
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioconversion of wastewater-derived cresols to methyl muconic acids for use in performance-advantaged bioproducts†
William R.
Henson‡
 ,
Nicholas A.
Rorrer‡
,
Alex W.
Meyers‡
,
Caroline B.
Hoyt
,
Heather B.
Mayes
,
Jared J.
Anderson
,
Brenna A.
Black
,
Lahiru
Jayakody
,
Rui
Katahira
,
William E.
Michener
,
Todd A.
VanderWall
,
Davinia
Salvachúa
,
Christopher W.
Johnson
and
Gregg T.
Beckham
*
,
Nicholas A.
Rorrer‡
,
Alex W.
Meyers‡
,
Caroline B.
Hoyt
,
Heather B.
Mayes
,
Jared J.
Anderson
,
Brenna A.
Black
,
Lahiru
Jayakody
,
Rui
Katahira
,
William E.
Michener
,
Todd A.
VanderWall
,
Davinia
Salvachúa
,
Christopher W.
Johnson
and
Gregg T.
Beckham
*
Renewable Resources and Enabling Sciences Center, National Renewable Energy Laboratory, Golden, CO 8040, USA. E-mail: gregg.beckham@nrel.gov
First published on 31st March 2022
Abstract
Catalytic fast pyrolysis of biomass is a promising technology to generate biofuel blendstocks. This process generates a carbon-rich wastewater, which represents a loss of carbon that could be converted to co-products. Here, we explored the biological conversion of methyl phenols (cresols), a major component of biomass pyrolysis wastewater, into 2-methyl and 3-methyl muconic acids for use as polymer building blocks and plasticizers. We engineered Pseudomonas putida KT2440 to convert all three cresol isomers, o-, m-, and p-cresol, into their methyl muconic acid counterparts via the heterologous aromatic hydroxylase DmpKLMNOP from Pseudomonas putida CF600. We optimized conversion of cresols by expressing a heterologous (methyl)catechol dioxygenase ClcA from Rhodococcus opacus 1CP, followed by proof-of-concept fed-batch bioreactor cultivations. Methyl muconic acids and the hydrogenated methyl adipic acids were incorporated into nylons and plasticizers to evaluate potential performance advantages relative to existing materials. Methyl muconic acids in nylon-6,6 analogs substantially reduced melting and glass transition temperatures and enable post-polymerization modifications, and incorporating methyl adipic acid into nylon-6,6 analogs leads to a slightly reduced glass transition temperature and a 12% reduction in water permeability relative to nylon-6,6. When methyl diacids were incorporated into plasticizers for poly(vinyl chloride), they exhibit lower glass transition temperatures at the same mass loadings as phthalic acid and adipic acid-based plasticizers. The methyl diacids were also predicted to exhibit reduced health and environmental risks compared to phthalic acid. Overall, this study encompasses the selection of a target product from an exemplary waste stream to the demonstration of multiple industrially relevant performance advantages relative to petroleum-derived analogs and highlights the potential for biological waste stream valorization.
Introduction
Catalytic fast pyrolysis (CFP) converts lignocellulosic biomass into a complex oil that can be catalytically converted to fuel blendstocks.1 This process, and pyrolysis processes generally, produces a carbon-rich aqueous stream (hereafter CFP wastewater) composed of acids, ketones, aldehydes, and aromatic compounds, including o-, m-, and p-cresol. The complexity and toxicity of this stream challenges traditional wastewater treatment, and accordingly, pyrolysis wastewaters are commonly slated for heat recovery in process models.2,3 While this approach recovers energy from biomass, techno-economic analysis indicates that this approach can increase the cost of biofuel blendstocks from pyrolysis, and further, valuable carbon in this stream goes unutilized.2–4 Thus, alternative approaches have been investigated to valorize this stream,2,3 including catalytic conversion of wastewater to fuel blendstocks5 and recovery of valuable compounds from these streams.4 However, the heterogeneity of CFP wastewater components remains a challenge. To this end, biological funneling is a promising approach for valorizing heterogeneous mixtures.6,7 This process harnesses microbial metabolism to convert mixtures of compounds to a single product. Some microbes have an impressive capacity to execute convergent chemistries on multiple substrates. Furthermore, the number and type of molecules that can be produced by microbes continues to grow as the development of improved tools enables improved engineering in a variety of microbial hosts.7–9Cresols and methyl catechols can comprise a substantial fraction of CFP wastewater (up to ∼40% in some cases),10 and microbial catabolism of these compounds has been studied in a variety of organisms. Of these pathways, the Dmp pathway in Pseudomonas putida CF600 has been characterized for cresol catabolism.11 The phenol monooxygenase DmpKLMNOP hydroxylates m-, o-, and p-cresols to methyl catechols for conversion by a meta-cleavage pathway into methyl muconic acids (MMs), which are further consumed for cellular growth.12,13 Of potential hosts for engineering for cresol catabolism, P. putida KT2440 (hereafter KT2440) is a promising choice as it natively consumes some aromatic compounds, and it has been engineered to produce multiple products.14–17 Cresols and alkylated catechols cannot be natively catabolized by KT2440, but metabolic engineering of aromatic lignin model compound catabolism in KT2440 suggests that this host could also be appropriate for cresol conversion.14,16–19
Hydroxylation of cresols followed by oxidative aromatic ring-opening would produce 2-methyl muconic acid or 3-methyl muconic acid (2MM, 3MM, respectively, Fig. 1). Two recent reports have examined the production of 3MM from m- and o-cresols in KT2440 and Amycolatopsis sp. ATCC 39116.14,20 However, to our knowledge, no reports have studied the properties of homopolymers or polymer additives based on 2MM and 3MM. Additionally, MMs could also be converted into other products, such as methyl valerolactone, which have been incorporated into polymers and they impart promising attributes to chemically recyclable materials.21–24
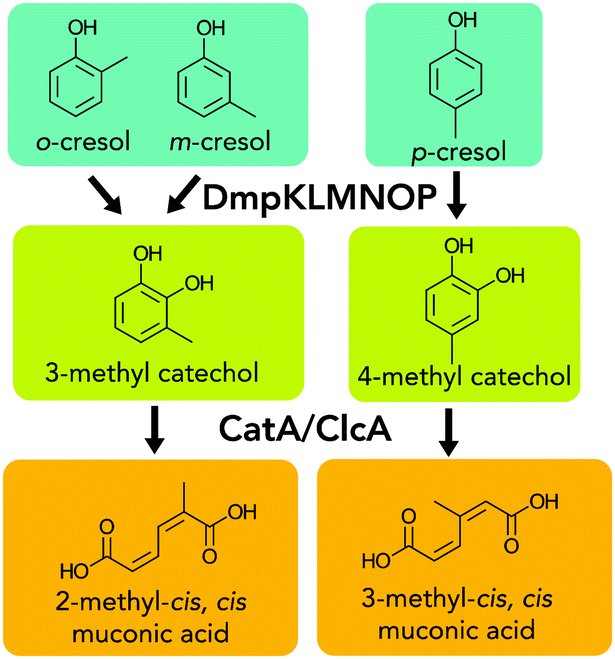 | ||
| Fig. 1 Catabolic pathway of o-, m-, and p-cresol to MMs. | ||
In this work, we engineer KT2440 to convert cresols to MMs and demonstrate production and purification of these compounds to high purities from bioreactor cultivations. 2MM, 3MM, and their hydrogenated methyl adipic acids (MA) were incorporated into nylons to evaluate the effect of the methyl side chain on polymer properties, resulting in significant reductions in melting and glass transition temperatures (Tm and Tg, respectively). Furthermore, we tune polymer properties using thiol–ene click chemistry to improve thermal degradation resistance and explore the use of methylated diacids in plasticizers for poly(vinyl chloride) (PVC). MM-derived plasticizers show performance advantages in Tg and reduced predicted human and environmental toxicity relative to two industry standard plasticizers, diethylhexyl phthalate (DEHP) and diethylhexyl adipate (DEHA).
