
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable, highly selective, and metal-free thermal depolymerization of poly-(3-hydroxybutyrate) to crotonic acid in recoverable ionic liquids†
Piotr
Jablonski
 a,
Dariush
Nikjoo
b,
Johan
Warna
c,
Knut
Irgum
a,
Jyri-Pekka
Mikkola
ac and
Santosh Govind
Khokarale
*a
a,
Dariush
Nikjoo
b,
Johan
Warna
c,
Knut
Irgum
a,
Jyri-Pekka
Mikkola
ac and
Santosh Govind
Khokarale
*a
aTechnical Chemistry, Department of Chemistry, Chemical-Biological Centre, Umeå University, S-90187 Umeå, Sweden. E-mail: santosh.khokarale@umu.se
bDivision of Materials Science, Luleå University of Technology, S-97187, Luleå, Sweden
cIndustrial Chemistry & Reaction Engineering, Department of Chemical Engineering, Johan Gadolin Process Chemistry Centre, Åbo Akademi University, FI-20500 Åbo-Turku, Finland
First published on 3rd May 2022
Abstract
Valorization of renewable and biodegradable biopolymers to value added chemicals and green fuels is currently considered as an important research topic aiming at reducing the dependency on fossil derived feedstocks as well as their negative consequences on the environment. In this report, we are introducing an ionic liquid (IL) mediated, sustainable, and green synthesis of crotonic acid (CA) from poly-(3-hydroxybutyrate, PHB), a biopolymer derived from microbial fermentation. In this actual case, imidazolium cation comprising ILs have been used in the synthesis, where the influence of various reaction parameters such as reaction temperature and types of ILs as well as the amount of polymer, water, and IL in the reaction mixture were examined. The conversion of PHB to CA in IL took place by a base catalyzed depolymerization with formation of crotonyl terminated polymeric entities as intermediates, a mechanism that was confirmed by NMR analysis of the reaction mixtures sampled when the reactions were carried out at various temperatures. The rate of CA formation via the IL mediated base catalyzed depolymerization increased with increasing temperature in the tested interval, and 97% yield of CA was obtained after 90 min at 140 °C. The [EMIM][AcO] IL applied as solvent and catalyst is capable of completely depolymerizing PHB to CA in 5 h at 120 °C up to a polymer loading of 40 wt%. At higher loadings the depolymerization became incomplete, which is attributed to a deactivation of the IL due to hydrogen bonding interactions with the in situ formed CA, confirmed by NMR and DSC techniques. Since the depolymerization is base catalyzed, the only tested ILs that were able to form CA were based on acetate anions, whereas the less basic or neutral [EMIM][Cl] IL was found to be inactive. Finally, more than 90% of CA as well as [EMIM][AcO] IL were recovered in high purity by solvent extraction with brine (saturated aqueous NaCl) and 2-methyl tetrahydrofuran (2-Me-THF). Most importantly, here we introduce a sustainable, metal free, and single solvent based reaction approach for selective depolymerization of PHB to industrially valuable CA in basic and recoverable ILs.
Introduction
Biopolymers are highly valued and sustainable sources for production of plastic materials, synthetic chemicals, and green fuels considering their biodegradable, biocompatible, and renewable nature. Biopolymers have therefore emerged as sustainable alternatives to petroleum derived materials and hence have an ability to reduce imminent environmental problems such as global warming and resultant climate change.1a,b Poly(hydroxyalkanoate) (PHA) is a newly emerged bio-plastic that thanks to its properties can be used for pharmaceuticals and therapeutic applications, but also as a substrate for production of various low molecular weight carboxylic acids and related chemicals.2a–d The current biotechnological approaches for the PHA production are oriented towards the use of underutilized carbon sources and waste-streams, in order to make the synthesis process economically more feasible, such as acidogenic fermentation of organic carbon in wastewater as well as microbial transformation of CO2 in biogas with methanotrophic bacteria.3a–d Recently, bioconversions of sugarcane bagasse as well as methane into PHA have also been successful using microbial processing.4a,bPoly-(3-hydroxybutyrate), PHB, is a well-studied and abundantly available bacterial polyester from the polyhydroxyalkanoate (PHA) family which has many industrial applications similar to conventional plastics and polymers such as polypropylene.2a–d Moreover, PHB can be valorized upon synthesis of industrially important and low molecular weight carboxylic acids such as 3-hydroxybutyric acid (3-HBA) as well as crotonic acid (CA) through hydrothermal or chemical treatments.5a–d Amongst these two carboxylic acids, CA is an industrially important unsaturated carboxylic acid with a wide range of applications and can be considered as a potential alternative to acrylic acid in polymer synthesis. Crotonic acid is also widely used in the synthesis of hydrophilic plastic materials for a variety of biomedical applications,6a and polymeric derivatives of CA are used in paints, insecticides, and as plasticizers, as well as softening agents in synthesis of synthetic rubber.6b Copolymers of CA and vinyl acetate, commercially known as Cevian, Gelva, Mowilith, and Vinac, are also used in cosmetics and hair styling products. Further, CA is also used as a precursor in the synthesis of acrylic acid, propene, n-butanol, maleic anhydride, and fumaric acid through various organic transformations.6d Recently, Strathmann et al. demonstrated that CA prepared from PHB via hydrothermal treatment can be directly converted to propene by a decarboxylation route.5a
Both pure PHB as well as its copolymers have been used in solvent-less and catalyst-free thermal conversion, i.e., pyrolysis, with the aim to synthesize CA. Zhu et al. proposed that thermal depolymerization of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) to CA and isopropyl-2-CA, initially proceeds through a six-membered β-elimination and α-deprotonation mechanism leading to random scission of the polymer.7a,b This further leads to the formation polymeric entities of low molecular weight having a crotonyl group at the terminal, producing CA and isopropyl-2-CA through repeated β-elimination and α-deprotonation. Recently, Iwata et al. also proposed a similar depolymerization mechanism for the thermal conversion of poly (3-hydroxybutyrate-co-4-hydroxybutyrate), PHB-copolymer, where the formation of cis- and trans-CA, 3-butenoic acid, and γ-valerolactone, as well as low molecular weight dimers and trimers, proceeds through the unzipping β-elimination route.8 Samori et al. synthesized CA from pure PHB as well as PHB-rich bacteria, whereby 92% selectivity for CA was achieved in the mixture of products after thermal distillation.5c Additionally, Ariffin et al. reported both catalytic and non-catalytic thermal conversion of PHB-homo-polymer in separate studies, where the formation of CA proceeds through repetitive β-elimination in both processes.5d,7c In this case the authors also reported that the MgO and Mg(OH)2 used as catalysts reduced both the activation energy and conversion temperature (by 40–50 °C) compared to the non-catalyzed thermal conversion approach, and increased the selectivity towards trans-CA.
