooxidative transformation to selectively afford the corresponding product 3g′ in good yield. Noteworthily, also a terminal alkyne was applicable, and gave minor amounts of the products 3h′ after double radical addition. In sharp contrast, no desired product was obtained by the reaction of alkyl alkynes except for the uncyclized products (3i’–3j′). The connectivity of product 3c′ was unambiguously confirmed by single-crystal X-ray analysis (Scheme 1).20a
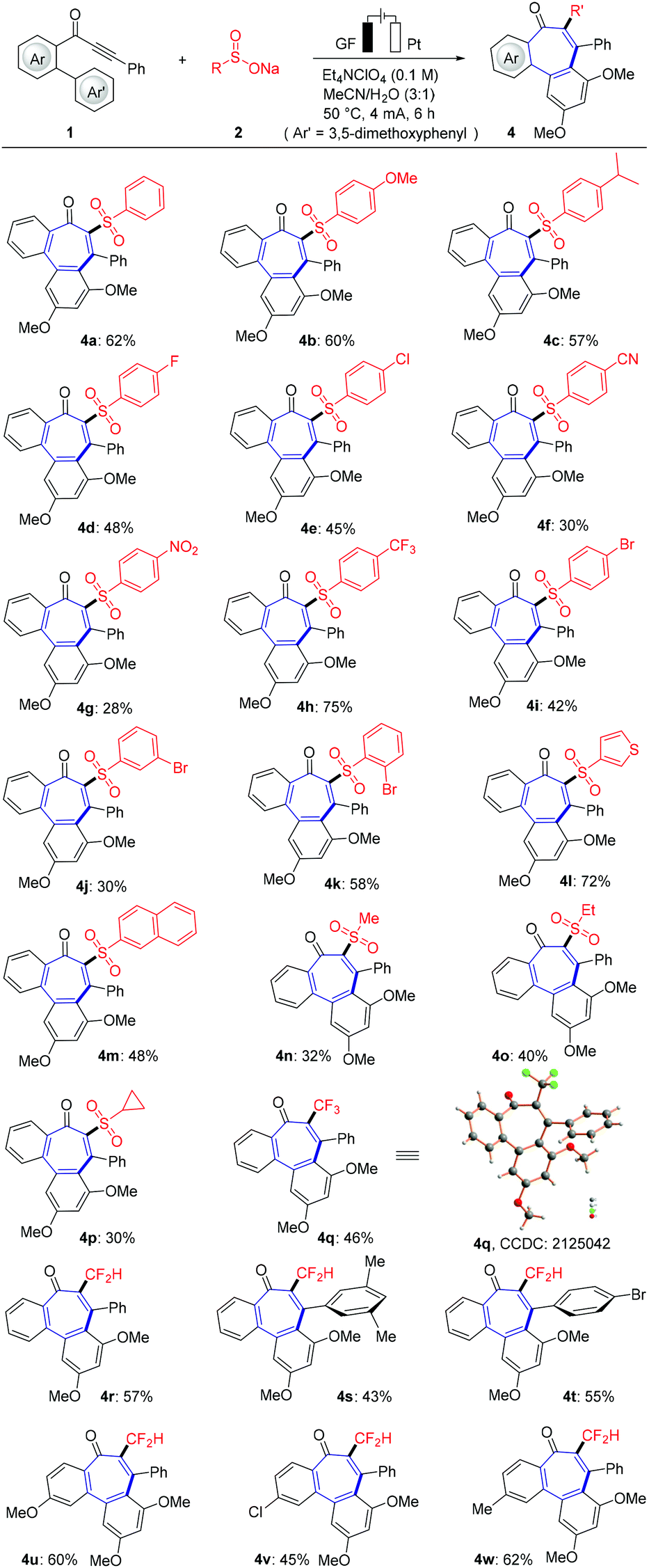
Next, the scope of various sodium sulfinates 2 with biarylynone 1 was examined to probe the efficacy of the present electrochemical domino cyclization (Scheme 2). To our delight, either common electron-donating substituents or electron-withdrawing functional groups (chloro, bromo, trifluoromethyl, nitro and cyano) showed good functional group tolerance by forming the desired product. In addition, naphthalene sulfonate and thiophene sulfonate also reacted well with substrate 1a to form tricyclic product in good yields (4l and 4m). Likewise, the mild electrooxidative radical cyclization approach was found