
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operando monitoring of mechanisms and deactivation of molecular catalysts
Katrin
Köhnke
 a,
Niklas
Wessel
a,
Jesús
Esteban
ab,
Jing
Jin
a,
Andreas J.
Vorholt
*a and
Walter
Leitner
ac
a,
Niklas
Wessel
a,
Jesús
Esteban
ab,
Jing
Jin
a,
Andreas J.
Vorholt
*a and
Walter
Leitner
ac
aMax-Planck-Institut für Chemische Energiekonversion, Stiftstraße 34-36, 45740 Mülheim an der Ruhr, Germany. E-mail: andreas-j.vorholt@cec.mpg.de
bDepartment of Chemical Engineering and Analytical Science, School of Engineering, The University of Manchester, Manchester M13 9PL, UK
cInstitut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
First published on 16th February 2022
Abstract
Observing and understanding the phenomena associated with the reaction mechanisms and catalyst deactivation in molecular catalysis is a very challenging task in green chemistry. This knowledge is crucial for applying and scaling catalyzed reactions as well as preventing misproduction at a very early point. Over the years, experimental arrangements have evolved towards analysis of catalysts and reaction products in the so-called operando setups. This contribution reflects on the potential of operando studies to elucidate reaction and deactivation mechanisms in homogeneous catalysis as well as the outstanding opportunities that arise from the application of operando experimental setups. Such setups mostly rely on spectroscopic analysis, optionally coupled with chromatographic techniques that monitor the reaction system. This in turn means that not only the evolution of the reaction substrates and products can be monitored, but also changes of the molecular catalyst species that may affect the catalytic performance. Therefore, this review focusses on techniques to monitor the catalyst under real conditions. In this review, different spectroscopic techniques relevant for monitoring molecular transition metal catalysts in solution are covered, followed by numerical methods used in the chemometrics literature to undertake the challenge of untangling the complex raw signals and allocating them to individual chemical species. Finally, two exemplary case studies of industrially relevant chemical reactions are presented, namely the hydroformylation and the asymmetric hydrogenation. These examples illustrate the significance of the application of both the experimental setups as well as data processing and signal resolution to have an insight into the deactivation of catalytic systems. The operando approach shows high potential for the increased use in future research to develop stable and more selective catalysts which can be applied in greener processes.
1. Introduction
Unravelling the mysteries of chemical reaction mechanisms and the deactivation of the catalytic species involved therein is a topic that has attracted the interest of industrial practice and research in catalysis.1 It is of paramount importance to understand catalytic cycles and how catalysts become inactive throughout the chemical reaction on a molecular level. In this way, researchers will be able to develop catalyst species or process designs that show optimal performance in terms of activity and selectivity as well as stability when applied in processes with adequate operating conditions. To detect a deviation on the molecular catalyst level can prevent misproduction and make the process greener and safer.Concerning these types of studies, the classic approach is to analyze the reaction kinetics and product mixtures ex situ in order to draw conclusions on the evolution of chemical reactions and deactivation mechanisms. The goal is to detect compounds associated with a deactivation process, such as a deactivated catalyst species.2 Although this strategy is limited to trace species which are stable at ex situ measuring conditions, very important knowledge can be gained.3–6In situ methods have been applied for several decades, allowing to perform spectroscopic measurements of the catalyst in a model setup or reactor under idealized and controlled conditions, such as an inert atmosphere. Later came the evolution to operando techniques, a term coined by Bañares and his collaborators.7,8 These feature the characterization of the used catalyst as well as the simultaneous detection and quantification of the substrates, intermediates and products of the reaction by real-time analytical techniques. Operando analytical methods run within the real operating reaction medium and conditions, thereby reducing sampling delay and avoiding air contacting, thus allowing the detection of unstable reaction intermediates that otherwise would deteriorate upon sample withdrawal and work up for analysis. In this way, a much more insightful relationship between the state of the catalyst, the reaction kinetics and the overall catalytic performance can be established.9
An ideal workflow of the operando analysis of catalytic chemical reaction starts by using an operando experimental setup featuring spectroscopic analytical methods to understand the events inside a catalytic process with many unknown elements. Depending on the complexity of the retrieved data, more or less complex data processing curve resolution techniques could be performed to identify individual chemical species. Such multivariate curve resolution methods can lead to the establishment of more accurate mechanisms by isolating the signals and identifying more species. Finally, with all the knowledge derived from this experimental arrangement and data analysis, catalytic and deactivation mechanisms can be elucidated.5
Different articles have illustrated that the analysis of reaction mechanisms and deactivation of the catalytic species is mainly performed by spectroscopic techniques. These may vary significantly depending on the type of catalysts being investigated. Wachs et al. have portrayed a thorough historical background on operando spectroscopic techniques with its prime focus on heterogeneous catalysis.9 More recently, a perspective article has described in situ and operando techniques applied for the characterization of single atom catalysts supported on heterogeneous materials.10 For heterogeneous catalysts, methods highlighting chemical and structural changes of the solid materials are of special relevance. Furthermore, recent work has used operando setups to conduct kinetic and mechanistic studies of molecular catalysts supported on solid materials.11 All these reviews show that the understanding of deactivation of active catalytic sites can lead to longer life time of the catalyst alongside stable selectivity.
In molecular transition metal catalysis,12–14 thus far research discussion has focused mostly on mechanistic studies, leaving the study of deactivation phenomena somewhat unattended, particularly under relevant reaction conditions.5 Molecular catalysis features milder reaction conditions and unique selectivity and reactivity in comparison to heterogeneous catalysis. The lack of operando studies of molecular catalysis can be seen in literature searches: searching the term “operando spectroscopy” together with “catalyst deactivation”, “catalyst degradation”, “catalyst decomposition” or “catalyst inhibition” in SciFinder® gives in total seven results which can be assigned to the field of homogeneous catalysis. In contrast, 116 references on heterogeneous catalysis and 39 on electrocatalysis can be found with the same search items. These results, without claiming completeness, clearly show an underrepresentation of molecular catalysis. Studies in this particular field of catalysis remain very challenging due to the sensitivity of the spectroscopic methods in relation to the usually very low concentration of the catalytic species. These studies would be of great importance in industrially established reactions like the hydroformylation of olefins,15 asymmetric hydrogenations,16 telomerizations17 or carbonylations.18
Anyhow, regardless of the type of catalysts to which the aforementioned spectroscopic techniques are applied, the complexity of the signals suggests that often times advanced mathematical techniques can provide a powerful tool towards a proper interpretation of the results. For this reason, several studies in the chemometrics literature have used different numerical approaches to face the challenging resolution of curves and its ascription to each individual chemical species involved.19–21
This review focusses on techniques which provide information of the catalyst species on a molecular level during a reaction. Emphasis is placed on metal organic compounds used as catalyst, even though most of these methods can also be applied to monitor organo-, photo- or electrocatalyzed reactions if the catalyst is in solution. The main focus and motivation of the article is to find direct proof of catalytic reaction and deactivation mechanisms.
Therefore, this work first describes the relevant operando spectroscopic techniques applicable to molecular catalysis, including their features and application in mechanistic studies. Then, an overview of numeric methods for multivariate curve resolution for the interpretation of the spectroscopic signal is presented. Last, reaction mechanisms and catalytic deactivation are explained in relation with operando studies for two exemplary case studies of well-established reactions in industry, namely hydroformylation and asymmetric hydrogenation. Overall, this review intends to highlight the importance of operando techniques and data treatment to shed light on the correct interpretation of homogeneously catalyzed reactions and especially their deactivation.
2. Operando techniques for molecular catalysis
In this section operando techniques that have been used for the study of molecular organometallic catalysts in solution, namely nuclear magnetic resonance, electron paramagnetic resonance, infrared spectroscopy, Raman spectroscopy, ultraviolet-visible spectroscopy, X-ray absorption spectroscopy and electrospray ionization mass spectrometry will be summarized. The use of a particular technique depends on the nature of the catalyst, the composition of the reaction medium, the characteristics of the analytical instruments, and of course the ease of device setting-up. The features of each technique and its application in kinetic and mechanistic operando studies will be discussed as well. Besides, chromatography techniques, such as online GC or HPLC are useful tools to quantify the conversion, selectivity, and kinetic study, which indirectly reflects the catalyst activity but will not be discussed here.2.1. Operando analytical techniques
Nuclear magnetic resonance spectroscopy (NMR), one of the most widely applied spectroscopic techniques in chemical research, provides detailed information about molecular structure as well as dynamic processes and allows the direct observation of chemical reactions.22 Jagadeesh and coworkers have investigated the reductive amination of carbonyl compounds to a variety of primary amines with a [RuCl2(PPh3)3] catalyst. With the help of in situ1H and 31P NMR spectroscopy, an appropriate reaction mechanism was proposed, clarifying the formed ruthenium hydride species to be the active catalytic species.23 In addition to the most common 1H NMR, multi-nuclear spectra supply more information for catalytic intermediates during reaction processing. Especially for transition metal catalyzed reactions, 13C NMR (for metal carbonyls, metal carbenes), 31P NMR (for phosphine ligands) and 19F NMR (for fluorous catalysts) are very useful approaches to observe the coordination mode and to identify the active intermediates, which eventually improves the understanding of the catalytic cycle.24–26Many molecular reactions, such as hydrogenation and hydroformylation, require high gas pressures. High pressure NMR technology has been performed for over 60 years.27 Limited by the volume of the NMR tube, in situ static NMR often lacks mixing, which might lead to inaccurate kinetic data for reactions involving gases.28 High pressure flow NMR cells were developed to improve the mass transfer. There are two typical designs. The first design keeps a constant stream of gas bubbles through the solution in the NMR tube.29,30 In order to reduce the degradation of spectral resolution caused by the gas bubbles, the gas flow has to be adjusted to a moderate rate and gas circulation has to be stopped during the excitation and acquisition periods. More recently, Dušan's group has developed a pure-shift method by removing all heteronuclear and homonuclear couplings of the selected signal, which can improve in the signal-to-noise (S/N) ratio remarkably. This method was then applied to monitor inhomogeneous reaction with gas sparging, and the singlets can be improved 8-fold in S/N.31 The other type of design continuously circulates the reaction solution from the reactor to the NMR probe. The liquid flow rate has to be optimized to allow enough residence time for sample magnetization and acquisition. A correction factor is necessary to quantify the integral data at flow conditions vs. static conditions.32 Recently, Hintermair's group reported a study using operando flow NMR spectroscopy to monitor the Rh/PPh3 catalyzed hydroformylation of 1-hexene with multi-nuclear 1H and 31P{1H} NMR spectroscopy.33 The reaction mixture was infused from high pressure vessels (up to 20 bar) to a high field NMR with a flow rate of 4 mL min−1 (about 30 s delay). To obtain quantitative data, correction factors of different species (reactant, catalyst and product) in the mixture were calculated with varied concentrations. The flow NMR setup allowed to monitor all the species and the corrected NMR data showed good agreement with the GC results, which makes it a promising method for further study of other catalytic hydroformylation systems.
High-field NMR instruments provide high sensitivity and large chemical-shift dispersion. However, large size and high cost prevent its wide application for operando studies. Instead, compact NMR spectrometers are an economic and operable alternative for real-time analysis on workbench.34–36 Danieli et al. reported a study on the hydrogenation of acetophenone with isopropanol catalyzed by iridium complexes in flow conditions in a low field NMR with 60 MHz. They considered that the field homogeneity and the kinetic rate could be studied as a function of the catalyst concentration and good agreement was found with the results obtained by gas chromatography.37 Compact NMR spectrometers have become an important analytical tool for both quality control and process monitoring. However, low sensitivity for reaction intermediate monitoring limits its application for chemical analysis. It is to be expected that the accuracy of compact NMR will be improved by device developments such as increasing magnetic field strengths and increasing line shape specifications.34
Electron paramagnetic resonance (EPR) is a magnetic resonance technique similar to NMR, but instead of measuring the nuclear transitions, it detects the transitions of unpaired electrons in an applied magnetic field. In situ EPR is widely used for studying transient radical-pairs in photo-excited systems, for instance for monitoring the primary photochemistry in reaction centers of photosystems I and II.38,39 Besides, it is a good tool for the characterization of catalysts involving paramagnetic centers, e.g. iridium, iron and chromium coordinated by N-heterocyclic carbene (NHC) or phosphine ligands.40,41 Brückner's group investigated photocatalyzed water reduction using iridium and iron complex catalysts.42In situ EPR enabled to monitor the paramagnetic radical intermediates. Coupled with Raman and IR spectroscopy, two detailed catalytic cycles were proposed. In this study, the reaction was carried out in a typical EPR tubing. The relaxation properties of transition metals are highly sensitive to temperature, which further influences the signal intensity of paramagnetic species.43 Hence operando EPR is preferably carried out under harsh conditions, for instance the above cases were measured below 200 K. However, Brückner et al. were able to perform operando EPR measurements of Cr in ethylene tetramerization at 40 °C and up to 14 bar ethylene pressure.44 These results gave hints to a deactivation pathway forming Cr(I) species.
Infrared spectroscopy (IR), concerns the study of molecular structure and properties from their vibrational transitions induced by infrared light. Compared to NMR, IR has a faster time scale and is more sensitive. Especially in the case where the catalytic species is a transition metal carbonyl complex, the strong CO vibrations provide an excellent analytical tool for studying intermediate species.12 Typical catalytic carbonylation reactions include alkene hydroformylation, methanol carbonylation and methoxycarbonylation, which employ high pressure and high temperature.45–48 In order to increase the concentration of gases in solution for faster reaction rates or beneficial shifts in chemical equilibria, high-pressure IR spectroscopic techniques have been developed.
