
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-free C–N bond formation under biocompatible reaction conditions†
Jeffrey
Goh
 a,
Seng Kheong
Ong
a,
Yan Sheng
Tan
a and
Teck-Peng
Loh
*ab
a,
Seng Kheong
Ong
a,
Yan Sheng
Tan
a and
Teck-Peng
Loh
*ab
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 637371 Singapore. E-mail: teckpeng@ntu.edu.sg
bHenan University of Technology, 100 Lianhua St, Zhongyuan District, Zhengzhou, Henan 450001, China
First published on 25th March 2022
Abstract
A C–N bond formation reaction under biocompatible conditions for the amination of allenic ketone compounds to access a diversity of β-keto enamines is developed. This reaction is atom economical, green, and highly regioselective and works well with many structurally important amines such as amino sugar and amino acid esters or peptides. A wide array of β-keto enamines was obtained in modest to excellent yields with wide functional group tolerance using this protocol. A gram-scale synthesis of an anti-microbial agent was also realized using this strategy under green reaction conditions.
The development of new, clean and biocompatible reactions for green pharmaceutical and biomolecule conjugate synthesis is one of the most challenging endeavors in organic synthesis. Accordingly, there has been much effort directed towards the development of new organic transformations that are highly chemoselective, and work in aqueous media and under mild reaction conditions. Among them, the carbon–nitrogen bond formation reaction has attracted the most attention since the amino group features widely in many pharmaceuticals and bioconjugates such as drugs,1,2 glycoconjugates, antibody conjugates, etc. (Fig. 1). Unfortunately, most of the reported methods for C–N bond forming reactions require the use of expensive metal catalysts, work under harsh reaction conditions, suffer from poor atom economy and are not environmentally friendly. As such, there is an urgent need to develop a metal-free and highly chemoselective C–N bond formation reaction that can work under biocompatible reaction conditions. If successful, this newly developed method will provide efficient and green access to many pharmaceuticals. In addition, bioconjugation with proteins can be potentially achieved where the tertiary structure of proteins can be preserved during the process of the reaction.
 | ||
| Fig. 1 Structurally crucial nitrogen compounds. | ||
Over the years, our group and others have contributed significantly to the green chemistry field through the development of catalyst-free water-based reactions. These include C–C bond forming reactions via the Mukaiyama-aldol strategy,3 and C–P and C–S bond reactions4,5b with allylic alcohols, using activated dichloro-acetophenones6 and 2H-azirines7 as linkers for disulphide bioconjugation. Inspired by our previous work on C–S bond formation reactions5 with allenic amide as an efficient linker for cysteine bioconjugation, we envisage that amines may react with highly reactive allenic carbonyl compounds under biocompatible conditions to yield the corresponding enaminones which are also versatile building blocks for pharmaceutical synthesis (Fig. 2).
