
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-heme manganese(II) complex-catalysed oxidative cleavage of 1,2-diols via alcohol-assisted O2 activation†
Zhiliang
Huang
 ,
Renpeng
Guan
,
Elliot L.
Bennett
and
Jianliang
Xiao
*
,
Renpeng
Guan
,
Elliot L.
Bennett
and
Jianliang
Xiao
*
Department of Chemistry, University of Liverpool, Liverpool L69 7ZD, UK. E-mail: j.xiao@liverpool.ac.uk
First published on 6th April 2022
Abstract
Bio-inspired oxidation strategies are gaining increasing interest in the synthesis of fine and commodity chemicals. In particular, the development of catalytic oxidation systems with bio-relevant metal complexes capable of activating molecular oxygen (O2) is of utmost interest. In this work, a well-defined non-heme Mn(II) complex is found to catalyse, under the irradiation of blue light, the efficient aerobic oxidative cleavage of 1,2-diols to afford valuable carbonyl compounds and five-membered heterocycles. The photo-promoted catalysis allows for aromatic, aliphatic as well as bio-derived 1,2-diols to be cleaved, while tolerating a broad range of functional groups. Preliminary mechanistic investigation suggests that the catalytic cycle features two sequential redox events, with the first involving one alcohol molecule reducing one oxygen atom of O2 to water while promoting the formation of an active Mn-oxo species, which cleaves a second alcohol molecule in the subsequent event.
Introduction
Manganese is one of the most abundant, inexpensive and environmentally friendly metals on Earth. With readily accessible oxidation states ranging from II to VII, manganese has a rich redox chemistry, which has been exploited by nature in a number of important enzymatic reactions,1 such as oxidation of water to form O2,2 disproportionation of toxic superoxide,3 oxidation of oxalate for seed germination4 and catabolism of xenobiotic aromatics.5 Inspired by nature, a great number of manganese-based homogeneous catalysts have been developed for oxidation reactions over the past several decades. However, the vast majority of these catalysts necessitate the use of activated oxidants, such as H2O2, NaOCl/Br, PhIO and its derivatives,6 eliciting cost, waste and/or environmental impact. Thus, manganese catalysts that enable more selective, milder and safer oxidation with cleaner and more economical oxidants are still highly sought after. Of particular interest is to develop bio-inspired Mn-catalysed oxidation strategies, in which the readily accessible, inexpensive, and clean O2 is used as the oxidant.Activation of O2 is generally required for aerobic oxidation. The ground state of O2 is a triplet diradical that is spin forbidden to react via common two-electron reaction mechanisms. Nature circumvents the spin state barrier and performs highly selective aerobic oxidations by employing redox-active metal ions (FeII, MnII, CuI, etc.) that are able to transfer a single electron to O2.7 Taking inspiration from nature, numerous biomimetic transition metal complexes bearing heme and non-heme polydentate ligands have been synthesized for studying O2 activation in the past four decades or so.7b,8 However, there are still relatively few well-defined Fe and Mn complexes that activate O2 in a controlled manner and particularly, the use of benign and inexpensive O2 for selective oxidation with such biologically relevant metal complexes remains “a significant challenge for the synthetic chemist”.7b The main challenging issues are: (1) O2 activation is not straightforward, as the redox potential for reduction of O2 to superoxide is unfavorable;7a so specific ligands must be employed to promote the reduction process and stabilize the resulting metal-superoxide; (2) the metal-superoxide intermediate is too reactive to engage in a selective oxidation process and more prone to poorly controlled radical-type pathways;9 (3) a reducing agent capable of donating two electrons at the right time is generally required to form high valent M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species for subsequent substrate oxidation.10 Few biomimetic metal complexes are known that can fulfil these roles, i.e. reduction of O2 while abstracting electrons from a reducing agent.11 Herein, we report a well-defined non-heme Mn(II) complex for oxidative cleavage of 1,2-diols via O2 activation promoted by visible light, in which one diol (or cheap methanol) provides electrons and protons, leading to its oxidative cleavage while initiating the cleavage of a second diol molecule.
O species for subsequent substrate oxidation.10 Few biomimetic metal complexes are known that can fulfil these roles, i.e. reduction of O2 while abstracting electrons from a reducing agent.11 Herein, we report a well-defined non-heme Mn(II) complex for oxidative cleavage of 1,2-diols via O2 activation promoted by visible light, in which one diol (or cheap methanol) provides electrons and protons, leading to its oxidative cleavage while initiating the cleavage of a second diol molecule.
Oxidative C–C bond cleavage of 1,2-diols is an important reaction, finding countless applications in organic chemistry since its discovery by Malaprade and Criegee around the 1930s.12 Although the original methods remain the most widely used for the cleavage of vicinal diols, the use of stoichiometric high-valent inorganic oxidants, mainly HIO4 and analogues, Pb(OAc)4 or KMnO4,12,13 brings about serious issues, such as high toxicity and copious amount of waste. Whilst the last three decades have witnessed the development of various catalytic methods and some significant advances in aerobic cleavage of 1,2-diols (Scheme 1),14 developing novel, cheap and green catalysts for this cleavage reaction that shows a broad scope with O2 or air under mild conditions remains highly necessary. In particular, the environmentally friendly, bio-relevant manganese has been much less studied for the aerobic oxidative cleavage of 1,2-diols, with the few reported Mn catalysts all showing limited substrate scope while requiring either a high temperature or a co-reductant (Scheme 1b).14m,n,15 In continuing our study of catalytic oxidation,6d,11b–d,16 this prompted us to develop an efficient, widely applicable non-heme Mn catalyst for this transformation (Scheme 1c).
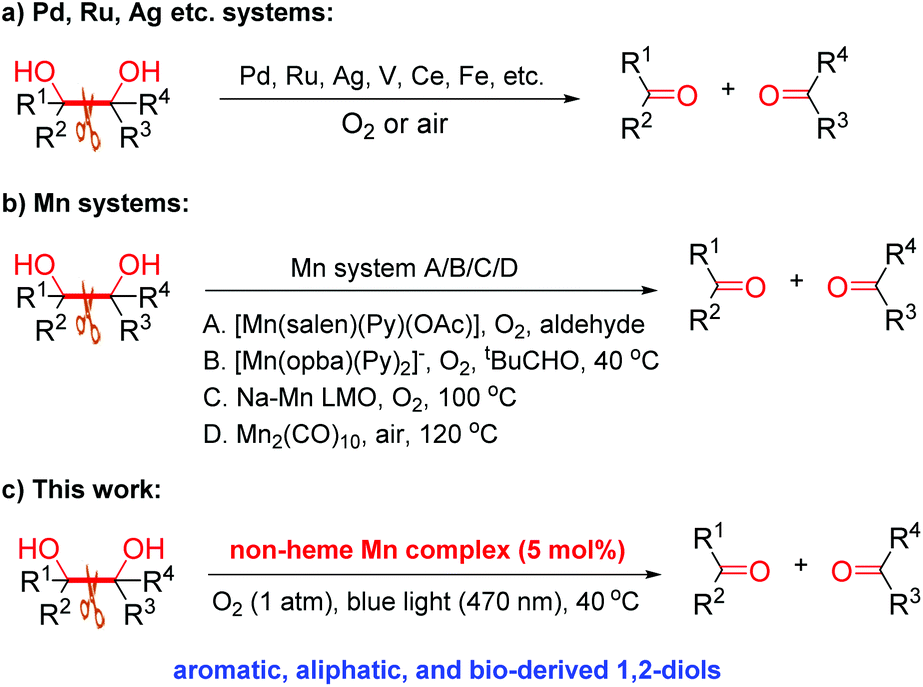 | ||
| Scheme 1 Oxidative cleavage of 1,2-diols with molecular O2. | ||
Results and discussion
Method development
We recently reported a novel photo-Mn catalytic system for the oxidative cleavage of alkenes, in which MeOH acts as the electron and proton donor in the process of O2 activation by Mn(II), leading to its oxidation to formaldehyde and the formation of a Mn(IV)O species capable of catalytically cleaving olefins in a dioxygenase manner [Scheme 2(1)].11b Interestingly, the active oxo species is stabilized by forming a more stable bis-μ-O2-Mn2 dimer, which is photoreversible, thus enhancing the catalyst lifetime. Building on this knowledge, we envisaged a substrate-promoted O2 activation strategy for the oxidative cleavage of 1,2-diols [Scheme 2(2)]. First, a 1,2-diol substrate acts as a two-electron and two-proton donor, reducing one oxygen atom of O2 to water while being cleaved, with the other oxygen atom oxidizing Mn(II) to a Mn(IV)-oxo species under blue light irradiation. As a highly reactive species, the oxo intermediate would then react with another 1,2-diol, leading to its cleavage. There are literature precedents regarding the cleavage of 1,2-diols by Mn-oxo species.15,17The overall process thus involves the oxidative cleavage of two diol molecules with one O2molecule via two sequential two-electron steps, with water as the only by-product. As indicated, methanol can also act as a reductant in forming the Mn(IV)O species.
