
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Separation of nickel from cobalt and manganese in lithium ion batteries using deep eutectic solvents†
Dana L.
Thompson
a,
Ioanna M.
Pateli
 ab,
Chunhong
Lei
a,
Abbey
Jarvis
c,
Andrew P.
Abbott
a and
Jennifer M.
Hartley
*a
ab,
Chunhong
Lei
a,
Abbey
Jarvis
c,
Andrew P.
Abbott
a and
Jennifer M.
Hartley
*a
aSchool of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: jmh84@le.ac.uk
bSchool of Chemistry, University of St Andrews, North Haugh, Fife, UK KY16 9ST
cSchool of Chemistry, University of Birmingham, Birmingham, UK B15 2TT
First published on 23rd May 2022
Abstract
A cornerstone of the decarbonisation agenda is the use of lithium ion batteries, particularly for electric vehicles. It is essential that effective recycling protocols are developed and this includes the ability to selectively digest and recover components of the cathode materials, most commonly including manganese, cobalt and nickel. This study shows a method by which nickel oxide can be efficiently separated from cobalt and manganese oxides using an oxalic acid-based deep eutectic solvent. The subsequent addition of water to the pregnant solution enables the co-precipitation of cobalt and manganese oxalates. This permits a route to the reformulation of the active materials from high cobalt and manganese content to high nickel content.
1. Introduction
Over the past 30 years, the demand for lithium ion batteries (LIBs) in electric vehicles (EVs) has increased significantly due to their role in reducing greenhouse gas emissions,1,2 with many countries banning the sale of new petrol or diesel vehicles from 2030.3,4 As of 2017, there were 3 million electric vehicles in the global stock, with a projected growth estimate predicting that if all EV targets are met then by 2030, there will be ca. 230 million electric vehicles on the road.5 The high content of critical metals such as cobalt and nickel means that at the end-of-life (EoL), these batteries will need to be recycled.Current processes include pyrometallurgy, which recovers cobalt, nickel, and copper in the form of a metal alloy, which can then be further processed to produce LiCoO2. While pyrometallurgy allows for the easy processing of different battery chemistries and cell types, the recycling efficiency is lower than hydrometallurgical processes due to the downcycling of manganese, aluminium, and lithium.6–8 Hydrometallurgical approaches are known for their ability to be highly selective towards different metals, but can involve complex multi-step recovery routes.9,10 The leaching of metals from spent LIBs has been reviewed many times,11–15 and several processes have been applied on a semi-commercial scale (1000–5000 t/a).10 The most common leaching agents are strong acids, such as HCl, HNO3, and H2SO4, with H2O2 often added as a reducing agent.10 Copper has also been used as a reducing agent of lithium cobalt oxide (LCO) in H2SO4 leaching, with the advantage that it is already present in battery waste.16 Alkaline solutions of NaOH or LiOH are also employed to dissolve the aluminium current collector prior to acidic leaching in order to improve the purity of the final products.17,18 An alternative method to improve the quality of the waste stream involves ultrasonic delamination of the active materials from the current collectors, which retains the current collectors in their metallic (and more easy to recycle) state.19 Separation of current collectors and active materials has also been achieved via binder dissolution using solvents such as N-methyl pyrrolidone (NMP), although it is expensive and toxic. More recently, alternative solvents such as dimethyl isosorbide (DMI) have been investigated due to their less toxic nature.20 Organic acids such as citric acid, maleic acid, ascorbic acid, L-tartaric acid and oxalic acid have also proven to be successful in extracting metals from spent LIBs,21 and can exhibit higher metal leaching selectivity.10,22,23 Bioleaching processes, in which fungi such as Aspergillus Niger and Penicillin simplicissimum produce these organic acids, have also been applied to LIB leaching. However; longer leaching times and sterilisation requirements limit their applications.22,24
An alternative route involves the use of ionometallurgy, where ionic liquids (ILs) or deep eutectic solvents (DESs) are employed instead of traditional aqueous processes. There have been numerous investigations into the dissolution of metal oxides in IL and DES media,25–29 including more industrially applicable studies on electric arc furnace dust,30 flue dust,31 and lamp phosphors.32 DESs formed from choline chloride with ethylene glycol, urea, or glycerol have recently been used to digest the cathode black mass from spent Ni–MH batteries,33 LCO, and lithium nickel manganese cobalt oxides (LiNMC), with a leaching efficiency of up to 99.3%.34 Peeters et al.9 reported the use of a citric acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :choline chloride system for the leaching of LCO with the presence of aluminium or copper as reducing agents. Non-aqueous solvent extraction, followed by stripping of the organic phase with aqueous oxalic acid solution resulted in a total recovery yield of 81% cobalt as the oxalate species, with a purity of 99.9%. The use of a neutral DES such as ethylene glycol:choline chloride has been reported with a hydrothermal reactor followed by solvent extraction to recover LiNMC.35 The use of a thiourea-based DES has also been investigated by Chen et al. for the dissolution of LCO.36 Hydrophobic DESs have also been investigated, as they can be used in place of common solvent extraction solvents.37 The advantages of using DESs in metal oxide dissolution include the possibility of recycling the DES, greater selectivity than traditional inorganic acids, less aqueous waste and avoiding emission of harmful gases.9,34,38,39 DESs used in conjunction with ultrasound have also been reported, with the advantage of increasing the reaction speed via increasing the metal oxide surface area and mixing.40
:choline chloride system for the leaching of LCO with the presence of aluminium or copper as reducing agents. Non-aqueous solvent extraction, followed by stripping of the organic phase with aqueous oxalic acid solution resulted in a total recovery yield of 81% cobalt as the oxalate species, with a purity of 99.9%. The use of a neutral DES such as ethylene glycol:choline chloride has been reported with a hydrothermal reactor followed by solvent extraction to recover LiNMC.35 The use of a thiourea-based DES has also been investigated by Chen et al. for the dissolution of LCO.36 Hydrophobic DESs have also been investigated, as they can be used in place of common solvent extraction solvents.37 The advantages of using DESs in metal oxide dissolution include the possibility of recycling the DES, greater selectivity than traditional inorganic acids, less aqueous waste and avoiding emission of harmful gases.9,34,38,39 DESs used in conjunction with ultrasound have also been reported, with the advantage of increasing the reaction speed via increasing the metal oxide surface area and mixing.40
Previous work showed that selective dissolution of metal oxides is possible in a range of choline chloride-based DESs, based on careful selection of the hydrogen bond donor (HBD) and solvent pH.38 Of special interest was the oxalic acid dihydrate:choline chloride (OxA:ChCl) system, which displayed the ability to rapidly dissolve very high concentrations of MnO2, MnO, Co3O4 and CoO (up to 0.1 mol dm−3 within the first hour) due to the low pH value of the solvent, whilst barely affecting NiO within the same timeframe. More specifically, the concentrations of dissolved MnO2 and Co3O4 were between 250 and 750 times higher than the concentration of NiO. It was also proposed that this behaviour could also be applied to the leaching of the active materials from end-of-life LIB cathodes and recovery as useful chemical precursors that can be returned to the supply chain. In addition, OxA has been employed in many recycling processes as both a leaching and precipitating agent,41,42 as the oxalate end-product can easily be transformed into an oxide upon heating at 950 °C.43 Therefore, in the present work, a protocol for the separation and recovery of the critical elements manganese, cobalt and nickel from LiNMC using a DES formed from a 1:1 molar ratio of OxA:ChCl is developed.