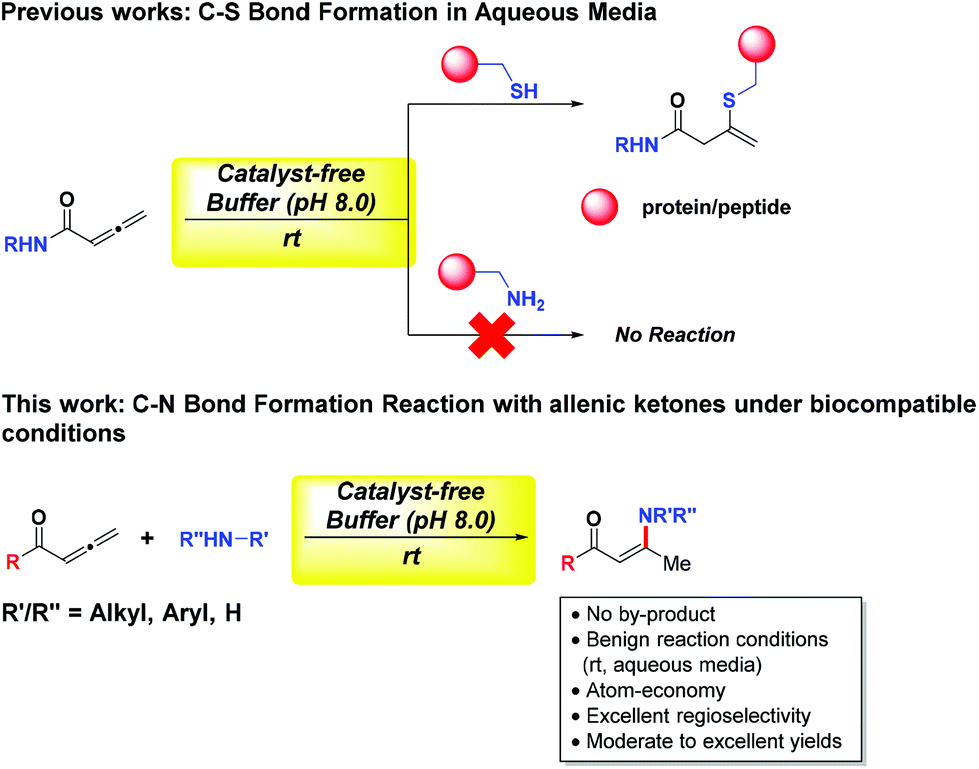 | ||
| Fig. 2 C–N bond formation reaction via allenic ketones and amines under biocompatible conditions. | ||
The amino functional group features in many bioconjugation reactions with biomolecules such as proteins. As such, there has been a considerable amount of attention paid to amino chemical conjugation strategies such as lysine conjugation. The most common lysine conjugation strategies are conjugation with the N-hydroxysuccinimide (NHS) ester, isocyanates, isothiocyanates, 4-azidobenzoyl fluoride (ABF) etc.8 While these strategies offer great selectivity and are carried out under biocompatible conditions, isolation of the desired product can prove to be cumbersome. Therefore, there is an urgent need to achieve a strategy that works under both green and biocompatible conditions while ensuring that the entire process is easy to operate, and is able to isolate the pure products without the need for liquid–liquid extraction or column chromatography.
1-Phenylbuta-2,3-dien-1-one (1a) and benzyl amine (2a) were chosen as model substrates for the aza-Michael reaction (Table 1). A variety of solvents were utilized to screen the reaction (refer to the ESI†), affording the desired product 3a in modest to excellent yields. Compound 3a was obtained in 35% yield when H2O was used as the solvent (Table 1, entry 6). The yields can be improved modestly (51–55%) when phosphate buffered saline (PBS) at pH 7.0 and 8.0 were employed as solvents for the reaction (Table 1, entries 7 and 8). Further investigations on the optimization of the reaction in PBS buffer were carried out extensively and the optimal conditions were determined (Table 1, entry 12). Remarkably, this reaction shows an atom economy of 100% and an E-factor of 58 based on green chemistry metrics. The results show that this strategy could potentially be employed for bioconjugation reactions with proteins or peptides. As with all other reported “in water” or “on water” reactions,9 the continuous and intense stirring proved to be vital for the organic reagents to interact efficiently during the process of the reaction.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Time (h) | Conversionb (%) | Yieldb (%) |
| a Experimental conditions: 1a (0.20 mmol) and 2a (0.30 mmol) in the specified solvent (2 mL) at room temperature. b Conversions and yields were determined by 1H NMR using CH2I2 as the internal standard. c 1.0 equiv. of 2a was used. d 2.0 equiv. of 2a was used. e Isolated yield. | ||||
| 1 | PhCF3 | 12 | 100 | 82 |
| 2 | Et2O | 12 | 100 | 70 |
| 3 | EtOAc | 12 | 100 | 80 |
| 4 | CH3CN | 12 | 100 | 75 |
| 5 | EtOH | 12 | 100 | 67 |
| 6 | H2O | 12 | 100 | 35 |
| 7 | PBS buffer (pH 7.0) | 12 | 100 | 51 |
| 8 | PBS buffer (pH 8.0) | 12 | 100 | 55 |
| 9c | PBS buffer (pH 8.0) | 12 | 100 | 58 |
| 10d | PBS buffer (pH 8.0) | 12 | 100 | 59 |
| 11d | PBS buffer (pH 8.0) | 1 | 100 | 79 |
| 12 | PBS buffer (pH 8.0) | 0.67 | 100 | 81 (70) |