Results
Expression of the DmpKLMNOP hydroxylase in KT2440 enables cresol conversion to MMs
Because KT2440 is unable to natively hydroxylate cresols, dmpKLMNOP, which encodes the promiscuous aromatic monooxygenase from P. putida CF600,11,12 was chromosomally integrated into KT2440 (Tables S1–S3†). The integration was performed so that the dmpKLMNOP operon was constitutively expressed by the tac promoter, and catBC was simultaneously deleted to enable muconic acid accumulation, as homologues of KT2440 CatB can lactonize MMs (Fig. 1).25 Furthermore, the native 1,2 catechol dioxygenase, catA, was constitutively expressed downstream of dmpKLMNOP operon, creating strain AM107. To determine if the engineered strain AM107 could hydroxylate m-cresol and o-cresol and catA could perform ring cleavage to produce 2MM, KT2440 and AM107 were grown for 36 hours in M9 minimal medium supplemented with 20 mM glucose as a carbon and energy source and either 4 mM m-cresol or 3.2 mM o-cresol for conversion to MM (Fig. 2A–D and Table S4†). AM107 completely consumed both m-cresol and o-cresol over 36 hours, and we observed 3-methyl catechol (3MC) as an intermediate along with the appearance of 2MM. As no commercial standards were available, a 2MM standard was synthesized and characterized by LC-MS3, 1H-NMR spectroscopy, and 13C-NMR spectroscopy (Fig. 2E–G, 3 and S1–S4†). 2MM was initially identified by retention time and fragmentation patterns using LC-MS3 (Fig. 2E–G). However, attempts to quantify 2MM were complicated by the differing response factors of the isomers of 2MM. Accordingly, 2MM was quantified using 1H NMR spectroscopy using trimethylsilylpropanoic acid as a proton standard. 1H NMR spectroscopy revealed that the 2MM standard was primarily the cis,cis form in DMSO-d6 and the cis,trans form in D2O (Fig. S3 and S4†).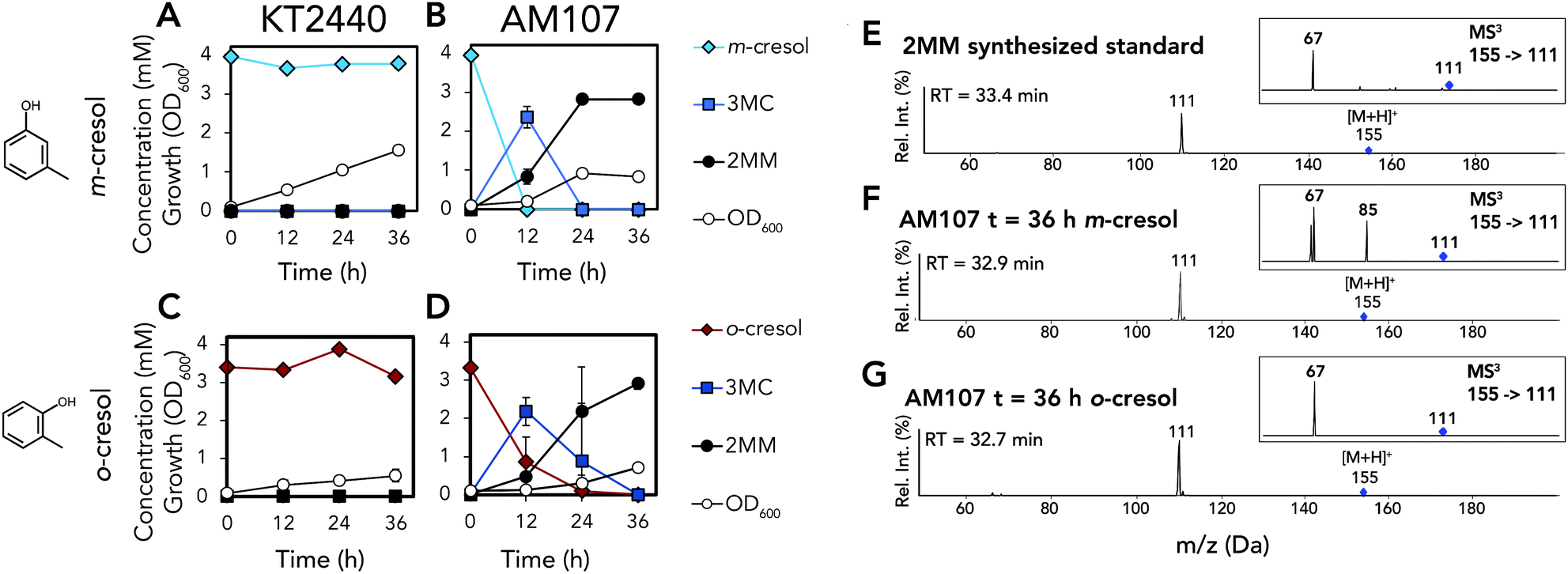 | ||
| Fig. 2 Biological conversion of o- and m-cresol to 2MM. (A) Growth and metabolite concentrations of P. putida KT2440 in the presence of 4 mM m-cresol. (B) Growth and metabolite concentrations of AM107 (P. putida KT2440 ΔcatBC::Ptac:dmpKLMNOP:catA) in the presence of 4 mM m-cresol. (C) Growth and metabolite concentrations of P. putida KT2440 in the presence of 3.2 mM o-cresol. (D) Growth and metabolite concentrations of AM107 in the presence of 3.2 mM o-cresol. (E) Positive ion mode MS2 and MS3 spectra of chemically synthesized 2MM standard. (F) Positive ion mode MS2 and MS3 spectra of putative 2MM (retention time = 32.9 min) from culture supernatant after 36 hours of AM107 grown in the presence of 4 mM m-cresol. (G) MS2 and MS3 spectra of putative 2MM (retention time = 32.7 min) from culture supernatant after 36 hours of AM107 grown in the presence of 3.2 mM o-cresol. For A–D, points represent the average of three replicates and error bars are ± SD. For A–G, cells were grown in 10 mL of M9 medium supplemented with 20 mM glucose and either 4 mM m-cresol or 3.2 mM o-cresol in 50 mL shake flasks. See Table S4† for raw data and ESI for more information. Abbreviations: 3MC, 3-methyl catechol; 2MM, methyl muconic acid; RT, retention time; Rel. Int., relative intensity. | ||
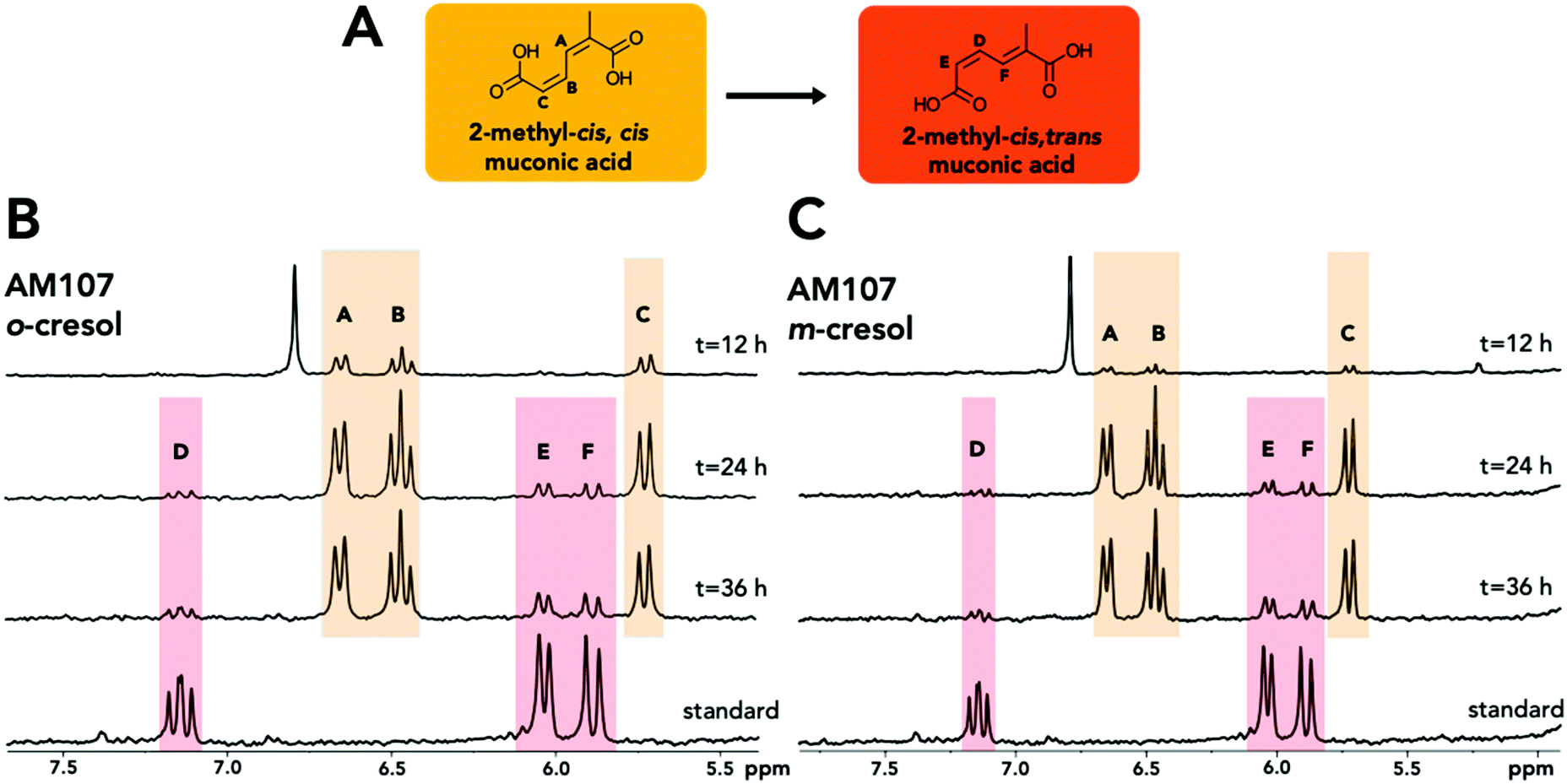 | ||
| Fig. 3 Structural analysis of 2MM isomers from biological cultivations. (A) Structure and conformational change of 2MM. (B) 1H-NMR spectra of the 2MM standard and the shake flask culture supernatant of AM107 (P. putida KT2440 ΔcatBC::Ptac:dmpKLMNOP:catA) after 12, 24, and 36 hours of growth in M9 minimal medium supplemented with 20 mM glucose and 3.2 mM o-cresol. (C) 1H-NMR spectra of the 2MM standard and the shake flask culture supernatant of AM107 after 12, 24, and 36 hours of growth in M9 minimal medium supplemented with 20 mM glucose and 4 mM m-cresol. Letters and colors indicate protons for each isomer. See ESI† for full spectra and additional information. | ||
When comparing the chemically synthesized 2MM to the biologically produced 2MM and quantifying 2MM production, we found that the biologically produced 2MM was a mixture of cis,cis and cis,trans isomers, and the cis,trans form appeared to increase over time (Fig. 3). After 36 hours, AM107 produced a total of 2.83 ± 0.06 mM 2MM (both cis,cis and cis,trans isomers) from m-cresol and 2.96 ± 0.18 mM 2MM (both cis,cis and cis,trans isomers) from o-cresol, with yields of 0.71 ± 0.02 (mol mol−1) and 0.93 ± 0.06 (mol mol−1), respectively (Fig. 2A–D and Table S4†). Complete consumption of o-cresol was observed after 36 hours, which was slightly slower than the m-cresol consumption rate (Fig. 2A–D, Table S4†). Neither 3MC nor 2MM were detected in KT2440 culture supernatants, indicating that wild-type KT2440 cannot natively hydroxylate m- and o-cresol, as expected. During the experiment, the culture medium for cultivations using both o- and m-cresol darkened, potentially due to oxidation of 3MC, which may have negatively impacted 2MM yields.