Two main sampling setups are in common use: (1) transmission and (2) attenuated total reflectance (ATR). Varied types of high-pressure transmission IR cells have been developed to facilitate real reaction conditions for operando studies.49 Due to the limited mechanical strength of the observation windows and optimum pathlength for different compounds, the volume of high-pressure transmission IR cells is usually relatively small. Hence, one type of design consists of an extra variable pathlength view-cell connected to an autoclave through which the reaction mixture is circulated. As the reaction is carried out in a big vessel, this design avoids the mass transfer issue but has to consider the delay time. A second type is a high-pressure vessel with two IR transparent windows wherein the reaction occurs. This realizes in situ testing but lacks mixing and pathlength variation. To minimize the delay time and mixing problem, van Leeuwen's group has developed a high-pressure IR autoclave with integrated IR cell and reaction vessel (50 cm3) via two connected chambers. A turbine rotor was employed to stir and circulate the reaction mixture from reaction vessel to IR cell in 33 ms at the quickest. This cell has been used to investigate the kinetics of CO ligand exchange between the rhodium complex and H2, which proved to be a key step in hydroformylation catalysis.50
Different from the transmission IR, attenuated total reflection infrared spectroscopy (ATR-IR) measures the optical absorption of evanescent waves at the interface between a sample and a transparent sensing element rather than through a very thin sample. The ATR setup can be mounted onto a benchtop instrument or implemented in an immersion probe, which is easier and more flexible than transmission IR setups.51,52 It is already commercially available in systems such as ReactIR53 and Arcoptix FTIR-FC.54 The evanescent wave of infrared radiation has a limited penetration depth which is larger in the case of longer wavelengths, causing a sensitivity change of the ATR spectrum in the different range of wavelengths. This is the main difference between ATR and transmission IR and also the reason for the low sensitivity of ATR spectra in short wavelength regions, which has been proved by our group in the study of the hydroformylation of 1-dodecene. It compared the ability of transmission and ATR IR setups for monitoring the active catalyst species in batch and continuous flow reactions. In the latter, transmission IR enables the detection of organometallic intermediates at a ppm level while ATR cannot.55 In conclusion, both transmission and ATR IR are adequate tools for operando study, and the individual choice certainly depends on the specific process constraints and the required specificity.
Raman spectroscopy is IR complimentary, as it can access IR inactive vibrations since Raman active vibrations cause a polarizability change, differing from IR active vibrations which change the dipole moment of a molecule.56 As a scattering technique, Raman signals are weaker than the ones obtained from IR spectroscopy. However, Raman spectra are generally sharp with rare overlapping, which is convenient for spectral analysis. Since water is a weak Raman scatterer, Raman spectroscopy is ideal for monitoring aqueous phase reactions. One application of operando Raman spectroscopy in aqueous phase was performed by Haumann et al., investigating ruthenium catalyzed methanol dehydrogenation.57 The reaction intermediate formate was detectable by Raman spectroscopy, which further proved the postulated mechanism.
UV-vis spectroscopy produces comparatively sensitive spectra, which can show the d–d* and charge transfer transitions of transition metal complexes and the formation of organic molecules via their n–p* or p–p* transitions.58,59 Due to the limited structural information UV-vis can provide mainly qualitative data. However, for those compounds having UV-vis chromophores, it is still a useful tool for supplementing information gained from other spectroscopic techniques. Schaub and coworkers investigated the chromium catalyzed dehydroperoxidation of cyclohexyl hydroperoxide to cyclohexanone, using in situ UV-vis spectroscopy to monitor the intermediate alkylperoxychromium(VI) complex formation and decomposition. Coupled with NMR spectroscopy and DFT calculations, a promising mechanism was proposed.59
X-ray absorption spectroscopy (XAS), measuring the energy-dependent fine structure of the X-ray absorption coefficient near the absorption edge of a particular element, is a powerful tool for probing the average local electronic and geometric structures of catalysts in the working state.60,61 It is widely used in heterogeneous operando studies, however, only few cases have been presented on molecular catalysis in situ analysis.62–66 Among one of the first in situ XAS studies is a study on the bromobenzene homocoupling with a [Ni(cod)(bpy)] by Tanaka's group.67 Using time-resolved XANES and EXAFS analysis they were able to monitor the reactant [Ni(cod)(bpy)], the intermediate [Ni(bpy)(Ph)Br(DMF)2] and the subproduct [Ni(bpy)Br2(DMF)]2. Recently, Bäckvall's group published a work of operando XAS to study the activation of a ruthenium racemization catalyst.68 X-ray absorption spectroscopy enabled the inspection of the stereostructure coordination environment around ruthenium during activation, as well as the proposed acyl intermediate and the activated alkoxide complexes for different molecular ruthenium catalysts, providing a valuable foundation for efforts to further catalyst development. Since XAS analysis requires structural models which hinder it to discriminate elements from the same periodic row, a combination with other structural and analytical techniques, such as NMR, can make it more effective.69
Electrospray ionization mass spectrometry (ESI-MS) is a soft ionization technique applicable particularly for solutions containing charged species, which yields little fragmentation products and proves to be an excellent tool for analyzing organometallic complexes.70,71 Compared to standard spectroscopy, it is relatively simple to analyze reaction mixtures applying ESI-MS, as each species in solution usually can be identified by a single peak in the mass spectrum. Therefore, this technique has been widely used to investigate the mechanisms of several classic and organocatalyzed reactions.72–75 In 2007, Santos et al. were able to detect three catalytically involved species of the Stille reaction via direct infusion ESI-MS, characterizing most of the major intermediates in the cycle for the first time.76 The same group reviewed the application of real-time ESI-MS analysis on the mechanistic study of Morita–Baylis–Hillman (MBH) reactions.77 Some neutral intermediates were expected to be in equilibrium with their protonated or cationized forms in methanolic solutions, and could therefore be detected and analyzed via ESI-MS. After testing several different catalysts, a dual mechanism was observed by online ESI-MS. Additionally, ionic liquids acting as additives as well as (thio)urea working as organocatalyst were monitored.
The typical way of continuous infusion of reaction mixture into a mass spectrometer is by using syringe pumps or HPCL pumps. In order to realize in situ conditions, instead of applying a pump, McIndoe et al. have connected the reaction vessel directly to a mass spectrometer via PEEK tubing. By slightly pressurizing the reactor, the reaction mixture was infused into the instrument. This simplified technique has been used for the kinetic study of the rhodium catalyzed hydrogenation of a charge-tagged alkyne, and the experimental data showed great agreement with numerical modeling.78
Chen combined in situ chemical synthesis and gas-phase ion molecular reaction in a tandem ESI-MS.79 This technique can achieve reaction, “purification”, and analysis in a single device, which makes it an extremely fast method for the assessment of catalytic activity relative to conventional studies. Reactions such as olefin metathesis and Ziegler Natta polymerization were screened using this setup. The results of the in situ study showed good agreement with the analysis of the separately conducted synthesis, which demonstrated that this technique is an effective and reliable method.
However, the use of ESI-MS for accurate quantification of a species has its limitations because numerous minor variations can affect the ionization efficiency of different compounds.80 Accurate ESI-MS quantification can only be achieved via repeatable sample preparation methods that provide reliable recoveries, for example the use of internal standards.81
2.2. Combination of multiple techniques
Multiple operando and in situ techniques applied to investigate the catalytic system from different perspectives allow us to bring all the information together and gain a detailed understanding of the catalyst performance. In Fig. 1, two examples are presented to show the currently available techniques for operando and in situ analysis of molecular catalysts. | ||
| Fig. 1 Examples for the application of multiple operando and in situ techniques. A: Investigation of the influence of activators on the performance of a chromium complex in ethylene oligomerizaion;82 B: exploration on the photoreduction of aryl halides.58 Chemical bonds and atoms are colored corresponding to the detecting spectroscopic technique listed in the leading entry. | ||
Example A shows the investigation on the impact of different activators (MMAO and AlR3) on the in situ formation of the depicted chromium complex and its performance in ethylene oligomerization to 1-octene.82 After mixing the chromium precursor (Cr(acac)3) 1, ligand (PNP) 2 and activator (MMAO or AlR3), operando EPR immediately detected the reduction of EPR active CrIII to EPR silent CrII, which indicates the formation of the active species (PNP)CrII(CH3)23. However, when using AlR3 activators, CrI species were also observed along with a reduced consumption of ethylene and low selectivity for 1-octene. Raising the bulkiness of the alkyl could promote the reduction to CrI. However, the ethylene consumption with AlEt3 and AliBu3 was the same and slightly higher with AlOct3. In order to elucidate the reasons for the decreased activity, in situ UV-vis, ATR-IR and XAS analysis were applied. Firstly, with AliBu3, the [acac]− ligand, which could facilitate further reduction of CrII to CrI, was detected using UV-vis spectroscopy, consistent with the operando EPR results. The ATR-IR results showed that in the case of AlR3 activators, a ligand exchange between Cr(acac)31 and the activator takes place followed by the formation of Al(acac)3 rather than the active CrII species, which was also observed by UV-vis. From the XAS results, it is evident that only with MMAO a bidentate coordination by two phosphorous atoms was observed, while with AlR3 the larger alkyl groups partially hindered the complete coordination of the PNP ligand 2 to the chromium center. Hence, with the help of operando EPR and in situ UV-vis, ATR-IR and XAS spectroscopy, the low conversion when using the AlR3 activator is found to be partially due to the formation of CrI instead of the active CrII species. However, the main influence on the chromium complex performance in this reaction system was proved to be the mode of PNP coordination rather than the CrI content.
It seems clear that approaches which integrate multiple techniques together can provide a more complete picture of the reaction system.83 Two or more techniques are applied simultaneously to supplement or contrast each other, improving the quality of the obtained information. Gschwind's group developed a novel fully automated setup that skillfully integrated illumination with operando NMR and UV-vis spectroscopy, which enables the simultaneous and time-resolved detection of paramagnetic and diamagnetic species.58 This setup was applied for the study of the photoreduction of aryl halides 4 with PDI 5 as photocatalyst (Example B, Fig. 1). During the illumination, operando UV-vis spectroscopy enabled the detection of PDI and the stable PDI˙− radical, while the proton NMR signals of the PDI core vanished completely. Therefore, the UV-vis data provided time-resolved information about the photocatalyst that was invisible to NMR spectroscopy. In the meantime, the operando NMR spectra supplied full quantitative and structural insight into the diamagnetic reactant and product 6.
In general, the selection of operando techniques is determined by the nature of the targeted species, reaction conditions and other specific issues caused by the particular reaction. Therefore, it is crucial to investigate the catalyst, reactants and reaction conditions by the optional techniques considered in advance. The most common technique for reaction monitoring is NMR, which can be applied under high pressure, but it is not applicable for paramagnetic species. Instead, EPR spectroscopy can only detect species with unpaired electrons. Raman and IR spectroscopy provide complementary information about the vibrational state of molecules, in particular when metal complexes are participating in catalytic reactions. UV-vis spectroscopy gives limited information about the molecular structure, but for those compounds having UV-vis chromophores, it is still a useful tool for supplementing information for other spectroscopy. XAS spectroscopy enables to inspect the stereo-structure coordination environment around a working state, but low sensitivity and discriminating elements from the same periodic row limit its information content. ESI-MS, which transfers ions directly from the solution to the gas phase with efficiency and gentleness, can provide proper snapshots of the ion composition of the reaction solutions. Repeatable sample preparation is required for reliable ESI-MS result.