to be generally applicable for aliphatic sodium sulfinates. CF3SO2Na (2r) and CF2HSO2Na (2s) were suitable substrates, furnishing the valuable tri- and difluoromethylated tricyclic 6–7–6 fused products (4q–4w). The structure of trifluoromethylated product 4q was unambiguously verified by X-ray crystallographic analysis.20b
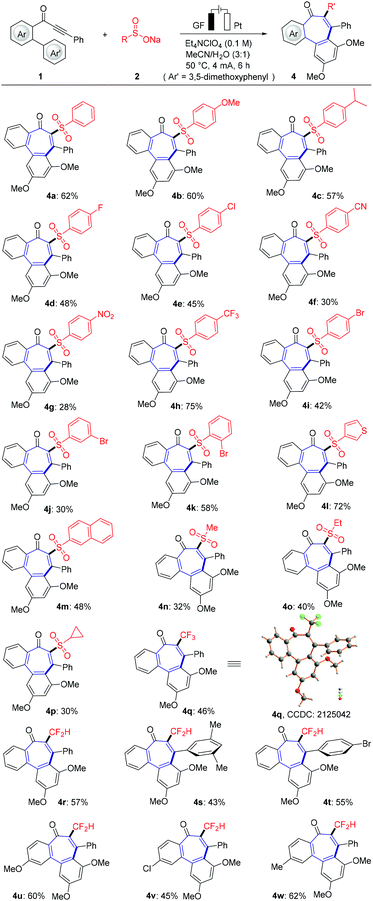 |
| | Scheme 2 Substrate scope of various sodium sulfonates 2. | |
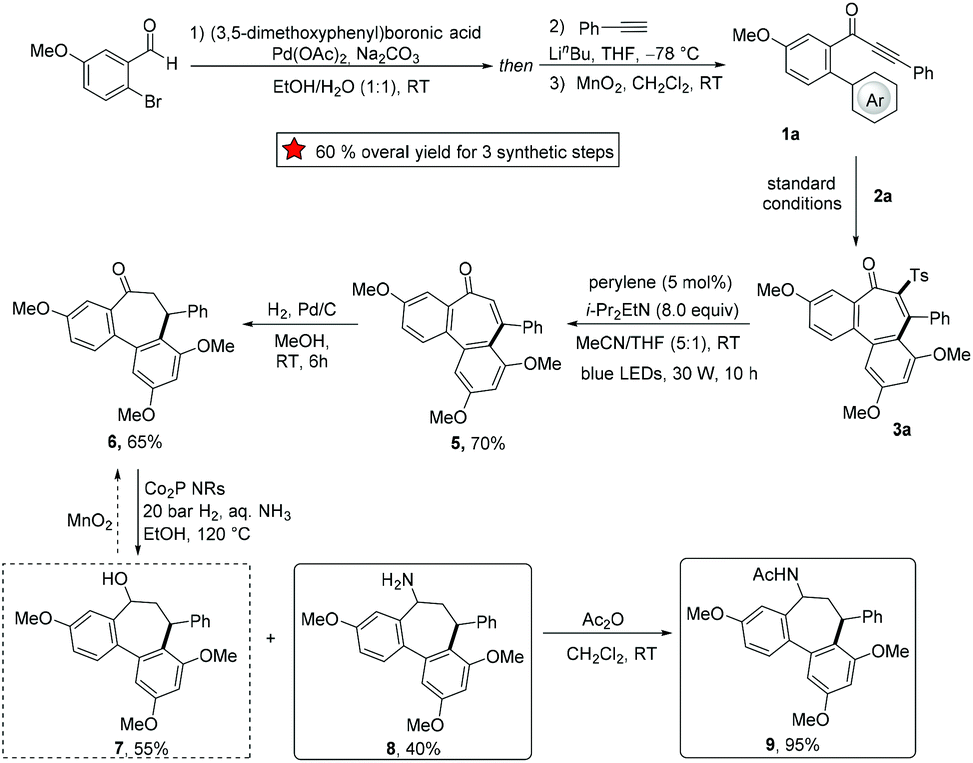
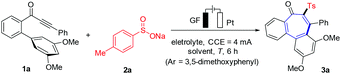
The synthetic utility of the developed domino strategy was further reflected by the efficient preparation of bioactive NSC 51046 analogues (Scheme 3). NSC 51046 is an analogue of the natural allocolchicine (Fig. 1a), which displays potent anticancer activity by inhibition of the tubulin polymerization.21 Due to the modularity of our electrosynthesis, this approach could be beneficial for preparing diverse derivatives of NSC 51046 from 3-phenyl-1-(3′,4,5′-trimethoxy-[1,1′-biphenyl]-2-yl)prop-2-yn-1-one 1a. Under the standard electrooxidative reaction conditions, the sulfonylation product 