2. Experimental
2.1 Solvent preparation
The deep eutectic solvent (DES) (referred to hereafter as OxA:ChCl) was synthesised by mixing choline chloride (ChCl) (Acros Organics, 99%) with oxalic acid dihydrate (OxA) (Alfa Aesar, 98%) in a 1:1 molar ratio at a temperature of 50 °C, until a colourless homogenous liquid had formed. The liquids were then used directly to avoid the time- and heat-related esterification that has been observed to take place with acidic DESs.44
2.2 Leaching and precipitation experiments
To make the synthetic LIB leachates for developing the recovery process, Co3O4 (Alfa Aesar, 99%) and MnO2 (Acros Organics, 99%) were dissolved in OxA:ChCl, using a solid-to-liquid (S:L) ratio of 1.98 g L−1 manganese and 1.83 g L−1 cobalt (0.030 mol kg−1 Mn and 0.026 mol kg−1 Co). This equates to the molar ratio of cobalt and manganese found in LiNMC-532, without saturation of the solvent, and will avoid any nickel contamination of the precipitates. The mixture was stirred at 80 °C for 48 hours in a sealed container, using a sand bath to ensure a constant temperature. To initiate precipitation, deionised water or aqueous solutions of 0.5, 1.0, and 1.8 mol dm−3 (1.5 mol kg−1) oxalic acid were added to the pregnant leaching solution (PLS) in a PLS:anti-solvent volume ratio of 1:1. The resulting precipitates were subsequently filtered, rinsed with deionised water (50 °C), and then dried in an oven at 50 °C overnight. Each experiment was carried out three times to ensure reproducibility.
To determine the optimal solid:liquid (S:L) ratio of synthetic LiNMC (98%, <0.5 μm particle size, Sigma Aldrich) and solvent for leaching, S:L ratios of 5, 15, 25, and 33 g L−1 were tested. The LiNMC was added to an 80 °C solution of OxA:ChCl and stirred at 300 RPM up to a maximum of 5 hours. Aliquots (1–3 mL) of the solution were taken at various time intervals (10, 20, 40, 60, 120 and 240 min) using luer-lock syringes (2 mL polypropylene, Fisherbrand), then filtered with Nylon syringe filters (0.2 μm, 30 mm, ThermoScientific) into a sample vial. An aliquot (10 μL) was immediately taken with a positive displacement pipette for ICP-MS analysis and stored in dil. 2% HNO3. This was to ensure that an accurate sample was taken before precipitation occurred upon cooling of the solution. It was assumed that the S:L ratio did not change in the pregnant leachate solution (PLS) with the removal of aliquots.
2.3 Instrumentation
The speciation of the metals in the pregnant leachate solutions were identified using UV-Visible spectroscopy (UV-Vis) on a Mettler Toledo UV5 Bio spectrometer (Fig. S1†). These spectra were then compared to the spectra of known species. Quartz cuvettes with path length 1 cm were used, except where the sample was very concentrated and dilution with the same DES resulted in a change of colour. In these instances, quartz slides with a path length of 0.1 mm or 0.5 mm were used instead. Calibration curves were constructed from solutions of CoCl2·6H2O in EG:ChCl, using concentrations from 0.001 to 0.1 mol dm−3 and plotted against the absorbance at the 694.6 nm maximum.
The pure LiNMC was characterised using inductively coupled plasma mass spectrometry (ICP-MS) using a Thermo Scientific iCAP Qc ICP-MS. The material was first digested in aqua regia at ambient conditions for 30 minutes. The resulting solutions were then diluted 1000 times in 2 vol% nitric acid (TraceMetal Grade, Fisher Scientific), with an additional 10 times dilution if needed. To determine the metal content and ratios in the different precipitates, approximately 10 mg of sample was digested in 2–3 mL of 1 mol dm−3 HCl at ambient conditions for 1 hour. The resulting solutions were then diluted 1000 times in 2 vol% nitric acid, with an additional 10 times dilution if needed. Calibration lines of each element between 10 and 3000 ppb were determined using dilutions of a multi-element reference solution 2A (SPEX CertiPrep, 99%), and the internal standards (spikes) in each solution were 0.1 ppm of both lanthanum (Fisher Scientific, 1000 μg mL−1 in 2–5% HNO3, SPEX CertiPrep™) and rhodium (Merck, 10 mg kg−1 Rh in HNO3).
To determine the identity of the compounds present in the precipitates, they were analysed using powder X-ray diffraction (XRD), infrared spectroscopy, scanning electron microscopy (SEM) and energy dispersive X-ray (EDX), and thermogravimetric analysis (TGA). XRD patterns were recorded using a Phillips model PW 1730 X-ray generator, and the species present were identified through comparison to literature data available in the associated DIFFRAC.EVA software database. The infrared spectra were measured using a Bruker Alpha II spectrometer with a Platinum ATR and OPUS software, over the range of 200–4000 cm−1, with 24 scans per sample and a resolution of 4 cm−1.
The thermal properties of the precipitates were investigated using a Mettler Toledo TGA/DSC1 machine with a resolution of ±1 μg and maximum temperature of 1100 °C, controlled by STARe software (version 12.10). The balance used to weigh the samples was a Mettler Toledo Semi-Micro Balance (MS105DU), with a resolution of 0.1 mg. Samples of ca. 5 mg were placed in 70 μL alumina (Al2O3) crucibles with no lid and packed down with a steel rod. The heating program used was from 25 °C to 550 °C, at a heating rate of 10 K min−1, and a gas flow of 50 mL min−1 of N2. In order to counter the buoyancy effect generated by heating the air inside the furnace, a background subtraction method was employed when testing the samples. All measurements were taken in triplicate to allow the calculation of experimental error.
The morphology and elemental composition of the precipitates and black mass were investigated with SEM and EDX with a FEI Quanta 650 FEG in backscattered electron mode at 20 kV and 5 nm spot size, with Aztec controlling software. The precipitates were first carbon coated using an Emitech K950X to ensure that a conductive layer was present to obtain the images.