In contrast to solvent-less pyrolysis, hydrothermal depolymerization of PHB is less selective towards CA, as it also involves a hydrolysis mechanism. Strathmann et al. hence showed that hydrothermal processing of PHB produces both CA and 3-HBA, where the ratio of these compounds formed in the reaction were significantly influenced by the pH of the reaction medium.5a Also in hydrothermal processing, the authors proposed that the carboxylate anion acted as a catalyst to facilitate the highly selective formation of CA through α-deprotonation and β-elimination. Similarly, Yu et al. also found that depolymerization of PHB in aqueous NaOH produces both CA and 3-HBA in ratios that vary with the pH and the duration of the reaction.9 Pyrolytic or hydrothermal conversion of PHB is therefore considered as potential future alternatives to current commercial routes of CA production starting with ethylene.
Ionic liquids (ILs) have emerged as a new solvent class due to their special properties such as thermal stability, low vapor pressure, and liquid state over wide temperature ranges. ILs can therefore be considered as “green” reaction media compared to volatile and often toxic conventional organic solvents.10 ILs are successfully applied as solvents in processing of lingocellulosic biomasses11a and are also used as catalysts in various organic synthesis approaches, where both basic as well as acidic ILs have been successfully applied.11b–e Recently, ILs have been also applied for depolymerization of poly(ethylene terephthalate).11f,g In this report, we demonstrate for the first time a highly selective and versatile process for depolymerization of PHB to CA in basic ILs with imidazolium cations under mild reaction conditions. The influence of key parameters such as the reaction temperature, ratio of PHB to IL, and types of ILs were examined. The reaction progress as well as the purities of the produced CA and the recovered ILs were confirmed by nuclear magnetic resonance (NMR) analysis of samples taken during the reaction. NMR analysis was also used to understand the reactions mechanisms of thermal treatment in ILs. The recovery of both CA and IL followed by a solvent extraction process, and it was found that the recoverable basic ILs performing the roles as solvent and catalyst were highly efficient in promoting complete conversion of PHB into CA selectively and under mild reaction conditions.
Experimental
Materials and methods
Kinetic models for the synthesis of CA from PHB, with and without catalysts deactivation parameter, were developed using the modelling and optimization software ModEst, where the kinetic parameters such as reaction rate constants (k), activation energy (Ea) and deactivation parameter (kd) were estimated by nonlinear regression using the Simplex and Levenberg–Marquardt methods.12a,b This software solves the system of ordinary differential equations forming the batch reactor model with the backward difference method. The mass balances for the starting material (PHB as moles of monomer units) and the product (CA) in a batch reactor are defined by the following differential equations, where mcat, c, and α are the catalyst mass, concentration, and catalyst activity, respectively while t is time.
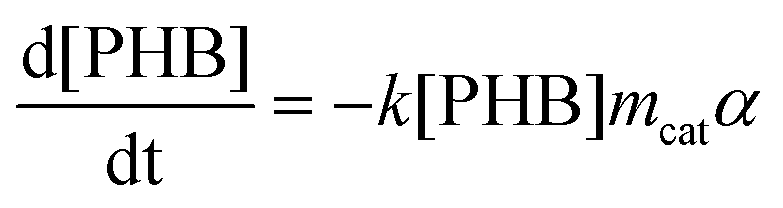 | (1) |
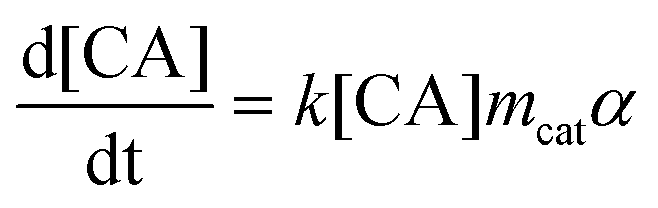 | (2) |
The rate constants were presumed to obey the Arrhenius law where the modified Arrhenius eqn (3) was used to suppress the correlation between the pre-exponential factor and the activation energy,
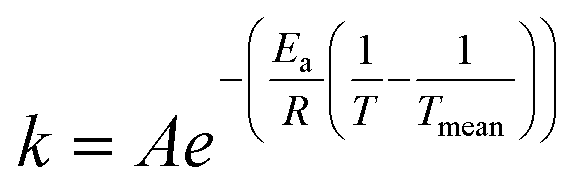 | (3) |
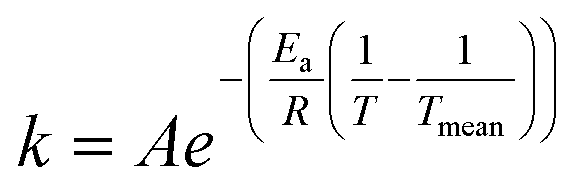 | (4) |
The objective function (Q) was set to minimize the degree of explanation between the experimental (Cexp) and calculated (Cest) values of concentrations (eqn (5)).
| Q = ∑(Cexp − Cest)2 | (5) |
The degree of explanation (R2) is defined with following equation,
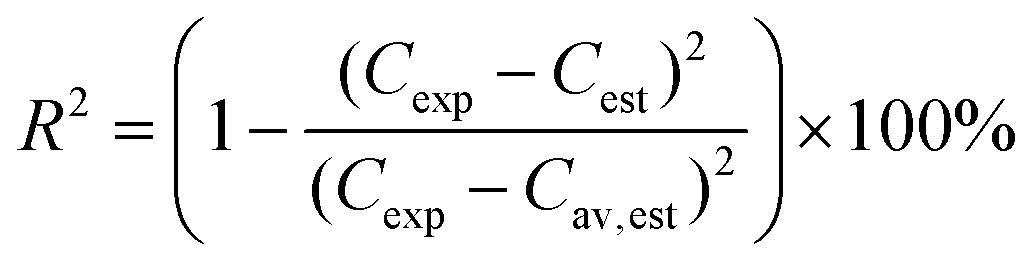 | (6) |
 | (7) |
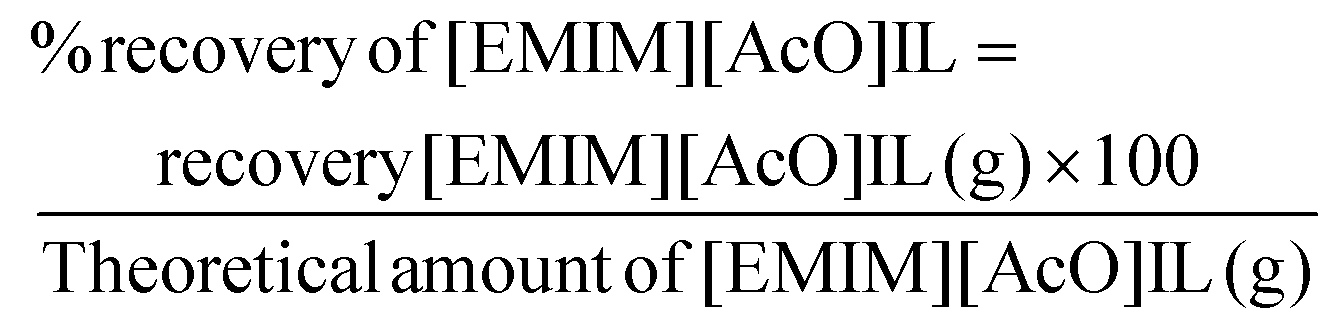 | (8) |
Results and discussion
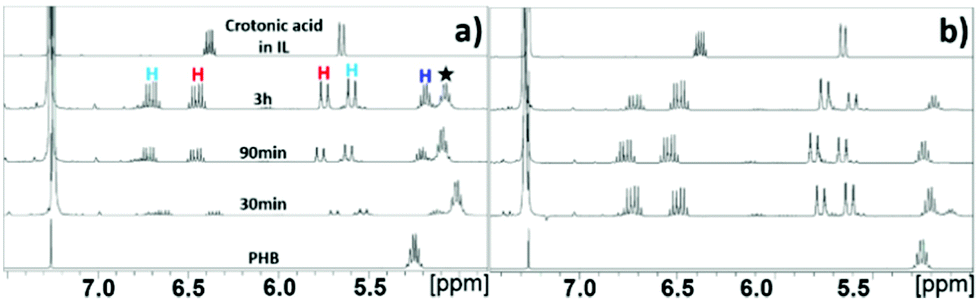
Synthesis of CA through depolymerization of poly-(3-hydroxy butyric acid) in ILs. The synthesis of CA was carried out in ionic liquid media by thermal depolymerization of commercially available PHB, where the influence of key reaction parameters such as the IL type, reaction temperature and time, polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IL ratio, and addition of water were studied. An initial experiment was carried out by reacting 20 wt% of PHB in [EMIM][AcO] IL at 120 °C for 5 h, monitoring changes in the composition of the reaction mixtures periodically by NMR spectroscopy. The 1H NMR and 13C NMR spectra after different reaction times are shown in Fig. 1 and S3,† respectively.
