
Identification and quantification of lignin monomers and oligomers from reductive catalytic fractionation of pine wood with GC × GC – FID/MS†
Hang
Dao Thi‡
a,
Korneel
Van Aelst‡
b,
Sander
Van den Bosch
 b,
Rui
Katahira
c,
Gregg T.
Beckham
c,
Bert F.
Sels
*b and
Kevin M.
Van Geem
*a
b,
Rui
Katahira
c,
Gregg T.
Beckham
c,
Bert F.
Sels
*b and
Kevin M.
Van Geem
*a
aLaboratory for Chemical Technology, Ghent University, Technologiepark 121, 9052 Ghent, Belgium. E-mail: kevin.vangeem@ugent.be
bCenter for Sustainable Catalysis and Engineering, KU Leuven, Celestijnenlaan 200F, Leuven 3001, Belgium. E-mail: bert.sels@kuleuven.be
cRenewable Resources and Enabling Sciences Center, National Renewable Energy Laboratory, Golden, CO 80401, USA
First published on 4th December 2021
Abstract
Thorough lignin characterization is vital to understand the physicochemical properties of lignin and to evaluate lignocellulose biorefinery processes. In this study, an in-depth characterization of lignin oil, obtained from reductive catalytic fractionation (RCF) of pine wood, was performed with quantitative GC × GC – FID analysis and qualitative GC × GC – MS. By utilizing high-temperature resistant column sets in the GC × GC system and by applying a derivatization step, unambiguous detection of lignin monomers, dimers, and trimers is enabled. In addition to confirm the identity of eleven monomers, corresponding to 34 wt% of the RCF lignin oil, thirty-six dimers (16 wt%) and twenty-one trimers (7 wt%) were comprehensively identified by analysis of their mass spectra and quantified by a FID, encompassing the identity of an additional 23 wt% of the RCF lignin oil. The proposed structures reveal the interlinkages present in the dimeric and trimeric oligomers, containing β-5, β-1, β–β, 5–5, and a minor fraction of β-O-4 and 4-O-5 bonds. Furthermore, aliphatic end-units in the dimeric and trimeric molecules were identified, consisting of various substituents at the C4 position, that have been previously observed in the RCF-derived lignin monomers. To reduce complexity for analysis, the RCF oil was separated into six fractions, prior to analysis. The structural motifs (inter-unit linkages and end-units) that are found in the different fractions vary significantly, such that the lignin fractions extracted in more polar solvents contained higher molecular weight fragments and more hydroxyl containing structural motifs. The identified structures of individual dimer and trimer molecules by GC × GC align well with and further complement the recent findings from 1H–13C HSQC NMR spectroscopy, demonstrating complementarity between both 2D techniques to obtain a holistic view on both the molecular structures and the distribution of bonds and end-units in RCF oil. The combination of these two techniques provides a powerful tool for future RCF and other lignin depolymerization research.
Introduction
The use of sustainable carbon sources for the production of chemicals and fuels has gained increased attention in the last decades.1,2 To this end, lignocellulose holds enormous potential due to its abundance, renewable nature, and composition. It is mainly composed of two polysaccharides, cellulose and hemicellulose, and the aromatic polymer lignin. The latter is formed by radical polymerization of p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) units, among others, in the plant cell wall, forming β-O-4 (β-aryl ether), β-5 (phenylcoumaran), 5–5 (dibenzodioxocin), 4-O-5 (diaryl ether), β-1 (spirodienone), and β–β (resinol) inter-unit linkages.3,4 Among them, the β-O-4 linkage is most abundant and is relatively labile, making it the target linkage for depolymerization processes to yield aromatic molecules with low molecular weights.5–7Many efforts have been made to convert lignin, obtained through various biorefining processes,4–6,8–10 into liquid fuels11,12 and valuable chemicals (e.g. phenol,13 polyurethane,14 phenol-formaldehyde (PF) resin,15 and epoxy resin16,17). One promising biorefining process that emerged notably in recent years is reductive catalytic fractionation (RCF) of lignocellulosic biomass.18–25 During RCF, lignin is extracted with protic solvents (e.g. MeOH,18 alcohol/H2O26) from lignocellulose, generating phenolic intermediates by selective cleavage of the labile β-O-4 linkages in lignin. Subsequently, these intermediates are stabilized by hydrogenation and hydrogenolysis with a heterogeneous redox catalyst (e.g. Pd/C) at elevated temperatures (150–250 °C) in a reductive environment (e.g. hydrogen atmosphere). As a result, the lignin macromolecules are depolymerized, yielding a mixture of phenolic monomers, dimers, and short oligomers.24,27–30 Full state of knowledge on RCF can be found in these reviews.23,24,30–34 Besides the monomers, there is little molecular understanding of the dimer and oligomer fractions of these RCF oils.30 Given the complex nature of the RCF oil in terms of composition, heterogeneity, and molecular size distribution, solvent-based fractionation can be used to provide relatively homogeneous lignin oil fractions (with regard to molecular weight and structure).6,35–37 Consequently, the resulting fractions can provide fruitful information on the molecular weight and its relationship with individual molecular structures (e.g. inter-phenolic linkages) and lignin properties such as the hydroxyl content, which are key properties to be considered for the development of the production of new materials and chemicals.17,37–39
Various analytical techniques have been developed and applied in the analysis of lignin-derived oil samples generated from lignin depolymerization,40–42 such as nuclear magnetic resonance (NMR) spectroscopy,43 Fourier-transform infrared resonance (FT-IR) spectroscopy,44 gel permeation chromatography (GPC),45,46 thermogravimetric analysis (TGA),47 and elemental analysis (EA).47–49 However, these analytical tools provide exclusively bulk information of the lignin depolymerization products.47–53 To separate and individually identify the compounds in (mostly) less/non-volatile oligomeric fractions of (lignin-derived) oil samples, high-pressure liquid chromatography (HPLC) or comprehensive two-dimensional liquid chromatography (LC × LC) combined with high-resolution multi-stage tandem mass spectrometry (HRMSn) is a common method of choice because this technique is not limited by the volatility of the analyte(s). Nonetheless, studies using this approach have focused on monomer identification, through analyzing their mass fragmentation patterns, but not substantially on the oligomers. The comprehensive quantification of oligomers is also inadequate due to the shortage of authentic standard compounds used to support the quantification of the oligomers.54–60 The most popular method to analyze RCF oils is gas chromatography (GC) coupled to mass spectrometry (MS) or a flame ionization detector (FID). However, this approach only allows identification and quantification of the volatile monomeric fractions and a small number of dimers after derivatization.18,20,27,46,52,61,62 Furthermore, due to the complex composition of lignin-derived samples, “co-elution” of components with similar physicochemical properties often occurs. As a result, the components can be incorrectly assigned and their quantification thus inaccurate.63,64
Alternatively, two-dimensional gas chromatography (GC × GC) can be used as it has a higher resolution, larger peak capacity, and higher sensitivity than conventional one-dimensional GC.48,53,63,65–71,72,73 Several studies have described the use of GC × GC coupled to a MS/FID detector for qualitative and semi-quantitative analyses of mainly monomers and some dimers in complex bio-oil samples.41,56,64,65 Until now, no work has been reported to our knowledge on the detailed molecular characterization of the phenolic oligomers in this complex matrix. The methods that were often applied for quantification consist of: (i) a quantification based on an external quantification method of selected compounds,7,67,72 (ii) a quantification in which response factors were calculated based on (modified) effective carbon number factors,66,74 or (iii) a relative quantification in which the relative response factors were measured through an internal standard.65,72,73,75
RCF literature also has focused mainly on the identification and quantification of the phenolic monomers, and only little on the identification of some phenolic dimers in the RCF lignin oil. Structural chemical information of the RCF lignin oligomers that comprise over 40% of the lignin oil has not been detailed, although it is recognized as critical.30,52 Recently, a thorough structural study of the pine wood RCF lignin oil was reported that combined solvent fractionation and a variety of classic chromatographic (GC, GC-MS, GPC) and spectroscopic (1D-, 2D-NMR) analyses. This study unambiguously assigned more than 80% of the structural molecular units within the RCF lignin oligomers, including β-5 γ-OH, β-1 γ-OH, β–β 2× γ-OH, β-5 ethyl, β-1 ethyl, β–β THF, and 5–5 inter-unit linkages. However, only monomers and some dimers were characterized individually.27 Here, high-temperature GC × GC-MS/FID was utilized to comprehensively reveal the individual structural features of the RCF lignin phenolic dimers and trimers from pine wood RCF, including their reliable quantification. Fractionation was used primarily to facilitate the analytical work and product identification. The results provide molecular insight of individual lignin oil components, revealing insight into their formation in the RCF process, and a better understanding of the lignin oil chemical reactivity, which is indispensable to direct further valorization efforts for RCF oil, including but not limited to materials such as polyurethanes,76,77 epoxy resins,17,78 and others,79–82 and chemicals such as antioxidants83 or antimicrobial agents.84
Materials and methods
Chemicals
All commercially purchased chemicals in this study were used without further purification. Guaiacol (2-methoxyphenol, 98%), 4-n-propylguaiacol (<99%), N-methyl-N-(trimethylsilyl)trifluoroacetamide (>98.5%), anhydrous pyridine (99.8%), 2-isopropylphenol (>98%), 4-propanolguaiacol (3-(4-hydroxy-3-methoxyphenyl)-1-propanol, >98%), 4-ethylguaiacol (98%), and isoeugenol (2-methoxy-4-propenylphenol, >98%) were purchased from Sigma Aldrich. Acetonitrile (99.9%) and methanol (99.9%) were purchased from ChemLab. 2-Phenoxy-1-phenyl ethanol (1), 1-(4-hydroxyphenyl)-2-phenoxy-1,3-propanediol (2), and 2-(2,6-dimethoxyphenoxy)-1-(4-hydroxy-3-methoxyphenyl)propane-1,3-diol (3), were synthesized as described in the ESI (S6, ESI†).Sample preparation
Pine wood was soxhlet extracted with an ethanol/toluene mixture (1/2; volume%) for 3 h to remove most extractives. The RCF oil is obtained from processing 150 g pre-extracted pine wood for 3 h at 235 °C in a 2 L batch reactor in the presence of 800 mL MeOH, 30 bar H2, and 15 g Pd/C as a catalyst, as described in a previous study.27 The entire RCF lignin oil (Foil) was sequentially fractionated using a binary solvent mixture of heptane (Hept) and ethyl acetate (EtOAc) with increasing polarity. The sequential fractionation steps resulted in 6 lignin oil fractions: FH100 (100 vol% Hept/0 vol% EtOAc), FH80 (80 vol% Hept/20 vol% EtOAc), FH60 (60 vol% Hept/40 vol% EtOAc), FH40 (40 vol% Hept/60 vol% EtOAc), FH20 (20 vol% Hept/80 vol% EtOAc), FEA100 (0 vol% Hept/100 vol% EtOAc). The detailed preparation of these fractions can be found in our previous study.27Subsequently, an internal standard (IS) was added to a weighed amount of the entire oil Foil sample and the FH100, FH80, FH60, FH40, FH20, FEA100 fractions, which were then derivatized before further analysis according to the following procedure: first a small amount of 2-isopropyl phenol (∼5 mg), used as an IS, was added into a GC-vial containing a weighted amount of lignin oil (∼50 mg). Subsequently, 0.5 mL of anhydrous pyridine, 0.5 mL of N-methyl-N-(trimethylsilyl)trifluoroacetamide, and 0.5 mL of anhydrous acetonitrile was added. The vial was sealed and put in an oven at 80 °C for 30 minutes. Then, the vial was removed from the oven and cooled to room temperature. Afterward, the sample was analyzed on Thermo Scientific TRACE GC × GC setup (Interscience, Belgium).