KT2440 and AM107 were also grown in M9 minimal medium supplemented with 20 mM glucose and 4 mM p-cresol to determine if DmpKLMNOP could hydroxylate this substrate and CatA could ring open 4-methyl catechol (4MC) to 3MM (Fig. 4A and B). Similar to 2MM, we synthesized a 3MM standard, which was characterized using LC-MS3, 1H NMR, and 13C-NMR (Fig. 4C–E and S5–S7†). Analysis using these methods indicated that the standard was primarily the trans,trans isomer in DMSO-d6 and D2O, and subsequent quantification was performed using HPLC (Fig. S5–S7†). After 12 hours, AM107 consumed 3.87 ± 0.44 mM of p-cresol and produced 1.45 ± 0.22 mM 4MC and 0.82 ± 0.07 mM 3MM. After 36 hours, neither p-cresol or 4MC was detected in culture supernatants, and 3MM titers reached 1.24 ± 0.08 mM (Fig. 4A, B and Table S4†). Interestingly, 3MM yields were 0.31 ± 0.02 (mol mol−1), which is significantly lower than 2MM yields from m- and o-cresol. Further analysis of culture supernatants by 1H NMR revealed the presence of 3-methyl muconolactone (3MML), which is generated via 3MM lactonization, and thus could contribute to reduced 3MM yields (Fig. 4F, S8 and Table S4†). We observed the production of 0.98 ± 0.18 mM of 3MML after 36 hours for a total yield of 3MM and 3MML of 0.54 ± 0.04 (mol mol−1) from p-cresol. Taking into account lactone production, this value is still lower than yields of 2MM from m- and o-cresol, and observed darkening of cultures also suggested that oxidation of 4MC may have occurred and negatively impacted 3MM yields. Lactonization of cis,cis-muconic acid has been previously observed in both biological and chemical systems at a pH below 7.0,26,27 and the yield of 0.24 ± 0.04 (mol mol−1) 3MML from p-cresol after 36 hours suggests that the additional methyl group on 3MM relative to muconic acid could promote lactonization.
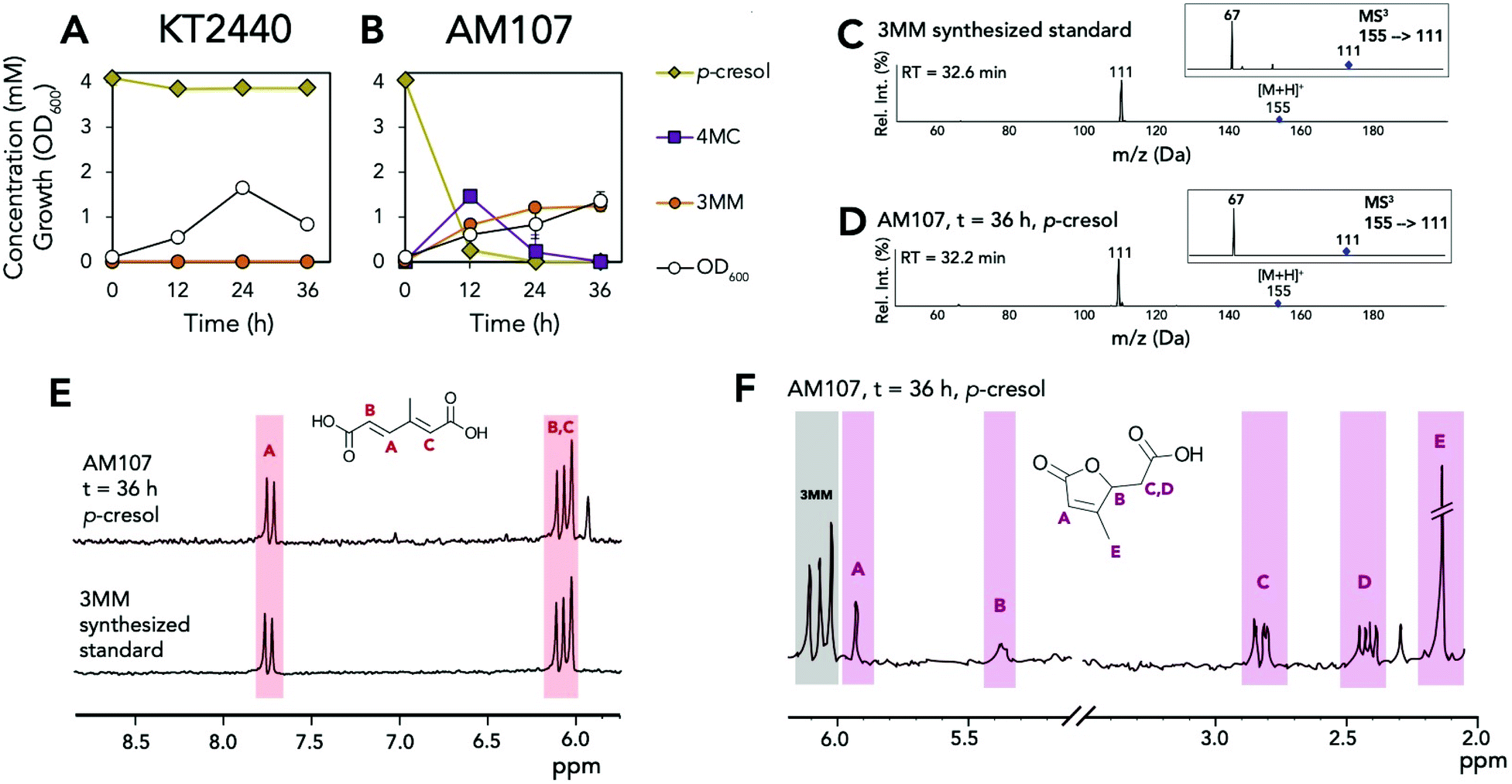 | ||
| Fig. 4 Biological conversion of p-cresol to 3MM and 3MML. (A) Growth and metabolite concentrations of P. putida KT2440 in the presence of 4 mM p-cresol. (B) Growth and metabolite concentrations of P. putida AM107 (P. putida KT2440 ΔcatBC::Ptac:dmpKLMNOP:catA) in the presence of 4 mM p-cresol. (C) Positive ion mode MS2 and MS3 spectra of the synthesized 3MM standard. (D) Positive ion mode MS2 and MS3 spectra of putative 3MM (RT = 32.2 min) from culture supernatants after 36 hours of growth in 20 mM glucose supplemented with 4 mM p-cresol. (E) 1H-NMR spectra of the 3MM synthesized standard and the shake flask culture supernatant of AM107 after 36 hours of growth in the presence of 4 mM p-cresol. (F) 1H-NMR spectrum of the shake flask culture supernatant of AM107 after 36 hours of growth in the presence of 4 mM p-cresol and putative identification of 3-methyl muconolactone. For A and B, cells were grown in 10 mL of M9 medium supplemented with 20 mM glucose and 4 mM p-cresol in 50 mL shake flasks. For A and B, points represent the average of three replicates and error bars are ±1 SD. For E and F, letters and colors indicate protons for each molecule. See Table S4† for raw data and ESI† for the full spectra and additional information. Abbreviations: 4MC, 4-methyl catechol; 3MM, 3-methyl muconic acid; RT, retention time; Rel. Int., relative intensity. | ||
Expression of ClcA dioxygenase improves MM production
Because we observed the darkening of the culture medium during shake flask cultivations (Fig. S9†), which suggested accumulation of methyl catechol (MC) intermediates, we explored the use of the chlorocatechol 1,2 dioxygenase, ClcA, from Rhodococcus opacus 1CP, which has been shown to exhibit higher activity on MCs relative to catechol.28,29clcA was codon optimized for KT2440 and chromosomally integrated downstream of dmpKLMNOP at the catBCA locus in AM107 to create AM111. To prevent competition between native and heterologous catechol dioxygenases, catA2 (PP_3166) was also deleted from both AM107 and AM111 to create AM112 and AM114, respectively. AM112 and AM114 and their parent strain, AM107, were tested with all three cresol isomers to determine if clcA overexpression and catA2 deletion reduced the accumulation of MCs and improved MM production (Fig. 5A–J and Table S4†). We observed similar titers and yields between strains AM112 and AM107 (Fig. 5J), suggesting that catA2 is either not expressed during growth in the presence of methyl phenols, that expression of catA is increased when catA2 is deleted, or CatA2 does not act on methyl phenols. Replacement of catA with clcA in AM114 resulted in 14 ± 4%, 16 ± 5%, and 17 ± 1% higher titers compared to AM112 for m-, o-, and p-cresol, respectively (P < 0.04 for all cases, Fig. 5A–J and Table S4†). We also observed significantly higher yields for all three cresols for AM114 compared to AM112 (P < 0.02 for all cases, Fig. 5J). Lower concentrations of 3MC and 4MC intermediates were also observed for AM114 for all three cresols compared to AM107 after 12 hours, but they did not meet the threshold for