
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On-demand, in situ, generation of ammonium caroate (peroxymonosulfate) for the dihydroxylation of alkenes to vicinal diols†
Benjamin J.
Deadman‡
 a,
Sarah
Gian
a,
Violet Eng Yee
Lee
a,
Luis A.
Adrio
a,
Klaus
Hellgardt
b and
King Kuok (Mimi)
Hii
*a
a,
Sarah
Gian
a,
Violet Eng Yee
Lee
a,
Luis A.
Adrio
a,
Klaus
Hellgardt
b and
King Kuok (Mimi)
Hii
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, 82 Wood Lane, London W12 0BZ, UK. E-mail: mimi.hii@imperial.ac.uk
bDepartment of Chemical Engineering, Imperial College London, Exhibition Road, South Kensington, London SW7 2AZ, UK
First published on 21st June 2022
Abstract
Using the dihydroxylation of alkenes as a benchmark, the reactivities of fresh and aged solutions of (NH4)2S2O8 (electrochemically generated) were compared to commercially-procured peroxydisulfate and Oxone. The study revealed that peroxymonosulfate (Caro's acid, PMS) is the active oxidant in such reactions. Using complementary redox colorimetry and in situ IR spectroscopy, the decomposition of peroxydisulfate in an acidic solution into PMS and H2O2 can be quantified for the first time. The new insights enabled the design and implementation of both batch and flow processes to maximise the concentration of active PMS oxidant. The utility of these oxidants for organic synthesis is demonstrated by the dihydroxylation of eight styrenes and seven alkyl alkenes, where the ammonium PMS solutions performed better than Oxone (counterion effect). Last but not least, a chromatography-free method for isolating and purifying the water-soluble diol product was developed.
Introduction
Oxidation is one of the most important and widely employed processes for upgrading petrochemical feedstocks to more valuable chemicals.1 A quintessential example is the conversion of alkenes to 1,2-diols (a.k.a. vicinal alcohols or glycols), which are a class of highly valuable chemicals widely used in the production of personal care, food and medicinal products. This can be achieved by the reaction of the alkene with a reactive oxidant to generate an epoxide intermediate, that can undergo ring-opening under acidic or basic conditions to the diol (Scheme 1).2,3 | ||
| Scheme 1 ‘Green’ synthetic routes for the dihydroxylation of alkenes. | ||
In principle, O2 gas is the ideal oxidant in terms of its availability, atom economy and sustainability. However, implementation of aerobic processes can have unique challenges, not least of all, the management of solubility and flammability limits of O2 gas in organic solvents.4 Conversely, solid (e.g. mCPBA, bromates, periodates, peroxysulfates) or liquid (e.g. perchloric acid, H2O2) oxidants are also frequently employed. However, these are inherently reactive materials which can be hazardous to transport, store and use, and have been responsible for laboratory accidents in recent years.5–7 In an analysis of accidents that occurred in the Chinese Chemical Industry between 2006–2017, it was reported that 340 (>8%) ‘casualty accidents’ were associated with oxidants and organic peracids.8 Explosions of organic peroxides at the Arkema plant in Texas during the tropical storm ‘Harvey’ in 20179 and the devastation caused by the explosion of ammonium nitrate in Beirut in 202010,11 have also highlighted, in a very public way, the dangers of stockpiling large volumes of oxidants. Even before these events, the ACS Green Chemistry Institute has championed the need for environmentally friendlier and safer oxidation methodologies avoiding organic peroxides and halogenated solvents,12 which, in turn, has generated considerable interest in the development of green oxidation methods.13 From a wider perspective, the post-reaction treatment is also a significant issue. In an authoritative review by Roger Sheldon and industrial co-authors,1 it was stated: “A truly holistic discussion, in which not only the nature of the oxidant, but also the workup of the product, the recycling of solvents and catalysts, and the disposal of byproducts and wastewater are considered, has very seldom been done in an academic context”.
Peroxysulfates are widely employed in advanced oxidation processes (AOPs) for environmental remediation. Compared to most oxidants that generate only ˙OH radicals, sulfate-based AOPs can be activated in different ways to generate a variety of oxidants (radical and non-radical), which can be used to target the treatment of specific pollutants.14 Two types of persulfate oxidants are commonly employed in organic synthesis: Peroxydisulfate (S2O82−, PDS), available as a sodium, potassium or ammonium salt, is capable of oxidizing virtually all functional groups, even hydrocarbons.15,16 Conversely, peroxymonosulfate (SO52−, PMS), supplied commercially as a triple salt of potassium (2KHSO5·KHSO4·K2SO4) under the tradename ‘Oxone’, is used for highly selective oxidative transformations in organic synthesis.17 The conjugate peroxymonosulfuric acid (a.k.a. Caro's acid, H2SO5), is known to be an extremely hazardous oxidant, prone to explode spontaneously.5
In 2008, Page, Marken and co-workers first described the use of electrochemically-generated percarbonate and persulfate oxidants for the asymmetric epoxidation of an alkene (1-phenylcyclohexene).18 Employing a boron-doped diamond (working) anode and a Pt cathode in an undivided electrochemical cell, a constant potential of 5 V was applied to convert sulfuric acid into PDS, which was deployed for the epoxidation reaction after adjusting the pH to between 7–9. It was suggested that PMS may be the active oxidant under these conditions.
