
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Digital light processing (DLP) 3D-fabricated antimicrobial hydrogel with a sustainable resin of methacrylated woody polysaccharides and hybrid silver-lignin nanospheres†
Luyao
Wang
a,
Qingbo
Wang
a,
Anna
Slita
b,
Oskar
Backman
a,
Zahra
Gounani
c,
Emil
Rosqvist
 d,
Jouko
Peltonen
d,
Stefan
Willför
a,
Chunlin
Xu
a,
Jessica M.
Rosenholm
b and
Xiaoju
Wang
*ab
d,
Jouko
Peltonen
d,
Stefan
Willför
a,
Chunlin
Xu
a,
Jessica M.
Rosenholm
b and
Xiaoju
Wang
*ab
aLaboratory of Natural Materials Technology, Faculty of Science and Engineering, Åbo Akademi University, Henrikinkatu 2, Turku FI-20500, Finland. E-mail: xiaoju.wang@abo.fi
bPharmaceutical Sciences Laboratory, Faculty of Science and Engineering, Åbo Akademi University, Tykistökatu 6A, Turku FI-20520, Finland
cPhysics, Faculty of Science and Engineering Åbo Akademi University, Henrikinkatu 2, Turku FI-20500, Finland
dLaboratory of Molecular Science and Engineering, Faculty of Science and Engineering, Åbo Akademi University, Henrikinkatu 2, Turku FI-20500, Finland
First published on 10th February 2022
Abstract
Lithography-based digital light processing (DLP) 3D printing has gained increasing interest in the fabrication of custom-designed hydrogel scaffolds. Current research development calls for the versatility of the bio-based resin formulations that deliver a suitable printability for fabricating biomedical hydrogel using DLP printing. Here, a bio-based antimicrobial resin was developed for DLP printing engaging the methacrylated O-acetyl-galactoglucomannan (GGMMA) as a photo-crosslinkable polymeric matrix and the nanocomposite lignin nanoparticles that are surface-embedded with silver nanoparticles (LNP@Ags) as a high-performance antimicrobial reagent. The alkali-resistance of LNPs as demanded for in situ reduction of silver nanoparticles (AgNPs) from silver ammonia solution (pH 11) was achieved by upgrading the technical lignin via a combination of lignin solvent fractionation with laccase-catalyzed polymerization. The dimension of the as-prepared LNPs (in the range of 120–290 nm) and the content of residual phenolic hydroxyl and methoxyl groups for reducing Ag+ could be modulated by tuning the laccase-catalyzed lignin polymerization degree. The encapsulation of nanocomposite LNP@Ag into the GGMMA hydrogel did not significantly alter the gelation rate of the GGMMA/LNP@Ag resin and the mechanical property of the resulted hydrogel, when the dosage of LNP@Ag was less than 0.1 wt% in 10 wt% GGMMA. The GGMMA/LNP@Ag hydrogel also possesses high antimicrobial activity due to the bactericidal ability of Ag+ that was leached out of the hydrogel in a sustained manner. Upon the layer-by-layer photocuring in DLP printing, the GGMMA/LNP@Ag resin allowed us to fabricate hydrogel constructs, including honeycomb and crosshatch with good structural stability and printing fidelity. This study proposes a green and effective fabrication route for LNP supported AgNPs as well as a novel case study for a sustainable resin suitable for DLP 3D hydrogel printing.
Introduction
Nanocomposite hydrogels have prevailed as extracellular matrix-mimicking biomaterials in various biomedical applications ranging from wound dressing to tissue engineering scaffolds.1 When biopolymers are used in building up such a category of functional materials, therapeutic-relevant bioactivities are desired to be incorporated in the matrix to provide equivalent or even outperformed functionalities in comparison to a large number of well-established synthetic materials. Natural polymers (cellulose, hemicellulose, and lignin) fractionated from various lignocellulosic biomass have established a valorization niche in producing high-value biomedical hydrogels owing to their biocompatibility, biodegradability, and versatility in chemical modifications.2–5 It has been demonstrated previously that integrating noble metal nanoparticles (NPs), such as gold (Au), silver (Ag), and platinum (Pt), into nanocomposite hydrogel is particularly feasible for biomedical uses due to their unique optoelectronic and antimicrobial properties.6–8 As a broad-spectrum antimicrobial agent, silver nanoparticles (AgNPs) are currently the most widely commercialized nanomaterial. They are increasingly used for surface and air sterilization, e.g., antimicrobial coatings for medical devices and household sterilizing sprays.9,10 However, the aggregation propensity of bare AgNPs often causes a large increase in local silver concentration, which has the risk of reaching the minimum lethal concentration of silver ions (Ag+) and further exerts toxicity to the organisms.11Various approaches have been used to fabricate noble metal NPs with durable dispersity in aqueous or organic medium, e.g., thiol-based functional capping,11,12in situ reduction and stabilization,8 and fabricating robust microsphere/nanosphere supports.13,14 The aforementioned methodologies encourage the development of biopolymer-based spherical support for in situ reduction and stabilization of noble metal NPs. Currently, the non-toxicity of lignin nanoparticles (LNPs) in combination with its antioxidant activity, ultraviolet (UV)-shielding property, and large specific surface area have attracted a heightened interest in engaging this natural polyphenol for biomaterials.15–18 Moreover, the presence of rich multi-functional groups, e.g., reductive phenolic hydroxyls (phenolic-OH) and methoxyl (–OCH3) in lignin would facilitate in situ reduction and stabilization of noble metal NPs on the nanosphere.8 In fact, in most of the earlier reported studies where the technical lignin was solely used as the reducing agent and the silver ammonia solution (pH 11) is commonly engaged as the silver precursor, the lignin was mainly in dissolved states in the synthesis of AgNPs. Eventually, lignin was remained as a thin capping layer to stabilize the AgNPs as well as a supporting matrix in the lignin-AgNPs complex.8,19–21 Nevertheless, to use spherical LNPs as the core for in situ reduction of AgNPs is sparingly investigated, ascribed to the low stability and dispersity of LNPs at extreme alkaline conditions. To improve the stability of LNPs in alkaline and organic solvents, lignin is generally demanded to be polymerized or condensed to present recalcitrant structures. For instance, Chen et al. synthesized lignin-containing phenolic resin (LPR) nanospheres using a hydrothermal polymerization method and these LPR nanospheres were successfully applied as reducing agent in in situ reduction of AgNPs to result the well-dispersed spherical nanocomposite of LPR@Ag.13 In our previous work, one bacteria-derived laccase that showed high catalytic activity under alkaline pH was proven to result in extensive polymerization of solvent fractionated hardwood alkaline lignin.22 Through this above-mentioned enzymatic treatment, polymerized lignin with enhanced thermal stability and chemical tolerance was obtained. This has inspired us to adapt these laccase-polymerized lignin fractions in resulting extreme alkali-resistant LNPs and to further decorate the as-synthesized spherical LNPs with surface-embedded silver nanoparticles (LNP@Ags).
Back to the fabrication methods to address the demand of controlled architecture and properties in producing biologically relevant hydrogel constructs, additive manufacturing has been established as a mainstream approach to deliver impressive outputs. Presently, several additive manufacturing techniques are applicable in the scenario of biomedical hydrogel fabrication and are advantageous to offer digitally designed geometry and complex spatial connectivity within the hydrogels as fabricated. For instance, recent advances in digital light processing (DLP)-based 3D printing technique have shown their promise for achieving this purpose by offering stable customized imaging as well as excellent spatial resolution and pattern fidelity with rapid fabrication speed.23,24 DLP additive manufacturing creates models in a layer-by-layer manner through photopolymerization via UV or visible light. To date, only a limited number of photo-crosslinkable biopolymers such as hyaluronic acid,25 gelatin,26,27 and silk fibroin28 that are chemically modified with such photoreactive moieties as methacryloyls, have been used as resins for DLP printing. The methacryloyls are known to be easily cross-linked by the photo-triggered free-radical polymerization to form an interpenetrating polymer network of the hydrogel.23 In our previous study, the major hemicellulose derived from softwood, O-acetyl-galactoglucomannan (GGM) was chemically modified with methacryloyls in an aqueous medium.29 This methacrylated GGM derivative (GGMMA) together with cellulose nanofibrils was successfully formulated as a feedstock ink for UV-aided extrusion-based 3D printing. In addition, the hydrogel scaffold printed with the GGMMA-based inks showed great cytocompatibility and mechanical properties. We hypothesized that GGMMA would also be an excellent biopolymer resin for DLP printing.
In this work, we developed a resin based on photo-crosslinkable GGMMA and all-lignin-supported LNP@Ag nanospheres for lithography-based DLP printing. The alkali-resistant LNP@Ag was also fabricated for the first time from laccase-polymerized lignin fractions. We evaluated the degree of laccase-catalyzed lignin polymerization and characterized the functionalities of laccase-polymerized lignin for silver reduction and physiochemical properties of the synthesized LNP@Ag, in terms of morphology, chemical composition, stability, and crystalline features. The printability of GGMMA/LNP@Ag resin was demonstrated via DLP printing by fabricating hydrogels with patterned structures. The printing fidelity and antimicrobial activity were further assessed.