A competitive study between different allenic carbonyl compounds (i.e. allenic ketones/estesr/amides) was first performed (Scheme 1). The reactions were performed in phosphate buffer (pH 8.0). The yields were determined by NMR analyses using CH2I2 as the internal standard. The results showed that the allenic ketone is the most reactive species under the reaction conditions; the substrate was fully converted in the reaction and the corresponding enaminone was obtained in 81% yield. In contrast, the allenic ester and amide variants failed to react under these conditions; this observation was expected due to the lower reactivity of these allenic carbonyl variants as compared to allenic ketones.
 | ||
Scheme 1 Competitive study between various allenes. Experimental conditions: 1 (0.20 mmol) and 2a (0.40 mmol) in PBS buffer (pH 8.0) (2 mL) at room temperature for 40 min. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Conversions and yields were determined by 1H NMR using CH2I2 as the internal standard. Conversions and yields were determined by 1H NMR using CH2I2 as the internal standard. | ||
The substrate scope of the amines was subsequently explored in the optimized phosphate buffer (pH 8.0) (Scheme 2). A variety of aryl and aliphatic amines were investigated. Aryl amines bearing electron-donating groups (3b–3i) reacted smoothly to afford the corresponding enaminones in good to excellent yields. Even electron-withdrawing substituents present on the aryl rings (3c and 3j) worked well under these conditions. In addition, aliphatic amines (3k–3m) also reacted with ease to give the enaminones in modest yields. Interestingly, a series of chiral amines (3o–3v) such as protected glucosamine (3p) and amino acid/peptide esters (3r–3v) also reacted well in this transformation (Scheme 3).
 | ||
| Scheme 2 Substrate scope of the amines. Experimental conditions: 1a (0.20 mmol) and 2 (0.40 mmol) in PBS buffer (pH 8.0) (2 mL) at room temperature for 40 min. Isolated yields. aReaction stirred overnight. bReaction carried out in EtOAc. | ||
 | ||
| Scheme 3 Substrate scope of chiral amines. Experimental conditions: 1a (0.20 mmol) and 2 (0.40 mmol) in PBS buffer (pH 8.0) (2 mL) at room temperature overnight. Isolated yields. aConditions: 1a (0.44 mmol) and 2 (0.2 mmol) in EtOAc (2 mL). bReaction carried out in EtOAc. The side product formed in 22% yield. | ||
Subsequently, the substrate scope of the allenic ketones was explored (Scheme 4). The aryl rings with electron-donating groups (3w–3z) reacted smoothly under the optimized conditions to give the desired enaminones in modest yields. Even with an electron-withdrawing substituent on the aryl ring, enaminone 3aa was obtained in 51% yield. Enaminone 3ab was obtained in 67% yield when a heterocyclic allenic ketone was employed in the reaction. The presence of an alkyne in the substrates (3ac and 3ad) did not appear to hinder the outcome of the reaction; the desired enaminones were obtained in 90% and 45% yields, respectively. Aliphatic allenic ketones (3ae–3ah) also reacted smoothly to afford the corresponding enaminones in modest to excellent yields.
