
Electric potential-determined redox intermediates for effective recycling of spent lithium-ion batteries†
Yunhui
Hua‡
ab,
Zhenghe
Xu
c,
Baojun
Zhao
bd and
Zuotai
Zhang
 *a
*a
aSchool of Environmental Science and Engineering, Southern University of Science and Technology (SUSTech), Shenzhen 518055, China. E-mail: zhangzt@sustech.edu.cn
bSchool of Chemical Engineering, The University of Queensland, St Lucia 4072, Brisbane, Australia
cDepartment of Materials Science and Engineering, Southern University of Science and Technology (SUSTech), Shenzhen 518055, China
dFaculty of Materials, Metallurgy and Chemistry, Jiangxi University of Science and Technology, Ganzhou 341000, China
First published on 30th March 2022
Abstract
Recycling of spent lithium-ion batteries (LIBs) by hydrometallurgy faces a major problem of consuming an excessive amount of acid and requiring different redox additives for effective metal leaching. Reducing chemical consumption in the recycling process is highly desirable for the environmentally friendly and sustainable development of renewable energy. In this study, an electrochemical approach for analyzing electric potentials was developed to evaluate redox abilities. Based on the results, a salt leaching method was proposed using water-soluble NH4Fe(SO4)2 as a redox intermediate for synergistic recovery of valuable metals from spent ternary lithium-ion batteries (NCM) and LiFePO4 batteries (LFP). More than 97% of the Li, Mn, Co and Ni from the mixed cathode can be leached under mild conditions (50 °C, 30 min), with PO43− being completely retained in residues. The first-step reaction between Fe3+ and LFP to release Fe2+ proceeded rapidly, whereas the following slower reaction between Fe2+ and NCM was the rate-controlling process. Thermodynamic analysis of leaching solutions was carried out systematically and shown to be feasible for designing a precipitate recovery process for both LFP and NCM battery systems, with the recovered products being used for regenerating new materials. The synergistic salt-leaching treatment of spent LFP and NCM batteries based on electrochemical principles helped achieve high efficiency and high selectivity with a great benefit to preserving the environment.
1 Introduction
A global tendency toward sustainable development is leading to a rapid increase in applications of mobile equipment, including electric vehicles (EVs) and other portable electronic devices. According to statistics from international agencies, the annual sales of EVs in 2021 still reached a growth of 98% over 2020 despite the worldwide impact of the COVID-19 pandemic.1,2 More countries are setting carbon-neutral policies and more electric transportation modes are expected in the future. Electric transportation mostly utilizes lithium-ion batteries (LIBs) as a power source because of their superior performance to those of lead, Ni–Cd, and Ni–MH batteries.3,4 However, after their life span, large amounts of spent batteries are generated, with the associated environmental pollution in the form of heavy metals and toxic organics.5,6 On the one hand, spent LIBs still have relatively high energy potential and may be able to go through second life, which may extend their lifetimes. The comprehensive use of spent LIBs can also reduce the environmental pressure by providing flexibility and additional time to establish the required recycling facilities for sustainable development.7 Spent LIBs that finally reach the end of their lifetime contain a high content of valuable metals such as Co, Ni and Li, which can be an important resource for sustainable production.8,9 Therefore, recycling of spent LIBs is of great importance from both economic and environmental perspectives.Most research on recycling of spent LIBs focuses on the recovery of valuable metals from cathodes, including LiCoO2 (LCO), LiNixCoyMnzO2 (NCM), LiNixCoyAlzO2 (NCA), LiMn2O4 (LMO) and LiFePO4 (LFP).10 As for LCO, NCM, NCA and LMO, these cathode materials are usually composed of high-valence, transition metal oxides. The methods to recover these valuable transition metals are primarily pyrometallurgy and hydrometallurgy, while other methods including direct recycling are still in early stages of development.11 Pyrometallurgy uses a carbothermic reduction or salt-roasting method at high temperatures to extract metals, which requires high energy consumption and generates greenhouse gases, toxic gases and other hazardous slags.12,13 The pyrometallurgical process therefore could impose a number of environmental risks including global warming and photochemical pollution.14 To attain the energy-saving and carbon-neutral objective for future development, hydrometallurgy is becoming more preferable.15 Hydrometallurgy uses inorganic acids (e.g., HCl16,17 and H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18,19) or organic acids (e.g., acetic acid,20 citric acid,21–23 and oxalic acid24,25) as leaching agents for metal recovery. However, various kinds of spent battery cathodes need to be separated first because they require different redox additives for metal recovery. For the spent NCM and LCO cathodes, for example, reductants (e.g., H2O2, NaHSO3, and ascorbic acid26–28) are usually used to facilitate the reduction and release of the metals; but for spent LFP cathodes, additional oxidants (e.g., H2O2 and Na2S2O8) are required to release Li for recovery.29,30 Moreover, excessive amounts of leaching reagents are often needed to achieve high leaching efficiency because some components, such as H2O2 in the reagent, are unstable and easily decomposed by the catalytic effect of the metal ions (e.g., Fe3+/Fe2+ (ref. 31 and 32)). It is desirable to investigate new processes to reduce the use of reductant and oxidant additives to achieve green and highly efficient recycling of spent LIBs.33
18,19) or organic acids (e.g., acetic acid,20 citric acid,21–23 and oxalic acid24,25) as leaching agents for metal recovery. However, various kinds of spent battery cathodes need to be separated first because they require different redox additives for metal recovery. For the spent NCM and LCO cathodes, for example, reductants (e.g., H2O2, NaHSO3, and ascorbic acid26–28) are usually used to facilitate the reduction and release of the metals; but for spent LFP cathodes, additional oxidants (e.g., H2O2 and Na2S2O8) are required to release Li for recovery.29,30 Moreover, excessive amounts of leaching reagents are often needed to achieve high leaching efficiency because some components, such as H2O2 in the reagent, are unstable and easily decomposed by the catalytic effect of the metal ions (e.g., Fe3+/Fe2+ (ref. 31 and 32)). It is desirable to investigate new processes to reduce the use of reductant and oxidant additives to achieve green and highly efficient recycling of spent LIBs.33
To save energy and resources, some studies have proposed “treating waste by waste”. For example, carbon black or aluminum foil from spent LIBs have been taken as reductants for spent LCO or NCM cathodes.34,35 Spent Ni–MH batteries have also been reported as reductants for LCO cathodes, and valuable metals from both batteries can be effectively recovered.36 LFP and NCM batteries have emerged as major battery types on the market and they are expected to occupy a greater market share in the future.37 Some studies have attempted to use sulfuric acid to recycle LFP, LCO and NCM synergistically.38,39 It was reported that the intrinsic redox reaction could limit the acid dosage and also reduce the addition of reductants or oxidants. However, the kinetics and mechanisms of the synergistic reactions remain unclear, and an in-depth study is needed to achieve efficient production. Moreover, sulfuric acid as a leaching agent does not have selectivity for every element. The phosphorus existing as PO43− (or H3PO4, H2PO4−, or HPO42−) in the leaching solution will inevitably bring impurities into an NCM or LCO system, and the iron from LFP may not be able to combine with PO43− effectively for complete precipitation. Therefore, a selective leaching system is needed for the synergistic treatment of spent LFP and NCM batteries.
To achieve such selectivity, we proposed in this study a salt leaching method to recycle valuable metals from spent LFP and LiNi0.6Co0.2Mn0.2O2 (NCM622) battery cathodes. The electrochemical measurements of electric potentials were conducted to evaluate redox ability. Based on this electrochemical study, the freely soluble ammonium ferric sulfate (NH4Fe(SO4)2) was taken as an intermediate leaching agent for metal extraction from both cathodes. It was revealed that the introduction of ferric ions into this synergistic treatment can effectively extract valuable metals and completely precipitate phosphorus as ferric phosphate. The amount of ferric salt needed for the process was close to theoretical calculations (stoichiometric amount), and there were no other impurities released throughout the process. A thermodynamic study on the leaching solution is shown to be suitable for designing product recovery. The selective salt leaching by Fe3+ is a feasible approach for the green and efficient recycling of spent LIBs.