Previously, we have described the design and construction of an electrochemical flow reactor to generate an aqueous solution of PDS from ammonium sulfate, which was deployed directly in a baffled reactor to convert styrene into the corresponding 1,2-diol.19 In contrast to the earlier work by Page and Marken, a divided cell was deployed to oxidize a mixture of sulfuric acid and ammonium hydrogen sulfate to (NH4)2S2O8 in the anodic chamber over boron-doped diamond. By performing the electrolysis in a batch-recycle flow process, higher current efficiencies can be achieved, as the flowing liquid replenishes the electroactive layer on the electrode's surface, whilst also purging it of O2 bubbles.20 This is an appealing approach for the development of a sustainable oxidation process. As the oxidant can be produced and deployed as needed (‘on-demand’), the amount of hazardous inventory can be minimized, thus eliminating explosive hazards associated with handling large amounts of the highly reactive oxidant.
In this paper, we report the results of a study on the identification of the oxidant(s) that are thermally generated from PDS in an acidic solution, and new protocols to improve the productivity and sustainability of the alkene epoxidation reaction using this oxidant, including a method for extracting the amphiphilic diol product from the reaction mixture, without using chromatography or a large quantity of organic solvents.
Results and discussion
The work began with a comparison of the reactivities of commercially-procured and electrochemically-generated (NH4)2S2O8, with Oxone (active oxidant SO52−), using the dihydroxylation of styrene as a model reaction (Scheme 1). Initial investigations were performed at 40 °C, in a round-bottomed flask fitted with an overhead mechanical stirrer to provide good mixing between the aqueous and organic phases. The reactions were initiated by the addition of the ‘neat’ alkene (without solvent) to the aqueous oxidant and the reaction progresses were monitored by HPLC analysis of extracted reaction aliquots (Scheme 2 and Fig. 1). | ||
| Scheme 2 Comparing PDS and PMS in the hydroxylation of styrene to 1-phenyl-1,2-diol. | ||
 | ||
| Fig. 1 Dihydroxylation of styrene by acidic solutions of (NH4)2S2O8, Oxone or thermally activated (NH4)2S2O8. Reaction conditions: Oxidant (50 mmol), styrene (25 mmol, 2.9 mL), aq. H2SO4 (2 M, 50 mL), 40 °C, 500 rpm. | ||
The comparison revealed Oxone as a more active oxidant than PDS in the dihydroxylation reaction (Fig. 1, triangle vs. square markers). Combining this observation with the presence of an induction period in the reaction using PDS, led us to speculate PMS as the active oxidant, which is known to be formed by the thermal decomposition of PDS.21
To test the hypothesis, an acidic solution of (NH4)2S2O8 was thermally treated at 40 °C overnight. The reaction performed using this ‘activated PDS’ solution was not only faster than that performed with the unactivated solution, but also outperform that conducted with Oxone without an induction period (Fig. 1, circle vs. square and triangle markers), thus confirming that the active oxidant is generated thermally.
Conversion of PDS to PMS: an in situ kinetic assessment
The aqueous chemistry of PDS has been extensively studied, largely for applications in wastewater treatment,14 where it is known that the peroxide bond in PDS can be activated by physical (e.g. thermal, UV light) or chemical (e.g. alkaline, transition metals) means.22 To date, however, the vast majority of these studies are conducted in either neutral or basic pH's, where PDS is known to dissociate homolytically into sulfate radical anions (SO4˙−), which act as a strong oxidant.In contrast, there are very few studies of the decomposition of PDS in strongly acidic solutions (pH < 3). One of the earlier reports dates back to 1951 (Scheme 3),23 when Kolthoff and Miller proposed that the protonolysis of the PDS lead to the disproportionation of PDS to hydrogensulfate (HSO4−) and an unstable sulfur tetroxide (SO4). The latter reacts rapidly with water to form Caro's acid, H2SO5 (eqn (1)–(3)), which will eventually break down to H2O2 and O2 (Scheme 2, eqn (4) and (5)). In the intervening seven decades, there had only been sporadic reports where the kinetics of the PDS decomposition to H2O2 and O2 were examined in an acidic medium, using polarography,23 redox titrations21,24 (most commonly by iodometry25,26), gasometry,27 and ESR.28 The involvement and the stability of the PMS intermediate was never discussed in detail in any of these earlier reports. This may be due to the lack of in situ analytical methods that can distinguish between the different oxidants (PDS, PMS and H2O2), especially when they are all present in a mixture.
 | ||
| Scheme 3 Proposed pathways for the generation and decomposition of PMS in an acidic aqueous solution. | ||
At this juncture, we proposed that PMS is the only active oxidant for the dihydroxylation reaction of styrene (Fig. 1). To prove this, we set out to delineate the kinetics of these reactions. In an earlier report, we have developed redox colorimetric methods to rapidly determine [PMS] and [H2O2] in aqueous solutions directly, where [PDS] is derived from the difference between a series of colorimetric tests.29 Subsequently, we found that IR spectroscopy can, indeed, be employed as an effective in situ analytical tool to study the decomposition of PDS to PMS directly, as each of these oxidants can be detected by their diagnostic IR absorbances at 1260 cm−1 (PDS) and 760 cm−1 (PMS), respectively (Fig. 2).