Analytical method
The GC × GC comprises an Mxt column (60 m × 0.25 mm × 0.25 μm) as the first dimension column connected to a ZB-35HT (2.2 m × 0.18 mm × 0.18 μm) as the second dimension column through a Sil Tite connection. The column set and a dual-state cryogenic modulator (liquid CO2) are placed in the same oven. The outlet of the second column is connected to an FID/MS detector. For the GC × GC – FID setup, the flow rates of H2, air, and N2 (make-up gas) were set at 35, 350, and 35 mL min−1, respectively. The FID temperature was set at 350 °C and the data acquisition rate was 100 Hz. Moreover, a PTV injector was used in these analyses with a programmed temperature injector from 40 °C to 370 °C (hold 25 minutes at 370 °C) to avoid discrimination in the injector. For the GC × GC – MS setup, the data acquisition rate was set at 30 spectra per s with the scanning range set from 150 to 1100 amu. The GC – MS interface (transfer line) temperature was set at 280 °C and the ion source temperature was set at 300 °C. The MS detector used electron ionization (70 eV). Helium was used as a carrier gas at a constant flow rate (2.1 ml min−1). The modulation period was optimized (10 s) to obtain a maximal resolution in the first dimension without causing wrap-around. The GC system was operated in programmed temperature conditions: 40 °C to 420 °C with a heating rate of 3 °C min−1.Data acquisition and quantification
Thermo Scientific's Chrom-Card data system was used for data acquisition and processing of the FID while Thermo Scientific's XCalibur software was applied for data acquired with the MS. The raw data of GC × GC – FID was exported to a .cdf file, subsequently processed by GC Image (Zoex Corporation, USA) for quantification. The tentative identification of the resulting peaks from GC × GC – FID was achieved by reproducing the analysis using the GC × GC – MS with the identical column combination and an optimized carrier gas flow. Thanks to the stability and linearity of the FID response, the quantification of the identified compounds was, therefore, conducted using the GC × GC – FID chromatogram.Results and discussion
General methodology
GPC results of the RCF lignin oil samples indicated that their composition contains monomers, dimers, and oligomers with varying distribution over the samples and thus varying molecular weights, ranging from 203 to 1771 g mol−1 (Fig. S1.1, see ESI†).27 This information reveals that many molecules in these fractions have a high boiling point. Consequently, the GC × GC setup for analyzing RCF lignin samples was equipped with two high-temperature columns with different polarity, allowing chromatographic separation up to 430 °C. Firstly, the use of this GC × GC – FID/MS setup was assessed by measuring an untreated Foil sample. The result of this test revealed that the GC × GC – FID/MS operating at high temperature could elute the monomers, dimers, and a small number of trimers according to three structural regions (Fig. S1.2, see ESI†). However, the eluted components often suffered from peak tailing and co-elution, likely due to interaction of hydroxyl groups of the phenolic compounds in the sample either with the glass material of the liner or with the stationary phase of the columns used.85 As a consequence, the components present in the sample could be incorrectly assigned and inaccurately quantified. To avoid this issue, the hydroxyl groups of the phenolic compounds were shielded, prior to analyzing on the GC × GC setup, via a derivatizing step using N-methyl-N-(trimethylsilyl)trifluoroacetamide as a reagent. The chromatographic result of the testing Foil sample after derivatization on the GC × GC – FID is illustrated in Fig. 1a.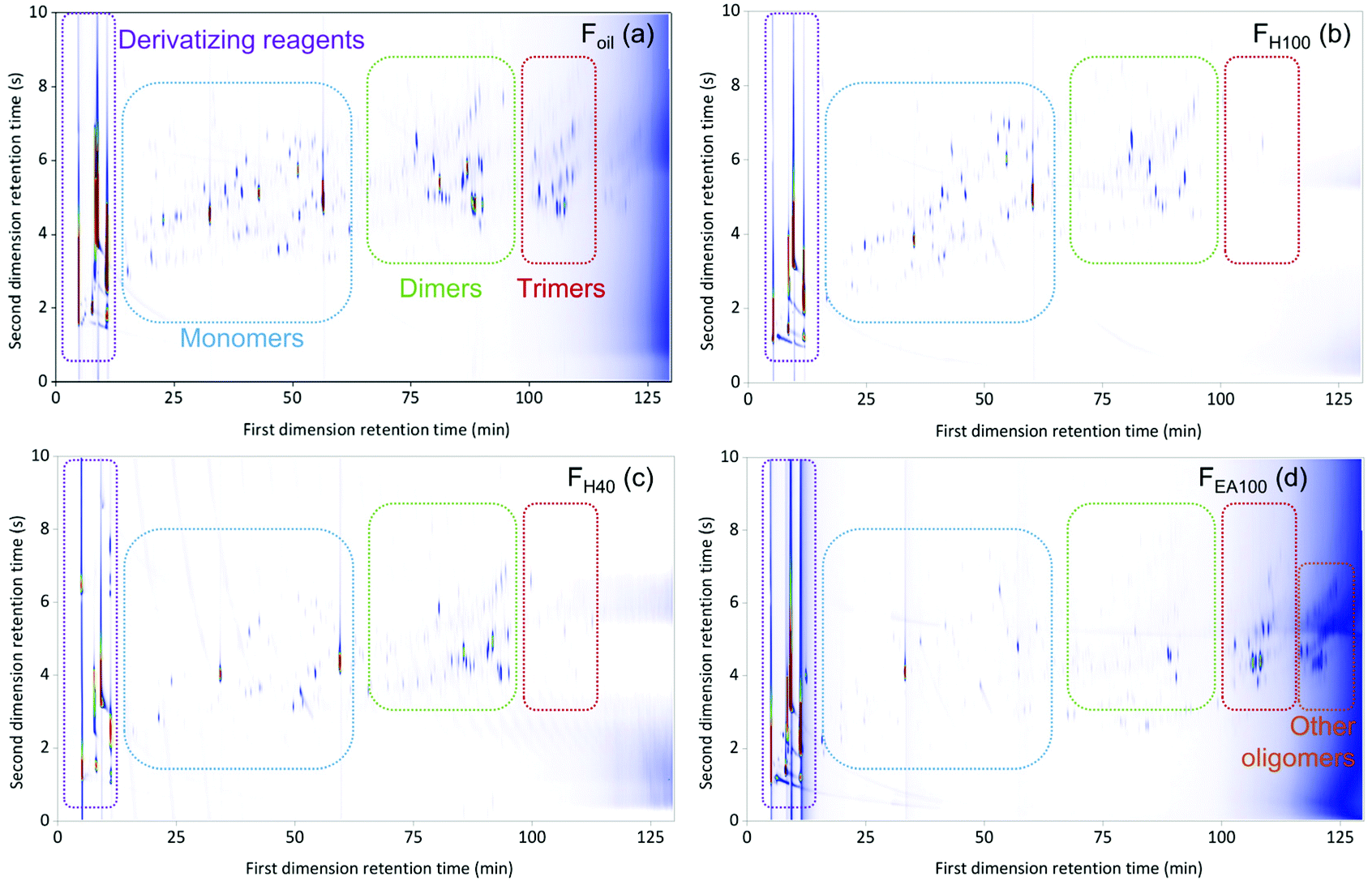 | ||
| Fig. 1 The GC × GC color plots of the derivatized entire Foil (a) sample and FH80 (b), FH40 (c), and FEA100 (d) fractions (Mxt as the first column × ZB35-HT as the second column). | ||
The derivatization step significantly improved the separation of the Foil sample. The phenolic compounds were eluted in the individual monomeric, dimeric, trimeric, and other oligomeric regions with well-defined peak shapes. With the key derivatization step in hand, the six fractions (i.e. FH100, FH80, FH60, FH40, FH20, and FEA100) were pretreated accordingly before analysis. The GC × GC chromatograms of FH80, FH40, and FEA100 are presented in Fig. 1b, c, and d, respectively, (the chromatogram of FH100, FH60, and FH20 fraction can be found in the ESI, Fig. S1.3–S1.5†). Thereby, the identification and calculation of the components present in the seven RCF oil samples will be based on chromatograms of derivatized samples.
Identification and quantification of lignin-derived phenolic monomers, dimers, and trimers in the RCF lignin oil
The identification of monomers in the RCF lignin oil samples was performed by comparing the deconvoluted mass spectra obtained from GC × GC – MS with the NIST library or with retention indices of authentic monomers. However, this approach could not be applied to the dimers and trimers due to the limitation of the NIST library and the lack of the authentic dimers and trimers. The dimeric and trimeric compounds have been assigned based on detailed analysis of their mass fragmentation patterns (see ESI S3 and ESI S4†).To accurately quantify the monomers, dimers, and trimers in the RCF oil fractions by GC × GC, it strictly requires individual response factors (RFs) between each analyte and the internal standard. In other words, a library of authentic compounds is required to attain the corresponding response factors. However, this quantitative approach can solely be applied to the available monomers, not to oligomers owing to the lack of reference standards. Thus, in this study, a calibration mixture of monomers and dimers having exact chemical structures (in the case of monomers) or similar chemical structures (in the case of dimers) of compounds in the real lignin oil fractions was prepared and measured in the same way as the actual samples (more information on calibration mixture can be found in S2 in the ESI†). The experimental RFs of individual components in the calibration mixture were used to determine the RFs of other components, based on the assumption that the response factor on GC-FID is a function that depends on the molecular weight of molecules and the number of carbon, hydrogen, oxygen atoms, and aromatic rings in their structures.75,86,87 With these RFs in hand, all the identified monomers, dimers, and trimers in the seven RCF lignin oil samples were individually quantified. The monomer quantification using this RF approach on the GC × GC – FID set up (Fig. 2) is in line with the results obtained in our previous study,27 for which their RFs have been determined based on external calibration of the authentic compounds on 1D-GC (the detailed results of the monomer quantification by GC × GC & 1D-GC can be found in Table S2.1., ESI.† Comparison of monomers determined by GC × GC and 1D-GC is presented in Fig. S2.1, see ESI†).
