
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDehydrogenative ester synthesis from enol ethers and water with a ruthenium complex catalyzing two reactions in synergy†
Sayan
Kar
 a,
Jie
Luo
a,
Michael
Rauch
a,
Yael
Diskin-Posner
b,
Yehoshoa
Ben-David
a and
David
Milstein
*a
a,
Jie
Luo
a,
Michael
Rauch
a,
Yael
Diskin-Posner
b,
Yehoshoa
Ben-David
a and
David
Milstein
*a
aDepartment of Molecular Chemistry and Materials Science, The Weizmann Institute of Science, Rehovot 76100, Israel. E-mail: david.milstein@weizmann.ac.il
bDepartment of Chemical Research Support, The Weizmann Institute of Science, Rehovot 76100, Israel
First published on 4th February 2022
Abstract
We report the dehydrogenative synthesis of esters from enol ethers using water as the formal oxidant, catalyzed by a newly developed ruthenium acridine-based PNP(Ph)-type complex. Mechanistic experiments and density functional theory (DFT) studies suggest that an inner-sphere stepwise coupled reaction pathway is operational instead of a more intuitive outer-sphere tandem hydration–dehydrogenation pathway.
Oxidation reactions play a central role in producing value-added fine and bulk chemicals in our society.1–3 The currently employed large-scale oxidations often involve excessive harsh oxidants such as peroxides, metal salts, or pure O2, which are energy-intensive to prepare, difficult to handle, and poorly tolerated by sensitive functional groups, and often generate large amounts of harmful byproducts.4 As a result, increasing attention is being paid to developing alternative oxidation procedures utilizing readily available and environment-friendly reagents such as water or air, while generating minimal side products.5–8 Among them, carrying out oxidation using water as the formal oxidant is highly desirable due to (i) its abundant availability, (ii) benign nature, and (iii) simultaneous formation of H2 gas as reaction by-product, which has been dubbed the “fuel of the future”.9,10 The overwhelming majority of water-based organics oxidation involves (photo)electrochemical methods, which can be challenging to understand, scale up and rationally improve.11–13 By contrast, examples of well-defined molecular complexes capable of thermally oxidizing various substrates utilizing water as a formal oxidant with H2 liberation are limited.
Using ruthenium and iridium-based pincer complexes, our group and others have reported catalytic primary alcohol oxidation to carboxylate salts under alkaline conditions using water while liberating H2 (Fig. 1a).14–17 Similarly, aqueous reforming of simple alcohols such as methanol,18–23 ethanol,24 glycol,25 glycerol26 is reported by Beller, us, and others. Goldberg and co-workers have reported molecular complexes that can catalyse aldehyde oxidation to carboxylic acids under neutral conditions using water as an oxidant (Fig. 1b).27,28 At the same time, using ruthenium acridine based PNP complexes, our group has demonstrated the oxidation of cyclic amines to lactams (Fig. 1c),29,30 alkenes to ketones (Fig. 1d),31 and amines to carboxylate salts (Fig. 1e).32 Notably, in these reports, the molecular complexes act as dehydrogenation catalysts while the water addition to substrate/intermediate happens spontaneously or is facilitated by additional Lewis acid or base catalysts. No further involvement of the molecular complex during the reaction is observed.
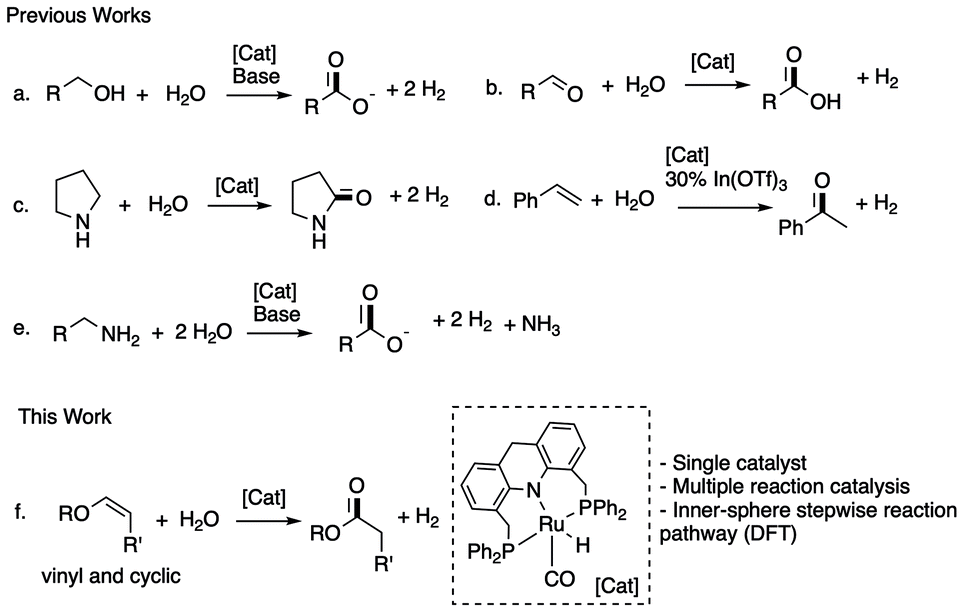 | ||
| Fig. 1 Previous water-based oxidation reactions catalyzed by molecular complexes (a–e) and this study (f). | ||
Here we report the dehydrogenative oxidation of vinyl and cyclic enol ethers to the corresponding esters using water as the oxidant. The reaction is selectively catalysed by a newly developed ruthenium acridine PNP(Ph) based complex, which catalyses both hydration and dehydrogenation steps, allowing the reaction to proceed under neutral additive-free conditions. Interestingly, DFT calculations revealed a possible stepwise inner-sphere coupled dehydrogenation–hydration mechanism instead of the intuitive outer-sphere tandem hydration–dehydrogenation route.33 Esters are widespread commodity chemicals with myriad applications, including odorants, lubricants, polymer precursors, and this method provides an alternative to access esters to the traditional Fischer–Speier esterification,34 and other advanced oxidative35–37 and dehydrogenative38–42 methods.