2.3. Operando techniques for monitoring molecular homogeneous electrocatalysis
With the rising relevance of the chemical conversion of renewable energy, the field of homogeneous electrocatalysis is increasingly gaining in importance, especially with regard to the activation of small molecules such as CO2, N2, O2 and H2O. Special analytical techniques are available to investigate electrocatalytic systems. They are tailored to the specific features of these reactions, such as the central role of electron transfer processes or the fact that electrocatalytic events mostly occur within a reaction diffusion layer at the vicinity of the electrode rather than in the bulk. The most relevant methods are shortly introduced below. For interested readers, recent reviews84–90 give a detailed overview about the potential of the application of these techniques in the in situ/operando context to help understand catalytic mechanisms and deactivation processes.Analytical voltammetries, among which cyclic voltammetry (CV) is the most popular, can be applied to investigate mechanistic aspects of catalytic systems involving electron transfer reactions. The energy needed to oxidize or reduce a molecule is measured by recording the current passing through an electrolyte containing the analyte as a function of an applied potential. Even though no direct structural information can be obtained from CV measurements, the resulting voltammograms provide information namely about the redox nature of the catalytic species, reaction kinetics regarding chemical and electrochemical (electron transfers) steps or catalyst deactivation.84,85,87,90
The field of operandospectroelectrochemistry (SEC)84–90 involves the utilization of the spectroscopic tools described above, often in specific setups adapted to the needs of electrocatalytic reactions. The most widespread are IR and UV-vis, as they are easily accessible in most laboratories. The development of dedicated cells and beamline setups have also made Raman, fluorescence, EPR, NMR and XAS available. A special technique within the SEC analysis of molecular catalyst is the application of sum-frequency generation (SFG), where two lasers operating at different frequencies generate a beam in a nonlinear optical interaction of the involved photons. It is extremely sensitive to molecular orientations near an electrode surface and therefore a particularly informative method.89 Transient absorption spectroscopy (TAS), where a the formation of an excited state is generated by a flash of light, triggering for example an electron or proton transfer, can detect short-lived intermediates and resolve reaction kinetics on picosecond scales.87
3. Mathematical methods for signal resolution in spectroscopy
Whilst providing great insights into the behavior of chemical systems, operando spectroscopic measurements retrieve responses of challenging interpretation. The overlapping of signals characteristic of spectroscopy requires chemometric approaches for the resolution of the signals and allocation to individual chemical species. Several authors have tackled the challenge of processing the entangled sets of data obtained from measurements, for which a number of advanced numerical approaches have been proposed.In general, multivariate curve resolution (MCR) analysis is based on the following matrix expression:
| D = CST + E | (1) |
| Classification | Method | Description | Application example | Ref. | |
|---|---|---|---|---|---|
| Description | Analytical techniques | ||||
| Soft-model/non-iterative | Window factor analysis (WFA) | The concentration profiles are obtained from the original data set and the window at concentration equals zero. This way, a vector with the spectral variation of the component of interest is calculated and, combining with the original data set, the concentration profile can be determined. | Resolution of 7 bismuth species in flow injection experiments | UV-vis | 92 |
| Soft-model/non-iterative | Subwindow factor analysis (SFA) | The method retrieves the pure response profile of each component using the knowledge of the concentration windows. The pure component spectra of each species are calculated as the intersection of two subspaces with only that compound in common. | Resolution of 4 components of a polyaromatic mixture | HPLC-DAD | 93 |
| Soft-model/non-iterative | Heuristic evolving latent projections (HELP) | It starts finding a selective concentration region for the component to be resolved. Such region provides directly the component spectrum and the concentration profile is recovered from this selective region as well as the zero-concentration window. The algorithm then recovers concentration profiles one at a time by subtraction of the contribution of the resolved compound. | Resolution of mixtures of drug isomers | HPLC-DAD | 94 |
| Soft model/iterative approach | Key set factor analysis (KSFA) | The strategy is to properly select pure wavelength columns applying principal component analysis (PCA) on the set of mixture spectra. A column of normalized PC values is obtained dividing the spectra by the length of the PCs at a wavelength value. This procedure emphasizes the purest wavelengths, from which the compounds can be determined in subsequent order. The main issue is dealing with spectral noise, since regions of low absorbances may yield large random signals. | Effect of solvent in 1H NMR shifts of substituted methanes; retention indices for a set of ethers in different stationary phases; resolution of mass spectra of mixtures of aromatic compounds | NMR, GC, HPLC, mass spectrometry | 95 |
| Soft model/iterative approach | Simple-to-use interactive self-modelling mixture analysis (SIMPLISMA) | The processing of the original spectra does not use PCA. The user must select a pure wavelength and an average spectrum is calculated from the mixture spectra. The difference between the average and each of the mixture spectra determines a standard deviation. Then, in subsequent iterations, pure spectra for the pure wavelengths are resolved depending on the magnitude of the standard deviation. As in KSFA, the main difficulty is setting a correct noise level, which affects resolution. | Elucidation of the reaction mechanism for the synthesis of 4-amino-3,5-dimethyl pyrazole | ATR-FTIR | 96 |
| Soft model/iterative approach | Interactive principal component analysis (IPCA) | Developed from a combination of KSFA and SIMPLISMA to determine the pure wavelengths and predict the components. This method gives a good estimate of background noise and improves the signal to noise ratio. However, for minor components, spectral predictions are improvable. | Esterification of 2-propanol and acetic anhydride; analysis and mixture of gases in natural gas | FTIR (mid IR data and near IR) | 97 |
| Soft model/iterative approach | Band target entropy minimization (BTEM) | It is based on singular value decomposition obtaining the resolved curves sequentially until no additional spectra can be retrieved. The method searches for spectral patterns with the least information entropy. An initial estimate of the observable species is not a prerequisite, it does not rely on the concept of pure wavelength and it can resolve minor species in the analyzed system. | Monitoring hydroformylation reactions with organometallics (Rh-based); spectral resolution of mixtures of highly overlapping organics; analysis of Raman images of kidney calculi | FTIR, mass spectrometry and Raman imaging | 21, 98 and 99 |
| Hard model | Complemental hard modeling (CHM) | It is an IHM method whose starting point is an incomplete nonlinear model to which additional peak functions are complemented rather than having a difference spectrum calculated. After each complementing step, both the partial model and all complemental peaks are regressed at the same time with a non-linear least squares fitting. This enables the incomplete model to interact with the unknown pure component model throughout the process and a correct allocation of the unknown spectrum to the mixture spectrum. | Resolution of chloroform, n-heptane and ethyl acetate mixtures | FTIR and Raman spectroscopy | 100 |
| Hard model | Hard modeling factor analysis (HMFA) | It can identify all pure spectra in a completely unknown mixture from a limited set of mixed spectra. The method first estimates peak functions, but requires the presence of a distinctive peak for every component, even if it overlaps with the spectra of other components. Advantageously, HMFA does not require initial guesses for concentration profiles or pure spectra. | Resolution of chloroform, n-heptane and ethyl acetate mixtures | FTIR and Raman spectroscopy | 100 |
A method is referred to as soft-model if there is no constraint to the profile shape. These models describe processes empirically without prior knowledge of the underlying phenomena, which avoids errors derived from wrong assumptions. Conversely, the drawback is the intensity and rotational ambiguity of the profiles, which becomes relevant when different chemical species show similar profiles.91,101
In non-iterative soft models, the sequential structure of concentration profiles is obtained combining subsets of information from the global and local ranks. Such subsets correspond to certain concentration windows or regions of the original data matrix with particular properties, such as the presence or absence of chemical species in the systems.19,20 Examples of these models include window factor analysis (WFA),92 subwindow factor analysis (SFA)93 or heuristic evolving latent projections (HELP).94 However, the use of these algorithms is currently in decline owing to the difficulty of setting the concentration or spectral windows in multicomponent datasets and non-sequential orders or with unstructured concentration directions.19,20
For their part, iterative approaches optimize initial estimates of concentration values or the corresponding spectra imposing a series of constraints until a convergence tolerance is reached. Typically, these models are referred to as multivariate curve resolution-alternating least squares (MCR-ALS).20,102 Among them are key set factor analysis (KSFA),95 simple-to-use interactive self-modelling mixture analysis (SIMPLISMA)96 or interactive principal component analysis (IPCA).97 They are preferred to non-iterative for not requiring a structured concentration direction and, more significantly, the fact that the implementation of constraints can be made in the optimization process. These constraints can contain chemical or mathematical information, such as non-negativity of profiles, unimodality, closure of mass-balances, correspondence of species, calibrations, etc. In turn, this means that soft iterative models open the door to the application of hybrid soft–hard models, particularly MCR-ALS.20,101 This is the case of the work by Cruz et al., who developed a MCR-ALS method with multiple hard constraints to elucidate kinetic paramaters of the homogenously catalyzed Heck reaction between iodobenzene and n-butyl acrylate catalyzed by Pd-based complexes measuring FTIR spectra.103
Despite the overall good performance of the methods addressed above, there are drawbacks to spectral resolution. These include the weak signals displayed by minor species, the presence of components with a high degree of spectral overlap and the possible non-linearity of the spectra. A different approach named band-target entropy minimization (BTEM) was developed to face these issues, with implementation in in situ spectroscopic investigations.98,104,105 BTEM is based on the subsequent reconstruction of pure component spectra by singular value decomposition of the original data set according to eqn (2):
| D = UΣVT | (2) |
Recently, a comparison between MCR-ALS and BTEM was published, where the authors implement the use of these techniques separately and jointly to mass and UV-Vis spectra as well as Raman images. The conclusion reached is that the two methods performed well in all cases, although MCR-ALS performed better for UV-vis spectra and was more robust to noise. On the other hand, BTEM led to more accurate profiles in Raman and mass spectra data sets as well as being better at reducing the rotational ambiguity of data.99 BTEM had previously been compared to MCR techniques to obtain the pure FTIR spectra in a mixture of six organic compounds. In this case, BTEM outperformed SIMPLISMA and IPCA not only in the ease of use (not requiring initial estimates), but also because the latter two methods resolved spectra including negative values for certain components.106 Nevertheless, there are few pieces of work that compare data sets of similar nature with these numerical methods, which makes it difficult to establish a much desirable direct relationship or identification between the suitable algorithms to be used depending on the spectroscopic technique used.
MCR methods often provide non-unique solutions. This in turn means that many authors have accepted the output of these algorithms without pondering over the actual significance of the results and whether there could be a range of other possible results. Characterizing an area of feasible solutions (AFS) for 2-component systems is relatively simple, but for systems comprising more components this calculation becomes far more complicated. For 3-component systems, different algorithms based on the variation of the area of polygons have been applied, including Borgen plots, the triangle-boundary-enclosure algorithm or the polygon-inflation methods.107,108 For 4-component mixtures the sliced-triangle-boundary-enclosure and the polyhedron-inflation algorithms have been reported.107,108 Finally, for higher number of components, the so-called ray casting method has been proposed.109 In this context, it is relevant to highlight the work by Sawall, Neymeyr and collaborators, who have implemented methods into the software FACPACK.107
Contrary to soft models, hard models rely on physicochemical constraints, thereby retrieving parameters with fundamental significance. These methods provide very insightful results if the proposed model is plausible, such as equilibrium or kinetic rate constants. On the other hand, their main weakness is precisely that prior knowledge of the chemical system is expected; otherwise, data regression can evolve into cumbersome trial and error. An additional disadvantage of these models is that they cannot be applied with unmodelled noise or spectral artifacts like a baseline drift.19,101
Marquardt et al. developed significant work on hard models, first introducing what they called indirect hard modeling (IHM). This consisted of a nonlinear spectral model, using the Voigt functions to describe the convolution of peaks, and a linear calibration to predict the concentration from pure component weights. IHM allows accounting for nonlinear effects such as peak variations or spectral shifts.110 Within the IHM methods, they developed complemental hard modeling (CHM), especially indicated to obtain a single unidentified pure component spectrum in a mixture where the rest of the components is known. Additionally, hard modeling factor analysis (HMFA) has been proposed. In this method, all pure component spectra can be deduced from unordered data with correlated concentration profiles if there is at least one peak per component that does not occur in any other pure component spectrum (although this distinctive peak can be highly overlapped with others).100
4. Cases of operando deactivation studies in hydroformylation and asymmetric hydrogenation
Operando spectroscopic techniques, optionally assisted by mathematical deconvolution of the complex spectral data, have been applied to various molecularly catalyzed reactions. As an example, the aerobic oxidation of alcohols can be mentioned. Multi-technique operando studies by the groups of Stahl on the Pd-catalyzed111 and Brückner on the Cu-catalyzed aerobic oxidation112–114 have contributed significantly to a deeper mechanistic understanding of the reaction.14 Other reactions that are relevant in industrial application and for the transformation towards a sustainable chemical economy, like hydroformylation and asymmetric hydrogenation, were likewise subjected to extensive studies, also to investigate deactivation mechanisms. In this section a comprehensive description of the previously described operando spectroscopic techniques used to unravel deactivation mechanisms in hydroformylation and asymmetric hydrogenation are given.A lot of knowledge about deactivation processes in these two reactions has been gained from experiments using classic ex situ analysis. The systematic variation of reaction conditions, the use of distinct substituents115 or isotope labelling studies116,117 are helpful tools to deepen the insights gained from this strategy.
Kinetic investigations of deactivation reactions can be performed in the same manner, taking samples during the reaction or repeating the same experiment with different reaction times.115 Another possibility is the measurement of uptake of a specific gas, e.g. O2118 or H2.119–121 The observation of colour changes during the reaction has also been used to draw conclusions about the time of formation of inactive species.115,119
In contrast, the in situ and operando technologies described above allow for direct spectroscopic measurements while catalyst species are forming or decomposing and therefore provide particularly realistic insights to reaction and deactivation mechanisms of molecular catalysts.7,8 Generally, these measurements can be conducted by connecting a suitable analytical device to a reactor122 or by the performance of chemical reactions directly in measuring cells such as NMR tubes.123,124 Some relevant practical applications are presented in the following sections.
4.1. Rh-Catalyzed hydroformylation
The hydroformylation of olefins (see Fig. 2) is one of the most prominent examples for molecularly catalyzed reactions, yielding more than 10 million tons of aliphatic aldehydes per year. Phosphine modified Rh catalysts allow for high activities as well as selectivities and operate under mild reaction conditions. Other features of the reaction, such as high atom efficiency, availability of efficient recycling concepts, applicability to the conversion of sustainable resources and the suitability to combine a hydroformylation step with other reactions in a tandem catalysis to form tailormade value products, make hydroformylation a highly interesting field of modern research with regard to sustainable chemistry.6 | ||
| Fig. 2 Rh-Catalyzed Hydroformylation of a terminal olefin. | ||
Deactivation processes in hydroformylation reactions have been extensively reviewed.3–6,125 It is noticeable that even in very recent publications on the topic,6 the majority of cited works are from the years before 1990 and result from ex situ analytical investigations.
The main focus of most operando spectroscopic studies in hydroformylation catalysis has been on the investigation of mechanistic and kinetic aspects. Some authors126–128 also describe the observation of deactivation processes. The use of operando technology for the targeted investigation of deactivation mechanisms in hydroformylation catalysis is, however, rare. Appropriate examples highlighting the potential of such studies are illustrated below. A graphic summary of most discussed deactivation products and their role in the catalytic hydroformylation cycle (species 8 to 10) can be found in Fig. 3. Infrared spectroscopy – being especially powerful when it comes to monitoring metal carbonyls – is the most frequently applied technology, but there are also examples for the use of NMR spectroscopy.