statistical significance for o-cresol and p-cresol (Fig. 5A–I). However, shake flask culture supernatants and cell pellets did not have significant culture darkening for AM114 compared to AM112, suggesting that there was reduced oxidation of MCs (Fig. S9†). For p-cresol shake flask experiments, similar concentrations of 3MML were observed between AM107 and AM112, but 44 ± 6% higher titers of 3MML were observed between AM112 and AM114, and final overall yields of 3MM and 3MML from p-cresol reached 0.72 ± 0.07 (mol mol−1). The ratio of 3MM to 3MML for AM107, AM112, and AM114 were 1.30 ± 0.27, 1.36 ± 0.01, and 1.20 ± 0.19, respectively, and no statistically significant differences were observed between strains (P > 0.05 for all cases), indicating that the lactonization process is independent of catA, catA2, and clcA expression. Together, these results show that clcA expression improves flux from cresols to their MM counterparts and improves MM titers and yields.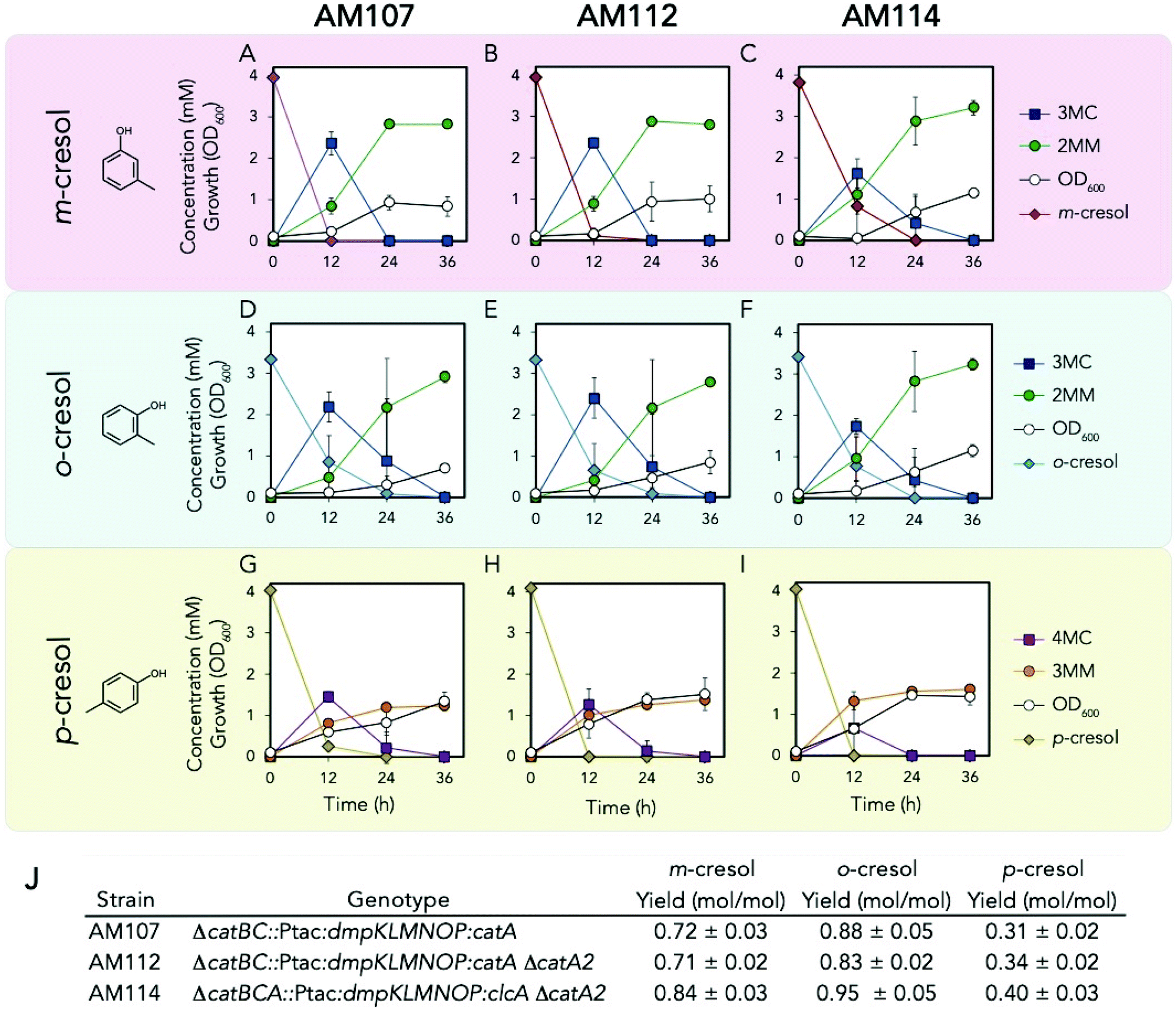 | ||
| Fig. 5 Biological conversion of p-cresol to 3MM. (A–C) Growth and metabolite concentrations of AM107, AM112, and AM114 in the presence of 4 mM m-cresol. (D–F) Growth and metabolite concentrations of AM107, AM112, and AM114 in the presence of 3.2 mM o-cresol. (G–I) Growth and metabolite concentrations of AM107, AM112, and AM114 in the presence of 4 mM p-cresol. (J) Genotypes and yields of engineered strains for each cresol isomer. For A–J, cells were grown in 10 mL of M9 medium supplemented with 20 mM glucose and either 4 mM m-cresol or 3.2 o-cresol in 50 mL shake flasks, and points represent the average of three replicates and error bars are ±1 SD. See Table S4† for raw data and ESI† for more information. Abbreviations: 3MC, 3-methyl catechol; 2MM, 2-methyl muconic acid; 4MC, 4-methyl catechol; 3MM, 3-methyl muconic acid. | ||
Bioreactor cultivations for MM production
To produce 2MM and 3MM for evaluation in polymer applications, bioreactor cultivations of AM114 were performed to convert m- and p-cresols to 2MM and 3MM, respectively (Fig. 6A, B and S10†). Because of the toxicity of cresols, we applied a dissolved oxygen (DO)-stat fed-batch approach to feed low concentrations (0.5 mM) of cresol in each feeding pulse. Furthermore, the feed solution contained glucose and cresol in a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, respectively, to provide carbon and energy for cell growth. We performed duplicate bioreactor runs for each cresol (Fig. 6A, B, S10 and Table S4†). For m-cresol cultivations, we achieved titers of 14.8 ± 2.8 mM (2.3 ± 0.4 g L−1) for 2MM and 13.4 ± 0.9 mM (2.1 ± 0.1 g L−1) for 3MM in 44–48 hours, representing yields of 35 ± 6% and 30 ± 2% for m-cresol and p-cresol, respectively. For both cresol substrates, yields were lower in bioreactor cultivations than in shake flask experiments (Fig. 6A, B and Table S4†). After 44–48 h, 2MM and 3MM production rates decreased significantly. However, the cultivations continued until oxygen consumption was negligible to maximize production for further downstream separations (Table S5†). We again observed darkening of the cultivations and the presence of the lactonized product 3MML using p-cresol as a substrate. Supernatants from one bioreactor run using p-cresol as a substrate were analyzed for 3MML production, and we observed titers of 12.4 mM 3MM (1.9 g L−1) and 6.78 mM 3MML (1.0 g L−1) and an overall yield of 3MM and 3MML from p-cresol of 0.44 (mol mol−1). Taken together, these results demonstrate that MMs can be produced by bioreactor cultivations, but additional optimization may be required to reach higher titers, rates, and yields of MMs.
:1 molar ratio, respectively, to provide carbon and energy for cell growth. We performed duplicate bioreactor runs for each cresol (Fig. 6A, B, S10 and Table S4†). For m-cresol cultivations, we achieved titers of 14.8 ± 2.8 mM (2.3 ± 0.4 g L−1) for 2MM and 13.4 ± 0.9 mM (2.1 ± 0.1 g L−1) for 3MM in 44–48 hours, representing yields of 35 ± 6% and 30 ± 2% for m-cresol and p-cresol, respectively. For both cresol substrates, yields were lower in bioreactor cultivations than in shake flask experiments (Fig. 6A, B and Table S4†). After 44–48 h, 2MM and 3MM production rates decreased significantly. However, the cultivations continued until oxygen consumption was negligible to maximize production for further downstream separations (Table S5†). We again observed darkening of the cultivations and the presence of the lactonized product 3MML using p-cresol as a substrate. Supernatants from one bioreactor run using p-cresol as a substrate were analyzed for 3MML production, and we observed titers of 12.4 mM 3MM (1.9 g L−1) and 6.78 mM 3MML (1.0 g L−1) and an overall yield of 3MM and 3MML from p-cresol of 0.44 (mol mol−1). Taken together, these results demonstrate that MMs can be produced by bioreactor cultivations, but additional optimization may be required to reach higher titers, rates, and yields of MMs.