Results and discussion
Design rationale of the sustainable-resin-based antimicrobial hydrogel
The GGMMA/LNP@Ag hydrogel was fabricated in two steps, as shown in Fig. 1a. First, LNP@Ag nanoparticles were synthesized through a redox reaction between the silver precursor [Ag(NH3)2]+ complex and the LNPs prepared from the laccase-polymerized lignin by a solvent shifting method. Second, the LNP@Ag dispersion was mixed into 10 wt% GGMMA solution and UV light was applied to induce free-radical polymerization of methacryloyls with the presence of lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) as a photoinitiator, resulting the cross-linked nanocomposite hydrogel of GGMMA/LNP@Ag. The lignin nanospheres with surface-embedded AgNPs can function as nanoantibiotics to leach Ag+ in aqueous solutions exposed to air,11 and therefore would endow the GGMMA/LNP@Ag hydrogel with antimicrobial activity.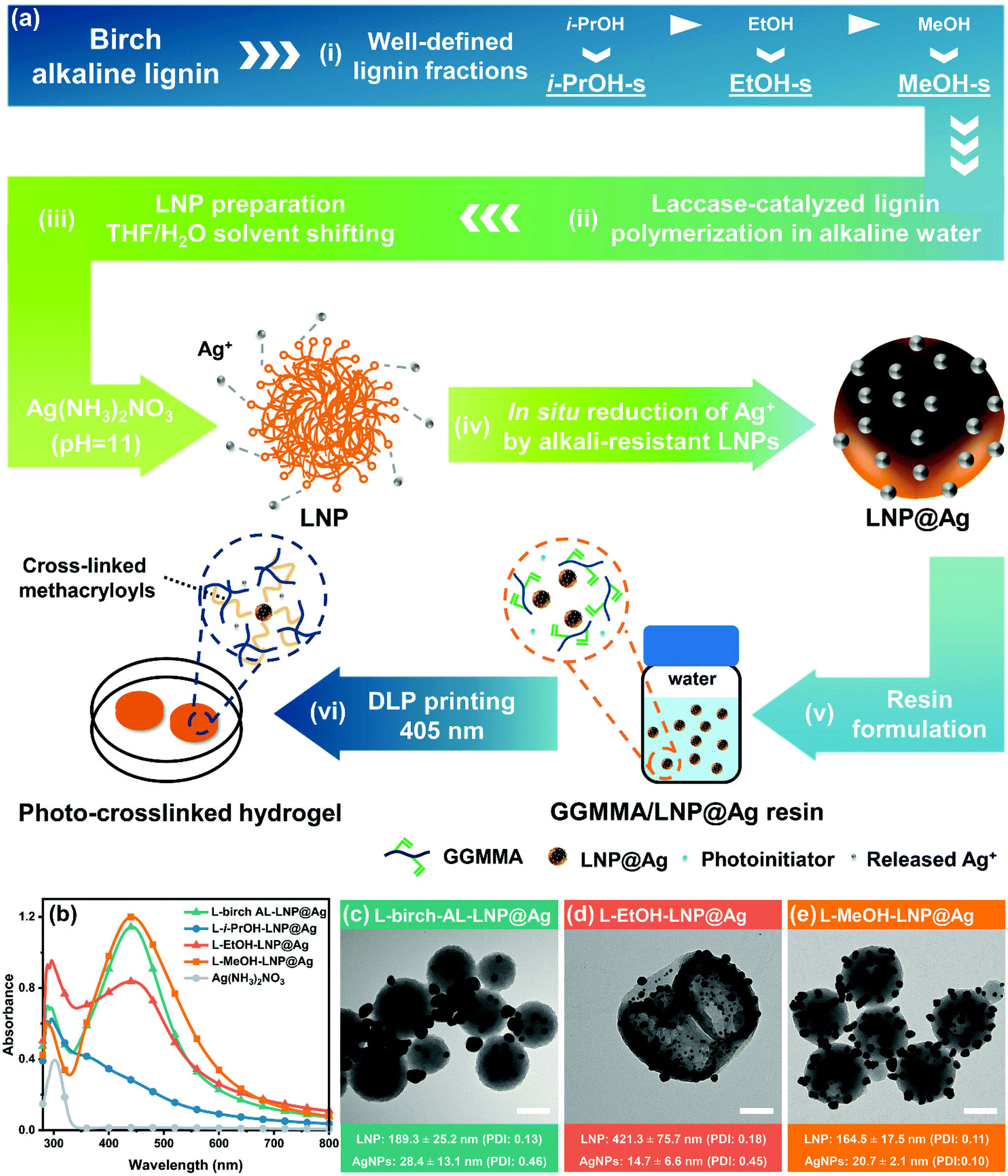 | ||
Fig. 1 (a) Design strategy for the sustainable resin and antimicrobial GGMMA/LNP@Ag hydrogel: (i and ii) Preparation of laccase-polymerized lignin in alkaline aqueous media from well-defined lignin fractions; (iii) fabrication of LNPs by the THF/H2O solvent shifting method followed by (iv) in situ reduction of Ag+ on the surface of alkali-resistant LNPs; (v and vi) Scheme of the molecular structure of photo-crosslinkable GGMMA/LNP@Ag resin and hydrogel. (b) Ultraviolet–visible (UV-vis) spectra of LNP@Ag aqueous dispersion. (c–e) TEM images of LNP@Ag from laccase-polymerized lignin with a scale bar of 100 nm and magnification of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. Note: x ± y in (c–e), x denotes mean value of particle size (nm) and y is the standard deviation value (nm) of x. 000×. Note: x ± y in (c–e), x denotes mean value of particle size (nm) and y is the standard deviation value (nm) of x. | ||
In this study, the laccase-polymerized lignin showed increased chemical tolerance and facilitated the preparation of alkali-resistant LNPs. First, birch alkaline lignin (AL) was fractionated by alcohols (i-PrOH, EtOH, and MeOH) with gradient polarity that resulted lignin fractions with well-defined characteristics in terms of the molar mass, molar mass dispersity (ĐM), and functional groups (e.g., phenolic-OH and –OCH3 groups) according to our previous study (Fig. 1a(i), Table 1).30 These molar-mass-dependent differences of birch AL fractions exerted tuneable lignin polymerization performance in the aspect of molar mass increase, i.e., L-i-PrOH-s (6.6-fold) > L-EtOH-s (2.9-fold) > L-birch AL (2.8-fold) > L-MeOH-s (2.2-fold) (Table 1). Second, the oxidation of phenolic-OH and demethylation of –OCH3 groups occurred simultaneously upon laccase-assisted lignin polymerization (Table 1). In addition, as revealed from our recent in-depth analysis to the structure–property-application relationship for the laccase-catalyzed alkaline lignin polymerization, the oxidative radical–radical coupling raised lignin condensation with the formation of robust interunit carbon–carbon linkages, which will further limit the dissolution of laccase-polymerized lignin in harsh conditions.22
| Lignins |
M
Wa (g mol−1) |
Đ
Mb |
Phenolic-OHc (mmol g−1) | –OCH3d (mmol g−1) |
|---|---|---|---|---|
| a Weight-average molar mass. b Molar mass dispersity = weight-average molar mass (Mw)/number-average molar mass (Mn). c Determined by quantitative 31P NMR.30 d Quantified by 1H NMR with 4-nitrobenzaldehyde as internal standard.22 e Lignin solution was incubated with laccase in alkaline condition (pH 10) at 39 °C with air (O2) circulation for 4 hours. | ||||
| Birch AL | 7200 | 2.8 | 3.3 | 16.8 |
| i-PrOH-s | 3700 | 1.4 | 3.8 | 18.1 |
| EtOH-s | 4900 | 1.5 | 3.4 | 12.2 |
| MeOH-s | 6200 | 1.6 | 3.0 | 9.6 |
| L-Birch ALe | 27000 |
8.9 | 2.5 | 11.9 |
| L-i-PrOH-se | 28000 |
4.5 | 1.5 | 11.1 |
| L-EtOH-se | 19000 |
2.3 | 2.1 | 9.1 |
| L-MeOH-se | 20000 |
2.3 | 2.0 | 7.8 |
Based on previous research, the most interesting water-miscible solvents used for LNP preparation were tetrahydrofuran (THF) and acetone, of which acetone is more environmentally benign and easier to recycle.31 Under the green chemistry principle, we compared the solubility of laccase-polymerized lignin in THF–H2O (9:1, v/v) and acetone–H2O (9:1, v/v) binary solvents, which resulted in a solubility yield of 92% and 70%, respectively (Fig. S1†). To represent the bulk property of the laccase-treated lignin, the colloidal LNPs used in this work were prepared by dissolving lignin in THF–H2O (9:1, v/v) and continuously introducing anti-solvent water into the lignin-THF–H2O solution. After being incubated with [Ag(NH3)2]+ (10 mg mL−1) for 4 hours, the colloids of LNP dispersions prepared from the laccase-polymerized birch AL, EtOH-s, and MeOH-s fractions showed the characteristic surface plasmon resonance (SPR) absorption peak of metallic silver at 440 nm, but this was not observed for the LNPs prepared from the laccase-treated i-PrOH-s fraction (Fig. 1b). This strongly indicates that the intensive oxidation of phenolic-OH and –OCH3 groups in L-i-PrOH-s has compromised its surface reactivity towards reducing Ag+ (Table 1), although the pronounced lignin–lignin condensation promoted its alkali stability. The transmission electron microscopy (TEM) micrographs further showed the morphological difference of the in situ reduced AgNPs with LNP based on the molecular difference of laccase-polymerized lignin fractions in Fig. 1c–e. As a control, the AgNPs were prone to form large aggregates (28.4 ± 13.1 nm) in the vicinity of the L-birch-AL-LNPs, instead of being surface-embedded (Fig. 1c). This is believed to be associated with the high molar mass dispersity (ĐM = 8.9) of the laccase-polymerized birch AL. The L-i-PrOH-LNP maintained the spherical morphology intact in silver ammonia solution but without forming AgNPs (Fig. S2†), which is consistent with the UV-vis results. Meanwhile, the L-EtOH-LNPs and AgNPs embedded on the surface were less homogenized than that of L-MeOH-LNPs as comparing the polydispersity index (PDI) of LNPs and AgNPs in Fig. 1d and e. Specifically, small and compact nanospheres and some particles with hollow circular regions were both derived from L-MeOH-s lignin, whereas large and hollow particles were obtained from L-EtOH-s lignin. The formation of hollow structures during solvent-shifting is possibly associated with a nanoemulsion formation prior to the self-assembly due to the presence of amphiphilic lignin molecules.32,33 Nevertheless, the hollow particles prepared from L-MeOH-s lignin were solid and different from the incomplete structure of L-EtOH-LNPs with penetrating holes and cracks. This is indicative of the presence of less amphiphilic structure in the latter lignin molecules during nanoemulsion formation, as a similar phenomenon has been observed and discussed by Xiong et al.32 and Pang et al.33 Above all, these results validated the feasibility of combining the laccase-catalyzed lignin polymerization with the sequential solvent lignin fractionation to fine-tune the structural features of lignin and to fabricate uniform surface-embedded hybrid morphology of LNP@Ag. In addition, this further confirms the significance of lignin fractionation in deriving lignin fractions with narrow ĐM and high-performance lignin nanomaterials. Unless stated otherwise, the LNP@Ag used in this work was synthesized by laccase-polymerized MeOH-s (LM) fraction.