Our investigation started by exploring the reactivity of ethyl vinyl ether with water under neutral conditions in the presence and absence of complex 1, which was previously reported by us for other water-based oxidations (Table 1).29–31 When a mixture comprising 0.30 mmol of ethyl vinyl ether and 0.1 mL of water (5.6 mmol) in 2.0 mL of 1,4-dioxane was heated at 120 °C for 16 h in a 25 mL Schlenk flask, with no added catalyst, no significant reaction was observed by GC and NMR spectroscopy (entry 1). This is in line with previous reports, which showed that hydrolysis of enol ethers to aldehydes and alcohols proceeds only slowly under neutral or basic conditions.43–46 Interestingly, when a similar reaction was carried out in the presence of 0.75 mol% complex of 1, ethyl acetate was observed as a product in 17% yield (entry 2), along with diethyl ether as a minor by-product (4%). The presence of H2 gas in the reactor headspace was detected by GC analysis (Fig. S16†). The formation of ethyl acetate from ethyl vinyl ether presumably proceeds via a hemiacetal-like intermediate upon initial water addition, followed by its dehydrogenation (Fig. S14†). The above results indicate that complex 1 can catalyse both enol ether hydration and dehydrogenation under the reaction conditions, ultimately leading to ester formation. At the same time, the liberated H2 hydrogenates, in the presence of 1, some of the starting material to form the ether side product. Increasing the catalyst loading to 1.5 mol%, water amount to 0.2 mL (11.1 mmol), reaction temperature to 125 °C, and reaction time to 24 h increased the ester yield to 31%, while also increasing the amount of ether by-product (19%; entry 3). Using a larger amount of water (1 mL, 56 mmol) was detrimental to the process, leading to significant hydrolysis of both the enol ether and ester (entry 4). Replacing the substrate with a higher boiling one, namely, butyl vinyl ether, increased conversion to 82%. The desired ester was obtained in 57% yield, along with 24% of the hydrogenated by-product ethyl butyl ether (entry 5). Among different solvents, tetrahydrofuran (THF) was almost equally effective as co-solvent as 1,4-dixoane (entry 6). On the other hand, when toluene was used as co-solvent, the ester yield significantly decreased, likely due to the limited solubility of water in toluene forming a biphasic reaction system (entry 7). Similarly, when only water was used as solvent with no co-solvent, only traces of ester formation were observed due to the sparing solubility of complex 1 in water (entry 8).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | Cat. (mol%) | H2O (mL) | T (°C)/t (h) | Conv.a (%) |
P1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (%) a (%) |
P2a (%) |
| Reaction conditions: Vinyl ether (0.30 mmol), 1,4-dioxane (2.0 mL), water, [Ru], oil bath temperature, and reaction time as specified.a Yields were determined by GC analysis with mesitylene as an internal standard.b The additional products were ethanol (40%), acetaldehyde (20%), and acetic acid (20%).c THF used as co-solvent.d Toluene used as co-solvent.e No 1,4-dioxane was used. | |||||||
| 1 | Me | — | 0.1 | 120/16 | 1 | 0 | 0 |
| 2 | Me | 1 (0.75) | 0.1 | 120/16 | 21 | 17 | 4 |
| 3 | Me | 1 (1.5) | 0.2 | 125/24 | 53 | 31 | 19 |
| 4 | Me | 1 (1.5) | 1.0 | 125/24 | 100b | 30 | 20 |
| 5 | n Pr | 1 (1.5) | 0.2 | 125/24 | 82 | 57 | 24 |
| 6c | n Pr | 1 (1.5) | 0.2 | 125/24 | 75 | 51 | 21 |