:IL ratio, and addition of water were studied. An initial experiment was carried out by reacting 20 wt% of PHB in [EMIM][AcO] IL at 120 °C for 5 h, monitoring changes in the composition of the reaction mixtures periodically by NMR spectroscopy. The 1H NMR and 13C NMR spectra after different reaction times are shown in Fig. 1 and S3,† respectively.
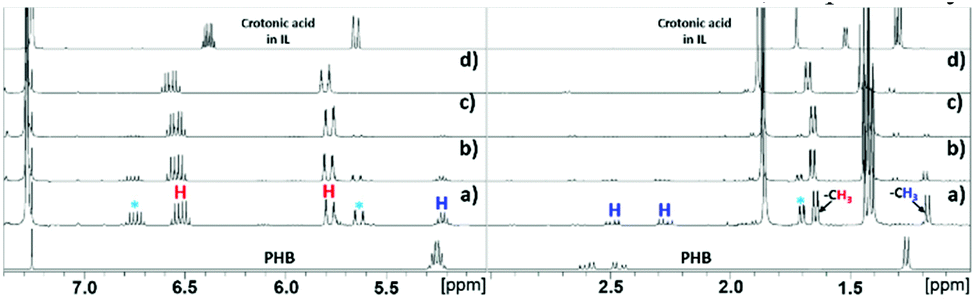 | ||
| Fig. 1 Relevant regions of 1H NMR spectra for the conversion of PHB to CA in [EMIM][AcO] IL at 120 °C for (a) 30 min, (b) 90 min (c) 3 h, (d) 5 h, PHB loading = 20 wt% in IL. | ||
The 1H NMR spectra reveal that the amount of PHB in the reaction mixture gradually decreased with time and was completely reacted after 5 h under the applied conditions. This was accompanied by a steady signal increase for the desired CA. Hence, according to these preliminary observations based on NMR analysis as well as the base catalyzed mechanism described previously, it can be concluded that the basic IL promoted depolymerization of PHB to CA.7,8 The mild conditions required for practically quantitative depolymerization and conversion of PHA into CA in [EMIM][AcO] IL corroborates a previous report, which demonstrates that carboxylate anions can promote depolymerization of PHA by acting as base catalysts.5a
Alongside the signals for the protons in PHB and CA, unidentified proton signals emerged at 1.68, 5.61, and 6.76 ppm (marked by turquoise asterisks in Fig. 1 and 2), which gradually decreased as the reaction progressed. In order to identify the cause of these new signals and to further confirm the existence of PHB and CA, two-dimensional (2D) correlation NMR analyses were made of the reaction mixture after 30 min. Representative heteronuclear single quantum coherence (HSQC) and heteronuclear multiple bond correlation (HMBC) 2D NMR spectra are shown in Fig. S4† and Fig. 2, respectively. The proton–carbon correlation peaks for both CA and PHB were all found in the HSQC NMR spectra in Fig. S4.† Besides that, the correlation peaks at chemicals shifts 1.68/17.72, 5.61/123.3, and 6.78/143.78 ppm, were also observed for the protons of the unknown chemical entities. The HMBC analysis in Fig. 2 showed that the protons associated with the unknown transient chemical entities producing 1H shifts 5.61 and 6.78 ppm correlated with the signals from the carbon atom in the carbonyl group at 165.9 ppm. The same carbonyl group is also in correlation with the proton of the –CH group (5.18 ppm) in PHB. This confirms that the unknown entities are not independent, but chemically bonded to the PHB. Furthermore, the 1H signals obtained with chemical shifts at 1.68, 5.61 and 6.78 ppm, respectively, are identical to the chemical shifts of the protons of the pure CA produced in the reaction. Considering these observations, it indicates that the 1H signals appearing at chemicals shifts 5.18, 2.23–2.53, and 1.14 ppm did not belong to PHB, but to the backbone in the crotonyl terminated oligomers. Hence, as reported previously in hydrothermal conversion of PHB5a a base catalyzed mechanism seemed to govern the depolymerization of PHB to CA in the basic IL, where random chain scissions caused by β-elimination and α-deprotonation7a,b initially led to a fast decrease in degree of polymerization with formation of crotonyl terminated oligomers as intermediates (Fig. 3b), followed by conversion to CA.8
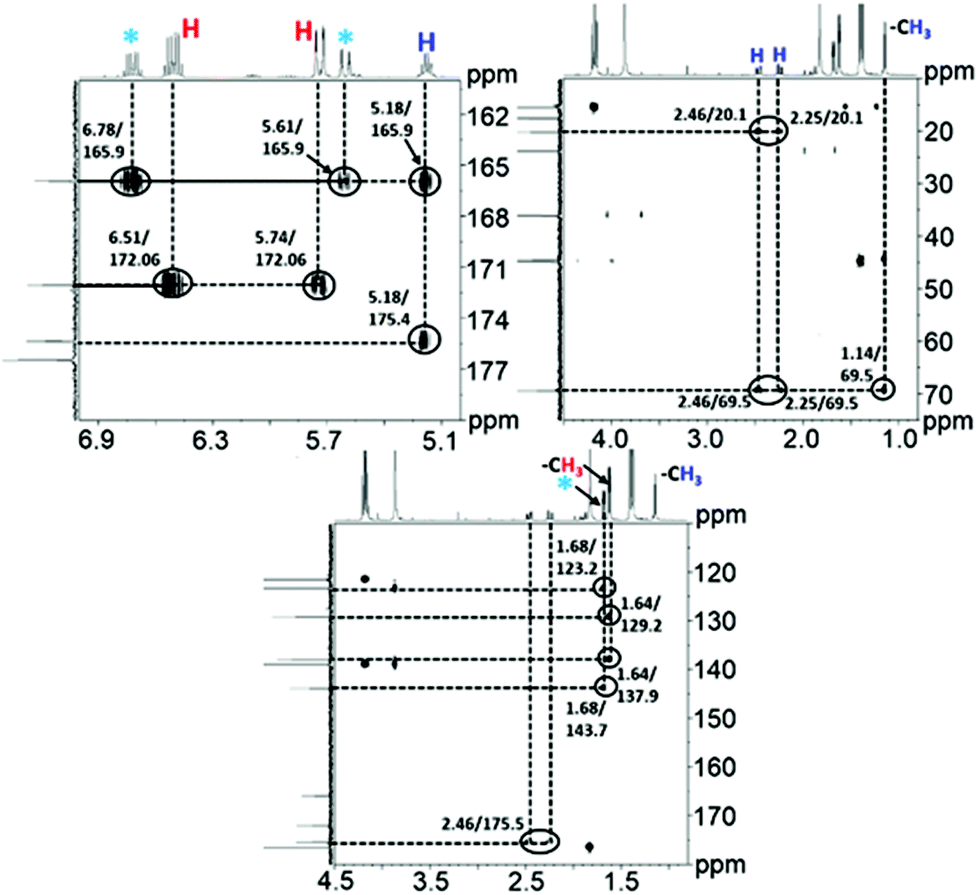 | ||
| Fig. 2 1H–13C HMBC 2D NMR spectra for the conversion of PHB to crotonic acid in [EMIM][AcO] IL at 120 °C for 30 min, PHB = 20 wt% in IL. | ||