Tuneable lignin reactivity by laccase-catalyzed polymerization dictating the properties of LNP and LNP@Ag
The functional groups (phenolic-OH and –OCH3) serving for Ag+ reduction in lignin were examined by quantitative 31P and 1H NMR. The total amount of phenolic-OH (for both C5-substituted and non-condensed phenolic-OH form) and –OCH3 groups in LM lignin progressively decreased with increasing laccase incubation time (corresponds to increased lignin polymerization/condensation degree and molar mass), i.e., from 12.6 mmol g−1 of birch MeOH-s fraction to 8.2 mmol g−1 of LM-6 lignin (Fig. 2a). This result is well rationalized by the laccase-induced chemical modifications of lignin, as previously reported by us and by the other research groups.22,34 The size and uniformity of the spherical LNPs are crucial metrics for assessing their properties and applications as nanomaterials. The intensity weighted average hydrodynamic diameter (Z-average) of the colloidal spheres from LM lignin was observed to be dependent on the oxidative polymerization/condensation degree of lignin (the duration of laccase treatment in experimental setup), ranging from 290 to 120 nm (Fig. 2b). More specifically, the lignin in higher polymerization/condensation degree favoured the formation of small-size LNPs. Similar phenomenon has been previously reported in the case of cellulolytic enzyme lignin derived from steam pre-treated biomass, and lignin condensation among aromatics occurred during steam pre-treatment.35 Likely, the formation of compact micelle nucleus during the self-assembling of lignin hydrophobic aromatics explains the decreasing size of LNPs as the covalent carbon–carbon linkages formed in the polymerized lignin dictates the lignin nucleation to a larger extent than the π–π and van der Waals interactions.36,37 Dynamic light scattering (DLS) results confirmed a relatively monodisperse (0.10 < PDI < 0.15) system for most of the as-prepared LNPs prepared from the LM lignin (Table S1†). Nevertheless, the polymerized lignin molecules became more heterogeneous as the duration of laccase treatment was extended, which in turn resulted in a deteriorated monodispersity, e.g., LM-6-NPs dispersion (PDI = 0.28, ĐM of LM-6 lignin was 3.78). The diverse lignin chemistries (molar mass, polymerization/condensation degree, and functional group distributions) resulted from laccase-catalyzed polymerization have motivated us to systematically investigate the impact of polymerized/condensed lignin characteristics on the formation of LNP@Ag and its nanomaterial properties. Apparently, the synthetic route as currently deployed for fabricating LNP@Ag would actually demand the LNP to have a sufficient alkali resistance in [Ag(NH3)2]+ silver precursor solution (pH 10.5–11.0). To address this perspective, the effect of lignin polymerization on the LNP alkali-resistance capacity was correlated in Fig. 2c. An enzymatic treatment duration prolonged over 4 hours was necessary in the polymerization of birch MeOH-s fraction to ensure the alkali resistance of the as-prepared LNPs in pH 11 solution (adjusted by 2 wt% ammonia solution). For these LM-4, LM-5, and LM-6 lignins, more than 80% LNPs could be retained as uniformly nanosized lignin under this alkaline pH (Fig. 2c, insert (i)). Nevertheless, LNPs prepared from MeOH-s, LM-1, LM-2, and LM-3 lignins were not able to keep the spherical morphology intact under the high alkaline condition. As it can be seen from the TEM images of MeOH-LNP@Ag, the LNPs collapsed and AgNP aggregates were entrapped by the lignin (Fig. S3†). | ||
| Fig. 2 Correlation of the Z-average and ζ-potential of LNP and its corresponding LNP@Ag with tailored lignin chemistry by laccase-catalyzed polymerization: (a) functional group distributions in LM lignin as a function of molar mass (the ĐM was shown above the molar mass data point), (b) Z-average of LM-NP dispersed in neutral deionized water, (c) alkali resistance of LM-NP to pH 11 ammonia solution and Z-average of LM-NP@Ag in neutral deionized water, and (d) ζ-potential of LM-NP and LM-NP@Ag. The inset (i) in (c) was the TEM image of LM-4-NP redispersed in pH 11 ammonia solution to demonstrate good alkali resistance. Note: (b) LM-x-NP, x denotes reaction time of lignin with laccase. | ||
The surface charge of nanoparticles dictates the colloidal stability in the cases of LNP dispersions. As noted in Fig. 2d, LNPs showed negative values of ζ-potential, which was partially due to the negative charges of the deprotonated phenolic-OH and possible carboxylic acid groups.36,38,39 The negative surface charge can stabilize the LNP dispersion via electrostatic repulsion. The as-synthesized LNPs and LNP@Ags were stable in aqueous dispersion and their surface charge is dependent on the polymerization/condensation degree and content of functional groups in LM lignins. As noted in Fig. 2d, a significant decrease in the absolute ζ-potential of LM-1-NP (−31 mV) relative to the MeOH-NPs (−44 mV) was observed, which could be ascribed to the loss of phenolic-OH groups during laccase-catalyzed lignin polymerization (Fig. 2a). Interestingly, with further extending the laccase treatment duration in preparation of the LM lignin, the LM-2-NPs, LM-3-NPs, LM-4-NPs, and LM-5-NPs showed slightly higher negative surface charge than LM-1-NPs. The redox reaction between the [Ag(NH3)2]+ complex and LM-NPs created more negative charges, which were presumably derived from the oxidation of hydroxyl groups to carboxylic acid groups.19 The generated carboxyl (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) was further confirmed by the high-resolution X-ray photoelectron spectroscopy (XPS) spectra of C 1s and O 1s, as shown in Fig. S4 and Table S3.† The relatively high negative surface charge of LNP@Ags, in the range from −40 to −50 mV, induces sufficient electrostatic repulsion to hinder particle aggregation.40 The colloidal dispersion of LNP@Ag was stable and rather insensitive to variations in pH from 2.5 to 12, with no significant change in the average particle size (Fig. 3a), although the absolute value of the ζ-potential dramatically decreased when the pH value was lower than 4 (Fig. 3b). Overall, these high-quality LNP@Ag dispersions are resulted from fine-tuning the lignin chemistry tailored by lignin fractionation and laccase-catalyzed polymerization. In this synthetic strategy, two aspects are important to underline for the hybrid LNP@Ag featured with hierarchical nano-dimensions as-reported: (i) to weigh the fractionation-dependent lignin polymerization kinetics out is critical to balance between the sufficient alkali resistance of the LNPs and the amount of residual functional groups on the LNP surface that are still available for Ag+ reduction; and (ii) with concomitant of an extended laccase-induced polymerization, the decreased content of phenolic-OH group and increased molar mass as well as the increased content of recalcitrant carbon–carbon linkages in laccase-polymerized MeOH-s fraction together contribute to the alkali stability of LNP and facilitate the formation of LNP@Ag with smaller nano-dimension.
O) was further confirmed by the high-resolution X-ray photoelectron spectroscopy (XPS) spectra of C 1s and O 1s, as shown in Fig. S4 and Table S3.† The relatively high negative surface charge of LNP@Ags, in the range from −40 to −50 mV, induces sufficient electrostatic repulsion to hinder particle aggregation.40 The colloidal dispersion of LNP@Ag was stable and rather insensitive to variations in pH from 2.5 to 12, with no significant change in the average particle size (Fig. 3a), although the absolute value of the ζ-potential dramatically decreased when the pH value was lower than 4 (Fig. 3b). Overall, these high-quality LNP@Ag dispersions are resulted from fine-tuning the lignin chemistry tailored by lignin fractionation and laccase-catalyzed polymerization. In this synthetic strategy, two aspects are important to underline for the hybrid LNP@Ag featured with hierarchical nano-dimensions as-reported: (i) to weigh the fractionation-dependent lignin polymerization kinetics out is critical to balance between the sufficient alkali resistance of the LNPs and the amount of residual functional groups on the LNP surface that are still available for Ag+ reduction; and (ii) with concomitant of an extended laccase-induced polymerization, the decreased content of phenolic-OH group and increased molar mass as well as the increased content of recalcitrant carbon–carbon linkages in laccase-polymerized MeOH-s fraction together contribute to the alkali stability of LNP and facilitate the formation of LNP@Ag with smaller nano-dimension.
 | ||
| Fig. 3 Effect of pH on (a) Z-average and (b) ζ-potential of LM-4-NP@Ag. The LM-4-NP@Ag were redispersed in distilled water with pH range from 2.0 to 12.5 at a concentration of 0.2 mg mL−1. At pH 12.5 the colloids of LNP@Ag were observed to start dissolving as the color of the dispersion turned yellowish. Note: (x ± y, z) in (a), x denotes Z-average (nm), y for standard deviation value of Z-average (nm), and z denotes PDI at each pH value. | ||
In several research paper as recently reported, the chemical stability of lignin nanomaterials under high pHs or in certain organic solvent has been highlighted as a desired property for efficient chemical integration of these bio-based nanomaterials in functional materials/systems.41,42 Hereby, we compared and discussed our approach to endow the lignin nanomaterials with alkali resistance with other existing approaches in the literature, as summarized in Table 2. Most commonly, in order to obtain harsh alkaline solution and organic solvent dispersed LNPs, covalent cross-linking is in need to govern the lignin solubility, such as intraparticular enzymatic polymerization,43 intraparticular cross-linking via phenolic resin curing13 as well as epoxy condensation.41,44 Compared with these studies, the synthesis of LNP@Ag in our approach relies solely on the biopolymer of lignin resulting inert nanomaterials as the carrier for in situ reduction of AgNPs, which mitigates the need of petroleum-based polymers/crosslinkers (e.g., phenol-formaldehyde or epoxy resin) in this type of application and is comparatively more benign and eco-friendly.