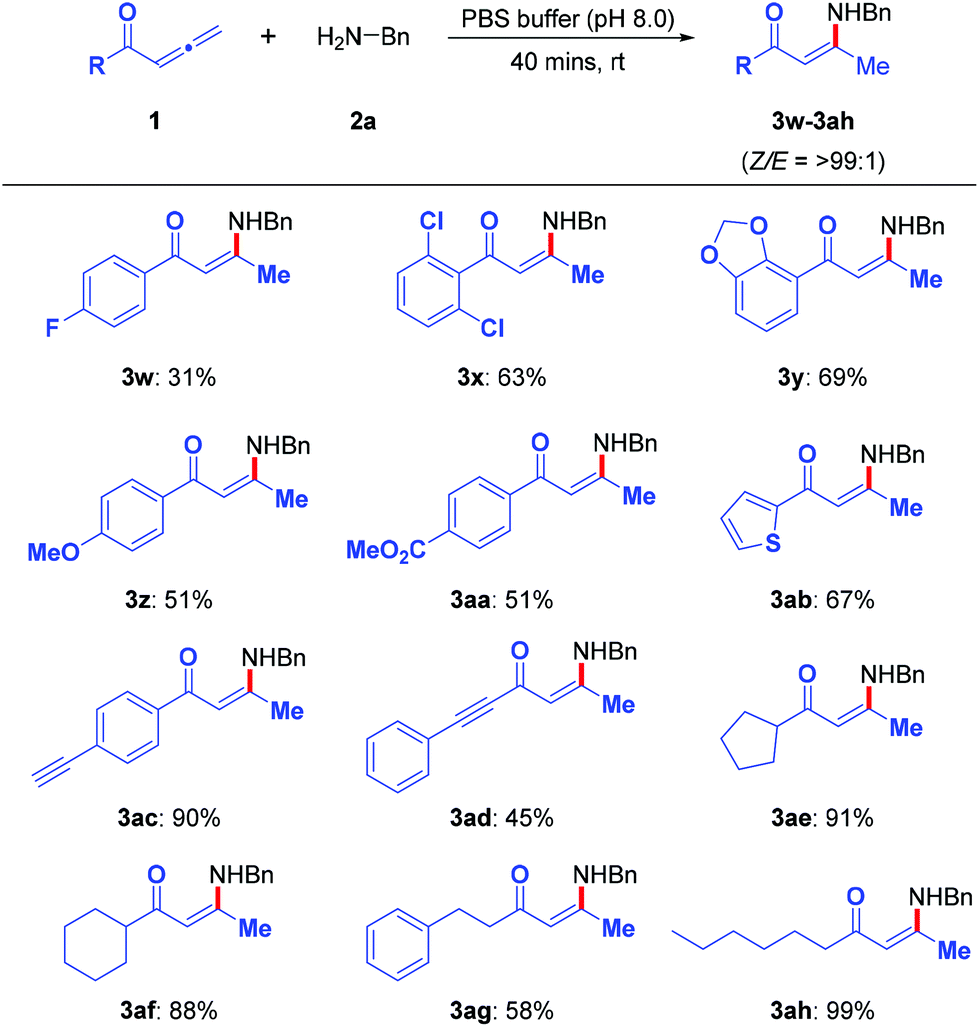 | ||
| Scheme 4 Substrate scope of the allenic ketones. Experimental conditions: 1 (0.20 mmol) and 2a (0.40 mmol) in PBS buffer (pH 8.0) (2 mL) at room temperature for 40 min. Isolated yields. | ||
The synthetic utility of the enaminones was subsequently explored (Fig. 3). Enaminone 3a was thiolated with Lawesson's reagent to afford enaminothione 4 in 59% yield.10 Treating enaminone 3a with the phenyl vinyl ketone gave the corresponding cyclohexa-1,3-diene 5 in 56% yield.11 A gram-scale synthesis of the anti-microbial drug 6 was realized under benign conditions, yielding the desired product in 90% yield by removing the solvent under reduced pressure. Enaminone 3ac can participate in a Cu(I)-catalyzed [3 + 2] cycloaddition12 with tosyl azide to give compound 7 in 88% yield. Enaminone 3ac can also undergo a click reaction5c with the anti-viral drug, Zidovudine,13 giving compound 8 in 27% yield.