3a was obtained in high yield. Subsequently, 3a was converted to the desulfonylation product enone 5 following a perylene-catalyzed photodesulfonylation procedure.22 Then, catalytic hydrogenation of alkene 5 led to the formation of dibenzocycloheptanone 6, which provided the allocolchicinoid 9 after cobalt phosphide nanorods (Co2P NRs) catalyzed reductive amination23 followed by acetylation. Notably, though the reductive amination showed poor selectivity between amine 8 and alcohol 7 (7 was also formed in about 50% yield), 7 could be converted into ketone 6 by simple oxidation. Hence, our strategy opened a new avenue to a versatile synthesis of allocolchicine analogues with readily available starting materials and high efficacy.
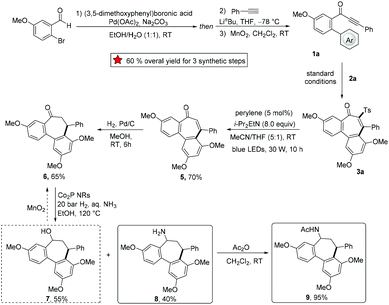 |
| | Scheme 3 NSC 51046 analogue synthesis (Ar = 3,5-dimethoxyphenyl). | |
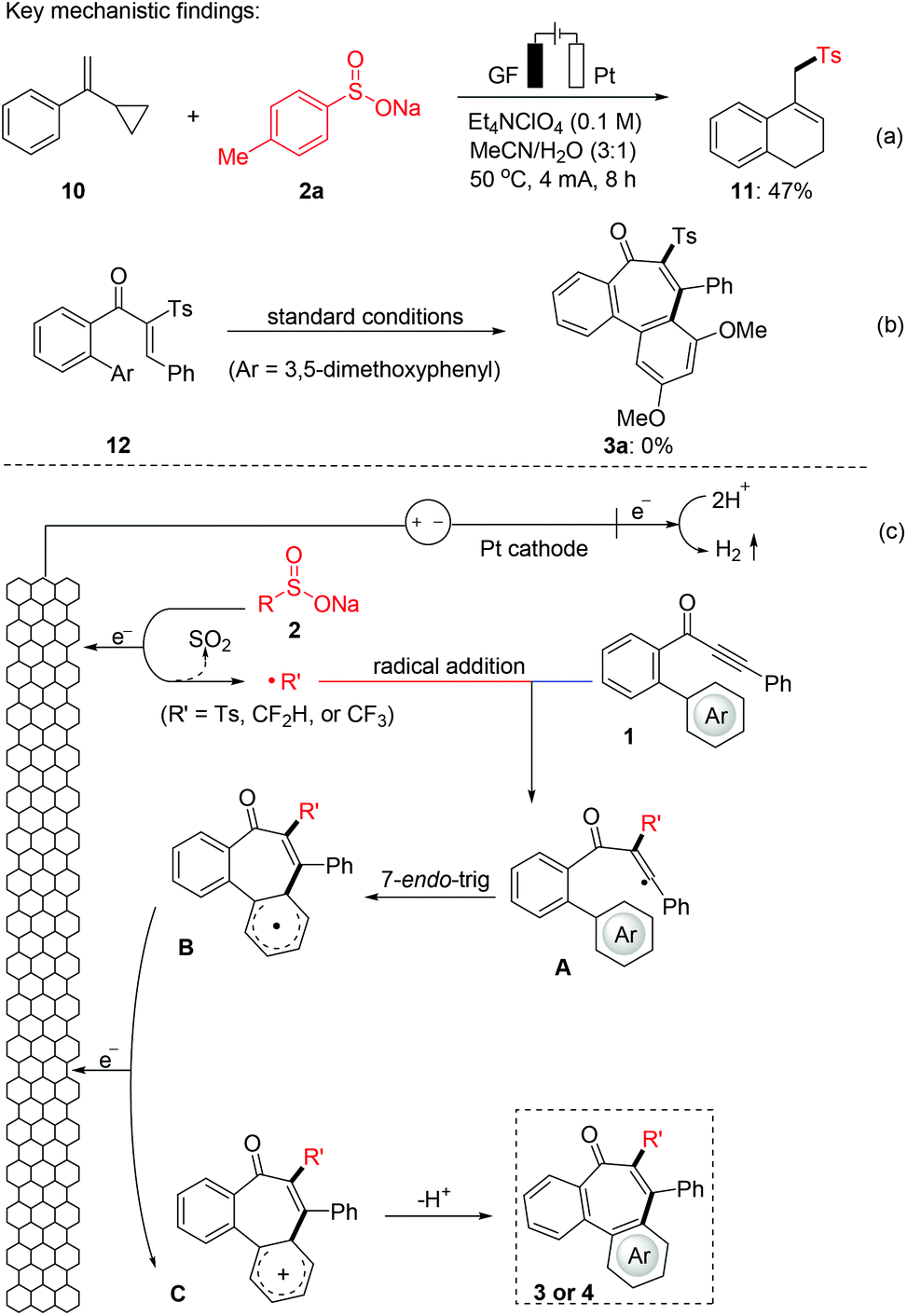
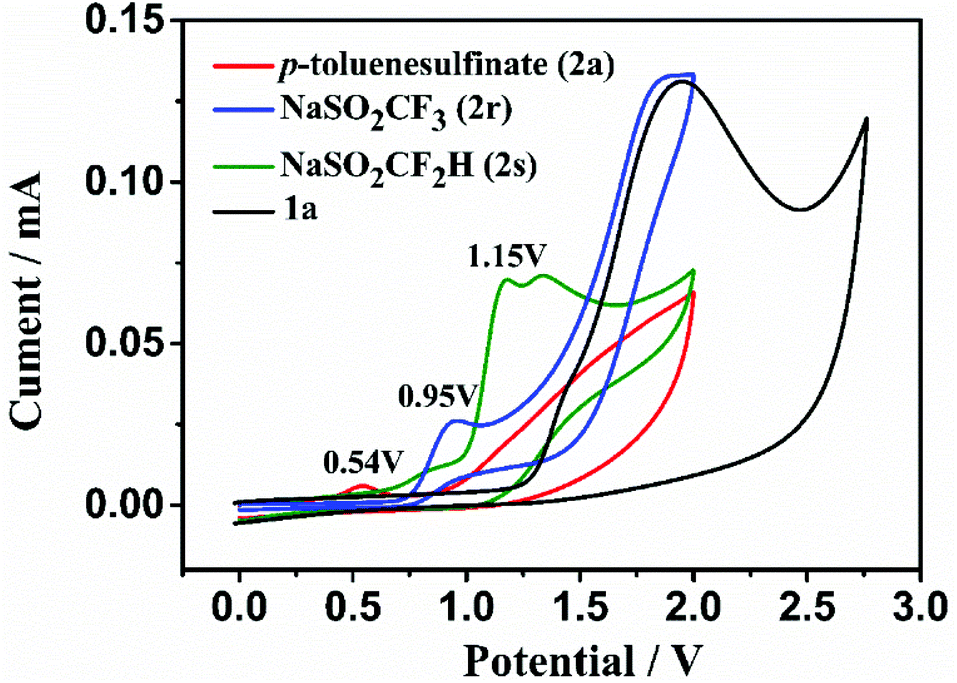
To gain mechanistic insight into this electrochemical radical addition/cyclization reaction, a radical clock reaction using (1-cyclopropylvinyl)benzene (10) provided the product 11 in 47% yield (Fig. 3a). In addition, when the direct addition product 12 (for detailed information, see the ESI†) was subjected to the standard electrochemical conditions, the cyclization product 3a was not obtained. Based on these experimental results and literature,24 a radical mechanism is proposed for this electrooxidative radical reaction as depicted in Fig. 3b. First, the S-radical, CF3-radical or CF2H-radical is generated from 2 through anodic oxidation. Selective radical addition of R′ to C–C triple bonds of 1 affords a vinyl radical A, which undergoes 7-endo-trig cyclization to intermediate B. Then, intermediate B undergoes further SET oxidation and deprotonation to form the product 3 or 4. In addition, we also tried to give the oxidation potential of the different radical sources in order to better understand the experimental results. Reactants 2a, 2r and 2s exhibit oxidation peaks at 0.54, 0.95 and 1.15 V vs. Ag/AgCl, respectively (Fig. 4 and Fig. S1 in the ESI†). These results indicate that these radical precursors are preferentially oxidized under anodic oxidation.