| Approach to improve alkali/solvent resistance of LNP | Reaction condition | Applications | Pros (+) and cons (−) | Ref. |
|---|---|---|---|---|
| Intraparticle crosslinking via lignin-containing phenol-formaldehyde curing | Heat: 65 °C for 1 h; 90 °C for 30 min; hydrothermal at 120 °C for 12 h | Nanosphere support for catalysis by AgNPs | (+) resistant to dissolution at pH 11 | 13 |
| (−) low lignin content in nanosphere (40 wt%) | ||||
| Intraparticle cross-linking via epoxy curing reaction | Heat: 105 °C for 4 h | Waterborne wood adhesive with covalent integration of LNPs | (+) alkali (pH 12) and organic solvent resistant, e.g., acetone-water (3:1) |
41 |
| (−) toxicity and leaching issues of the cross-linker | ||||
| Epichlorohydrin cross-linking resulting lignin supracolloidal structure | Heat: 55 °C for 40 min | LNPs stabilize Pickering emulsions as nanoreactor for in situ AgNPs reduction | (+) tunable size distribution; resistant to dissolution at pH 13 | 44 |
| (−) most of the silver reduced resides inside the LNP | ||||
| Surface and intraparticle cross-linking via enzyme-catalyzed oxidation | Ambient temperature, overnight | LNP decolorization | (+) improved colloidal stability in organic solvent, e.g., THF | 43 |
| (−) low concentration (0.1–0.3 mg mL−1) | ||||
| Bacterial-laccase assisted lignin polymerization prior to LNP preparation | Heat: 39 °C for 4 h | Sole LNP supported AgNP as a nanocomposite antimicrobial agent | (+) high Ag loading; holistic water-phase synthesis | This work |
| (−) less environmentally friendly THF was used |
Characterizations of LNP@Ag hybrid nanoparticles
The kinetic curves of LNPs impregnated in [Ag(NH3)2]+ solution were studied by UV-vis spectrum, and the SPR bands of AgNPs were monitored as a function of time. As illustrated in Fig. 4a, the absorption peaks from lignin were observed at around 289 and 295.8 nm, which belong to the characteristics of π–π* transition of the benzene structural units in lignin and extended conjugation (e.g., carbonyl groups and/or double bonds) on the benzene rings, respectively.45,46 The UV-vis absorption peaks of lignin were maintained in [Ag(NH3)2]+ solution at pH 11 during the reaction (Table S2†). Noteworthy, the peak position of the maximum plasmon absorption band is progressively red-shifted from 361 nm (160 min) to 432 nm (360 min), and the relative intensity between it and lignin at 289 nm and/or 295.8 nm increased with reaction time until 6 hours (Table S2†). The results are indicative of the formation and continuous growth of AgNPs on the surface of LNPs, which was also evident from the particle size analysis of AgNPs by TEM (Fig. S5†). In addition, there is a change in the color of the LNP-[Ag(NH3)2]+ aqueous dispersion from colorless to yellowish, followed by getting into brown yellow (Fig. 4b). These changes were evidence of oxidation and the associated reaction between LNPs and [Ag(NH3)2]+. FTIR spectroscopy was further used to probe the chemical composition of LM-4-NP and LM-4-NP@Ag (Fig. S6†). The non-conjugated CO stretching of carbonyl/carboxyl groups at 1720 cm−1 in LM-4-NP@Ag was obviously stronger than that of the LM-4-NP, while the peaks belonged to the aromatic –OCH3 groups (e.g., 2936 and 2841 cm−1) decreased in intensity. Furthermore, the peak belonged to the syringyl ring breathing with C–O stretching at 1215 cm−1 shifted to a higher wavenumber in the FTIR spectrum of LM-4-NP@Ag. Hereby, we propose that the hydroxyl groups and -OCH3 in lignin are oxidized to chromophores, such as quinone and α-carbonyl groups during the redox reaction that can absorb UV light in the vicinity of 250–400 nm, and possibly even reach into visible region of the spectrum.47,48 In the meantime, XPS analysis corroborated the formation of carbonyl and carboxyl groups after the redox reaction (Fig. S4, Table S3†), which is in agreement with the XPS results reported elsewhere.19
 | ||
| Fig. 4 (a) UV-vis absorption spectra of aqueous dispersion containing LM-4-NPs and [Ag(NH3)2]+ solution (10 mg mL−1). 0–120 min, no silver SPR band formation, also no absorption peak of ionized phenolic structures at 350–400 nm, indicating the alkali resistance of the LM-4-NPs; 120–150 min, the silver reduction reaction is starting; 160–220 min, the peak position of the maximum silver SPR band is red-shifted from 361.2 nm to 419.2 nm presenting the formation and rapid growth of AgNPs; 230–360 min, the peak position of the maximum silver SPR band is red-shifted from 421 nm to 431.8 nm suggesting a continuous growth of AgNPs, but the reaction rate slowed down. (b) The photograph of the LNP-[Ag(NH3)2]+ aqueous dispersion over time. TEM images of (c) LM-4-NP@Ag-3, (d) LM-4-NP@Ag-5, and (e) LM-4-NP@Ag-10 with a scale bar of 100 nm and magnification of 80000×, and their particle size histograms (f–i). | ||
The morphology and size of the LNP@Ag were examined by TEM (Fig. 4c–e) and scanning electron microscope (SEM) measurements (Fig. S7†). Spherical/quasi-spherical shape of AgNPs was surface-embedded on LNP support and the hybrid LNP@Ag displayed a nanoparticle size of 164.5 ± 17.5 nm (Fig. 4f), which was consistent with DLS results (Fig. 3a). As well recognized, the antimicrobial activity of AgNPs is size-dependent, influencing the Ag+ release (smaller AgNPs release more Ag+) and particle-specific biological effects, e.g., nanoparticles/bacteria interaction.11,49,50 In the present synthesis, it is confirmed that the size of the AgNPs formed on the LNP spheres was dependent on the concentration of silver precursor solution. As illustrated in Fig. 4c–e and their corresponding histograms of AgNPs size distribution (Fig. 4g–i), an increase in the concentration of [Ag(NH3)2]+ solution from 3 to 5 mg mL−1 led to a significant increase in the average size of the AgNPs from 8 to approx. 16 nm. It can be assumed that compared with Ag+, a higher lignin content can result in faster reducing and stabilizing of the formed AgNPs, thus small-size AgNPs form. However, the average size of AgNPs became less significant with further increase in the [Ag(NH3)2]+ concentration from 5 to 10 mg mL−1, suggesting the amount of Ag+ in the medium was excessive during the reaction. In addition, the uniform distribution of AgNPs on LNP spheres was further ascertained with narrow PDI (0.10–0.15) derived from the AgNPs size-distribution histograms (Fig. 4g–i).
The crystalline structure of LNP@Ag was investigated by X-ray diffraction (XRD), as shown in Fig. 5a. The broad diffraction peak at around 2θ = 21° was attributed to the amorphous phase of lignin. Five characteristic diffraction peaks at 2θ = 38.1°, 44.1°, 64.4°, 77.4°, and 81.5°, corresponding to the (111), (200), (220), (311) and (222) lattice planes, could be readily indexed to the face-centered-cubic crystalline (FCC) structure of metallic Ag confirmed by JCPDS (no. 89-3722). The key parameters, including the lattice constant (a0) and crystallite size of AgNPs were also calculated from the XRD patterns of LNP@Ag (see details in ESI†). It was found that there was only a 0.10–0.15% deviation between the a0 (4.0910 Å) calculated from AgNPs and the standard a0 of Ag (4.0857 Å) with FCC crystal structure (Table S4†). In addition, the characteristic diffraction peaks of silver oxides (e.g., Ag2O and AgO) were not found in the XRD patterns of LNP@Ag, supporting the conclusion that lignin is an effective reducing agent for the formation of metallic AgNPs with a well-defined crystalline structure.13,51 The average crystallite size of Ag (i.e., individual crystallite) calculated from the XRD pattern of LNP@Ag using Scherrer equation was around 3.5–4.0 nm (see details in ESI, Table S5†),52 which was smaller than the particle size of AgNPs (8.1–20.7 nm) estimated from TEM measurement. This difference suggests the presence of Ag clusters in the TEM images composed of several Ag particles. In addition, the presence of incoherent regions of lignin capping layer on the crystallite core of Ag could also partially account for the above-mentioned size difference (Fig. S8†). The elemental composition of the near-surface layer of the LNP@Ag was evaluated by XPS, where Ag present 3.4% atomic percentage, besides 75.1% of C and 21.5% of O from lignin (Table S6†). XPS was then used to further investigate the valence state of Ag on the surface of LNP. The bands observed at binding energies of 368.8 and 374.9 eV in the Ag 3d high-resolution spectrum are attributed to Ag 3d5/2 and Ag 3d3/2 of Ag0, respectively (Fig. 5b and c). It should be noted here that the binding energy of Ag 3d5/2 for LNP@Ag sample shifted to a higher value compared with those of the pure AgNPs reduced by sodium borohydride and the bulk Ag0, which has a standard binding energy of Ag 3d5/2 at about 368.2 eV.53 This suggests that the electron density of Ag atoms decreased, which might be ascribed to the embedding of AgNPs on the surface of lignin nanosphere. This observation is consistent with the results reported by Zhang et al.53
 | ||
| Fig. 5 (a) XRD patterns of the LM-4-NP and LM-4-NP@Ag. (b) XPS survey spectra of LM-4-NP and LM-4-NP@Ag and (c) is the Ag 3d high-resolution spectrum of LM-4-NP@Ag. | ||
The loading amount of AgNPs on LNP@Ag spheres fabricated in different concentrations (3, 5, and 10 mg mL−1) of [Ag(NH3)2]+ solution was respectively determined as 30.6, 46.8, and 47.2 wt%, by the inductively coupled plasma-optical emission spectroscopy (ICP-OES) measurements. In comparison to a maximum loading value of 21.6 wt% for AgNP on the LPR nanospheres as earlier reported by Chen et al.,13 this again confirms a high efficacy of the laccase-polymerized LNP for reducing Ag+ as AgNP. Under the principles of “greenness” and economic feasibility, two metrics for measuring the resource efficiency and environmental impact of the LNP@Ag manufacturing process were also evaluated in Table S7,† besides the yield of LNP@Ag. It was found that the Ag(NH3)2NO3 reagent loss is the major source of waste, and the E factor – mass of waste/mass of product as well as the reaction mass efficiency – mass of product/mass of total input maintained at a good level when increasing the concentration of Ag(NH3)2NO3 from 3 to 5 mg mL−1. However, further increasing the Ag(NH3)2NO3 concentration to 10 mg mL−1 and extending the reaction time from 4 to 6 hours, the E factor increased and the reaction mass efficiency was impaired, although the yield of LNP@Ag had a slight increase from 143 to 150 wt%. Therefore, in industrial practice, LNP@Ag was recommended to be synthesized in Ag(NH3)2NO3 solution with intermediate concentration (e.g., 3–5 mg mL−1) and reaction time to maximize resource efficiency and minimize waste.