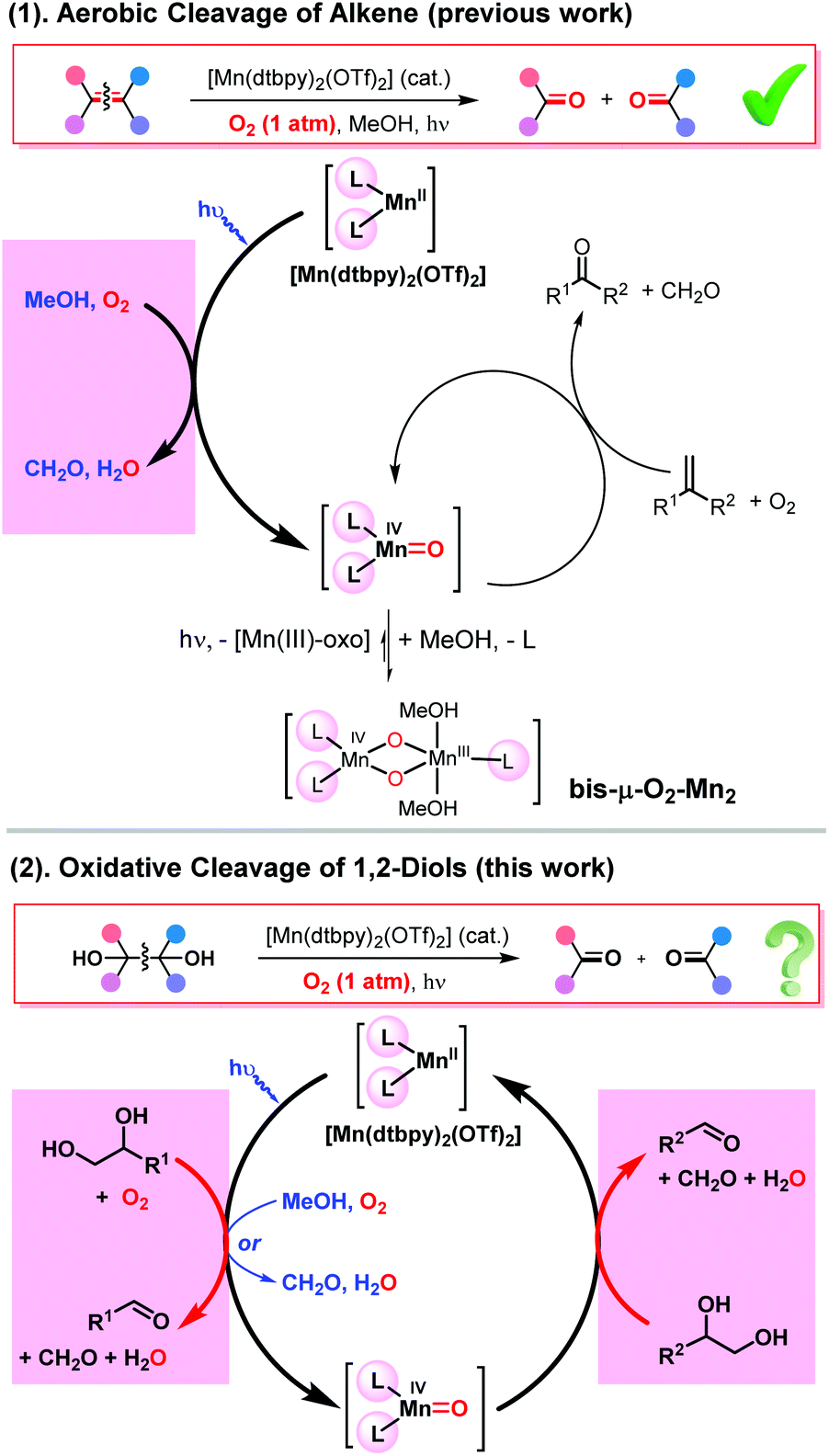 | ||
| Scheme 2 (1) MeOH-promoted O2 activation for the aerobic oxidation of alkenes by a non-heme Mn(II) complex; (2) schematic showing of a substrate or MeOH-promoted O2 activation strategy for the aerobic cleavage of 1,2-diols (L = dtbpy). | ||
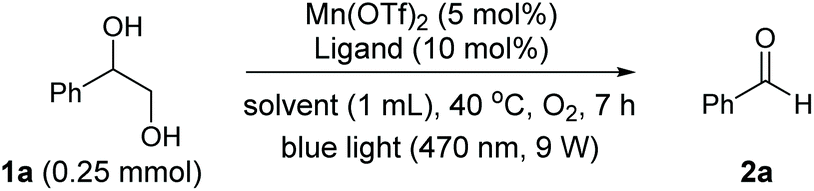
We set out to examine the oxidation of 1,2-diols by employing 1-phenyl-1,2-ethanediol (1a) as the model substrate (Table 1). As expected, under the irradiation of blue light (470 nm, 9 W), 1a was successfully cleaved by O2 with 5 mol% [Mn(dtbpy)2(OTf)2] as the precatalyst in DCE, affording the desired carbonyl product 2a in 67% yield (Table 1, entry 1). The [Mn(dtbpy)2(OTf)2] prepared in situ by combining Mn(OTf)2 and 4,4′-di-tert-butyl-2,2′-bipyridine (L1) displayed a similar activity, which could afford 2a in 69% yield (Table 1, entry 2). As is clear, both the ligand and blue light play important roles in this transformation; in their absence, a dramatically decreased yield or no target product was observed (Table 1, entries 3 and 22, also see Fig. S3 in the ESI†). The effect of the ligand was further explored by testing a range of substituted bipyridines (L2–L9), revealing L1 to give the best yield (Table 1, entries 4–11). A variety of solvents, including those that are considered green18 (Table 1, entries 12–19), were then screened. All of them afforded the desired product in good yields. In particular, for 1-propanol (PrOH), which is environmentally safe in the industrial SSGs and is at the top of the list of green chemicals,19 a high yield of 84% was obtained. These results indicate that this reaction has excellent solvent compatibility. Furthermore, when the oxidation was performed at a 10 mmol scale, 46% isolated yield of 2a could be obtained with 2 mol% Mn catalyst in propanol (4 mL) under an O2 balloon at room temperature for 24 h, indicating the robustness of this protocol (see the ESI for more details†). A somewhat higher yield was obtained when the reaction was performed in a mixed solvent (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 DCE/tBuOH, Table 1, entries 20 and 21). Our subsequent study was therefore focused on the following reaction conditions: Mn(OTf)2 (5 mol%) as the precatalyst, L1 (10 mol%) as the ligand, O2 as the oxidant in DCE/tBuOH (1:3) at 40 °C with blue light irradiation. We note, however, that the oxidative cleavage reactions could well be carried out in a green solvent, such as PrOH (see the ESI† for examples).
:3 DCE/tBuOH, Table 1, entries 20 and 21). Our subsequent study was therefore focused on the following reaction conditions: Mn(OTf)2 (5 mol%) as the precatalyst, L1 (10 mol%) as the ligand, O2 as the oxidant in DCE/tBuOH (1:3) at 40 °C with blue light irradiation. We note, however, that the oxidative cleavage reactions could well be carried out in a green solvent, such as PrOH (see the ESI† for examples).
|
|
||||
|---|---|---|---|---|
| Entry | [Mn] | Ligand | Solvent | Yield of 2a |
a Yield determined by 1H NMR with mesitylene as the internal standard; isolated yield in parentheses.
b Without blue light.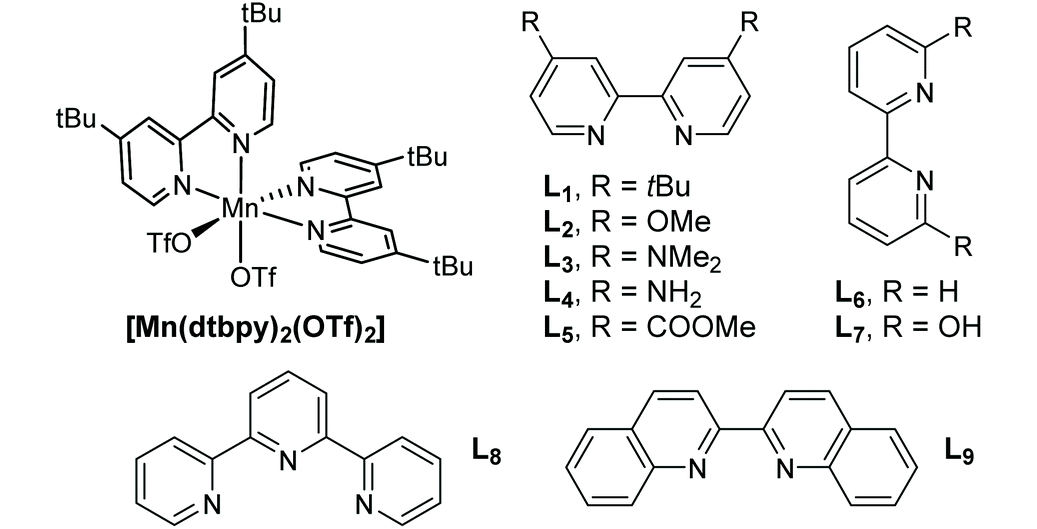
|
||||
| 1 | [Mn(dtbpy)2(OTf)2] | — | DCE | 67% |
| 2 | Mn(OTf)2 | L1 | DCE | 69% |
| 3 | Mn(OTf)2 | — | DCE | 8% |
| 4 | Mn(OTf)2 | L2 | DCE | 58% |
| 5 | Mn(OTf)2 | L3 | DCE | 46% |
| 6 | Mn(OTf)2 | L4 | DCE | 10% |
| 7 | Mn(OTf)2 | L5 | DCE | 34% |
| 8 | Mn(OTf)2 | L6 | DCE | 59% |
| 9 | Mn(OTf)2 | L7 | DCE | 35% |
| 10 | Mn(OTf)2 | L8 | DCE | 36% |
| 11 | Mn(OTf)2 | L9 | DCE | 21% |
| 12 | Mn(OTf)2 | L1 | tBuOH | 76% |
| 13 | Mn(OTf)2 | L1 | PrOH | 84% |
| 14 | Mn(OTf)2 | L1 | EtOH | 60% |
| 15 | Mn(OTf)2 | L1 | Acetone | 72% |
| 16 | Mn(OTf)2 | L1 | EA | 67% |
| 17 | Mn(OTf)2 | L1 | 2-Me-THF | 82% |
| 18 | Mn(OTf)2 | L1 | CH3CN | 70% |
| 19 | Mn(OTf)2 | L1 | TFE | 83% |
| 20 | Mn(OTf) 2 | L 1 |
DCE/tBuOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :3) :3)
|
92% (90%) |
| 21 | [Mn(dtbpy)2(OTf)2] | — | DCE/tBuOH (1:3) |
90% |
| 22b | Mn(OTf)2 | L1 | DCE/tBuOH (1:3) |
0% |
Substrate scope
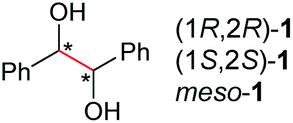
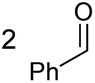
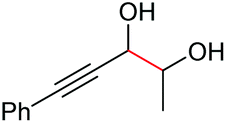
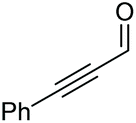
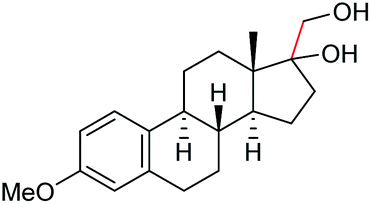
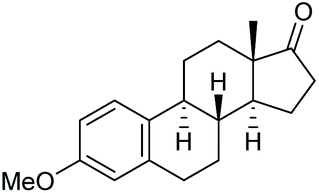
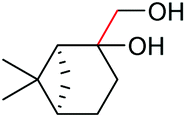
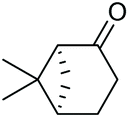
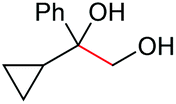
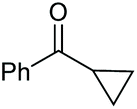
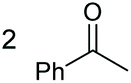