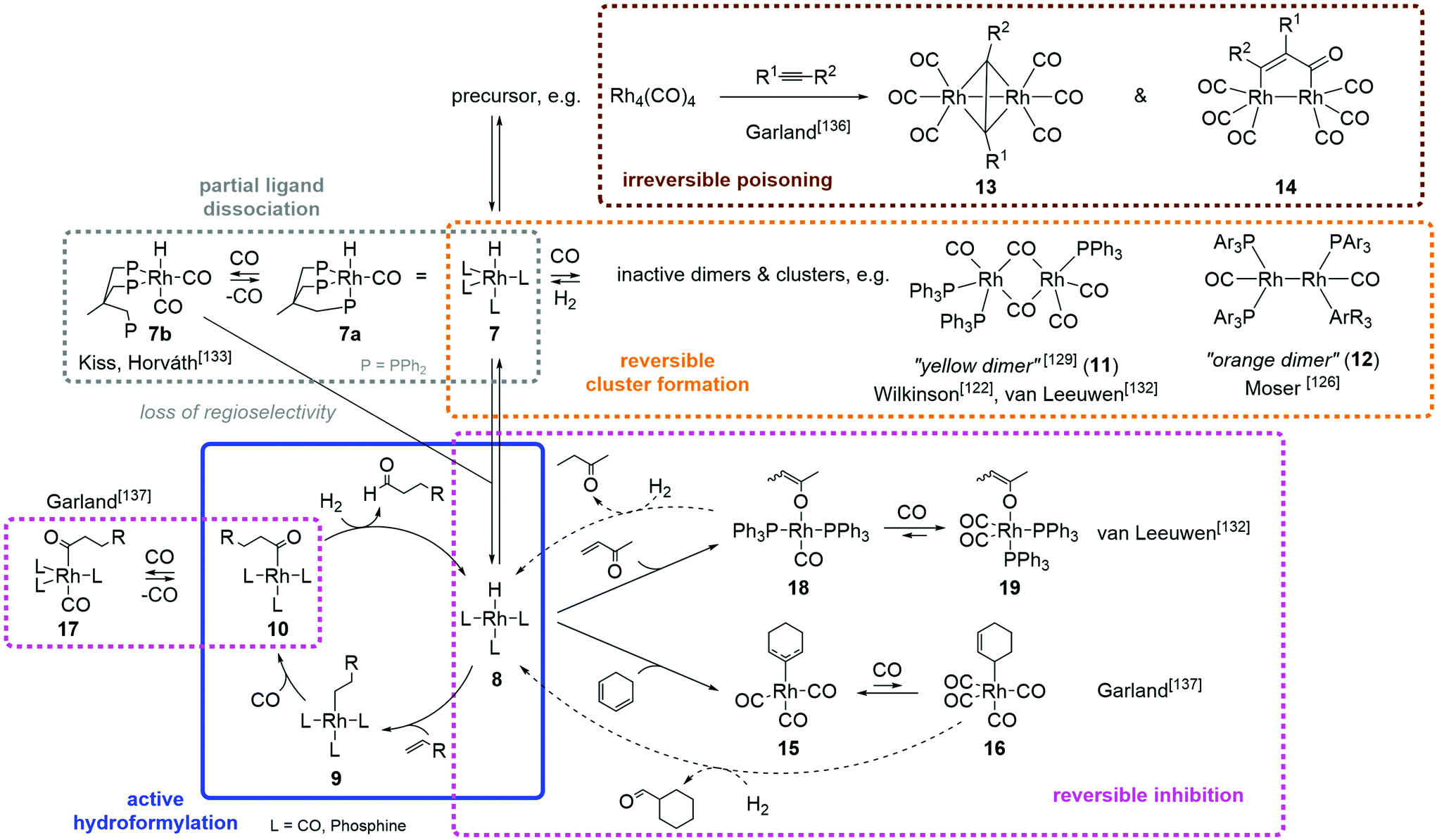 | ||
| Fig. 3 Examples of deactivated catalyst species and their formation pathways in the Rh catalyzed alkene hydroformylation observed by operando/in situ spectroscopic methods. The ligand L can be CO or different phosphines, in different ratios for every compound and varying for each system. | ||
One of the first studies using in situ spectroscopy to investigate hydroformylation catalysts was published in 1968 by the group of Wilkinson.122 They were able to identify species formed from RhH(CO)(PPh3)3 that are only stable under CO atmosphere by connecting an IR cell to an autoclave containing stoichiometric concentrations of the catalyst under atmospheric CO and/or H2 pressure. Among these species was a carbonyl-bridged dimeric Rh carbonyl phosphine species, the so-called “yellow dimer” (11).129 The formation of this species was reversible upon the addition of H2.
Moser placed a cylindrical internal reflection (CIR) crystal into a stainless-steel autoclave, allowing for measurements directly in the reactor.130 He was able to observe the formation of an inactive dimer 12 under 1-hexene hydroformylation conditions leading to deactivation of the system.126 Moderately high catalyst concentrations of 0.01 M rhodium were applied in this setup. The formation of inactive dimers and clusters has also been observed by other groups106,131,132 using operando transmission infrared spectroscopy.
Kiss and Horvath combined high-pressure IR and NMR spectroscopy to study a Rh catalyst system modified with the tripodal triphos ligand (CH3C(CH2PPh2)3). At typical hydroformylation conditions, they showed a reversible dissociation of one phosphine arm of the tripodal ligand in RhH(CO)[η3-CH3C(CH2PPh2)3] (7a) to form RhH(CO)2[η2-CH3C(CH2PPh2)3] (7b) under high pressure of CO. This process can lead to a loss of n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :iso selectivity in triphos-modified hydroformylation catalysis.133
:iso selectivity in triphos-modified hydroformylation catalysis.133
The group of Garland has greatly contributed to demonstrate the power of chemometrics in the field of operando spectroscopic monitoring of molecular catalyst by the development of the BTEM algorithm (cf. Section 3) and also the provision of illustrative application examples thereof.105,134,135 With regard to catalyst deactivation, the group investigated the influence of alkynes136 and dienes137 on the unmodified Rh4(CO)12 hydroformylation catalyst. Being more reactive towards the catalyst, these typical alkene feedstock impurities block the catalytic activity for alkene hydroformylation. In a first study,136 Rh4(CO)12 was reacted with 20 structurally varying terminal and internal alkynes under CO pressure. The forming catalyst species were monitored by connecting a high-pressure infrared transmission cell to the autoclave. The deconvolution of multicomponent spectra was achieved using Gauss–Lorenz areas as basic functions. It showed only two repeating absorbance patterns for the different alkynes, which were assigned to the dinuclear complexes 13 and 14 where R1 and R2 represent the substituents of the respective alkyne. Both complexes showed high stability in the presence of H2 gas, so that the catalyst deactivation can be interpreted as irreversible poisoning.136
In a similar work, Rh4(CO)12 was exposed to a series of conjugated dienes both in non-competitive situations and as impurities under cis-oct-4-ene hydroformylation conditions. Spectral deconvolution was carried out using a precursor version of BTEM, suggesting the presence of at least three mononuclear species which do not occur in the hydroformylation of uncontaminated alkenes. The authors suggested to assign them to an η3-allyl rhodium tricarbonyl species 15, a σ-allyl rhodium tetracarbonyl species 16 and an acyl rhodium tetracarbonyl species R'CORh(CO)4 (17, R' = alkenyl and/or formylalkyl).137
The group of van Leeuwen has developed a special autoclave to conduct operando transmission infrared spectroscopic measurements of fast catalytic reactions.47 In 2003, the group used this cell to investigate the deactivation of a Rh(acac)(CO)2/PPh3 hydroformylation catalyst system by alkynes, dienes (Fig. 4) and, in particular, enones.132 Just like alkynes and dienes, enones have a higher affinity to the rhodium catalyst than olefins, blocking its active hydroformylation sites. In phosphine modified catalyst systems, the aforementioned compounds undergo slow hydroformylation themselves, so that the catalyst deactivation is only temporary; it is reactivated as soon the impurity has been completely converted. Besides the formation of the inactive “yellow dimer”122 mentioned earlier, the formation of an inactive carboalkoxyrhodium species 19 formed from the reaction of RhH(PPh3)2(CO) (8) with 3-buten-2-one could be shown.
 | ||
| Fig. 4 Reversible inhibition of hydroformylation catalyst RhH(CO)2(PPh3)2 (7c) in the presence of 1,3-pentadiene monitored by operando HP-IR.132 Reproduced with permission from John Wiley & Sons, Ltd.138 | ||
Supplementary in situ1H, 13C and 31P NMR experiments conducted at low temperatures (80 °C to 20 °C) allowed for the identification of several intermediates of the deactivation process, also assisted by the simulation of spectra.132
The monitoring of catalyst deactivation in continuous flow was realized by our own group.55 A comparative study of different setups implementing both high pressure transmission and attenuated total reflection (ATR) IR spectroscopy in a batch setup and a continuously operated miniplant was presented. All four resulting setups were tested under authentic reaction conditions for the hydroformylation of 1-dodecene with a Rh/BiPhePhos catalyst. A special challenge in the continuous setup proved to be the realization of a very precise dosing strategy which is necessary for the spectral deconvolution via BTEM (cf. Section 3). While the sensitivity of the ATR-IR probe turned out not to be sensitive enough to monitor catalyst in the continuous setup, a spectral estimate for a metal organic species was detected with the other three setups. The comparison with simulated spectra obtained from DFT calculations suggested that it is actually caused by two or more metal organic phosphine species. The decay of these species can be connected with the simultaneous loss of regioselectivity of the hydroformylation reaction on the continuously operated miniplant, as can be seen in Fig. 5.
 | ||
| Fig. 5 Single component spectral estimates for the hydroformylation of 1-dodecene catalyzed by Rh/BiPhePhos monitored by operando high pressure transmission IR spectroscopy (left) and corresponding concentration profiles (right). The decline in regioselectivity occurs simultaneously with the loss of metal organic species 20.55 Reproduced and adapted with permission from the American Chemical Society.139 | ||
An illustrative example demonstrating the potential to enhance catalyst performance by in-depth understanding of the system's deactivation pathways and corresponding rational adaption was provided by the group of Börner. Phosphites represent a particularly active class of ligands in the n-regioselective hydroformylation. Unfortunately, they are prone to hydrolysis by nucleophilic cleavage of the P–O bond. To study the hydrolysis reactions of hybrid bidentate phosphite ligand 21 (Fig. 6), in situ NMR spectroscopy was performed directly in an NMR tube under elevated temperatures. The acylphosphite unit (red) of the ligand proved very sensitive towards water, while the phenolphosphite part (blue) is more stable. The detailed understanding of the hydrolysis pathway of 21 allowed for a rational modification of the backbone of the acyl unit, giving a ligand 22 of much higher hydrolysis stability.124 In a separate study, the group investigated the effect of substitution patterns on the reactivity and hydrolysis stability, showing the stabilizing effect of substituents in ortho-position, especially tert-butyl substituents.140 An additional surprising discovery was that the coordination to rhodium seemed to stabilize monodentate ligands, while bidentate ligands such as 22 were destabilized towards hydrolysis.124
 | ||
| Fig. 6 In situ 31P NMR spectra of the hydrolysis of 21 (100 equiv. of water, 120 °C) in the tetravalent oxide phosphorus (PV) region (left) and structure of modified and more stable ligand 22 (right).124 Reproduced and adapted with permission from the American Chemical Society.141 | ||
4.2. Asymmetric hydrogenation
The asymmetric hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO and CN bonds is considered the most relevant enantioselective reaction,142 awarded with a Nobel prize for Knowles143 and Noyori144 in 2001. Instead, the use of catalysis for asymmetric hydrogenation has made it possible to form chiral compounds through its inherent atom efficiency without creating waste. The high enantioselectivity further reduces the need of complex separation processes for racemic mixtures contributing to the sustainable nature of such processes.145
C, CO and CN bonds is considered the most relevant enantioselective reaction,142 awarded with a Nobel prize for Knowles143 and Noyori144 in 2001. Instead, the use of catalysis for asymmetric hydrogenation has made it possible to form chiral compounds through its inherent atom efficiency without creating waste. The high enantioselectivity further reduces the need of complex separation processes for racemic mixtures contributing to the sustainable nature of such processes.145
The first highly enantioselective catalysts used for the hydrogenation of CC bonds were cationic rhodium complexes containing chiral chelating diphosphine ligands, such as Diop (2,3-O-isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane).146 The mechanism of this reaction was deeply investigated in the late 1970's and early 1980's by Brown147–152 and Halpern153–155 using dehydroamino acids or esters as substrates.
In 1980 Halpern found that the main product of the hydrogenation of ethyl-(Z)-α-acetamidocinnamate with [Rh(chiraphos)][BF4] is N-acetyl-(R)-phenylalanine ethyl ester even though the major complex occurring in the reaction would yield N-acetyl-(S)-phenylalanine ethyl ester (Fig. 7). Later, Halpern and Landis studied the kinetics of the hydrogenation of methyl-(Z)-α-acetamidocinnamate using dipamp as ligand to discover the origin of the enantioselectivity. Besides classic ex situ measurements, they performed reactions in NMR tubes to measure the equilibrium of the diastereomers and to monitor the hydrogenation reaction of the substrate adduct. A stopped-flow UV-vis apparatus helped determining the association rates of the substrates and measurement of the H2 gas uptake under constant pressure was used for the determination of the catalytic hydrogenation rates. It was found that the reactivity of the diastereomers towards hydrogen is the origin of the enantioselectivity.155
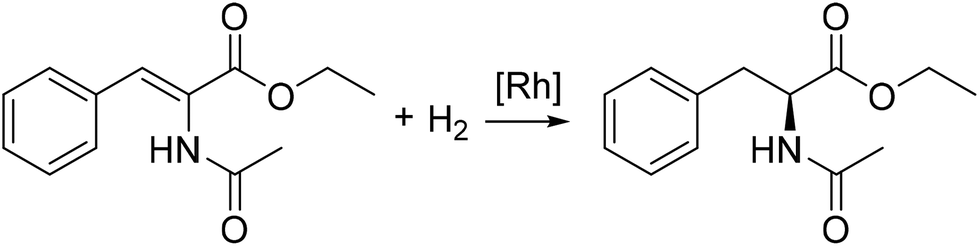 | ||
| Fig. 7 Asymmetric hydrogenation of ethyl-(Z)-α-acetamidocinnamate to the amino acid derivate N-acetyl-(S)-phenylalanine ethyl ester. | ||
Besides mechanistic investigations, overcoming catalyst deactivation was a major issue early on in the development of asymmetric hydrogenation reactions. Examples are the use of iridium as catalyst metal119 or the realization of the technical production of the herbicide (S)-metolachlor, which is nowadays the most important enantioselective process with a capacity of 10000 tons per year.2
Known deactivation mechanisms have been recapped in review literature.4,5 Next to classic ex situ investigations, measuring kinetics by monitoring gas uptake has helped to identify inhibition and deactivation processes.119,121 In particular, Heller et al. monitored the hydrogen uptake to investigate the induction period needed for the activation of catalyst precursors. In asymmetric hydrogenations precursors are commonly stabilized by diolefins like norbornadiene and cyclooctadiene. In a catalytic reaction with in situ formation of the active species, these diolefins need to be hydrogenated to form the active species. By monitoring the gas uptake in a reaction it was found that this induction period not only takes longer than assumed but also that it differs for the stabilizing diolefin used.121,156
In order to further examine these findings Heller et al. performed a Rh-catalyzed hydrogenation of a cyclooctadiene and norbornadiene mixture with a diop ligand in situ in an NMR cell designed for monitoring gas–liquid reactions. In this cell, constant bubbling of the reaction mixture with hydrogen is ensured without disturbance of the NMR signal by cutting a PTFE inlet pipe directly above the measurement region. Alternating 1H and 31P measurements were conducted to monitor both the formation of the monoalkenes and the complexes present. They showed that norbornadiene complexes (24) are formed first and afterwards, after hydrogenation of norbornadiene, the cyclooctadiene complexes (25) emerge (Fig. 8).123 In a subsequent study Heller et al. used the same set-up to investigate the effect in the asymmetric hydrogenation of prochiral olefins using five-membered diphosphine Rh-catalysts. With this type of catalyst the difference in reactivity can be up to three orders of magnitude due to very slow hydrogenation of cyclooctadiene.157 This difference has to be considered in the evaluation of catalyst activity of different catalysts since differences in TOFs might be caused by longer induction periods, not lower activity.