GGMMA/LNP@Ag resin: photo-crosslinking kinetics and hydrogel mechanical properties
The photo-crosslinkable resin was formulated with 10 (w/v)% GGMMA, 0.25 (w/v)% LAP photoinitiator, and varied LNP@Ag contents, ranging from 0.02 to 0.5 wt%. The photo-crosslinking kinetics of GGMMA/LNP@Ag resins were studied by photorheology. Fig. 6a shows that the cross-linking was initiated immediately after shedding upon UV405 (start from 60 s), and the crosslinking process was as short as within 30 s to reach a levelling-off of the storage modulus (G′) in all investigated formulations. Noteworthy, the value of the maximum G′ decreased as LNP@Ag dosage increased from 0.1 to 0.5 wt%. To further ascertain the effect of LNP@Ag content on the mechanical properties of GGMMA-based hydrogel, the compressive stress–strain profile of the UV405-cast GGMMA/LNP@Ag discs was compared in Fig. 6b. It was clear that the addition of LNP@Ag in GGMMA up to 0.1 wt% did not dramatically affect the mechanical stiffness of the resulted GGMMA-based hydrogels. However, the stress at break decreased significantly as the doping amount of LNP@Ag increased from 0.1 to 0.25 wt% (Fig. 6b). To achieve desirable mechanical and DLP printing performance, a demand is that the formulated resins are homogeneous. Consequently, the distribution of the LNP@Ag within the polymeric matrix was examined after processing the composite hydrogel as the lyophilized cryogel by the SEM imaging. The result showed that 0.1 wt% dosage of LNP@Ag was distributed uniformly inside the hydrogel (Fig. 6c). However, when the dosage of LNP@Ag increased from 0.1 to 0.25%, the distribution of LNP@Ag in the GGMMA matrix was less homogenous with presence of larger-sized aggregates of LNP@Ag in the polymeric matrix, as revealed by the atomic force microscopy (AFM) measurement (Fig. 6d and e). It is inferred that a high amount of LNP@Ag can deteriorate the network connectivity of the cross-linked GGMMA matrix. Other factors taken into consideration for the deteriorated crosslinking with a high LNP@Ag doping also include the severe UV405 absorption by AgNPs. | ||
| Fig. 6 (a) Photo-crosslinking profiles of the formulated GGMMA/LNP@Ag resin during UV405 irradiation. (b) Stress–strain curves of GGMMA/LNP@Ag hydrogel discs with varied LNP@Ag contents. (c) SEM image of polymeric walls observed in the cross-sectional aerogel of 10GGMMA/0.1LNP@Ag. AFM images of (d) 10GGMMA/0.1LNP@Ag and (e) 10GGMMA/0.25LNP@Ag spin-coated films. | ||
The printability of biomass-based GGMMA/LNP@Ag resin via DLP printing
GGMMA/LNP@Ag hydrogel was fabricated via DLP additive manufacturing using 10GGMMA/0.1LNP@Ag resin, including 0.25 (w/v)% LAP photoinitiator. Honeycomb and crosshatch scaffolds as well as ultra-thin pattern (490 μm thickness) were printed as demos on the compatibility of this bio-based resin in use for DLP hydrogel printing (Fig. 7a). The printed scaffold had small pores of 500 μm reflecting their computer-aided design (CAD) images (Fig. S9†). To assess the pattern fidelity of DLP printing using GGMMA-based resin, we measured the actual pore size of the honeycomb via microscope right after printing and determined the deviation by comparing the designed size. Deteriorated lateral and axial resolution and fidelity (Fig. 7b(i)) were found in the DLP printed 10GGMMA honeycomb hydrogel, namely, the geometry size was not identical to the targeted printing size. More specifically, the blurred projected pattern (over-curing layers beyond the focus plane, indicated by arrows) and excess crosslinking in the honeycomb indicated the problem of excessive “light trespassing” owing to the weak light absorption of optically clear 10GGMMA resin. In a reference resin of 10GGMMA/0.1LNP, the lateral fidelity of the DLP printing was improved, although axial fidelity was not satisfactory by the excessive light penetration. Very interesting, well-defined projected patterns mirroring their CAD images were fabricated with the resin formulation doping of 0.1wt% LNP@Ag to the 10%GGMMA. These results implied that AgNPs in 10GGMMA/0.1LNP@Ag resin served to mitigate excess light penetration into the honeycombs, performing as a “photo-absorber” since the SPR peak absorbance of LNP@Ag (380–420 nm) encompasses the UV405 light source. Consequently, high-quality DLP-printed structures were fabricated with desired lateral and axial fidelity using GGMMA/LNP@Ag resin as demonstrated in (Fig. 7b(ii)) with 1 mm pore size and 35 μm layer thickness. Similarly, AuNPs were also found for attenuating light in lithography-based 3D printing.23,54 The dimensional changes of DLP printed 10GGMMA, 10GGMMA/0.1LNP, and 10GGMMA/0.1LNP@Ag hydrogels (4 × 4 mm in cross-section and 3 mm in height) were measured over time at PBS buffer at 37 °C. It was found that the printed cubic hydrogels using 10%GGMMA-based resin could maintain their shape structural stability and no swollen was observed over 7 days at physiological conditions presumably owing to the high cross-linking density. | ||
| Fig. 7 Fidelity of 10GGMMA/0.1LNP@Ag resin using DLP printer. (a) Fabrication of 3D objects by projection lithography. Light patterns that corresponding to a 3D model are projected into a bath containing photocurable liquid resin. Upon layer-by-layer photopatterning, 3D scaffolds with distinct structures were obtained. From left to right: Honeycomb, crosshatch, and ultra-thin layer. (b) The desired pattern was designed through CAD software. Microscopic images; (i) from left to right: DLP printed honeycomb scaffold (1 mm in height) using 10GGMMA, 10GGMMA/0.1LNP, and 10GGMMA/0.1LNP@Ag resin, respectively. (ii) From left to right: DLP printed honeycomb scaffold using 10GGMMA/0.1LNP@Ag resin with height of 1 mm, 3 mm, and its zoom in microscopic image demonstrating high resolution. | ||
Ag+ release and antimicrobial properties of the GGMMA/LNP@Ag hydrogel
As a proof-of-concept, the potential applications of GGMMA/LNP@Ag hydrogel are further demonstrated as antimicrobial hydrogel. The antibacterial activity of silver containing hydrogels is commonly dependent on the release of Ag+ from the hydrogel to the pathogenic environment, where Ag0 was oxidized in an aqueous solution exposed to air, resulting in the progressive release of Ag+.11 The release profile of Ag+ from GGMMA/LNP@Ag hydrogels exhibits a sustained release kinetics without burst release (Fig. 8a), which could be exerted in prolonged therapeutic effects.55 It is worth noting that the total amount of Ag+ released upon 11 days of soaking in PBS (pH 7.4) media did not exceed 3 wt% of the Ag loading content, indicating the majority of the AgNPs remained in the hydrogel. As expected, the GGMMA/LNP@Ag hydrogels showed strong antibacterial activities against both Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus) as revealed by the diameter of inhibition zone in disk diffusion test and bactericidal ratio (%) from quantitative analysis. The diameter of the inhibition zone reflects the magnitude of susceptibility of microorganisms, while the strains susceptible to antibiotic compounds exhibit a larger diameter of inhibition zone, whereas resistant strains exhibit a smaller inhibition zone diameter. As noted in Fig. S10,† the agar plates seeded with GGMMA/LNP@Ag hydrogels exhibited a clear inhibition zone, whereas pristine GGMMA and GGMMA/LNP hydrogels had no measurable effect on E. coli and S. aureus, and the diameter of the inhibition zone increased with increasing content of LNP@Ags. We can speculate that the Ag+ released from the surface-embedded AgNPs on spherical LNPs endows the antibacterial activities to the polysaccharide-based hydrogels. Quantitative analysis results indicate that the microbial survivors on GGMMA/LNP@Ag hydrogels were appreciably less than that on control group (bacterial suspension without hydrogel) and GGMMA and GGMMA/LNP hydrogels only upon 1-hour incubating with E. coli (Fig. 8c and d) and S. aureus (Fig. 8e and f), which is in agreement with the results of inhibition zone test. In addition, the bactericidal ratios were significantly upregulated upon integrating higher concentrations of LNP@Ag in GGMMA hydrogels. For instance, upon incubating with E. coli for 1 hour, the bactericidal ratios increased in the order of 10GGMMA/0.1LNP@Ag (53%), 10GGMMA/0.25LNP@Ag (81%), and 10GGMMA/0.5LNP@Ag (92%). When the incubation time of hydrogel with bacteria was prolonged to 4 hours, almost all the bacteria were inactivated by the GGMMA/LNP@Ag hydrogels regardless of the concentration of entrapped LNP@Ags and the bacterial strains. We correlate the antibacterial activity of GGMMA/LNP@Ag mainly associated with the Ag+ release and that even a low dosage of LNP@Ag in hydrogel can be accounted for its antibacterial response. Another possible explanation is that the increase in hydrophobicity of GGMMA surface after doping with LNP@Ag limited the binding with bacteria, therefore displaying an antibacterial effect (Fig. 8b). Table S8† compares the bactericidal ratio of our 10GGMMA/0.1LNP@Ag hydrogel with recently reported studies that engaged lignin-capped AgNPs as the antimicrobial component in the fabrication of a hydrogel or film.8,50,56–58 The results show that the LNP@Ag prepared in this work is of high efficacy with respect to less loading of lignin-silver nanocomposites in achieving effective antibacterial activity. | ||
| Fig. 8 Evaluation of antimicrobial activity of the GGMMA/LNP@Ag hydrogels. (a) In vitro release profiles of Ag+ from GGMMA/LNP@Ag hydrogels. The inset was 10GGMMA/0.1LNP@Ag hydrogels upon 11 days immersing in PBS presenting good mechanical integrity. (b) Water contact angle of photocurable resin spin-coated films. Viability assays of (c and d) E. coli and (e and f) S. aureus on GGMMA-based hydrogels. Bar = mean ± STDEV; N = 9; compared with 10GGMMA hydrogel, the statistical difference in bactericidal ratio was considered significant at p ≤ 0.05. Not significant difference in bactericidal ratio was observed at some 10GGMMA/0.1LNP hydrogels, as indicated by one asterisk (*). Two asterisks (**) represent significant increase in bactericidal ratio (p < 0.01). The (d) and (f) are the agar plate cultured with survival colonies upon 1-hour incubation. | ||
In therapeutic strategy, local delivery yields higher local concentrations of the therapeutic agents, which increases the effectiveness at the site. Supported by the high-efficacy antimicrobial activity, this fast-photocurable resin of GGMMA/LNP@Ag is foreseen with a premise in use for therapeutic interfacing in a biomedical device for treatment of infections, namely referring to an implantable dressing for impaired wound healing or as an antimicrobial bioadhesive hydrogel for the treatment of peri-implant diseases in orthopaedic or dental implants, for instance. Still, the cytocompatibility of GGMMA/LNP@Ag has then to be assessed further with specific cell lines that are associated with the selected therapeutic application in future work. Importantly, the current work has also demonstrated that engaging DLP printing to print the resin formulation of GGMMA/LNP@Ag into robust hydrogel supports personalized fabrication with precise control over the microarchitecture and fast manufacturing speed. This is a starting point to propose a new frontier of utilizing these bio-based nanomaterials and biopolymers in high value-added valorizations.