2 Experimental
2.1 Electrochemical test of reaction potentials
The reactants in this study include solid powders and aqueous solutions. Each gram of solid powder was mixed with 0.1 g carbon black, 0.25 g ethyl cellulose, 19 ml ethanol and 1.5 ml terpineol. The mixture was put in a beaker and stirred for 12 h, and then kept still for another 12 h. The supernatant of the mixture was spin-coated on a conductive glass, followed by a thermal treatment at 200 °C for 2 hours. The aqueous solution was prepared by mixing the tested salt with sodium sulfate (Na2SO4) in water. The concentration of each tested reactant was set at 0.1 M, with the Na2SO4 at 0.5 M to improve the electro-conductivity.The electrochemical test was conducted with an electrochemical work station (produced by Corretest, Wuhan, China). Coated conductive glass was used as the working electrode and a platinum foil electrode as the auxiliary electrode. A Hg(s)|Hg2SO4(aq.)| and Sat. K2SO4(aq.) electrode (reference potential: 0.64 V vs. NHE) was used as the reference electrode. The electric potential was obtained by measuring the open circuit potential of each system and then calculated according to eqn (1):
| E = Em + Ere | (1) |
2.2 Process for metal leaching and recovery
The flow process for leaching and recovery of metals from spent cathode materials is given in Scheme 1. Spent NCM and LFP cathode powders were mixed with the selected salt in a glass flask. Water was added at a solid to liquid ratio of 50 g L−1. The leaching reaction was conducted in a water bath under different conditions. After the reaction, the solution and residue were separated in a centrifuge tube. In one treatment, the leaching residue was put into an H3PO4/NH4H2PO4 buffer solution to regenerate FePO4·2H2O. The pH was adjusted according to theoretical calculations using Visual Minteq software. After the reaction, the solid was heated at 600 °C under an air atmosphere for 1 h to burn carbon and other impurities.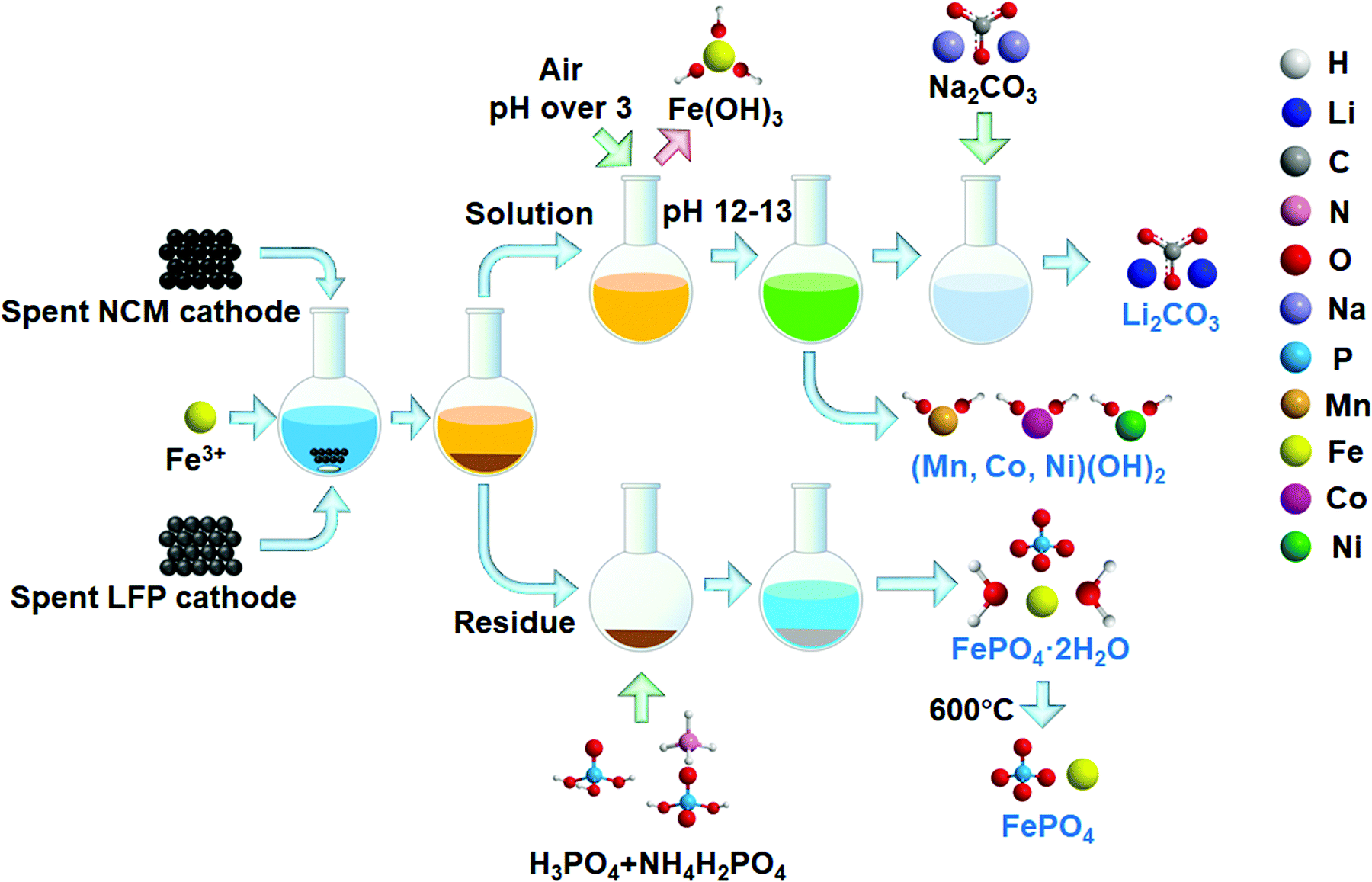 | ||
| Scheme 1 Flow process for recycling of valuable components in NCM and LFP with Fe3+ as an intermediate. | ||
In the other treatment, the solution was adjusted to a pH of over 3 and pumped with air to fully oxidize and precipitate the remaining Fe3+/Fe2+; it was then filtered with a 0.22 μm pore size filter. After filtration, NaOH was added to adjust the pH for Mn, Co and Ni precipitation. The precipitation pH was also designed based on thermodynamic calculations. The precipitates were separated and heated at 900 °C under an air atmosphere for 1 h to obtain metal oxides, while Na2CO3 was added into the supernatant to collect Li2CO3. The remaining alkaline solution can be used in gas control technology to absorb some acidic greenhouse or hazardous gases, including CO2, SOx and HCl to reduce their negative environmental impact.
2.3 Metal leaching efficiency analysis
To determine the composition of the spent NCM622 and LFP cathodes, the cathode powder was digested with aqua regia (composed of hydrochloric acid and nitric acid with a volume ratio of 3:1) using digestion apparatus, and then diluted with deionized water. The metal concentrations in the leachate were analyzed by inductively coupled plasma mass spectrometry (ICP-MS) using an Agilent Technologies 7700X ICP-MS. The leaching efficiency was calculated using eqn (2):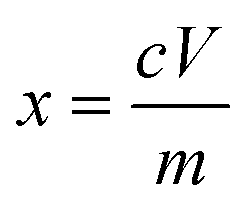 | (2) |
2.4 Characterization of materials and products
Materials and products at different stages were ground into a powder and loaded on a silicon wafer and then characterized by X-ray diffraction (XRD) using a Rigaku Smartlab XRD operating at 45 kV and 200 mA. The scanning angle 2θ varied from 10 to 80° at a scanning rate of 5° per minute.The morphology and element distribution of products were analyzed using a scanning electron microscope (SEM) equipped with an energy-dispersive X-ray spectrometer (EDX) and a Zeiss Merlin SEM. The scanning voltage for SEM and EDX mapping was set at 15 kV.
Elements and their valences in the products were analyzed by X-ray photoelectron spectroscopy (XPS) using a PHI 5000 Versaprobe III XPS. MultiPak software was used to assist in the identification of elements and their valences. The carbon binding energy was set at 284.8 eV for standard calibration.
3 Results and discussion
3.1 Design of the leaching system by reaction potentials
The synergistic reaction involves two parts, including reducing high valence Mn, Co and Ni into corresponding Mn2+, Co2+ and Ni2+,30,40 and oxidizing LiFePO4 into FePO4.41,42 NCM622 contains multiple transition metals with different valences. To understand the reduction reaction in the NCM622 cathode, the XPS analysis of its transition metals is given in Fig. 1a–c.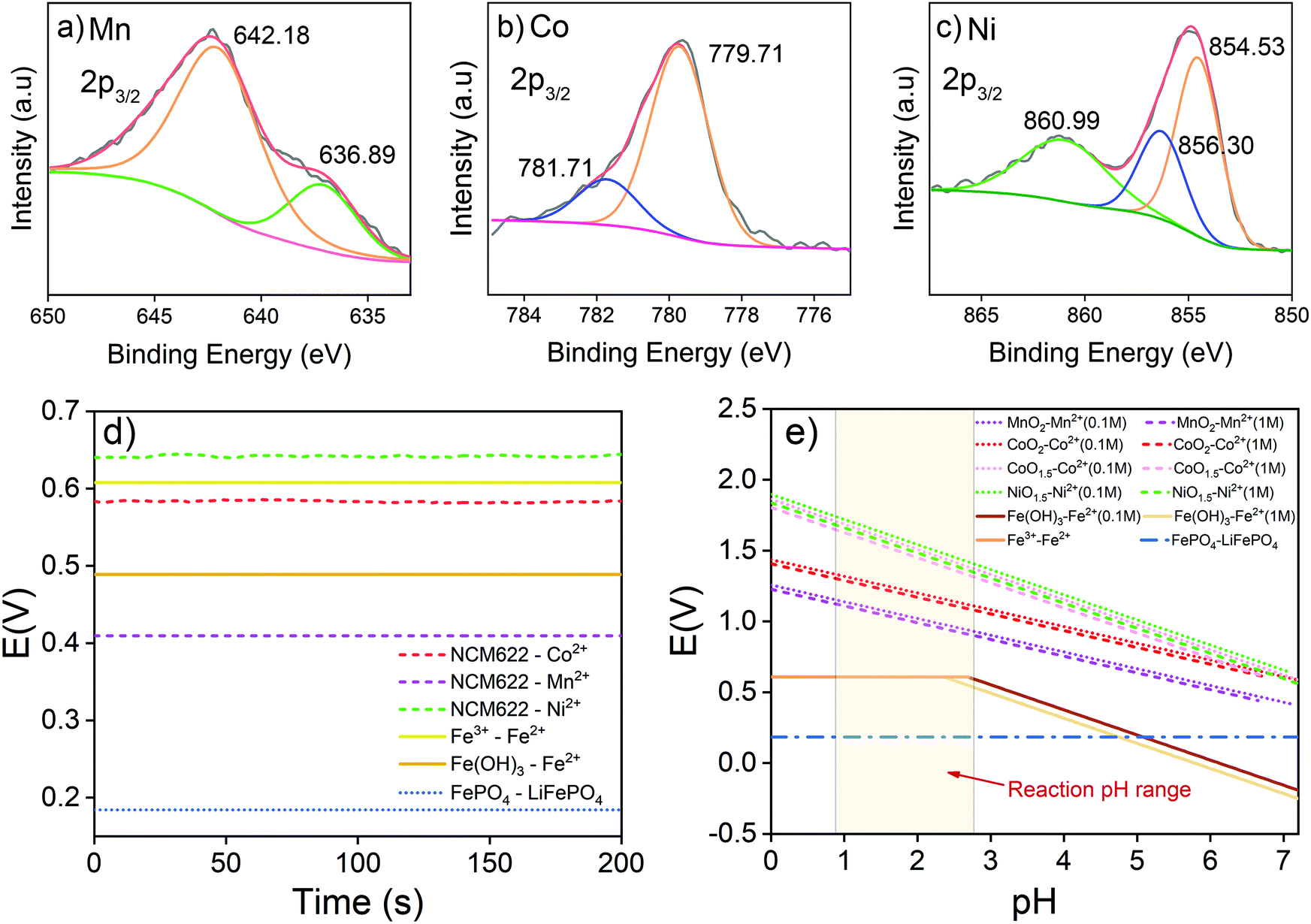 | ||
| Fig. 1 XPS spectra of (a) Mn, (b) Co, and (c) Ni from the spent NCM622 cathode material; (d) reaction potentials of redox couples; and (e) calculated reaction potentials of redox couples at different pH values according to the Nernst equation. | ||
The main peak at 642.18 eV is assigned to Mn(IV), and the small peak at 636.89 eV is an Auger peak of Mn.43–45 As for Co, the main peak at 779.71 eV and the weaker peak at 781.7 eV are assigned to Co(III) and Co(IV), respectively. The Ni in the cathode mainly exists as Ni(II) (854.53 eV) and Ni(III) (856.30 eV).44–48 The satellite peak at 860.99eV is also assigned to Ni(II).43,48 XPS peaks for Mn(III) in NCM111 or NCM523 are not detected.47–49 Based on the results from XPS analysis, transition metals in LiNi0.6Co0.2Mn0.2O2 can be expressed as NiO, NiO1.5, CoO1.5, CoO2, and MnO2. The reduction reaction involving electron transfer in LiNi0.6Co0.2Mn0.2O2 expressed in eqn (3) can also be separated and written as eqn (4) (MOx represents NiO, NiO1.5, CoO1.5, CoO2, and MnO2).