3. Results and discussion
3.1 Optimum dissolution parameters for LiNMC in OxA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :ChCl
:ChCl
The temperature, water content, solid-to-liquid (S:L) ratio, and time for digesting LiNMC in oxalic acid dihydrate:choline chloride (OxA:ChCl) were optimised initially using just the pure powdered material to avoid any issues related to encapsulation caused by the polymer binder present in LIB active materials. Fig. 1a shows the effect of temperature on the dissolution of LiNMC powder after 24 h. An increased temperature increases metal oxide dissolution due to improved mass transport from lower viscosity (149 mm2 s−1 at 40 °C, vs. 28 mm2 s−1 at 100 °C),45 and also because of a general increase in the reaction rate constant, as described by the Arrhenius equation. It can be seen that temperatures of ca. 80 °C are required for complete leaching of cobalt and manganese, while the leaching of lithium also benefits from an increase in temperature. Nickel, however, has poor leaching behaviour across all temperatures. This could be due to poor solubility of the nickel oxide species in OxA:ChCl, or alternatively that dissolved nickel ions immediately precipitate out as insoluble nickel oxalates.38 It was not possible to confirm the identity of this residual solid, as washing it with water, alcohol (ethanol, methanol) or oxalic acid solution resulted in immediate precipitation of mixed cobalt/manganese species from the residual PLS. Non-polar organic solvents were found to be immiscible with the DES. While this precipitation is inconvenient for the purposes of recovering and characterising the undigested material, it does, however, present interesting options for the recovery of cobalt and manganese from solution via the use of an anti-solvent to induce precipitation.
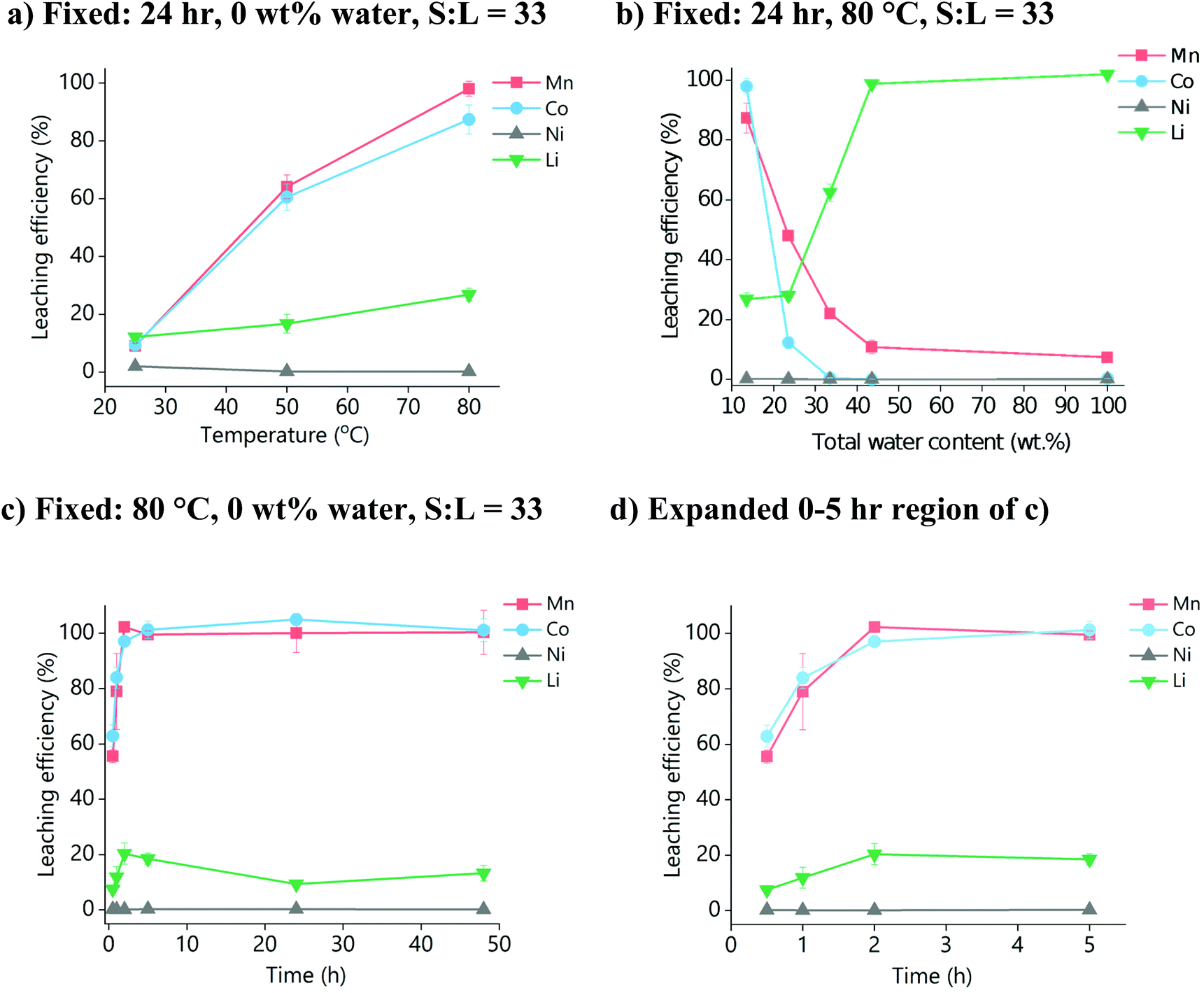 | ||
| Fig. 1 Dissolution of LiNMC powder in OxA:ChCl with respect to variable: (a) temperature, (b) total water content and (c) and (d) time. For (a) and (b), the leaching time was 24 h. For (b) and (c), the temperature was 80 °C. For (a) and (c) no extra water was added. For all systems, S:L = 33 g L−1 and the stirring rate was 500 rpm. | ||
The effect of water content on LiNMC dissolution was investigated, as it is known that an esterification reaction between the carboxylic acid and the alcohol group of the choline can occur.44 This increased water content will decrease the viscosity of the DES, hence improving mass transport, but may also reduce the solubility of the metal ions by forming metal oxalates. Fig. 1b shows the effect of an additional 10, 20 and 30 wt% water (corresponding to a total water content of 23.5, 33.5 and 43.5 wt%, respectively) on LiNMC dissolution. The leaching behaviour of LiNMC in an aqueous 1.8 mol dm−3 solution of oxalic acid is also compared. It can be seen that Ni dissolution is unaffected by water content, whereas the leaching of lithium increased to 100% when greater than 30 wt% water was added.46 The solubility of cobalt and manganese declined sharply upon addition of water, through the formation of insoluble precipitates. This decrease is more significant for cobalt than for manganese, with a decrease from almost 100% solubilisation to ca. 15% with the addition of only 10 wt% water. The decrease in solubility for manganese only changes from ca. 90% to 50% soluble species. Therefore, it is most optimal for leaching to be carried out in OxA:ChCl with no additional water.
The effect of leaching time is shown in Fig. 1c, for OxA:ChCl at 80 °C with no additional water. It can be seen that the maximum concentrations of all elements were reached within 5 hours, with manganese and cobalt being fully leached. Therefore, the optimal dissolution conditions are proposed to be: 80 °C for up to a maximum of 5 h, with no added water.