Conclusions
A novel hybrid antimicrobial nanocomposite comprising an inert spherical LNP carrier and surface-embedded AgNPs was fabricated using facile in situ reduction method. We addressed the insufficient alkali-resistance challenge of conventional LNPs in the silver ammonia solution (pH 11) by applying a laccase-catalyzed lignin polymerization strategy. The in situ reduced AgNPs were homogeneously decorated on the surface of LNP spheres in resulting hybrid LNP@Ag. Successful fabrication of the LNP@Ag nanomaterials relies on fine-tuning the laccase-catalyzed lignin polymerization to balance the fractionation-dependent lignin polymerization kinetics in gaining the alkali resistance but meanwhile preserving the amount of residual functional groups available for Ag+ reduction. For instance, 4 hours enzymatic duration was optimized in obtaining LNPs from laccase-polymerized birch-MeOH-s lignin with high alkali-resistance in pH 11 alkaline solution. Meanwhile, 30.6–47.2 wt% of AgNP loading could be achieved depending on the concentration of silver precursor solution (3–10 mg mL−1). The LNP@Ag showed high aqueous dispersion stability. Upon subsequent integration with photo-crosslinkable polysaccharides, a novel sustainable resin of 10 wt% GGMMA and 0.1 wt% LNP@Ag was presented for lithography-based DLP printing. In contrast to the poor printing resolution of GGMMA hydrogel, the doping of LNP@Ag improved the lateral and axial printing fidelity and structural stability owing to the photo-absorbing effect of metallic silver. Also, the GGMMA/LNP@Ag hydrogel has bactericidal ability, which is a promising material suitable for biomedical applications. Overall, this work provides a new frontier in lithography-based DLP printing research focusing on natural-polymer-based resins.Experimental section
Materials
Birch AL was provided by CH-Bioforce Oy (Espoo, Finland). It was prepared by BLN biorefinery process, namely a pressurized hot water extraction in combination with a mild alkali pulping process under oxygen-starved conditions.59,60 The birch AL used in this work was purified by acid water (pH 2.5, 2 M HCl) washing and methyl-t-butyl ether extraction.30 Birch AL fractions were derived from the purified birch AL by three-step solvent fractionation according to the protocol established in our previous study.30 Laccase (MetZyme® product) was supplied by MetGen Oy (Kaarina, Finland). The enzyme activity of this laccase was 357 U mL−1 (on ABTS at pH 4.2, 25 °C) and the protein concentration was 9.7 mg mL−1 determined using Coomassie (Bradford) protein assay kit (ThermoFisher Scientific™). GGMMA was synthesized by reacting GGM (Norway spruce, Mn of 9 kDa) with methacrylic anhydride in mild alkaline (pH 8) aqueous solution following the protocol reported in our previous work.29 The Mn of the GGMMA was 14 kDa and degree of substitution of the methacryloyls was 0.25 determined by quantitative 13C NMR according to the integral comparison of double bond carbons (δ 126 and 135 ppm) based on anomeric carbon (δ 95–105 ppm).29 All chemicals were purchased from Sigma-Aldrich and used as received.Preparation of polymerized lignin by laccase
To prepare laccase-polymerized lignin, 200 mg of lignin were completely dissolved in 20 mL of sodium hydroxide solution (NaOH, pH 10) at 25 °C. 560 μL of laccase solution (MetZyme®) was then added following heating up the lignin solution to 39 °C. This laccase amount corresponded to enzyme activity of 1 U mg−1 of lignin. Then, the solubilized lignin was incubated with laccase at 39 °C for 0 to 6 hours with ambient air (O2) circulation under gentle agitation (500 rpm). The laccase-treated lignin was precipitated by HCl (2 M) and was collected in centrifuge tubes by centrifugation at 10000 rpm for 10 min, washed using distilled water, and then freeze-dried in a lyophilizer prior to their use for the synthesis of LNP@Ag.
Preparation of colloidal lignin nanospheres supported Ag nanoparticles (LNP@Ag)
The laccase-treated lignin without post chemical modification was used to fabricate the LNPs via solvent shifting method. For instance, 100 mg lyophilized laccase-treated MeOH-s lignin was dissolved in 100 mL THF/H2O medium (9:1, v/v). The mixtures were stirred at 500 rpm for 3 hours and then filtered through a 0.2 μm PTFE syringe filter to remove undissolved lignins. 200 mL of distilled water was added dropwise into the lignin solution under rapid magnetic stirring (1000 rpm), and then a large amount of water (around 200 mL) was added and stirred for 20 min. Thereafter, the THF was removed from the water medium by rotary evaporation, and the LNP dispersion was concentrated to 10 mL by centrifugation (10000 rpm, 10 min). LNP@Ag was prepared according to the procedure reported in previous literature.13 Silver ammonia solution (10 mg mL−1) was obtained by adding 1–2 drops of 0.1 M NaOH and subsequently adding aqueous ammonia solution (2 wt%) to 10 mL of 10 mg mL−1 AgNO3 solution until all of the brown precipitates were just dissolved. 4 mL of the silver ammonia solution (10 mg mL−1) was added to the above 2 mL of LNPs dispersion (8 mg mL−1, the yield of production of LNP from laccase-treated MeOH-s lignin was around 80 wt%, mass balance analysis in Table S9†), and the mixture was kept in an orbital shaker (Stuart S1600) at 30 °C for 4 hours under orbital shaking motion (200 rpm). After the in situ reduction, the LNP@Ag solid was collected by centrifugation (10000 rpm, 10 min) and washed with distilled water, followed by redispersing LNP@Ag in distilled water. Considering the environmental impact, the supernatant with residual Ag+ was collected and precipitated by HCl (0.5 M) for recycling. Consequently, the LNP@Ag-10 dispersion was obtained. The same sample preparation procedure was carried out to obtain the LNP@Ag-3 and LNP@Ag-5 nanospheres by using 3 mg mL−1 and 5 mg mL−1 silver ammonia solution, respectively. The LNP@Ag yield obtained from L-birch AL, L-i-PrOH, L-EtOH, and L-MeOH lignin were 126, 88, 112, and 150 wt%, respectively (Table S9†), which were calculated based on the mass balance analysis (Fig. S11†).
Preparation of photo-crosslinked hydrogel incorporated with LNP@Ag
The photocurable resin of 10GGMMA (10 w/v%) was prepared by placing in a 3 mL syringe 100 mg of freeze-dried GGMMA powder, 950 μL of distilled water, and 50 μL of LAP aqueous solution (50 mg mL−1), after which the mixture was mixed at 50 °C using a vortex mixer (FINEPCR, Korea) until GGMMA was completely dissolved to produce a homogeneous liquid resin. This LAP amount corresponded to a photoinitiator consistency of 0.25 w/v% of resin. The preparation of photocurable resin incorporated with LNP@Ag was performed with procedures similar to that of 10GGMMA. The compositional weight ratios between GGMMA and LNP@Ag were 100:1, 100:2.5, and 100:5 w/w. For example, the 10GGMMA/0.1LNP@Ag resin was prepared firstly by mixing 125 μL of LNP@Ag dispersion (8 mg mL−1) and 50 μL of LAP aqueous solution with 825 μL of distilled water and then with 100 mg GGMMA powder. Cast disc hydrogels were prepared for compression (8 mm in diameter and 4.2 mm in height) and antimicrobial tests (16 mm in diameter and 1 mm in height). The uniform discs were cast by first injecting formulated resins into a plastic disc mold followed by exposing to UV-LED (bluepoint LED eco, Hönle group, Germany) irradiation operating at 405 nm for 120 s.