 | ||
| Fig. 3 Synthetic utility and functionalization of enaminones. | ||
The proposed mechanism for the formation of the enaminone is illustrated in Fig. 4. The initial aza-Michael attack by the nucleophilic benzylamine regioselectively at the β-position of the allenic ketone 1a afforded an enol intermediate. This enol tautomer was proposed to be stabilized through a six-membered transition state via intramolecular hydrogen bonding, which readily tautomerized to yield (Z)-enaminone 3a exclusively as the desired product.
 | ||
| Fig. 4 Proposed mechanism for the enaminone formation. | ||
In summary, we have developed a practical and efficient strategy for the amination of allenic ketones in a green and biocompatible manner. The reaction is carried out in a phosphate buffer system (pH 7–8) under catalyst-free conditions and at room temperature. The special features of this method are: (1) no protection of sensitive functional groups is required; (2) applicable for glycoconjugation; (3) dual-linker functionality; (4) gram scale synthesis of an anti-microbial agent with no usage of metals and organic solvents and no column chromatography is required for its purification. With this novel strategy, we have once again taken a step towards increasing our toolbox of green synthetic and bioconjugation methods.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the Distinguished University Professor grant (Nanyang Technological University, Singapore), AcRF Tier 1 grants from the Ministry of Education of Singapore (RG107/19, RG11/20 and RT14/20), and the Agency for Science, Technology and Research (A*Star) under its MTC Individual Research Grant (IRG). We would also like to express our gratitude to Dr Li Yongxin for elucidating the X-ray crystal structure.Notes and references
- (a) W. Masocha, S. B. Kombian and I. O. Edafiogho, Evaluation of the antinociceptive activities of enaminone compounds on the formalin and hot plate tests in mice, Sci. Rep., 2016, 6, 21582 CrossRef CAS PubMed; (b) J. Apraku and C. O. Okoro, Design, synthesis and anticonvulsant evaluation of fluorinated benzyl amino enaminones, Bioorg. Med. Chem., 2019, 27, 161–166 CrossRef CAS PubMed.
- (a) M. Bhat, M. Al-Omar, A. Naglah and A. Khan, Enaminone-derived pyrazoles with antimicrobial activity, J. Chem., 2019, 2467970 CAS; (b) I. J. Amaye, R. D. Haywood, E. M. Mandzo, J. J. Wirick and P. L. Jackson-Ayotunde, Enaminones as building blocks in drug development: Recent advances in their chemistry, synthesis, and biological properties, Tetrahedron, 2021, 83, 131984 CrossRef CAS.
- (a) T.-P. Loh, L.-C. Feng and L.-L. Wei, Water-Accelerated Aldol Reaction of Ketene Silyl Acetals with Carbonyl Compounds, Tetrahedron, 2000, 56, 7309–7312 CrossRef CAS; (b) J. Alam, T. H. Keller and T.-P. Loh, Functionalization of Peptides and Proteins by Mukaiyama Aldol Reaction, J. Am. Chem. Soc., 2010, 132, 9546–9548 CrossRef CAS PubMed; (c) J. Xiao, Y.-L. Liu and T.-P. Loh, The Organocatalytic Enantioselective Michael Addition of Aldehydes to Vinyl Sulfones in Water, Synlett, 2010, 2029–2032 CAS.
- P. Xie, W. Fu, Y. Wu, X. Cai, Z. Sun, S. Li, C. Gao, X. Yang and T.-P. Loh, Allylic Phosphorus Ylides Directly Generated from Alcohols with Water as the Only Byproduct, Org. Lett., 2019, 21, 4168–4172 CrossRef CAS PubMed.