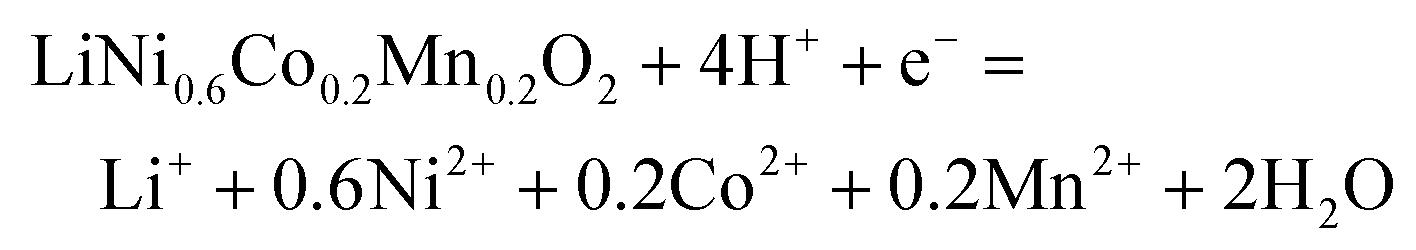 | (3) |
| MOx + 2xH+ + (2x − 2)e− = M2+ + xH2O | (4) |
The measured reaction potentials of transition metal reductions in LiNi0.6Co0.2Mn0.2O2 are given in Fig. 1d and Table 1. The oxidation potential of LiFePO4 into FePO4 (eqn (5)) was also determined.
| FePO4 + Li+ + e− = LiFePO4 | (5) |
| Redox couple | E (V) | pH |
|---|---|---|
| LiNi0.6Co0.2Mn0.2O2/Ni2+ | 0.6419 | 7.08 |
| LiNi0.6Co0.2Mn0.2O2/Co2+ | 0.5833 | 7.22 |
| LiNi0.6Co0.2Mn0.2O2/Mn2+ | 0.4094 | 7.17 |
| FePO4/LiFePO4 | 0.1841 | 8.10 |
| Fe3+/Fe2+ | 0.6077 | 2.30 |
| Fe(OH)3/Fe2+ | 0.4890 | 3.36 |
It is noticed that the reaction conditions of the electrochemical test (pH and ion concentrations) were not the same as the metal leaching conditions. Therefore, the potentials are converted into the actual pH and concentration by the Nernst equation as given in eqn (6).
| aox + ne− → bred |
 | (6) |
In this study, reactions on NCM622 involved the participation of H+; therefore, the redox reaction is affected by the acidity of the system. The Nernst equation is expressed as eqn (7) (MOx = NiO, NiO1.5, CoO1.5, CoO2, and MnO2):
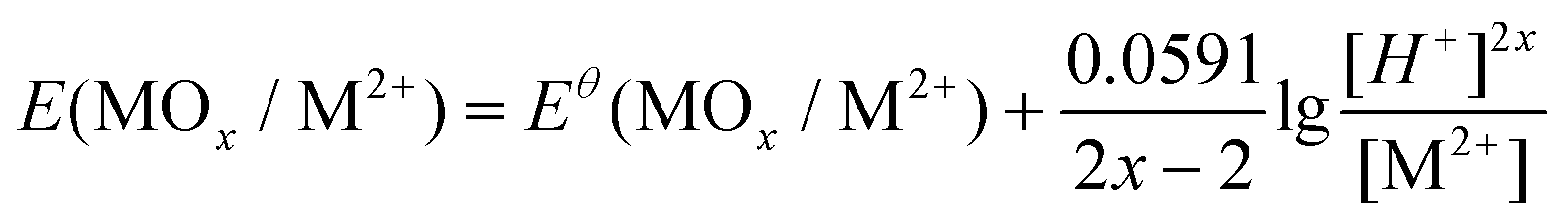 | (7) |
The oxidation of LiFePO4 into FePO4 does not involve the acidity or the concentration of the redox couple. Therefore, the potential remains unchanged under the test conditions. The variation of calculated reaction potentials with pH and the concentration of redox couples is given in Fig. 1e. In this study, the solid-to-liquid ratio (S/L) was set at 50 g L−1. Concentrations of metal elements in the solution ranged from 0.1 to 1 M. However, as revealed in Fig. 1e, the concentration differences do not have as much influence as pH on redox reaction potentials. The acidity of the solution is the dominant factor for the redox reactions of NCM and LFP, and a lower pH is preferable. NCM622 and LFP are both insoluble solids, and a water-soluble intermediate is needed for effective reaction. To avoid introducing more elements into the system, the Fe3+ was assumed to be a good candidate of intermediates.
The ionic Fe3+ can only exist at low pH, whereas at pH higher than 3, it will be transferred into Fe(OH)3 precipitates. Redox potentials for an Fe3+/Fe2+ couple and an Fe(OH)3/Fe2+ couple are given in Fig. 1d and Table 1. The redox reactions are represented by eqn (8) and (9), with their corresponding redox reaction potentials at different concentrations and pH being calculated (eqn (10) and (11)) and shown in Fig. 1e.
| Fe3+ + e− = Fe2+ | (8) |
| Fe(OH)3 + 3H+ + e− = Fe2+ + 3H2O | (9) |
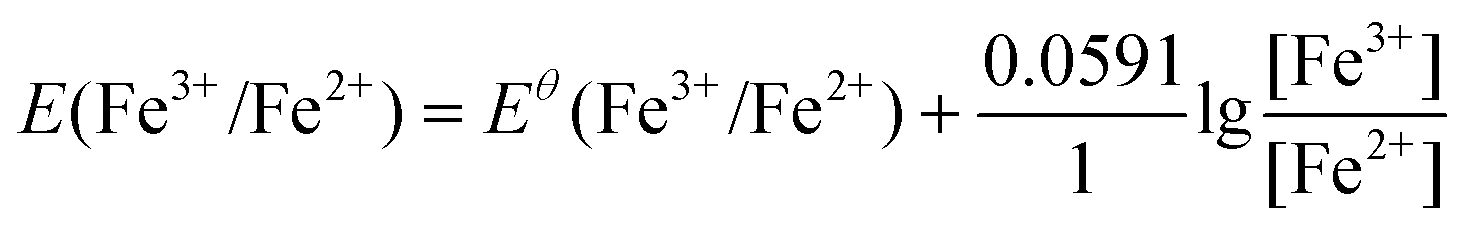 | (10) |
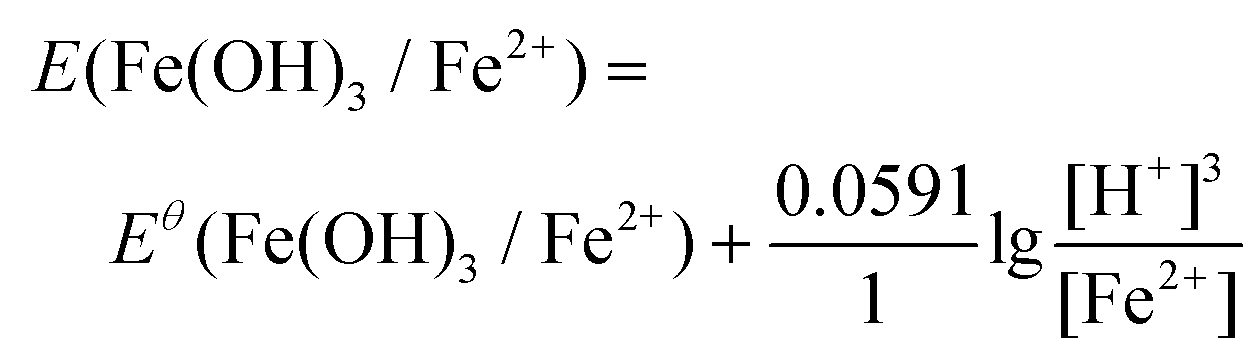 | (11) |
When Fe(III) exists as ionic Fe3+, the electric potential remains unchanged with pH, but when Fe(III) exists as Fe(OH)3, the potential decreases with increasing pH. On the one hand, the electric potential of Fe(III)/Fe2+ is always lower than those of Mn, Co, and Ni from NCM622, which indicates that NCM622 can be reduced by Fe2+. On the other hand, it is predictable that at pH values higher than 5, Fe(OH)3 will no longer be able to oxidize LiFePO4 into FePO4. According to these analyses, Fe3+ should be an effective intermediate for the synergistic treatment of NCM and LFP at low pH. Since Fe3+ can already provide an acidic environment, additional acid should not be needed for the metal extraction reaction. In conclusion, an electrochemical study is shown to be a convenient approach to design redox additives.