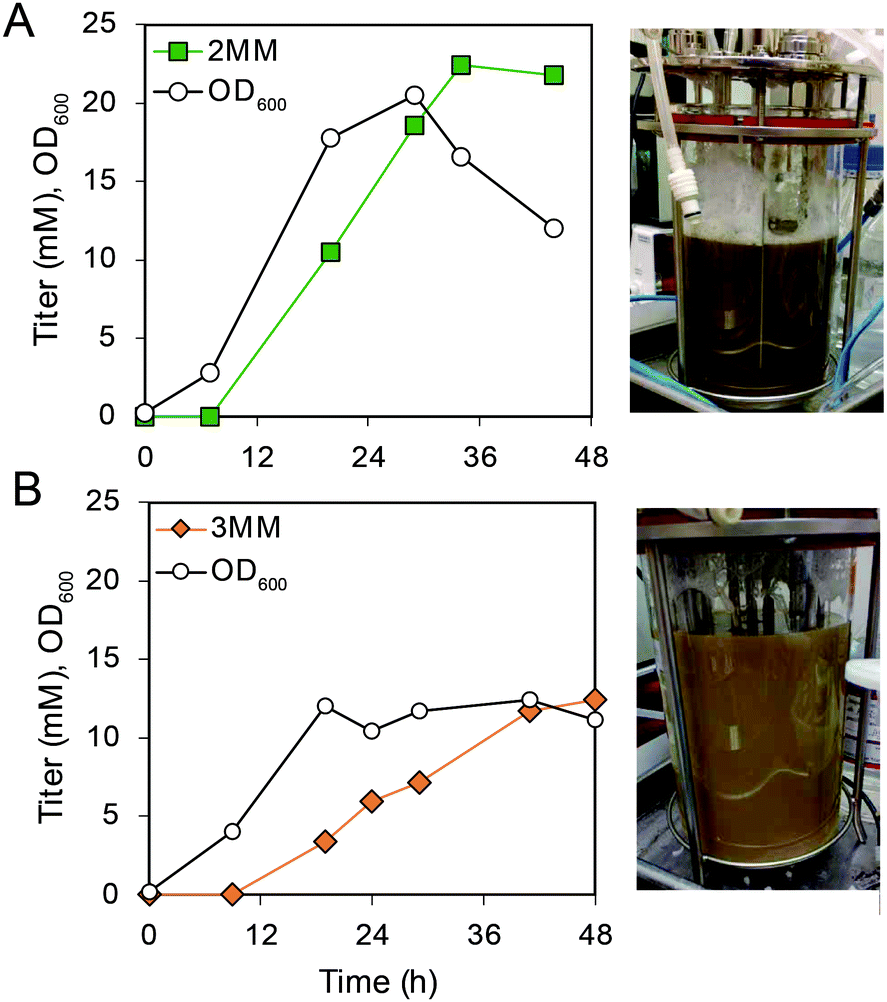 | ||
| Fig. 6 Bioreactor cultivations using AM114 for conversion of m- and p-cresol to 2MM and 3MM. (A) Growth and metabolite concentrations of AM114 during m-cresol feeding following a DO-stat fed-batch strategy with image of the bioreactor at the end of the cultivation. (B) Growth and metabolite concentrations of AM114 during p-cresol feeding following a DO-stat fed-batch strategy with image of the bioreactor at the end of the cultivation. Plots show a single representative bioreactor run. See Table S4† for raw data and ESI (Fig. S10†) for more information. | ||
After bioreactor cultivations to produce MMs, we isolated these compounds from the cultivation broth using previously described procedures for muconic acid, resulting in an off-white powder30 (bioreactor and isolation yields are provided in Table S5†). After separation, the MM isomers were determined to be isomerically pure via1H-NMR spectroscopy (i.e., either the trans,trans isomer of 2MM or 3MM) (Fig. S11 and S12†). Additionally, the MMs were converted to methyl adipic acids (MAs) via hydrogenation of the MMs at room temperature with Pd/C and H2 gas.30,31 Following separation, these bio-derived diacids were evaluated as precursors for performance-advantaged nylons and plasticizers. Melting points and purities of the alkylated muconates and adipates are provided in Table S6.†
MM and MA nylons
MMs and MAs were evaluated in nylon applications. Due to the structural similarity of 2MA and 3MA to adipic acid, the MMs were converted to MAs and all diacids were polymerized with hexamethylene diamine (HMDA) to produce nylon-6,6 analogs. Polymerization was conducted via salt polymerization, in which HMDA and the diacids were separately dissolved into water, combined, and then precipitated.32,33 The salt was subsequently dried and melted at 300 °C. Nylon-6,6, the base case polymer in this work, exhibits a Tg of 62 ± 3 °C and a Tm of 262 ± 5 °C (Fig. 7). Because CFP wastewater contains mixtures of different cresol isomers, we also tested 1:1 molar ratios of 2MM and 3MM and 2MA and 3MA as precursors. Relative to nylon-6,6, the polymers synthesized from the trans,trans isomers of 2MM and 3MM exhibited extreme plasticization, as observed by a 107 ± 11 °C Tg (averaged across the different isomers) reduction and reduction of the Tm to 142 ± 5 °C (the 3MM polymer is presented in Fig. 7 while the other polymers are presented in Table S7†). Importantly, there are no discernable differences between the bio-derived methyl muconates, used for most of this work, and the methyl muconates standard prepared from the chemo-catalytic conversion of methyl catechols based on thermal properties (Table S7†). The lack of difference is attributed to the use of the salt polymerization procedure, which further purifies the monomers prior to polymerization and ensures proper stoichiometry.
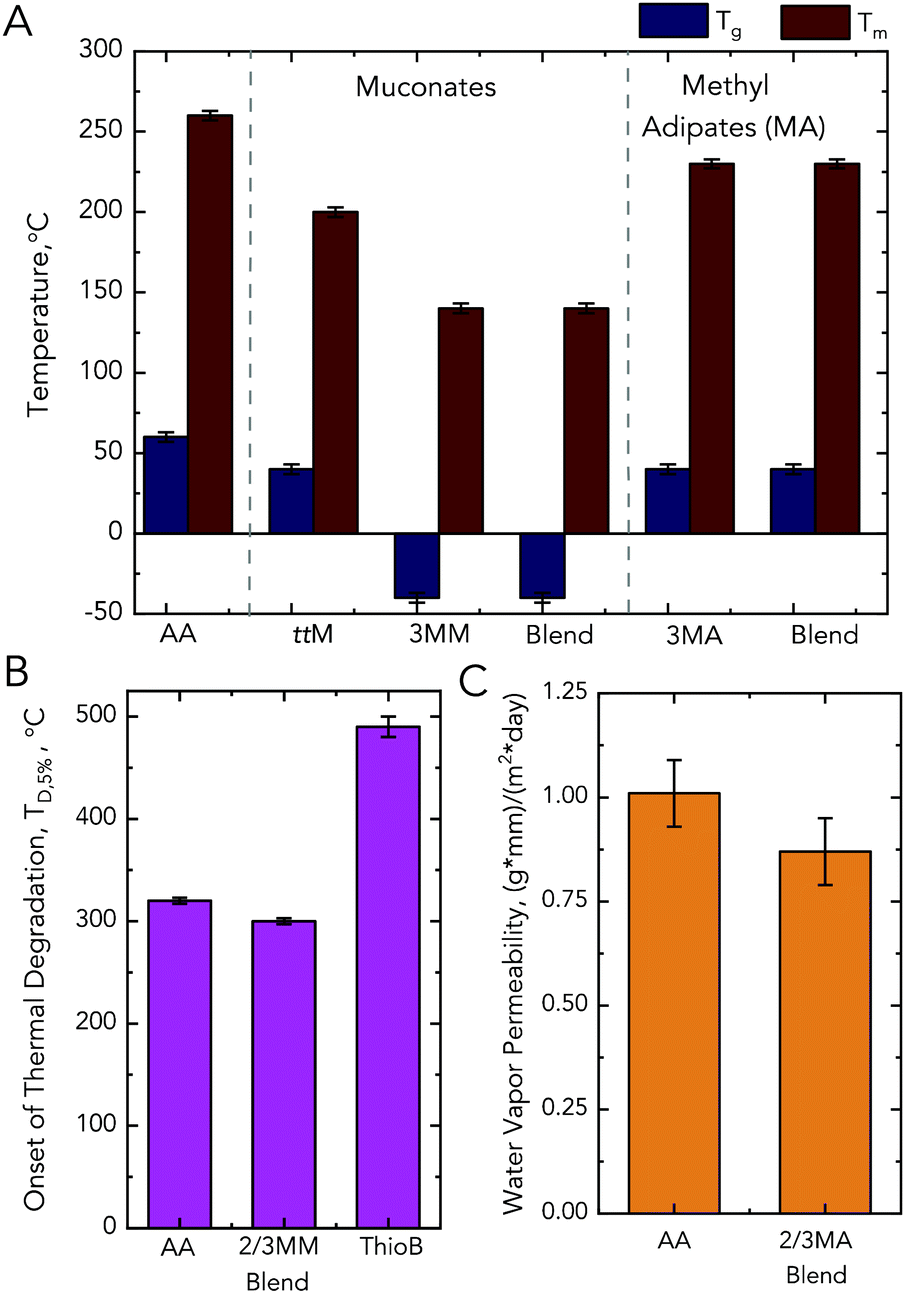 | ||
| Fig. 7 Performance advantaged bioproducts from MMs. (A) Thermal properties of the nylons synthesized from different diacids with HMDA, where the starting diacid is listed on the x-axis. (B) Onset of thermal degradation (TD,5%) for the synthesized nylons. (C) Water vapor transmission rate for the MA polymers relative to nylon-6,6. Associated error with thermal analysis is ±3 °C for Tg, ±5 °C for Tm, and ±10 °C for Td,5%. Associated equipment error with water vapor transmission rate is 10%. Blend indicates a 1:1 molar ratio of 2-methyl and 3-methyl isomers. See Tables S7 and S8† for raw data and ESI† for more information. Abbreviations: thiobenzene, ThioB; adipic acid, AA; trans,trans muconic acid, ttMA; 3-methyl muconic acid, 3MM; 3-methyl adipic acid, 3MA. | ||
Conversely, when trans,trans muconic acid or the 2- and 3MA isomers are used, only a 20 ± 3 °C, 19 ± 3 °C, and 22 ± 3 °C reduction in Tg was observed, respectively. Blends of MM and MA isomers performed similarly to 3MM and 3MA. As we observed significant reductions in the Tm for MM and MA-based polymers, co-polymers with adipic acid and the alkylated diacids were also synthesized. These co-polymers were synthesized by mixing the alkylated diacid–HMDA salts with the adipic acid–HMDA salt at a 1:1:1 molar ratio. In all cases, the degree of plasticization was reduced; in the case of the MMs, the reduction was 16 ± 3 °C compared to the 107 ± 11 °C of the homopolymer (Table S7†).
To explore polymers resulting from other chemistries, we exploited the double bonds in the backbone of the MM-containing polymers and investigated if the methyl group in the MA homopolymers led to other advantaged properties. For the MMs, we performed post-polymerization modification by thiol–ene click chemistry to increase the degradation temperature (TD,5%) of the polymers. To do so, the MM-containing nylons were stirred in N-methyl-2-pyrrolidone with thiobenzene overnight. This modification minimally changed the Tg and Tm but increased the polymer's TD,5%, which enables a wider processing window (Fig. 7B and Tables S7, S8†). For the MAs, we aimed to investigate if the methyl group provided a reduction in water permeability relative to an adipic acid precursor. Compared to nylon-6,6, the MA-blended polymer exhibited a 12% ± 5% lower water permeability. The large error is associated with the equipment employed; however, this result suggests that, following further investigation, MAs may be promising for applications where reduced water permeability is desired (Fig. 7C and Tables S7, S8†).