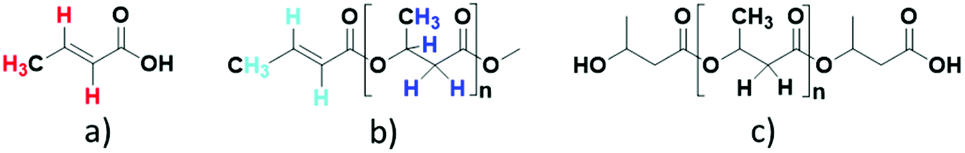 | ||
| Fig. 3 Possible chemical entities formed in the depolymerization of PHB in [EMIM][AcO] IL (a) crotonic acid, (b) crotonyl group terminated polymer and (c) poly(3-hydroxybutyrate). Colour coding of the protons in (a) and (b) correspond to markings in the 1H NMR spectra. | ||
After successful thermal depolymerization of PHB to CA in [EMIM][AcO] IL had been confirmed at 120 °C, the same process was studied at reaction temperatures 80, 100, and 140 °C with the PHB loading kept at 20 wt%. The 1H NMR spectra in Fig. 4 and S5† show that the rate of CA formation increased steadily with temperature from 80 to 140 °C. Complete utilization of the polymer had taken place in 90 min at 140 °C, whereas a substantial amount of polymeric matter remained unconverted in the reaction mixture after 90 min at 80 °C. Similarly, after 3 h of reaction, the rate of formation of CA was found to be higher at 120 °C compared to 100 °C. As observed at 120 °C, CA and oligomeric chemical entities with terminal crotonyl groups were also observed at 80, 100, and 140 °C, respectively.
 | ||
| Fig. 4 1H NMR spectra of samples taken from the conversion of PHB to CA in [EMIM][AcO] IL at (a) 80 and (b) 100 °C, PHB = 20 wt% in IL. | ||
However, according to the NMR analysis of the reaction carried out at 80 °C, signals from new unknown chemical entities appeared at 1.1, 2.30, 2.43, and 5.48 ppm (Fig. 4a, denoted by a filled star), in addition to the signals for the CA and crotonyl terminated oligomeric entities. As the reaction progressed, the signals from these unknown chemical entities decreased continuously with a concomitant increase in the signals of both CA and crotonyl group terminated oligomeric entities. However, these unidentified signals were not observed at higher temperatures, except at 100 °C and only after 30 min reaction time.
In order to confirm the chemical entities observed at 80 °C, HSQC and HMBC NMR analyses of the reaction mixture after 3 h were carried out, with corresponding correlation NMR spectra shown in Fig. S6 and S7,† respectively. As in the HSQC NMR analysis discussed above for the reaction at 120 °C for 30 min, correlation signals for the proton and carbon in CA and crotonyl group terminated oligomeric entities were also observed in the mixture reacted at 80 °C for 3 h. In addition, correlation signals for new unknown chemical entities were observed at 1.1/20.14, 2.30/40.5, 2.43/40.7, and 5.08/67.4 ppm (Fig. S6†). The HMBC NMR analysis featured similar correlation signals for CA, crotonyl terminated oligomers, and an unknown chemical entity (represented by a filled star, Fig. S7†). Unlike the 5.18 ppm proton in the crotonyl terminated oligomers, the protons at 2.30, 2.43, and 5.08 ppm belonging to the unknown entity, all showed independent correlation signals with the carbonyl carbon atom at 169.05 ppm. The signals from these unknown chemical entities are identical to pure PHB. This means that the unknown chemical entities observed at 80 °C must belong either to unreacted PHB, or, more likely, to oligomers thereof. The NMR data from this temperature study thus showed that the PHB was initially converted to its low molecular weight forms and crotonyl terminated oligomeric entities via a base catalyzed mechanism, i.e., β-elimination in a basic IL (Scheme 1). Further, these low molecular weight PHB entities followed a scheme with repetitive β-elimination and depolymerization to yield more crotonyl terminated oligomers, eventually resulting in transformation of all the PHB to monomeric CA.