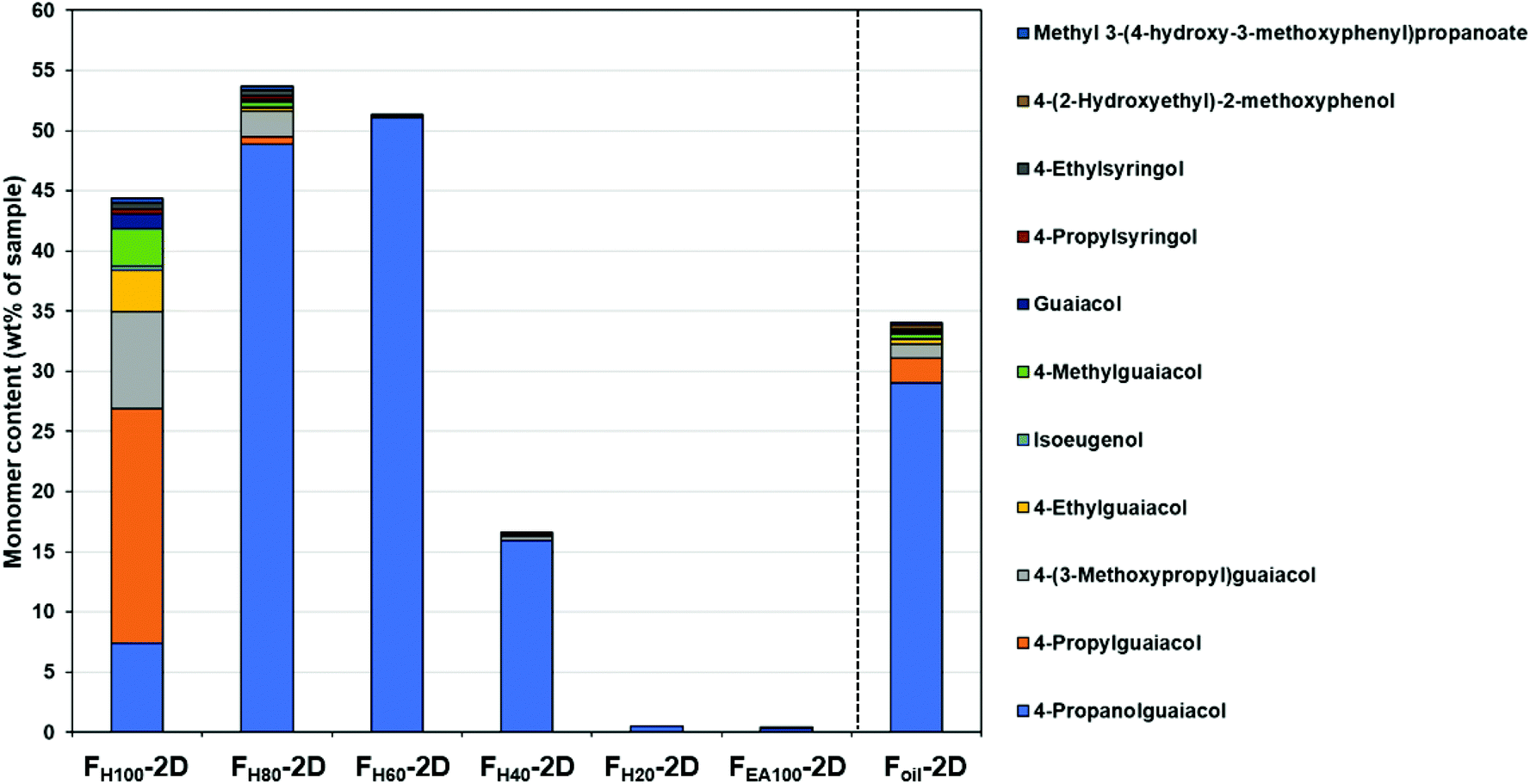 | ||
| Fig. 2 Observable monomers determined by GC × GC – FID. The detailed results of the monomer quantification can be found in Table S2.1.† | ||
Fig. 2 shows that the monomer content in the first three fractions (FH100, FH80, and FH60) was enhanced in comparison with the entire Foil sample. Furthermore, in the pure heptane fraction (FH100) the non-polar phenolic monomer (4-propylguaiacol) consists of up to 19.5 wt% of the total monomeric mass fraction. However, 4-propanolguaiacol is the primary monomer in fractions FH80 and FH60 (48.9 wt% and 51.0 wt%, respectively). Furthermore, the number of monomers decreases significantly in the FH40 fraction, whereas negligible amounts were observed in fractions FH20 and FEA100. Sequential fractionation by increasing slightly the polarity of solvent thus influences both the distribution and type of monomers in each fraction.
The use of the high-temperature GC × GC setup provided a better monomer separation and detection than 1D-GC (more detailed information found in Table S2.1, ESI†). In particular, the monomers guaiacol (M7), 4-propylsyringol (M8), 4-ethylsyringol (M9), 4-(2-hydroxyethyl)-2-methoxyphenol (M10), and methyl 3-(4-hydroxy-3-methoxyphenyl)propanoate (M11) were separated and detected on the GC × GC chromatogram while not clearly visible on the chromatogram of 1D-GC. Fig. 3 illustrates the main monomers found in the Foil sample using the GC × GC. It should be noted that in all seven RCF lignin oil samples, the G-type monomers were found in a significantly higher content, as expected from using softwood feedstock, but also S-type monomers were observed due to the higher resolution of GC × GC system, that were not detected on 1D-GC in earlier work.27
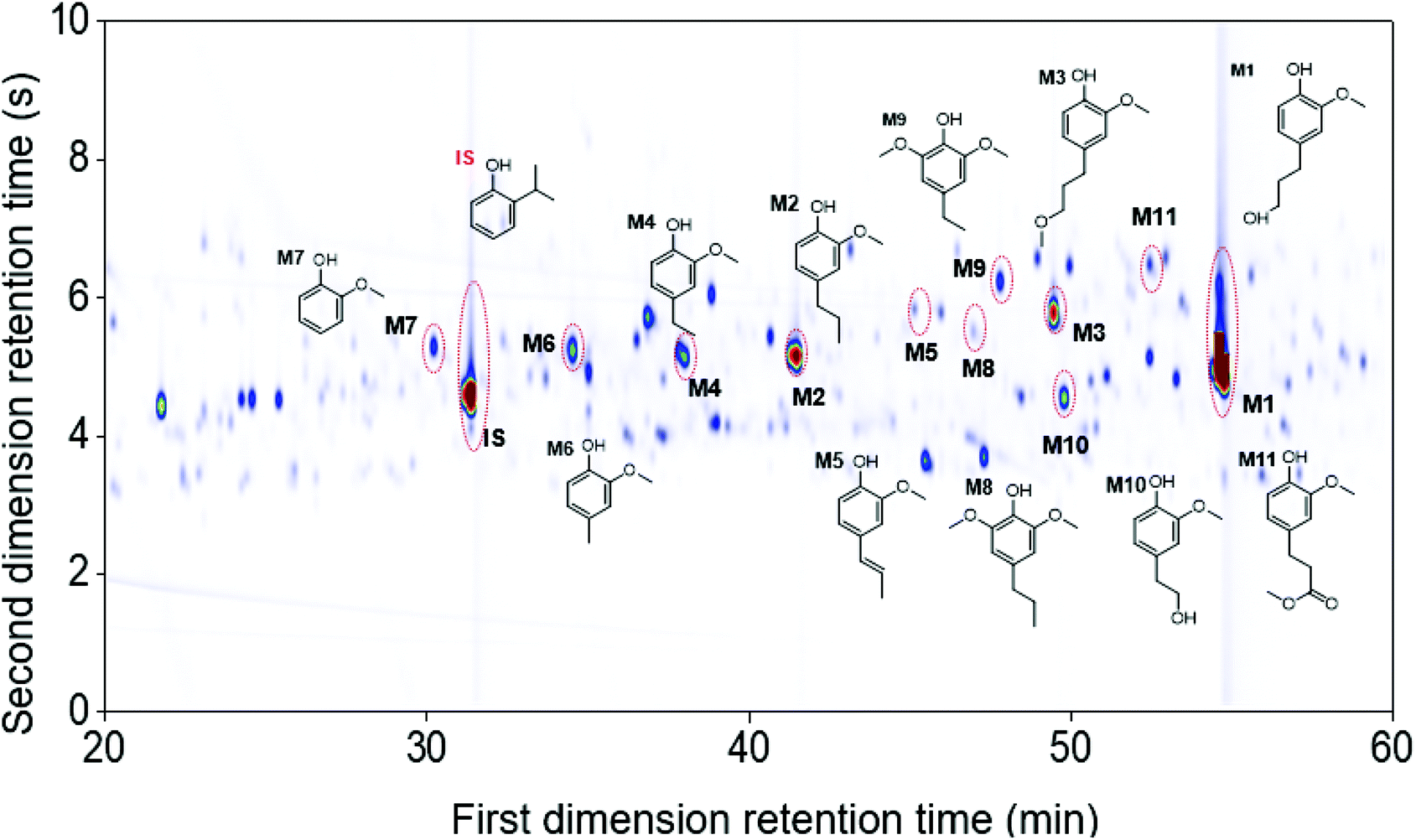 | ||
| Fig. 3 The GC × GC chromatogram of the monomeric region in the Foil sample. | ||
In addition to the monomers, analysis of GC × GC data identified thirty-six phenolic dimers in the RCF lignin fractions, of which only twelve dimers have been previously reported.7,18,27,52 Furthermore, twenty-one trimers were also determined for the first time in these fractions (MS information can be found in the ESI, for dimers (Fig. S3.1–S3.36†) and trimers (Fig. S4.1–S4.21†)). It is worth noting that not only monomers, dimers, and trimers were detected by using a high peak capacity GC × GC setup, but also other larger oligomers could be eluted (Fig. 1). The presence of such oligomers was apparent in the most polar fractions FH20 and FEA100 (Fig. S1.5† and Fig. 1d). However, due to the inherent limitation of the GC × GC – MS and the low concentration of these oligomers, this study only focused on the identification and quantification of the dimers and trimers. Fig. 4 and 5 summarize the chemical structures.
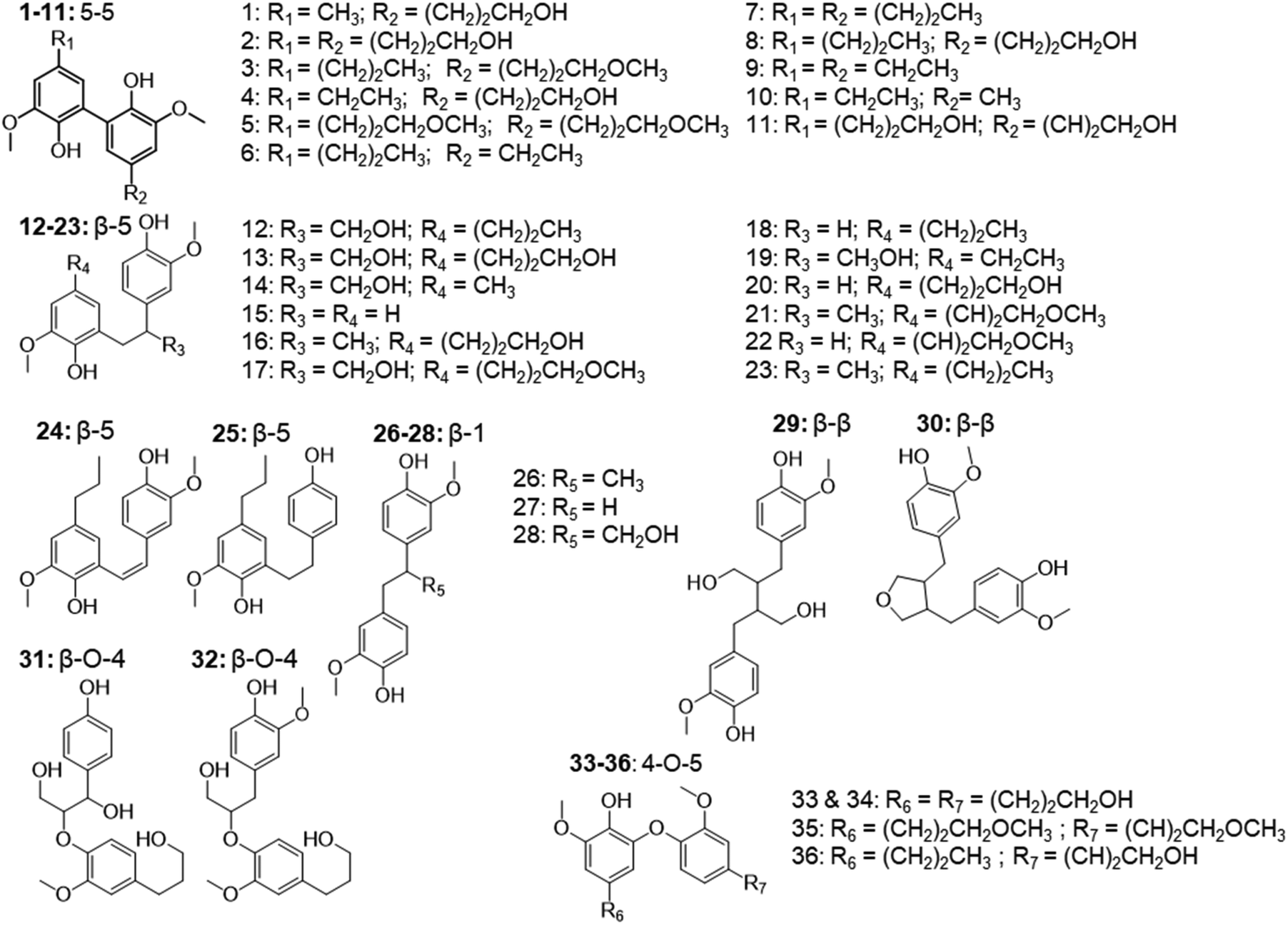 | ||
| Fig. 4 The structure of observed dimers in the RCF oil samples, derived from the MS spectra using high-temperature GC × GC. | ||
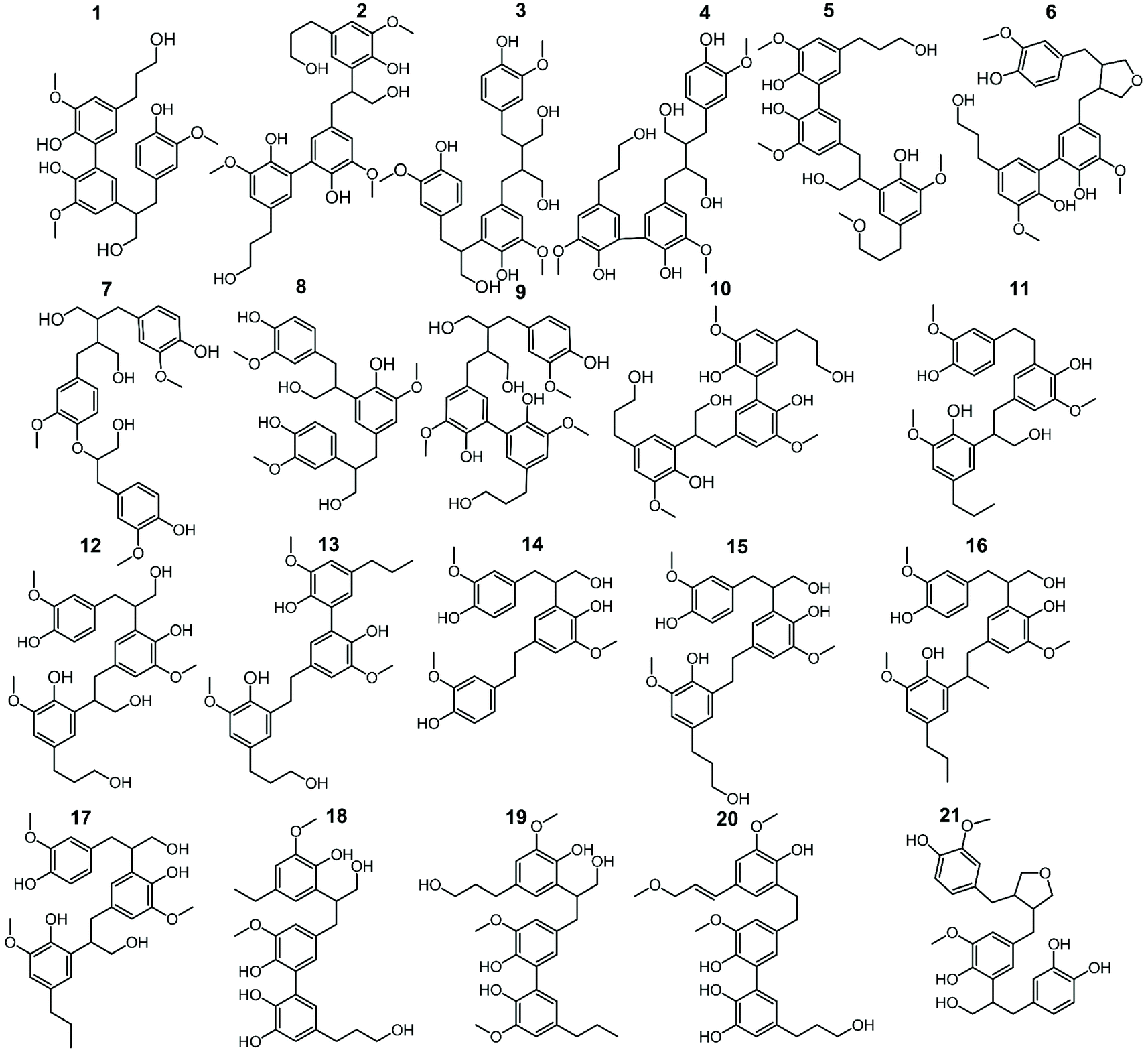 | ||
| Fig. 5 The structure of observed trimers in the RCF oil samples, derived from the MS spectra using high-temperature GC × GC. | ||
These dimers and trimers consist of different G units coupled mainly via C–C inter-unit linkages, including β-5 (β-5 γ-OH, β-5 ethyl, and β-5 propyl), β-1 (β-1 γ-OH, β-1 ethyl, and β-1 propyl), β–β (β–β 2× γ-OH and β–β), and 5–5. Furthermore, only a minor number of β-O-4 and 4-O-5 inter-linkages were observed in these RCF lignin oil fractions. Most of the inter-unit linkages align well with the bulk information from 1H–13C HSQC NMR spectroscopy.27 However, the 1H–13C HSQC NMR technique could only assign the inter-unit linkages present of compounds with relatively high concentrations in the entire lignin oil. The inter-unit linkages with low abundance such as β-5 propyl, β-1 propyl, and 4-O-5 could not be observed properly by the 2D NMR approach, due to its inherent moderate detection limit. High Temperature-GC × GC (HT-GC × GC) can also identify the aliphatic end-units for the different molecules in the fractionated RCF lignin oil samples. Fig. 4 and 5 shows that the aliphatic end-units in the dimeric and trimeric molecules consist of 4-propanol (4-P-γ-OH), 4-propyl (4-P), 4-ethyl (4-E), 4-(3-methoxypropyl) (4-P-γ-OMe), 4-methyl (4 M), 4-propenol, and 4-(3-methoxyprop-1-en-1-yl) as an end-unit. Presence of the two methoxy substituted end-groups indicates some RCF solvent incorporation in the final products of the RCF biorefinery.
The identified dimers and trimers are all composed of similar structural units through the same inter-unit linkages. This indicates that they have been subject of the same chemistry during RCF processing; almost all inter-unit ether linkages (i.e. β-O-4) are cleaved, whereas the lignin-original C–C inter-unit linkages remain intact. Furthermore, detailed inspection of the end-units of the oligomers also reveals strong structural resemblance with those of the monomers.
Subsequently, the identified dimers and trimers were quantified according to the RF approach explained above. Fig. 6 presents the product distribution and total mass of the monomers, dimers, and trimers in the seven RCF lignin oil fractions (additional information can be found in Table S2.2, ESI†).
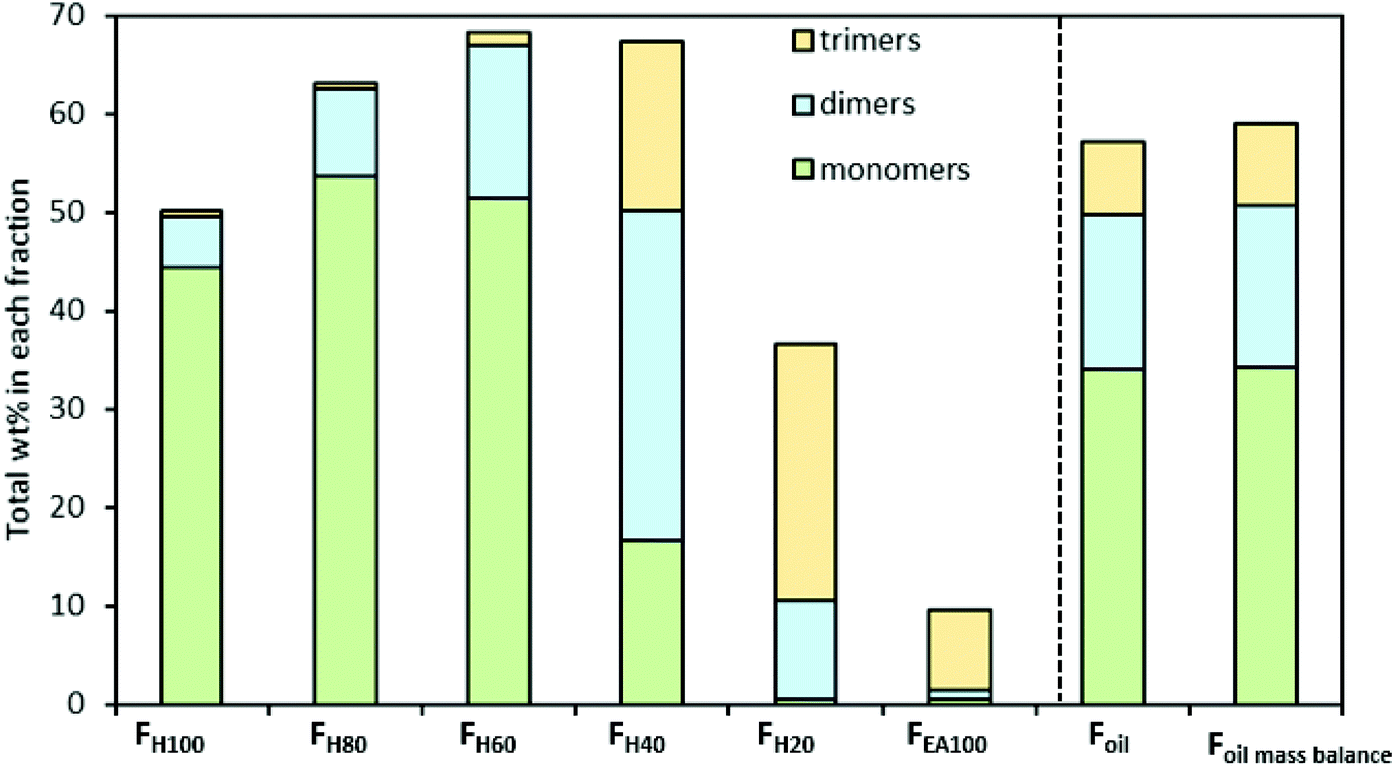 | ||
| Fig. 6 Distribution and total mass of monomers, dimers, and trimers in the RCF oil samples. Detailed results can be found in Table S2.2† and Table 1. | ||
The quantitative analysis shows that the entire RCF lignin oil (Foil) consists of more monomers (34.03 wt%, of which 29.01 wt% is 4-propanolguaiacol) than dimers (15.79 wt%) and trimers (7.26 wt%), respectively. Fig. 6 also shows that only a small number of dimers are found in the less polar fraction (FH100), whereas the largest amount is found in FH40. Trimers are primarily observed in 2 fractions (FH40 and FH20) and are negligibly present in the less polar fractions (FH100, FH80, and FH60). Furthermore, almost all monomers, dimers, and trimers were extracted in the FH100, FH80, FH60, FH40, and FH20 fractions, corresponding to 50.11, 62.91, 68.23, 64.63, and 33.15 wt% of the respective samples. Only 9.64 wt% of the pure ethyl acetate fraction (FEA100) could be identified. Moreover, the accumulated mass balance of the monomers, dimers, and trimers over each individual fraction (Fig. 5, Foil mass balance) is nearly identical to that of Foil, showing the reliability of the analysis. Clearly, the sequential fractionation of the entire RCF oil (Foil) by using a solvent mixture (Hept/EtOAc) can separate the complicated entire lignin oil Foil into relatively more homogeneous fractions in terms of molecular weight, in line with the GPC result in earlier studies,88,89 and structural functionality, as revealed here.