MM and MA plasticizers
Due to the plasticization effect of the MMs relative to adipic acid in nylons, we also explored the application of the alkylated diacids as PVC plasticizers in comparison to the commonly used and petroleum-based adipic acid and phthalic acid-based plasticizers. Plasticizers were synthesized via the reaction of the diacid (in the case of the MM the trans,trans isomers) variants with 2-ethylhexanol, and post purification, they were blended with PVC at a 10 wt% loading. Thermal analysis revealed greater reductions in the PVC Tg for all the alkylated diacids relative to plasticizers from phthalic and adipic acid.Unplasticized PVC exhibited a Tg of 92 ± 3 °C, and a 10 wt% loading of either DEHP or DEHA reduced Tg values to 32 ± 3 °C and 25 ± 3 °C, respectively (Fig. 8 and Table S9†). At the same loading, plasticizers derived from either MM isomer or a 1:1 molar blend of isomers resulted in a Tg of 11 °C ± 3 °C, and the same loading of the MA-based plasticizers reduced the Tg to 18 °C ± 3 °C. MM- and MA-based plasticizers demonstrated a 60 ± 4% and 32 ± 4% lower Tg compared to DEHA and 69 ± 4% and 47 ± 4% lower Tg compared to DEHP, respectively. Taken together, the superior performance of MM- and MA-based plasticizers to two industry relevant plasticizers illustrate the potential of MMs and MAs for plasticizer applications.
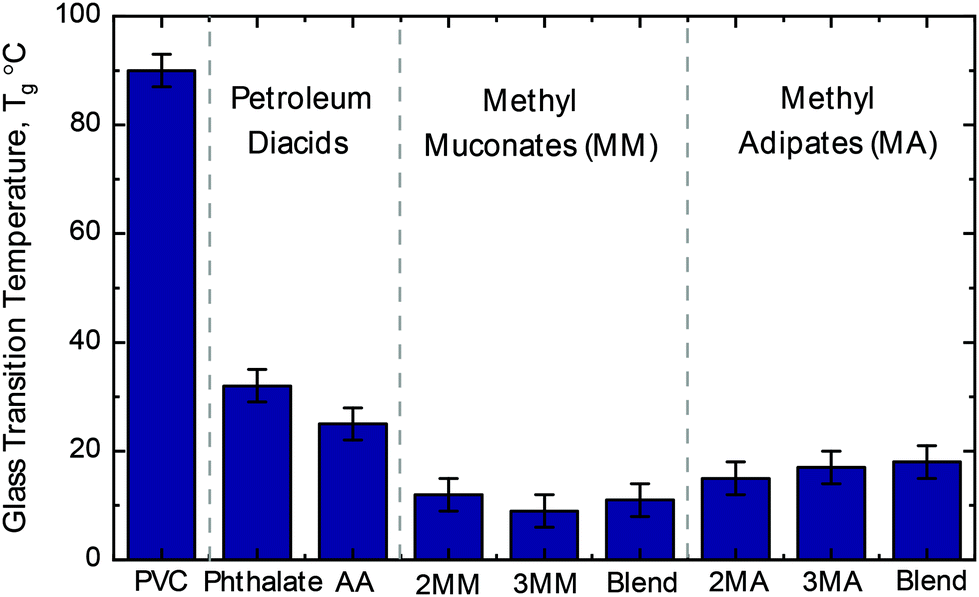 | ||
| Fig. 8 Glass transition temperature of PVC with different plasticizers. All glass transition temperatures are reported for the 2-ethylhexyl di-ester of the respective diacid at a 10 wt% loading relative to PVC. Associated DSC error of ±3 °C. Blend indicates a 1:1 molar ratio of 2-methyl and 3-methyl isomers. See Table S9† for raw data. Additionally, Table S9† reports a control reaction with non-bio-derived methyl muconates showing no difference between the bio-derived and standard starting material which is attributed to the plasticizer workup. Abbreviations: adipic acid, AA; 2-methyl muconic acid, 2MM; 3-methyl muconic acid, 3MM; 2-methyl adipic acid, 2MA; 3-methyl adipic acid, 3MA. | ||
Toxicity of precursors and plasticizers
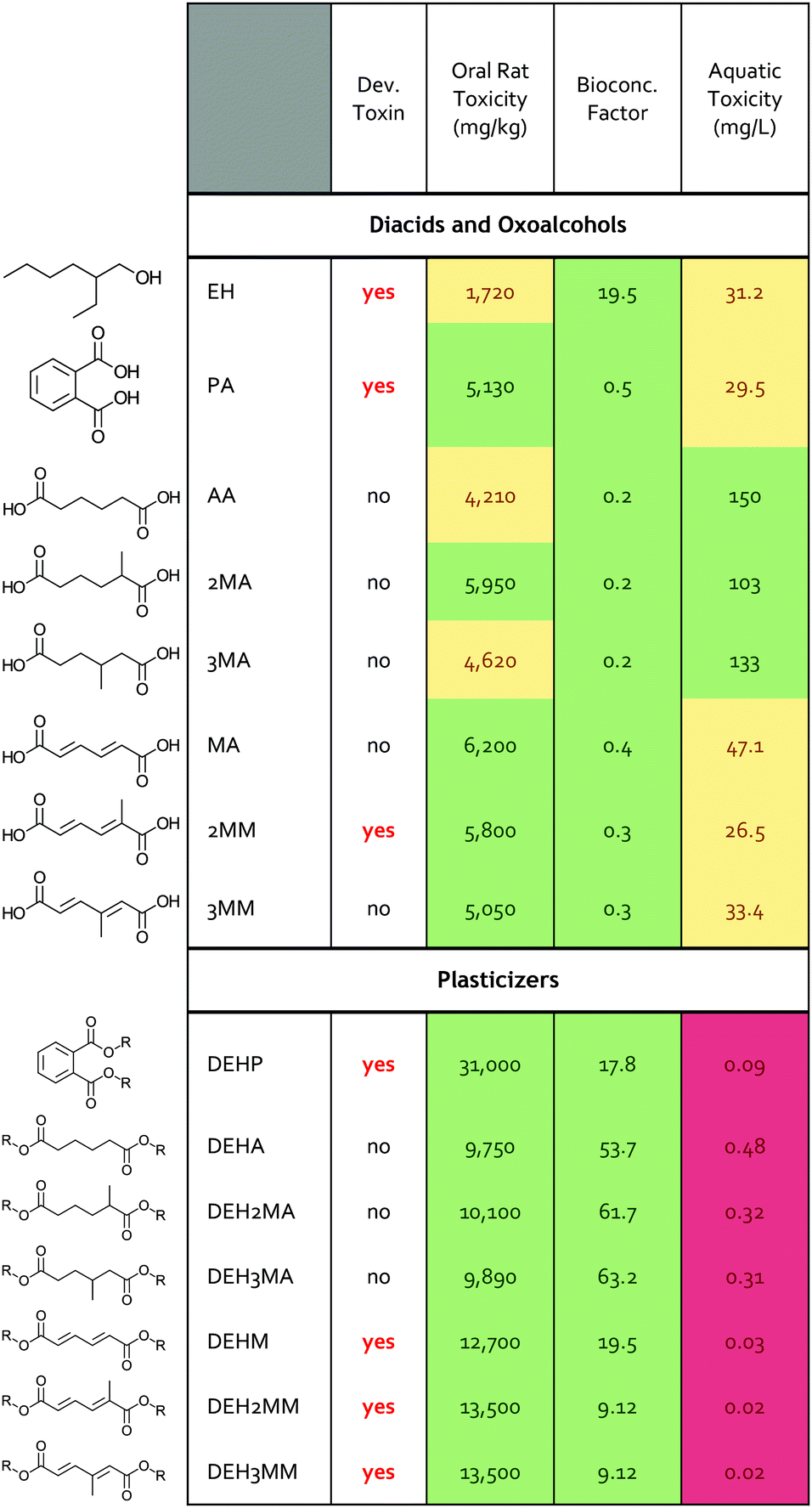
Bio-based products and their derivatives can also provide a performance advantage through reduced human and ecological toxicity, and the toxicity of precursors can play a role in manufacturing practices and guidelines. To explore this dimension for MMs and MAs, we implemented the Environmental Protection Agency (EPA) toxicity estimation software tool (TEST) to compare industrial nylon and plasticizer monomers and their derivatives across multiple human and environmental factors.34 The TEST software predicts human health impacts by determining a molecule's potential for estrogen receptor binding, developmental toxicity, Ames mutagenicity, and oral rat acute toxicity. For environmental impacts, TEST assesses the bioconcentration factor, defined as the ratio of the concentration in biota to the concentration in water, and the acute toxicity for three freshwater aquatic organisms: fathead minnow, Daphnia magna, and Tetrahymena pyriformis.34 Each prediction has an associated hazard classification, with ‘I’ being the most toxic to ‘IV’ being the least toxic (Table S10†).35–37 Using the TEST program, we compared the predicted toxicity of phthalic acid, adipic acid, muconic acid, both MM isomers, both MA isomers, ethylhexanol, and the plasticizers created by esterifying the diacids with ethylhexanol.The MAs and MMs and their ethylhexanol derivatives were predicted to have reduced toxicity profiles relative to the commercial, petroleum-based plasticizers (Table 1 and Table S11†). None of the MMs, MAs, or their ethylhexanol derivatives were predicted to have estrogen receptor binding or mutagenic activity in contrast to phthalic acid (Table S11†). The building blocks 2-ethylhexanol, phthalic acid, and 2MM as well as the plasticizers of DEHP and all diethylhexyl MMs were predicted to be developmental toxins. Interestingly, the MA-derived plasticizers were not predicted developmental toxins and had higher thresholds for predicted aquatic toxicity, indicating reduced toxicity to aquatic life and suggesting they may present a lower risk to humans and the environment. The MMs and MAs were predicted to have similar oral rat and bioconcentration factors to phthalic acid, but MAs also showed higher thresholds for predicted toxicity and were not predicted as aquatic hazards. To mechanistically understand the differences between molecules from the TEST tool, the mammalian metabolism of the molecules was examined with the EPA's Chemical Transformation Simulator.38 The results highlight that plasticizer toxicity is governed by the component oxoalcohols and diacids (Fig. S15†). The adipates, muconates, and 2-ethylhexanol are predicted to be metabolized, unlike phthalic acid (Fig. S16†), whereas MAs do not possess the functional groups (i.e. double bonds) necessary for these transformations. In summary, these computational tool predictions suggest that the MMs and MAs may have reduced toxicity in comparison to precursors for current industry standard plasticizers, and MA-based plasticizers may be promising alternatives to industry standard plasticizers based on multiple human and environmental factors.