The S:L ratio is important as it controls the cost of the process. The effect of S:L ratio using the optimal dissolution conditions of 80 °C, up to a maximum of 2 h, with no added water was investigated, using S:L ratios of 5, 15, 25, and 33 g L−1 (Fig. 2). It was found that at least 80% leaching efficiency for cobalt and manganese in all four systems was reached within 2 hours. However, lithium leaching was more dependent on the S:L ratio, with higher amounts of solid resulting in poorer dissolution. This is probably due to the species that lithium is forming in solution, as it will require at least 4 molar equivalents of water or oxalic acid to properly form the first hydration shell.47 Therefore, for maximum leaching efficiency in the minimum sensible amount of solvent, we propose the use of: a S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :L ratio of ca. 15 g L−1.
:L ratio of ca. 15 g L−1.
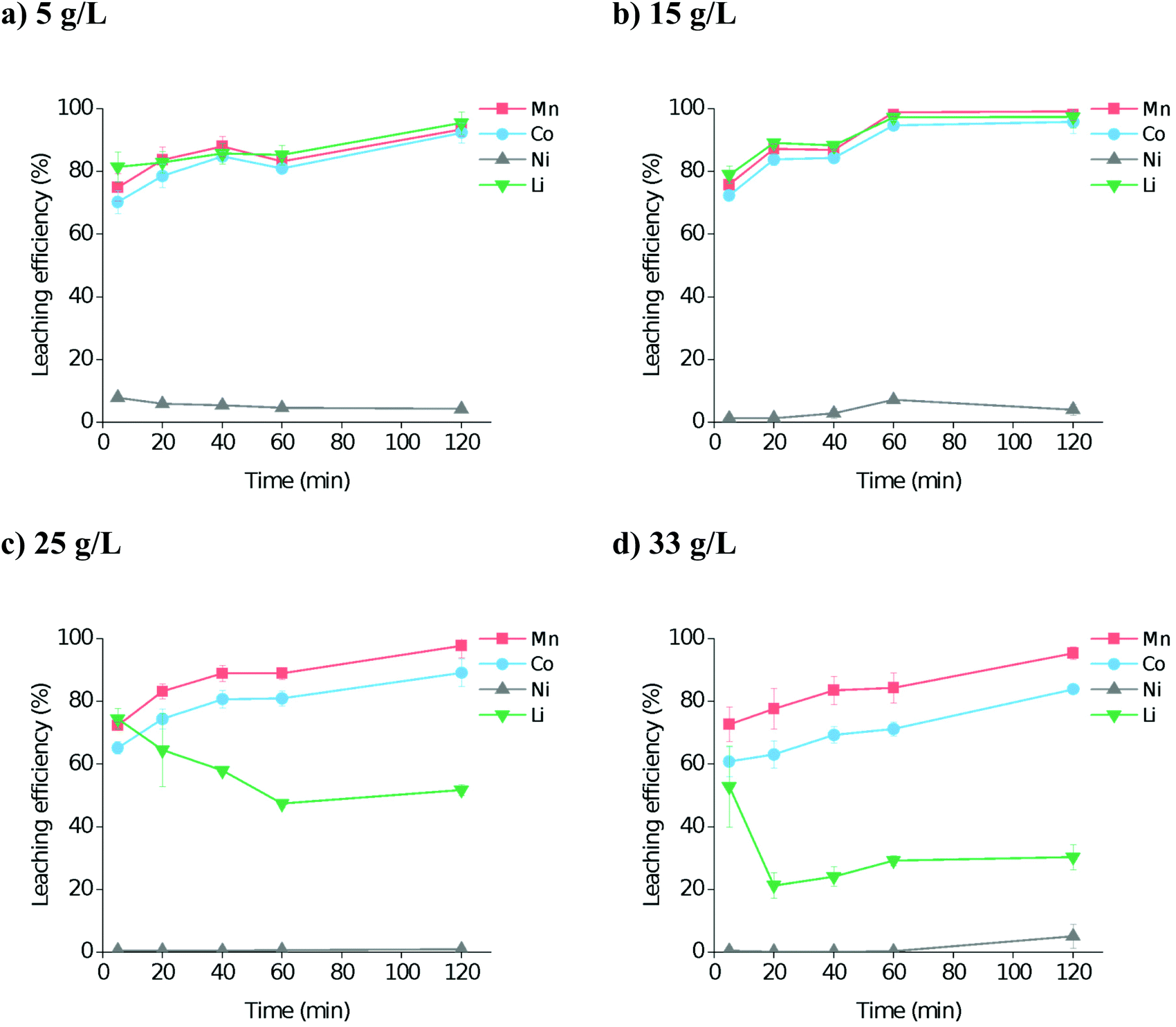 | ||
| Fig. 2 Dissolution of LiNMC synthetic powder in OxA:ChCl at 80 °C as a function of time, at S:L ratios of (a) 5 g L−1, (b) 15 g L−1, (c) 25 g L−1, and (d) 33 g L−1 with a stirring rate of 300 rpm. | ||
To alter the material composition from an NMC ratio of 1:1:1 to 8:1:1, 70% of the Co and Mn needs to be removed. As it was not possible to directly analyse the residual solids due to contamination with precipitated metal oxalates during washing stages, leaching efficiency (and residue composition) was instead determined from ICP measurements of the PLS. Fig. 2 shows the leaching efficiency of metals into solution, and hence shows that the necessary 70% leaching of Co and Mn can be achieved within 5 minutes, even under relatively mild reaction conditions. Although in industrial processes with mixed feedstock materials it would be preferable to fully leach all materials, it could be beneficial to adapt a continuous flow process for a feedstock of NMC 1:1:1 as this will significantly reduce the processing times. The remaining black mass could then be calcined with lithium hydroxide or carbonate to form LiNMC 8:1:1, whilst the leachate containing Mn and Co is treated separately to produce precursor chemicals for other industries or cathode blends.