 | ||
| Fig. 2 Monitoring the decomposition of PDS at 40 °C over 20 h, using in situ IR spectrometry. The decreasing peak at 1260 cm−1 is attributed to PDS while the increasing peak at 760 cm−1 is attributed to PMS. | ||
Thermal stability of PDS
Consequently, IR spectroscopy and redox colorimetry can be deployed in a complementary manner to study the decomposition of PDS in an acidic solution at 40, 50 and 60 °C (Fig. 3 and Fig. S1, ESI†). At 40 °C, the decomposition of 0.94 M PDS proceeded slowly over 20 h, reaching a maximum concentration, [PMS]max, of 0.94 M. It is interesting to note that PMS is relatively stable at this temperature; an effective [PMS] of >0.87 M can be maintained, even after 46 h. By raising the temperature to 50 °C, a [PMS]max of 0.92 M can be attained after 5.5 h. The thermal stability of PMS observed at 40 and 50 °C is particularly notable as it has never been reported before. Further elevating the temperature to 60 °C, however, triggered the competitive decomposition of PMS to H2O2. In this case, an optimal [PMS]max of 0.89 M was achieved in just over 2 h (123 min), beyond which it started to decompose, such that only <0.15 M PMS and 0.41 M H2O2 remained in a solution after 23 h (Fig. S1, ESI†). The decomposition is also visible by the formation of O2 gas bubbles within the reaction vessel. At higher pH's (>6), the decomposition of Caro's acid into O2 is known to occur spontaneously via a radical mechanism.30,31 In this case, the thermal decomposition of PDS to O2 in an acidic solution is believed to occur via H2O2 (eqn (4) and (5), Scheme 2), which was detected as an intermediate in these experiments. | ||
| Fig. 3 The conversion of PDS (Δ) to PMS (○) in 2 M H2SO4 solutions at (A) 40 °C, (B) 50 °C and (C) 60 °C, by in situ IR spectroscopy. The [PDS] + [PMS] plots (■) indicates the presence of an unknown transient intermediate. | ||
The ability to monitor the reaction in situ also allowed us to detect the presence of another intermediate, revealed by the delayed onset of PMS formation following the consumption of PDS. The evolution of this intermediate can be tracked by plotting the sum of [PDS] and [PMS], which was observed to decrease in the early stages of the reaction, before recovering to the expected value (Fig. 3, filled squares). This appears to support the involvement of sulfur tetroxide (Scheme 2, eqn (2)), although currently we have no means of identifying the structure of this transient species.
Subsequently, the decomposition of PDS was modelled as a pseudo first-order reaction, fitted to the experimental data to obtain rate constants (Table 1 and Fig. S2, ESI†). The resultant Arrhenius plot provided an activation energy of 99.2 ± 0.2 kJ mol−1, which is slightly lower than reported values. This is attributed to the strongly acidic conditions, as well as the lower stability of the ammonium salt of PDS, compared to the corresponding potassium and sodium salts,32 which were previously reported in the range of 100–110 kJ mol−1 (Table 2).
| Temperature (°C) | k/min−1 | RMS of fit |
|---|---|---|
| 40 | 2.81 × 10−3 | 0.01102 |
| 50 | 9.61 × 10−3 | 0.01949 |
| 60 | 2.82 × 10−2 | 0.04840 |
Generation of active PMS oxidant in continuous flow (residence time control)
The kinetic information allows us to design a process to optimize the thermal conversion of PDS to PMS. A tubular flow reactor system was chosen for the residence-time sensitive process, with a high surface area-to-volume ratio to enable efficient heat transfer; this was subsequently constructed from an HPLC pump, PTFE tubing and a back-pressure regulator (Fig. 4). The conversion to PMS was achieved by pumping the acidic PDS solution (1 M) through the coiled tube reactor held at 70, 80 or 90 °C (maintained by immersion in an oil bath). The residence time was varied between 1 min and 1 h by adjusting the flow rate of the feed pump. To ensure rapid heat removal after the reaction, a heat exchanger was created by wrapping a second coil of PTFE tubing around a water condenser (Fig. 4). Samples were collected across the residence time range, and immediately analyzed by redox colorimetry (Table 3).29 | ||
| Fig. 4 Design of the continuous flow reactor for effective residence time control. | ||
| Temperature (°C) | τ (min) | [PMS] (±0.05 M) | [H2O2] (±0.01 M) | [Ox]tot (±0.02 M) |
|---|---|---|---|---|
| 70 | 20 | 0.82 | 0.03 | 0.85 |
| 80 | 10 | 0.75 | 0.05 | 0.83 |
| 90 | 5 | 0.77 | 0.05 | 0.74 |
Eventually, a [PMS]max of 0.82 M can be obtained by treatment at 70 °C with a residence time of 20 min. Operating the flow reactor at 80 and 90 °C reduced the residence time to 10 and 5 min respectively, with a corresponding reduction in [PMS] and total oxidant concentration ([Ox]tot). During these reactions, the formation of O2 bubbles can be observed in the reaction channels; suggesting significant competitive decomposition of PMS at these elevated temperatures.