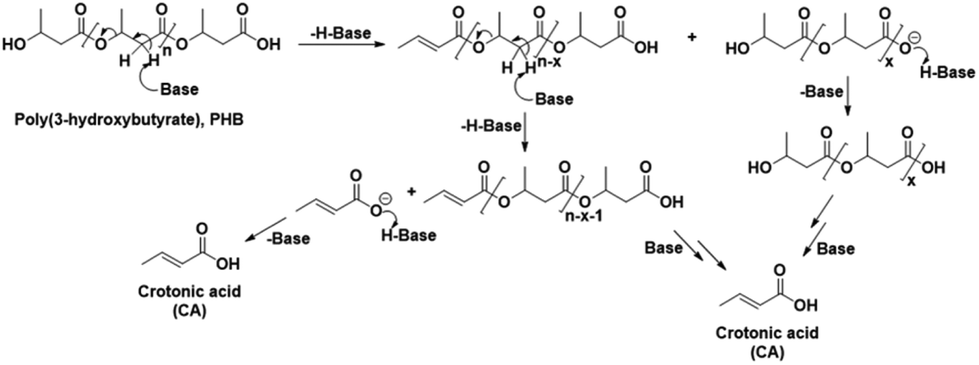 | ||
| Scheme 1 Schematic representation of the base catalyzed conversion of poly(3-hydroxybutyrate) to crotonic acid. | ||
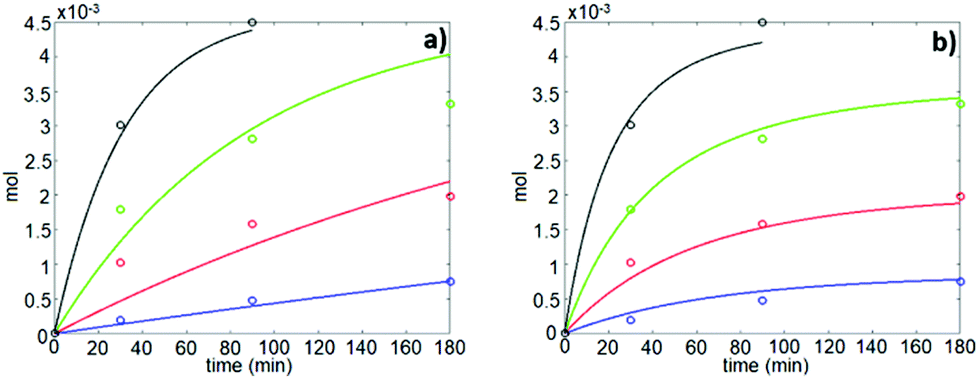
The quantification study for the formation of CA at varying temperature showed that the rate of formation of CA from PHB was significantly influenced by the reaction temperature. As the temperature was increased from 80 °C to 100 and 120 °C, the yield of CA after 3 h of reaction also increased from 15 to 39 and 73 wt%, respectively, while at 140 °C, it reached 97 wt% in 90 min (Fig. 5a, shown by circle). In this case, besides the important role as catalyst, the IL also acted as a thermally stable solvent medium, which facilitated the synthesis of CA at higher temperatures to achieve a high yield.
 | ||
| Fig. 5 Yield of CA vs. time for experimental (circle) and calculated (line) data from depolymerization of PHB to CA in [EMIM][AcO] IL at varying temperature and time, fitted (a) without and (b) with the catalysts deactivation parameter. Color coding: 80 °C, blue; 100 °C, red; 120 °C, green; 140 °C, black. | ||
In Fig. 5, the estimated and observed data for the concentration of CA are shown, with the kinetic curves represented without and with consideration of the catalysts deactivation parameter, kd in the calculations. In Fig. 5a, the calculated amount of CA (moles) deviated from the experimental data as a result of omitting the catalysts deactivation parameter in the calculation, and the degree of explanation, R2 reached 93.0%. However, as shown in Fig. 5b, after the catalyst deactivation parameter was introduced, the deviation between the calculated and the experimental values decreased significantly and the R2 value improved to 98.8%. The good correspondence between calculated and experimental data shows that the applied kinetic model is suitable for this study. The correlation matrix shows low correlation between parameters except for the frequency factor and the deactivation parameter, but the MCMC sensitivity calculation allowed us to conclude that correlation still remains at an acceptable level since all parameters arrive at clear optima (Fig. 6). The estimated kinetic parameters before and after taking catalyst deactivation into consideration are presented in Tables 1 and 2, respectively.
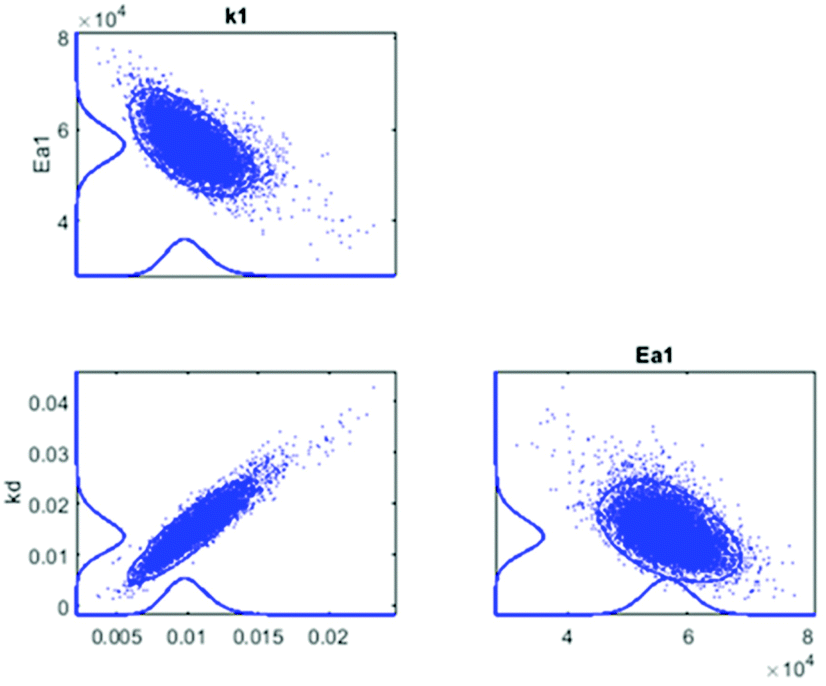 | ||
| Fig. 6 The correlation matrix of the parameters using the MCMC method. | ||
| Parameters | Value ± standard error | Estimated std. error | Relative std. error (%) |
|---|---|---|---|
| k 1 (mol min−1 g−1) | 0.48 × 10−2 ± 0.050 × 10−2 | 10.3 | 9.7 |
| E a1 (K mol−1) | 0.70 × 105 ± 0.062 × 105 | 8.9 | 11.3 |
| Parameters | Value ± standard error | Estimated std. error | Relative std. error (%) |
|---|---|---|---|
| k 1 (mol min−1 g−1) | 0.97 × 10−2 ± 0.097 × 10−2 | 10.0 | 10.0 |
| E a1 (K mol−1) | 0.57 × 105 ± 0.028 × 105 | 5.0 | 20.2 |
| k d (mol min−1 g−1) | 0.13 × 10−1 ± 0.022 × 10−1 | 16.4 | 6.1 |
Further, as the applied kinetic model demonstrated high degree of explanation after introducing the catalysts deactivation parameter, this means that the catalyst, i.e., [EMIM][AcO] IL, is deactivated during the course of the reaction under applied reaction conditions, as explained in the next section.