3.2 Analysis of the leaching process
The NCM and LFP cathodes are both insoluble solid powders. Therefore, a water-soluble Fe(III) salt is needed to act as an intermediate for an effective reaction. FeCl3 and Fe(NO3)3 were not suitable here because they have potential to generate hazardous Cl2 or NOx.16 Therefore, ferric sulfate including Fe2(SO4)3 and NH4Fe(SO4)2 (which existed as NH4Fe(SO4)2·12H2O) were selected for the metal leaching test. The comparison of the leaching effect between Fe2(SO4)3 and NH4Fe(SO4)2 is given in Fig. S1.† The results reveal a better leaching performance of NH4Fe(SO4)2 than that of Fe2(SO4)3. The superior performance of NH4Fe(SO4)2·12H2O is attributed to its higher dissolution rate and hence more Fe3+ released into the solution in a shorter time. In contrast, the dissolution of Fe2(SO4)3 into water is a slow process, and thus the whole reaction rate is limited. The limit of slow dissolution of Fe2(SO4)3 was confirmed by conducting an additional test by using Fe2(SO4)3 solutions as the intermediate in the leaching test. The results in Fig. S2† show a significantly improved leaching efficiency by using Fe2(SO4)3 aqueous solutions as compared with the addition of Fe2(SO4)3 powders. In fact, leaching using Fe2(SO4)3 aqueous solutions could achieve similar (Li and Mn) or better (Co and Ni) efficiencies as compared with the case of using NH4Fe(SO4)2·12H2O in the powder as the intermediate. This finding suggests a great benefit of using aqueous solutions rather than powder forms of the intermediates, which will be further tested as an important parameter in the upcoming scale up tests and reported in a separate communication. Furthermore, an overdose of Fe2(SO4)3 to fully extract the transition metals from NCM622 inevitably leads to some extra Fe2+ in the liquid that needs to be oxidized after leaching. Higher pH is desirable for oxidation and removal of Fe2+ (Fig. 1e) but it may also lead to the precipitation of other transition metals. The ammonium present can form a complex with Mn, Co and Ni, preventing them from precipitating at an early stage and providing more flexibility for treating the remaining Fe2+. Therefore, NH4Fe(SO4)2·12H2O was selected as a leaching intermediate in this study.The influence of the mass ratio between LFP and NCM622 is given in Fig. 2a and Fig. S3.† LFP and NCM622 theoretically react at a molar ratio of 1:1. The mass ratio is therefore calculated to be LFP/NCM622 = 1.63. Leaching results show that LFP/NCM622 > 1.8 is suitable for high-efficiency metal extraction where over 97% valuable metals can be extracted from both cathodes. For a higher mass ratio, the leaching efficiency of Li, Mn, Co, and Ni does not change markedly, but excessive Fe2+ will be generated, which needs additional oxidation by air or another oxidant to separate it from other transition metals. Therefore, LFP/NCM622 = 1.8 is a suitable composition.
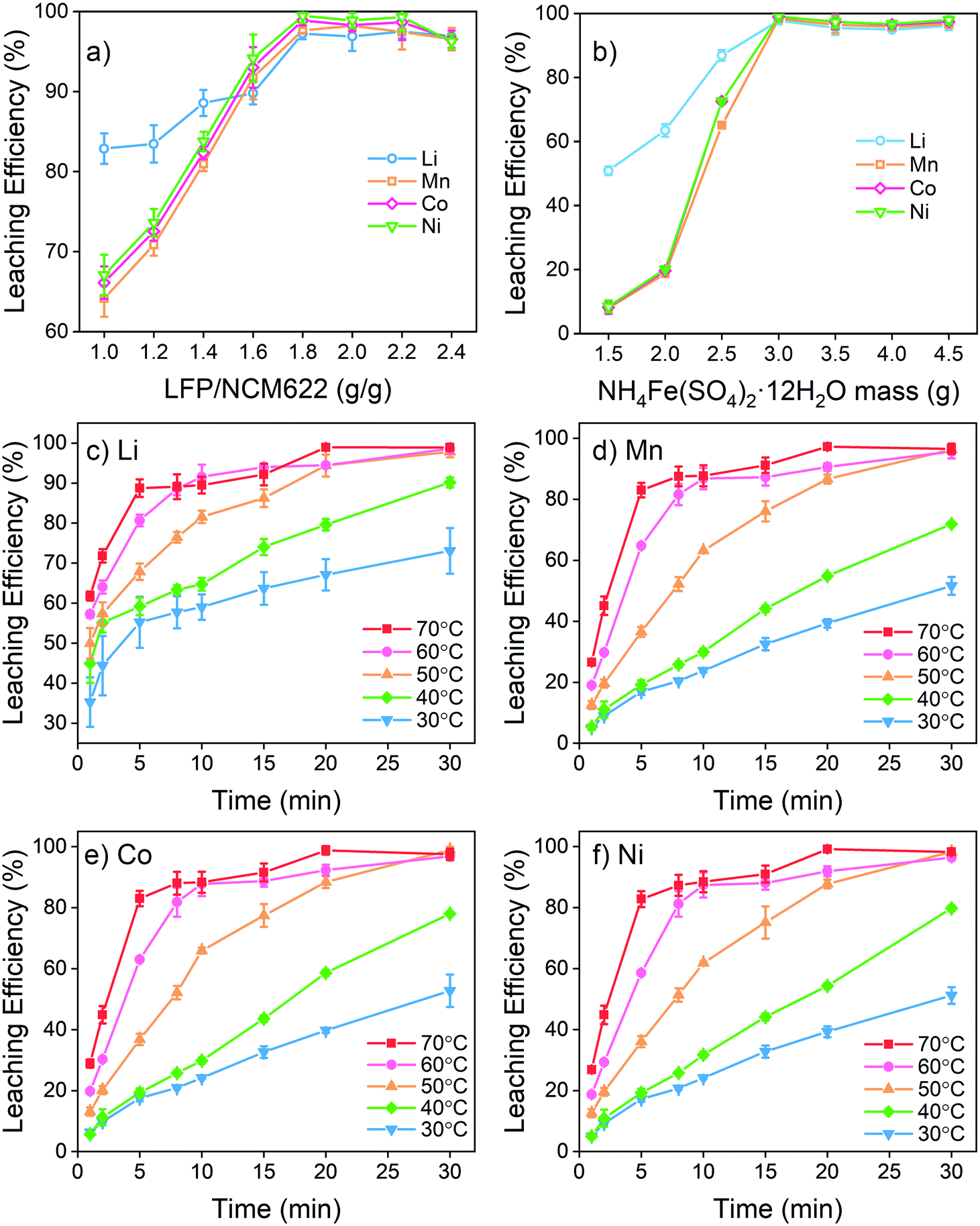 | ||
| Fig. 2 (a) Leaching efficiency of metals at different LFP/NCM622 mass ratios (g g−1) with NH4Fe(SO4)2·12H2O/mixed cathode powder = 3:1 (g g−1) at 50 °C for 30 min; (b) leaching efficiencies of metals with different amounts of NH4Fe(SO4)2·12H2O with LFP/NCM622 = 1.8 (g g−1) at 50 °C for 30 min; and (c–f) leaching efficiencies of metals at different temperatures and times with LFP/NCM622 = 1.8 (g g−1) and NH4Fe(SO4)2·12H2O/mixed cathode powder = 3:1 (g g−1). | ||
The amount of NH4Fe(SO4)2·12H2O added is also important for full metal extraction. According to the reaction function, it is theoretically predicted that 2.56 g of NH4Fe(SO4)2·12H2O is needed for complete metal extraction from 1 g of the mixed powders. It is revealed in Fig. 2b that at least 3 g of NH4Fe(SO4)2·12H2O is needed to fully extract the metals. Excessive Fe3+ introduced into the solution does not have any more positive influence on leaching efficiency or other process behaviors. Therefore, 3 g of NH4Fe(SO4)2·12H2O to treat 1 g of mixed powder (LFP/NCM622 = 1.8) is proposed to be the optimized internal condition. Under the optimized internal conditions, the system pH is found to be 0.91 at the beginning and 2.88 after the reaction is completed (as colored in Fig. 1e). According to the electrochemical study discussed earlier, such a pH range is assumed to be suitable for the redox reaction between LiNi0.6Co0.2Mn0.2O2 and LiFePO4 with the assistance of the intermediate Fe3+. The leaching efficiency of phosphate is given in Fig. S4 and S5.† PO43− is not extracted in all tests, which is different from the leaching with a strong inorganic acid. With the addition of Fe3+ into the system, the phosphate remains as residue. Previous studies also showed that PO43− remains as a FePO4 solid in such a pH range.50,51 The high-efficiency selective leaching demonstrates that the proposed electrochemical test and calculation method are applicable for treating mixed spent LIB cathodes with different redox properties.
The external reaction conditions including temperature and time were also investigated, and the results are given in Fig. 2c–f. Higher temperatures and longer reaction times were shown to improve the leaching efficiencies. With increasing temperatures, the leaching time for full extraction is reduced gradually. According to the experimental results, 50 °C and 30 min are regarded as suitable conditions. During the leaching process, Li is extracted from both LiNi0.6Co0.2Mn0.2O2 and LiFePO4, featuring a faster leaching rate than transition metals. At 30 °C, for example, over 50% of Li is already released into the solution within 5 min. Mn, Co and Ni are known to be uniformly distributed in the cathode structure. Therefore, even though they have different redox potentials, they eventually have similar leaching characteristics and are simultaneously released into the solution.
In summary, the optimal leaching conditions are determined to be LFP/NCM622 = 1.8 (g g−1) and LiNi0.6Co0.2Mn0.2O2/mixing cathode = 3 (g g−1) at 50 °C and 30 min. Under these conditions, valuable metals from both LFP and NCM622 cathodes can be effectively extracted, while the Fe2+ and Fe3+ that need further treatment are minimized.