Under the chosen reaction conditions, the scope of diols for the oxidative cleavage was investigated. As shown in Table 2, terminal aromatic 1,2-diols bearing electron-withdrawing or electron-donating groups are all suitable substrates, affording the corresponding aldehydes in good to excellent yields (2b–2i). Internal aromatic 1,2-diols, such as 1-phenylpropane-1,2-diol, (S,S)-(−)-hydrobenzoin, (R,R)-(+)-hydrobenzoin, and meso-hydrobenzoin were also tolerated in this transformation to give 2a in 50%–80% yield (Table 2, entries 9–12). Notably, an unsaturated C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond is compatible with this oxidation process, as is seen from the oxidation leading to 2j in 62% yield. Two alkenyl 1,2-diols were also tested under the standard conditions, both of which could produce the desired cleavage product successfully (2q and 2r). However, lower yields were obtained, which might be caused by the oxidation of the unsaturated CC double bond. Furthermore, a 1,2-diol with an amide functional group was cleaved to afford the amido ketone 2k in 83% yield. In addition, the natural product derivatives, 1-(hydroxymethyl)-4-phenylcyclohexan-1-ol, 2,10-pinanediol, and 17-hydroxy-3-methoxyestra-1,3,5(10)-triene-17-methanol, are all viable for this transformation, affording the ketone products with good to excellent yields (2l–2n). In the oxidation of a cyclopropyl-containing substrate, the cyclopropyl ring remained intact (2o). The viability of oxidative cleavage of tetra-substituted diols is demonstrated by the formation of 2p.
C triple bond is compatible with this oxidation process, as is seen from the oxidation leading to 2j in 62% yield. Two alkenyl 1,2-diols were also tested under the standard conditions, both of which could produce the desired cleavage product successfully (2q and 2r). However, lower yields were obtained, which might be caused by the oxidation of the unsaturated CC double bond. Furthermore, a 1,2-diol with an amide functional group was cleaved to afford the amido ketone 2k in 83% yield. In addition, the natural product derivatives, 1-(hydroxymethyl)-4-phenylcyclohexan-1-ol, 2,10-pinanediol, and 17-hydroxy-3-methoxyestra-1,3,5(10)-triene-17-methanol, are all viable for this transformation, affording the ketone products with good to excellent yields (2l–2n). In the oxidation of a cyclopropyl-containing substrate, the cyclopropyl ring remained intact (2o). The viability of oxidative cleavage of tetra-substituted diols is demonstrated by the formation of 2p.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yieldb |
| a All of the reactions were performed with in situ prepared [Mn(dtbpy)2(OTf)2] and 1 (0.5mmol) in DCE/tBuOH (1/3, 2 mL) at 40 °C under O2 (1 atm) and blue light for 13 h. b Yield of the isolated product. | |||
| 1 |
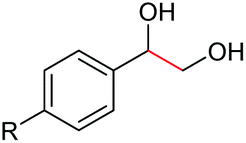
|
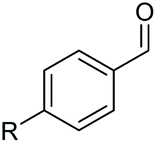
|
2b, R = Cl, 82% |
| 2 | 2c, R = F, 85% | ||
| 3 | 2d, R = CF3, 90% | ||
| 4 | 2e, R = COOMe, 81% | ||
| 5 | 2f, R = OMe, 86% | ||
| 6 |
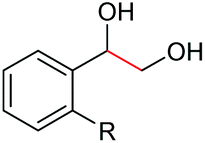
|
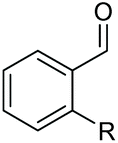
|
2g, R = Cl, 86% |
| 7 | 2h, R = Br, 87% | ||
| 8 |
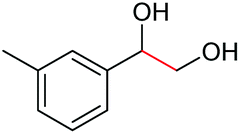
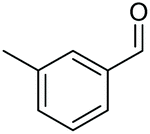
|
|
2i, 84% |
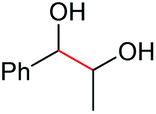
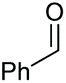
| 9 |
|
|
2a, 51% |
| 10 |
|
|
2a, 77% |
| 11 | 2a, 80% | ||
| 12 | 2a, 78% | ||
| 13 |
|
|
2j, 62% |
| 14 |
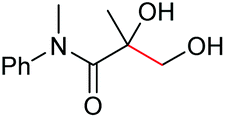
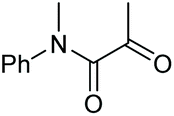
|
|
2k, 83% |
| 15 |
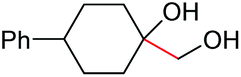
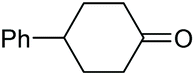
|
|
2l, 86% |
| 16 |
|
|
2m, 76% |
| 17 |
|
|
2n, 92% |
| 18 |
|
|
2o, 54% |
| 19 |
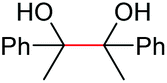
|
|
2p, 59% |
| 20 |
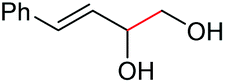
|

|
2q, 25% |
| 21 |
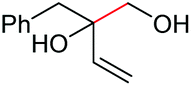
|
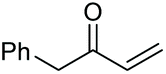
|
2r, 26% |
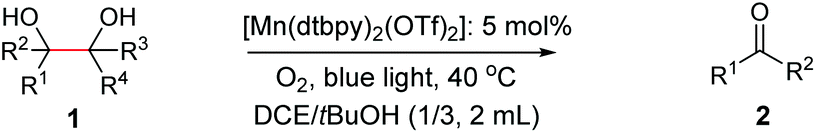
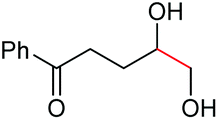
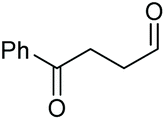
As proposed in Scheme 2, two diols are cleaved in one catalytic cycle. Since the Mn(II) and Mn(IV) species are expected to have different catalytic activities, diols of different reactivities could be cleaved in one-pot. Thus, as exemplified in Table 3, the oxidation of a 1:1 mixture of 1a with 1-(3,4-dimeth-oxyphenyl)ethane-1,2-diol, 2-phenylpropane-1,2-diol, 1-phenylcyclohexane-1,2-diol, cyclooctane-1,2-diol, or 4,5-dihydroxy-1-phenylpentan-1-one afforded 2a and the other corresponding cleavage products in a one-pot, highly selective manner (Table 3, entries 1, 2 and 5–7). Note that only a trace amount of 2s, 2p and 2t and none of 2u and 2v were observed when those oxidation reactions were performed in the absence of 1a under the same conditions. Meanwhile, the product yield from 2,3-diphenylbutane-2,3-diol and 1-cyclopropyl-1-phenylethane-1,2-diol was also improved in the presence of 1a (c.f.Table 3, entries 3, 4 with Table 2, entries 18 and 19). These observations show that diols of lower reactivities can be oxidatively cleaved with a more active diol in a one-pot fashion. The oxidation of the latter presumably activates O2, giving rise to a highly active Mn(IV)-oxo species that cleaves the former.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate 1 | Product 2b (yield) | Yield of 2ac | |
| a All of the reactions were performed with in situ prepared [Mn(dtbpy)2(OTf)2] (5 mol%), 1a (0.5 mmol), and 1 (0.5 mmol) in DCE/tBuOH (1/3, 2 mL) at 40 °C under O2 (1 atm) and blue light for 13 h. b Yield of isolated product. c Yield was obtained by 1H NMR with mesitylene as internal standard. d Without 1a. | ||||