3.2 Recovery of Co and Mn through precipitation
As Ni has low solubility in OxA:ChCl, a synthetic leaching system containing 1.98 g L−1 manganese and 1.83 g L−1 cobalt was prepared and used to test different precipitation parameters. The anti-solvents used were deionised water, and aqueous solutions of 0.5, 1.0, and 1.8 mol dm−3 OxA, all at room temperature. The effect of solid oxalic acid dihydrate was also tested, and it was found that the oxalic acid remained as a solid and did not induce precipitation of metal oxalate complexes. Upon the addition of each of the anti-solvents to the blue PLS in a volume ratio of 1:1 PLS-to-anti-solvent, a pink solution developed within seconds, along with the formation of a pink precipitate (Fig. 3). After 1 hour under static conditions, all solutions contained pale pink precipitates with no trace of the blue cobalt tetrachloride solution species. It was observed that the higher the oxalic acid dihydrate content, the slower the anti-solvent mixed with the PLS, and the more gelatinous the resulting precipitate.
 | ||
| Fig. 3 Images of the mixed element solutions showing the effect of anti-solvent on the PLS, after (a) t = 0 min, and (b) t = 24 h. The full time series can be seen in Fig. S2.† | ||
X-ray diffraction (XRD) analysis of these precipitates showed the presence of Mn(C2O4)·2H2O and Co(C2O4)·2H2O in all cases (Fig. S3†), although overlapping peaks suggests a mixed oxalate rather than two separate oxalate phases. SEM images in Fig. S4† show their morphology, with analysis via EDX confirming the idea of a mixed oxalate, showing a distribution of Mn and Co in the same places (Fig. S5†). The relative amounts of Co and Mn detected in these precipitates obtained from use of water as the anti-solvent were 60.9 and 39.1 atomic%, respectively. The precipitates were also analysed via infrared spectroscopy (IR) to confirm their structure (Fig. S6 and Table S2†), which showed the presence of C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and O–C–O stretches typical of oxalate species at approximately 1360 and 1612, 1315, and 820 cm−1, respectively. The presence of OH2 asymmetric stretches at 3362 cm−1 indicates a hydrated oxalate species. Mn–O and Co–O–H librations were assigned to values of 494 and 732 cm−1, respectively, suggesting coordination of the oxalate moiety to the metals.48,49
O, and O–C–O stretches typical of oxalate species at approximately 1360 and 1612, 1315, and 820 cm−1, respectively. The presence of OH2 asymmetric stretches at 3362 cm−1 indicates a hydrated oxalate species. Mn–O and Co–O–H librations were assigned to values of 494 and 732 cm−1, respectively, suggesting coordination of the oxalate moiety to the metals.48,49
The hydration number of the precipitates were confirmed via thermogravimetric analysis (TGA) (Fig. S7 and Table S3†). For monoclinic α-Mn(C2O4)·2H2O and Co(C2O4)·2H2O, the dehydration typically occurs in one step at 130 °C and is associated with a ca. 20% mass loss.50,51 For complexes with higher hydration numbers, e.g. Mn(C2O4)·3H2O, dehydration occurs at a lower temperature (80 °C) and is a three-step process. The precipitates produced from the OxA:ChCl systems showed a loss of ca. 18–19 wt% at 145–150 °C, indicating the loss of 2 moles of water per mole of metal oxalate complex. Comparison of these values to the TGA of pure Mn(C2O4)·2H2O and Co(C2O4)·2H2O confirms the hydration number, with mass losses of 19.9(1) and 18.5(2)%, respectively. TGA also supports the hypothesis that the formed precipitate is a mixed oxalate rather than two separate oxalates, as only two mass loss steps are present. Previous studies on two separate oxalates that have been mechanically mixed shows four mass loss steps, as the onset decomposition temperature is related to the electronegativity of the central metal ion.52,53 Since Co is more electronegative than Mn, the TGA curve from the precipitates resemble that of Co(C2O4)·2H2O more than Mn(C2O4)·2H2O. This suggests a mixed oxalate has been formed with the structure of MnxCoy(C2O4)·2H2O.