The detailed quantification of individual dimers (D) and trimers (T) identified in each lignin oil fraction is presented in Table 1. The result shows that D2, D13, D20, and D28 are the primary dimeric molecules found in the entire RCF lignin oil (Foil), corroborating earlier suggestions.27 These dimers contain the same 4-propanol end-group as observed in the monomers, and consist of 5–5, β-5 γ-OH, β-5 E, and β-1 γ-OH inter-phenolic linkages, respectively. They account for 2.14, 2.81, 1.96, and 2.32, wt% of the total 15.79 wt% identified dimers in Foil. Because of the sequential fractionation of the entire Foil, the occurrence of these dimers varied between the fractions. Similarly, T1, T7, T4, and T2 are the most occurring trimers, corresponding to 0.94, 0.81, 0.80, and 0.72 wt% of the total 7.26 wt% of the identified trimers in the entire lignin oil (Foil). The structural motifs of these trimers consist of 4-propanol end-group, similar to the observed monomers, and contain 5–5 & β-1 γ-OH (T1), β-O-4 & β–β 2 × γ-OH (T7), β–β 2 × γ-OH & 5–5 (T4), and β-5 γ-OH & 5–5 (T2) inter-unit linkages.
| Dimer | F oil | F H100 | F H80 | F H60 | F H40 | F H20 | F EA100 | Trimer | F oil | F H100 | F H80 | F H60 | F H40 | F H20 | F EA100 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D1 | 0.27 | 0.00 | 0.08 | 0.51 | 0.51 | 0.06 | 0.00 | T1 | 0.94 | 0.00 | 0.39 | 0.00 | 1.97 | 2.52 | 0.60 |
| D2 | 2.14 | 0.00 | 0.04 | 0.31 | 6.80 | 4.09 | 0.40 | T2 | 0.72 | 0.00 | 0.00 | 0.00 | 0.60 | 3.86 | 1.95 |
| D3 | 0.08 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | T3 | 0.68 | 0.00 | 0.00 | 0.00 | 0.86 | 2.60 | 0.64 |
| D4 | 0.19 | 0.00 | 0.18 | 0.31 | 0.25 | 0.00 | 0.00 | T4 | 0.80 | 0.00 | 0.00 | 0.00 | 0.98 | 5.23 | 2.57 |
| D5 | 0.10 | 0.00 | 0.04 | 0.00 | 0.16 | 0.03 | 0.00 | T5 | 0.62 | 0.00 | 0.00 | 0.00 | 1.24 | 1.40 | 0.44 |
| D6 | 0.00 | 0.09 | 0.06 | 0.00 | 0.00 | 0.00 | 0.00 | T6 | 0.48 | 0.00 | 0.00 | 0.00 | 1.63 | 1.02 | 0.52 |
| D7 | 0.00 | 0.20 | 0.08 | 0.00 | 0.00 | 0.00 | 0.00 | T7 | 0.81 | 0.00 | 0.00 | 0.00 | 1.04 | 2.16 | 0.77 |
| D8 | 0.31 | 0.00 | 0.19 | 0.60 | 0.33 | 0.00 | 0.00 | T8 | 0.21 | 0.00 | 0.00 | 0.00 | 0.33 | 0.00 | 0.00 |
| D9 | 0.00 | 0.13 | 0.13 | 0.00 | 0.00 | 0.00 | 0.00 | T9 | 0.17 | 0.00 | 0.00 | 0.00 | 0.85 | 0.38 | 0.00 |
| D10 | 0.00 | 0.07 | 0.06 | 0.00 | 0.00 | 0.00 | 0.00 | T10 | 0.17 | 0.00 | 0.00 | 0.00 | 0.38 | 0.16 | 0.00 |
| D11 | 0.09 | 0.00 | 0.00 | 0.09 | 0.24 | 0.15 | 0.02 | T11 | 0.19 | 0.00 | 0.00 | 0.00 | 0.00 | 0.28 | 0.00 |
| D12 | 0.25 | 0.00 | 0.32 | 0.48 | 0.15 | 0.00 | 0.00 | T12 | 0.36 | 0.00 | 0.00 | 0.00 | 0.52 | 1.05 | 0.36 |
| D13 | 2.81 | 0.00 | 0.12 | 1.28 | 9.06 | 3.18 | 0.00 | T13 | 0.10 | 0.46 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| D14 | 0.06 | 0.11 | 0.25 | 0.00 | 0.00 | 0.00 | 0.00 | T14 | 0.32 | 0.00 | 0.14 | 0.38 | 1.13 | 0.27 | 0.00 |
| D15 | 0.00 | 0.00 | 0.03 | 0.00 | 0.00 | 0.00 | 0.00 | T15 | 0.41 | 0.00 | 0.00 | 0.82 | 0.63 | 0.48 | 0.18 |
| D16 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | T16 | 0.11 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| D17 | 0.54 | 0.00 | 0.10 | 0.77 | 1.04 | 0.10 | 0.02 | T17 | 0.15 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| D18 | 0.55 | 2.97 | 1.26 | 0.00 | 0.00 | 0.00 | 0.00 | T18 | 0.00 | 0.00 | 0.00 | 0.00 | 0.30 | 0.20 | 0.00 |
| D19 | 0.07 | 0.00 | 0.08 | 0.13 | 0.05 | 0.00 | 0.00 | T19 | 0.00 | 0.00 | 0.00 | 0.00 | 0.39 | 0.00 | 0.00 |
| D20 | 1.96 | 0.12 | 1.01 | 3.57 | 2.79 | 0.14 | 0.00 | T20 | 0.00 | 0.00 | 0.00 | 0.00 | 0.55 | 0.39 | 0.00 |
| D21 | 0.08 | 0.00 | 0.05 | 0.07 | 0.09 | 0.00 | 0.00 | T21 | 0.00 | 0.00 | 0.00 | 0.00 | 1.15 | 0.80 | 0.23 |
| D22 | 0.23 | 0.00 | 0.10 | 0.30 | 0.33 | 0.00 | 0.00 | Total | 7.26 | 0.46 | 0.54 | 1.20 | 14.55 | 22.78 | 8.26 |
| D23 | 0.04 | 0.15 | 0.06 | 0.00 | 0.00 | 0.00 | 0.00 | ||||||||
| D24 | 0.21 | 0.00 | 0.15 | 0.30 | 0.25 | 0.00 | 0.00 | ||||||||
| D25 | 0.00 | 0.00 | 0.14 | 0.00 | 0.00 | 0.00 | 0.00 | ||||||||
| D26 | 0.17 | 0.30 | 0.54 | 0.18 | 0.00 | 0.00 | 0.00 | ||||||||
| D27 | 0.51 | 0.50 | 1.60 | 0.61 | 0.06 | 0.00 | 0.00 | ||||||||
| D28 | 2.32 | 0.11 | 0.67 | 3.22 | 4.68 | 0.38 | 0.08 | ||||||||
| D29 | 0.85 | 0.00 | 0.06 | 0.56 | 2.85 | 0.84 | 0.33 | ||||||||
| D30 | 0.33 | 0.08 | 0.33 | 0.47 | 0.28 | 0.00 | 0.00 | ||||||||
| D31 | 0.10 | 0.00 | 0.00 | 0.00 | 0.24 | 0.14 | 0.00 | ||||||||
| D32 | 0.56 | 0.00 | 0.09 | 0.50 | 1.52 | 0.29 | 0.04 | ||||||||
| D33 | 0.52 | 0.00 | 0.07 | 0.50 | 1.38 | 0.47 | 0.06 | ||||||||
| D34 | 0.46 | 0.04 | 0.44 | 0.94 | 0.39 | 0.00 | 0.00 | ||||||||
| D35 | 0.00 | 0.22 | 0.18 | 0.00 | 0.00 | 0.00 | 0.00 | ||||||||
| D36 | 0.00 | 0.16 | 0.15 | 0.00 | 0.00 | 0.00 | 0.00 | ||||||||
| Total | 15.79 | 5.28 | 8.68 | 15.68 | 33.47 | 9.87 | 0.95 | ||||||||
Inter-unit linkages and end-groups of dimers and trimers in the pine wood RCF lignin oil
The quantitative GC × GC results of the dimers and trimers show that the structural motifs of these molecules consist of inter-unit linkages including β-5, β-1, β–β, 5–5, β-O-4, 4-O-5 and aliphatic end-units including 4-P-γ-OH, 4-P, 4-E, 4-P-γ-OMe, 4-M, 4-propenol, and 4-(3-methoxyprop-1-en-1-yl). A fraction-dependent distribution between these structural motifs and the increasing polarity of the extraction solvent can be observed in Fig. 7. These relative distributions are calculated based on the individual mol% of the dimers and trimers relative to the total mol% of dimers and trimers. Furthermore, the individual distribution of β-5, β-1, and β–β inter-unit linkages in the RCF lignin oil, based on the dimer and trimer molecular identification, is also depicted in Fig. 8.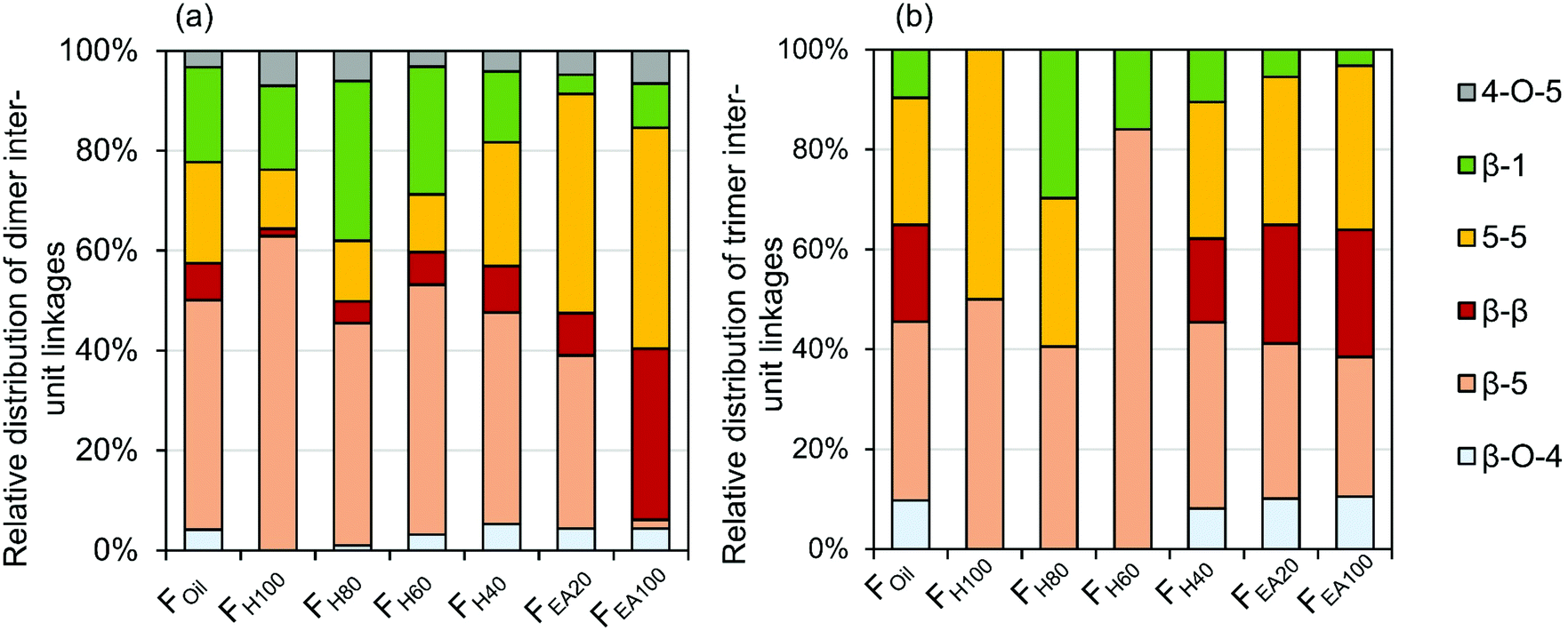 | ||