- (a) A. Abbas, B. Xing and T.-P. Loh, Allenamides as Orthogonal Handles for Selective Modification of Cysteine in Peptides and Proteins, Angew. Chem., Int. Ed., 2014, 53, 7491–7494 CrossRef CAS PubMed; (b) P. Xie, J. Wang, Y. Liu, J. Fan, X. Wo, W. Fu, Z. Sun and T.-P. Loh, Water-promoted C-S bond formation reactions, Nat. Commun., 2018, 9, 1321 CrossRef PubMed; (c) J. Goh, M. Maraswami and T.-P. Loh, Synthesis of Vinylic Sulfones in Aqueous Media, Org. Lett., 2021, 23, 1060–1065 CrossRef CAS PubMed.
- L.-H. Wu, S. Zhou, Q.-F. Luo, J.-S. Tian and T.-P. Loh, Dichloroacetophenone Derivatives: A Class of Bioconjugation Reagents for Disulfide Bridging, Org. Lett., 2020, 22, 8193–8197 CrossRef CAS PubMed.
- Y. Chen, W. Yang, J. Wu, W. Sun, T.-P. Loh and Y. Jiang, 2H-Azirines as Potential Bifunctional Chemical Linkers of Cysteine Residues in Bioconjugate Technology, Org. Lett., 2020, 22, 2038–2043 CrossRef CAS PubMed.
- M. S. Kang, T. W. S. Kong, J. Y. X. Khoo and T.-P. Loh, Recent developments in chemical conjugation strategies targeting native amino acids in proteins and their applications in antibody–drug conjugates, Chem. Sci., 2021, 12, 13613–13647 RSC.
- (a) A. Chanda and V. V. Fokin, Organic Synthesis “On Water”, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed; (b) R. N. Butler and A. G. Coyne, Water: Nature's Reaction Enforcer-Comparative Effects for Organic Synthesis “In-Water” and “On-Water”, Chem. Rev., 2010, 110, 6302–6337 CrossRef CAS PubMed; (c) S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, “On Water”: Unique Reactivity of Organic Compounds in Aqueous Suspension, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed; (d) Y. Yang, L. Tang, S. Zhang, X. Guo, Z. Zha and Z. Wang, Catalyst-free sulfonylation of activated alkenes for highly efficient synthesis of mono-substituted ethyl sulfones in water, Green Chem., 2014, 16, 4106–4109 RSC; (e) G. Lu, C. Cai, F. Chen, R. Ye and B. Zhou, Facile Sulfa-Michael Reactions with Sodium Arylsulfinates in Water: The Promotion of Water on the Reaction, ACS Sustainable Chem. Eng., 2016, 4, 1804–1809 CrossRef CAS.
- Z. Liu, P. Wu, Y. He, T. Yang and Z. Yu, [4 + 1] Cycloaddition of Enaminothiones and Aldehyde N-Tosylhydrazones Toward 3-Aminothiophenes, Adv. Synth. Catal., 2018, 360, 4381–4392 CrossRef CAS.
- T. Feng, M. Tian, X. Zhang and X. Fan, Tunable Synthesis of Functionalized Cyclohexa-1,3-dienes and 2-Aminobenzophenones/Benzoate from the Cascade Reactions of Allenic Ketones/Allenoate with Amines and Enones, J. Org. Chem., 2018, 83, 5313–5322 CrossRef CAS PubMed.
- T. Miura, T. Nakamuro, K. Hiraga and M. Murakami, The stereoselective synthesis of α-amino aldols starting from terminal alkynes, Chem. Commun., 2014, 50, 10474–10477 RSC.
- V. Gogineni, R. F. Schinazi and M. T. Hamann, Role of Marine Natural Products in the Genesis of Antiviral Agents, Chem. Rev., 2015, 115, 9655–9706 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2159225. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2gc00027j |
| This journal is © The Royal Society of Chemistry 2022 |