A comparison of cathode material leaching from NCM and LFP is given in Table 2. In most studies, inorganic acids or organic acids are used as leaching agents to extract metals from the cathode. Additional reductants for LCO and NCM, or oxidants for LFP, are also necessary to improve the leaching efficiency. Compared with the results from previous studies, the leaching process developed in this study does not require the addition of an acid. Moreover, the intrinsic redox reaction between LiNi0.6Co0.2Mn0.2O2 and LiFePO4 also reduces the additional reductant or oxidant to a large extent. Synergistic leaching is a feasible method for treating mixed cathodes from spent LIBs.
| Cathode | Leaching agent | Conditions | Efficiency | Ref. |
|---|---|---|---|---|
| NCM, LFP | 0.3 M NH4Fe(SO4)2 | 50 g L−1, 50 °C, 30 min | 97.8% Li, 98.3% Mn, 98.9% Co, 99.0% Ni | This study |
| NCM, LFP | 0.25 M H2SO4 | 32 g L−1, 80 °C, 4 h | >96% Li, Mn, Co, Ni | 38 |
| LCO, LFP | 0.5 M H2SO4 | 30 g L−1, 20 min | 99% Li, Fe, P, 92.4% Co | 39 |
| LCO, LMO, NCM, LFP | 4 M H2SO4, 30% H2O2 | 70–80 °C, 2–3 h | Li, Mn, Co, Ni dissolved, LFP partly dissolved | 51 |
| LCO | 2 M H2SO4, 6% H2O2 | 100 g L−1, 60 °C, 1 h | 97% Li, 98% Co | 19 |
| LCO | 1.25 M citric acid, 1% H2O2 | 20 g L−1, 90 °C, 30 min | 100% Li, >90%Co | 21 |
| NCM | 2.5 M H2SO4, 0.8 M NH4Cl | 100 g L−1, 80 °C, 1 h | 99.11% Li, 97.34% Mn, 97.55% Co, 97.49% Ni | 52 |
| NCM | 3 M formic acid, 6% H2O2 | 50 g L−1, 60 °C, 2 h | 98.22% Li, 99.95% Mn, 99.96% Co, 99.96% Ni | 53 |
| LFP | 0.3 M H2SO4, H2O2/Li = 2.07, H2SO4/Li = 0.57 | 60 °C, 2 h | 96.85% Li | 29 |
| LFP | 0.8 M acetic acid, 6% H2O2 | 120 g L−1, 50 °C, 30 min | 95.05% Li | 54 |
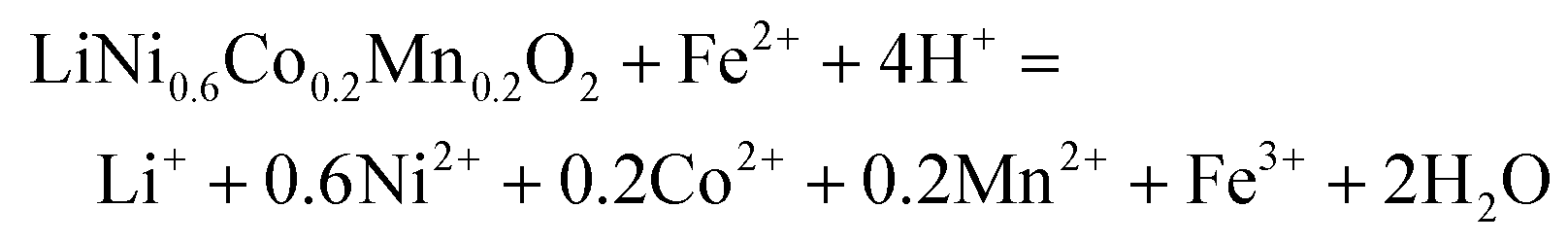 | (12) |
| Fe3+ + 3H2O ↔ Fe(OH)3 + 3H+ | (13) |
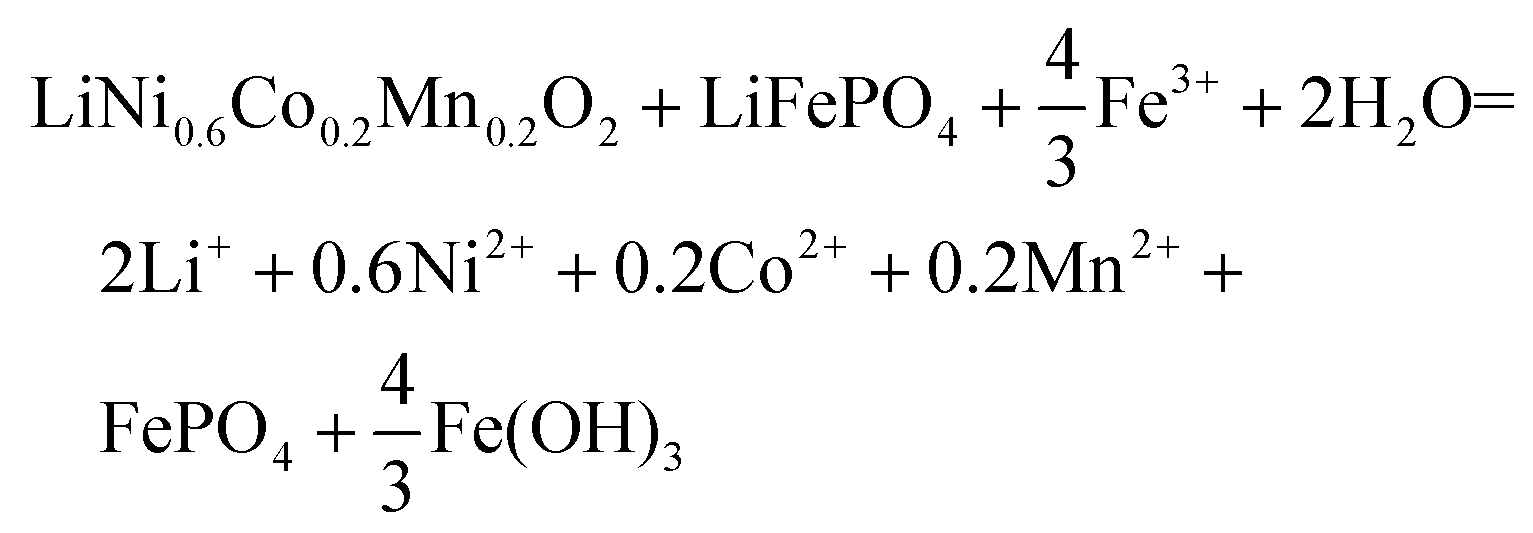 | (14) |
The reaction kinetics was studied in terms of various solid–liquid reaction models. In general, the solid–liquid reaction is controlled by the following three factors: mass transfer in the liquid boundary layer (eqn (15)), chemical reaction on the surface (eqn (16)) and diffusion in the solid surface layer (eqn (17)).53,55
| x = k1t | (15) |
| 1 − (1 − x)1/3 = k2t | (16) |
| 1 − 3(1 − x)2/3 + 2(1 − x) = k3t | (17) |
 | (18) |
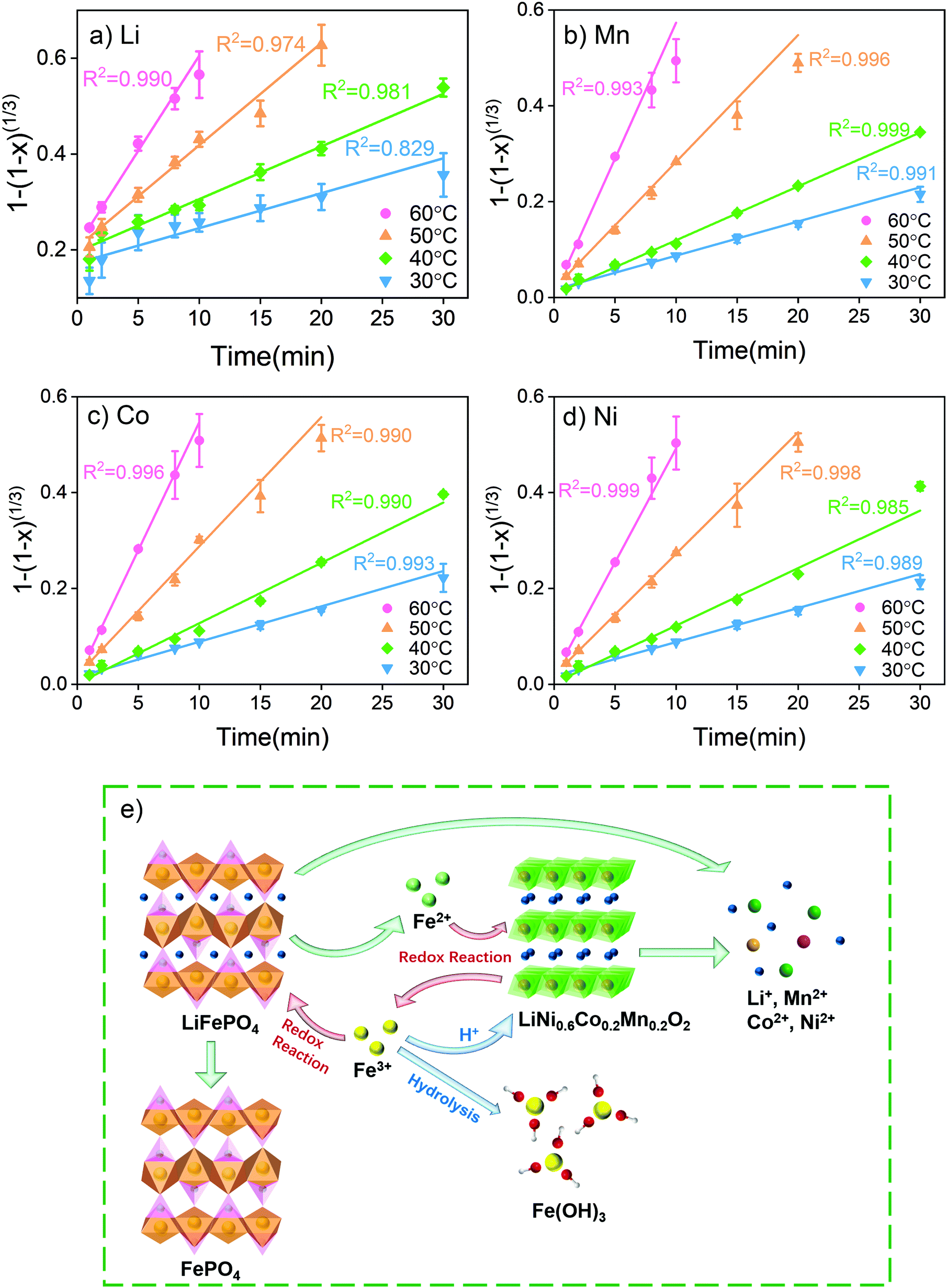 | ||
| Fig. 3 (a) Fitting results of (a) Li, (b) Mn, (c) Co and (d) Ni, according to a surface chemical reaction model, and (e) reaction mechanisms of synergistic metal extraction from NCM and LFP using Fe3+ as an intermediate. | ||
According to the fitting results, the leaching of transition metals Mn, Co and Ni is mainly controlled by the surface chemical reactions. At a relatively low leaching temperature of 30 °C and 40 °C, the reaction is also influenced by mass transfer. Lower leaching temperatures led to slow precipitation of FePO4 and Fe(OH)3 with the precipitates being dispersed in solution to increase the viscosity of the system. The increasing leaching temperature increases the thermal energy and hence weakens interactions among the molecules in solution, which decreases the system viscosity. At the same time, increasing the leaching temperature also increases the rate of FePO4 and Fe(OH)3 precipitation. Under these conditions, the influence of mass transfer becomes weaker. In this study, it was found that surface diffusion does not have a noticeable impact on transition metal leaching. Finally, the fitting of activation energy given in Fig. S10† shows an increase in apparent activation energies of 47.47, 53.21, 55.39 and 58.03 kJ mol−1 for Li, Ni, Co and Mn, respectively.