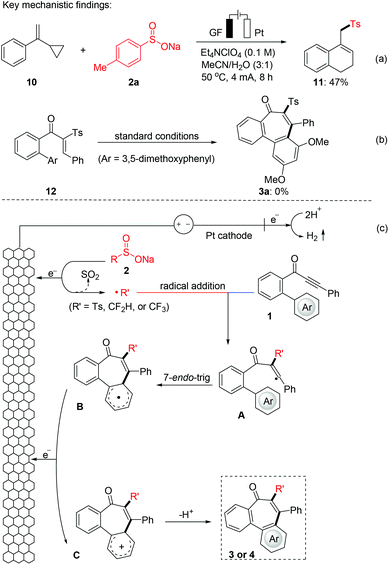 |
| | Fig. 3 Mechanistic investigation on the radical annulation (a) radical trapping experiment, (b) control experiment, and (c) proposed mechanism. | |
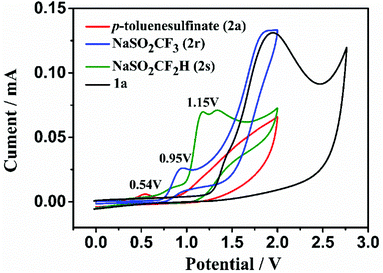 |
| | Fig. 4 Cyclic voltammetry studies. Conditions: cyclic voltammogram of target molecule (10 mM) in acetonitrile (10.0 mL containing 0.05 M n-Bu4NPF6) at room temperature, using a glassy carbon working electrode, a platinum wire counter electrode, and a Ag/AgCl reference electrode at a scan rate of 100 mV s−1. [black line for 1a, red line for 2a, blue line for 2r and green line for 2s]. | |
Conclusions
In summary, we have developed an electrochemical strategy for the construction of tricyclic 6–7–6-system through a domino radical addition of biarylynones with various radical sources. This environmentally-friendly approach showed high regioselectivity, ample substrate scope and high functional group compatibility. It is worth mentioning that the introduced sulfonyl radical fragment of the products was easily removed in the presence of photocatalyst to give the corresponding allocolchicine analogues. The developed electrooxidative strategy represents a rare 7-endo-trig vinyl radical cyclization processes and is a useful method for the concise assembly of a variety of novel and drug-type fused molecules bearing valuable 6–7–6-scaffolds.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Generous support by the Natural Science Foundation of Zhejiang Province (LY22B020001, LY20B020006), the National Natural Science Foundation of China (No. 21702188), the ERC and the DFG (Gottfried-Wilhelm-Leibniz award to LA) is gratefully acknowledged. We thank Dr Takato Mitsudome (Osaka University) for providing the catalyst Co2P NRs.
Notes and references
-
(a) I. Shiina, Chem. Rev., 2007, 107, 239–273 CrossRef CAS PubMed;
(b) M. Inoue, Chem. Rev., 2005, 105, 4379–4405 CrossRef CAS PubMed.
-
(a) T. Graening and H.-G. Schmalz, Angew. Chem., Int. Ed., 2004, 43, 3230–3256 CrossRef CAS PubMed;
(b) B. Pérez-Ramírez, M. J. Gorbunoff and S. N. Timasheff, Biochemistry, 1998, 37, 1646–1661 CrossRef PubMed;
(c) A. Brossi, J. Med. Chem., 1990, 33, 2311–2319 CrossRef CAS PubMed.
-
(a) F. Büttner, S. Bergemann, D. Guénard, R. Gust, G. Seitz and S. Thoret, Bioorg. Med. Chem., 2005, 13, 3497–3511 CrossRef PubMed;
(b) Q. Shi, K. Chen, X. Chen, A. Brossi, P. Verdier-Pinard, E. Hamel, A. T. McPhail, A. Tropsha and K.-H. Lee, J. Org. Chem., 1998, 63, 4018–4025 CrossRef CAS.
-
(a) N. S. Sitnikov and A. Y. Fedorov, Russ. Chem. Rev., 2013, 82, 393–411 CrossRef;
(b) P. D. Davis, G. J. Dougherty, D. C. Blakey, S. M. Galbraith, G. M. Tozer, A. L. Holder, M. A. Naylor, J. Nolan, M. R. L. Stratford, D. J. Chaplin and S. A. Hill, Cancer Res., 2002, 62, 7247–7253 CAS;
(c) Q. Shi, K. Chen, A. Brossi, P. Verdier-Pinard, E. Hamel, A. T. McPhail and K.-H. Lee, Helv. Chim. Acta, 1998, 81, 1023–1037 CrossRef CAS;
(d) O. Boyé, A. Brossi, H. J. C. Yeh, E. Hamel and B. Wegrzynski, Can. J. Chem., 1992, 70, 1237–1249 CrossRef.
-
(a) T. Beckers and S. Mahboobi, Drugs Future, 2003, 28, 767–785 CrossRef CAS;
(b) R. L. Hood, Colchicine poisoning, J. Emerg. Med., 1994, 12, 171–177 CrossRef CAS PubMed;
(c) S. T. Milne and P. D. Meek, Am. J. Emerg. Med., 1998, 16, 603–608 CrossRef CAS PubMed;
(d) R. M. Naidus, R. Rodvien and C. H. Mielke, Arch. Intern. Med., 1977, 137, 394–396 CrossRef CAS PubMed.
- F.-D. Boyer and I. Hanna, Org. Lett., 2009, 9, 715–718 CrossRef PubMed.
- A. V. Vorogushin, A. V. Predeus, W. D. Wulff and H.-J. Hansen, J. Org. Chem., 2003, 68, 5826–5831 CrossRef CAS PubMed.
- M. Leblanc and K. Fagnou, Org. Lett., 2005, 7, 2849–2852 CrossRef CAS PubMed.
-
(a) S. Djurdjevic, F. Yang and J. R. Green, J. Org. Chem., 2010, 75, 8241–8251 CrossRef CAS PubMed;
(b) S. Djurdjevic and J. R. Green, Org. Lett., 2007, 9, 5505–5508 CrossRef CAS PubMed.
- G. Besong, K. Jarowicki, P. J. Kocienski, E. Sliwinski and F. T. Boyle, Org. Biomol. Chem., 2006, 4, 2193–2207 RSC.
- W. M. Seganish and P. DeShong, Org. Lett., 2006, 8, 3951–3954 CrossRef CAS PubMed.
-
(a) The 6–7–6 tricyclic motif can also be achieved by the radical annulation process using excess Mn(OAc)3, see: L. Zhou, Y. Xia, Y.-Z. Wang, J.-D. Fang and X.-Y. Liu, Tetrahedron, 2019, 75, 1267–1274 CrossRef CAS;
(b) Y. Chen, C. Huang, X. Liu, E. Perl, Z. Chen, J. Namgung, G. Subramaniam, G. Zhang and W. H. Hersh, J. Org. Chem., 2014, 79, 3452–3464 CrossRef CAS PubMed.
- For reviews on radical chemistry, see:
(a) H. Xiao, Z. Zhang, Y. Fang, L. Zhu and C. Li, Chem. Soc. Rev., 2021, 50, 6308–6319 RSC;
(b) H. Zhou, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, ACS Catal., 2021, 11, 7978–7986 CrossRef CAS;
(c) M. Latrache and N. Hoffmann, Chem. Soc. Rev., 2021, 50, 7418–7435 RSC;
(d) D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS PubMed;
(e) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed;
(f) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed;
(g) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed;
(h) M.-C. Belhomme, T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2015, 21, 12836–12865 CrossRef CAS PubMed;
(i) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 114, 2587–2693 CrossRef PubMed;
(j) F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed;
(k) U. Wille, Chem. Rev., 2013, 113, 813–853 CrossRef CAS PubMed;
(l) P. Chen and G. Liu, Synthesis, 2013, 2919–2939 CAS.
- Y. Zhang, Z. Cai, J. Struwe, C. Ma, W. Zeng, X. Liao, M. Xu and L. Ackermann, Chem. Sci., 2021, 12, 15727–15732 RSC.
-
A. Scheremetjew, T. H. Meyer, Z. Lin, L. Massignan and L. Ackermann, Fundamental Principles of Organic Electrochemistry, in Science of Synthesis: Electrochemistry in Organic Synthesis, ed. L. Ackermann, Thieme, Stuttgart, 2021, pp. 3–32. DOI:10.1055/sos-SD-236-00002.
- For reviews on electrochemical synthesis, see:
(a) P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed;
(b) C. Xu, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C.-C. Zeng, CCS Chem., 2022, 4, 1120–1152 CrossRef;
(c) C. Ma, P. Fang, D. Liu, K.-J. Jiao, P.-S. Gao, H. Qiu and T.-S. Mei, Chem. Sci., 2021, 12, 12866–12873 RSC;
(d) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed;
(e) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed;
(f) L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed;
(g) G. M. Martins, G. C. Zimmer, S. R. Menders and N. Ahmed, Green Chem., 2020, 22, 4849–4870 RSC;
(h) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed;
(i) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed;
(j) Q.-L. Yang, P. Fang and T.-S. Mei, Chin. J. Chem., 2018, 36, 338–352 CrossRef CAS;
(k) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC;
(l) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS;
(m) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed;
(n) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC;
(o) H.-C. Xu, J. M. Campbell and K. D. Moeller, J. Org. Chem., 2014, 79, 379–391 CrossRef CAS PubMed;
(p) A. Jutand, Chem. Rev., 2008, 108, 2300–2347 CrossRef CAS PubMed.
- Y. Zhang, J. Struwe and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 15076–15080 CrossRef CAS PubMed.
-
(a) Y. Zhang, C. Ma, J. Struwe, J. Feng, G. Zhu and L. Ackermann, Chem. Sci., 2021, 12, 10092–10096 RSC;
(b) Y. Zhang, Z. Lin and L. Ackermann, Chem. – Eur. J., 2021, 27, 242–246 CrossRef CAS PubMed.
-
(a) P. Xiong, H.-H. Xu, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 2460–2464 CrossRef CAS PubMed;
(b) Y. Li and J.-H. Li, Org. Lett., 2018, 20, 5323–5326 CrossRef CAS PubMed.
-
(a) CCDC 2125043;†;
(b) CCDC 2125042.†.
- S. G. Davies, A. M. Fletcher, P. M. Roberts, J. E. Thomson and A. Yeung, J. Nat. Prod., 2019, 82, 2659–2663 CrossRef CAS PubMed.
- H. Watanabe, M. Takemoto, K. Adachi, Y. Okuda, A. Dakegata, T. Fukuyama, I. Ryu, K. Wakamatsu and A. Orita, Chem. Lett., 2020, 49, 409–412 CrossRef CAS.
- M. Sheng, S. Fujita, S. Yamaguchi, J. Yamasaki, K. Nakajima, S. Yamazoe, T. Mizugaki and T. Mitsudome, JACS Au, 2021, 1, 501–507 CrossRef CAS PubMed.
-
(a) J. Liu, M. Wang, L. Li and L. Wang, Green Chem., 2021, 23, 4733–4740 RSC;
(b) V. K. K. Pampana, V. P. Charpe, A. Sagadevan, D. K. Das, C.-C. Lin, J. R. Hwu and K. C. Hwang, Green Chem., 2021, 23, 3569–3574 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all new compounds and details for DFT calculations. CCDC 2125042 and 2125043. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2gc00684g |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Chanchan
Ma
a,
Zhenzhi
Cai
a,
Julia
Struwe
b,
Shengjie
Chen
a,
Jinming
Xu
a,
Shiyin
Li
a,
Wangyu
Zeng
a and
Lutz
Ackermann
*a,
Chanchan
Ma
a,
Zhenzhi
Cai
a,
Julia
Struwe
b,
Shengjie
Chen
a,
Jinming
Xu
a,
Shiyin
Li
a,
Wangyu
Zeng
a and
Lutz
Ackermann