ICP analysis of the stripped solutions showed that up to 97% of all cobalt and manganese ions were precipitated out of solution (Fig. 4a). ICP analysis of the precipitates showed that the ratio of Mn:Co was approximately 1:1.4 mol% for each anti-solvent (Fig. 4b), indicating that the anti-solvents used here have little effect on selectivity. The recovered precipitates were calculated to contain ca. 30 wt% Mn(C2O4)·2H2O and ca. 45 wt% Co(C2O4)·2H2O, along with 25 wt% of solid from the DES, suggesting that x = 0.4 and y = 0.6 for the compound MnxCoy(C2O4)·2H2O. Therefore, the most optimal anti-solvent is the one which is most cost-effective, i.e. deionised water. To regenerate the DES, one potential method would be to evaporate off the excess water, and the OxA content can be adjusted through simple addition, based on how many moles were removed in the precipitate.
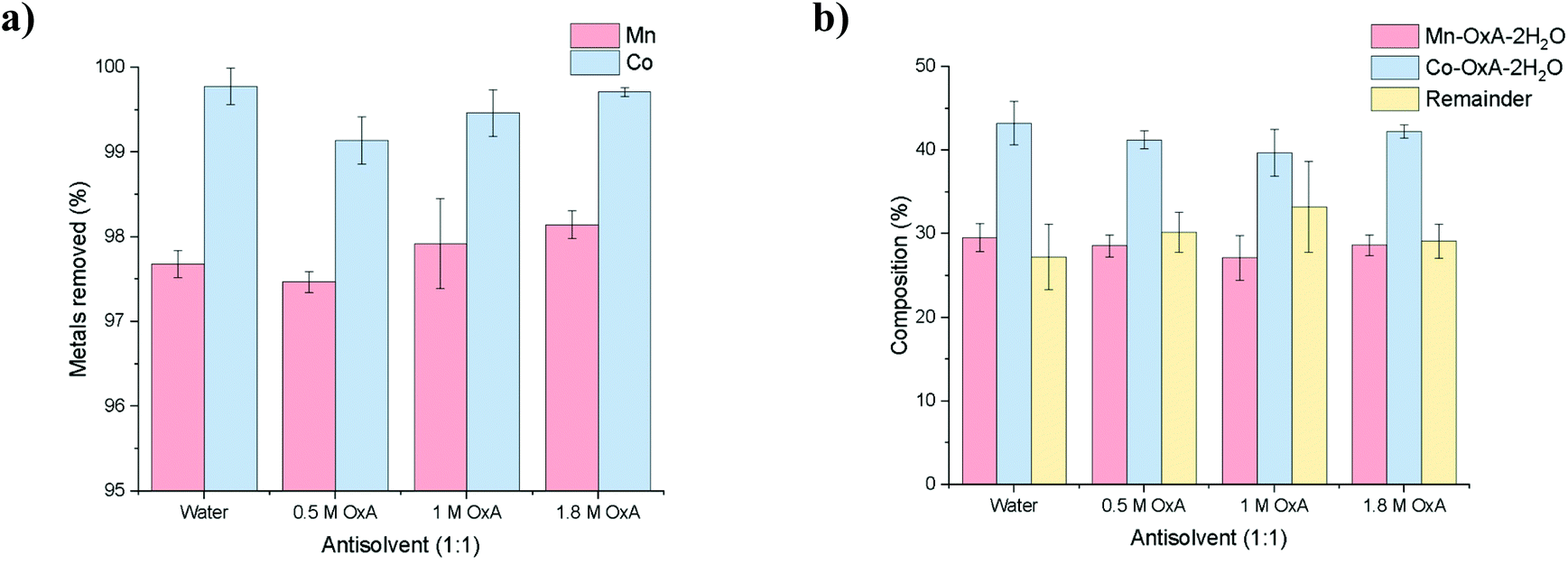 | ||
| Fig. 4 ICP data for: (a) the stripped solution, showing metal removal, and (b) Co and Mn content of the precipitates obtained from addition of various anti-solvents. Elemental compositions are in mol%. | ||
3.3 Proposed flowsheet
Historically, LiCoO2 and LiMn2O4 have been used as cathode active materials in EV LIBs, although over the last decade there have been developments into different materials such as LiFePO4 (LFP), LiNiCoAlO2 (NCA) and LiNiMnCoO2 (LiNMC), in which the ratios of nickel, manganese, and cobalt can vary depending on both the manufacturer and also within a manufacturer's fleet.54,55 This variability in cathode chemistry will result in a waste stream that will have different LiNMC compositions, and it is likely that even in the future there will be mixed cathode chemistries needed to be recycled because of EoL car batteries being diverted into second life applications, or the continuance of EVs in the second hand car market.56,57While this variability in waste stream composition is likely to be problematic for the direct recycling of LIB cathodes, with the subsequent requirements for variable amounts of processing chemicals for each batch of electrodes, the ability to enrich a nickel-bearing phase is important because manufacturers are shifting towards the use of LiNMC-811 as the active cathode material.58–60 By selectively extracting cobalt and manganese from the black mass through the use of OxA:ChCl, the nickel content of the undissolved material is enriched, allowing it to be selectively recovered. Cobalt and manganese can then be precipitated out of solution as mixed metal oxalates in a 1:1 ratio using water as an antisolvent. These mixed oxalates can either be used as a feedstock for LIB manufacture via high temperature processing with the requisite nickel and lithium complexes in the desired ratios or converted to mixed Mn/Co oxides for use in alkaline fuel cell catalysts.61–64
We therefore propose the following flowsheet (Fig. 5) for processing cathode black mass from LIBs. First, the battery must be disassembled and the cathodes must be separated from the anodes. The active materials will then be removed from the electrodes via an ultrasonic process.19 Digestion of the LiNMC black mass will take place in OxA:ChCl at 80 °C, leaving behind a nickel-enriched solid phase which can then be recovered via filtration. To recover cobalt and manganese from the PLS, water will be used as an antisolvent. This has previously been carried out on a large scale (200 kg) for the recovery of zinc chloride from a process to recycle metals from electric arc furnace dust, where the DES was shown to be recovered by evaporation of the water and this was demonstrated on 5 repeat runs without significant loss of material.30
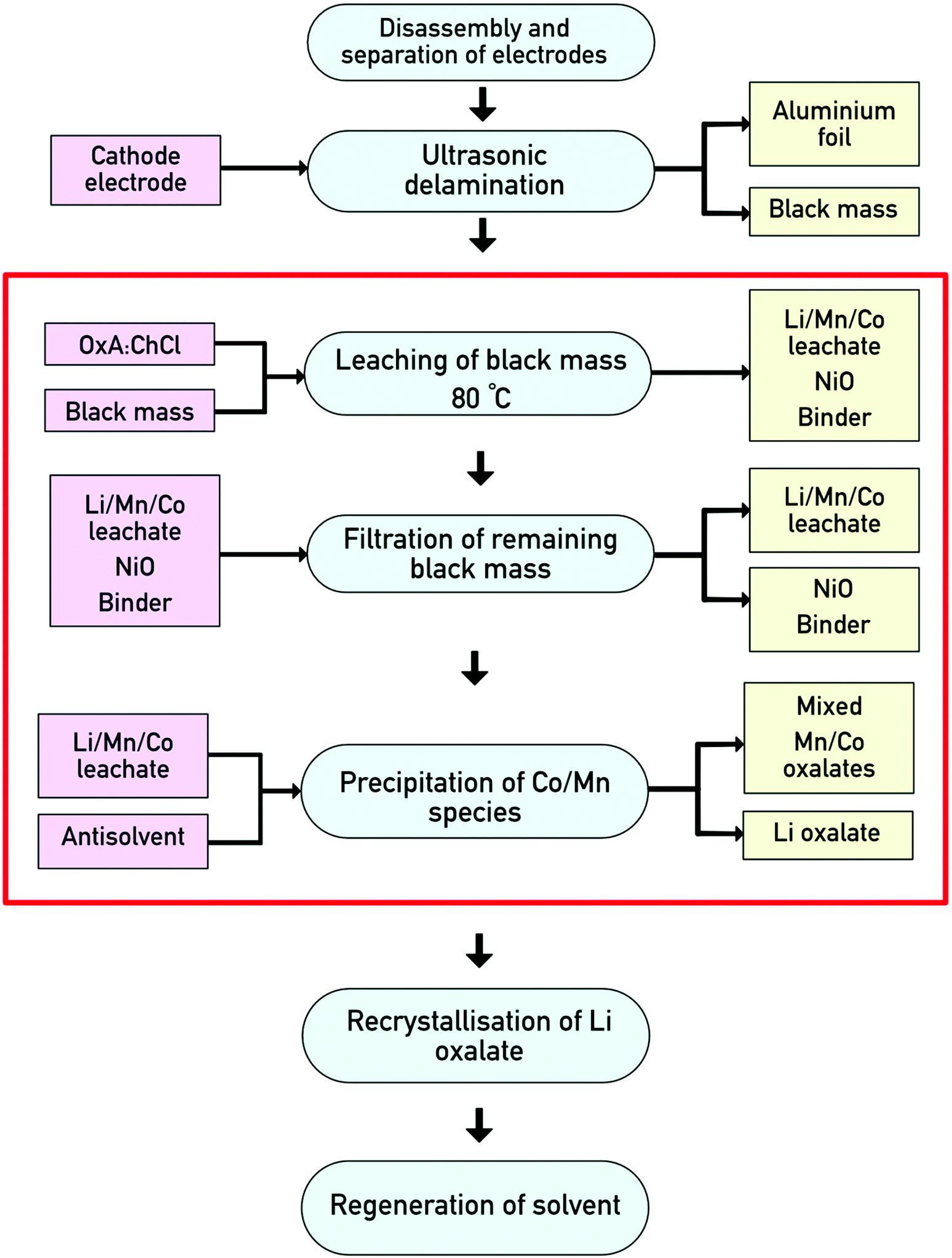 | ||
| Fig. 5 Proposed flow sheet for the processing of LIB cathodes. The red box highlights the processes discussed in the present work. | ||
The recovery of lithium from LiCoO2 in OxA:ChCl has been demonstrated by Lu et al. by cooling of the leachate to form LiHC2O4 and CoC2O4.42 The solubilities of LiHC2O4 and CoC2O4 in water are 80 g L−1 and 0.037 g L−1 respectively, allowing for efficient separation when washed with water.65,66 LiHC2O4·4H2O was then recovered by recrystallisation. In this study, LiHC2O4·4H2O will be present in the antisolvent after the precipitation of manganese and cobalt due to its high solubility in water, and can be recovered via recrystallisation. Since the cathode will be subjected to ultrasound for delamination prior to dissolution, there is the possibility of lithium dissolution at this stage depending on the solvent used. Subjecting the cathode to ultrasound using the same electrodes and conditions as Lei et al. (1250 W in 0.1 mol dm−3 NaOH) for 5 seconds was found to result in 10–11% Li removal.19 This could be increased by prolonging the time the cathode spends in contact with the solvent, depending on how the user wishes to design the flowsheet.