| 7d | n Pr | 1 (1.5) | 0.2 | 125/24 | 35 | 17 | 7 |
| 8e | n Pr | 1 (1.5) | 2.2 | 125/24 | 86 | 1 | 1 |
| 9 | n Pr | 2 (1.5) | 0.2 | 125/24 | 7 | 0 | 0 |
| 10 | n Pr | 3 (1.5) | 0.2 | 125/24 | 2 | 0 | 0 |
| 11 | n Pr | 4 (1.5) | 0.2 | 125/24 | 3 | 0 | 0 |
| 12 | n Pr | 5 (1.5) | 0.2 | 125/24 | 12 | 9 | 0 |
| 13 | n Pr | 6 (1.5) | 0.2 | 125/24 | 67 | 58 | 3 |
| 14 | n Pr | 6 (1.5) | 0.25 | 125/36 | 91 | 81 | 3 |
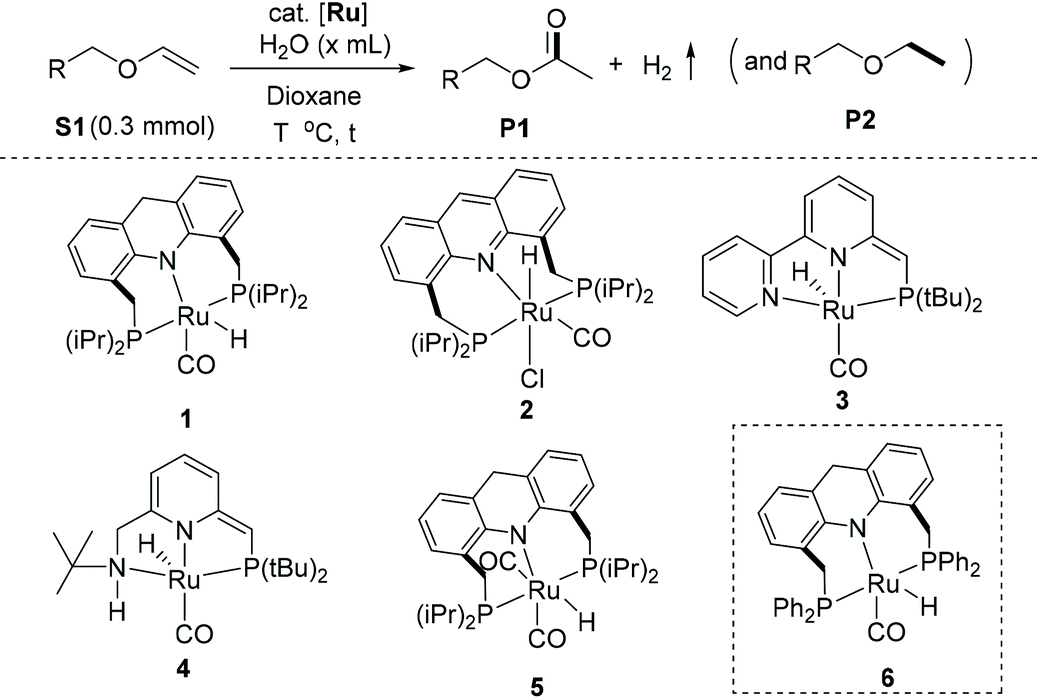
Among other complexes, the acridine-based hydrido-chloride complex 2 did not promote the reaction to any significant extent (entry 9). Similarly, the dearomatized bipyridine-based complex 3 and the dearomatized aminopyridine-based (PNNH) complex 4, showed no catalytic activities (entries 10–11).14,47 The acridine-based dicarbonyl complex 5 showed only minor catalytic activity (entry 12), highlighting the importance of a vacant coordination site for the reaction to proceed. Quite interestingly, upon decreasing the electron density at the ruthenium centre, as in the newly synthesized phenyl-substituted analog of complex 1, complex 6 (synthesis and characterization details in the ESI†), similar ester yield (58%) was observed, but with much better selectivity (only 3% of the respective olefin hydrogenation product) (entry 13). The ester yield increased to 81% upon increasing water amount to 0.25 mL and the reaction time to 36 h, without compromising selectivity (entry 14).
Substrate scope
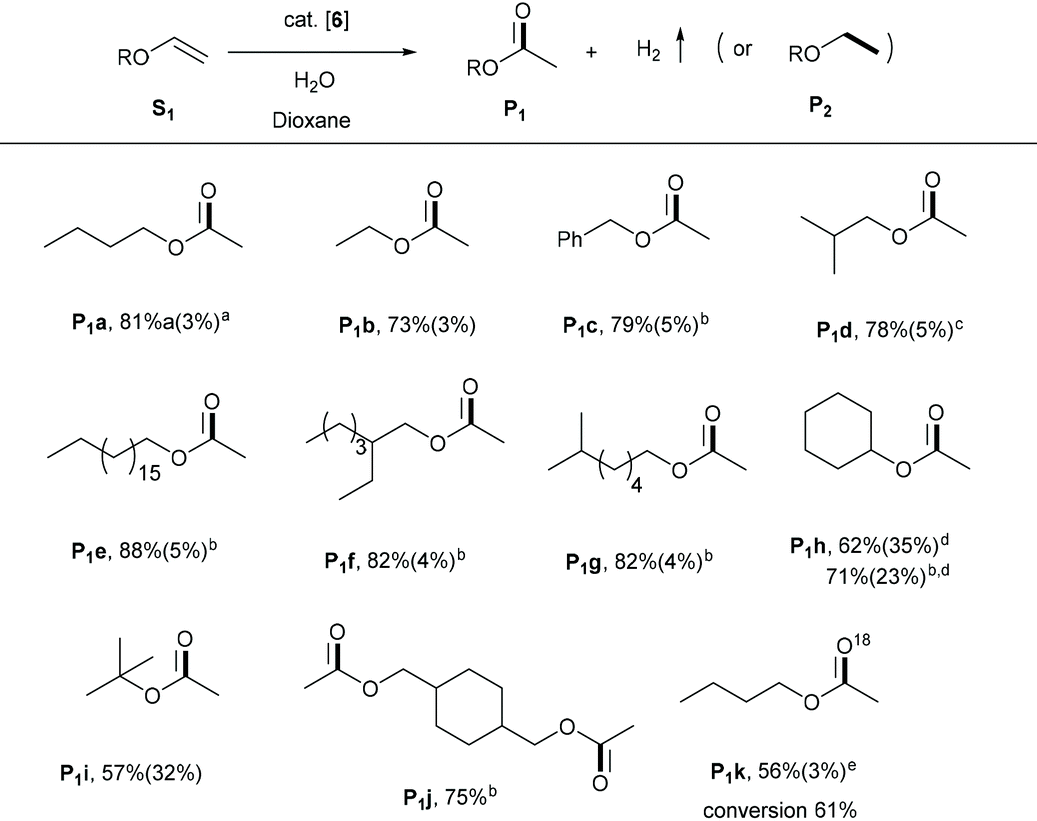
We subsequently explored the substrate scope of the process using the active and selective Ru-Acr(Ph) cat 6. Vinyl ethers were most reactive among various enol ethers—their reactions proceeded efficiently at 125 °C (Table 2). Under the conditions, vinyl ethers with ethyl, n-butyl, isobutyl, iso-octyl, octadecyl, and benzyl groups were converted to their corresponding esters, P1a–1g, in high yields, i.e., 73–88%, with minimal hydrogenation products generation (5% or less). The reactions with low boiling substrates (P1a–1b, P1d, P1i) were carried out in closed flasks, whereas for high boiling substrates, an open reaction system with argon flow was used, allowing the generated H2 gas to escape. With cyclohexyl vinyl ether as substrate, lower reaction selectivity was observed (P1h, 62%) in a closed system with significant hydrogenation product (P2h, 35%). Carrying out the reaction in an open system improved ester yield to 71%. Similar selectivity was also observed in the conversion of t-butyl vinyl ether into its respective ester (P1i). In addition to monoesters, a diester product, P1j, was also efficiently obtained from the corresponding divinyl ether. When the reaction was carried out in the presence of H218O, the method also allowed easy access to the 18O-labelled ester (P1k) with high selectivity (Fig. S33†). Notably, when phenyl vinyl ether, vinyl acetate or vinyl benzoate were used as substrates, no significant reaction was observed, likely due to the reduced alkene electron density owing to conjugation which pulls electron density away from the double bond (Fig. S15†).| Reaction conditions: Vinyl ether (0.3 mmol), 6 (1.5 mol%), H2O (0.25 mL), 1,4-dioxane (2 mL), 125 °C (oil bath temperature), 36 h, closed 25 mL Schlenk flask.a Ester yields were determined by GC or 1H NMR spectroscopy, using mesitylene as an internal standard; yields of the corresponding hydrogenated products (P2) are in parentheses.b Reflux in an open system under argon flow.c 48 h.d 24 h.e H218O, 0.1 mL. |
|---|
|
Compared to the vinyl ether substrates, cyclic enol ethers were less reactive, requiring harsher reaction conditions (1 mL water, 150 °C) to yield lactones via this method (Scheme 1). Under these reaction conditions, dihydrofuran reacted selectively to produce γ-butyrolactone in 89% yield (Scheme 1a), with 90% H2 yield. Dihydropyran showed slightly lower reactivity compared to dihydrofuran, and 72% of the corresponding lactone was observed after similar reaction conditions (Scheme 1b).48 In contrast to the vinyl and cyclic enol ethers, the internal linear enol ether, 1-butenyl ethyl ether, did not react under the reaction conditions in the presence of either complex 6 or 1 as catalyst (Scheme 1c). This is likely due to its increased steric bulk and thermodynamic stability, disfavouring its coordination to the metal centre of the catalysts. A similar reactivity pattern is observed in other organometallic reactions involving alkene coordination, including hydrogenation with Wilkinson's and Vaska's catalysts.49 The addition of a Lewis acid cocatalyst such as LiBF4 resulted in the hydrolysis of the starting material to butanal and ethanol with no significant ester formation.
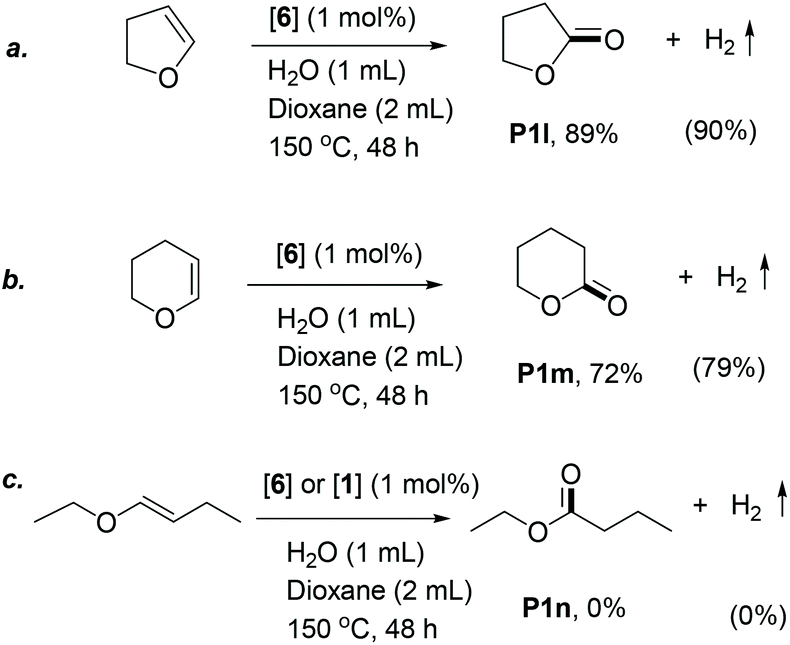 | ||
| Scheme 1 Oxidation of cyclic enol ethers by water. | ||
Mechanistic investigations
We further focused on the reactivity of the ruthenium acridine-9H based complexes to understand the reaction mechanism (Scheme 2). Preliminary NMR observations with 6 in the presence of butyl vinyl ether/water substrates revealed that the Ru-Acr(Ph) system is highly fluxional with several possible close-energy conformers, making it difficult to follow its reactivity due to peak broadening in NMR. As complexes 1 and 6 displayed similar catalytic reactivities for ester formation (Table 1), reactivity of complex 1 was explored as a substitute. Complex 1 has a vacant coordination site trans to the hydride ligand, where a vinyl ether (butyl vinyl ether) or water molecule can reversibly coordinate to form the adduct complexes 1a or 1b, respectively, as observed by NMR (Scheme 2a and b; Fig. S34†). In the presence of both butyl vinyl ether and water in the solution, preferential binding of vinyl ether to the ruthenium centre was observed, generating complex 1a (Scheme 2c).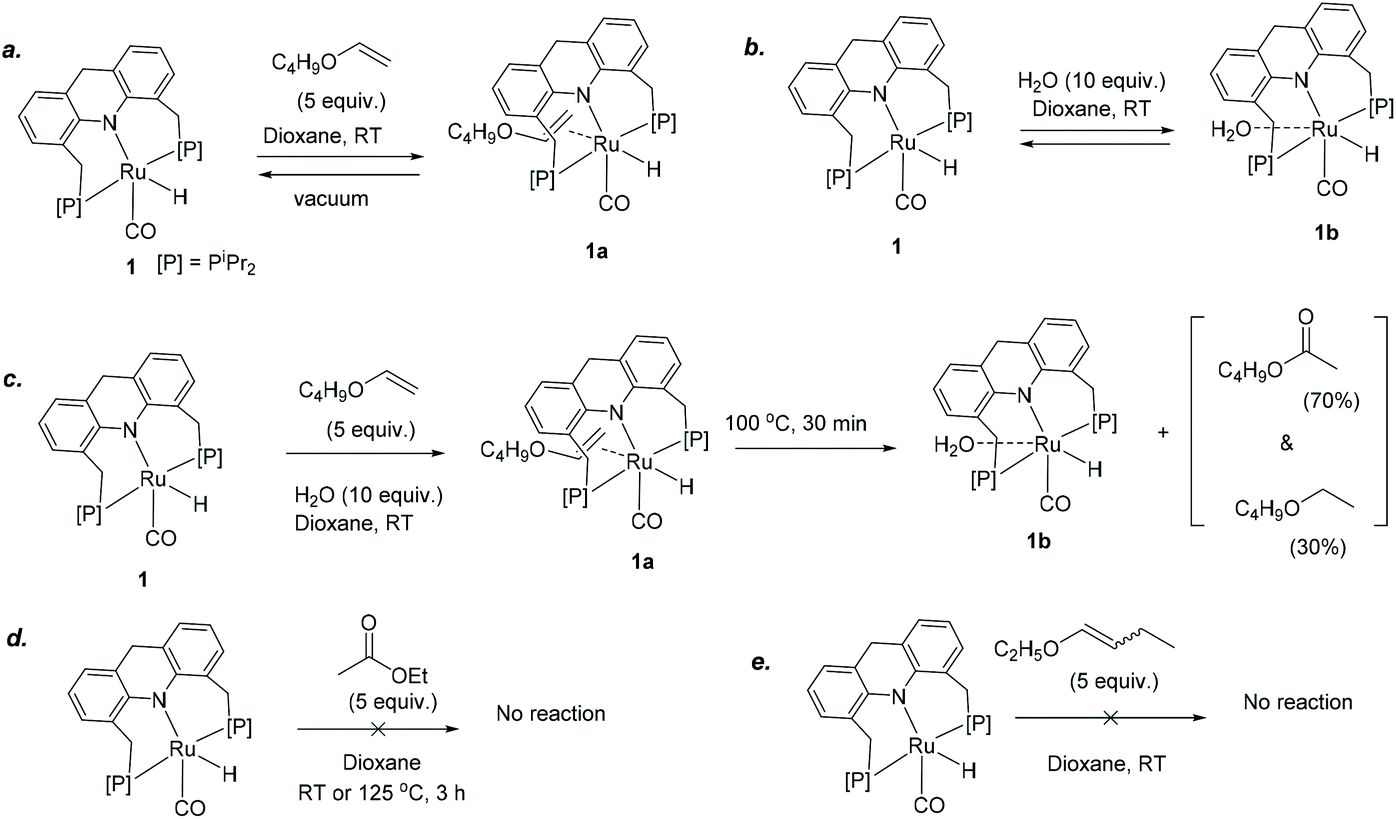 | ||
| Scheme 2 Reactivity of 1 with enol ethers and water. | ||
When a 1,4-dioxane solution containing complex 1, butyl vinyl ether (5 equiv. to 1), and water (10 equiv. to 1) was heated at 100 °C in a J Young NMR tube, formation of ester was observed by 1H NMR, although the 31P NMR revealed only the presence of complex 1a in solution. The reaction was complete after 30 min, forming butyl acetate (70%) and butyl ethyl ether (30%) as products, with the aqua complex 1b present afterward in solution following the consumption of all available vinyl ether (Scheme 2c, Fig. S34†). Thus, complex 1a seemingly acts as the resting state during active catalysis, and other catalytic species formed during reaction are high-energy intermediates, not observed by NMR. Formation of the ester product is irreversible with no observed reactivity of complex 1 with the ester (ethyl acetate), even at elevated temperatures (Scheme 2d).
Similar to butyl vinyl ether, dihydrofuran also coordinated to the vacant site of 1. On the contrary, no interaction was observed when ethyl butenyl ether was added to a solution of 1 (Scheme 2e). This lack of coordination may explain why no reaction is observed for internal enol ethers under the reaction conditions (Scheme 1). The relative binding propensities of different substrates were explored by NMR through exchange experiments at room temperature in C6D6, and among different substrates, butyl vinyl ether coordinated to the metal centre of 1 most strongly, followed by dihydrofuran and water, respectively.
Although no catalytic intermediates were observed in mechanistic investigations, based on previously observed reactivities of the ruthenium acridine system by us and others,30,50–55,63 we surmise that the reaction proceeds via the fac isomer of acridine complex 1/6, with the mer isomers being off-cycle intermediates. (For other selected examples of fac isomers of pincer complexes, see ref. 15, 56–61). Density functional theory (DFT) calculations were carried out to gain further insight regarding the reaction mechanism, with ethyl vinyl ether as the model substrate (Fig. 2). To account for the reaction conditions, the structures were optimized in a 1,4-dioxane continuum following the method reported by Truhlar and co-workers,62 and free energies were computed at 125 °C with standard state corrections. Under the conditions, the fac isomer of 6 was calculated to be close in energy to the mer isomer (5.6 kcal mol−1 uphill) (Fig. 2). However, the outer-sphere water addition to the bound vinyl ether was found quite energy demanding (ΔG 36.1 kcal mol−1), with the relevant transition state associated with simultaneous H2 liberation, akin to a previous report with imines (Fig. 2, in red).29,30 At the same time, outer-sphere water addition to vinyl ether facilitated by the mer isomer of 6 acting as Lewis acid catalyst, similar to a mechanism proposed for alkene hydration with the stronger Lewis acid In(OTf)3,31 is also unlikely, with the relevant Zwitter ionic intermediate not found by computation (ESI sec 6.2†). Interestingly, an inner-sphere stepwise mechanism from the aqua complex 6b-fac involving initial dehydrogenation,30,32 followed by hydroxide insertion to cis-bound vinyl ether, and β-hydride elimination was computed to have an overall low activation barrier (26.0 kcal mol−1 from 6a-fac) (Fig. 2, in blue). In this mechanism, initial dehydrogenation of the aqua complex is the most energy demanding step (6TS1, ΔG: 22.4 kcal mol−1 with respect to 6-mer), with subsequent hydroxide migration (6TS2) and β-H elimination (6TS3) transition states being relatively lower in energy (ΔG: 17.2 and 15.2 kcal mol−1, respectively). The likelihood of the stepwise inner-sphere mechanism is further supported by the observations that – (i) changing the reactant from water to D2O slowed down the ester formation significantly (KIE kH/kD: 2.3), suggesting the involvement of O–H(D) bond in the slowest steps (ESI, sec 3.1†) and (ii) the overall energy barrier of the pathway (26.0 kcal mol−1) is in accordance to the reaction temperature (125 °C). A similar energy profile was found with complex 1 with an overall activation barrier of 28.6 kcal mol−1 (ESI sec 6.3†). Notably, another reaction mechanism involving spontaneous water addition across vinyl ether followed by Ru catalysed dehydrogenation can be eliminated based on experimental observation (Table 1, entry 1) and computational studies (Ea: 51.1 kcal mol−1; ESI sec 6.1†).
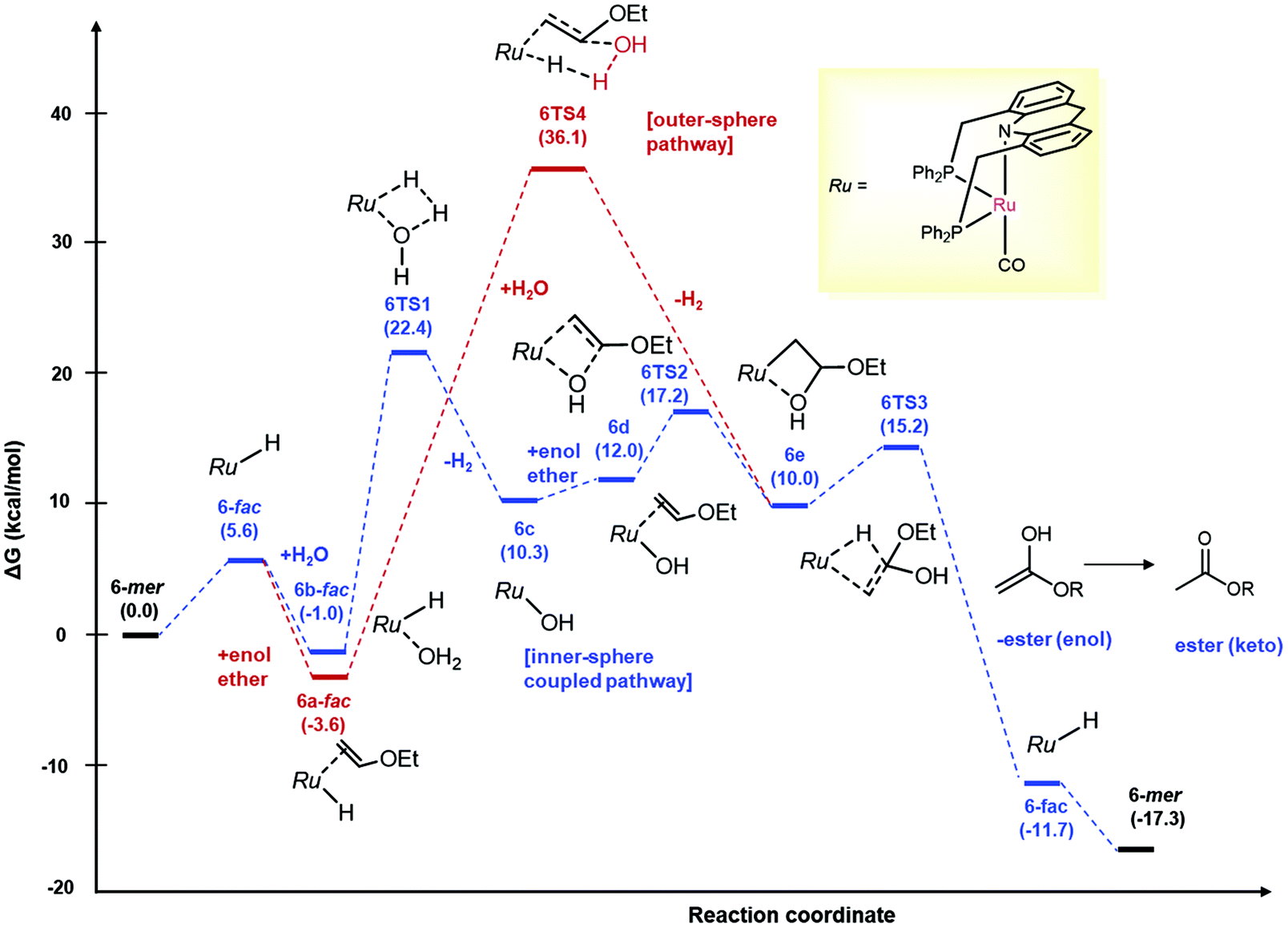 | ||
| Fig. 2 Free energy pathway for the synthesis of ethyl acetate from ethyl vinyl ether with water catalyzed by 6. Free energies (kcal mol−1) were calculated at 398.15 K relative to 6-mer + water + ethyl vinyl ether and are calculated in a 1,4-dioxane continuum (all solutes are 1 M except for H2 and H2O, which are at 1 atm and 6.17 M, respectively). Mass balance is ensured throughout. The CO and acridine-based ligands are omitted for clarity. | ||
Conclusions
In summary, we report the direct synthesis of esters by oxidation of the corresponding vinyl and cyclic enol ethers using water as the formal oxidant. The reaction is highly atom-economical, generating H2 as the only by-product, which itself is a valuable commodity. Selective oxidation is catalysed by a novel Ru-Acr(Ph) complex 6, which simultaneously catalyses the reaction's hydration and dehydrogenation steps. DFT calculations indicate an inner-sphere coupled dehydrogenation–hydration reaction pathway at the ruthenium centre rather than outer-sphere tandem hydration–dehydrogenation mechanisms. While electron-rich and sterically uncongested alkenes such as vinyl ethers or cyclic enol ethers are required for this additive-free reaction, it might also be applied in the future to other unsaturated functionalities by increasing the Lewis acidity and decreasing the steric crowding around the metal centre. Investigations toward this direction are ongoing in our lab.Author contributions
DM and SK conceived the project and designed the experiments. SK performed the experiments, analyzed the data, and performed computational studies. JL developed the synthesis of complex 6 and performed selected experiments. MR provided crucial insights regarding computation and reaction mechanism. YD-P performed experiment and analyzed data for the X-ray structure determination. YB-D synthesized the ligands used in this study. SK and DM prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the European Research Council (ERC AdG 692775). DM holds the Israel Matz Professorial Chair of Organic Chemistry. SK acknowledges the Sustainability and Energy Research Initiative (SAERI) of the Weizmann Institute of Science for a research fellowship. JL is thankful to the Feinberg Graduate School, Weizmann Institute of Science, for a Senior Postdoctoral Fellowship. MR acknowledges the Zuckerman STEM Leadership insights Program for a research fellowship. We thank Dr Michael Montag for carefully proofreading the manuscript and providing important insights.Notes and references
- J.-M. Brégeault, Dalton Trans., 2003, 3289–3302, 10.1039/B303073N.
- B. W. Michel, L. D. Steffens and M. S. Sigman, in Organic Reactions, 2014, pp. 75–414, DOI:10.1002/0471264180.or084.02.
- S. Valange and J. C. Védrine, Catalysts, 2018, 8, 483 CrossRef.
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed.
- Modern Oxidation Methods, ed. J.-E. Bäckvall, Wiley, 2004 Search PubMed.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed.
- S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756–1766 CrossRef CAS PubMed.
- N. Gunasekaran, Adv. Synth. Catal., 2015, 357, 1990–2010 CrossRef CAS.
- M. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Renewable Sustainable Energy Rev., 2021, 146, 111180 CrossRef.
- J. O. Abe, A. P. I. Popoola, E. Ajenifuja and O. M. Popoola, Int. J. Hydrogen Energy, 2019, 44, 15072–15086 CrossRef CAS.
- H. Zhao, D. Lu, J. Wang, W. Tu, D. Wu, S. W. Koh, P. Gao, Z. J. Xu, S. Deng, Y. Zhou, B. You and H. Li, Nat. Commun., 2021, 12, 2008 CrossRef CAS PubMed.
- C. Y. Toe, C. Tsounis, J. Zhang, H. Masood, D. Gunawan, J. Scott and R. Amal, Energy Environ. Sci., 2021, 14, 1140–1175 RSC.
- Y. Yao, X. Gao, Z. Li and X. Meng, Catalysts, 2020, 10, 335 CrossRef CAS.
- E. Balaraman, E. Khaskin, G. Leitus and D. Milstein, Nat. Chem., 2013, 5, 122–125 CrossRef CAS PubMed.
- H.-M. Liu, L. Jian, C. Li, C.-C. Zhang, H.-Y. Fu, X.-L. Zheng, H. Chen and R.-X. Li, J. Org. Chem., 2019, 84, 9151–9160 CrossRef CAS PubMed.
- D. R. Pradhan, S. Pattanaik, J. Kishore and C. Gunanathan, Org. Lett., 2020, 22, 1852–1857 CrossRef CAS PubMed.
- V. Cherepakhin and T. J. Williams, ACS Catal., 2018, 8, 3754–3763 CrossRef CAS PubMed.
- M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85 CrossRef CAS PubMed.
- R. E. Rodríguez-Lugo, M. Trincado, M. Vogt, F. Tewes, G. Santiso-Quinones and H. Grützmacher, Nat. Chem., 2013, 5, 342–347 CrossRef PubMed.
- J. Kothandaraman, S. Kar, A. Goeppert, R. Sen and G. K. S. Prakash, Top. Catal., 2018, 61, 542–559 CrossRef CAS.
- E. Alberico, P. Sponholz, C. Cordes, M. Nielsen, H.-J. Drexler, W. Baumann, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14162–14166 CrossRef CAS PubMed.
- P. Hu, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2014, 4, 2649–2652 CrossRef CAS.
- J. Luo, S. Kar, M. Rauch, M. Montag, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2021, 143, 17284–17291 CrossRef CAS PubMed.
- P. Sponholz, D. Mellmann, C. Cordes, P. G. Alsabeh, B. Li, Y. Li, M. Nielsen, H. Junge, P. Dixneuf and M. Beller, ChemSusChem, 2014, 7, 2419–2422 CrossRef CAS PubMed.
- Y.-Q. Zou, N. von Wolff, M. Rauch, M. Feller, Q.-Q. Zhou, A. Anaby, Y. Diskin-Posner, L. J. W. Shimon, L. Avram, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2021, 27, 4715–4722 CrossRef CAS PubMed.
- Y. Li, M. Nielsen, B. Li, P. H. Dixneuf, H. Junge and M. Beller, Green Chem., 2015, 17, 193–198 RSC.
- T. P. Brewster, W. C. Ou, J. C. Tran, K. I. Goldberg, S. K. Hanson, T. R. Cundari and D. M. Heinekey, ACS Catal., 2014, 4, 3034–3038 CrossRef CAS.