After having confirmed the reaction pathway for the formation of CA in [EMIM][AcO] IL, and in view of the kinetic study, we also examined the influence of the amount of PHB loaded in the reaction mixture on the reaction progress. The amount of crotonyl terminated oligomeric entities remaining unreacted in the reaction mixture after 30 min of reaction time increased as the PHB loading was increased from 10 to 40 wt% (Fig. S8†). Moreover, in the reaction mixtures where the PHB loading was 50 and 60 wt%, unreacted PHB was also observed along with unreacted crotonyl terminated polymer. This was further confirmed by HMBC NMR analysis of the reaction mixture with 60 wt% PHB loading, where a substantial amount of the polymer was found to remain unreacted after reaction for 30 min (Fig. S9†). When the reaction time was extended to 5 h, the complete conversion was achieved up to 40 wt% of PHB in the reaction mixture. In case of 50 and 60 wt%. PHB loading, the conversion of PHB was still not complete and detectable amounts of polymer remained unconverted in the reaction mixture (Fig. S9†).
The CA which is gradually formed in the reaction mixture will likely deactivate the [EMIM][AcO] IL through acid–base interaction between the basic [AcO−] anion in the IL and the acidic proton of the carboxylate group of CA (Fig. 7), which have essentially the same aqueous pKa. This interaction is possibly equivalent to the hydrogen bonding interactions between hydrogen bond donor (HBD, CA) and hydrogen bond acceptor (HBA, IL) which usually form deep eutectic solvents (DESs).13
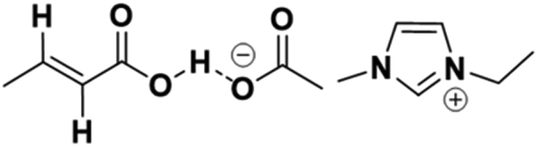 | ||
| Fig. 7 Possible hydrogen bonding interaction between crotonic acid and [EMIM][AcO] IL. | ||
The 1H NMR spectra of the physical mixtures of CA and IL at varying CA/IL molar ratio in Fig. 8a show changes in the chemicals shifts of protons of both CA and IL with molar ratio in the mixtures. It was observed, that an increase in the CA/IL ratio resulted in the decrease in chemical shift of the C2 proton of the imidazolium cation. Moreover, the distance between the signals of the C4 and C5 imidazolium protons decreased with increased loading of CA. The complete H-bonds of acetate anion with the acidic proton of crotonic acid at 1.5:1 and 2:1 CA/IL ratios provide weaker anion–cation interactions in IL. This reduces the H-bonding ability of most acidic C2 protons with the acetate anions. This weaker H-bonding of C2 protons occurs because of the weaker ion pair interactions which further induce a shielding of the C2 proton and smaller gap of C4 and C5 protons.14 Hence, the NMR analysis showed that strong chemical interactions were established between the CA and the IL, which led to changes in the chemical environment of both molecules.
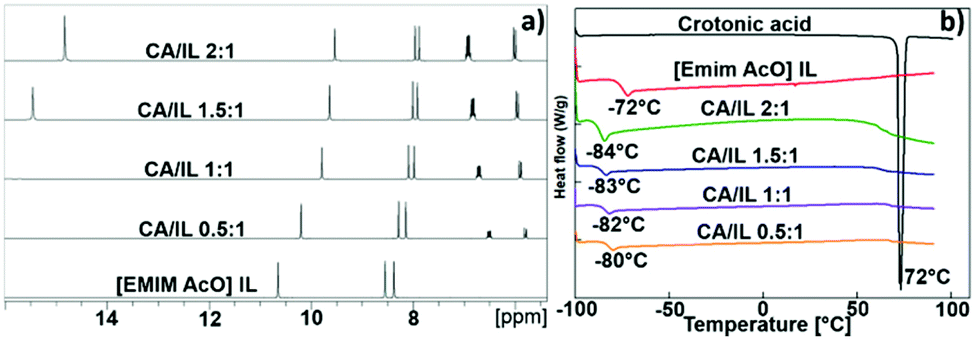 | ||
| Fig. 8 (a) 1H NMR and (b) DSC study of the mixtures of crotonic acid (CA) and [EMIM][AcO] IL at varying CA/IL molar ratios 0.5:1, 1:1, 1.5:1 and 2:1. | ||
The DSC curves for the CA and [EMIM][AcO] IL mixtures at varying compositions are shown in Fig. 8b. The [EMIM][AcO] IL showed a glass transition temperature (Tg) at −72 °C, in agreement with a previous report.15 Additionally, in the case of pure CA, a sharp and intense signal was observed at its melting point at 72 °C. Along with an increase in the CA/IL molar ratio from 0.5:1 to 2:1, the Tg values for the combinations shifted below −80 °C whereas the distinct melting point signal for the of CA disappeared, in spite of its concentration being increased to twice the concentration of IL. Based on the DSC study, mixing of CA and IL appeared to result in the formation of a low temperature transition mixture (LTTM), since no intense signals for the individual components were seen, especially for CA, which again is a characteristic feature of deep eutectic mixtures.15
The NMR and DSC analyses hence confirmed that strong hydrogen bonding interactions were established between the CA and the IL, which led to the formation of a LTTM. The catalytic depolymerization of PHB to CA in [EMIM][AcO] IL appears to be a base catalyzed process, and since hydrogen bonding interactions can be established between the IL and CA, the presence of CA can reduce the catalytic activity of IL. Loadings from 10 to 60 wt% of PHB in IL were tested in steps of 10 wt%, and assuming complete conversion, the theoretical, CA/IL molar ratios in the reaction mixtures will be 0.22, 0.49, 0.84, 1.31, 1.98, and 2.96, respectively. Hence, at a PHB loading approaching 35 wt%, complete conversion will produce CA at an amount that is equivalent the [AcO−] in the IL, which could potentially lead to complete deactivation of the catalytic activity by hydrogen bonding interaction. However, as shown in Fig. S8,† complete conversion of PHB still took place. A significant fraction of the PHB was furthermore converted to CA in 5 h when the PHB loadings were 50 and 60 wt%. Hence, this demonstrates that the acetate ion is capable of catalyzing the depolymerization and conversion of PHB into CA, even when more than one monomer equivalent of PHB is present in the reaction mixture per mole of IL.
In order to confirm that excess CA reduced the catalytic activity of the IL, reaction media were prepared where CA was added to the IL at CA/IL molar ratios of 1:1, 2:1, and 4:1. These were used in attempts to react PHB at 20 wt% loading. This addition of the CA product to the IL before the reaction reduced the catalytic ability of the IL, and when the CA/IL molar ratio was higher than 1:1 a substantial amount of PHB remained unreacted after reaction at 120 °C for 3 h (Fig. S10†).
After the influence of the temperature and the amount of PHB on the rate of CA formation had been examined, we continued to investigate the influence of water in the reaction mixture. In this case, the reaction was carried out at 120 °C for 3 h with 20 wt% PHB loading, varying the weight percentage of water in the IL while keeping the total solvent amount in the reaction composition at 2 g. The rate of conversion of PHB to CA remained unchanged with 2.5 wt% of water in the solvent mixture, which indicates that the process does not require anhydrous conditions (Fig. S11†). However, with higher water content (25 and 50 wt%) the conversion of PHB was reduced significantly. This is likely because addition of water reduces the basicity of acetate anion of the IL, therefore its overall ability to depolymerize PHB decreases.16b
Various ILs based on imidazolium cations with different functional groups and acetate anion were also tested, as was [EMIM][Cl] IL to evaluate the influence of different IL anions on the reaction progress. Fig. 9 shows the types of ILs used and the NMR spectra for the progress of CA synthesis in these ILs.
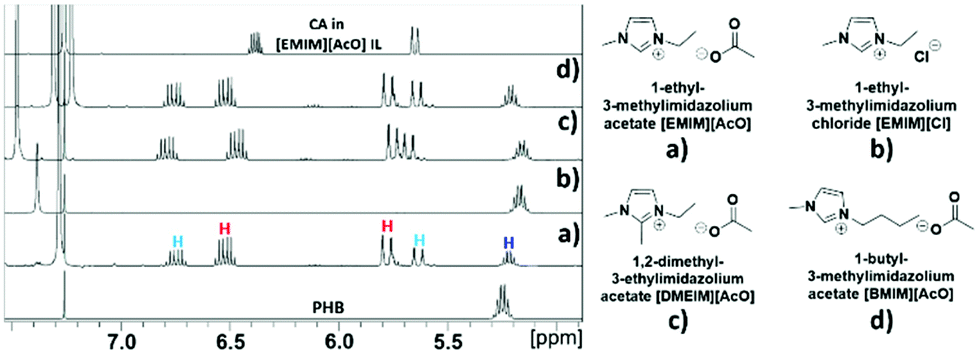 | ||
| Fig. 9 Types of ILs used and 1H NMR spectra for the attempted conversion of 0.4 g PHB to crotonic acid in 1.2 g of each of these ILs at 120 °C for 30 min; (a) [EMIM][AcO], (b) [EMIM][Cl], (c) [DMEIM][AcO] and, (d) [BMIM][AcO]. | ||
As shown in Fig. 9, all three tested ILs comprised of acetate as anion with imidazolium cations carrying different alkylation, [EMIM][AcO], [DMEIM][AcO], and [BMIM][AcO], were capable of depolymerizing PHB with accompanying formation of CA. On the other hand, although [EMIM][Cl] IL dissolved PHB, neither depolymerization nor formation of CA were observed. As described previously, synthesis of CA from PHB proceeds through a base catalyzed β-elimination mechanism. Hence, as seen in Fig. 9, all three tested ILs based on acetate anions appear to be sufficiently basic to act as catalyst in depolymerizing PHB and forming CA, whereas the [EMIM][Cl] IL fails in both these tasks due to its considerably less basic chloride anion.16a,b This further confirms that the depolymerization of PHB to CA is a base catalyzed process.
Recovery of CA and IL was performed by using reaction mixture with 20 wt% PHB treated with IL at 140 °C for 90 min. The separation of [EMIM][AcO] IL and CA was carried out with biorenewable 2-Me-THF and water as extracting solvents, where the purpose of adding 2-Me-THF was to extract CA from the aqueous reaction mixture. When the extraction was carried out without use of brine, CA was not effectively extracted by 2-Me-THF. A possible explanation could be formation of a LTTM through hydrogen bonding interactions, preventing separation of IL and CA from each other. However, by adding brine, any hydrogen bonding interactions between IL and CA should be broken, facilitating the transfer of CA from the aqueous phase into the organic 2-Me-THF phase. After recovery of the IL and the CA, NMR and gravimetric measurements were used to reveal their purity and recovery percentage, respectively. Both the IL and CA were recovered in a high purity using this solvent extraction technique. Recovery levels of CA and [EMIM][AcO] IL were 89–91 and 92–95 wt%, respectively, when the recovery experiments were carried out three times with different reaction mixtures of identical compositions (Fig. S12†).
The reported study describes all important aspects of the depolymerization of bacterial polyester, PHB to CA in basic ILs. Without use of metal-dependent catalysts or co-catalysts, this applied IL mediated process is capable of producing CA with higher selectivity (>99%), yield, and purity under mild reaction conditions compared to previously reported schemes. Since the applied method for the depolymerization of PHB to CA in IL is highly selective and efficient under mild reaction conditions, it should also be useful for chemical recycling of PHA containing materials. Moreover, the method can be applied to produce CA directly from pellets of microorganisms containing intracellular PHB granules. This is the subject of a current study, which includes biomass from a variety of microorganisms containing variable amounts of PHB in the dry cell matter.
Conclusions
Basic ILs with imidazolium cations and acetate anions have for the first time been used for efficient and highly selective depolymerization of PHB to CA under mild conditions, converting 20 wt% PHB in IL to CA with 97% yield in 90 min at 140 °C. NMR analysis revealed that the reaction proceeded by repeated α-deprotonation and β-elimination under base catalysis mediated by the acetate anion of the IL, with crotonyl terminated oligomeric PHB entities as intermediates. Conversion of PHB was complete up to 40 wt% PHB loading, while small quantities of PHB remained unreacted under otherwise identical conditions if >40 wt% of PHB was loaded. NMR and DSC analyses revealed, when combined with a kinetic parameter study, that partial deactivation of the IL was caused by product inhibition due to hydrogen bond interactions between the acetate anion of the IL and the carboxylate proton of the CA product. Presence of elevated levels of water was detrimental in terms of yield, likely due to decreased solubility of PHB at higher water admixture. Base catalysis mediated by the acetate ion of the IL is crucial for this process, evident from the absence of PHB depolymerization when [EMIM][Cl] was used as solvent/catalyst instead of [EMIM][AcO]. Finally, extraction with biorenewable 2-Me-THF as solvent assisted by brine allowed efficient separation of CA and [EMIM][AcO] IL with >90 wt% of both the IL and the CA recovered at high purity. This single solvent process based on metal free catalysis is sustainable and greener compared to previously reported methods.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by grants from the Swedish Research Council for Sustainable Development (MISTRA project 2016-02011), part of the ERA-NET Marine Biotechnology framework, and by The Bio4Energy program. Wallenberg Wood Science Center under auspices of Alice and Knut Wallenberg Foundation are gratefully acknowledged for funding part of this work. This work is also a part of the activities of the Johan Gadolin Process Chemistry Centre at Åbo Akademi University.References
- (a) Y. F. Tsang, V. Kumar, P. Samadar, Y. Yang, J. Lee, Y. S. Ok, H. Song, K.-H. Kim, E. E. Kwon and Y. J. Jeon, Environ. Int., 2019, 127, 625–644 CrossRef CAS PubMed; (b) W. Y. Chia, D. Y. Y. Tang, K. S. Khoo, A. N. K. Lup and K. W. Chew, Environ. Sci. Ecotechnology, 2020, 4, 100065–100074 CrossRef; (c) H. Shaghaleh, X. Xu and S. Wang, RSC Adv., 2018, 8, 825–842 RSC.