4. Conclusions
The dissolution of LiNMC was investigated in a deep eutectic solvent formed from oxalic acid dihydrate and choline chloride. In a system with no added water, manganese and cobalt could be fully leached within 5 hours at a temperature of 80 °C, whereas only 20% lithium was leached. Higher water contents decreased the cobalt and manganese leaching efficiency, but improved lithium leaching, potentially due to the formation of insoluble Co/Mn mixed oxalate species. The optimal leaching parameters were found to be 80 °C with a S:L ratio of 15 g L−1 for 2 hours, with no added water. In all systems, nickel remained in the solid phase as either an oxide or an oxalate and could be recovered by filtration. The cobalt and manganese were precipitated from the DES as mixed metal oxalates through the simple addition of water. The lithium could be recovered via recrystallisation from the remaining PLS. The DES could theoretically be regenerated by evaporating off the excess water and adjusting the oxalic acid content by an amount stoichiometric to the moles of metal oxalates removed. This approach enables selective leaching of metals from LiNMC-based cathode materials and enables the composition of recycled materials to be tuned so that the current trends for high nickel content materials such as LiNMC-811 can be achieved.
Author contributions
Dana Thompson: Investigation, writing – original draft, writing – review & editing, visualization. Ioanna Pateli: Investigation, writing – original draft, visualization. Chunhong Lei: Investigation. Abbey Jarvis: Investigation. Andrew Abbott: Conceptualization, writing – original draft, writing – review & editing, supervision, funding acquisition. Jennifer Hartley: Investigation, writing – original draft, writing – review & editing, visualization.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the Faraday Institution (grant codes FIRG005 and FIRG006) for funding (Project website https://relib.org.uk). This research also received funding from the European Commission's H2020 – Marie Sklodowska Curie Actions (MSCA) − Innovative Training Networks within the SOCRATES project under the grant agreement no. 721385 (Project website: https://etn-socrates.eu).References
- H. M. Government, The Road To Zero, Department for Transport, London, 2018 Search PubMed.
- C. Helbig, A. M. Bradshaw, L. Wietschel, A. Thorenz and A. Tuma, J. Cleaner Prod., 2018, 172, 274–286 CrossRef CAS.
- Government takes historic step towards net-zero with end of sale of new petrol and diesel cars by 2030, Department for Transport, Office for Low Emission Vehicles, Department for Business, Energy & Industrial Strategy, The Rt Hon Alok Sharma MP, and The Rt Hon Grant Shapps MP, UK, 2020.
- European Commission: Secretariat General, ‘Fit for 55′: delivering the EU's 2030 Climate Target on the way to climate neutrality, European Comission, Brussels, 2021 Search PubMed.
- International Energy Agency, Prospects for electric vehicle deployment, International Energy Agency, Paris, 2021 Search PubMed.
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich, S. Lambert, A. Abbott, K. Ryder, L. Gaines and P. Anderson, Nature, 2019, 575, 75–86 CrossRef CAS PubMed.
- H. E. Melin, The lithium-ion battery end-of-life market 2018–2025, Circular Energy Storage, United Kingdom, 2018 Search PubMed.
- C. Ekberg and M. Petranikova, in Lithium Process Chemistry, ed. A. Chagnes and J. Swiatowska, Elsevier, 2015, vol. 1, ch. 7, pp. 233–267 Search PubMed.
- N. Peeters, K. Binnemans and S. Riaño, Green Chem., 2020, 22, 4210–4221 RSC.
- F. Larouche, F. Tedjar, K. Amouzegar, G. Houlachi, P. Bouchard, G. P. Demopoulos and K. Zaghib, Materials, 2020, 13, 801 CrossRef CAS PubMed.
- X. Zheng, Z. Zhu, X. Lin, Y. Zhang, Y. He, H. Cao and Z. Sun, Engineering, 2018, 4, 361–370 CrossRef CAS.
- W. Lv, Z. Wang, H. Cao, Y. Sun, Y. Zhang and Z. Sun, ACS Sustainable Chem. Eng., 2018, 6, 1504–1521 CrossRef CAS.
- L. Li, X. Zhang, M. Li, R. Chen, F. Wu, K. Amine and J. Lu, Electrochem. Energy Rev., 2018, 1, 461–482 CrossRef CAS.
- J. Ordoñez, E. J. Gago and A. Girard, Renewable Sustainable Energy Rev., 2016, 60, 195–205 CrossRef.
- J. Neumann, M. Petranikova, M. Meeus, J. D. Gamarra, R. Younesi, M. Winter and S. Nowak, Adv. Energy Mater., 2022, 2102917 CrossRef CAS.
- J. Partinen, P. Halli, S. Helin, B. P. Wilson and M. Lundström, Hydrometallurgy, 2022, 208, 105808 CrossRef CAS.
- J. Nan, D. Han and X. Zuo, J. Power Sources, 2005, 152, 278–284 CrossRef CAS.
- J. Nan, D. Han, M. Yang, M. Cui and X. Hou, Hydrometallurgy, 2006, 84, 75–80 CrossRef CAS.
- C. Lei, I. Aldous, J. M. Hartley, D. L. Thompson, S. Scott, R. Hanson, P. A. Anderson, E. Kendrick, R. Sommerville, K. S. Ryder and A. P. Abbott, Green Chem., 2021, 23, 4710–4715 RSC.
- O. Buken, K. Mancini and A. Sarkar, RSC Adv., 2021, 11, 27356–27368 RSC.
- P. Meshram, A. Mishra, Abhilash and R. Sahu, Chemosphere, 2020, 242, 125291 CrossRef CAS PubMed.
- R. Golmohammadzadeh, F. Faraji and F. Rashchi, Resour. Conserv. Recycl., 2018, 136, 418–435 CrossRef.
- W. Gao, J. Song, H. Cao, X. Lin, X. Zhang, X. Zheng, Y. Zhang and Z. Sun, J. Cleaner Prod., 2018, 178, 833–845 CrossRef CAS.
- T. Or, S. W. D. Gourley, K. Kaliyappan, A. Yu and Z. Chen, Carbon Energy, 2020, 2, 6–43 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, K. J. McKenzie and S. U. Obi, J. Chem. Eng. Data, 2006, 51, 1280–1282 CrossRef CAS.
- A. P. Abbott, G. Frisch, J. Hartley and K. S. Ryder, Green Chem., 2011, 13, 471–481 RSC.
- P. Cen, K. Spahiu, M. S. Tyumentsev and M. Foreman, Phys. Chem. Chem. Phys., 2020, 22, 11012–11024 RSC.