- A. S. Phearman, J. M. Moore, D. D. Bhagwandin, J. M. Goldberg, D. M. Heinekey and K. I. Goldberg, Green Chem., 2021, 23, 1609–1615 RSC.
- J. R. Khusnutdinova, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2014, 136, 2998–3001 CrossRef CAS PubMed.
- U. Gellrich, J. R. Khusnutdinova, G. M. Leitus and D. Milstein, J. Am. Chem. Soc., 2015, 137, 4851–4859 CrossRef CAS PubMed.
- S. Tang, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2020, 142, 5980–5984 CrossRef CAS PubMed.
- S. Tang, M. Rauch, M. Montag, Y. Diskin-Posner, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2020, 142, 20875–20882 CrossRef CAS PubMed.
- A. Comas-Vives, A. Stirling, A. Lledós and G. Ujaque, Chem. – Eur. J., 2010, 16, 8738–8747 CrossRef CAS PubMed.
- W. P. Weber and G. W. Gokel, in Phase Transfer Catalysis in Organic Synthesis, ed. W. P. Weber and G. W. Gokel, Springer Berlin Heidelberg, Berlin, Heidelberg, 1977, pp. 85–95, DOI:10.1007/978-3-642-46357-0_6.
- K.-M. Choi, T. Mizugaki, K. Ebitani and K. Kaneda, Chem. Lett., 2003, 32, 180–181 CrossRef CAS.
- Y. Kon, T. Chishiro, D. Imao, T. Nakashima, T. Nagamine, H. Hachiya and K. Sato, Tetrahedron Lett., 2011, 52, 6739–6742 CrossRef CAS.
- Y. Kon, S. Tanaka, T. Nakashima, K. Sato and H. Shimada, J. Chin. Chem. Soc., 2014, 61, 749–756 CrossRef CAS.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- A. Sølvhøj and R. Madsen, Organometallics, 2011, 30, 6044–6048 CrossRef.
- S. Shahane, C. Fischmeister and C. Bruneau, Catal. Sci. Technol., 2012, 2, 1425–1428 RSC.
- K. Paudel, B. Pandey, S. Xu, D. K. Taylor, D. L. Tyer, C. L. Torres, S. Gallagher, L. Kong and K. Ding, Org. Lett., 2018, 20, 4478–4481 CrossRef CAS PubMed.
- U. K. Das, Y. Ben-David, G. Leitus, Y. Diskin-Posner and D. Milstein, ACS Catal., 2019, 9, 479–484 CrossRef CAS.
- C. E. Schildknecht, A. O. Zoss and C. McKinley, Ind. Eng. Chem. Res., 1947, 39, 180–186 CrossRef CAS.
- T. H. Fife, J. Am. Chem. Soc., 1965, 87, 1084–1089 CrossRef CAS PubMed.
- A. J. Kresge and Y. Chiang, J. Chem. Soc. B, 1967, 53–57, 10.1039/J29670000053.
- L. R. Fedor and J. McLaughlin, J. Am. Chem. Soc., 1969, 91, 3594–3597 CrossRef CAS PubMed.
- S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron and D. Milstein, Science, 2009, 324, 74–77 CrossRef CAS PubMed.
- Minor amounts of lactone hydrolysis products were observed (7%) in this case also in the final reaction solution in 1H NMR spectrum.
- M. E. Vol'pin and I. S. Kolomnikov, Homogeneous Hydrogenation, Russ. Chem. Rev., 1969, 38, 273–289 CrossRef.
- Y.-Q. Zou, N. von Wolff, A. Anaby, Y. Xie and D. Milstein, Nat. Catal., 2019, 2, 415–422 CrossRef CAS PubMed.
- J. Luo, M. Rauch, L. Avram, Y. Diskin-Posner, G. Shmul, Y. Ben-David and D. Milstein, Nat. Catal., 2020, 3, 887–892 CrossRef CAS.
- M. Rauch, J. Luo, L. Avram, Y. Ben-David and D. Milstein, ACS Catal., 2021, 11, 2795–2807 CrossRef CAS PubMed.
- S. Kar, M. Rauch, G. Leitus, Y. Ben-David and D. Milstein, Nat. Catal., 2021, 4, 193–201 CrossRef CAS.
- C. Gunanathan, B. Gnanaprakasam, M. A. Iron, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 14763–14765 CrossRef CAS PubMed.
- X. Ye, P. N. Plessow, M. K. Brinks, M. Schelwies, T. Schaub, F. Rominger, R. Paciello, M. Limbach and P. Hofmann, J. Am. Chem. Soc., 2014, 136, 5923–5929 CrossRef CAS PubMed.
- M. Wilklow-Marnell, W. W. Brennessel and W. D. Jones, Polyhedron, 2018, 143, 209–214 CrossRef CAS.
- X. He, Y. Li, H. Fu, X. Zheng, H. Chen, R. Li and X. Yu, Organometallics, 2019, 38, 1750–1760 CrossRef CAS.
- V. Tegethoff, T. Lübbering, C. Schulte to Brinke, B. Schirmer, J. Neugebauer and F. E. Hahn, Organometallics, 2021, 40, 606–617 CrossRef CAS.
- Y. Shimizu, S. Fukui, T. Oi and H. Nagao, Bull. Chem. Soc. Jpn., 2008, 81, 1285–1295 CrossRef CAS.
- P. L. Chiu and H. M. Lee, Organometallics, 2005, 24, 1692–1702 CrossRef CAS.
- F. Laurent, E. Plantalech, B. Donnadieu, A. Jiménez, F. Hernández, M. n. Martínez-Ripoll, M. Biner and A. Llobet, Polyhedron, 1999, 18, 3321–3331 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. Kar, Q.-Q. Zhou, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2022, 144(3), 1288–1295 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2079903. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1gc04574a |
| This journal is © The Royal Society of Chemistry 2022 |