DLP printing
The used DLP printing system (M-One Pro 30, Makex Co., Ltd, China) consists of three major components: UV Digital Micromirror Device (DMD) capable of 1920 × 1080 pixel resolution (related to an x–y nominal printing resolution of 33 μm, Texas Instruments, Dallas, USA), industrial HD UV-LED (405 nm) light engine, and a lens module with two UV-grade biconvex lenses. The 3D digital models were created with Autodesk Fusion 360 and sliced with X-maker V2.7.1. Printing parameters were used as follows: sample loading, 0.5 mL of photocurable liquid resin; exposure time, 80 s; layer thickness, 35 μm. After the printing process, the photo-crosslinked hydrogel was detached from the printing platform and soaked in distilled water in order to wash away the unreacted resin.Characterizations
The TEM analysis was performed using JEM-1400 in bright-field mode with an acceleration voltage of 80 kV (see details in ESI†). The Z-average, PDI, and ζ-potential of the LNP@Ag aqueous dispersion (0.2 mg mL−1) were measured by using a Zetasizer Nano instrument (Malvern Instruments) (see details in ESI†). The silver loading of samples was evaluated using ICP-OES, combined with microwave digestion pretreatment. Molar mass characterization was conducted by a gel permeation chromatography (GPC) system equipped with a differential refractive index (RI) concentration detector and a multiangle light scattering detector (MALS), following the protocol reported previously.61 The eluent for GPC analysis was DMSO with 0.05 M LiBr. The crystal structure of freeze-dried LNP@Ag sample was measured on a Bruker D8 ADVANCE X-ray diffractometer (XRD, Bruker-AXS, Karlsruhe, Germany) with CuKα radiation (λ = 1.54184 Å) under 40 kV accelerating voltage and 30 mA emission current. The diffraction angle 2θ was from 5 to 100° with a step size of 0.013°. FTIR spectra of the freeze-dried LNP and LNP@Ag nanoparticles were recorded on a Nicolet iS50 FT-IR Spectrometer (ThermoFisher Scientific™) using potassium bromide as substrate (see details in ESI†). XPS data were obtained from an ESCA Lab220i-XL electron spectrometer (ThermoFisher Scientific™) operating with a monochromated Al Ka X-ray source (300 W). The C 1s signal (at 284.8 eV) was used as the internal standard for calibration of the binding energy. Gaussian–Lorentzian Product functions (70% Gaussian and 30% Lorentzian) were used to approximate the line shapes. In addition, the full width at half maximum (FWHM) was constrained during the deconvolution of all core-level regions of XPS scans (see details in Table S3†). The photo-crosslinking kinetics of the formulated resins were measured using a rheometer (MCR 702 Multidrive, Anton Paar, Austria) under oscillation mode with a gap distance of 0.1 mm at a constant angular frequency and strain of 10 rad s−1 and 1%, respectively. The testing resins were irradiated by a UV-LED (405 nm, bluepoint LED eco, Hönle Group, Germany) after 60 s and the G′ was recorded. The compression tests were performed on cast discs using a dynamic mechanical analyzer (DMA, MCR 702 Multidrive, Anton Paar, Austria). The microscopic images of the DLP-printed hydrogels were taken using a Nikon ECLIPSE E200 optical microscope. The cross-sectional morphology of the cast disc was imaged by SEM (LEO 1530). The sample for SEM measurement was prepared through lyophilisation by first freezing it with liquid nitrogen. Topographical features of the GGMMA/LNP@Ag dry film were characterized by a MultiMode 8 AFM equipped with a Nanoscope V controller (Bruker, Santa Barbara, CA). AFM samples were obtained by spin-coating of thin GGMMA/LNP@Ag film onto a glass wafer. The films were imaged in intermittent contact mode in air at room temperature conditions (T = 22.3 ± 2 °C, relative humidity (RH) = 60 ± 5%) using gold-coated silicon probes (NSG03 AFM probe, NT-MDT, Russia). Images captured were of 5 μm × 5 μm size with a 1024 × 1024 resolution. Shown images have been plane fitted after scanning, and their z-axis aspect-ratios exaggerated by a factor of 3 for clear visualization of the data. The static water contact angle of the GGMMA-based resin was measured with a KSV CAM200 instrument (KSV Instruments Ltd, Finland) using a sessile drop method. The samples were prepared by spin-coating the photocurable resin onto a silicon wafer at 2000 rpm for 90 s and curing by UV irradiation for 5 s.Ag+ release from GGMMA/LNP@Ag hydrogel and antimicrobial tests
UV405 cast hydrogel discs with an overall dimension of 16 mm in diameter and 1 mm in height (201 mm3) were soaked in 15 mL of phosphate-buffered saline (PBS, pH 7.4) at 37 °C to examine Ag+ release, and the tests were carried out in triplicate. At predetermined intervals (6, 12 hours, 1, 3, 5, 7, 11 days), 1 mL of the release media was collected and replaced with 1 mL of fresh PBS. The Ag+ concentration of the release media was quantified using an ICP-OES at 328 nm. The antimicrobial activity of the GGMMA/LNP@Ag hydrogels was evaluated using E. coli and S. aureus as reported previously.62 Three parallel samples of each group were used for the antibacterial test. First, E. coli and S. aureus were respectively inoculated in Luria–Bertani (LB) broth and Trypticase–Soy (TS) broth for 18 h at 37 °C with shaking, and then diluted in corresponding broth media to a concentration of 105 colony-forming unit (CFU mL−1). Next, 300 μL of the GGMMA, GGMMA/LNP, and GGMMA/LNP@Ag resins were placed and UV405-crosslinked in a 24-well plate, and 200 μL of bacterial suspension (105 CFU mL−1) was seeded onto the hydrogel surface. In addition, each plate carried a negative control (200 μL bacterial suspension, 105 CFU mL−1) and gentamicin (0.0125 mg mL−1) as a positive control. The 24-well plates were incubated at 37 °C for 1, 2, and 4 hours in an incubator, and 2 mL of PBS was added to each well to resuspend the viable bacteria. A series of 100-fold dilutions of bacterial suspensions (100 μL) from each well were respectively plated onto LB and TS agar, and then incubated at 37 °C for 24 h. The number of bacterial colonies was recorded, and data were expressed as bactericidal ratio calculated based on the numbers of CFU on agar plate according to eqn (1): | (1) |
The statistical differences in bactericidal ratio between two groups were calculated with the two-tailed unpaired t test.
Author contributions
L. Wang, C. Xu, and X. Wang conceived and initiated the work. All authors contributed to experimental design, planning, execution, and data analysis. L. Wang carried out the main experiment work and drafted the manuscript. Q. Wang carried out the lab work on the synthesis of GGMMA as well as mainly contributed to the DLP printing, photorheology, mechanical analysis, and microscope analyses. A. Slita contributed to the antimicrobial analysis of the hydrogel samples. O. Backman developed model designs and parameters for the DLP printing. Z. Gounani and E. Rosqvist contributed to the AFM measurement. J. Peltonen, S. Willför, C. Xu, and J. M. Rosenholm provided critical revision to the manuscript. X. Wang is the main principal scientist who supervised the work. The manuscript has been approved by all the coauthors for submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Both Ms Luyao Wang and Mr Qingbo Wang would like to acknowledge the financial support from the China Scholarship Council (Student ID 201804910639 for L. Wang and Student ID 201907960002 for Q. Wang respectively) to their doctoral study at ÅAU, Finland. X. Wang would like to thank Academy of Finland (333158) as well as Jane and Aatos Erkko Foundation for their funds to her research at ÅAU. C. Xu would like to acknowledge funding from Business Finland Project (43674/31/2020). E. Rosqvist acknowledges Jane and Aatos Erkko Foundation for their funding of his work. J. M. Rosenholm acknowledges funding from the Sigrid Jusélius Foundation, Medicinska Understödsföreningen Liv och Hälsa r.f. and Magnus Ehrnrooth Foundation. Electron microscopy samples were processed and analyzed in the Electron Microscopy Laboratory, Institute of Biomedicine, University of Turku, which receives financial support from Biocenter Finland. Petri Ihalainen and Liji Sobhana Seleenmary Sobhanadhas (MetGen Oy, Finland) are acknowledged for providing the enzyme used in this study.Notes and references
- I. Armentano, M. Dottori, E. Fortunati, S. Mattioli and J. M. Kenny, Polym. Degrad. Stab., 2010, 95, 2126–2146 CrossRef CAS.
- V. K. Thakur and M. K. Thakur, Int. J. Biol. Macromol., 2015, 72, 834–847 CrossRef CAS PubMed.
- W. Xu, X. Wang, N. Sandler, S. Willför and C. Xu, ACS Sustainable Chem. Eng., 2018, 6, 5663–5680 CrossRef CAS PubMed.
- J. Yang, X. An, L. Liu, S. Tang, H. Cao, Q. Xu and H. Liu, Carbohydr. Polym., 2020, 250, 116881 CrossRef CAS PubMed.
- H. Liu, T. Chen, C. Dong and X. Pan, Curr. Med. Chem., 2020, 27, 4647–4659 CrossRef CAS PubMed.
- R. R. Arvizo, S. Bhattacharyya, R. A. Kudgus, K. Giri, R. Bhattacharya and P. Mukherjee, Chem. Soc. Rev., 2012, 41, 2943–2970 RSC.
- M. Biondi, A. Borzacchiello, L. Mayol and L. Ambrosio, Gels, 2015, 1, 162–178 CrossRef PubMed.
- D. Gan, W. Xing, L. Jiang, J. Fang, C. Zhao, F. Ren, L. Fang, K. Wang and X. Lu, Nat. Commun., 2019, 10, 1487 CrossRef PubMed.
- M. E. Quadros and L. C. Marr, Environ. Sci. Technol., 2011, 45, 10713–10719 CrossRef CAS PubMed.
- X. Chen and H. J. Schluesener, Toxicol. Lett., 2008, 176, 1–12 CrossRef CAS PubMed.
- Z. M. Xiu, Q. B. Zhang, H. L. Puppala, V. L. Colvin and P. J. J. Alvarez, Nano Lett., 2012, 12, 4271–4275 CrossRef CAS PubMed.
- H. Hiramatsu and F. E. Osterloh, Chem. Mater., 2004, 16, 2509–2511 CrossRef CAS.
- S. Chen, G. Wang, W. Sui, A. M. Parvez and C. Si, Green Chem., 2020, 22, 2879–2888 RSC.
- G. Wang, T. Pang, S. Chen, W. Sui, C. Si and Y. Ni, Green Chem., 2020, 22, 8594–8603 RSC.