Thus, two different procedures have been developed for the generation of peroxymonosulfate (PMS): Using a batch reactor, the thermal treatment of the PDS solution at 50 °C for 6 h results in very high conversion of PDS to PMS (0.92 M). Alternatively, the PMS solution can be produced, continuously, from PDS at 70 °C with a controlled residence time of 20 min. Using the tubular flow reactor, up to 0.82 M of PMS can be produced at a rate of 24.6 mmol h−1. The ammonium PMS solutions prepared by these methods is up to 10 times more concentrated than that which can be generated from the less soluble potassium salt,33 and yet is stable and safe to be stored, as an aqueous solution, at 4 °C over several weeks without discernible degradation.
Dihydroxylation of alkenes
Peroxymonosulfate (PMS) is the active ingredient in commercially available Oxone. Aqueous solutions of Oxone react with alkenes to produce epoxides at near neutral or buffered solutions.34,35 The reaction is believed to proceed via an electrophilic O-transfer (Scheme 4).36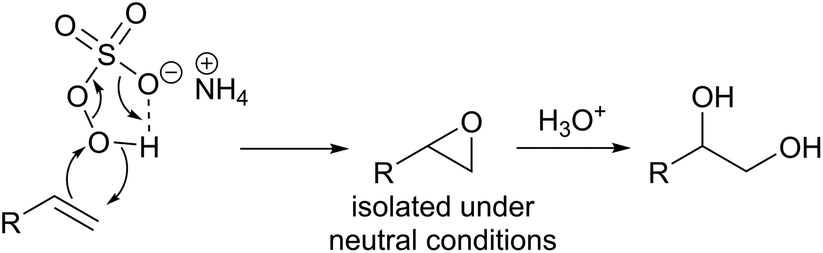 | ||
| Scheme 4 Electrophilic O-transfer to produce an epoxide and subsequent ring-opening to form 1,2-diols. | ||
As the ammonium PMS solution was generated in a strongly acidic solution, it is expected to produce 1,2-diols, without a separate hydrolysis step. To demonstrate this, the activated PMS solution was utilized in the dihydroxylation of a number of aryl-conjugated alkenes (Table 4). The experiments were conducted on a small scale in sealed vials, where the addition of acetonitrile co-solvent was necessary to improve the emulsification of the organic substrate with the oxidant present in the aqueous phase. The alkenes are converted to polar 1,2-diols (epoxides were not observed), the biphasic system often turned homogeneous, providing an added visual aid to reaction progress. With this in mind, the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values of the precursors are also included in the comparisons of the results, to identify any correlation between reaction yield and lipophilicity of the substrates.
P values of the precursors are also included in the comparisons of the results, to identify any correlation between reaction yield and lipophilicity of the substrates.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | logPb |
t (h) | Yieldc (%) |
| a Reactions were conducted in sealed glass vials: aq. PMS solution (5 mL, 4.7 mmol active oxidant), alkene (2.35 mmol), acetonitrile (1.66 mL), 50 °C. b Pubchem (release 2019.06.18).37 c Isolated yields after purification. | ||||
| 1 |

|
2.9 | 2.5 | 80 |
| 2 | Z = CH3, 1b | 3.6 | 3 | 91 |
| 3 | Z = OMe, 1c | 3.1 | 3 | 87 |
| 4 | Z = Cl, 1d | 3.7 | 5 | 72 |
| 5 | Z = F, 1e | 2.8 | 3 | 71 |
| 6 |

|
3.4 | 6 | 84 |
| 7 |
|
3.5 | 3 | 84 |
| 8 |

|
2.9 | 21 | 83 |
| 9 |

|
4.8 | 21 | 0a |
| 10 |

|
4.8 | 5 | 0a |

Under the specified conditions, the dihydroxylation of the model styrene substrate can be achieved with 80% yield (Table 4, entry 1). In line with the expectations for electrophilic (ep)oxidation reactions, the presence of electron-donating groups enhances the yield (entries 2, 3 and 6), while electron-withdrawing groups has the opposite effect (entries 4 and 5). Likewise, the dihydroxylation of the 1,1-disubstituted α-methylstyrene produced the corresponding 1,2-diol 2g in good yield (entry 7), and the oxidation of the cyclic indene (1h) can be achieved with a good yield, albeit requiring 21 h (entry 8). In this case, only the trans-diol product was obtained; consistent with a reaction pathway that proceeds via an epoxide intermediate, which hydrolyses in the acidic solution, in an SN2 fashion, to the trans-1,2-diol. In contrast, attempted dihydroxylation of trans- (entry 10) and cis-stilbenes (entry 9) only afforded recovered starting material in both cases.
With this set of substrates, there appears to be no clear correlation between the lipophilicity (logP) of the alkene with reaction yield. To examine this in a more systematic way, the dihydroxylation of a homologous series of linear and cyclic aliphatic alkenes were subsequently performed (Table 5).
| Entry | Substrate | logPb |
Aq.-MeCN ratio | T (h) | Yieldc (%) |
|---|---|---|---|---|---|
| 1 |
|
3.4 | 3:1 |
23 | 67 |
| a Unless otherwise indicated, reactions were conducted as described in footnote a of Table 4. b Pubchem (release 2019.06.18).37 c Isolated yields after purification. d Unreacted alkene recovered. | |||||
| 2 | R = n-C6H13, 1l | 4.6 | 3:1 |
23 | 85 |
| 3 | R = n-C8H17, 1m | 5.7 | 3:1 |
18 | 32 |
| 4 | R = n-C10H21, 1n | 6.8 | 3:1 |
23 | 2 |
| 5 | R = n-C10H21, 1n | 6.8 | 1.4:1 |
23 | 11 |
| 6 | R = n-C12H25, 1o | 7.9 | 3:1 |
26 | 0d |
| 7 |

|
3.7 | 3:1 |
5 | 63 |
| 8 |

|
2.9 | 3:1 |
2.5 | 62 |
| 9 |

|
3.5 | 3:1 |
23 | 14 |

Compared to the styrene derivatives, dihydroxylation reactions of the aliphatic alkenes are undoubtedly more sluggish. Nevertheless, a clear trend between the lipophilicity of the alkene and the oxidation outcome can be observed: With the exception of the volatile 1-hexene, the yield of diol declines dramatically as the alkyl chain length increases from C6 to C14, which is commensurate with rising logP values (entries 1–4, 6). Interestingly, the dihydroxylation of (E)-2-octene (Table 5, entry 7) was complete in 5 h, compared to the more lipophilic terminal 1-octene (23 h, entry 2), suggesting that the nucleophilicity of the alkene is a dominant effect. Similarly, the dihydroxylation of cyclohexene (logP 2.9, entry 8) was found to proceed rapidly to provide moderate yields (>60%) of the rac-cyclohexane-1,2-diol, while the more lipophilic cycloctene (entry 9), with a similar logP value to 1-hexene and 2-octene (entries 1 and 7), afforded a significantly lower yield of the diol, reflecting the general lower activity of cyclic alkenes in epoxidation reactions.
The successful dihydroxylation of 1-hexene and 1-octene (1k and 1l) is particularly noteworthy, as they produce highly valuable products. Being hygroscopic and non-toxic, glycols 2k and 2l possess broad anti-microbial properties that are widely exploited in the formulation of personal care products and pharmaceuticals as humectants, and as safer replacements of traditional preservatives (such as toxic parabens).38
At this juncture, it is clear that the immiscibility of the reacting phases is causing reactions to be slow. This may be mitigated somewhat by adding acetonitrile, which had a moderate effect on the formation of 1,2-dodecanediol, from trace levels to 11% (Table 5, entries 4 vs. 5). Other possible strategies for improving mixing of the biphasic mixture include the use of phase-transfer reagents, or by mechanical means. Favouring the latter approach, we have previously demonstrated the effectiveness of the approach by deploying a baffled flow reactor.19 In this work, however, we were also able to achieve a satisfying outcome using a batch reactor, equipped with a mechanical stirrer and a cross-bladed paddle, to provide vigorous mixing between the inorganic and organic components. Under these conditions, the dihydroxylation of styrene 1a can be achieved without using any organic solvents, providing over 3 g of 2a in 88% yield in 90 minutes (Scheme 5vs.Table 4, entry 1). For the dihydroxylation of the aliphatic 1-decene, the addition of acetonitrile was necessary to overcome the extremely hydrophobic nature of this substrate. Nonetheless, over 3 g of 1,2-decanediol can be obtained in a 70% yield in 20 h, which is an improvement compared to the reaction conducted in a reaction vial (Table 5, entry 3).
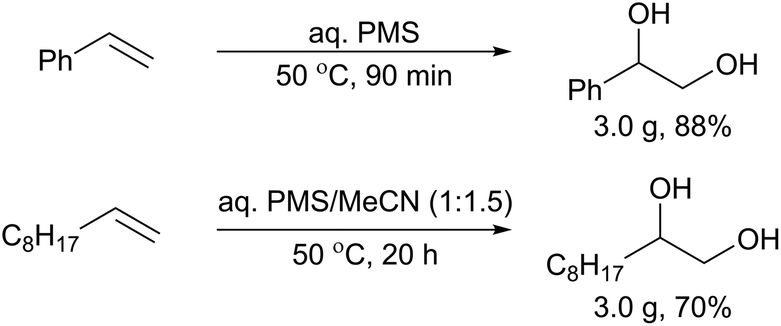 | ||
| Scheme 5 Gram-scale dihydroxylation of styrene and 1-decene using thermally-activated PMS solution, using a batch reactor. | ||
Counterion effect
In earlier experiments on the dihydroxylation of styrene (Scheme 1, Fig. 1), the activated ammonium peroxysulfate solution appeared to be more reactive than the commercial potassium PMS triple salt (Oxone). This was examined in greater detail by a direct comparison between these two oxidants, in the dihydroxylation of 1-hexene and 1-octene (Table 6). In both cases, the yields of the glycols afforded by the ammonium oxidant were double of that achieved by using Oxone: from 38% to 67%, and 43% to 85% for 2k and 2l, respectively. The dramatic improvement in yield is attributed to the lesser propensity of the ammonium cation to ‘salting-out’ than the potassium cation (particularly when the latter is a triple salt),39 therefore permitting better miscibility between the biphasic solutions. The NH4+ cation can also further enhance the reaction by acting as a phase-transfer agent.Further improvement in sustainability (workup)
For the dihydroxylation of alkenes, product separation is particularly challenging: While the reaction began with the hydrophobic alkene precursor, the biphasic mixture transforms into a homogeneous solution over the course of the reaction, as the hygroscopic diol product is formed. This will usually require the use of a large volume of solvent (typically dichloromethane) in multiple liquid–liquid extractions to retrieve it from the aqueous layer, which is deleterious for both product yield and process mass intensity. In the last part of this work, we also addressed the sustainability of the downstream processes (workup, product extraction).40Subsequently, a resin-capture-release strategy was developed, where a non-polar polymeric resin (Amberlite XAD4) was deployed to trap and separate the 1,2-diols from unreacted alkene and sulfate by-products. The procedure utilizes only aqueous solutions for loading and rinsing, apart from the final step, where methanol was used to release the diol from the resin. While the process is not entirely solvent-free, it does, nevertheless, substantially reduce the volume of organic solvents that will be otherwise be required for solvent extractions and column chromatography. The workup procedure was applied to the gram-scale synthesis of 1-phenyl-1,2-diol and 1,2-decanediol (Scheme 3), with good yields.
Conclusions
This work is a part of a wider research programme to utilise inorganic salt precursors for sustainable, green and scalable oxidation reactions. Previously, we have shown that ammonium PDS can be generated electrochemically from non-hazardous ammonium sulfate and used for the dihydroxylation of styrene in a baffled flow reator.19,20 In this article, we studied the thermal conversion of PDS, in batch and continuous flow, into the active oxidant (PMS) for alkene dihydroxylation reactions. The sulfate byproduct can potentially be recovered and reused, achieving a high level of material conservation for the overall process (Scheme 6). | ||
| Scheme 6 Sustainable generation of active oxidant ‘on-demand’ for the dihydroxylation of alkenes to 1,2-diols. | ||
During this work, in situ IR spectroscopy was employed as an in situ tool, to monitor the sequential decomposition of peroxydisulfate to peroxymonosulfate anion (PMS) and H2O2 in a strongly acidic solution. The thermally-activated PMS was found to be more effective than the corresponding potassium salt in the reactions with olefins (Fig. 1 and Table 6). This suggests that PDS can be not just a more atom-economical, but also a more effective replacement of Oxone in organic transformations. Last but not least, we have shown the use of a reusable non-ionic resin in the post-reaction workup of the highly hydrophilic diol products, minimizing the need for large amounts of organic solvents otherwise needed for extractions and chromatography.
Experimental
Reaction progress of the dihydroxylation of styrene by different oxidants (Fig. 1)
Studies of the rate of dihydroxylation of styrene were performed in a 100 mL jacketed reactor with overhead stirring (Mettler Toledo EasyMax 102), and fitted with a programmable, retractable sampling pocket probe (Mettler Toledo EasySampler™). The rate of stirring was set at 500 rpm during the reaction, except during sampling when stirring was reduced to 400 rpm for 1 min.41In a typical experiment, the reactor was initially charged with the oxidant (50 mmol) while the jacket temperature was maintained at 20 °C. Aqueous H2SO4 (2 M, 50 mL) was then added to the reactor and stirred slowly. With solid oxidants, the reactor was monitored visually for the disappearance of particles. Once dissolution was complete, the reactor temperature was raised to 40 °C. After allowing the temperature to stabilise, the reactor was then charged with styrene (25 mmol, 2.9 mL) and stirring was adjusted to 500 rpm.
Reaction aliquots (20 μL) were collected at preset intervals using the sampling probe, which were automatically diluted with methanol/water (1:3 v/v, 5 mM 2-phenylethanol standard, 180 μL) for HPLC analysis.
Electrochemical generation of peroxysulfate solution
Peroxysulfate solutions were generated electrochemically using the methods previously reported by our group:20 The electrochemical reactor (equipped with boron doped diamond anode and a stainless steel cathode) was operated in batch recirculation mode. The anolyte chamber and reservoir were maintained below 15 °C.Ammonium sulfate (53 g, 400 mmol) was dissolved in aqueous sulfuric acid (2 M, 200 mL) and divided evenly between the anolyte and catholyte reservoirs. Applying a constant current of 1.5 A, the electrolytes were recirculated (200 mL min−1) through the electrolysis cell, and the oxidant composition was periodically determined by redox colorimetry.29 The electrochemical reaction was stopped once the oxidant concentration was greater than 1 M. The electrochemically generated oxidant solution could be stored at 4 °C for several months without loss of oxidant.
Monitoring the oxidant transformation by in situ IR spectroscopy
The EasyMax reactor was fitted with a Mettler-Toledo ReactIR 15 with a diamond window insertion probe, to monitor the transformation of peroxysulfate solutions. A spectral region of 1350 to 700 cm−1 was scanned at a resolution of 8 cm−1, and with 1024 scans per data point. Spectral data was processed without solvent subtraction. A baseline offset at 1350 cm−1 was applied to the spectral data. Peroxydisulfate (PDS) was measured by the height of a strong shoulder peak (1297–1245 cm−1) relative to a two point baseline (1320–1230 cm−1). Peroxymonosulfate (PMS) was measured by the height of a weaker peak (783–730 cm−1) relative to a two-point baseline (801–725 cm−1).The IR signals were calibrated by monitoring the sequential dilution of oxidant solutions of known composition, with portions of aqueous sulfuric acid (2 M). In subsequent experiments where the calibrated IR signals were used to follow changes in the oxidant composition, samples were also analysed by redox colorimetry.29
Activation of the peroxysulfate oxidant
Small scale dihydroxylation experiments (Tables 4 and 5)
Activated peroxysulfate solution (5 mL, ca. 4.7 mmol PMS) and acetonitrile (1.66 mL) were combined in a 7 mL screw-top vial to form a monophasic system. The alkene (2.35 mmol) was added to the vial, causing the aqueous oxidant phase to separate from the organic phase. The reaction vial was sealed with a screw cap with septa, and the mixture was stirred at 50 °C. The reaction progress was followed by TLC or HPLC (for styrene) of aliquots, extracted by a needle through the septum. Where the diol product is hydrophilic, the reaction can also be observed by the formation of a monophasic solution.Once the alkene was consumed, an excess of solid NaHCO3 was added in portions to neutralise the reaction mixture, before it was extracted three times with EtOAc. The combined organic extracts were dried with anhydrous Na2SO4, concentrated by rotary evaporation and dried under high vacuum to give the corresponding diols.
Multigram synthesis of 1-phenyl-1,2-diol (Scheme 5)
The Easymax 102 reactor was charged with 1 M solution of PDS in 2 M H2SO4 (50 mL). The peroxysulfate solution was activated by stirring it at 50 °C for 6 h, with the reaction progress followed by in situ IR spectroscopy. Once the [PMS] had reached a maximum, styrene (2.9 mL, 25 mmol) was added to the reaction, and the stirring rate increased to 500 rpm. Over 90 min the styrene droplets were observed to disappear, and a monophasic orange solution formed.A chromatographic column containing XAD-4 resin (50 mm Ø × 750 mm) was pre-saturated with water. The reaction solution was passed through the resin column twice. The column was washed with a saturated solution of NaHCO3 (50 mL) with the eluent being collected. Additional solid NaHCO3 was added to the eluted solution until it was pH neutral. The eluted NaHCO3 solution was then filtered through the column a second time to capture any remaining diol, before it was flushed with compressed air to remove excess NaHCO3 solution. The diol was then released from the column with methanol (200 mL). After removal of the methanol under reduced pressure a white solid (containing NaHCO3) was obtained. This solid was triturated with ethyl acetate (3 × 10 mL), filtered, and evaporated to afford the diol product as a pale yellow solid (3.0 g, 88% yield).
Multigram synthesis of 1,2-decanediol (Scheme 5)
The active PMS oxidant was generated in the EasyMax 102 reactor as described above. A solution of 1-decene (3.5 g, 25 mmol) in acetonitrile was then added over 1 min, and the stirring rate increased to 500 rpm. The biphasic mixture was allowed to react for 20 h before being cooled to 25 °C. The acetonitrile was removed by rotary evaporation to obtain a white suspension. The product was purified using the XAD-4 resin as described above. The solid collected from the column was triturated with toluene (3 × 10 mL), filtered and evaporated to afford the diol as a waxy-white solid (3.0 g, 70% yield).Conflicts of interest
There are no conflicts to declare.Author contributions
The project was conceived by K. H. and K. K. H., who secured the funding, supervised the project and participated in the experimental design as well as data analysis and interpretation. The experiments were performed by B. J. D., V. E. Y. L., S. G and L. A. A.; who also collected and analysed the data. The manuscript draft was prepared by K. K. H. and B. J. D. and submitted to the other co-authors for review and comment prior to submission.Acknowledgements
This work is supported by a research grant awarded by the UK's Engineering and Physical Sciences Research Council (EPSRC), titled: “Manufacturing in Flow: Controlled Multiphase Reactions on Demand” (CoMRaDe, EP/L012278/1). The authors thank Dr K. N. Loponov, Prof. C. B. Rielly and Prof. R. Holdich (University of Loughborough), and Dr J. Zhu (Imperial College) for the advice and contributions they provided to the project. We are grateful to Dr Henry Dubina (Mettler Toledo Autochem), for a loan of in situ reaction tools (EasyMax reactor, ReactIR, EasySampler) for this project.Notes and references
- J. H. Teles, I. Hermans, G. Franz and R. A. Sheldon, in Ullmann's Encylcopedia of Industrial Chemistry, Wiley-VCH, Weinhem, Germany, 2005, DOI:10.1002/14356007.a18_261.pub2.
- C. Wang, Asian J. Org. Chem., 2018, 7, 509–521 CrossRef CAS.
- M. J. Rawling and N. C. O. Tomkinson, Org. Biomol. Chem., 2013, 11, 1434–1440 RSC.
- A. Gavriilidis, A. Constantinou, K. Hellgardt, K. K. Hii, G. J. Hutchings, G. L. Brett, S. Kuhn and S. P. Marsden, React. Chem. Eng., 2016, 1, 595–612 RSC.
- Hazards of persulfates: B. Zhang, L. Zhang, H. Wang and X. Wang, ACS Chem. Health Saf., 2021, 28, 244–249 CrossRef CAS; H. G. Schmidt, ACS Chem. Health Saf., 2022, 29, 53–61 CrossRef.
- Hazards of perchlorates: W. R. Robinson, J. Chem. Educ., 1985, 62, 1001 CrossRef CAS.
- Hazards of peroxides: D. Wu, X. Qian, L. Liu and N. Zang, J. Loss Prev. Process Ind., 2019, 57, 34–40 CrossRef CAS.
- L. Zhao, Y. Qian, Q.-M. Hu, R. Jiang, M. Li and X. Wang, Sustainability, 2018, 10, 2935 CrossRef.
- https://en.wikipedia.org/wiki/2017_Arkema_plant_explosion , accessed 27 April 2022.
- L. R. Boeck and P. W. Mahan, Process Saf. Prog, 2021, Early Access, DOI:10.1002/prs.12322.
- S. Sivaraman and S. Varadharajan, J. Loss Prev. Process Ind., 2021, 69, 104387 CrossRef CAS.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- Green Oxidation in Organic Synthesis, ed. N. Jiao and S. S. Stahl, Wiley, 2019 Search PubMed.
- J. Lee, U. von Gunten and J.-H. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS PubMed.
- F. Minisci, A. Citterio and C. Giordano, Acc. Chem. Res., 1983, 16, 27–32 CrossRef CAS.
- S. Mandal, T. Bera, G. Dubey, J. Saha and J. K. Laha, ACS Catal., 2018, 8, 5085–5144 CrossRef CAS.
- H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329–3371 CrossRef CAS PubMed.
- P. C. B. Page, F. Marken, C. Williamson, Y. Chan, B. R. Buckley and D. Bethell, Adv. Synth. Catal., 2008, 350, 1149–1154 CrossRef CAS.
- K. N. Loponov, B. J. Deadman, J. Zhu, C. Rielly, R. G. Holdich, K. K. Hii and K. Hellgardt, Chem. Eng. J., 2017, 329, 220–230 CrossRef CAS.
- J. Zhu, K. K. Hii and K. Hellgardt, ACS Sustainable Chem. Eng., 2016, 4, 2027–2036 CrossRef CAS.
- S. Fronaeus, Acta Chem. Scand. A, 1986, 40, 572–578 CrossRef.
- J. L. Wang and S. Z. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- I. M. Kolthoff and I. K. Miller, J. Am. Chem. Soc., 1951, 73, 3055–3059 CrossRef CAS.
- F. Hippauf, S. Doerfler, R. Zedlitz, A. Vater and S. Kaskel, Electrochim. Acta, 2014, 147, 589–595 CrossRef CAS.
- R. L. Johnson, P. G. Tratnyek and R. O. B. Johnson, Environ. Sci. Technol., 2008, 42, 9350–9356 CrossRef CAS PubMed.
- M. J. A. Abualreish, Am. J. Chem., 2012, 2, 214–217 CrossRef CAS.
- N. M. Beylerian, L. R. Vardanyan, R. S. Harutyunyan and R. L. Vardanyan, Macromol. Chem. Phys., 2002, 203, 212–218 CrossRef CAS.
- N. M. Beylerian, R. M. Hagopyan, R. A. Asaturyan, R. S. Harutyunyan, V. V. Grigoryan, L. R. Vardanyan and E. R. Saroukhanian, Oxid. Commun., 2004, 27, 71–80 CAS.
- B. J. Deadman, K. Hellgardt and K. K. Hii, React. Chem. Eng., 2017, 2, 462–466 RSC.
- D. L. Ball and J. O. Edwards, J. Am. Chem. Soc., 1956, 78, 1125 CrossRef CAS.
- X. Y. Lou, C. L. Fang, Z. N. Geng, Y. M. Jin, D. X. Xiao, Z. H. Wang, J. S. Liu and Y. G. Guo, Chemosphere, 2017, 173, 529–534 CrossRef CAS PubMed.
- E. J. Behrman and J. O. Edwards, Rev. Inorg. Chem., 1980, 2, 179 CAS.
- Potassium peroxydisulfate has a maximum solubility of 0.17 M in water.
- R. Bloch, J. Abecassis and D. Hassan, J. Org. Chem., 1985, 50, 1544–1545 CrossRef CAS.
- W. M. Zhu and W. T. Ford, J. Org. Chem., 1991, 56, 7022–7026 CrossRef CAS.
- J. N. Moorthy and K. N. Parida, J. Org. Chem., 2014, 79, 11431–11439 CrossRef CAS PubMed.
- T. J. Cheng, Y. Zhao, X. Li, F. Lin, Y. Xu, X. L. Zhang, Y. Li, R. X. Wang and L. H. Lai, J. Chem. Inf. Model., 2007, 47, 2140–2148 CrossRef CAS PubMed.
- P. Werle, M. Morawietz, S. Lundmark, K. Sörensen, E. Karvinen and J. Lehtonen, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2008, DOI:10.1002/14356007.a01_305.pub2.
- A. M. Hyde, S. L. Zultanski, J. H. Waldman, Y. L. Zhong, M. Shevlin and F. Peng, Org. Process Res. Dev., 2017, 21, 1355–1370 CrossRef CAS.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- Achieved by using a Hastelloy C-22 pitched blade stirrer with four impeller blades (45° pitch). At a stirring rate of 500 rpm or higher, the sampling is hampered by the creation of a vortex well where the sampling pocket is located. The reduced stirring rate of 400 rpm provided sufficient agitation to maintain the reaction emulsion for the duration of sampling.
Footnotes |
| † Electronic supplementary information (ESI) available: Further experimental details, characterization data of 1,2-diols, additional figures and schemes. See DOI: https://doi.org/10.1039/d2gc00671e |
| ‡ Current affiliation: Centre for Rapid Online Analysis of Reactions (ROAR), Imperial College London, Molecular Sciences Research Hub, 82 Wood Lane, London W12 0BZ, UK |
| This journal is © The Royal Society of Chemistry 2022 |