 | ||
| Fig. 8 Formation of the solvate complex which serves as starting point in the asymmetric hydrogenation and which was studied by Heller et al. (a). 31P NMR spectra of the complexes 24 and 25 which are formed consecutively (b).156 Reproduced and adapted by permission from John Wiley & Sons, Ltd.158 | ||
The studies by the group of Heller show how measurements of the gas uptake and conducting reactions in specifically designed NMR cells contributed to a deeper understanding of the nature of catalyst activation. Meanwhile, another operando study regarding the deactivation of Noyori's [(mesitylene)((R,R)-TsDPEN)-RuCl] catalyst (25) in the asymmetric transfer hydrogenation of acetophenone with isopropanol (Fig. 9a, blue) can serve as an example for the use of 1H NMR and selective excitation techniques.159 The Noyori type catalyst has many advantages, among them a high enantioselectivity at high reaction rates, and can be used with a variety of substances and auxiliary bases. It is, however, also known for deactivation.5,160
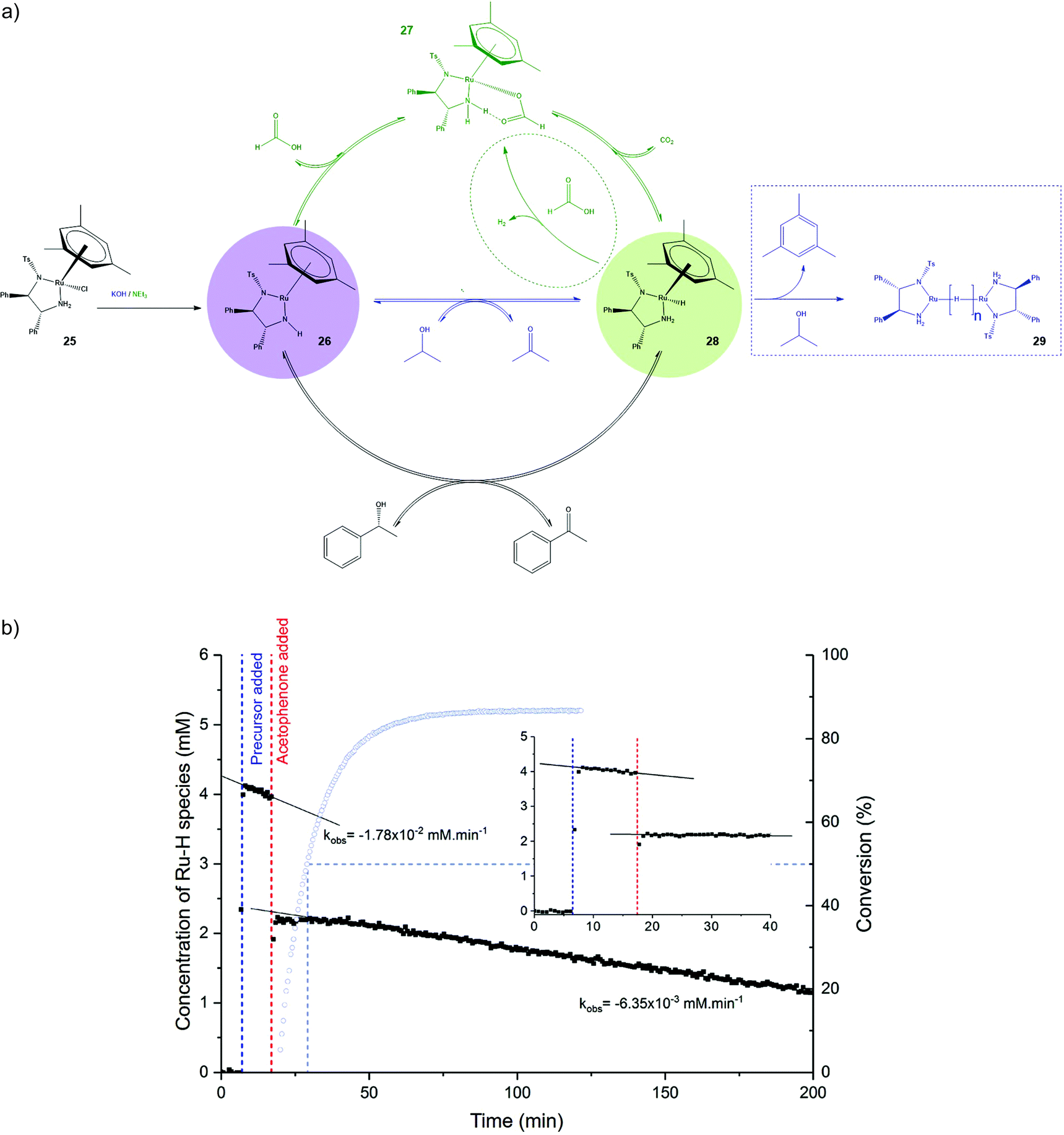 | ||
| Fig. 9 (a) Catalytic cycle of the asymmetric transfer hydrogenation of acetophenone with isopropanol (blue) and formic acid (green) as H-donor including the proposed deactivation pathways (dashed), (b) concentration of in-cycle Ru–H species (28) (black) and conversion (blue) over time. The colored circles refer to the colour code introduced in Fig. 1.159,162 Reproduced and adapted with permission from the American Chemical Society.163 | ||
For this reason, Hintermair and coworkers used an established FlowNMR operando setup32 for the investigation of transfer hydrogenation of acetophenone with this catalyst.159 Starting point of the investigation was a Variable Time Normalization Analysis (VTNA)161 on catalyst concentration which showed deviations in a range between 50% conversion and equilibrium conversion, indicating catalyst deactivation at this part of the reaction. In order to gain insight into the deactivation mechanism, Hintermair et al. used a selective excitation method to monitor the concentration of the known in-cycle Ru–H species 28. By these means, its signal was amplified by >1000 times while allowing quantitative evaluation by linking the receiver gain to the integral of the hydride.
Following the course of the reaction by normal 1H NMR measurements while tracking the amount of species 28 showed a decline in its concentration before addition of acetophenone and again a smaller decay when 50% conversion were reached (Fig. 9b), which corresponds to the results of the VTNA. The decay before addition of substrate indicates deactivation occurring from 28.
Conducting the hydrogenation reaction in deuterated solvents a liberation of mesitylene could be observed. As the rate of formation of free mesitylene corresponds to the decline of concentration of the Ru–H species and the chemical shifts and T1 values of the deactivation product are in accordance with hydride-bridged dimers, Hintermair et al. propose that the formation of hydride-bridged dimers (29) by loss of mesitylene ligand is a major deactivation process of Noyori's type catalysts (Fig. 9a).
While this shows the power of a flow NMR setup in combination with signal enhancement techniques for operando studies, a consecutive work by the same group can be used as a practical example of the use of multiple complementing techniques.162
The authors investigated the same reaction but using formic acid as H-donor instead of isopropanol. The flow NMR setup from the previous study was expanded by a HPLC, a UV-vis spectrometer and a gas phase mass-spectrometer installed on the reactor. These techniques made a complete monitoring of the reactants possible. The gas phase mass spectrometer allowed tracking of the formation of CO2 and H2, the HPLC was used for measuring the enantiomeric excess, 1H NMR for the determination of conversion, dissolved H2164 and again the Ru–H species 28. UV–vis spectroscopy allowed tracking of the intermediate 26 by a characteristic absorbance band.162
The authors found a continuous formation of H2 throughout the reaction and figured out that the formate species 27 is the major, stable intermediate in the reaction with formic acid as species 26 and 28 make up less than 2% and maximum 40% respectively of the total catalyst amount. Using this information about the catalytic species in the reaction system Hintermair et al. report that three reactions compete from species 28: a re-insertion of CO2 forming species 27, hydrogen transfer to form the desired product and protonation to H2, which consumes the substrate formic acid and is thus a side reaction lowering the reaction performance.162
5. Conclusions and outlook
In this review, key techniques have been summarized to study molecular catalyst under reaction conditions in an operando approach in order to develop green processes. Spectroscopic methods like NMR, IR, EPR, Raman, UV-Vis or ESI-MS can individually give access to specific structural data that is important to gain information on the low concentrated molecular catalyst in a reaction system. To harvest the full information out of the spectroscopic data, different mathematical tools were introduced and compared. Most of them were used to give mechanistic insights of catalytic systems but can also be applied to give information on deactivation mechanisms also in continuously operated systems. These mathematical models differ mostly on initial constraints and therefore hard modelling methods need more prior knowledge of the system.In two cases, the state of the art of operando studies in the investigation of deactivation in hydroformylation and asymmetric hydrogenation were summarized. In hydroformylation, mostly IR and NMR studies have been published to show resting states and dormant species. Also poisoning effects for the molecular rhodium catalyst have been investigated under reaction conditions. Deactivation processes have been described for the hydroformylation in a miniplant setup. The asymmetric hydrogenation was studied in detail by NMR techniques to detect the activation and deactivation of the molecular catalyst by tracing ligand structures.
These examples show the importance and potential of these studies to describe apart from the ideal catalytic cycle also the fate of the molecular catalyst under reaction conditions. The activity and the lifetime of a catalyst is crucial for scaling and applicability in a process. Also, the design of more stable catalysts due to the operando data is a rare exercise in the molecular catalysis community. This technique will contribute to design sustainable processes by allowing to apply stable and selective molecular catalyst at mild reaction conditions.
The data of operando experiments highly depend on finding the best analytical method to detect the catalyst features at real reaction conditions. A challenge, in addition to high temperatures and pressures, is the usually low catalyst concentration that causes relatively small signals in comparison to substrates, products, solvents etc. Therefore, mathematical tools are handy to detect even traces by assigning single component spectra if needed.
In the future, the merging of the broad variety of spectroscopic techniques has to be applied to provide a deeper insight into the molecular catalyst at work. This data will show deactivation mechanisms that are at the moment only theory or are unknown. If these combined data are fed to algorithms, new insights can be gained and stable catalyst systems can be designed in the future. The development of data formats for operando data and wide while cheap availability of spectroscopic techniques play an important role to push these techniques into the production field. Also the ever improving processing of these vast data amounts makes it more attractive for the industry to work and learn from this approach.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Max Planck Society is gratefully acknowledged for financial support. The authors gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – Exzellenzcluster 2186 “The Fuel Science Center” ID: 39091983. Katrin Köhnke thanks the networking program ‘Sustainable Chemical Synthesis 2.0’ (SusChemSys 2.0) for the support and fruitful discussions across disciplines, and the Fonds der Chemischen Industrie and the NEUMAN & ESSER Stiftung der Familie Peters for financial support. Open Access funding provided by the Max Planck Society.References
- P. C. J. Kamer, D. Vogt and J. W. Thybaut, Contemporary Catalysis: Science, Technology, and Applications, RSC Publishing, Cambridge, 2017 Search PubMed.
- A. Behr and P. Neubert, Applied Homogeneous Catalysis, Wiley-VCH, Weinheim, Germany, 2012 Search PubMed.
- P. W. N. M. van Leeuwen, Decomposition Pathways of Homogeneous Catalysts, Appl. Catal., A, 2001, 212(1–2), 61–81 CrossRef CAS.
- P. W. N. M. Van Leeuwen and J. C. Chadwick, Homogeneous Catalysts: Activity – Stability – Deactivation, Wiley-VCH, Weinheim, 2011 Search PubMed.
- R. H. Crabtree, Deactivation in Homogeneous Transition Metal Catalysis: Causes, Avoidance, and Cure, Chem. Rev., 2015, 115(1), 127–150 CrossRef CAS PubMed.
- A. Börner and R. Franke, Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis, Wiley-VCH, Weinheim, Germany, 2016 Search PubMed.
- M. A. Bañares, M. O. Guerrero-Pérez, J. L. G. Fierro and G. G. Cortez, Raman spectroscopy during catalytic operations with on-line activity measurement (operando spectroscopy): a method for understanding the active centres of cations supported on porous materials, J. Mater. Chem., 2002, 12(11), 3337–3342 RSC.
- M. A. Bañares and I. E. Wachs, Molecular structures of supported metal oxide catalysts under different environments, J. Raman Spectrosc., 2002, 33(5), 359–380 CrossRef.
- A. Chakrabarti, M. E. Ford, D. Gregory, R. R. Hu, C. J. Keturakis, S. Lwin, Y. D. Tang, Z. Yang, M. H. Zhu, M. A. Bañares and I. E. Wachs, A decade plus of operando spectroscopy studies, Catal. Today, 2017, 283, 27–53 CrossRef CAS.
- X. Li, X. Yang, J. Zhang, Y. Huang and B. Liu, In Situ/Operando Techniques for Characterization of Single-Atom Catalysts, ACS Catal., 2019, 9(3), 2521–2531 CrossRef CAS.
- N. Weder, B. Probst, L. Severy, R. J. Fernandez-Teran, J. Beckord, O. Blacque, S. D. Tilley, P. Hamm, J. Osterwalder and R. Alberto, Mechanistic insights into photocatalysis and over two days of stable H-2 generation in electrocatalysis by a molecular cobalt catalyst immobilized on TiO2, Catal. Sci. Technol., 2020, 10(8), 2549–2560 RSC.
- O. Diebolt, P. W. N. M. van Leeuwen and P. C. J. Kamer, Operando Spectroscopy in Catalytic Carbonylation Reactions, ACS Catal., 2012, 2(11), 2357–2370 CrossRef CAS.
- D. Selent and D. Heller, In situ Techniques for Homogeneous Catalysis, in Catalysis: From Principles to Applications, ed. M. Beller, A. Renken and R. A. van Santen, Wiley-VCH, Weinheim, 2012, pp. 465–492 Search PubMed.
- R. Chung and J. E. Hein, The More, The Better: Simultaneous In Situ Reaction Monitoring Provides Rapid Mechanistic and Kinetic Insight, Top. Catal., 2017, 60(8), 594–608 CrossRef CAS.
- R. Franke, D. Selent and A. Börner, Applied Hydroformylation, Chem. Rev., 2012, 112(11), 5675–5732 CrossRef CAS PubMed.
- G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Catalytic (de)hydrogenation promoted by non-precious metals – Co, Fe and Mn: recent advances in an emerging field, Chem. Soc. Rev., 2018, 47(4), 1459–1483 RSC.
- T. A. Faßbach, A. J. Vorholt and W. Leitner, The Telomerization of 1,3-Dienes – A Reaction Grows Up, ChemCatChem, 2019, 11(4), 1153–1166 CrossRef.
- C. Zhu, J. Liu, M.-B. Li and J.-E. Bäckvall, Palladium-catalyzed oxidative dehydrogenative carbonylation reactions using carbon monoxide and mechanistic overviews, Chem. Soc. Rev., 2020, 49(2), 341–353 RSC.
- A. de Juan and R. Tauler, Multivariate Curve Resolution (MCR) from 2000: Progress in Concepts and Applications, Crit. Rev. Anal. Chem., 2006, 36(3–4), 163–176 CrossRef CAS.
- A. de Juan, J. Jaumot and R. Tauler, Multivariate Curve Resolution (MCR). Solving the mixture analysis problem, Anal. Methods, 2014, 6(14), 4964–4976 RSC.
- M. Garland, Combining operando spectroscopy with experimental design, signal processing and advanced chemometrics: State-of-the-art and a glimpse of the future, Catal. Today, 2010, 155(3), 266–270 CrossRef CAS.
- I. del Río, C. Claver and P. W. N. M. van Leeuwen, On the Mechanism of the Hydroxycarbonylation of Styrene with Palladium Systems, Eur. J. Inorg. Chem., 2001, 2719–2738 CrossRef.
- T. Senthamarai, K. Murugesan, J. Schneidewind, N. V. Kalevaru, W. Baumann, H. Neumann, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Simple ruthenium-catalyzed reductive amination enables the synthesis of a broad range of primary amines, Nat. Commun., 2018, 9(1), 4123 CrossRef.
- V. de la Fuente, M. Waugh, G. R. Eastham, J. A. Iggo, S. Castillón and C. Claver, Phosphine Ligands in the Palladium-Catalysed Methoxycarbonylation of Ethene: Insights into the Catalytic Cycle through an HP NMR Spectroscopic Study, Chem. – Eur. J., 2010, 16(23), 6919–6932 CrossRef CAS PubMed.
- J. Wolowska, G. R. Eastham, B. T. Heaton, J. A. Iggo, C. Jacob and R. Whyman, The effect of mechanistic pathway on activity in the Pd and Pt catalysed methoxycarbonylation of ethene, Chem. Commun., 2002, 2784–2785 RSC.
- A. L. Casado and P. Espinet, On the Configuration Resulting from Oxidative Addition of RX to Pd(PPh3)4 and the Mechanism of the cis-to-trans Isomerization of [PdRX(PPh3)2] Complexes (R = Aryl, X = Halide), Organometallics, 1998, 17(5), 954–959 CrossRef CAS.
- I. T. Horvath and J. M. Millar, NMR under high gas pressure, Chem. Rev., 1991, 91(7), 1339–1351 CrossRef CAS.
- D. A. Foley, A. L. Dunn and M. T. Zell, Reaction monitoring using online vs tube NMR spectroscopy: seriously different results, Magn. Reson. Chem., 2016, 54(6), 451–456 CrossRef CAS PubMed.
- J. A. Iggo, D. Shirley and N. C. Tong, High pressure NMR flow cell for the insitu study of homogeneous catalysis, New J. Chem., 1998, 22(10), 1043–1045 RSC.
- N. J. Beach, S. M. M. Knapp and C. R. Landis, A reactor for high-throughput high-pressure nuclear magnetic resonance spectroscopy, Rev. Sci. Instrum., 2015, 86(10), 104101 CrossRef CAS PubMed.
- A. B. Jones, G. C. Lloyd-Jones and D. Uhrin, SHARPER Reaction Monitoring: Generation of a Narrow Linewidth NMR Singlet, without X-Pulses, in an Inhomogeneous Magnetic Field, Anal. Chem., 2017, 89(18), 10013–10021 CrossRef CAS PubMed.
- A. M. R. Hall, J. C. Chouler, A. Codina, P. T. Gierth, J. P. Lowe and U. Hintermair, Practical aspects of real-time reaction monitoring using multi-nuclear high resolution FlowNMR spectroscopy, Catal. Sci. Technol., 2016, 6(24), 8406–8417 RSC.
- A. Bara-Estaún, C. L. Lyall, J. P. Lowe, P. G. Pringle, P. C. J. Kamer, R. Franke and U. Hintermair, Multi-Nuclear, High-pressure, Operando FlowNMR Spectroscopic Study of Rh/PPh3 – Catalysed Hydroformylation of 1-Hexene, Faraday Discuss., 2020, 229, 422–442 RSC.
- B. Blümich and K. Singh, Desktop NMR and Its Applications From Materials Science To Organic Chemistry, Angew. Chem., Int. Ed., 2018, 57(24), 6996–7010 CrossRef PubMed.
- F. Dalitz, M. Cudaj, M. Maiwald and G. Guthausen, Process and reaction monitoring by low-field NMR spectroscopy, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 60, 52–70 CrossRef CAS PubMed.
- K. Singh and B. Blümich, NMR spectroscopy with compact instruments, TrAC, Trends Anal. Chem., 2016, 83, 12–26 CrossRef CAS.
- E. Danieli, J. Perlo, A. L. L. Duchateau, G. K. M. Verzijl, V. M. Litvinov, B. Blümich and F. Casanova, On-Line Monitoring of Chemical Reactions by using Bench-Top Nuclear Magnetic Resonance Spectroscopy, ChemPhysChem, 2014, 15(14), 3060–3066 CrossRef CAS.
- W. Lubitz, F. Lendzian and R. Bittl, Radicals, Radical Pairs and Triplet States in Photosynthesis, Acc. Chem. Res., 2002, 35(5), 313–320 CrossRef CAS PubMed.
- J. Messinger, J. H. A. Nugent and M. C. W. Evans, Detection of an EPR Multiline Signal for the S0* State in Photosystem II, Biochemistry, 1997, 36(37), 11055–11060 CrossRef CAS PubMed.
- E. Carter and D. M. Murphy, The Role of Low Valent Transition Metal Complexes in Homogeneous Catalysis: An EPR Investigation, Top. Catal., 2015, 58(12), 759–768 CrossRef CAS.
- M. Goswami, A. Chirila, C. Rebreyend and B. de Bruin, EPR Spectroscopy as a Tool in Homogeneous Catalysis Research, Top. Catal., 2015, 58(12–13), 719–750 CrossRef CAS.
- D. Hollmann, F. Gärtner, R. Ludwig, E. Barsch, H. Junge, M. Blug, S. Hoch, M. Beller and A. Brückner, Insights into the Mechanism of Photocatalytic Water Reduction by DFT-Supported In Situ EPR/Raman Spectroscopy, Angew. Chem., Int. Ed., 2011, 50(43), 10246–10250 CrossRef CAS PubMed.
- T. Risse, D. Hollmann and A. Brückner, In situ electron paramagnetic resonance (EPR) – a unique tool for analysing structure and reaction behaviour of paramagnetic sites in model and real catalysts, in Catalysis, 2015, ch. 1, pp. 1–32 Search PubMed.
- J. Rabeah, M. Bauer, W. Baumann, A. E. C. McConnell, W. F. Gabrielli, P. B. Webb, D. Selent and A. Brückner, Formation, Operation and Deactivation of Cr Catalysts in Ethylene Tetramerization Directly Assessed by Operando EPR and XAS, ACS Catal., 2012, 3(1), 95–102 CrossRef.
- A. Haynes, The Use of High Pressure Infrared Spectroscopy to Study Catalytic Mechanisms, in Mechanisms in Homogeneous Catalysis, Wiley-VCH, Weinheim, Germany, 2005, pp. 107–150 Search PubMed.
- R. Whyman, In Situ Spectroscopic Studies in Homogeneous Catalysis, in Homogeneous Transition Metal Catalyzed Reactions, American Chemical Society, 1992, vol. 230, pp. 19–31 Search PubMed.
- P. C. J. Kamer, A. van Rooy, G. C. Schoemaker and P. W. N. M. van Leeuwen, In situ mechanistic studies in rhodium catalyzed hydroformylation of alkenes, Coord. Chem. Rev., 2004, 248(21–24), 2409–2424 CrossRef CAS.
- T. Ghaffar, H. Adams, P. M. Maitlis, A. Haynes, T. Ghaffar, H. Adams, G. J. Sunley and M. J. Baker, Spectroscopic identification and reactivity of [Ir(CO)3I2Me], a key reactive intermediate in iridium catalysed methanol carbonylation, Chem. Commun., 1998, 1023–1024 RSC.
- J. J. Bravo-Suárez and P. D. Srinivasan, Design characteristics of in situ and operando ultraviolet-visible and vibrational spectroscopic reaction cells for heterogeneous catalysis, Catal. Rev., 2017, 59(4), 1–151 CrossRef.
- L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, New Phosphacyclic Diphosphines for Rhodium-Catalyzed Hydroformylation, Organometallics, 1999, 18(23), 4765–4777 CrossRef CAS.
- Y. Raichlin and A. Katzir, Fiber-Optic Evanescent Wave Spectroscopy in the Middle Infrared, Appl. Spectrosc., 2008, 62(2), 55A–72A CrossRef CAS PubMed.
- C. B. Minnich, P. Buskens, H. C. Steffens, P. S. Bäuerlein, L. N. Butvina, L. Küpper, W. Leitner, M. A. Liauw and L. Greiner, Highly Flexible Fibre-Optic ATR-IR Probe for Inline Reaction Monitoring, Org. Process Res. Dev., 2007, 11(1), 94–97 CrossRef CAS.
- Mettler Toledo ReactIR In-situ Reaction Analysis, https://www.mt.com/in/en/home/products/L1_AutochemProducts/ReactIR.html, (accessed June 2021).
- ARCoptix Compact FTIR for operation with a fiber output and input, http://www.arcoptix.com/ftir_fiber_coupled.htm, (accessed June 2021).
- J. M. Dreimann, E. Kohls, H. F. W. Warmeling, M. Stein, L. F. Guo, M. Garland, T. N. Dinh and A. J. Vorholt, In Situ Infrared Spectroscopy as a Tool for Monitoring Molecular Catalyst for Hydroformylation in Continuous Processes, ACS Catal., 2019, 9(5), 4308–4319 CrossRef CAS.
- T. A. Hamlin and N. E. Leadbeater, Raman spectroscopy as a tool for monitoring mesoscale continuous-flow organic synthesis: Equipment interface and assessment in four medicinally-relevant reactions, Beilstein J. Org. Chem., 2013, 9, 1843–1852 CrossRef PubMed.
- V. Strobel, J. J. Schuster, A. S. Braeuer, L. K. Vogt, H. Junge and M. Haumann, Shining light on low-temperature methanol aqueous-phase reforming using homogeneous Ru-pincer complexes – operando Raman-GC studies, React. Chem. Eng., 2017, 2(3), 390–396 RSC.
- A. Seegerer, P. Nitschke and R. M. Gschwind, Combined In Situ Illumination-NMR-UV/Vis Spectroscopy: A New Mechanistic Tool in Photochemistry, Angew. Chem., Int. Ed., 2018, 57(25), 7493–7497 CrossRef CAS PubMed.
- J. N. Hamann, M. Hermsen, A.-C. Schmidt, S. Krieg, J. Schießl, D. Riedel, J. H. Teles, A. Schäfer, P. Comba, A. S. K. Hashmi and T. Schaub, Selective Decomposition of Cyclohexyl Hydroperoxide using Homogeneous and Heterogeneous CrVI Catalysts: Optimizing the Reaction by Evaluating the Reaction Mechanism, ChemCatChem, 2018, 10(13), 2755–2767 CrossRef CAS.
- C. S. Schnohr and M. C. Ridgway, Introduction to X-Ray Absorption Spectroscopy, in X-Ray Absorption Spectroscopy of Semiconductors, ed. C. S. Schnohr and M. C. Ridgway, Springer, Berlin, Heidelberg, Germany, 2015, pp. 1–26 Search PubMed.
- K. Nomura, T. Mitsudome, K. Tsutsumi and S. Yamazoe, Solution XAS Analysis for Exploring the Active Species in Homogeneous Vanadium Complex Catalysis, J. Phys. Soc. Jpn., 2018, 87(6), 061014 CrossRef.
- S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Reactivity of Surface Species in Heterogeneous Catalysts Probed by In Situ X-ray Absorption Techniques, Chem. Rev., 2013, 113(3), 1736–1850 CrossRef CAS PubMed.
- D. Koziej and S. DeBeer, Application of Modern X-ray Spectroscopy in Chemistry—Beyond Studying the Oxidation State, Chem. Mater., 2017, 29(17), 7051–7053 CrossRef CAS.
- S. N. MacMillan and K. M. Lancaster, X-ray Spectroscopic Interrogation of Transition-Metal-Mediated Homogeneous Catalysis: Primer and Case Studies, ACS Catal., 2017, 7(3), 1776–1791 CrossRef CAS.
- J. N. Jaworski, C. V. Kozack, S. J. Tereniak, S. M. M. Knapp, C. R. Landis, J. T. Miller and S. S. Stahl, Operando Spectroscopic and Kinetic Characterization of Aerobic Allylic C–H Acetoxylation Catalyzed by Pd(OAc)2/4,5-Diazafluoren-9-one, J. Am. Chem. Soc., 2019, 141(26), 10462–10474 CrossRef CAS PubMed.
- G. J. Sherborne and B. N. Nguyen, Recent XAS studies into Homogeneous metal catalyst in fine chemical and pharmaceutical syntheses, Chem. Cent. J., 2015, 9, 37 CrossRef PubMed.
- H. Asakura, T. Shishido and T. Tanaka, In situ time-resolved XAFS study of the reaction mechanism of bromobenzene homocoupling mediated by [Ni(cod)(bpy)], J. Phys. Chem. A, 2012, 116(16), 4029–4034 CrossRef CAS PubMed.
- K. P. J. Gustafson, A. Guðmundsson, É. G. Bajnóczi, N. Yuan, X. Zou, I. Persson and J.-E. Bäckvall, In Situ Structural Determination of a Homogeneous Ruthenium Racemization Catalyst and Its Activated Intermediates Using X-Ray Absorption Spectroscopy, Chem. – Eur. J., 2020, 26(15), 3411–3419 CrossRef CAS PubMed.
- R. Ortega, A. Carmona, I. Llorens and P. L. Solari, X-ray absorption spectroscopy of biological samples. A tutorial, J. Anal. At. Spectrom., 2012, 27(12), 2054–2065 RSC.
- J. S. McIndoe and K. L. Vikse, Assigning the ESI mass spectra of organometallic and coordination compounds, J. Mass Spectrom., 2019, 54(5), 466–479 CrossRef CAS PubMed.
- K. L. Vikse, Z. Ahmadi and J. Scott McIndoe, The application of electrospray ionization mass spectrometry to homogeneous catalysis, Coord. Chem. Rev., 2014, 279, 96–114 CrossRef CAS.
- L. P. E. Yunker, Z. Ahmadi, J. R. Logan, W. Wu, T. Li, A. Martindale, A. G. Oliver and J. S. McIndoe, Real-Time Mass Spectrometric Investigations into the Mechanism of the Suzuki–Miyaura Reaction, Organometallics, 2018, 37(22), 4297–4308 CrossRef CAS.
- S. M. Jackson, D. M. Chisholm, J. S. McIndoe and L. Rosenberg, Using NMR and ESI-MS to Probe the Mechanism of Silane Dehydrocoupling Catalyzed by Wilkinson's Catalyst, Eur. J. Inorg. Chem., 2011, 2011(3), 327–330 CrossRef.
- M. A. O. Volland, C. Adlhart, C. A. Kiener, P. Chen and P. Hofmann, Catalyst Screening by Electrospray Ionization Tandem Mass Spectrometry: Hofmann Carbenes for Olefin Metathesis, Chem. – Eur. J., 2001, 7(21), 4621–4632 CrossRef CAS.
- L. S. Santos, Online Mechanistic Investigations of Catalyzed Reactions by Electrospray Ionization Mass Spectrometry: A Tool to Intercept Transient Species in Solution, Eur. J. Org. Chem., 2007, 235–253 Search PubMed.
- L. S. Santos, G. B. Rosso, R. A. Pilli and M. N. Eberlin, The mechanism of the Stille reaction investigated by electrospray ionization mass spectrometry, J. Org. Chem., 2007, 72(15), 5809–5812 CrossRef CAS PubMed.
- V. Carrasco-Sanchez, M. J. Simirgiotis and L. S. Santos, The Morita-Baylis-Hillman Reaction: Insights into Asymmetry and Reaction Mechanisms by Electrospray Ionization Mass Spectrometry, Molecules, 2009, 14(10), 3989–4021 CrossRef CAS PubMed.
- J. Luo, A. G. Oliver and J. S. McIndoe, A detailed kinetic analysis of rhodium-catalyzed alkyne hydrogenation, Dalton Trans., 2013, 42(31), 11312–11318 RSC.
- P. Chen, Electrospray Ionization Tandem Mass Spectrometry in High-Throughput Screening of Homogeneous Catalysts, Angew. Chem., Int. Ed., 2003, 42(25), 2832–2847 CrossRef CAS PubMed.
- W. W. Christie and X. Han, Quantification of lipid molecular species by electrospray ionization mass spectrometry, in Lipid Analysis, Elsevier, 2012, pp. 365–392 Search PubMed.
- G. Loos, A. Van Schepdael and D. Cabooter, Quantitative mass spectrometry methods for pharmaceutical analysis, Philos. Trans. R. Soc., A, 2016, 374(2079), 20150366 CrossRef PubMed.
- R. Grauke, R. Schepper, J. Rabeah, R. Schoch, U. Bentrup, M. Bauer and A. Brückner, Impact of Al Activators on Structure and Catalytic Performance of Cr Catalysts in Homogeneous Ethylene Oligomerization – A Multitechnique in situ/operando Study, ChemCatChem, 2020, 12(4), 1025–1035 CrossRef CAS.
- S. J. Tinnemans, J. G. Mesu, K. Kervinen, T. Visser, T. A. Nijhuis, A. M. Beale, D. E. Keller, A. M. J. van der Eerden and B. M. Weckhuysen, Combining operando techniques in one spectroscopic-reaction cell: New opportunities for elucidating the active site and related reaction mechanism in catalysis, Catal. Today, 2006, 113(1–2), 3–15 CrossRef CAS.
- J. M. Savéant, Molecular catalysis of electrochemical reactions. Mechanistic aspects, Chem. Rev., 2008, 108(7), 2348–2378 CrossRef PubMed.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Evaluation of homogeneous electrocatalysts by cyclic voltammetry, Inorg. Chem., 2014, 53(19), 9983–10002 CrossRef CAS PubMed.
- K. S. Joya and X. Sala, In situ Raman and surface-enhanced Raman spectroscopy on working electrodes: spectroelectrochemical characterization of water oxidation electrocatalysts, Phys. Chem. Chem. Phys., 2015, 17(33), 21094–21103 RSC.
- K. J. Lee, N. Elgrishi, B. Kandemir and J. L. Dempsey, Electrochemical and spectroscopic methods for evaluating molecular electrocatalysts, Nat. Rev. Chem., 2017, 1(5), 0039 CrossRef CAS.
- R. Francke, B. Schille and M. Roemelt, Homogeneously Catalyzed Electroreduction of Carbon Dioxide-Methods, Mechanisms, and Catalysts, Chem. Rev., 2018, 118(9), 4631–4701 CrossRef CAS PubMed.
- C. W. Machan, Recent advances in spectroelectrochemistry related to molecular catalytic processes, Curr. Opin. Electrochem., 2019, 15, 42–49 CrossRef CAS.
- E. Boutin, L. Merakeb, B. Ma, B. Boudy, M. Wang, J. Bonin, E. Anxolabéhère-Mallart and M. Robert, Molecular catalysis of CO2 reduction: recent advances and perspectives in electrochemical and light-driven processes with selected Fe, Ni and Co aza macrocyclic and polypyridine complexes, Chem. Soc. Rev., 2020, 49(16), 5772–5809 RSC.
- A. de Juan, M. Maeder, M. Martínez and R. Tauler, Combining hard- and soft-modelling to solve kinetic problems, Chemom. Intell. Lab. Syst., 2000, 54(2), 123–141 CrossRef CAS.
- E. R. Malinowski, Window factor analysis: Theoretical derivation and application to flow injection analysis data, J. Chemom., 1992, 6(1), 29–40 CrossRef CAS.
- R. Manne, H. Shen and Y. Liang, Subwindow factor analysis, Chemom. Intell. Lab. Syst., 1999, 45(1), 171–176 CrossRef CAS.
- Y. Z. Liang, O. M. Kvalheim, H. R. Keller, D. L. Massart, P. Kiechle and F. Erni, Heuristic evolving latent projections: resolving two-way multicomponent data. 2. Detection and resolution of minor constituents, Anal. Chem., 1992, 64(8), 946–953 CrossRef CAS.
- E. R. Malinowski, Obtaining the key set of typical vectors by factor analysis and subsequent isolation of component spectra, Anal. Chim. Acta, 1982, 134, 129–137 CrossRef CAS.
- X. Gao, J. Ma, F. Ruan, T. Zhang and H. Li, In situ ATR-FTIR combined with SIMPLISMA algorithm to investigate the synthesis mechanism of 4-amino-3,5-dimethyl pyrazole, Chem. Res. Chin. Univ., 2016, 32(6), 985–991 CrossRef CAS.
- D. Bu and C. W. Brown, Self-Modeling Mixture Analysis by Interactive Principal Component Analysis, Appl. Spectrosc., 2000, 54(8), 1214–1221 CrossRef CAS.
- E. Widjaja, C. Li, W. Chew and M. Garland, Band-Target Entropy Minimization. A Robust Algorithm for Pure Component Spectral Recovery. Application to Complex Randomized Mixtures of Six Components, Anal. Chem., 2003, 75(17), 4499–4507 CrossRef CAS PubMed.
- C. G. Bertinetto and A. de Juan, Systematic comparison and potential combination between multivariate curve resolution–alternating least squares (MCR-ALS) and band-target entropy minimization (BTEM), J. Chemom., 2018, 32(6), e3000 CrossRef.
- E. Kriesten, D. Mayer, F. Alsmeyer, C. B. Minnich, L. Greiner and W. Marquardt, Identification of unknown pure component spectra by indirect hard modeling, Chemom. Intell. Lab. Syst., 2008, 93(2), 108–119 CrossRef CAS.
- M. Garrido, F. X. Rius and M. S. Larrechi, Multivariate curve resolution–alternating least squares (MCR-ALS) applied to spectroscopic data from monitoring chemical reactions processes, Anal. Bioanal. Chem., 2008, 390(8), 2059–2066 CrossRef CAS PubMed.
- C. Ruckebusch and L. Blanchet, Multivariate curve resolution: A review of advanced and tailored applications and challenges, Anal. Chim. Acta, 2013, 765, 28–36 CrossRef CAS PubMed.
- S. C. Cruz, G. Rothenberg, J. A. Westerhuis and A. K. Smilde, Estimating kinetic parameters of complex catalytic reactions using a curve resolution based method, Chemom. Intell. Lab. Syst., 2008, 91(2), 101–109 CrossRef CAS.
- M. Garland, Processing Spectroscopic Data, in Mechanisms in Homogeneous Catalysis, Wiley-VCH, Weinheim, Germany, 2010, pp. 151–193 Search PubMed.
- M. Garland and C. Li, A Review of BTEM Analysis for Catalytic Studies and a Recent Homogeneous Catalytic Example, Top. Catal., 2009, 52(10), 1334–1341 CrossRef CAS.
- E. Widjaja, C. Li and M. Garland, Semi-Batch Homogeneous Catalytic In-Situ Spectroscopic Data. FTIR Spectral Reconstructions Using Band-Target Entropy Minimization (BTEM) without Spectral Preconditioning, Organometallics, 2002, 21(9), 1991–1997 CrossRef CAS.
- A. Golshan, H. Abdollahi, S. Beyramysoltan, M. Maeder, K. Neymeyr, R. Rajkó, M. Sawall and R. Tauler, A review of recent methods for the determination of ranges of feasible solutions resulting from soft modelling analyses of multivariate data, Anal. Chim. Acta, 2016, 911, 1–13 CrossRef CAS PubMed.
- M. Sawall, A. Jürß, H. Schröder and K. Neymeyr, On the Analysis and Computation of the Area of Feasible Solutions for Two-, Three-, and Four-Component Systems, in Data Handling in Science and Technology, ed. C. Ruckebusch, Elsevier, Amsterdam, Netherlands, 2016, ch. 5, vol. 30, pp. 135–184 Search PubMed.
- M. Sawall and K. Neymeyr, A ray casting method for the computation of the area of feasible solutions for multicomponent systems: Theory, applications and FACPACK-implementation, Anal. Chim. Acta, 2017, 960, 40–52 CrossRef CAS PubMed.
- E. Kriesten, F. Alsmeyer, A. Bardow and W. Marquardt, Fully automated indirect hard modeling of mixture spectra, Chemom. Intell. Lab. Syst., 2008, 91(2), 181–193 CrossRef CAS.
- B. A. Steinhoff, I. A. Guzei and S. S. Stahl, Mechanistic Characterization of Aerobic Alcohol Oxidation Catalyzed by Pd(OAc)2/Pyridine Including Identification of the Catalyst Resting State and the Origin of Nonlinear [Catalyst] Dependence, J. Am. Chem. Soc., 2004, 126(36), 11268–11278 CrossRef CAS PubMed.
- J. Rabeah, U. Bentrup, R. Stößer and A. Brückner, Selective Alcohol Oxidation by a Copper TEMPO Catalyst: Mechanistic Insights by Simultaneously Coupled Operando EPR/UV-Vis/ATR-IR Spectroscopy, Angew. Chem., Int. Ed., 2015, 54(40), 11791–11794 CrossRef CAS PubMed.
- S. Adomeit, J. Rabeah, A. E. Surkus, U. Bentrup and A. Brückner, Effects of Imidazole-Type Ligands in Cu I /TEMPO-Mediated Aerobic Alcohol Oxidation, Inorg. Chem., 2017, 56(1), 684–691 CrossRef CAS PubMed.
- J. Rabeah, V. Briois, S. Adomeit, C. La Fontaine, U. Bentrup and A. Brückner, Multivariate Analysis of Coupled Operando EPR/XANES/EXAFS/UV–Vis/ATR–IR Spectroscopy: A New Dimension for Mechanistic Studies of Catalytic Gas–Liquid Phase Reactions, Chem. – Eur. J., 2020, 26(33), 7395–7404 CrossRef CAS PubMed.
- A. G. Abatjoglou, E. Billig and D. R. Bryant, Mechanism of Rhodium-Promoted Triphenylphosphine Reactions in Hydroformylation Processes, Organometallics, 1984, 3(6), 923–926 CrossRef CAS.
- C. Larpent, R. Dabard and H. Patin, Rhodium(I) Production during the Oxidation by Water of a Hydrosoluble Phosphine, Inorg. Chem., 1987, 26(17), 2922–2924 CrossRef CAS.
- G. W. Parshall, W. H. Knoth and R. A. Schunn, Ligand-Metal Hydrogen-Transfer Reactions in Triphenyl Phosphite and Triphenylphosphine Complexes, J. Am. Chem. Soc., 1969, 91(18), 4990–4995 CrossRef CAS.
- J. Halpern and A. L. Pickard, Mechanism of Tris(Triphenylphosphine)Platinum(0)-Catalyzed Oxidation of Triphenylphosphine, Inorg. Chem., 1970, 9(12), 2798–2800 CrossRef CAS.
- R. H. Crabtree, H. Felkin and G. E. Morris, Cationic iridium diolefin complexes as alkene hydrogenation catalysts and the isolation of some related hydrido complexes, J. Organomet. Chem., 1977, 141(2), 205–215 CrossRef CAS.
- S. R. Patil, D. N. Sen and R. V. Chaudhari, Hydrogenation of allylic compounds using homogeneous RuCl2(PPh3)3 complex catalyst, J. Mol. Catal., 1983, 19(2), 233–241 CrossRef CAS.
- D. Heller, K. Kortus and R. Selke, Kinetische Untersuchungen zur Ligandenhydrierung in Katalysatorvorstufen für die asymmetrische Reduktion prochiraler Olefine, Liebigs Ann., 1995, 1995(3), 575–581 CrossRef.
- D. Evans, G. Yagupsky and G. Wilkinson, The reaction of hydridocarbonyltris(triphenylphosphine)rhodium with carbon monoxide, and of the reaction products, hydridodicarbonylbis(triphenylphosphine)rhodium and dimeric species, with hydrogen, J. Chem. Soc. A, 1968, 2660–2665 RSC.
- W. Baumann, S. Mansel, D. Heller and S. Borns, Gas bubbles in the NMR tube: an easy way to investigate reactions with gases in the liquid phase, Magn. Reson. Chem., 1997, 35(10), 701–706 CrossRef CAS.
- B. Zhang, H. Jiao, D. Michalik, S. Kloß, L. M. Deter, D. Selent, A. Spannenberg, R. Franke and A. Börner, Hydrolysis Stability of Bidentate Phosphites Utilized as Modifying Ligands in the Rh-Catalyzed n-Regioselective Hydroformylation of Olefins, ACS Catal., 2016, 6(11), 7554–7565 CrossRef CAS.
- P. E. Garrou, Transition-Metal-Mediated Phosphorus Carbon Bond-Cleavage and its Relevance to Homogeneous Catalyst Deactivation, Chem. Rev., 1985, 85(3), 171–185 CrossRef CAS.
- W. R. Moser, C. J. Papile, D. A. Brannon, R. A. Duwell and S. J. Weininger, The mechanism of phosphine-modified rhodium-catalyzed hydroformylation studied by CIR-FTIR, J. Mol. Catal., 1987, 41(3), 271–292 CrossRef CAS.
- C. Bianchini, W. Oberhauser, A. Orlandini, C. Giannelli and P. Frediani, Operando High-Pressure NMR and IR Study of the Hydroformylation of 1-Hexene by 1,1′-Bis(Diarylphosphino)metallocene-Modified Rhodium(I) Catalysts, Organometallics, 2005, 24(15), 3692–3702 CrossRef CAS.
- Y. Jiao, M. S. Torne, J. Gracia, J. W. Niemantsverdriet and P. W. N. M. van Leeuwen, Ligand effects in rhodium-catalyzed hydroformylation with bisphosphines: steric or electronic?, Catal. Sci. Technol., 2017, 7(6), 1404–1414 RSC.
- A. S. C. Chan, H.-S. Shieh and J. R. Hill, A new synthesis and X-ray molecular structure of di-μ-carbonyltricarbonyltris(triphenylphosphine)dirhodium, J. Chem. Soc., Chem. Commun., 1983, 688–689 RSC.
- W. Moser, Cylindrical internal reflectance: A new method for high-pressure in situ catalytic studies, J. Catal., 1985, 95(1), 21–32 CrossRef CAS.
- A. Castellanos-Páez, S. Castillón, C. Claver, P. W. N. M. van Leeuwen and W. G. J. de Lange, Diphosphine and Dithiolate Rhodium Complexes: Characterization of the Species under Hydroformylation Conditions, Organometallics, 1998, 17(12), 2543–2552 CrossRef.
- E. B. Walczuk, P. C. J. Kamer and P. W. N. M. van Leeuwen, Dormant states of rhodium hydroformylation catalysts: Carboalkoxyrhodium complex formed from enones in the alkene feed, Angew. Chem., Int. Ed., 2003, 42(38), 4665–4669 CrossRef CAS PubMed.
- G. Kiss and I. T. Horvath, Reversible arm-off dissociation of the tripodal 1,1,1-tri[(diphenylphosphino)methyl]ethane in rhodium complex carbonylhydrido[1,1,1-tris[(diphenylphosphino)methyl]ethane]rhodium under hydroformylation conditions, Organometallics, 1991, 10(11), 3798–3799 CrossRef CAS.
- C. Li, L. Chen, E. Widjaja and M. Garland, The catalytic binuclear elimination reaction: Confirmation from in situ FTIR studies of homogeneous rhodium catalyzed hydroformylation, Catal. Today, 2010, 155(3–4), 261–265 CrossRef CAS.
- S. Cheng, C. Li, L. Guo and M. Garland, Determining the pure component spectra of trace organometallic intermediates by combined application of in situ Raman spectroscopy and band-target entropy minimization analysis, Vib. Spectrosc., 2014, 70, 110–114 CrossRef CAS.
- G. Liu and M. Garland, Liquid-Phase Reaction of Monosubstituted and Disubstituted Alkynes with Tetrarhodium Dodecacarbonyl under CO and CO/H2 Mixtures. In-Situ IR Spectroscopic Characterization of 20 New Alkyne−Rhodium Complexes, Organometallics, 1999, 18(17), 3457–3467 CrossRef CAS.
- G. Liu and M. Garland, The competitive and non-competitive hydroformylation of conjugated dienes starting with tetrarhodium dodecacarbonyl. An in-situ high-pressure infrared spectroscopic study, J. Organomet. Chem., 2000, 608(1–2), 76–85 CrossRef CAS.
- Figure reproduced and adapted with permission from John Wiley & Sons, Ltd. from https://onlinelibrary.wiley.com/doi/10.1002/anie.200351884. Further permissions related to the material excerpted should be directed to the publisher.
- Figure reproduced and adapted with permission from the American Chemical Society from https://pubs.acs.org/doi/10.1021/acscatal.8b05066. Further permissions related to the material excerpted should be directed to the ACS.
- S. Kloß, D. Selent, A. Spannenberg, R. Franke, A. Börner and M. Sharif, Effects of Substitution Pattern in Phosphite Ligands Used in Rhodium-Catalyzed Hydroformylation on Reactivity and Hydrolysis Stability, Catalysts, 2019, 9(12), 1036 CrossRef.
- Figure reproduced and adapted with permission from the American Chemical Society from https://pubs.acs.org/doi/10.1021/acscatal.6b02185. Further permissions related to the material excerpted should be directed to the ACS.
- B. Cornils, W. A. Herrmann, M. Beller and R. Pacciello, Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Four Volumes, Wiley-VCH, Weinheim, Germany, 2018 Search PubMed.
- W. S. Knowles, Asymmetric Hydrogenations (Nobel Lecture), Angew. Chem., Int. Ed., 2002, 41(12), 1998–2007 CrossRef CAS.
- R. Noyori, Asymmetric Catalysis: Science and Opportunities (Nobel Lecture), Angew. Chem., Int. Ed., 2002, 41(12), 2008–2022 CrossRef CAS PubMed.
- H. U. Blaser, F. Spindler and M. Studer, Enantioselective catalysis in fine chemicals production, Appl. Catal., A, 2001, 221(1–2), 119–143 CrossRef CAS.
- H. B. Kagan and T.-P. Dang, Asymmetric catalytic reduction with transition metal complexes. I. Catalytic system of rhodium(I) with (-)-2,3-0-isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane, a new chiral diphosphine, J. Am. Chem. Soc., 1972, 94(18), 6429–6433 CrossRef CAS.
- J. M. Brown and P. A. Chaloner, The mechanism of asymmetric hydrogenation catalysed by rhodium(I) di pamp complexes, Tetrahedron Lett., 1978, 19(21), 1877–1880 CrossRef.
- J. M. Brown and P. A. Chaloner, Mechanism of asymmetric hydrogenation catalysed by rhodium(I)-trans-4,5-bis(diphenylphosphinomethyl)-2,2-dimethyldioxolan (DIOP) complexes, J. Chem. Soc., Chem. Commun., 1978, 321–322 RSC.
- J. M. Brown and P. A. Chaloner, Asymmetric homogeneous hydrogenation catalysed by rhodium complexes; the binding modes of enamides defined by 13C n.m.r. spectroscopy, J. Chem. Soc., Chem. Commun., 1979, 613–615 RSC.
- J. M. Brown and P. A. Chaloner, Structural characterisation of a transient intermediate in rhodium-catalysed asymmetric homogeneous hydrogenation, J. Chem. Soc., Chem. Commun., 1980, 344–346 RSC.
- J. M. Brown and P. A. Chaloner, The mechanism of asymmetric homogeneous hydrogenation. Rhodium(I) complexes of dehydroamino acids containing asymmetric ligands related to bis(1,2-diphenylphosphino)ethane, J. Am. Chem. Soc., 1980, 102(9), 3040–3048 CrossRef CAS.
- J. M. Brown, P. A. Chaloner, A. G. Kent, B. A. Murrer, P. N. Nicholson, D. Parker and P. J. Sidebottom, The mechanism of asymmetric homogeneous hydrogenation. Solvent complexes and dihydrides from rhodium diphosphine precursors, J. Organomet. Chem., 1981, 216(2), 263–276 CrossRef CAS.
- A. S. C. Chan and J. Halpern, Interception and characterization of a hydridoalkylrhodium intermediate in a homogeneous catalytic hydrogenation reaction, J. Am. Chem. Soc., 1980, 102(2), 838–840 CrossRef CAS.
- A. S. C. Chan, J. J. Pluth and J. Halpern, Identification of the enantioselective step in the asymmetric catalytic hydrogenation of a prochiral olefin, J. Am. Chem. Soc., 1980, 102(18), 5952–5954 CrossRef CAS.
- C. R. Landis and J. Halpern, Asymmetric hydrogenation of methyl (Z)-α-acetamidocinnamate catalyzed by [1,2-bis(phenyl-o-anisoyl)phosphino)ethane]rhodium(I): kinetics, mechanism and origin of enantioselection, J. Am. Chem. Soc., 1987, 109(6), 1746–1754 CrossRef CAS.
- D. Heller, S. Borns, W. Baumann and R. Selke, Kinetic Investigations of the Hydrogenation of Diolefin Ligands in Catalyst Precursors for the Asymmetric Reduction of Prochiral Olefins, II[1], Chem. Ber., 1996, 129(1), 85–89 CrossRef CAS.
- H.-J. Drexler, W. Baumann, A. Spannenberg, C. Fischer and D. Heller, Part III, COD versus NBD precatalysts. Dramatic difference in the asymmetric hydrogenation of prochiral olefins with five-membered diphosphine Rh-hydrogenation catalysts, J. Organomet. Chem., 2001, 621(1), 89–102 CrossRef CAS.
- Figure reproduced and adapted by permission from John Wiley & Sons, Ltd. from https://pubs.rsc.org/en/content/articlelanding/2019/fd/c9fd00060g. Further permissions related to the material excerpted should be directed to the publisher.
- A. M. R. Hall, P. Dong, A. Codina, J. P. Lowe and U. Hintermair, Kinetics of Asymmetric Transfer Hydrogenation, Catalyst Deactivation, and Inhibition with Noyori Complexes As Revealed by Real-Time High-Resolution FlowNMR Spectroscopy, ACS Catal., 2019, 9(3), 2079–2090 CrossRef CAS.
- R. Noyori, M. Yamakawa and S. Hashiguchi, Metal−Ligand Bifunctional Catalysis: A Nonclassical Mechanism for Asymmetric Hydrogen Transfer between Alcohols and Carbonyl Compounds, J. Org. Chem., 2001, 66(24), 7931–7944 CrossRef CAS PubMed.
- C. D. T. Nielsen and J. Burés, Visual kinetic analysis, Chem. Sci., 2019, 10(2), 348–353 RSC.
- D. B. G. Berry, A. Codina, I. Clegg, C. L. Lyall, J. P. Lowe and U. Hintermair, Insight into catalyst speciation and hydrogen co-evolution during enantioselective formic acid-driven transfer hydrogenation with bifunctional ruthenium complexes from multi-technique operando reaction monitoring, Faraday Discuss., 2019, 220, 45–57 RSC.
- Figure reproduced and adapted with permission from the American Chemical Society from https://pubs.acs.org/doi/10.1021/acscatal.8b03530. Further permissions related to the material excerpted should be directed to the ACS.
- J. Y. Buser and A. D. McFarland, Reaction characterization by flow NMR: quantitation and monitoring of dissolved H(2) via flow NMR at high pressure, Chem. Commun., 2014, 50(32), 4234–4237 RSC.
| This journal is © The Royal Society of Chemistry 2022 |