| Fig. 7 Relative distribution of inter-unit linkages based on wt% of the corresponding dimers (a) and trimers (b) in the RCF lignin oil. | ||
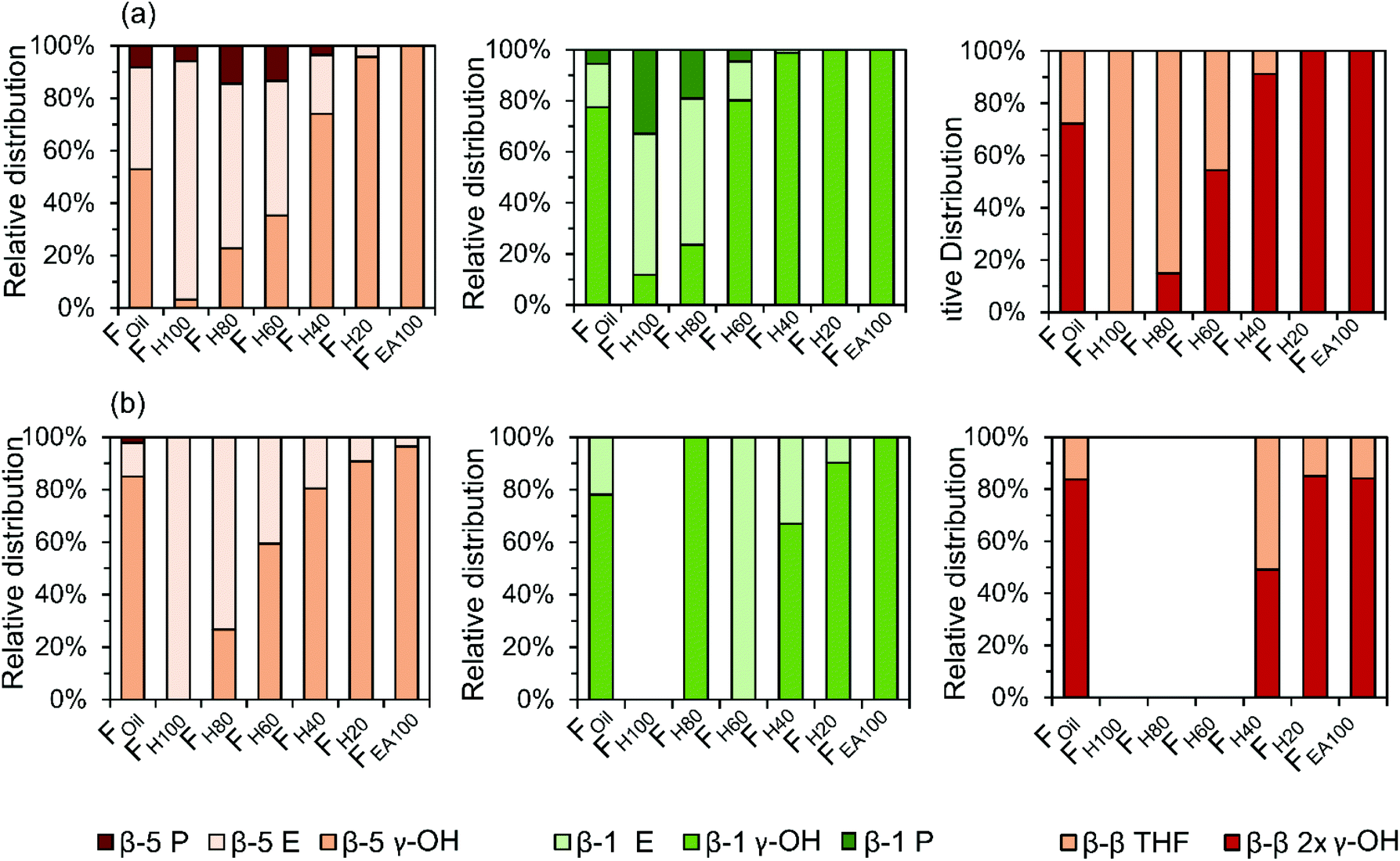 | ||
| Fig. 8 Individual distribution of β-5, β-1, and β–β inter-unit linkages of dimers (a) and trimers (b) in the RCF lignin oil. | ||
The first inter-unit linkage discussed herein is the β-5 inter-unit linkage, which is composed of β-5 γ-OH (e.g.D13), β-5 ethyl (e.g.D16), and β-5 propyl (e.g.D16) analogs, originating from the native β-5 phenylcoumaran structure. Over 45 wt% of the identified dimers in the entire RCF oil (Foil) contain a β-5 inter-unit linkage and over 59 wt% of the identified trimers in Foil contain at least one β-5 inter-unit linkage in the structure. Given always two inter-unit linkages are present in trimers, approximately 34% of all inter-unit linkages in trimers contains the β-5 structure (Fig. 7). Among these β-5 inter-unit linkages, β-5 γ-OH is most abundant in both dimers and trimers (relative amount of 51% and 84%, respectively; Fig. 8). Furthermore, the distribution of β-5 analogs of dimers and trimers varied between fractions. For example, β-5 dimers are predominant (63%) in the fraction FH100 (Fig. 7a), with mostly the β-5 E linkage (Fig. 8a). Only a small amount of these β-5 units (1.7%) is present in the most polar fraction FEA100, with sole contributor β-5 γ-OH. Overall, the high β-5 γ-OH occurrence increases at the expense of β-5 E (Fig. 8); this accords with the solvent polarity.
The β-1 motifs, consisting of β-1 γ-OH (e.g.D28), β-1 ethyl (e.g.D27), and β-1 propyl (e.g.D26) analogs, are a second group of inter-unit linkages present in the RCF lignin oil. Approximately 19 wt% of the identified dimers in Foil and over 20 wt% of the identified trimers in Foil have one β-1 inter-unit linkage, indicating that approximately 10% of the inter-unit linkages in the identified trimers holds a β-1 structure (Fig. 7). The β-1 γ-OH analog predominates in both dimers and trimers, showing a relative occurrence of 77% and 78%, respectively (Fig. 8). Presence of β-1 γ-OH increases with solvent polarity, similarly as observed for β-5 (Fig. 8). Moreover, presence of β-1 decreases with solvent polarity (Fig. 7). These observations of less β-1 in the trimers, compared to the dimers, and less β-1 in the more polar (higher molecular weight containing) fractions, is likely the consequence of the native-lignin structure. That is, in the β-1 spirodienone structure, one of the two phenolics of the β-1 linkage is a quinone methide, remaining unsubstituted on its phenolic and 5-position.3 Thus, only a linkage to a third phenolic group can be made through the second phenolic moiety. Given the high chance of this being a β-O-4 linkage in accordance with lignin formation mechanisms,90 relatively more β-1 inter-unit linkages are present in a dimer form, as observed in Fig. 7.
The β–β linkages are the third group of inter-unit linkages discussed herein. They originate from the native β–β resinol structure, and after subjecting to the RCF process this resinol structure is converted to β-β 2× γ-OH (e.g.D29) and β–β THF (e.g.D30). Around 7 wt% of the identified dimers and over 37 wt% of the trimers contain a β–β structure in Foil (Fig. 7), indicating that approximately 20% of the inter-unit linkages in trimers has a β–β structure (Fig. 7). The β–β 2 × γ-OH linkage in both dimers and trimers is more abundant than β–β THF (Fig. 8). Fig. 7 and 8 also shows that the relative number of β–β linkages and β–β 2 × γ-OH's presence in both dimers and trimers increases with increasing polarity of the extraction solvent, while β–β THF is mainly present in the less polar fractions (e.g. dimer fraction FH100 and FH80) (Fig. 8a).
It can be concluded that a significant amount of the γ-OH functional group in the inter-unit linkages of β-5, β-1, and β–β units in both dimers and trimers is observed in the more polar, higher molecular weight fractions (e.g. FH40, FH20, and FEA100), compared to the inter-unit linkages without γ-OH group. Thus, the oligomers containing the polar inter-unit linkages will be mainly extracted in more polar solvents, and they are, conversely, less soluble in the non/less-polar solvents. This observation supports the bulk results obtained from 1H–13C HSQC NMR spectroscopy.27
The fourth group is the biphenyl (5–5) inter-unit linkage, originating from the dibenzodioxocin inter-unit linkages in native lignin. Approximately 20 wt% of the detected dimers and 51 wt% of the detected trimers in Foil contain this 5–5 linkage, and thus 25% of the inter-unit linkages in the identified trimers have a 5–5 structure. Dimers containing 5–5 are present in considerably higher amounts in the more polar (higher molecular weight) fractions (Fig. 7a). An increasing trend of 5–5 containing trimers in the FH40, FH20, and FEA100 fractions with the higher polarity is also apparent in Fig. 7b. However, Fig. 7b shows that the 5–5 trimers account for up to 50% of inter-unit linkages in the non-polar fraction FH100. The reason for this is that only one trimer is detected in FH100, of which one of the inter-unit linkages has the 5–5 structure. This is because this non-polar extraction solvent impedes the solubility and extraction of trimers.