- (a) M. Winnacker, Eur. J. Lipid Sci. Technol., 2019, 121, 1900101–1900109 CrossRef CAS; (b) J. M. G. Alcantara, F. Distante, G. Storti, D. Moscatelli, M. Morbidelli and M. Sponchionia, Biotechnol. Adv., 2020, 42, 107582–107590 CrossRef PubMed; (c) M. Koller, Molecules, 2018, 23, 362–371 CrossRef PubMed; (d) Z. Li, J. Yang and X. J. Loh, NPG Asia Mater., 2016, 8, 265–284 CrossRef; (e) T. Iwata, Angew. Chem., Int. Ed., 2015, 54, 3210–3215 CrossRef CAS PubMed.
- (a) A. A. Amadu, S. Qiu, S. Ge, G. N. D. Addico, G. K. Ameka, Z. Yu, W. Xia, A. W. Abbew, D. Shao, P. Champagne and S. Wang, Sci. Total Environ., 2021, 756, 143729–143748 CrossRef CAS PubMed; (b) D. Ha, L. Nachtergaele, F. M. Kerckhof, D. Rameiyanti, P. Bossier, W. Verstraete and N. Boon, Environ. Sci. Technol., 2012, 46, 13425–13431 CrossRef PubMed; (c) J. M. G. Alcantara, F. Distante, G. Storti, D. Moscatelli, M. Morbidelli and M. Sponchioni, Biotechnol. Adv., 2020, 42, 107582–107590 CrossRef PubMed; (d) V. Perez, C. R. Mota, R. Munoz and R. Lebrero, Chemosphere, 2020, 255, 126929–126937 CrossRef CAS PubMed.
- (a) R. G. Saratale, S. K. Cho, G. D. Saratale, G. S. Ghodake, R. N. Bharagava, D. S. Kim, S. Nair and H. S. Shin, Bioresour. Technol., 2021, 324, 124673–124678 CrossRef CAS PubMed; (b) L. Y. Liu, G. J. Xie, D. F. Xing, B. F. Liu, J. Ding and N. Q. Ren, Environ. Sci. Ecotechnology, 2020, 2, 100029–100036 CrossRef.
- (a) Y. Li and T. J. Strathmann, Green Chem., 2019, 21, 5586–5597 RSC; (b) C. A. Mullen, A. A. Boateng, D. Schweitzer, K. Sparks and K. D. Snell, J. Anal. Appl. Pyrolysis, 2014, 107, 40–45 CrossRef CAS; (c) A. Parodi, A. Jorea, M. Fagnoni, D. Ravelli, C. Samori, C. Torri and P. Galletti, Green Chem., 2021, 23, 3420–3427 RSC; (d) M. Rahimi, Z. Mamat, H. Ariffin, M. A. Hassan and M. A. K. M. Zahari, J. Cleaner Prod., 2014, 83, 463–472 CrossRef.
- (a) E. Karadag, O. B. Uzum, D. Saraydin and O. Guven, Int. J. Pharm., 2005, 301, 102–111 CrossRef CAS PubMed; (b) J. Blumenstein, J. Albert, R. P. Schulz and C. Kohlpaintner, Crotonaldehyde and Crotonic Acid in Ullmann's Encyclopedia of Industrial Chemistry, Wiley VCH, Weinheim, 2015 Search PubMed; (c) H. Ariffin, H. Nishida, Y. Shirai and M. A. Hassan, Polym. Degrad. Stab., 2008, 93, 1433–1439 CrossRef CAS; (d) J. Van Walsem, E. Anderson, J. Licata, K. A. Sparks, C. Mirley and S. M. Sivasubramanian, US Pat. Appl20120315681A1, 2012 Search PubMed.
- (a) H. Xiang, X. Wen, X. Miu, Y. Li, Z. Zhou and M. Zhu, Prog. Nat. Sci.: Mater. Int., 2016, 26, 58–64 CrossRef CAS; (b) H. Abe, Macromol. Biosci., 2006, 6, 469–486 CrossRef CAS PubMed; (c) H. Ariffin, H. Nishida, Y. Shirai and M. A. Hassan, Polym. Degrad. Stab., 2010, 95, 1375–1381 CrossRef CAS.
- T. Omura, T. Goto, A. Maehara, S. Kimura, H. Abe and T. Iwata, Polym. Degrad. Stab., 2021, 183, 109460–109468 CrossRef CAS.
- (a) J. Yu, D. Plackett and L. X. L. Chen, Polym. Degrad. Stab., 2005, 89, 289–299 CrossRef CAS.
- (a) S. K. Singh and A. W. Savoy, J. Mol. Liq., 2020, 297, 112038–112060 CrossRef CAS; (b) T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- (a) A. Brandt, J. P. Hallett, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2010, 12, 672–679 RSC; (b) B. M. Matsagar and P. L. Dhepe, Catal. Sci. Technol., 2015, 5, 531–539 RSC; (c) E. J. G. Suarez, S. G. Khokarale, O. N. van Buu, R. Fehrmann and A. Riisager, Green Chem., 2014, 16, 161–166 RSC; (d) S. Hu, Z. Zhang, J. Song, Y. Zhou and B. Han, Green Chem., 2009, 11, 1746–1749 RSC; (e) R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed; (f) A. S. Amarasekara, J. A. Gonzalez and V. C. Nwankwo, J. Ionic Liquids, 2022, 2, 100021–100025 CrossRef; (g) A. M. Al-Sabagh, F. Z. Yehia, A. M. F. Eissa, M. E. Moustafa, G. Eshaq, A. M. Rabie and A. E. ElMetwally, Ind. Eng. Chem. Res., 2014, 53, 18443–18451 CrossRef CAS.
- (a) V. N. Sapunov, A. A. Stepacheva, E. M. Sulman, J. Warna, P. M. Arvela, M. G. Sulman, A. I. Sidorov, B. D. Stein, D. Y. Murzin and V. G. Matveeva, J. Ind. Eng. Chem., 2017, 46, 426–435 CrossRef CAS; (b) D. Ruiz, A. Aho, T. Saloranta, K. Eranen, J. Wärna, R. Leino and D. Y. Murzin, J. Chem. Eng., 2017, 307, 739–749 CrossRef CAS.
- Q. Zhang, K. O. Vigier, S. Royer and F. Jerome, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- M. Lee, Z. Niu, C. Slebodnick and H. W. Gibson, J. Phys. Chem. B, 2010, 114, 7312–7319 CrossRef CAS PubMed.
- M. Franciscoa, A. Bruinhorst, L. F. Zubeir, C. J. Peters and M. C. Kroon, Fluid Phase Equilib., 2013, 340, 77–84 CrossRef.
- (a) T. V. Doherty, M. Mora-Pale, S. E. Foley, R. J. Linhardt and J. S. Dordick, Green Chem., 2010, 12, 1967–1975 RSC; (b) M. Kudoua, Y. Kubota, N. Nakashimaa, F. Okazakia, K. Nakashimac, C. Ogino and A. Kondo, J. Mol. Catal. B: Enzym., 2014, 104, 17–22 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc00621a |
| This journal is © The Royal Society of Chemistry 2022 |