The leaching rate of Li is higher than those of transition metals and kinetic analysis reveals that the reaction of Li is influenced by both surface chemical reaction and diffusion. However, at 30 °C, the leaching result of Li does not fit well with any of the models. In the first minute, a large amount (more than 30%) of Li was leached out rapidly into the solution. After 2 minutes of leaching, Li exhibited a similar leaching trend to other transition metals. The analysis of the reaction mechanisms is given in Fig. 3e. Li comes from both LiFePO4 and LiNi0.6Co0.2Mn0.2O2. LiFePO4 has an olivine crystal structure and Li stays in octahedral voids. With a small ion radius, Li+ is easy to transfer without changing the structure of LiFePO4.
Similar to the working mechanism of LFP, the oxidation of LiFePO4 by Fe3+ simply involves the electron transfer and release of Li+, while the whole olivine structure of LiFePO4 is not destroyed during the reaction.54,56 The reaction is simple and can proceed at a fast rate, which explains the high leaching efficiency of Li in the first few minutes. The redox reaction between Fe2+ and LiNi0.6Co0.2Mn0.2O2 on the other hand is more complicated. The LCO and NCM cathodes have layered structures formed by Mn, Co, Ni and oxygen. Li exists as interlaid ions in the voids between the layers. During the redox reaction, electron transfer occurs between Fe2+ from the solution and Mn, Co and Ni from the cathode. The H+ generated by Fe3+ hydrolysis needs to break chemical bonds in the layer structure to take away the oxygen. Mn, Co and Ni can only be released after the structure is destroyed.57,58 Therefore, compared with the oxidation of LiFePO4, the reduction of LiNi0.6Co0.2Mn0.2O2 has a slower reaction rate and becomes the controlling process of the whole reaction.
3.3 Product recovery and analysis
The degradation of LIBs is usually caused by a number of processes, including structural failure, aging and decomposition of components, and overgrowth of the solid electrolyte interphase.13,59,60 Most of the metals are preserved in the battery cell. The recovered metals are proposed to generate new materials after separation and proper treatment. In this study, the leaching system includes components from both LFPs and NCMs. The study aimed at separating various components into the Fe–P system and Ni–Co–Mn system as in the form of original batteries. In the current case, the precursors FePO4, NiCoMn oxides and Li2CO3 were proposed to be synthesized. To obtain pure products, thermodynamic calculations on solution systems were conducted to assist in determining the experimental conditions.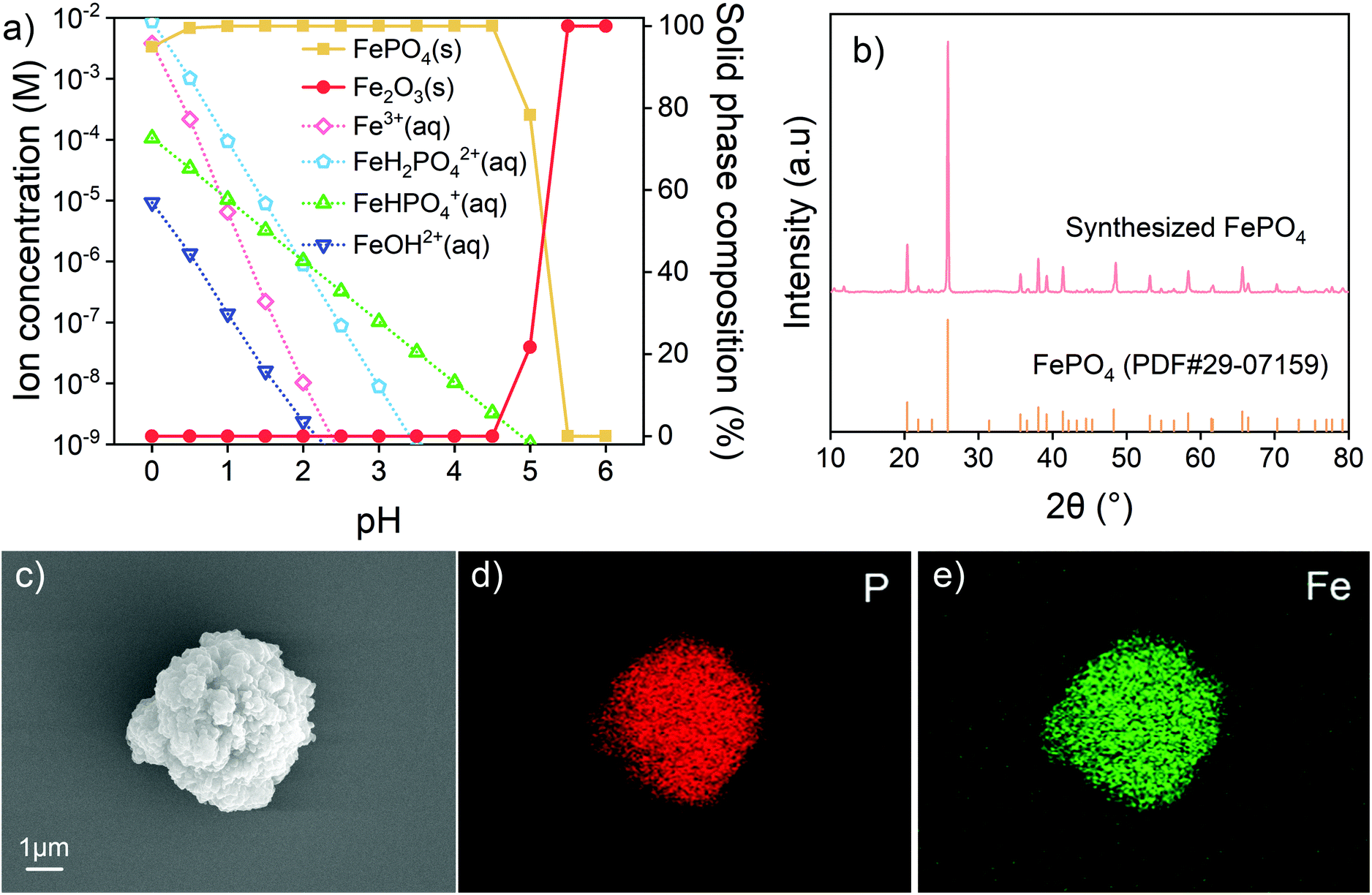 | ||
| Fig. 4 (a) Calculated ion concentrations and solid phase compositions of Fe(III)-PO43− at different pH values; (b) XRD pattern and (c) SEM image of synthesized FePO4 after 600 °C calcination; EDX mapping images of (d) P and (e) Fe from synthesized FePO4 after 600 °C calcination. | ||
Extra water-soluble H3PO4 and NH4H2PO4 were added to adjust pH and to balance Fe. Species with concentrations below 10−6 M in the calculated range were considered negligible in the solution. At pH below 1, there are still considerable amounts of different Fe(III) ion species including Fe3+ and FeH2PO42+ in solution. The solid phase is simply FePO4, in equilibrium with the corresponding soluble species. At low pH, the ionization of H+ from H3PO4 is known to be impeded, which limits the combination of Fe3+ and PO43−. All Fe(III) ion species decrease in concentration with increasing pH. Fe(III) is predicted to be completely precipitated out at pH above 2. However, when the pH exceeds 4.5, the FePO4 precipitates are converted into Fe(OH)3. Therefore, in this study, the leaching residue was washed with an H3PO4–NH4H2PO4 solution at pH 2–3. The product after washing is verified to be ferric phosphate hydrate (FePO4·2H2O) with two kinds of crystal structures (Fig. S11†). To clarify the component and eliminate the possible impurities, the product was calcined under an air atmosphere at 600 °C. As given in Fig. 4b, the anhydrous FePO4 is obtained after the calcination. The SEM image and EDX mapping in Fig. 4c–e show that P and Fe distribute uniformly in the product. Other metal elements (Ni, Co or Mn) from NCM cathodes are not detected in the recovered FePO4. The results suggest that such a method is feasible for FePO4 recovery and the product can be used to produce a second LFP cathode material.