|
Discussion and conclusions
In this work, we demonstrated the bioconversion of cresols to alkylated dicarboxylic acids and evaluated several bioproducts incorporating these molecules. Furthermore, we compared the predicted toxicities of these new molecules to petroleum-based counterparts using several computational tools. These results comprise the identification, production, and characterization of performance-advantaged bioproducts from a bio-derived waste stream.Cresols and MCs represent ∼40% of the total carbon in CFP wastewater in some processes.10 Many of the additional compounds are toxic and could interfere with the bioconversion of cresols and MCs to MMs and accordingly, separations is a key component of any waste stream valorization process. Separations could be implemented to purify the aromatic substrate before biological conversion, and separation of specific components has been previously explored.4,39 An alternative approach to additional separation is biological funneling and catabolism of additional wastewater components followed by upgrading of the aromatic compounds. Our previous efforts demonstrated metabolic engineering of KT2440 to consume the majority of CFP stream components without supplementation at an extent of conversion of 89%, and demonstrated that aromatic compounds, including cresols, can be converted to a mixture of MMs at a 90% yield.40 Here we demonstrate the feasibility of upgrading CFP wastewater streams to several products and applications of these molecules. As CFP wastewater streams are expected to contain a mixture of aromatic compounds, the final composition from CFP wastewater will likely be a mixture of methylated and non-methylated muconic acids. Our work with blends of MMs along with muconic acid suggests that this composition will be of use as a plasticizer or as a component of nylons. While we used purified MMs from bioreactor experiments using pure cresols as a substrate, industrial processes will use a much more complex wastewater solution as a substrate, and uncatabolized components of wastewater could remain after diacid purification that could affect polymer properties. Additional study of contaminants from CFP wastewaters and their effects on downstream applications will require further investigation.
Bioconversion of cresols to MMs could be improved by several means. The accumulation of MC intermediates in shake flask experiments and low titers for fed-batch cultivations suggests that other strategies such as optimizing feeding rates of glucose41 and adaptive laboratory evolution (ALE) using cresols as a sole carbon source or tolerance ALE could improve titers, rates, and yields of MMs.40,42,43 The darkening of cultures, which was reduced upon replacement of CatA with ClcA, suggests that increasing the activity of ClcA could reduce oxidative losses from MCs. Deleting regulators related to catabolite repression could also improve conversion.44 Other strategies to improve bioprocess performance include determining energy requirements for engineered strains45 and addressing lactonization of 3MM. Our results using different solvents for NMR indicate that the addition of a methyl group changes the isomerization dynamics of MMs relative to muconic acid.26,27
While we targeted nylons and plasticizers from MMs for performance advantages relative to fossil carbon-based incumbent products, other products could be derived from MMs. MMs and MAs could be substituted in any product manufactured using adipic acid to determine if they introduce performance advantages. Previous work demonstrated that muconic acid can be used as a replacement for maleic acid in unsaturated polyesters, and it is possible that the MM could also be used in similar applications.46,47 Additionally, the diene in the backbone of the MM can undergo Diels–Alder condensation to form a wider suite of monomers, such as substituted terephthalates, for further materials development.48
When incorporating both MM and MA into the backbones of nylons, we postulated that the C6 backbone would maintain existing polymer properties while the presence of the methyl group could lead to a reduction in water permeability. Interestingly, we observed no differences in the thermal properties between materials incorporating the 2- and 3-substituted monomers, but the thermal properties of the MAs and MMs materials were substantial. The similarity of the 2- and 3MMs and MAs could be attributed to the lack of stereochemical control in the polymerization procedure, as diacids can be added in a random spatial distribution of the methyl groups. In both the MA and MM, plasticization occurs relative to nylon-6,6; however the plasticization imparted by the MMs is greater than the MA and is attributed to a synergistic effect between the lack of mobility of the muconic acid, which has been observed previously in polyesters,49 and the free-volume of the methyl group.
Nylons are an ideal target for incorporating performance-advantaged monomers, as they have a large market size (7.7 MMT per year as of 201950) and are used in a wide variety of textile, electronics, and automotive applications. Critically for bio-based products, they also demand higher selling prices than commodity thermoplastics.50 Structurally, nylons are semicrystalline polyamides with high melting points (Tm > 200 °C), and moderate glass transition temperatures (Tg ∼ 0–100 °C). At the degree of plasticization observed in this work, MMs may be able to find use as a co-monomer for nylon production to reduce the Tg, Tm, and crystallinity to reduce processing burdens (Tables S5 and S6†). MA co-monomers also demonstrated reduced water permeability without detrimentally sacrificing other properties, which could be useful for incorporation into current manufacturing practices for a variety of applications.
In comparison to new polymers, polymer additives have potential for more rapid market adoption, and also exhibit large market sizes (7.5 MMT per year as of 2019).50 Here, we demonstrated that alkylated diacids could be used as performance-advantaged plasticizers. Plasticizers are used at variable weight percents (10 wt% to 40 wt% in PVC) to aid in processing or reduce Tg, enabling polymers to be employed in both rigid and flexible applications.51,52 Compared to a new polymer formulation, an additive in a large commodity polymer requires less material to achieve the same performance and validate at scale, which could enable a faster path to market. Additionally, additives for PVC cost more than PVC itself, thus the use of less additive to achieve the same effect can be desirable depending on final cost.53 This is beneficial in several ways as using less additive can lower the cost of the entire resin and lower additive loadings may reduce leakage of small molecules during the lifetime of a product.
Further performance advantages include offering a safer alternative to phthalates, as their risks to human health and the environment are greater than the hazards captured by the EPA hazard prediction tool.54–56 As studies continue to report serious health and environmental concerns and regulatory action around phthalates increases,57,58 demand for alternatives is also likely to increase.59–61 Our predictions suggest that the toxicity of MMs and MAs is likely to be lower than phthalates, which could make them a safer, renewable alternative.
Other waste streams have high concentrations of unused or under-utilized carbon that could bring value to their parent process and coupling characterization of these streams along with metabolic engineering and chemical catalysis strategies could prove valuable to other areas outside of biomass pyrolysis.7 While additional process optimization will be required, this study provides direction for development of pyrolysis wastewater valorization and applications for target molecules.
Authorship contributions
WRH: investigation, methodology, visualization, writing – original draft. NAR: conceptualization, supervision, investigation, methodology, visualization, writing – original draft. AWM, CBH, HBM: investigation, methodology, visualization. JJA, BAB, LJ, RK, WEM, TVW: investigation, methodology. DS: investigation, methodology, supervision. CWJ: conceptualization, methodology, project administration, visualization, supervision, writing – original draft. GTB: conceptualization, funding acquisition, methodology, project administration, visualization, supervision, writing – original draft.Conflicts of interest
NAR, CBH, and GTB have filed a patent application on polymers and additives from alkylated muconic acids.Acknowledgements
This work was authored by National Renewable Energy Laboratory, operated by the Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding was provided by the U.S. DOE Office of Energy Efficiency and Renewable Energy Bioenergy Technologies Office. The authors thank Victoria Shingler at the Department of Molecular Biology at Umeå University for the generous gift of P. putida CF600 and Marc Hillmyer for helpful conversations.References
- M. B. Griffin, K. Iisa, H. Wang, A. Dutta, K. A. Orton, R. J. French, D. M. Santosa, N. Wilson, E. Christensen, C. Nash, K. M. Van Allsburg, F. G. Baddour, D. A. Ruddy, E. C. D. Tan, H. Cai, C. Mukarakate and J. A. Schaidle, Energy Environ. Sci., 2018, 11, 2904–2918 RSC.
- A. Dutta, A. H. Sahir, E. Tan, D. Humbird, L. J. Snowden-Swan, P. A. Meyer, J. Ross, D. Sexton, R. Yap and J. Lukas, PNNL Technical Report PNNL-23823, 2015.
- A. Dutta, M. K. Iisa, M. Talmadge, C. Mukarakate, M. B. Griffin, E. C. Tan, N. Wilson, M. M. Yung, M. R. Nimlos, J. A. Schaidle, H. Wang, M. Thorson, D. Hartley, J. Klinger and H. Cai, NREL Technical Report TP-5100-76269, 2020.
- A. N. Wilson, A. Dutta, B. A. Black, C. Mukarakate, K. Magrini, J. A. Schaidle, W. E. Michener, G. T. Beckham and M. R. Nimlos, Green Chem., 2019, 21, 4217–4230 RSC.
- C. Mukarakate, R. J. Evans, S. Deutch, T. Evans, A. K. Starace, J. ten Dam, M. J. Watson and K. Magrini, Energy Fuels, 2017, 31, 1600–1607 CrossRef CAS.
- J. G. Linger, D. R. Vardon, M. T. Guarnieri, E. M. Karp, G. B. Hunsinger, M. A. Franden, C. W. Johnson, G. Chupka, T. J. Strathmann, P. T. Pienkos and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12013 CrossRef CAS PubMed.
- A. J. Borchert, W. R. Henson and G. T. Beckham, Curr. Opin. Biotechnol., 2022, 73, 1–13 CrossRef CAS PubMed.
- P. Calero and P. I. Nikel, Microbiol. Biotechnol., 2019, 12, 98–124 CrossRef CAS PubMed.
- S. Y. Lee, H. U. Kim, T. U. Chae, J. S. Cho, J. W. Kim, J. H. Shin, D. I. Kim, Y.-S. Ko, W. D. Jang and Y.-S. Jang, Nat. Catal., 2019, 2, 18–33 CrossRef CAS.
- B. A. Black, W. E. Michener, K. J. Ramirez, M. J. Biddy, B. C. Knott, M. W. Jarvis, J. Olstad, O. D. Mante, D. C. Dayton and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 6815–6827 CrossRef CAS.
- V. Shingler, J. Powlowski and U. Marklund, J. Bacteriol., 1992, 174, 711–724 CrossRef CAS PubMed.
- V. Shingler, F. C. Franklin, M. Tsuda, D. Holroyd and M. Bagdasarian, J. Gen. Microbiol., 1989, 135, 1083–1092 CAS.