| 1 |
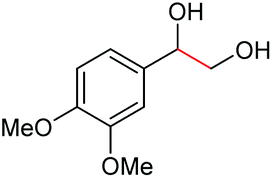
|
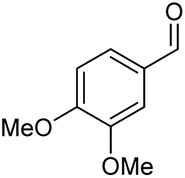
|
(2s, trace)d | 2a, 93% |
| (2s, 93%) | ||||
| 2 |
|
|
(2p, trace)d | 2a, 65% |
| (2p, 75%) | ||||
| 3 |
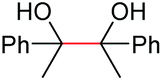
|
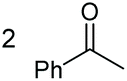
|
(2p, 88%) | 2a, 87% |
| 4 |
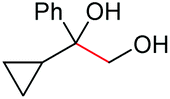
|
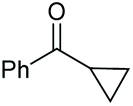
|
(2o, 87%) | 2a, 64% |
| 5 |
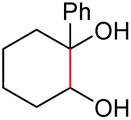
|
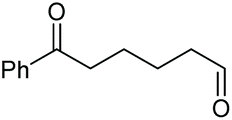
|
(2t, trace)d | 2a, 80% |
| (2t, 83%) | ||||
| 6 |
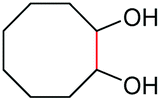
|
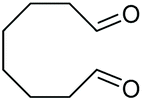
|
(2u, 0%)d | 2a, 51% |
| (2u, 46%) | ||||
| 7 |
|
|
(2v, 0%)d | 2a, 71% |
| (2v, 49%) | ||||
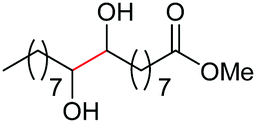
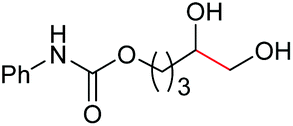
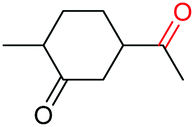
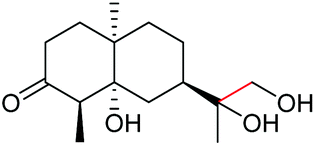
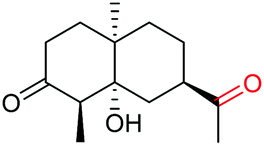
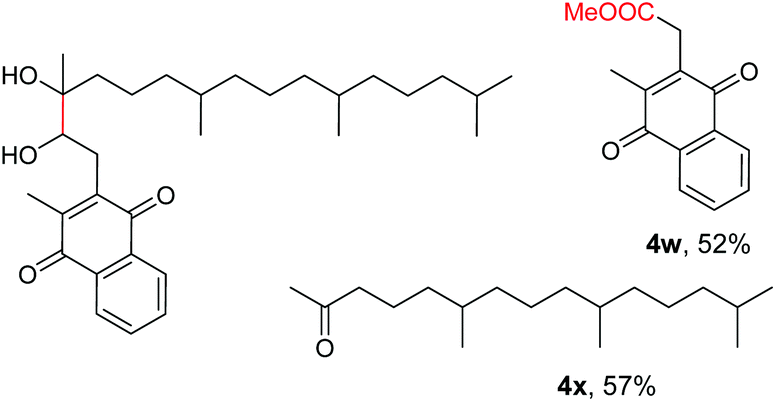
The more challenging aliphatic diols can be more easily dealt with when the oxidation is carried out in methanol, where the resulting more active carbonyls are protected, being converted in situ into the corresponding acetals. This strategy allows acetals as latent aldehydes to be readily synthesized through the oxidative cleavage of diols. As shown in Table 4, this aerobic oxidation method shows high chemoselectivity as well as an excellent functional group compatibility. For example, internal diols derived from methyl oleate, methyl cis-13-docosenoate and methyl 12-hydroxy-9-octadecenoate are viable substrates, producing the corresponding protected mono and di-carbonyl compounds in good yields (Table 4, entries 1–3). Thus, this method could be applied, in a two-step fashion, to the cleavage of unsaturated fatty acids that are challenging, i.e. oxidation to a diol followed by oxidative cleavage of the diol to the desired carbonyls. We notice that the oxidative cleavage of the CC double bonds in unsaturated fatty acids into carbonyls is a reaction of current interest in biomass valorization.20 Those carbonyl products from fatty acids, currently produced on an industrial scale by means of ozonolysis, are value-added commodity chemicals, such as plasticizers and polymer precursors. 1,2-Diols bearing acid, tert-butyldiphenylsilyl (TBDPS), ester, carbamate, urea or ketone functionalities are all suitable, affording the desired products in moderate to good yields (4e and 4h–4l). Both benzylic C–H bonds and isolated alcohol units, which are prone to oxidation, were also tolerated during the oxidation (4f, 4g, 4o and 4p), showing the high chemoselectivity of this oxidation protocol. Meanwhile, long chain 1,2-diols without other functional groups were also oxidatively cleaved to furnish the target carbonyls in good yields (Table 4, entries 12, 13, 16 and 17). Note that the relatively active tertiary C–H bonds in 4m remained intact. Chlorphenesin, which is a preservative and cosmetic biocide, could be oxidized selectively to 1-chloro-4-(2,2-dimethoxyethoxy)benzene 4t in 81% NMR yield. The acetal 4t appears to be unstable in a silica gel column, undergoing decomposition during isolation to afford 2-(4-chlorophenoxy)acetaldehyde in 71% yield. Several other natural product-derived diols, such as sulcatone, citronellylnitrile, dihydrocarvone, α-cyperone and vitamin K1 derivatives, were also studied and shown to be suitable, giving the corresponding carbonyl compounds in moderate to excellent yields (4r, 4s and 4u–4x).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yieldb |
| a All of the reactions were performed with in situ prepared [Mn(dtbgpy)2 (OTf)2] (5 mol%) and 3 (0.5 mmol) in MeOH (2 mL) at 40 °C under O2 (1 atm) and blue light (470 nm, 9 W) for 13 h. b Yield of the isolated product. c 24h. d TFA (30 mol%) was added as the co-catalyst. e Yield was determined by 1H NMR with mesitylene as the internal standard. | |||
| 1c |
|
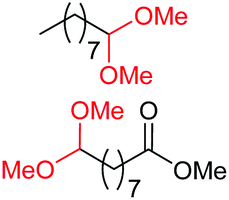
|
4a, 76% |
| 4b, 70% | |||
| 2c |
|
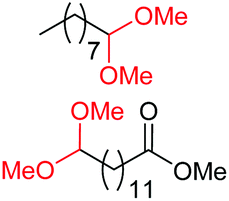
|
4a, 60% |
| 4c, 72% | |||
| 3c |
|
|
4d, 51% |
| 4b, 73% | |||
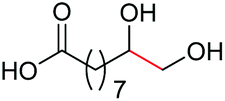
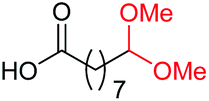
| 4 |
|
|
4e, 83% |
| 5 |
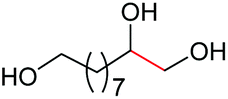
|
|
4f, 84% |
| 6 |
|
|
4g, 55% |
| 7 |
|
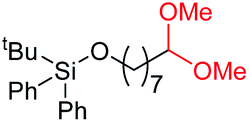
|
4h, 61% |
| 8 |
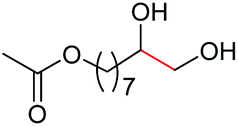
|
|
4i, 75% |
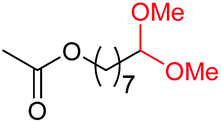
| 9 |
|
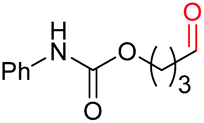
|
4j, 58% |
| 10 |
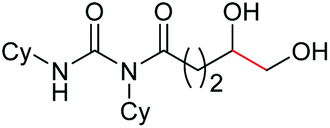
|
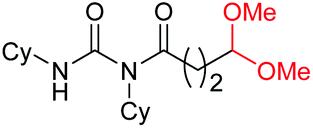
|
4k, 76% |
| 11 |
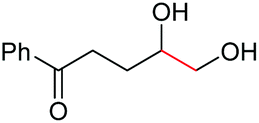
|
|
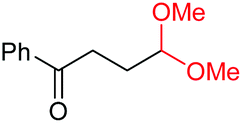
4l, 74% |
| 12 |
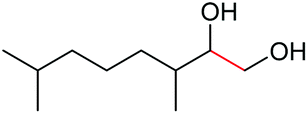
|
|
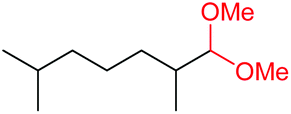
4m, 66% |
| 13 |
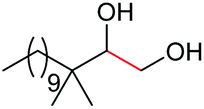
|
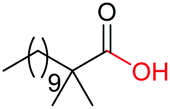
|
4n, 53% |
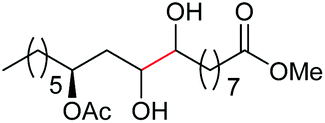
| 14d |
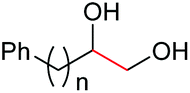
|
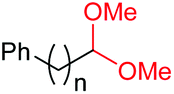
|
4o, n = 1, 70% |
| 15d | 4p, n = 2, 73% | ||
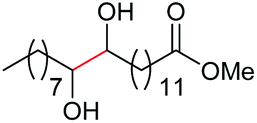
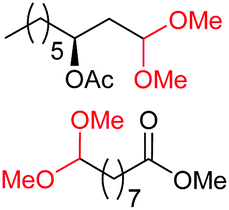
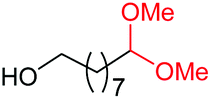
| 16d |
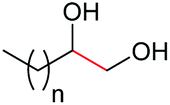
|
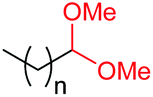
|
4a, n = 7, 74% |
| 17d | 4q, n = 13, 70% | ||
| 18 |
|
|
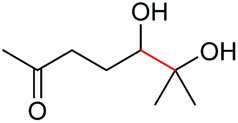
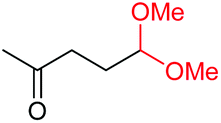
4r, 53% |
| 19 |
|
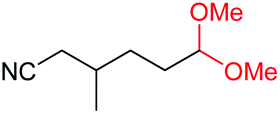
|
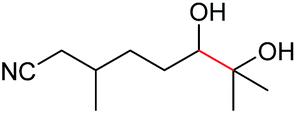
4s, 37% |
| 20 |
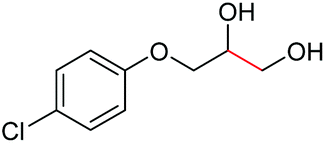
|
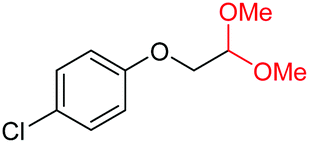
|
4t, 81%e |
| 21 |
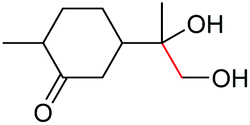
|
|
4u, 47% |
| 22 |
|
|
4v, 92% |
| 23 |
|
||
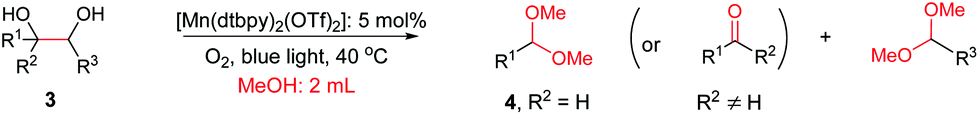
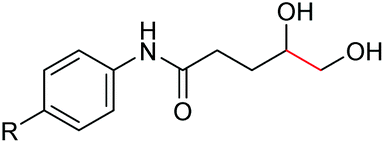
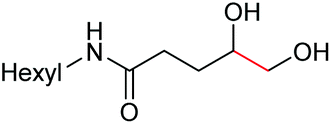
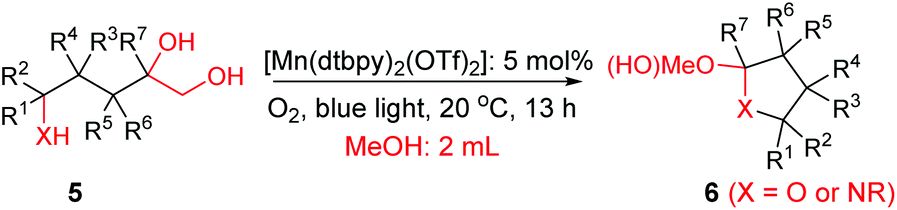
Interestingly, during the examination of the substrate scope, we noticed that a cyclization reaction occurred in nearly quantitative yield after the expected C–C bond cleavage process, when 4,5-dihydroxy-N-phenylpentanamide was employed (Table 5, entry 1). The formation of cyclic 5-methoxy-1-phenylpyrrolidin-2-one (6a) prompted us to further investigate the substrate scope, since 2-pyrrolidinones are important building blocks for a range of heterocyclic compounds,21 as well as the key subunit in many pharmaceuticals and bioactive compounds.22 As can be seen in Table 5, both N-aryl and N-alkyl substituted 4,5-dihydroxypentanamides are viable, giving the corresponding 2-pyrrolidinones in good to excellent yields (6a–6d). Notably, both benzylic C–H and CC triple bonds, usually sensitive to oxidation conditions, were well tolerated (6e and 6f). Amino acid, urea, and carbamate-containing diols could be oxidized selectively as well (6g–6i), showing potential applications in bio-conjugate chemistry. 4,5-Dihydroxy-2-methyl-N-phenylpentanamide, 4,5-dihydroxy-2-methyl-N,3-diphenylpentanamide and 5-dihydroxy-N-(4-(trifluoromethyl)phenyl)pentanamide were also viable for this transformation (6j, 6k and 6l). In the case of the latter two, the analogous 5-hydroxy-functionalized cyclic amides were isolated (Table 5, entries 11 and 12). Furthermore, N-(2-(2,3-dihydroxypropyl)phenyl)acetamide and 2-methyl-5-phenylhexane-1,2,5-triol could successfully be oxidized, furnishing important indoline and tetrahydrofuran products (6m and 6n).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yieldb |
| a All of the reactions were performed with in situ prepared [Mn(dtbpy)2 (OTf)2] (5 mol%) and 5 (0.5 mmol) in MeOH (2 mL) at 20 °C under O2 (1 atm) and blue light for 13 h. b Yield of the isolated product. | |||
| 1 |
|
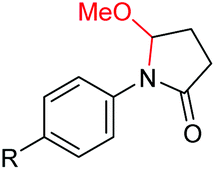
|
6a, R = H, 99% |
| 2 | 6b, R = OMe, 74% | ||
| 3 | 6c, R = CN, 88% | ||
| 4 |
|
|
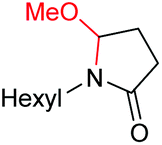
6d, 60% |
| 5 |
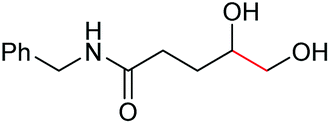
|
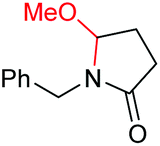
|
6e, 74% |
| 6 |
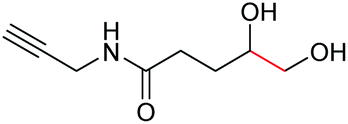
|
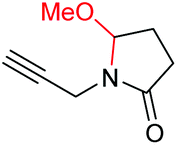
|
6f, 85% |
| 7 |
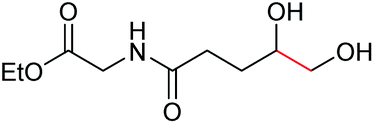
|
|
6g, 65% |
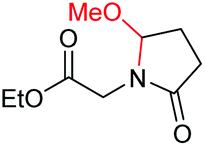
| 8 |
|
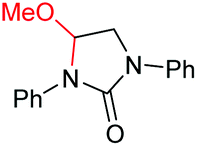
|
6h, 48% |
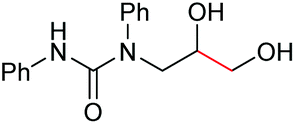
| 9 |
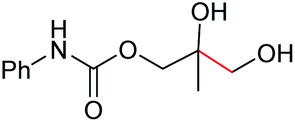
|
|
6i, 60% |
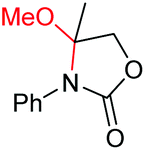
| 10 |
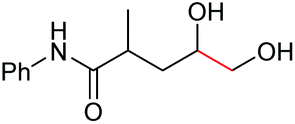
|
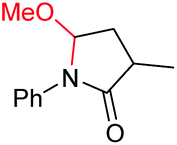
|
6j, 97% |
| 11 |
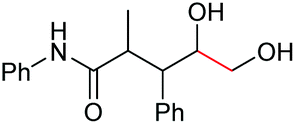
|
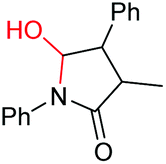
|
6k, 92% |
| 12 |
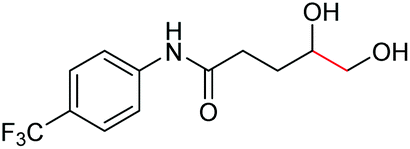
|
|
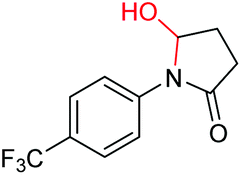
6l, 62% |
| 13 |
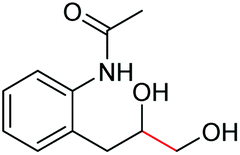
|
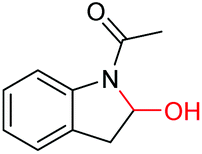
|
6m, 30% |
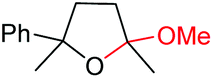
| 14 |
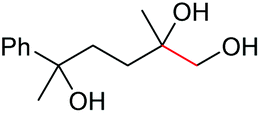
|
|
6n, 78% |
It is worth pointing out that although both aromatic and aliphatic 1,2-diols can be cleaved selectively with the reported catalysts in the presence of oxygen or air (Scheme 1a and b),14 most of those methods are limited to substrates without any functional groups, especially functionalities usually sensitive to oxidation conditions. To the best of our knowledge, there appears to be only two reports in which a few functionalized diols feature using 3 equiv. of base14a or high temperature (130 °C).14h Our protocol overcomes the disadvantage of functionality limitation under mild conditions, which should make it more applicable in organic synthesis. For example, alkyne, amide, acid, TBDPS, ester, carbamate, urea, ketone and even alcohol functionalities, which are often presented in pharmaceuticals and natural products, are tolerated in our protocol, but are barely seen in the previous reports of aerobic oxidation.14
Preliminary mechanistic investigation