Small amounts of β-O-4 inter-unit linkages that remained after RCF processing are also detected in dimers and trimers of the entire oil (Foil) (Fig. 7). Strikingly, the majority of detected β-O-4 structure underwent a α-dehydroxylation, yielding a reduced form of the native β-O-4 structure (e.g.Fig. 5, T7). This α-dehydroxylation reaction product has been previously observed in minor amounts when using Pd/C catalysis on β-O-4 model compounds. It was suggested to be a side product of the concerted catalytic β-O-4 cleavage.91 The occurrence of β-O-4 linkages increases in the high molecular weight fractions of dimers and trimers. Given their low occurrence – relative to the other inter-unit linkages – most of the β-O-4 linkages were effectively cleaved during the RCF process. Moreover, most of the non-cleaved β-O-4 structures has undergone a reductive reaction, yielding a reduced form of β-O-4.
Lastly, in 3 wt% of the dimers (in the entire oil, Foil), a 4-O-5 inter-unit linkage was found, while absent in the structure of the trimers. It should be noted that these 4-O-5-linked structures have never been detected by NMR techniques in the previous studies on RCF lignin. This is possibly due to the low concentration level of these units in the lignin oil. This example illustrates the high sensitivity of the GC × GC-FID/MS method as compared to that of the 2D NMR technique.27,92,93
Next to the inter-unit linkages, end-units resulting from β-O-4 cleavage and the reductive chemistry during RCF processing are another important structural motif. These groups consist of 4-P-γ-OH, 4-P, 4-E, 4-P-y-OMe, 4-M, 4-propenol, and 4-(3-methoxyprop-1-en-1-yl) units. Among them, the 4-P-γ-OH and 4-P end-units are found at high amounts in the various RCF lignin oil fractions (Fig. 9). The other end-units, including 4-E, 4-P-γ-OMe, 4-M, 4-propenol, and 4-(3-methoxyprop-1-en-1-yl) are detected with relatively low abundancy in the dimer and trimer structures, and therefore they are combined as “Others” in Fig. 9.
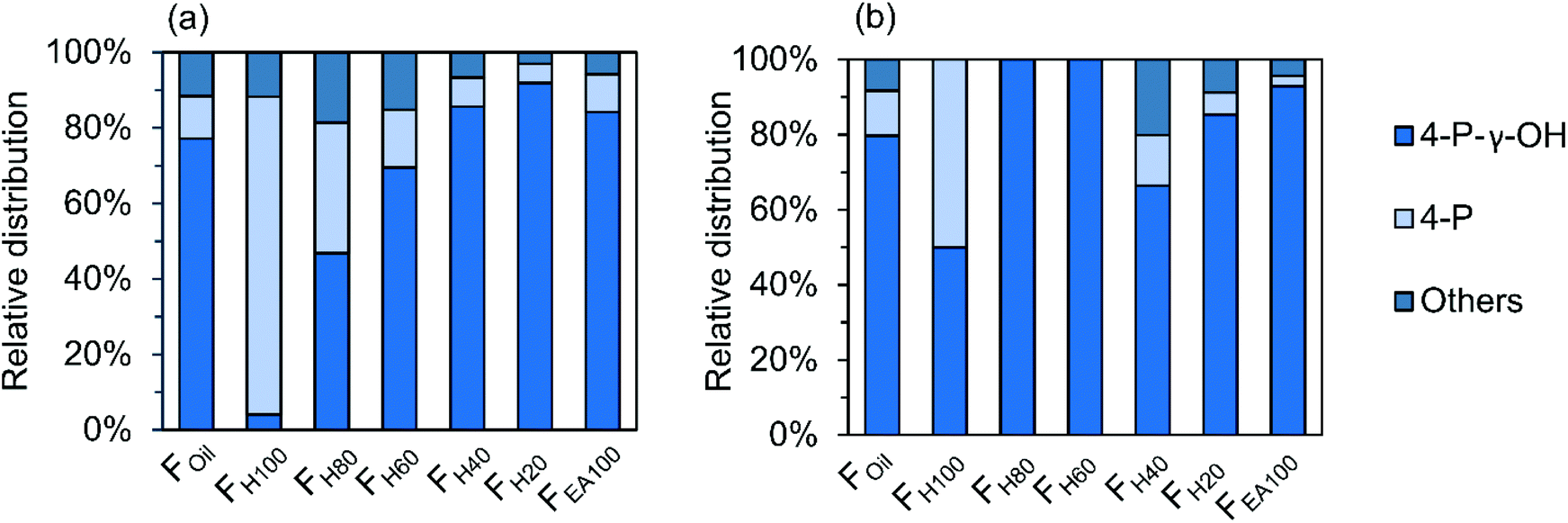 | ||
| Fig. 9 Relative distribution of end-unit groups in dimers (a) and trimers (b) of the RCF lignin oil. | ||
Around 80% of 4-P-γ-OH end-unit is found in both dimers and trimers, whilst only approximately 10% of the 4-P unit is observed in both dimers and trimers of the Foil fraction (Fig. 9). This is the consequence of the reduction chemistry with Pd catalysis, showing (as in the monomer fraction) large quantities of propanol end groups due to its low oxophilic character.10 It is also recognized that the presence of the P-γ-OH end-unit in dimers increases steadily with the increasing polarity of the fractions (Fig. 9a). A similar observation can be made for the trimers in FH40–EA100. Fig. 9 also indicates that the 4-P end-unit is most prevalent in the non-polar fraction (FH100) of both dimers and trimers. Obviously, this is the result of the favorable extraction of less polar 4-P substitution in pure heptane, while the more polar P-γ-OH end-unit is preferably extracted in the polar solvents.
GC × GC – NMR correlation
Similar solvent fractionation was used (for sample preparation) in an earlier study presenting structure elucidation of RCF lignin oil using 1H–13C HSQC NMR spectroscopy. The advantage of this particular method is that a specific end-unit or inter-unit linkage only has a limited amount of C–H correlation signals, which are independent of the individual molecular structure. For example, a dimer containing a β-5 ethyl (β-5 E) inter-unit linkage will have the same β-5 E C–H correlation pairs as a trimer also containing β-5 E. Hence, the total relative distribution of a specific inter-unit linkage or end-unit can be quantified for the entire RCF lignin oil. A general disadvantage of this spectroscopic method is that only bulk information of these molecular structures is obtained. Accordingly, it is challenging to investigate differences in the distribution of specific structures in specific classes (viz. monomers, dimers, trimers, etc.). Besides, the technique has sensitivity limits, as known for NMR spectroscopy. The high-resolution GC × GC method developed herein solves this latter issue, providing more detailed structural information, also of the minor compounds in the RCF lignin oil. To investigate if large differences in distribution can be observed be(in mol%; monomers, dimers, and trimers tween these classes and the entire lignin oil, the structural relative distributions obtained by GC × GC) are compared with the relative distributions obtained by 1H–13C HSQC NMR spectroscopy by analysis of the entire sample (in mol%, total).As shown in Fig. 10, the effect of the extraction solvent has a clear influence on the distribution of end-units, as discussed earlier. For almost all fractions, the relative occurrence of a specific end-unit obtained by the 1H–13C HSQC NMR spectroscopy lies between that of the specific fractions (i.e. monomer, dimer, and trimer) of a certain sample, validating the GC × GC analysis. Thus, in addition to the insight in chemical structure (from MS), the accumulated quantified information from GC × GC – FID accords with the bulk information delivered by 1H–13C HSQC NMR spectroscopy.
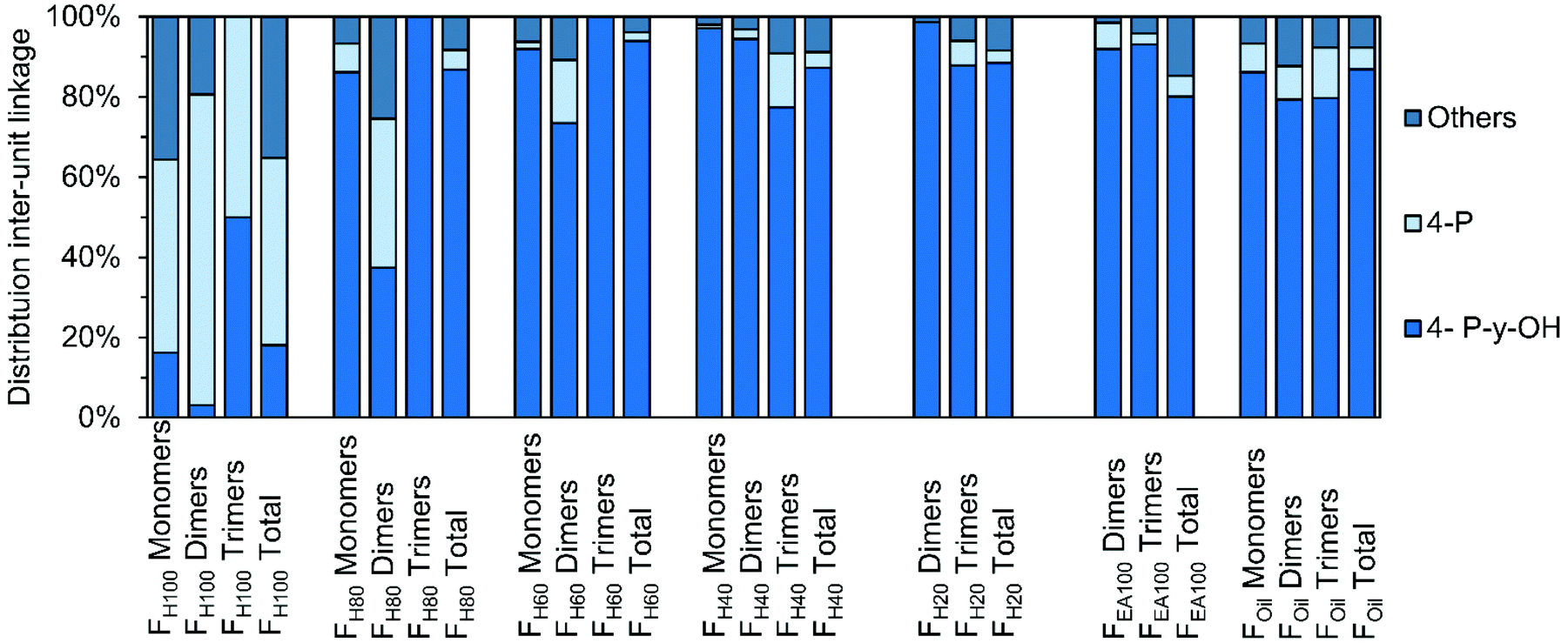 | ||
| Fig. 10 Comparison of distribution of end-units, divided in 4-P-γ-OH, 4-P, and “Others” in the different RCF lignin fractions. The monomer, dimer, and trimer distribution is obtained by GC × GC. The distribution of each total fraction is obtained by 1H–13C HSQC NMR spectroscopy.9 | ||
Just as for the end-units, the distribution differences for the various inter-unit linkages are shown in Fig. S5† (Fig. S5.1–S5.3, see ESI†) and similar trends can be observed. That is, the occurrence of the typical RCF β-5, β-1, and β–β structures found by GC × GC and NMR analysis are comparable for fraction Foil. One noticeable exception is the very low β-5 E substitution in the entire lignin oil's trimer fraction, compared to the two times higher β-5 E substitution observed in the entire oil. The reason for this might lie either in a reactivity difference during the RCF process (i.e. lower reactivity to form β-5 E in trimers) or in the GC × GC detection. Indeed, while an even amount of dimers bearing the β-5 E or β-5 γ-OH group are identified, four times more β-5 γ-OH structures are identified in the trimers as compared to the β-5 E. Since not all trimer signals were identified and quantified, the possible lower catalytic selectivity to the β-5 E linkage might thus simply be enhanced by the low number of identified β-5 E containing signals.