- S. Riaño, M. Petranikova, B. Onghena, T. Vander Hoogerstraete, D. Banerjee, M. R. S. Foreman, C. Ekberg and K. Binnemans, RSC Adv., 2017, 7, 32100–32113 RSC.
- N. Rodriguez Rodriguez, L. Machiels and K. Binnemans, ACS Sustainable Chem. Eng., 2019, 7, 3940–3948 CrossRef CAS.
- A. P. Abbott, J. Collins, I. Dalrymple, R. C. Harris, R. Mistry, F. Qiu, J. Scheirer and W. R. Wise, Aust. J. Chem., 2009, 62, 341–347 CrossRef CAS.
- P. Zürner and G. Frisch, ACS Sustainable Chem. Eng., 2019, 7, 5300–5308 CrossRef.
- I. M. Pateli, A. P. Abbott, K. Binnemans and N. Rodriguez Rodriguez, RSC Adv., 2020, 10, 28879–28890 RSC.
- M. Landa-Castro, J. Aldana-González, M. G. Montes de Oca-Yemha, M. Romero-Romo, E. M. Arce-Estrada and M. Palomar-Pardavé, J. Alloys Compd., 2020, 830, 154650 CrossRef CAS.
- M. K. Tran, M.-T. F. Rodrigues, K. Kato, G. Babu and P. M. Ajayan, Nat. Energy, 2019, 4, 339–345 CrossRef CAS.
- K. Wang, T. Hu, P. Shi, Y. Min, J. Wu and Q. Xu, ACS Sustainable Chem. Eng., 2021, 10(3), 1149–1159 CrossRef.
- Y. Chen, Y. Lu, Z. Liu, L. Zhou, Z. Li, J. Jiang, L. Wei, P. Ren and T. Mu, ACS Sustainable Chem. Eng., 2020, 8, 11713–11720 CrossRef CAS.
- G. Zante and M. Boltoeva, Sustainable Chem., 2020, 1, 238–255 CrossRef.
- I. M. Pateli, D. Thompson, S. S. M. Alabdullah, A. P. Abbott, G. R. T. Jenkin and J. M. Hartley, Green Chem., 2020, 22, 5476–5486 RSC.
- K. Binnemans and P. T. Jones, J. Sustain. Metall., 2017, 3, 570–600 CrossRef.
- O. M. Gradov, I. V. Zinov'eva, Y. A. Zakhodyaeva and A. A. Voshkin, Metals, 2021, 11, 1964 CrossRef CAS.
- A. Verma, R. Kore, D. R. Corbin and M. B. Shiflett, Ind. Eng. Chem. Res., 2019, 58, 15381–15393 CrossRef CAS.
- Q. Lu, L. Chen, X. Li, Y. Chao, J. Sun, H. Ji and W. Zhu, ACS Sustainable Chem. Eng., 2021, 9, 13851–13861 CrossRef CAS.
- D. Dupont and K. Binnemans, Green Chem., 2015, 17, 856–868 RSC.
- N. Rodriguez Rodriguez, A. van den Bruinhorst, L. J. B. M. Kollau, M. C. Kroon and K. Binnemans, ACS Sustainable Chem. Eng., 2019, 7, 11521–11528 CrossRef CAS.
- A. P. Abbott, E. I. Ahmed, R. C. Harris and K. S. Ryder, Green Chem., 2014, 16, 4156–4161 RSC.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed. Engl., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- H. H. Loeffler and B. M. Rode, J. Chem. Phys., 2002, 117, 110–117 CrossRef CAS.
- H. G. M. Edwards and P. H. Hardman, J. Mol. Struct., 1992, 273, 73–84 CrossRef CAS.
- H. L. Barazorda-Ccahuana, M. Nedyalkova, I. Kichev, S. Madurga, B. Donkova and V. Simeonov, ACS Omega, 2020, 5, 9071–9077 CrossRef CAS PubMed.
- B. Donkova and D. Mehandjiev, Thermochim. Acta, 2004, 421, 141–149 CrossRef CAS.
- S. Majumdar, I. G. Sharma, A. C. Bidaye and A. K. Suri, Thermochim. Acta, 2008, 473, 45–49 CrossRef CAS.
- C.-H. Zheng and D.-L. Fang, Mater. Res. Bull., 2008, 43, 1877–1882 CrossRef CAS.
- C. Laberty, P. Alphonse, J. J. Demai, C. Sarda and A. Rousset, Mater. Res. Bull., 1997, 32, 249–261 CrossRef CAS.
- A. Accardo, G. Dotelli, M. L. Musa and E. Spessa, Appl. Sci., 2021, 11, 1160 CrossRef CAS.
- Cathode Active Materials, https://www.targray.com/li-ion-battery/cathode-materials/cathode-active-materials, (accessed 01/12/2021).
- L. Pickett, J. Winnett, D. Carver and P. Bolton, Electric vehicles and infrastructure, House of Commons Library, 2021 Search PubMed.
- H. E. Melin, State-of-the-art in reuse and recycling of lithium-ion batteries – A research review, Circular Energy Storage, London, 2019 Search PubMed.
- NMC Battery Material, https://www.targray.com/li-ion-battery/cathode-materials/nmc, (accessed 04/01/2022).
- N. Campagnol, M. Erriquez, D. Schwedhelm, J. Wu and T. Wu, Building better batteries: Insights on chemistry and design from China, McKinsey & Company, 2021 Search PubMed.
- S. Gifford, Lithium, Cobalt and Nickel: The Gold Rush of the 21st Century, The Faraday Institution, UK, 2020 Search PubMed.
- Y. Yang, Y. Xiong, M. E. Holtz, X. Feng, R. Zeng, G. Chen, F. J. DiSalvo, D. A. Muller and H. D. Abruna, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 24425–24432 CrossRef CAS PubMed.
- Y. Wang, D. Y. C. Leung, J. Xuan and H. Wang, Renewable Sustainable Energy Rev., 2017, 75, 775–795 CrossRef CAS.
- O. A. Bulavchenko, T. N. Afonasenko, A. R. Osipov, A. A. Pochtar, A. A. Saraev, Z. S. Vinokurov, E. Y. Gerasimov and S. V. Tsybulya, Nanomaterials, 2021, 11, 988 CrossRef CAS PubMed.
- Y. Z. Zhang, J. Zhao, J. Xia, L. Wang, W. Y. Lai, H. Pang and W. Huang, Sci. Rep., 2015, 5, 8536 CrossRef CAS PubMed.
- S. Chandran, R. Paulraj and P. Ramasamy, J. Cryst. Growth, 2017, 468, 68–72 CrossRef CAS.
- W. M. Haynes, D. R. Lide and T. J. Bruno, CRC handbook of chemistry and physics: a ready-reference book of chemical and physical data, CRC Press, 2016, pp. 1–139 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc00606e |
| This journal is © The Royal Society of Chemistry 2022 |