While the detailed mechanism of the oxidation remains largely speculative, we have performed a range of experiments, aiming to gain further understanding of this transformation. As we proposed above (Scheme 2), Mn-oxygen species from O2 activation, instead of singlet oxygen (1O2) which is known to cleave diol derivatives,23 is likely to be the active species for the oxidative cleavage of 1,2-diol. Our control experiments appear to support this conjecture. The 1O2 trap, 9,10-diphenylanthracene (DPA), is known to react rapidly with 1O2 to give an endoperoxide product (k ≈ 1.3 × 106 M−1 s−1).24 When 1a was subjected to the standard oxidation conditions but in the presence of DPA as a 1O2 trap, no endoperoxide was detected, and importantly, the expected cleavage product 2a was formed in 74% yield (Scheme 3, eqn. (1)). In addition, when three well-known photosensitizers, eosin Y disodium salt, [Ru(bpy)3·6H2O], and [Ir(dFppy)3], which are all known to be capable of producing 1O2 under blue light irradiation,25 were used individually as a replacement catalyst for the oxidative cleavage of 1a, none were found to catalyse the reaction under the conditions employed (Scheme 3, eqn. (2)). These results support the proposition above.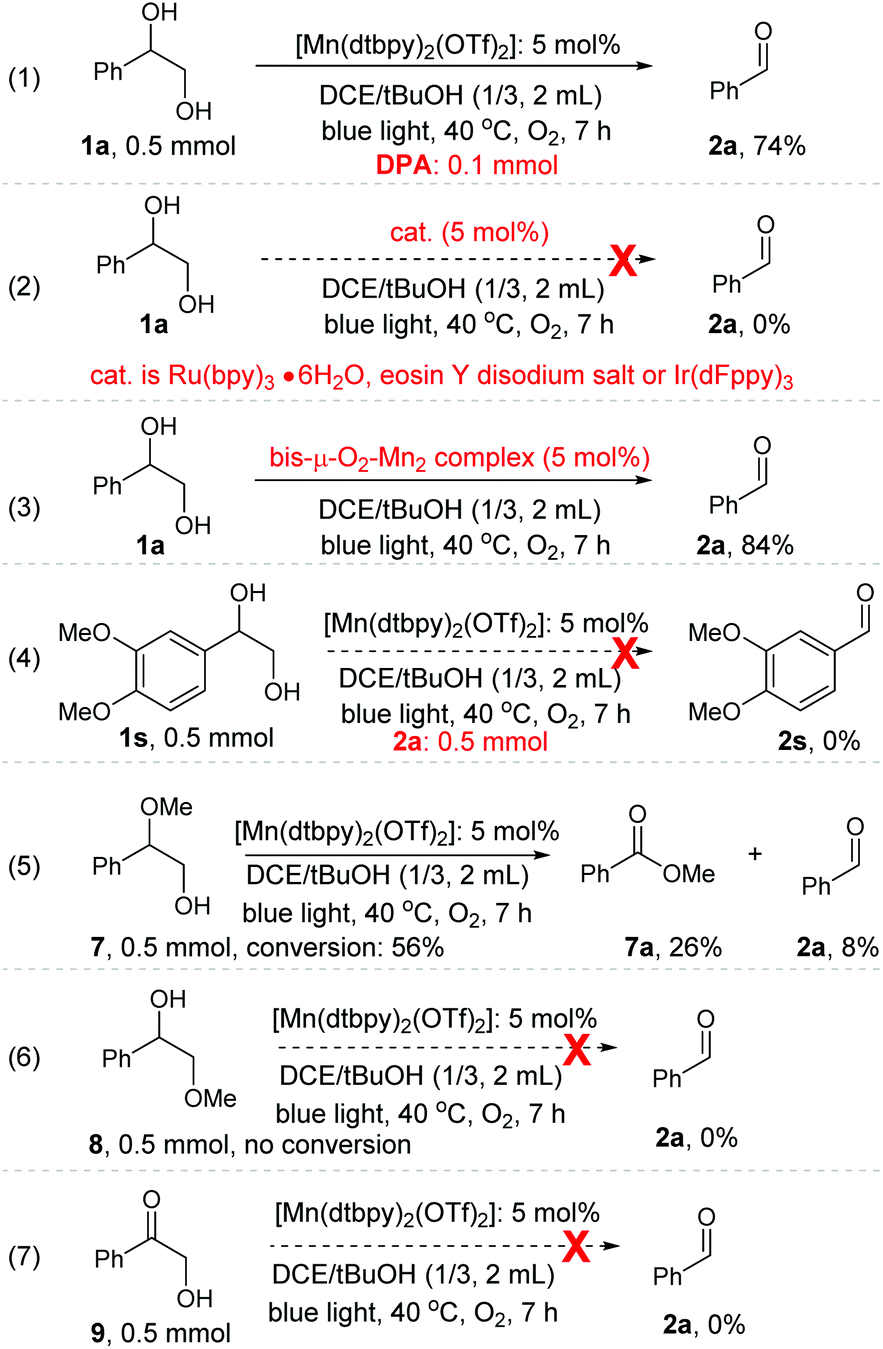 | ||
| Scheme 3 Control experiments to shed light on the mechanism. | ||
The bis-μ-O2-Mn2 complex turned out to be active, catalysing the oxidation of 1a to 2a in 84% yield in the mix solvent of DCE/tBuOH (Scheme 3, eqn. (3)). However, light was still required; in its omission, the oxidation stalled. This is not surprising, as the oxo dimer is an off-cycle species.11b Aldehyde 2a, which could be autoxidized to produce a peroxide under O2, was unable to replace 1a to promote the oxidation of less reactive 1s (Scheme 3, eqn. (4); also see Table 3, entry 1). In fact, when 1a was subjected to the standard conditions but in the presence of peroxides like mCPBA, TBHP, DTBP, H2O2, or benzoyl peroxide as the oxidant, only a small amount of 2a was obtained (see Table S2 in the ESI†). These observations make the possibility of an autoxidation pathway involving peroxides unlikely. When one of the –OH groups in 1a was protected or pre-oxidized, the desired C–C bond cleavage process became difficult as well as less selective (Scheme 3, eqn. (5), (6) and (7)). These results appear to indicate that the cleavage of the C–C bond of diols proceeds via a concerted, rather than stepwise, process.
Further insight was gained by following the kinetic time course of the oxidation of 1a, 1s and their mixture. As shown in Scheme 4, 1a could be smoothly oxidized to 2a by O2 with [Mn(dtbpy)2(OTf)2] as the catalyst in DCE/tBuOH, whilst 1s was inactive under the same conditions. However, when an equal molar 1a and 1s were mixed together and subjected to the same oxidation conditions, 2a and 2s were formed in an identical yield, and remarkably, by the same rate, suggesting that 1a and 1s are oxidized in the same catalytic cycle. As proposed in Scheme 2(2), 1a could promote O2 activation by Mn(II) to form 2a as well as a more active Mn(IV)-oxo species, which then oxidizes the less reactive 1s to 2s while regenerating Mn(II). Unfortunately, we failed to detect the possible Mn(IV)-oxo species or the oxo dimer via UV-Vis spectroscopy under the conditions employed. Only the signals of [Mn(dtbpy)2(OTf)2] were observed during the oxidation (Fig. S1A in the ESI†). Indeed, when the bis-μ-O2-Mn2 complex was mixed with 1a or 1s under blue light, the greenish brown solution of bis-μ-O2-Mn2 immediately changed to colorless, and the UV-Vis absorption of bis-μ-O2-Mn2 disappeared concomitantly (Fig. S1B in the ESI†).
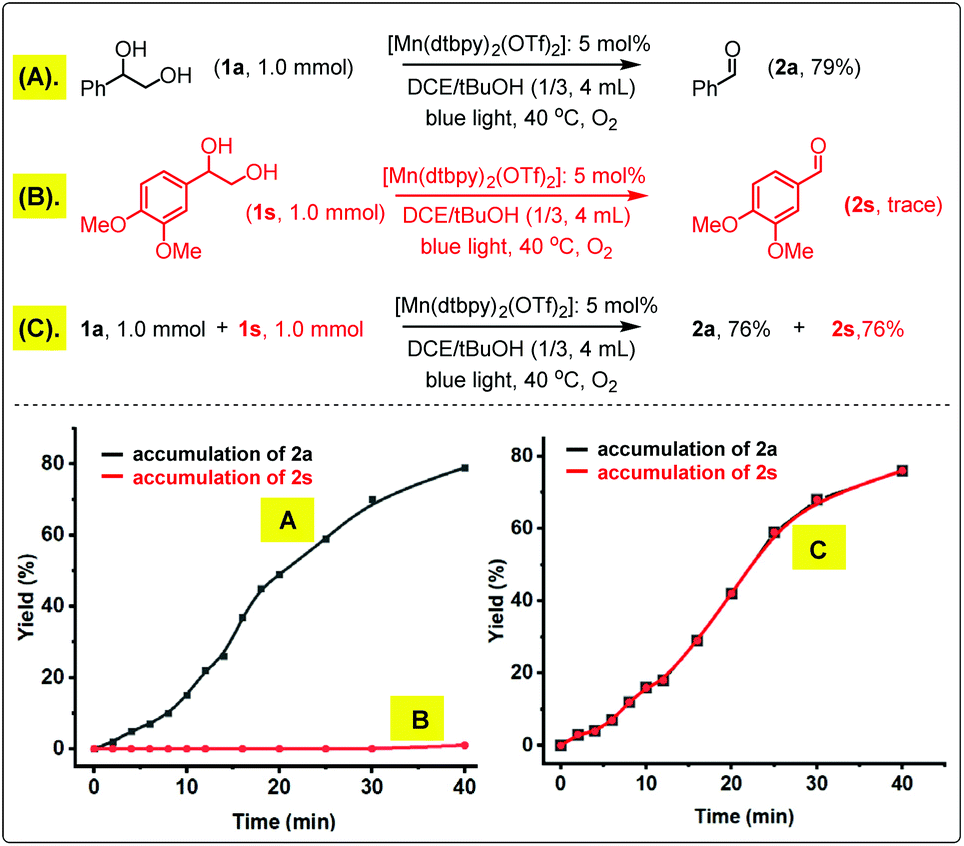 | ||
| Scheme 4 Kinetic behaviors of the oxidation of 1a, 1s and their mixture. | ||
We then followed the oxidation of 9,10-dihydroxydecanoic acid (3e), decane-1,2,10-triol (3f) and 3-phenylpropane-1,2-diol (3o) in MeOH by UV-Vis spectroscopy. Similar to the oxidation of 1a in DCE/tBuOH, no color change was observed during the reaction. As shown in Fig. S2,† although ca. 15% of the desired cleavage product was detected in the oxidation of 3e or 3o after 4 h, no obvious UV-Vis absorption arising from possible Mn-oxygen species was observed, which is in line with the observation of no color change. However, after the completion of the oxidation of 3e, 3f, or 3o, an identical color change to greenish brown was noted, and revealingly, all of three solutions showed a UV-Vis absorption at around 537 nm [Fig. 1(A)], which can be assigned to the MeOH-coordinated bis-μ-O2-Mn2 complex generated from O2 activation by [Mn(dtbpy)2(OTf)2] in MeOH [Fig. 1(A) and 1(B)].11b Of further note is that when the bis-μ-O2-Mn2 complex was mixed with 3o under blue light, the greenish brown solution immediately turned to colorless, accompanied with the disappearance of the absorption at 537 nm [Fig. 1(B)]. These observations indicate that the active Mn-oxygen species are too reactive to be observed by UV-Vis spectroscopy during the diol oxidation in either DCE/tBuOH or MeOH, although the more stable oxo dimer was observable in MeOH.