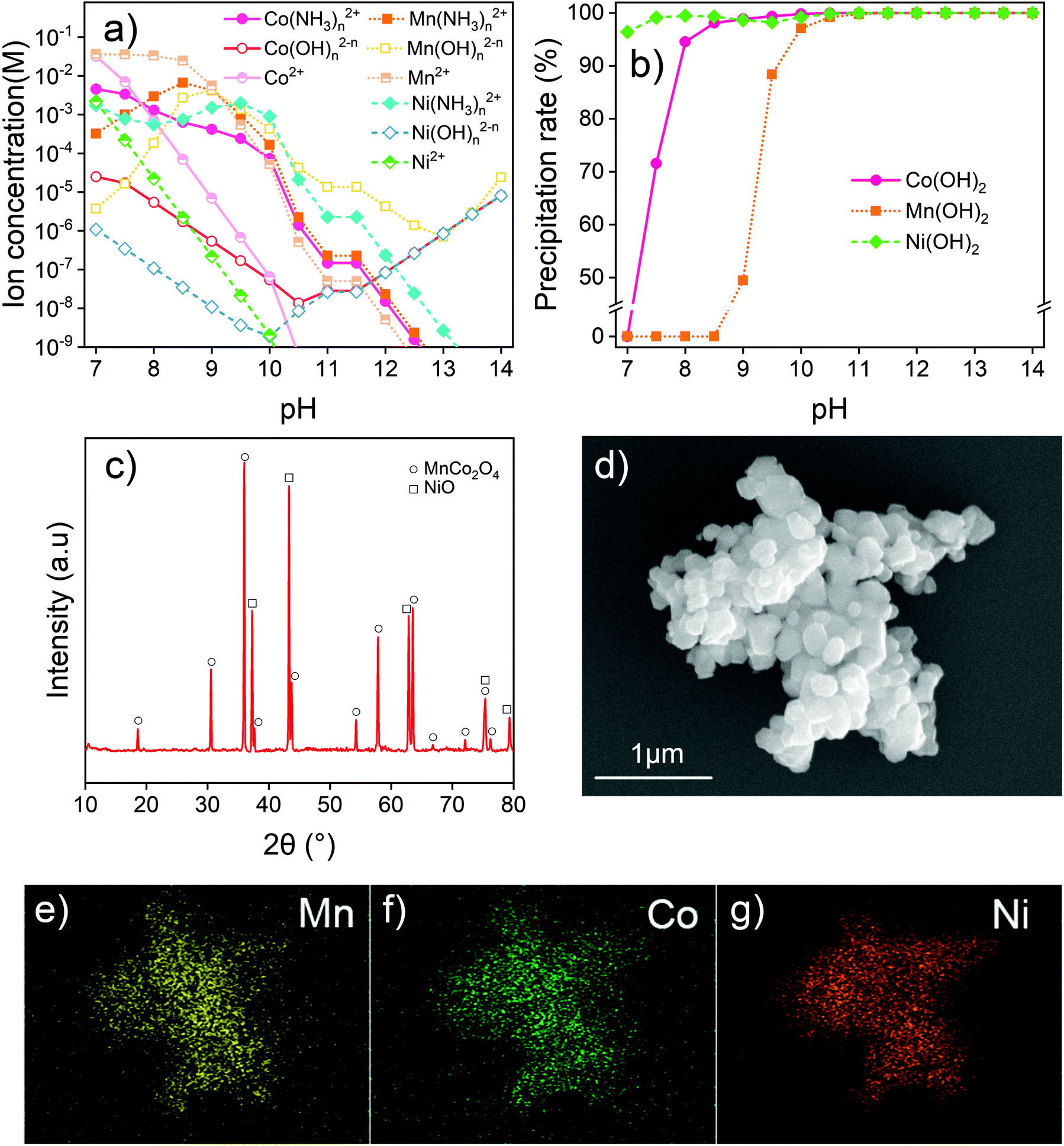 | ||
| Fig. 5 (a) Calculated ion concentrations of Mn(II), Co(II), and Ni(II) at different pH values. (b) Precipitation rate of Mn(OH)2, Co(OH)2, and Ni(OH)2 at different pH values. (c) XRD pattern and (d) SEM image of the synthesized MnCoNi oxide after 900 °C calcination. EDX mapping image of (e) Mn, (f) Co, and (g) Ni from the synthesized MnCoNi oxide after 900 °C calcination. | ||
Mn, Co and Ni can also combine with OH− to form complex ions in a strongly alkaline environment. Concentrations of Mn(OH)n2−n, Co(OH)n2−n and Ni(OH)n2−n follow a similar trend where they first decrease and then increase with increasing pH. Fig. 5b also shows the influence of pH on the recovery of the transition metal precipitate. With increasing basicity, Ni(OH)2 is first precipitated and fully recovered, followed by Co(OH)2 and Mn(OH)2. This finding indicates that the pH should be higher than 11.5 to fully precipitate the transition metals. To fully recover the transition metals as hydroxides, the system pH of 12–13 is considered suitable.
After filtration and separation, the remaining alkaline solution needs to be neutralized to reduce its potential negative impact on the environment. It can be used to absorb acidic greenhouse gases or industrial off gas including CO2, SOx and HCl which may bear implications of carbon neutralization or reduction in emissions of other toxicities into the environment. Normally, the acidic off gas from industrial production requires neutralization by an additional alkali (e.g., CaO and NaOH). During such treatment with the remaining solution, ammonium and sodium salts can be obtained, with the water being reused in the process and air pollution reduced. The recovered transition metal hydroxides were dried and calcined at 900 °C for full decomposition to obtain metal oxides. Fig. 5c shows the solids of NiO and MnCo2O4 crystal structures after the calcination, indicating the presence of transition metals from the NCM cathode. The XPS analysis of the calcined metal oxides is given in Fig. S12.† The valences of transition metals in the recovered solids were found to be the same as that in the raw material, which provides the possibility of regeneration of new materials. The SEM and EDX mapping images of the calcined mixed oxides are given in Fig. 5d–g, showing the calcined products as fine particles with little agglomeration. Ni, Co and Mn have a nearly uniform distribution. Other elements, such as Fe or P from LFPs, are not detected. The analysis of the product indicates that not only is the selective leaching method by Fe3+ suitable for the effective recovery of transition metals from NCM, but also the recovered product can be used to regenerate new materials with proper precipitation and calcination.
In short, the salt leaching by Fe3+ selected from the electric potential measurements can achieve synergistic treatment of spent NCMs and LFPs. Li, Mn, Co and Ni can all be fully extracted while FePO4 is completely separated and preserved in residues. Therefore, it is anticipated that this salt leaching method will be effective for treating mixed spent battery cathodes with high efficiency and selectivity. Moreover, the product recovery is designed based on thermodynamic calculations and the experimental results are consistent with the predictions. Therefore, it can be concluded that a theoretical study is feasible for designing reaction conditions for a solution system and such a method is expected to have a wide applicability in designing metal solution reactions.
4 Conclusions
Reducing the consumption of acid and redox additives in hydrometallurgical recycling of valuable metals from spent LIBs is the desired objective. In this study, we proposed the application of an electrochemical method by measuring the electric potential to determine the proper intermediate salt for synergistically treating spent LFPs and NCMs. Based on the test results, Fe3+ was found to be a suitable intermediate, and the theoretical predictions were verified by the results of leaching experiments. The water-soluble NH4Fe(SO4)2 was proven to be suitable as a leaching agent. By adjusting the leaching parameters, Li and transition metals from NCM cathodes can be effectively extracted under mild conditions, while Fe and PO43− from LFPs remain in solid residues. The kinetic study indicates that the reaction rate is mainly controlled by a surface chemical reaction between Fe2+ and the NCM cathode, which is explained by the structure destruction of the NCM cathode. A systematic thermodynamic study on the solution was conducted to design the recovery strategy of metals, and the method was verified to be applicable and is expected to have a wide range of applications. The intrinsic redox reaction between LFP and NCM avoids the need for additional oxidants or reductants, while the salt leaching by Fe3+ reduces the consumption of strong acids. The synergistic salt leaching method is expected to achieve recycling efficiencies of valuable metals from mixed spent LIB cathodes, with high efficiency, high selectivity and great environmental benefits.Author contributions
Yunhui Hua: Concept proposed, methodology, formal analysis, data analysis, writing – original draft preparation, formal analysis, and investigation. Zhenghe Xu: Review and editing and methodology. Baojun Zhao: Methodology, review and editing, and supervision. Zuotai Zhang: Concept proposed, methodology, review and editing, supervision, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Shenzhen Peacock Plan (KQTD20160226195840229) and the support from the Shenzhen Science and Technology Innovation Committee (JCYJ20200109141437586, JCYJ20200109141642225, and JCYJ20200109141227141). We are also grateful to the Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme 2018.Notes and references
- Global EV Outlook 2021 – Analysis – IEA, https://www.iea.org/reports/global-ev-outlook-2021, (accessed 13 December 2021).
- EV-Volumes – The Electric Vehicle World Sales Database, https://www.ev-volumes.com/country/total-world-plug-in-vehicle-volumes/, (accessed 13 December 2021).
- Y. Hua, X. Liu, S. Zhou, Y. Huang, H. Ling and S. Yang, Resour., Conserv. Recycl., 2021, 168, 105249 CrossRef.
- M. J. Esfandyari, V. Esfahanian, M. R. H. Yazdi, H. Nehzati and O. Shekoofa, Energy, 2019, 176, 505–520 CrossRef.
- P. Meshram, A. Mishra, Abhilash and R. Sahu, Chemosphere, 2020, 242, 125291 CrossRef CAS PubMed.
- C. M. Costa, J. C. Barbosa, R. Gonçalves, H. Castro, F. J. D. Campo and S. Lanceros-Méndez, Energy Storage Mater., 2021, 37, 433–465 CrossRef.
- M. Shahjalal, P. K. Roy, T. Shams, A. Fly, J. I. Chowdhury, M. R. Ahmed and K. Liu, Energy, 2022, 241, 122881 CrossRef CAS.
- M. Sethurajan and S. Gaydardzhiev, Resour., Conserv. Recycl., 2021, 165, 105225 CrossRef.