- M. H. Sipponen, H. Lange, M. Ago and C. Crestini, ACS Sustainable Chem. Eng., 2018, 6, 9342–9351 CrossRef CAS PubMed.
- Y. Wu, Y. Qian, H. Lou, D. Yang and X. Qiu, ACS Sustainable Chem. Eng., 2019, 7, 15966–15973 CrossRef CAS.
- M. Farooq, T. Zou, G. Riviere, M. H. Sipponen and M. Österberg, Biomacromolecules, 2019, 20, 693–704 CrossRef CAS PubMed.
- M. H. Sipponen, H. Lange, C. Crestini, A. Henn and M. Österberg, ChemSusChem, 2019, 12, 2039–2054 CrossRef CAS PubMed.
- Y. Xue, X. Qiu, Z. Liu and Y. Li, ACS Sustainable Chem. Eng., 2018, 6, 7695–7703 CrossRef CAS.
- Y. N. Slavin, K. Ivanova, J. Hoyo, I. Perelshtein, G. Owen, A. Haegert, Y. Lin, S. Lebihan, A. Gedanken, O. H. Urs, T. Tzanov and H. Bach, ACS Appl. Mater. Interfaces, 2021, 13, 22098–22109 CrossRef CAS PubMed.
- Z. Shen, Y. Luo, Q. Wang, X. Wang and R. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 16147–16155 CrossRef CAS PubMed.
- L. Wang, L. Tan, L. Hu, X. Wang, R. Koppolu, T. Tirri, B. van Bochove, P. Ihalainen, L. S. Seleenmary Sobhanadhas, J. V. Seppälä, S. Willför, M. Toivakka and C. Xu, ACS Sustainable Chem. Eng., 2021, 9, 8770–8782 CrossRef CAS.
- C. Yu, J. Schimelman, P. Wang, K. L. Miller, X. Ma, S. You, J. Guan, B. Sun, W. Zhu and S. Chen, Chem. Rev., 2020, 120, 10695–10743 CrossRef CAS PubMed.
- E. M. Maines, M. K. Porwal, C. J. Ellison and T. M. Reineke, Green Chem., 2021, 23, 6863–6897 RSC.
- M. Müller, J. Becher, M. Schnabelrauch and M. Zenobi-Wong, Biofabrication, 2015, 7, 35006 CrossRef PubMed.
- K. S. Lim, R. Levato, P. F. Costa, M. D. Castilho, C. R. Alcala-Orozco, K. M. A. van Dorenmalen, F. P. W. Melchels, D. Gawlitta, G. J. Hooper and J. Malda, Biofabrication, 2018, 10, 034101 CrossRef PubMed.
- B. Grigoryan, S. J. Paulsen, D. C. Corbett, D. W. Sazer, C. L. Fortin, A. J. Zaita, P. T. Greenfield, N. J. Calafat, J. P. Gounley, A. H. Ta, F. Johansson, A. Randles, J. E. Rosenkrantz, J. D. Louis-Rosenberg, P. A. Galie, K. R. Stevens and J. S. Miller, Science, 2019, 364, 458–464 CrossRef CAS PubMed.
- S. H. Kim, Y. K. Yeon, J. M. Lee, J. R. Chao, Y. J. Lee, Y. B. Seo, M. T. Sultan, O. J. Lee, J. S. Lee, S. Il Yoon, I. S. Hong, G. Khang, S. J. Lee, J. J. Yoo and C. H. Park, Nat. Commun., 2018, 9, 1–14 CrossRef PubMed.
- W. Xu, X. Zhang, P. Yang, O. Långvik, X. Wang, Y. Zhang, F. Cheng, M. Österberg, S. Willför and C. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 12389–12400 CrossRef CAS PubMed.
- L. Wang, L. Lagerquist, Y. Zhang, R. Koppolu, T. Tirri, I. Sulaeva, S. von Schoultz, L. Vähäsalo, A. Pranovich, T. Rosenau, P. C. Eklund, S. Willför, C. Xu and X. Wang, ACS Sustainable Chem. Eng., 2020, 8, 13517–13526 CrossRef CAS.
- J. D. Zwilling, X. Jiang, F. Zambrano, R. A. Venditti, H. Jameel, O. D. Velev, O. J. Rojas and R. Gonzalez, Green Chem., 2021, 23, 1001–1012 RSC.
- F. Xiong, Y. Han, S. Wang, G. Li, T. Qin, Y. Chen and F. Chu, ACS Sustainable Chem. Eng., 2017, 5, 2273–2281 CrossRef CAS.
- T. Pang, G. Wang, H. Sun, L. Wang, Q. Liu, W. Sui, A. K. Parvez and C. Si, ACS Sustainable Chem. Eng., 2020, 8, 9174–9183 CrossRef CAS.
- L. Munk, A. K. Sitarz, D. C. Kalyani, J. D. Mikkelsen and A. S. Meyer, Biotechnol. Adv., 2015, 33, 13–24 CrossRef CAS PubMed.
- D. Tian, J. Hu, R. P. Chandra, J. N. Saddler and C. Lu, ACS Sustainable Chem. Eng., 2017, 5, 2702–2710 CrossRef CAS.
- Z. H. Liu, N. Hao, S. Shinde, Y. Pu, X. Kang, A. J. Ragauskas and J. S. Yuan, Green Chem., 2019, 21, 245–260 RSC.
- Y. Qian, Y. Deng, X. Qiu, H. Li and D. Yang, Green Chem., 2014, 16, 2156–2163 RSC.
- M. Lievonen, J. J. Valle-Delgado, M. L. Mattinen, E. L. Hult, K. Lintinen, M. A. Kostiainen, A. Paananen, G. R. Szilvay, H. Setälä and M. Österberg, Green Chem., 2016, 18, 1416–1422 RSC.
- A. P. Richter, B. Bharti, H. B. Armstrong, J. S. Brown, D. Plemmons, V. N. Paunov, S. D. Stoyanov and O. D. Velev, Langmuir, 2016, 32, 6468–6477 CrossRef CAS PubMed.
- C. Frangville, M. Rutkevičius, A. P. Richter, O. D. Velev, S. D. Stoyanov and V. N. Paunov, ChemPhysChem, 2012, 13, 4235–4243 CrossRef CAS PubMed.
- T. Zou, M. H. Sipponen, A. Henn and M. Österberg, ACS Nano, 2021, 15, 4811–4823 CrossRef CAS PubMed.
- W. Jiang, S. Liu, C. Wu, Y. Liu, G. Yang and Y. Ni, Green Chem., 2020, 22, 8734–8744 RSC.
- M. L. Mattinen, J. J. Valle-Delgado, T. Leskinen, T. Anttila, G. Riviere, M. Sipponen, A. Paananen, K. Lintinen, M. Kostiainen and M. Österberg, Enzyme Microb. Technol., 2018, 111, 48–56 CrossRef CAS PubMed.
- T. E. Nypelö, C. A. Carrillo and O. J. Rojas, Soft Matter, 2015, 11, 2046–2054 RSC.
- B. Wang, D. Sun, H. M. Wang, T. Q. Yuan and R. C. Sun, ACS Sustainable Chem. Eng., 2019, 7, 2658–2666 CrossRef CAS.
- N. Ratanasumarn and P. Chitprasert, Int. J. Biol. Macromol., 2020, 153, 138–145 CrossRef CAS PubMed.
- H. Sadeghifar and A. Ragauskas, Polymers, 2020, 12, 1134 CrossRef CAS PubMed.
- Y. Qian, Y. Deng, H. Li and X. Qiu, Ind. Eng. Chem. Res., 2014, 53, 10024–10028 CrossRef CAS.
- J. R. Morones, J. L. Elechiguerra, A. Camacho, K. Holt, J. B. Kouri, J. T. Ramírez and M. J. Yacaman, Nanotechnology, 2005, 16, 2346–2353 CrossRef CAS PubMed.
- A. G. Morena, I. Stefanov, K. Ivanova, S. Pérez-Rafael, M. Sánchez-Soto and T. Tzanov, Ind. Eng. Chem. Res., 2020, 59, 4504–4514 CrossRef CAS.
- G. A. Sotiriou, A. Teleki, A. Camenzind, F. Krumeich, A. Meyer, S. Panke and S. E. Pratsinis, Chem. Eng. J., 2011, 170, 547–554 CrossRef CAS PubMed.
- J. S. J. Hargreaves, Catal., Struct. React., 2016, 33–37 CAS.
- L. Zhang, H. Lu, J. Chu, J. Ma, Y. Fan, Z. Wang and Y. Ni, ACS Sustainable Chem. Eng., 2020, 8, 12655–12663 CrossRef CAS.
- S. Kumar, J. Aaron and K. Sokolov, Nat. Protoc., 2008, 3, 314–320 CrossRef CAS PubMed.
- K. Yang, Q. Han, B. Chen, Y. Zheng, K. Zhang, Q. Li and J. Wang, Int. J. Nanomed., 2018, 13, 2217–2263 CrossRef CAS PubMed.
- M. Li, X. Jiang, D. Wang, Z. Xu and M. Yang, Colloids Surf., B, 2019, 177, 370–376 CrossRef CAS PubMed.
- S. Shankar, J. W. Rhim and K. Won, Int. J. Biol. Macromol., 2018, 107, 1724–1731 CrossRef CAS PubMed.
- S. Shankar and J. W. Rhim, Food Hydrocolloids, 2017, 71, 76–84 CrossRef CAS.
- S. von Schoultz, WO 2015/104460A1, 2015.
- S. von Schoultz, WO 2014/009604A1, 2014.
- G. Zinovyev, I. Sulaeva, S. Podzimek, D. Rössner, I. Kilpeläinen, I. Sumerskii, T. Rosenau and A. Potthast, ChemSusChem, 2018, 11, 3259–3268 CrossRef CAS PubMed.
- A. Sun, X. He, L. Li, T. Li, Q. Liu, X. Zhou, X. Ji, W. Li and Z. Qian, NPG Asia Mater., 2020, 12, 25 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03841a |
| This journal is © The Royal Society of Chemistry 2022 |