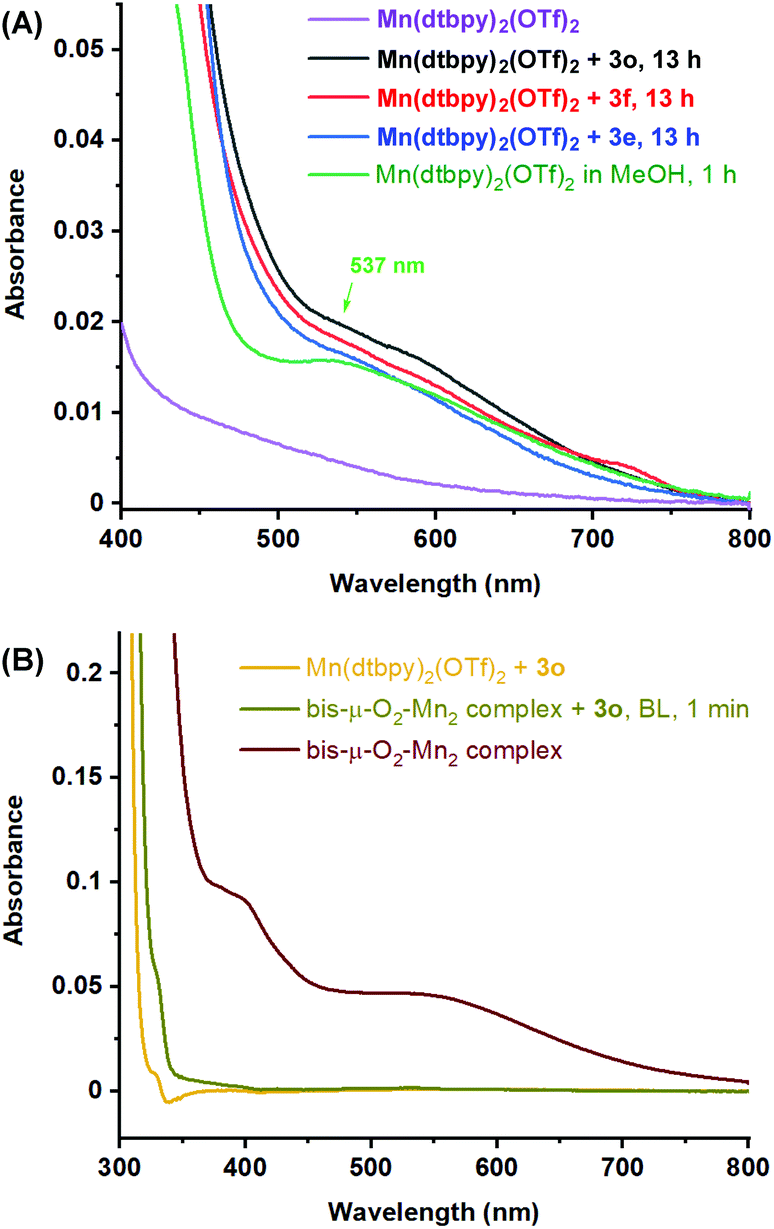 | ||
| Fig. 1 (A) UV-Vis spectra of [Mn(dtbpy)2(OTf)2], the reaction mixture of [Mn(dtbpy)2(OTf)2] with 3e/3f/3o in MeOH under O2 after blue light (BL) irradiation for 13 h, and MeOH solution of [Mn(dtbpy)2(OTf)2] under O2 after BL irradiation for 1 h; (B) UV-Vis spectra of the bis-μ-O2-Mn2 complex, the mixture of bis-μ-O2-Mn2 and 3o under N2 after BL irradiation for 1 min, and the mixture of [Mn(dtbpy)2(OTf)2] and 3o in the absence of BL. The concentration of [Mn] for UV-Vis measurement was 0.25 mM in MeOH. | ||
Conclusions
In conclusion, a well-defined, biologically relevant Mn(II) complex has been identified as an efficient catalyst for the aerobic oxidative cleavage of a wide range of diverse 1,2-diols under irradiation of visible light. Both aromatic and aliphatic diols are shown to be viable substrates, affording valuable carbonyl compounds and five-membered heterocycles with a broad range of functional groups tolerated in this oxidase-like system. Preliminary mechanistic investigations suggest that the reaction follows the pathway outlined in Scheme 2(2), in which two alcohol molecules are sequentially oxidized in one turnover, with the first alcohol reducing one oxygen atom to water while promoting the formation of an active Mn-oxo species that cleaves the second alcohol (also see Scheme S1 in the ESI† for a more detailed proposal). Further mechanistic studies and application of the catalytic system are underway in our laboratory and will be reported in the future.Author contributions
J.X. conceptualized the project. Z.H., R.G., and E.B. performed the experiments, analyzed the data, and discussed the results. Z.H. and J.X. wrote the paper. Z.H. wrote the ESI† and contributed to other related materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Engineering and Physical Sciences Research Council (EP/R009694/1) for funding, the China Scholarship Council and University of Liverpool for a PhD studentship (RPG), and the Analytical Services of the Department of Chemistry of the University of Liverpool for product analysis. We also thank Mr. Mark Norman and his team in the EEE Department Electronics Workshop and Mr. Gordon Bostock and Chemistry Electronics Workshop for help with the design and fabrication of our photoreactor.Notes and references
- (a) F. A. Armstrong, Philos. Trans. R. Soc., B, 2008, 363, 1263 CrossRef CAS PubMed; (b) G. C. Dismukes, Chem. Rev., 1996, 96, 2909 CrossRef CAS PubMed; (c) N. A. Law, M. T. Caudle and V. L. Pecoraro, Adv. Inorg. Chem., 1998, 46, 305 CrossRef CAS.
- (a) J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455 CrossRef CAS PubMed; (b) J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175 CrossRef CAS PubMed.
- (a) Y. Sheng, I. A. Abreu, D. E. Cabelli, M. J. Maroney, A.-F. Miller, M. Teixeira and J. S. Valentine, Chem. Rev., 2014, 114, 3854 CrossRef CAS PubMed; (b) E. C. Chang and D. J. Kosman, J. Biol. Chem., 1989, 264, 12172 CrossRef CAS PubMed.
- S. Alejandro, S. Höller, B. Meier and E. Peiter, Front. Plant Sci., 2020, 11, 300 CrossRef PubMed.
- P. K. Arora, Microbial Metabolism of Xenobiotic Compounds, Springer, Singapore, 2019 Search PubMed.
- (a) R. Ottenbacher, E. Talsi and K. Bryliakov, Molecules, 2016, 21, 1454 CrossRef PubMed; (b) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727 CrossRef CAS PubMed; (c) J. R. Clark, K. Feng, A. Sookezian and M. C. White, Nat. Chem., 2018, 10, 583 CrossRef CAS PubMed; (d) K. Wang, J. Zhou, Y. Jiang, M. Zhang, C. Wang, D. Xue, W. Tang, H. Sun, J. Xiao and C. Li, Angew. Chem., Int. Ed., 2019, 58, 6380 CrossRef CAS PubMed; (e) P. Saisaha, J. W. de Boer and W. R. Browne, Chem. Soc. Rev., 2013, 42, 2059 RSC; (f) W. Sun and Q. Sun, Acc. Chem. Res., 2019, 52, 2370 CrossRef CAS PubMed; (g) E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418 CrossRef CAS; (h) J. T. Groves, W. J. Kruper and R. C. Haushalter, J. Am. Chem. Soc., 1980, 102, 6375 CrossRef CAS; (i) M. Cianfanelli, G. Olivo, M. Milan, R. J. M. Klein Gebbink, X. Ribas, M. Bietti and M. Costas, J. Am. Chem. Soc., 2020, 142, 1584 CrossRef CAS PubMed; (j) G. Li, P. A. Kates, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, ACS Catal., 2019, 9, 9513 CrossRef CAS; (k) M. Guo, H. Dong, J. Li, B. Cheng, Y.-q. Huang, Y.-q. Feng and A. Lei, Nat. Commun., 2012, 3, 1190 CrossRef PubMed.
- (a) T. D. H. Bugg, Chem. Biol., 2014, 21, 168 CrossRef CAS PubMed; (b) S. Sahu and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 11410 CrossRef CAS PubMed.
- (a) D. B. Rice, A. A. Massie and T. A. Jackson, Acc. Chem. Res., 2017, 50, 2706 CrossRef CAS PubMed; (b) M. Guo, Y.-M. Lee, R. Gupta, M. S. Seo, T. Ohta, H.-H. Wang, H.-Y. Liu, S. N. Dhuri, R. Sarangi, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2017, 139, 15858 CrossRef CAS PubMed; (c) R. L. Shook, S. M. Peterson, J. Greaves, C. Moore, A. L. Rheingold and A. S. Borovik, J. Am. Chem. Soc., 2011, 133, 5810 CrossRef CAS PubMed; (d) C. Deville, S. K. Padamati, J. Sundberg, V. McKee, W. R. Browne and C. J. McKenzie, Angew. Chem., Int. Ed., 2016, 55, 545 CrossRef CAS PubMed; (e) C. J. Weschler, B. M. Hoffman and F. Basolo, J. Am. Chem. Soc., 1975, 97, 5278 CrossRef CAS PubMed; (f) J. A. Kovacs, Acc. Chem. Res., 2015, 48, 2744 CrossRef CAS PubMed; (g) S. Fukuzumi, Y.-M. Lee, J. Jung and W. Nam, Green Chem., 2018, 20, 948 RSC; (h) M. Guo, Y.-M. Lee, S. Fukuzumi and W. Nam, Coord. Chem. Rev., 2021, 435, 213807 CrossRef CAS; (i) M. Guo, J. Zhang, L. Zhang, Y.-M. Lee, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2021, 143, 18559 CrossRef CAS PubMed; (j) B. Xiong, X. Zeng, S. Geng, S. Chen, Y. He and Z. Feng, Green Chem., 2018, 20, 4521 RSC; (k) Q. Liu, P. Wu, Y. Yang, Z. Zeng, J. Liu, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 4666 CrossRef CAS PubMed.
- (a) A. B. Sorokin, Chem. Rev., 2013, 113, 8152 CrossRef CAS PubMed; (b) M. W. Grinstaff, M. G. Hill, J. A. Labinger and H. B. Gray, Science, 1994, 264, 1311 CrossRef CAS PubMed; (c) V. A. Larson, B. Battistella, K. Ray, N. Lehnert and W. Nam, Nat. Rev. Chem., 2020, 4, 404 CrossRef CAS.
- (a) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329 CrossRef CAS PubMed; (b) E. Roduner, W. Kaim, B. Sarkar, V. B. Urlacher, J. Pleiss, R. Gläser, W.-D. Einicke, G. A. Sprenger, U. Beifuß, E. Klemm, C. Liebner, H. Hieronymus, S.-F. Hsu, B. Plietker and S. Laschat, ChemCatChem, 2013, 5, 82 CrossRef CAS.
- (a) H. M. Neu, J. Jung, R. A. Baglia, M. A. Siegler, K. Ohkubo, S. Fukuzumi and D. P. Goldberg, J. Am. Chem. Soc., 2015, 137, 4614 CrossRef CAS PubMed; (b) Z. Huang, R. Guan, M. Shanmugam, E. L. Bennett, C. M. Robertson, A. Brookfield, E. J. L. McInnes and J. Xiao, J. Am. Chem. Soc., 2021, 143, 10005 CrossRef CAS PubMed; (c) S. Riaño, D. Fernández and L. Fadini, Catal. Commun., 2008, 9, 1282 CrossRef; (d) A. Gonzalez-de-Castro and J. Xiao, J. Am. Chem. Soc., 2015, 137, 8206 CrossRef CAS PubMed.
- (a) R. Criegee, Ber. Dtsch. Chem. Ges., 1931, 64, 260 CrossRef; (b) L. Malaprade, Bull. Soc. Chim. Fr., 1934, 3, 833 Search PubMed.
- (a) M. Frigerio and M. Santagostino, Tetrahedron Lett., 1994, 35, 8019 CrossRef CAS; (b) J. N. Moorthy, N. Singhal and K. Senapati, Org. Biomol. Chem., 2007, 5, 767 RSC; (c) C. Stark and A.-K. Schmidt, Synthesis, 2014, 3283 CrossRef; (d) K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646 CrossRef CAS PubMed; (e) C. Venturello and M. Ricci, J. Org. Chem., 1986, 51, 1599 CrossRef CAS.
- Selected references on Pd, Ag, Ru, V, Ce, Fe, etc. catalysed aerobic cleavage of 1,2-diols: (a) Z.-z. Zhou, M. Liu, L. Lv and C.-J. Li, Angew. Chem., Int. Ed., 2018, 57, 2616 CrossRef CAS PubMed; (b) H. Luo, L. Wang, S. Shang, J. Niu and S. Gao, Commun. Chem., 2019, 2, 17 CrossRef CAS; (c) J. Schwarz and B. König, Chem. Commun., 2019, 55, 486 RSC; (d) E. Amadio, J. González-Fabra, D. Carraro, W. Denis, B. Gjoka, C. Zonta, K. Bartik, F. Cavani, S. Solmi, C. Bo and G. Licini, Adv. Synth. Catal., 2018, 360, 3286 CrossRef CAS; (e) Y. Ito, K. Kunimoto, S. Miyachi and T. Kako, Tetrahedron Lett., 1991, 32, 4007 CrossRef CAS; (f) A. Wang and H. Jiang, J. Org. Chem., 2010, 75, 2321 CrossRef CAS PubMed; (g) E. Takezawa, S. Sakaguchi and Y. Ishii, Org. Lett., 1999, 1, 713 CrossRef CAS PubMed; (h) B. Guicheret, E. Da Silva, R. Philippe, A. Favre-Reguillon, L. Vanoye, P. Blach, Y. Raoul, C. De Bellefon, E. Métay and M. Lemaire, ACS Sustainable Chem. Eng., 2020, 8, 13167 CrossRef CAS; (i) L. Meng, W. Li, P. Guo, S. Wang and X. Tong, Catal. Commun., 2021, 154, 106305 CrossRef CAS; (j) J.-n. Teng, R. Zhu, X. Li and Y. Fu, ChemCatChem, 2021, 13, 4355 CrossRef CAS; (k) W. Chen, X. Xie, J. Zhang, J. Qu, C. Luo, Y. Lai, F. Jiang, H. Yu and Y. Wei, Green Chem., 2021, 23, 9140–9146 RSC; (l) R. Zhu, G. Zhou, J.-n. Teng, X. Li and Y. Fu, ChemSusChem, 2020, 13, 5248 CrossRef CAS PubMed; selected references on Mn-catalysed aerobic cleavage of 1,2-diols: (m) V. Escande, C. H. Lam, P. Coish and P. T. Anastas, Angew. Chem., Int. Ed., 2017, 56, 9561 CrossRef CAS PubMed; (n) S.-S. Meng, L.-R. Lin, X. Luo, H.-J. Lv, J.-L. Zhao and A. S. C. Chan, Green Chem., 2019, 21, 6187 RSC.
- (a) V. A. Larson, B. Battistella, K. Ray, N. Lehnert and W. Nam, Nat. Rev. Chem., 2020, 4, 404 CrossRef CAS; (b) S. Barroso, G. Blay, I. Fernández, J. R. Pedro, R. Ruiz-García, E. Pardo, F. Lloret and M. C. Muñoz, J. Mol. Catal. A: Chem., 2006, 243, 214 CrossRef CAS.
- (a) Y. Liu, C. Wang, D. Xue, M. Xiao, C. Li and J. Xiao, Chem. – Eur. J., 2017, 23, 3051 CrossRef CAS PubMed; (b) A. Gonzalez-de-Castro, C. M. Robertson and J. Xiao, Chem. – Eur. J., 2019, 25, 4345 CrossRef CAS PubMed; (c) Y. Liu, D. Xue, C. Li, J. Xiao and C. Wang, Catal. Sci. Technol., 2017, 7, 5510 RSC.
- S. Fukuzumi, T. Mizuno and T. Ojiri, Chem. – Eur. J., 2012, 18, 15794 CrossRef CAS PubMed.
- E. Yilmaz and M. Soylak, in New Generation Green Solvents for Separation and Preconcentration of Organic and Inorganic Species, eds. M. Soylak and E. Yilmaz, Elsevier, 2020, pp. 207 Search PubMed.
- J. M. Kokosa, Trends Anal. Chem., 2019, 118, 238 CrossRef CAS.
- P. Spannring, P. C. A. Bruijnincx, B. M. Weckhuysen and R. J. M. Klein Gebbink, Catal. Sci. Technol., 2014, 4, 2182 RSC.
- (a) F. Souquet, W. Drici, S. A. Fayssal, I. Lazouni, S. Thueillon and J. Pérard-Viret, Synthesis, 2020, 2970 CAS; (b) D. Baudelet, A. Daïch, B. Rigo, E. Lipka, P. Gautret, G. Homerin, C. Claverie, J. Rousseau, C.-M. Abuhaie and A. Ghinet, Synthesis, 2016, 2226 CAS.
- (a) M. H. Abdi, P. J. Beswick, A. Billinton, L. J. Chambers, A. Charlton, S. D. Collins, K. L. Collis, D. K. Dean, E. Fonfria, R. J. Gleave, C. L. Lejeune, D. G. Livermore, S. J. Medhurst, A. D. Michel, A. P. Moses, L. Page, S. Patel, S. A. Roman, S. Senger, B. Slingsby, J. G. A. Steadman, A. J. Stevens and D. S. Walter, Bioorg. Med. Chem. Lett., 2010, 20, 5080 CrossRef CAS PubMed; (b) T.-Y. Tsai, T.-K. Yeh, X. Chen, T. Hsu, Y.-C. Jao, C.-H. Huang, J.-S. Song, Y.-C. Huang, C.-H. Chien, J.-H. Chiu, S.-C. Yen, H.-K. Tang, Y.-S. Chao and W.-T. Jiaang, J. Med. Chem., 2010, 53, 6572 CrossRef CAS PubMed.
- C. Crestini and M. D'Auria, Tetrahedron, 1997, 53, 7877 CrossRef CAS.
- Y. You, Org. Biomol. Chem., 2018, 16, 4044 RSC.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and procedures, optimization studies, mechanistic experiments, and spectral data for all compounds. See DOI: https://doi.org/10.1039/d2gc00460g |
| This journal is © The Royal Society of Chemistry 2022 |