- K. M. Winslow, S. J. Laux and T. G. Townsend, Resour., Conserv. Recycl., 2018, 129, 263–277 CrossRef.
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich, S. Lambert, A. Abbott, K. Ryder, L. Gaines and P. Anderson, Nature, 2019, 575, 75–86 CrossRef CAS PubMed.
- X. Zhang, L. Li, E. Fan, Q. Xue, Y. Bian, F. Wu and R. Chen, Chem. Soc. Rev., 2018, 47, 7239–7302 RSC.
- B. Makuza, Q. Tian, X. Guo, K. Chattopadhyay and D. Yu, J. Power Sources, 2021, 491, 229622 CrossRef CAS.
- E. Fan, L. Li, Z. Wang, J. Lin, Y. Huang, Y. Yao, R. Chen and F. Wu, Chem. Rev., 2020, 120, 7020–7063 CrossRef CAS PubMed.
- W. Mrozik, M. A. Rajaeifar, O. Heidrich and P. Christensen, Energy Environ. Sci., 2021, 14, 6099–6121 RSC.
- Y. Yao, M. Zhu, Z. Zhao, B. Tong, Y. Fan and Z. Hua, ACS Sustainable Chem. Eng., 2018, 6, 13611–13627 CrossRef CAS.
- R. Wang, Y. Lin and S. Wu, Hydrometallurgy, 2009, 99, 194–201 CrossRef CAS.
- Z. Takacova, T. Havlik, F. Kukurugya and D. Orac, Hydrometallurgy, 2016, 163, 9–17 CrossRef CAS.
- L. Sun and K. Qiu, J. Hazard. Mater., 2011, 194, 378–384 CrossRef CAS PubMed.
- J. Kang, G. Senanayake, J. Sohn and S. M. Shin, Hydrometallurgy, 2010, 100, 168–171 CrossRef CAS.
- W. Gao, J. Song, H. Cao, X. Lin, X. Zhang, X. Zheng, Y. Zhang and Z. Sun, J. Cleaner Prod., 2018, 178, 833–845 CrossRef CAS.
- L. Li, J. Ge, F. Wu, R. Chen, S. Chen and B. Wu, J. Hazard. Mater., 2010, 176, 288–293 CrossRef CAS PubMed.
- R. Golmohammadzadeh, F. Rashchi and E. Vahidi, Waste Manage., 2017, 64, 244–254 CrossRef CAS PubMed.
- X. Chen, C. Luo, J. Zhang, J. Kong and T. Zhou, ACS Sustainable Chem. Eng., 2015, 3, 3104–3113 CrossRef CAS.
- X. Zhang, Y. Bian, S. Xu, E. Fan, Q. Xue, Y. Guan, F. Wu, L. Li and R. Chen, ACS Sustainable Chem. Eng., 2018, 6, 5959–5968 CrossRef CAS.
- X. Zeng, J. Li and B. Shen, J. Hazard. Mater., 2015, 295, 112–118 CrossRef CAS PubMed.
- Z. Zheng, M. Chen, Q. Wang, Y. Zhang, X. Ma, C. Shen, D. Xu, J. Liu, Y. Liu, P. Gionet, I. O'Connor, L. Pinnell, J. Wang, E. Gratz, R. Arsenault and Y. Wang, ACS Sustainable Chem. Eng., 2018, 6, 13977–13982 CrossRef CAS.
- P. Meshram, B. D. Pandey and T. R. Mankhand, Chem. Eng. J., 2015, 281, 418–427 CrossRef CAS.
- Y. Hua, Y. Sun, F. Yan, S. Wang, Z. Xu, B. Zhao and Z. Zhang, Chem. Eng. J., 2022, 436, 133200 CrossRef CAS.
- H. Li, S. Xing, Y. Liu, F. Li, H. Guo and G. Kuang, ACS Sustainable Chem. Eng., 2017, 5, 8017–8024 CrossRef CAS.
- J. Zhang, J. Hu, Y. Liu, Q. Jing, C. Yang, Y. Chen and C. Wang, ACS Sustainable Chem. Eng., 2019, 7, 5626–5631 CrossRef CAS.
- T. Tsuneda and T. Taketsugu, Phys. Chem. Chem. Phys., 2018, 20, 24992–24999 RSC.
- C. M. Lousada, M. Yang, K. Nilsson and M. Jonsson, J. Mol. Catal. A: Chem., 2013, 379, 178–184 CrossRef CAS.
- J. Xiao, J. Li and Z. Xu, Environ. Sci. Technol., 2020, 54, 9–25 CrossRef CAS PubMed.
- J. Li, G. Wang and Z. Xu, J. Hazard. Mater., 2016, 302, 97–104 CrossRef CAS PubMed.
- W. Wang, Y. Zhang, X. Liu and S. Xu, ACS Sustainable Chem. Eng., 2019, 7, 12222–12230 CAS.
- F. Liu, C. Peng, A. Porvali, Z. Wang, B. P. Wilson and M. Lundström, ACS Sustainable Chem. Eng., 2019, 7, 16103–16111 CrossRef CAS.
- C. Xu, Q. Dai, L. Gaines, M. Hu, A. Tukker and B. Steubing, Commun. Mater., 2020, 1, 99 CrossRef.
- Y. Jiang, X. Chen, S. Yan, S. Li and T. Zhou, Chem. Eng. J., 2021, 426, 131637 CrossRef CAS.
- Y. Liu, W. Lv, X. Zheng, D. Ruan, Y. Yang, H. Cao and Z. Sun, ACS Sustainable Chem. Eng., 2021, 9, 3183–3194 CrossRef CAS.
- E. A. Dalini, G. Karimi, S. Zandevakili and M. Goodarzi, Miner. Process. Extr. Metall. Rev., 2020, 42, 451–472 CrossRef.
- Q. Jing, J. Zhang, Y. Liu, C. Yang, B. Ma, Y. Chen and C. Wang, J. Phys. Chem. C, 2019, 123, 14207–14215 CrossRef CAS.
- J. Kumar, R. R. Neiber, J. Park, R. A. Soomro, G. W. Greene, S. A. Mazari, H. Y. Seo, J. H. Lee, M. Shon, D. W. Chang and K. Y. Cho, Chem. Eng. J., 2022, 431, 133993 CrossRef CAS.
- Z. Chen, J. Wang, D. Chao, T. Baikie, L. Bai, S. Chen, Y. Zhao, T. C. Sum, J. Lin and Z. Shen, Sci. Rep., 2016, 6, 25771 CrossRef CAS PubMed.
- T. Nshizirungu, M. Rana, Y. T. Jo and J. H. Park, J. Hazard. Mater., 2021, 414, 125575 CrossRef CAS PubMed.
- Y. Yang, G. Huang, M. Xie, S. Xu and Y. He, Hydrometallurgy, 2016, 165, 358–369 CrossRef CAS.
- X. Jiang, Y. Wei, X. Yu, P. Dong, Y. Zhang, Y. Zhang and J. Liu, J. Mater. Sci.: Mater. Electron., 2018, 29, 15869–15877 CrossRef CAS.
- Y. Zhang, W. Wang, Q. Fang and S. Xu, Waste Manage., 2020, 102, 847–855 CrossRef CAS PubMed.
- J. Li, S. Xiong, Y. Liu, Z. Ju and Y. Qian, Nano Energy, 2013, 2, 1249–1260 CrossRef CAS.
- Y. Shi, G. Chen, F. Liu, X. Yue and Z. Chen, ACS Energy Lett., 2018, 3, 1683–1692 CrossRef CAS.
- L. Yang, Y. Feng, C. Wang, D. Fang, G. Yi, Z. Gao, P. Shao, C. Liu, X. Luo and S. Luo, Chem. Eng. J., 2022, 431, 133232 CrossRef CAS.
- H. Zou, E. Gratz, D. Apelian and Y. Wang, Green Chem., 2013, 15, 1183–1191 RSC.
- W. Lv, Z. Wang, H. Cao, X. Zheng, W. Jin, Y. Zhang and Z. Sun, Waste Manage., 2018, 79, 545–553 CrossRef CAS PubMed.
- W. Gao, X. Zhang, X. Zheng, X. Lin, H. Cao, Y. Zhang and Z. Sun, Environ. Sci. Technol., 2017, 51, 1662–1669 CrossRef CAS PubMed.
- Y. Yang, X. Meng, H. Cao, X. Lin, C. Liu, Y. Sun, Y. Zhang and Z. Sun, Green Chem., 2018, 20, 3121–3133 RSC.
- X. Xiao, B. W. Hoogendoorn, Y. Ma, S. A. Sahadevan, J. M. Gardner, K. Forsberg and R. T. Olsson, Green Chem., 2021, 23, 8519–8532 RSC.
- J. Kumar, X. Shen, B. Li, H. Liu and J. Zhao, Waste Manage., 2020, 113, 32–40 CrossRef CAS PubMed.
- L. Li, J. B. Dunn, X. X. Zhang, L. Gaines, R. J. Chen, F. Wu and K. Amine, J. Power Sources, 2013, 233, 180–189 CrossRef CAS.
- L. Xing, J. Bao, S. Zhou, Y. Qiu, H. Sun, S. Gu and J. Yu, Chem. Eng. J., 2021, 420, 129593 CrossRef CAS.
- C. R. Birkl, M. R. Roberts, E. McTurk, P. G. Bruce and D. A. Howey, J. Power Sources, 2017, 341, 373–386 CrossRef CAS.
- X. Han, L. Lu, Y. Zheng, X. Feng, Z. Li, J. Li and M. Ouyang, eTransportation, 2019, 1, 100005 CrossRef.
- Y. Qi, F. Meng, X. Yi, J. Shu, M. Chen, Z. Sun, S. Sun and F. Xiu, J. Cleaner Prod., 2020, 251, 119665 CrossRef CAS.
- C. Wang, S. Wang, F. Yan, Z. Zhang, X. Shen and Z. Zhang, Waste Manage., 2020, 114, 253–262 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S12. See DOI: https://doi.org/10.1039/d2gc00331g |
| ‡ First author of the article. |
| This journal is © The Royal Society of Chemistry 2022 |