- I. Nordlund, J. Powlowski and V. Shingler, J. Bacteriol., 1990, 172, 6826–6833 CrossRef CAS PubMed.
- M. Kohlstedt, S. Starck, N. Barton, J. Stolzenberger, M. Selzer, K. Mehlmann, R. Schneider, D. Pleissner, J. Rinkel and J. S. Dickschat, Metab. Eng., 2018, 47, 279–293 CrossRef CAS PubMed.
- E. Martínez-García and V. de Lorenzo, Curr. Opin. Biotechnol., 2019, 59, 111–121 CrossRef PubMed.
- P. I. Nikel and V. de Lorenzo, Metab. Eng., 2018, 50, 142–155 CrossRef CAS PubMed.
- A. Weimer, M. Kohlstedt, D. C. Volke, P. I. Nikel and C. Wittmann, Appl. Microbiol. Biotechnol., 2020, 104, 7745–7766 CrossRef CAS PubMed.
- C. W. Johnson, D. Salvachúa, N. A. Rorrer, B. A. Black, D. R. Vardon, P. C. St. John, N. S. Cleveland, G. Dominick, J. R. Elmore, N. Grundl, P. Khanna, C. R. Martinez, W. E. Michener, D. J. Peterson, K. J. Ramirez, P. Singh, T. A. VanderWall, A. N. Wilson, X. Yi, M. J. Biddy, Y. J. Bomble, A. M. Guss and G. T. Beckham, Joule, 2019, 3, 1523–1537 CrossRef CAS.
- P. I. Nikel, E. Martínez-García and V. de Lorenzo, Nat. Rev. Microbiol., 2014, 12, 368–379 CrossRef CAS PubMed.
- N. Barton, L. Horbal, S. Starck, M. Kohlstedt, A. Luzhetskyy and C. Wittmann, Metab. Eng., 2018, 45, 200–210 CrossRef CAS PubMed.
- D. K. Schneiderman, M. E. Vanderlaan, A. M. Mannion, T. R. Panthani, D. C. Batiste, J. Z. Wang, F. S. Bates, C. W. Macosko and M. A. Hillmyer, Macro Lett., 2016, 5, 515–518 CrossRef CAS.
- J. Zhang, T. Li, A. M. Mannion, D. K. Schneiderman, M. A. Hillmyer and F. S. Bates, Macro Lett., 2016, 5, 407–412 CrossRef CAS.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 CrossRef CAS.
- M. Xiong, D. K. Schneiderman, F. S. Bates, M. A. Hillmyer and K. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8357 CrossRef CAS PubMed.
- M. D. Vollmer, H. Hoier, H.-J. Hecht, U. Schell, J. Gröning, A. Goldman and M. Schlömann, Appl. Environ. Microbiol., 1998, 64, 3290 CrossRef CAS PubMed.
- A. E. Settle, L. Berstis, S. Zhang, N. A. Rorrer, H. Hu, R. M. Richards, G. T. Beckham, M. F. Crowley and D. R. Vardon, ChemSusChem, 2018, 11, 1768–1780 CrossRef CAS PubMed.
- J. M. Carraher, T. Pfennig, R. G. Rao, B. H. Shanks and J.-P. Tessonnier, Green Chem., 2017, 19, 3042–3050 RSC.
- I. Matera, M. Ferraroni, M. Kolomytseva, L. Golovleva, A. Scozzafava and F. Briganti, J. Struct. Biol., 2010, 170, 548–564 CrossRef CAS PubMed.
- M. Ferraroni, M. Kolomytseva, A. Scozzafava, L. Golovleva and F. Briganti, J. Struct. Biol., 2013, 181, 274–282 CrossRef CAS PubMed.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 RSC.
- D. R. Vardon, N. A. Rorrer, D. Salvachúa, A. E. Settle, C. W. Johnson, M. J. Menart, N. S. Cleveland, P. N. Ciesielski, K. X. Steirer, J. R. Dorgan and G. T. Beckham, Green Chem., 2016, 18, 3397–3413 RSC.
- N. Hernandez, M. Yan, E. W. Cochran, J. E. Matthiesen and J.-P. Tessonnier, US Pat., 10793673, 2020 Search PubMed.
- M. Suastegui, J. E. Matthiesen, J. M. Carraher, N. Hernandez, N. Rodriguez Quiroz, A. Okerlund, E. W. Cochran, Z. Shao and J.-P. Tessonnier, Angew. Chem., Int. Ed., 2016, 55, 2368–2373 CrossRef CAS PubMed.
- T. M. Martin, U.S. Environmental Protection Agency, 2020 Search PubMed.
- Protection of Environment, Code of Federal Regulations, Title 40, Chapter I, Subchapter E, Part 156, Subpart D, § 156.62.
- U. S. E.P.A., Persistent Bioaccumulative Toxic (PBT) Chemicals; Lowering of Reporting Thresholds for Certain PBT Chemicals; Addition of Certain PBT Chemicals; Community Right-to-Know Toxic Chemical Reporting, 1999, 40, CFR Part 372.
- United Nations, Globally Harmonized System of Classification and Labelling of Chemicals (GHS), 2015, ST/SG/AC.10/30/Rev.6.
- U. S. EPA, Chemical Transformation Simulator (CTS), Version 1.0, 2019.
- A. N. Wilson, M. J. Grieshop, J. Roback, S. Dell'Orco, J. Huang, J. A. Perkins, S. Nicholson, D. Chiaramonti, M. R. Nimlos, E. Christensen, K. Iisa, K. Harris, A. Dutta, J. R. Dorgan and J. A. Schaidle, Green Chem., 2021, 23, 10145–10156 RSC.
- W. R. Henson, A. W. Meyers, L. N. Jayakody, A. DeCapite, B. A. Black, W. E. Michener, C. W. Johnson and G. T. Beckham, Metab. Eng., 2021, 68, 14–25 CrossRef CAS PubMed.
- S. Notonier, A. Z. Werner, E. Kuatsjah, L. Dumalo, P. E. Abraham, E. A. Hatmaker, C. B. Hoyt, A. Amore, K. J. Ramirez, S. P. Woodworth, D. M. Klingeman, R. J. Giannone, A. M. Guss, R. L. Hettich, L. D. Eltis, C. W. Johnson and G. T. Beckham, Met. Eng., 2021, 65, 111–122 CrossRef CAS PubMed.
- E. T. Mohamed, A. Z. Werner, D. Salvachúa, C. A. Singer, K. Szostkiewicz, M. Rafael Jiménez-Díaz, T. Eng, M. S. Radi, B. A. Simmons, A. Mukhopadhyay, M. J. Herrgård, S. W. Singer, G. T. Beckham and A. M. Feist, Metab. Eng. Commun., 2020, 11, e00143 CrossRef PubMed.
- T. E. Sandberg, M. J. Salazar, L. L. Weng, B. O. Palsson and A. M. Feist, Met. Eng., 2019, 56, 1–16 CrossRef CAS PubMed.
- L. Wirebrand, A. W. K. Madhushani, Y. Irie and V. Shingler, Environ. Microbiol., 2018, 20, 186–199 CrossRef CAS PubMed.
- D. Salvachúa, C. W. Johnson, C. A. Singer, H. Rohrer, D. J. Peterson, B. A. Black, A. Knapp and G. T. Beckham, Green Chem., 2018, 20, 5007–5019 RSC.
- N. A. Rorrer, D. R. Vardon, J. R. Dorgan, E. J. Gjersing and G. T. Beckham, Green Chem., 2017, 19, 2812–2825 RSC.
- N. A. Rorrer, S. Nicholson, A. Carpenter, M. J. Biddy, N. J. Grundl and G. T. Beckham, Joule, 2019, 3, 1006–1027 CrossRef CAS.
- A. E. Settle, L. Berstis, N. A. Rorrer, Y. Roman-Leshkóv, G. T. Beckham, R. M. Richards and D. R. Vardon, Green Chem., 2017, 19, 3468–3492 RSC.
- N. A. Rorrer, J. R. Dorgan, D. R. Vardon, C. R. Martinez, Y. Yang and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 6867–6876 CrossRef CAS.
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Joule, 2021, 5, 673–686 CrossRef CAS.
- IHS Markit, Chemical Economics Handbook: Plasticizers, https://ihsmarkit.com/products/plasticizers-chemical-economics-handbook.html, accessed June 2021.
- IHS Markit, Chemical Economics Handbook: Polyvinyl Chloride (PVC) Resins, https://ihsmarkit.com/products/polyvinyl-chloride-resins-chemical-economics-handbook.html, accessed June 2021.
- R. Babinsky, Plast. Addit. Compd., 2006, 8, 38–40 CrossRef.
- U.S. Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry (ATSDR), 2019, Toxicological profile for Di(2-ethylhexyl)phthalate (DEHP).
- U.S. Environmental Protection Agency, National Center for Environmental Assessment, Office of Research and Development, 1988, Integrated Risk Information System (IRIS) on Di(2-ethylhexyl)phthalate).
- C. S. Giam, H. S. Chan, G. S. Neff and E. L. Atlas, Science, 1978, 199, 419–421 CrossRef CAS PubMed.
- EU Registration, Authorisation, and Restriction of Chemicals (REACH), Directive 2005/84/EC, https://data.europa.eu/eli/reg/2006/166/oj, accessed October 21, 2021.
- Consumer Product Safety Commission, 2017, Prohibition of Children's Toys and Child Care Articles Containing Specified Phthalates, 16 CFR 1307, Docket No. (CPSC-2014-0033).
- D. Kim, R. Cui, J. Moon, J. I. Kwak and Y.-J. An, Chemosphere, 2019, 216, 387–395 CrossRef CAS PubMed.
- L. Hermabessiere, A. Dehaut, I. Paul-Pont, C. Lacroix, R. Jezequel, P. Soudant and G. Duflos, Chemosphere, 2017, 182, 781–793 CrossRef CAS PubMed.
- L. Trasande, B. Liu and W. Bao, Environ. Pollut., 2022, 292, 118021 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1gc04590c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |