
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient synthesis of 2,6-bis(hydroxymethyl)pyridine using whole-cell biocatalysis†
Tsvetan
Kardashliev
*,
Sven
Panke
and
Martin
Held
 *
*
Department of Biosystems Science and Engineering, ETH Zurich, Mattenstrasse 26, 4058 Basel, Switzerland. E-mail: tsvetank@ethz.ch; heldma@ethz.ch
First published on 21st April 2022
Abstract
We demonstrate a novel one-pot biocatalytic process for the preparation of a versatile chemical intermediate, 2,6-bis(hydroxymethyl)pyridine, from naturally-occurring 2,6-lutidine using recombinant microbial whole cells as a catalysts. After scale up, the bioconversion enabled titers exceeding 12 g L−1 with a space–time yield of 0.8 g L−1 h−1. This biocatalytic route offers a simpler and more sustainable alternative to multistep organic synthesis protocols.
Biocatalytic C–H oxyfunctionalisations hold a great promise especially for sustainable synthesis of fine and specialty chemicals, and active pharmaceutical ingredients.1,2 Hydroxylating enzymes exhibit chemo-, regio-, and stereoselectivities that are difficult to achieve chemically and thereby complement chemo-catalytic methods.3 Despite their attractiveness, the degree of acceptance of these enzymes for chemical synthesis remains modest, perhaps due to operational challenges such as the requirement for costly cofactor (most commonly, NAD(P)H) and oxygen for efficient catalysis.4 The application of hydroxylating enzymes as part of whole-cell biocatalysts can alleviate many issues as intact cells can regenerate the required cofactors and at the same time provide a stabilizing microenvironment. On the downside, the cell membrane can limit the mass transfer of compounds into the cytosol where most enzyme activities typically reside.5
2,6-Bis(hydroxymethyl)pyridine 6 is a valuable chemical precursor used for the preparation of metal complexes and catalysts,6–8 biopolymers,9 and active pharmaceutical ingredients.10,11 This chemical can be synthesized via 6-methyl-2-pyridine methanol 2 from a naturally-occurring aromatic heterocycle, 2,6-lutidine 1. A patented organic synthesis method involves oxidation of 2,6-lutidine to dipicolinic acid with potassium permanganate in water followed by reduction of the acid with sodium borohydride/iodine in tetrahydrofuran to yield 2,6-bis(hydroxymethyl)pyridine.12 This multistep route delivers unsatisfactory yields (64%) and imposes an environmental burden because strong oxidizing and reducing reagents are needed in molar excess to the pyridine reactants. We investigated whether the synthesis of 6 from 1 can be streamlined by utilizing enzyme catalysis.
Xylene monooxygenase (XMO) from Pseudomonas putida is known to catalyse the oxidation of the methyl group of m-xylene,13 a structural analogue of 1. XMO has been extensively researched13–16 and employed in its native host for the preparative synthesis of fine chemicals, e.g. for the synthesis of nicotinic acid from 3-methylpyridine.17 Furthermore, Maruyama et al. showed that both methyl groups in m-xylene were oxidized to alcohol and aldehyde functionalities when XMO was expressed in Escherichia coli.18 We reasoned that this enzyme represents a promising catalyst for hydroxylation of the methyl groups of 1. To test this hypothesis, we employed E. coli NEBxpress® (for short, E. coli NEB) transformed with a previously described XMO expression plasmid, pSPZ3,19 and propagated the cells in a defined minimal medium with glucose as the sole carbon source. Biotransformation of 1 (5 mM) was initiated 90 minutes after induction of recombinant protein production in early exponential phase and samples were collected in regular intervals over 300 minutes. The reaction was followed by UV-HPLC and the products of the biotransformation were identified and quantified against neat analytical standards (Fig. S1†). The starting material 1 was depleted after approximately 150 minutes and its disappearance was accompanied by accumulation of 2. After compound 1 had diminished, the formation of further oxidation products including 6-methyl-2-pyridinecarbaldehyde 3, 6-(hydroxymethyl)pyridine-2-carbaldehyde 5 (which could not be baseline separated from 3 using the employed HPLC protocol and column), 6-methyl-2-pyridinecarboxylic acid 4 and 6 was observed (Fig. 1B and S2A†). At the end of the biotransformation, the alcohol and the acid product were present whereas compound 6 represented more than 90% of the total product formed. When compound 2 was used as starting material (Fig. 1C and S2B†), intermediates 3 and 5 were formed first before 4 and 6 accumulated as side and main products, respectively. Our observations are consistent with the findings of Maruyama et al.18 that XMO forms the diol via an aldehyde intermediate, rather than by direct hydroxylation of the monohydroxylated intermediate 2 and that the terminal biocatalytic step is mediated by endogenous dehydrogenases. Another important observation is that the reaction products and intermediates could be measured directly in the nutrient broth. Thus, mass transfer of the heterocyclic compounds did not appear to limit the whole-cell biotransformation. This observation is supported by earlier findings that the E. coli envelope allows small, aromatic and water soluble molecules to traverse 1–6.20
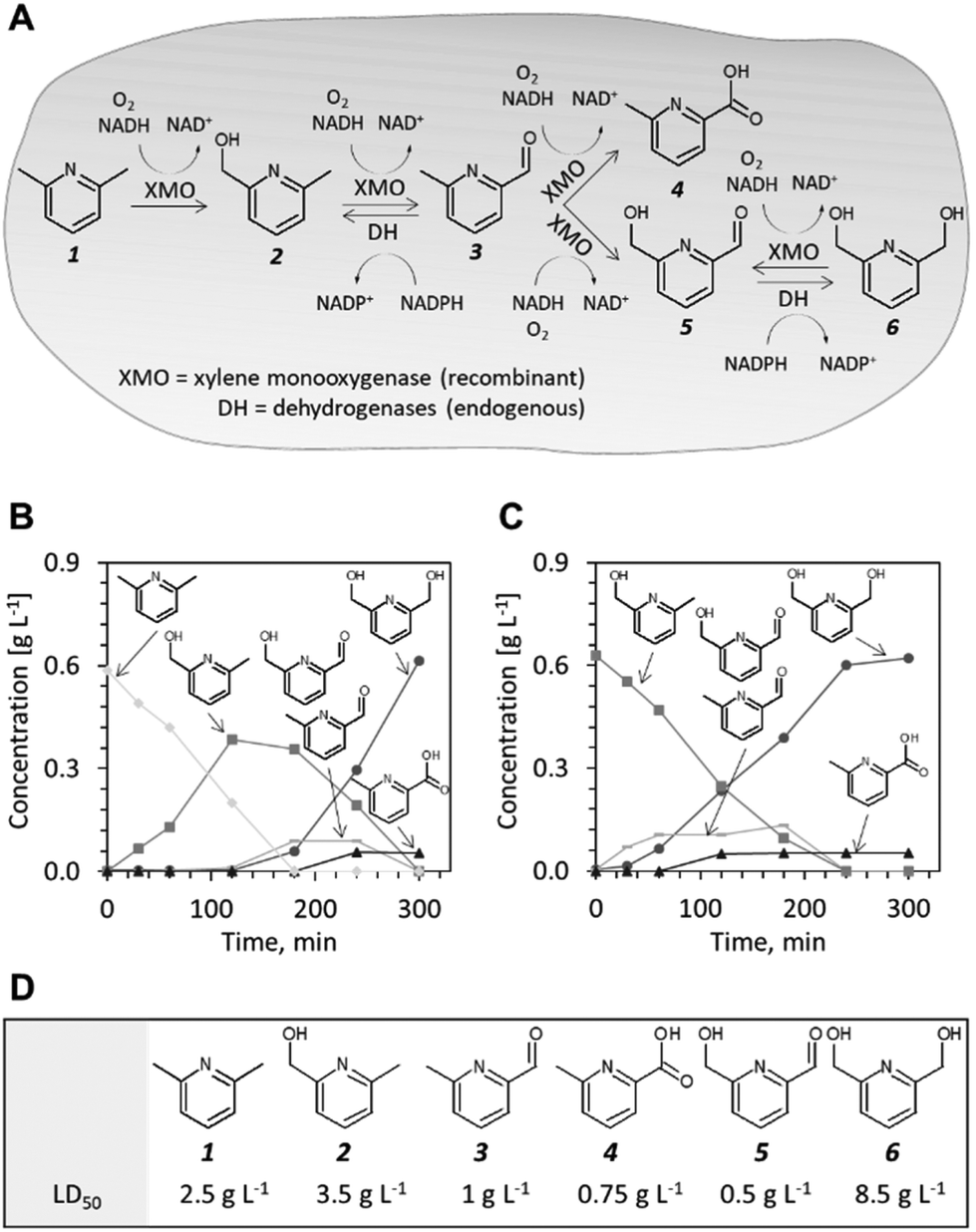 | ||
| Fig. 1 Pilot studies of XMO as a catalyst for the production of 2,6-bis(hydroxymethyl)pyridine 6. (A) Probable reaction scheme as deduced from sequential appearance of intermediates 2, 3 and 5, and target product 6. (B) Time course of the bioconversions of 2,6-lutidine 1. (C) Time course of the bioconversions starting from 6-methyl-2-pyridinemethanol 2 (D) Toxicity of compounds 1–6 to E. coli. LD50 is the concentration of compound necessary to inhibit cell growth by 50% relative to a non-treated culture. | ||
To identify the endogenous enzymes that contribute to the reaction cascade, we introduced plasmid pSPZ3 in an E. coli K-12 derivative with reduced aromatic aldehyde reduction capacity (E. coli RARE).21 The strain harbours six deletions of genes from the aldo-keto reductase and alcohol dehydrogenase superfamilies (namely, dkgB, yeaE, dkgA, yqhD, yjgB, and yahK). Biotransformations of 1 and 2 using E. coli RARE as host for XMO resulted in premature termination of the reaction sequence before the reduction of 5 to 6. An unknown product, likely derived from aldehydes 3 and/or 5 was also detected in the latter biotransformations. Because the unknown product was formed to a detectable level only when this specific E. coli strain was used as a host, the chemical authenticity of the latter was not investigated. Using plasmids from the ASKA collection,22 the individual reductase activities were reconstituted one-by-one in the E. coli RARE strain expressing XMO. The co-expression of any of the six enzymes restored the capacity of the cell to reduce 5 to 6 (Fig. S3†). Thus, aromatic aldehyde reduction activity in E. coli is mediated by several endogenous enzyme activities indispensable to the reaction sequence.
The conversion of 1 to compound 6 by E. coli NEB/pSPZ3 (Fig. 1B) proceeded with satisfactory initial activity (∼0.4 g gCDW−1 h−1) but an increase of the initial substrate load impaired the biotransformation suggesting toxification of the whole-cell biocatalyst (Fig. S4 and S6†). Thus, we investigated the influence of compounds 1–6 on the growth of E. coli NEB/pSPZ3. Our analysis showed that all six compounds exert a growth-inhibiting effect on E. coli with varying degrees of severity. Aldehydes 3 and 5 and side product 4 were the most harmful and incurred growth retardation at or below 1 g L−1. The starting material 1 stalled cell growth completely when its concentration exceeded 5 g L−1. Compounds 2 and 6 had the least detrimental effect on growth whereas cells could tolerate more than 16 g L−1 of target product 6 before growth was abolished completely. We also observed a pronounced difference in the tolerance to 6 of cells that expressed XMO and cells that were not induced and therefore did not express XMO. The former were growth-inhibited at lower concentrations of 6 (Fig. S4A†). Although it appears to be the least preferred substrate among the pyridine compounds, 6 can be also oxidized by XMO yielding the highly toxic compound 5 while compound 4 did not serve as a suitable substrate by XMO (Fig. S7†).
Given the promising yields observed from batch experiments, we aimed to develop a bioprocess for the gram-scale synthesis of 6. Pilot experiments (1 L scale) in a 2.5 L bioreactor were performed using a defined growth medium and glucose as carbon source. After consumption of 10 g L−1 glucose in batch mode, a glucose-limited fed-batch allowing for a growth rate of the whole-cell biocatalysts of ∼0.3 h−1 was initiated and maintained until 17.5 gCDW L−1 (OD600 of ∼50) had been formed. Recombinant enzyme production was induced 90 minutes before reaching the target biomass concentration. The biotransformation of 4.7 g L−1 compound 1 was initiated by supplying 1 with a syringe pump at a rate of 0.93 g L−1 for one minute, followed by 78 mg L−1 for 60 minutes. Adding 1 gradually ensured that its concentration did not reach highly cytotoxic levels. Fig. 2 shows the reaction progression over time. Compound 1 was rapidly converted to 2 within the first 120 minutes. Once depleted, it took an additional 300 minutes for conversion to the end products, namely 6 as the main product and 4 as a minute side product. The reaction yield exceeded 90% with an estimated 1–3% loss of the volatile starting material 1 through gas stripping. The production of 4.8 g L−16 was realized within 7 hours of biotransformation with a near quantitative conversion of the starting material to the desired dihydroxylation product at a space–time yield of 0.69 g L−1 h−1.
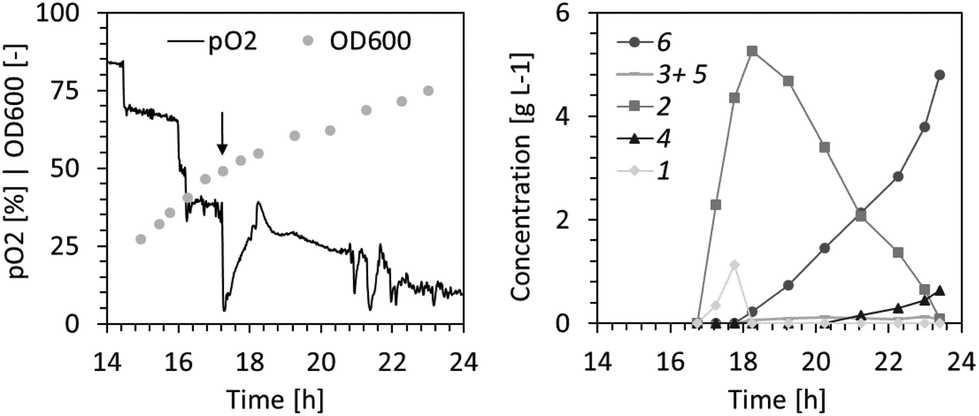 | ||
| Fig. 2 High cell density fermentation and biocatalysis with glucose as carbon source and continuous supply of substrate 1. Dissolved oxygen (pO2) level and biomass concentration (OD600) during fed-batch phase are shown in the left panel. The closed arrow indicates the point at which the addition of substrate 1 was initiated. The concentration courses of substrate, intermediates and final products are presented in the right panel. | ||
The surplus demand for oxygen during the biotransformation at high cell density (Fig. 2) resulted in periods of low dissolved oxygen (<5%). This may have caused deterioration of the cofactor regeneration capacity of cells and lowered co-factor availability for the XMO-catalyzed reactions.23 Thus, in the following fed-batch biotransformation we opted to reduce the effective biomass concentration by growing E. coli NEB/pSPZ3 to only 7 gCDW L−1 (OD600 of ∼20) before initiating substrate addition. Indeed, the dissolved oxygen level remained above 50% throughout the biotransformation. Additionally, we fed glycerol instead of glucose as a carbon source. As a by-product of the biodiesel and bioethanol industry, glycerol represents an attractive carbon source for bacterial biotransformations.24,25 Due to its higher degree of reduction compared to glucose, glycerol assimilation is superior to fuel co-factor demanding reactions.26,27 Lastly, an optimized substrate supply enabled titers exceeding 10 g L−1 of the target product 6. A primary consideration in devising the substrate supply scheme was the toxicity of the starting material (Fig. 1D). We reasoned that 1 should be provided in portions, such that its concentration does not exceed 2.5 g L−1 at all times (Fig. S6†). Further additions of fresh 1 must take place immediately after full conversion to 6 to prevent undesired formation of 5 from 6 by XMO (Fig. 1). Cell poisoning was accompanied by a colour change of the cell suspension from milky to greyish and an increase of dissolved oxygen level. This scenario was repeatedly observed during development of an optimized feeding profile, and an exemplary case is shown in Fig. 3A. Here, depletion of the starting material between hours 16 and 17 resulted in grey-colored biomass accompanied by an increase of dissolved oxygen level which was not associated with depletion of the carbon source. Note that the other spikes seen in the pO2 signal were triggered by the operator by intermittently pausing the carbon feed in order to ensure that the carbon is not overfed. Cell poisoning as a result of the reduction of 6 to 5 could be mitigated by immediate addition of 1, thereby providing a higher affinity substrate than 6 for XMO. Accurate execution of the substrate supply scheme (Fig. 3B) allowed for production of 12.5 g L−1 of the target compound 6 in 14 h of biotransformation. This corresponds to an average space–time yield of 0.8 g L−1 h−1. We also recorded the amount of residual glycerol and overflow metabolites derived thereof in the course of the latter fermentative biotransformations (Fig. S5†). Significant accumulation of acetate in the course of the biotransformation was observed. This may be explained by the high demand for NADH in the early hours of the biotransformation perhaps forcing cells to bypass the TCA cycle and regenerate ATP via fermentative pathways.28
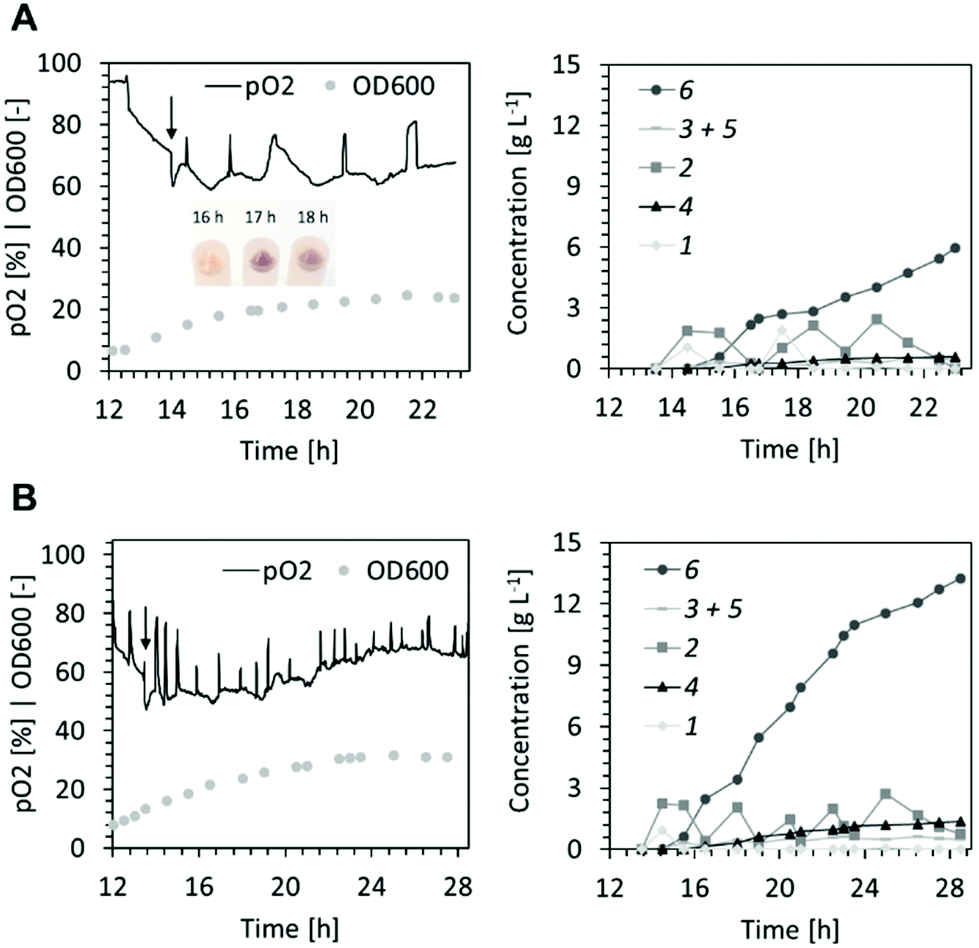 | ||
| Fig. 3 Lower cell density fermentation/biocatalysis using glycerol as carbon source and an intermittent feeding of 1. (A) Dissolved oxygen (pO2), optical density (left panel) and concentrations of reactant and products (right panel) of the biotransformation where the second dose of substrate 1 was not provided in time hence triggering the formation of aldehyde 5 leading to poisoning of the biocatalyst and a colour change of the cell pellet. (B) Dissolved oxygen (pO2), optical density (left panel) and concentrations of reactant and products (right panel) of the biotransformation with optimised substrate supply. | ||
Conclusions
We demonstrate a multi-gram, one-pot biocatalytic synthesis of 2,6-bis(hydroxymethyl)pyridine 6 from readily available 2,6-lutidine 1. The process is characterized by high productivity and yield, enabling more efficient and perhaps more cost-competitive synthesis than classical chemical routes. Significant improvements in terms of sustainability can be realized by adoption of enzyme catalysis as the need for strong oxidizing and reducing agents and non-aqueous solvent is eliminated. The reaction can be carried out entirely in aqueous solution at ambient temperatures and atmospheric pressure and the waste products (biomass and wastewater) could be treated in standard waste treatment facilities.As a final remark, this work exemplifies the potential of hydroxylating enzymes especially when they are employed as whole-cell biocatalysts to avoid the shortcomings associated with their in vitro use.
Author contributions
Tsvetan Kardashliev: Conceptualization, methodology, investigation, data curation, funding-acquisition, writing-original draft preparation. Martin Held: Supervisor, conceptualization, writing-reviewing/editing, funding-acquisition. Sven Panke: Supervisor, conceptualization, writing-reviewing/editing, funding-acquisition.Conflicts of interest
The authors declare competing financial interests.Notes and references
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- J. Dong, E. Fernández-Fueyo, F. Hollmann, C. Paul, M. Pasic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS PubMed.
- S. Chakrabarty, Y. Wang, J. C. Perkins and A. R. H. Narayan, Chem. Soc. Rev., 2020, 49, 8137–8155 RSC.
- B. Lin and Y. Tao, Microb. Cell Fact., 2017, 16, 106 CrossRef PubMed.
- M. Kadisch, M. K. Julsing, M. Schrewe, N. Jehmlich, B. Scheer, M. von Bergen, A. Schmid and B. Bühler, Biotechnol. Bioeng., 2017, 114, 874–884 CrossRef CAS PubMed.
- X. Zhang and M. Luo, CN106243015A, 2016.
- S. Winter, W. Seichter and E. Weber, J. Coord. Chem., 2004, 57, 997–1014 CrossRef CAS.
- A. Kundu, S. Saikia, M. Majumder, O. Sengupta, B. Bhattacharya, G. Chandra De and S. Ghosh, ACS Omega, 2019, 4(3), 5221–5232 CrossRef CAS PubMed.
- A. Pellis, S. Weinberger, M. Gigli, G. M. Guebitz and T. J. Farmer, Eur. Polym. J., 2020, 130, 109680 CrossRef CAS.
- T. Gaines, D. Camp, R. Bai, Z. Liang, Y. Yoon, H. Shim and S. R. Mooring, Bioorg. Med. Chem., 2016, 24, 5052–5060 CrossRef CAS PubMed.
- R. Hovland, C. Gløgård, A. J. Aasen and J. Klaveness, Org. Biomol. Chem., 2003, 1, 644–647 RSC.
- D. Qi and X. Wang, CN105646334A, 2014.
- B. Bühler, A. Schmid, B. Hauer and B. Witholt, J. Biol. Chem., 2000, 275, 10085–10092 CrossRef PubMed.
- R. N. Austin, K. Buzzi, E. Kim, G. J. Zylstra and J. T. Groves, J. Biol. Inorg. Chem., 2003, 8, 733–740 CrossRef CAS PubMed.
- J. Shanklin, E. Whittle and B. G. Fox, Biochemistry, 1994, 33, 12787–12794 CrossRef CAS PubMed.
- B. Bühler, B. Witholt, B. Hauer and A. Schmid, Appl. Environ. Microbiol., 2002, 68, 560–568 CrossRef PubMed.
- H. G. Kulla, Chimia, 1991, 45, 81–85 CrossRef CAS.
- T. Maruyama, H. Iida and H. Kakidani, J. Mol. Catal. B: Enzym., 2003, 21, 211–219 CrossRef CAS.
- S. Panke, A. Meyer, C. M. Huber, B. Witholt and M. G. Wubbolts, Appl. Environ. Microbiol., 1999, 65(6), 2324–2332 CrossRef CAS PubMed.
- Y. Ni and R. R. Chen, Biotechnol. Bioeng., 2004, 87, 804–811 CrossRef CAS PubMed.
- A. M. Kunjapur, Y. Tarasova and K. L. J. Prather, J. Am. Chem. Soc., 2014, 136(33), 11644–11654 CrossRef CAS PubMed.
- M. Kitagawa, T. Ara, M. Arifuzzaman, T. Ioka-Nakamichi, E. Inamoto, H. Toyonaga and H. Mori, DNA Res., 2005, 12, 291–299 CrossRef CAS PubMed.
- S. Shalel-Levanon, K.-Y. San and G. N. Bennett, Metab. Eng., 2005, 7, 364–374 CrossRef CAS PubMed.
- G. P. da Silva, M. Mack and J. Contiero, Biotechnol. Adv., 2009, 27, 30–39 CrossRef PubMed.
- V. K. Jain, B. Divol, B. A. Prior and F. F. Bauer, J. Ind. Microbiol. Biotechnol., 2011, 38(9), 1427–1435 CrossRef CAS PubMed.
- Z. Chen and D. Liu, Biotechnol. Biofuels, 2016, 9, 205 CrossRef PubMed.
- K. Martínez-Gómez, N. Flores, H. M. Castañeda, G. Martínez-Batallar, G. Hernández-Chávez, O. T. Ramírez, G. Gosset, S. Encarnación and F. Bolivar, Microb. Cell Fact., 2012, 11, 46 CrossRef PubMed.
- M. Szenk, K. A. Dill and A. M. R. de Graff, Cell Syst., 2017, 5, 95–104 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc00333c |
| This journal is © The Royal Society of Chemistry 2022 |