Besides comparing product distribution of the RCF process, as ascertained by GC × GC analysis and 1H–13C HSQC NMR spectroscopic analysis, the overall yield of the specific molecular structures obtained by GC × GC analysis can also be constructed and compared with the 1H–13C HSQC NMR spectroscopic results. This furthers the insight into the distribution of a certain molecular structure in the monomers, dimers, and trimers relative to the entire oil. As the 1H–13C HSQC NMR spectroscopic results are expressed in relative percentage per guaiacyl unit, the GC × GC results were recalculated according to formulas in Note S5.4 (see ESI†).
Before going into detail, a few important remarks on the interpretation of these results must be made. First, the results of two powerful analytical techniques are combined, each with their advantages and disadvantages. The obvious advantage of GC × GC is the identification of individual molecular structures. However, compounds which are only present in a very small amount are hard and laborious to detect, identify and quantify. Consequently not all molecules, containing a specific molecular structure are taken into account in this calculation, negatively effecting molecular structures with a low abundance; viz. there are still unassigned trimers. Besides, by recalculating the GC × GC results (in wt% or mol%) to the ‘relative abundancy vs. G-units’, the assumption has been made that 100% of each sample's mass is lignin. Whereas this is evidently more correct for the more polar samples (because of the almost closure of some balances), this is less correct for the less polar samples, likely the consequence of the presence of some non-polar extractives. One major disadvantage of 1H–13C HSQC NMR spectroscopy is that its quantification is only on a relative scale (vs. the aromatic part) and that the spectroscopic related issues (such as the JC–H dependency or relaxation effects) might play a large role in comparing these relative quantitated structures with the absolute quantitated structures by GC × GC. Despite these barriers, the results of these product distributions still contain various relevant trends, as shown in Fig. 11.
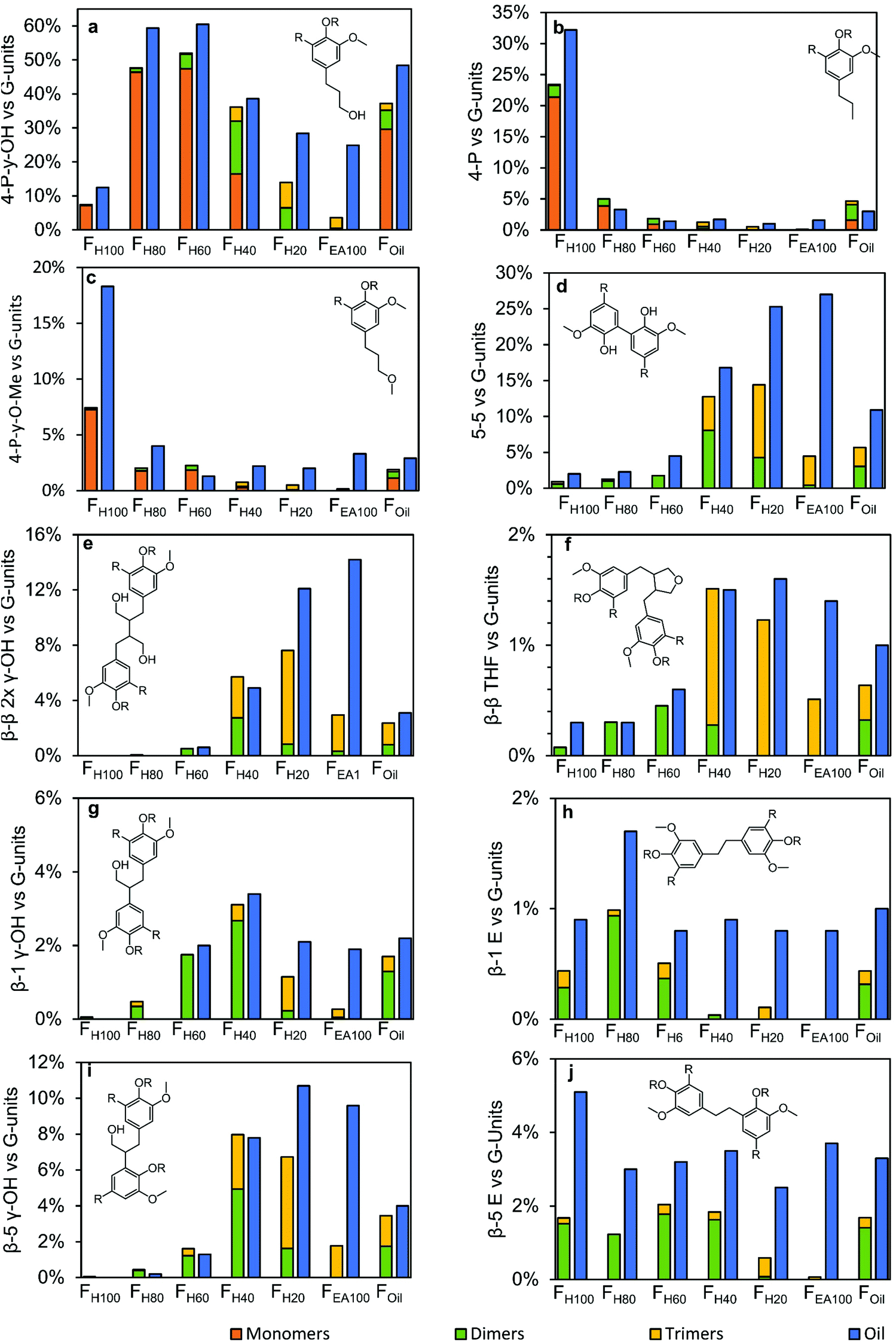 | ||
| Fig. 11 Distribution of the end-units and inter-unit linkages found in the monomers, dimers and trimers in the different fractions and compared to their amounts found in the entire sample. The monomer, dimer and trimer distribution is quantified according to Note S5.4 (see ESI†). The results of the oil are obtained by 1H–13C HSQC NMR spectroscopy.9 | ||
Firstly, it is obvious from Fig. 11a–c that the monomers account for by far the largest amount of end-units. Yet, in certain fractions from intermediate polarity (viz. FH40 and FH20), also the dimers and trimers make up for a large part of the end-units (Fig. 11a). Overall, in the results obtained by 1H–13C HSQC NMR spectroscopy, almost in all cases more end-units have been observed in the fractions. In FH100, FH80, and FH60, this can likely be ascribed to the reasons noted above, viz. presence of non-lignin molecules, analytical complexity, since only a minor amount of RCF lignin trimers are present in these fractions, excluding the possibility of higher molecular weight structures – which is also in correspondence with the GPC result (Fig. S1†). However, in FH40, FH20, and FEA100, the higher amount of end-units observed by 1H–13C HSQC NMR spectroscopy is likely the consequence of the undetected dimers and trimers, and the presence of higher molecular weight structures, such as RCF lignin-derived tetramers, pentamers, etc., which cannot be analyzed by the GC × GC technique due to too high evaporation temperatures of the products.
Secondly, between 40–80% of a specific inter-unit linkage quantified in 1H–13C HSQC NMR spectroscopy can be accounted for by the observed dimers and trimers (Fig. 11d–j), indicating that a considerable amount of the RCF lignin inter-unit linkages are present in the observed RCF dimers and trimers. More detailed interpreting of the distribution of specific inter-unit linkages has to be done with caution, due to the above described barriers arising from the construction of these figures. This is likely the consequence of the low number of identified trimers bearing these inter-unit linkages.
Conclusion
This study shows both the comprehensive identification and quantification of the dimeric and trimeric phenolic oligomers in the RCF lignin oil of pine wood. The successful combination of the high-temperature GC × GC – FID and GC × GC – MS, besides fractionation of lignin oil suing varying solvent polarity, allows to unambiguously assign molecular structures of thirty-six dimers and twenty-one trimers in the RCF lignin oil samples. Derivatization of these lignin samples was critical to prevent peak-tailing and co-eluting effects. The detailed structural information in terms of inter-unit linkages and aliphatic end-units of the dimeric and trimeric oligomers was revealed. The similar structural motifs (i.e. inter-unit linkages and end-units) of these dimers and trimers disclose that they are subjected to the same chemical transformation during the RCF process, a claim that can tentatively also be transferred to larger oligomers. Accumulation of the GC × GC quantified products with regard to end groups and inter-linkages agrees with recently acquired bulk 1H–13C HSQC NMR spectroscopic information. This study demonstrates a methodology using GC × GC in combination with recent 1H–13C HSQC NMR spectroscopic findings to advance the molecular structural information of RCF lignin. Thus, we encourage future RCF lignin related research to implement such combined analysis as to maximize their insights. Future GC × GC FID/MS dedicated research should focus on identifying more dimers and trimers by analyzing mass spectra, possibly in combination with NMR, organic synthesis, and purification strategies. Besides it might also be helpful to characterize lignin repolymerization products, not only in RCF lignin oil but also in other lignin types. Ultimately, the molecular information will enable the community to more rigorously assess the chemical transformations that lignin undergoes during RCF biorefinery processing as well as to steer further research in downstream lignin oil usage, functionalization and separations, and corresponding application development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the Catalisti-SBO Project NIBCON. B.S, S. V. D. B., and K. V. A. also acknowledge funding through FWO-SBO project BioWood and acknowledge that this project had received funding from the Bio-based Industries Joint Undertaking under the European Union's Horizon 2020 Research and Innovation Programme under grant agreement no 837890 (SMARTBOX) and from the National EoS (BIOFACT) and Flemish iBOF (NextBioRef) programs. S. V. D. B. acknowledges Flanders Innovation & Entrepreneurship (Innovation Mandate). Furthermore, Kevin M. Van Geem is holder of the ERC Grant OPTIMA (Process Intensification and Innovation in Olefin Production by Multiscale Analysis and Design) with the grant agreement ID 818607. This work was authored in part by the National Renewable Energy Laboratory, operated by the Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding was provided to RK and GTB by the U.S. DOE Office of Energy Efficiency and Renewable Energy Bioenergy Technologies Office. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344(6185) DOI:10.1126/science.1246843.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van Den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- J. Zhang, Y. Jiang, L. F. Easterling, A. Anstner, W. Li, K. Z. Alzarieni, X. Dong, J. Bozell and H. I. Kenttämaa, Green Chem., 2021, 23, 983–1000 RSC.
- C. Zhao, Z. Hu, L. Shi, C. Wang, F. Yue, S. Li, H. Zhang and F. Lu, Green Chem., 2020, 22, 7366–7375 RSC.
- F. Yue, F. Lu, M. Regner, R. Sun and J. Ralph, ChemSusChem, 2017, 10, 830–835 CrossRef CAS.
- A. De Santi, M. V. Galkin, C. W. Lahive, P. J. Deuss and K. Barta, ChemSusChem, 2020, 13, 4468 CrossRef CAS.
- A. Kumar and B. Thallada, Sustainable Energy Fuels, 2021, 5, 3802–3817 RSC.
- J. Zhu, C. Yan, X. Zhang, C. Yang, M. Jiang and X. Zhang, Prog. Energy Combust. Sci., 2020, 76, 100788 CrossRef.
- X. Dou, W. Li, C. Zhu and X. Jiang, Appl. Catal., B, 2021, 287, 119975 CrossRef CAS.
- F. Wang, D. Ouyang, Z. Zhou, S. J. Page, D. Liu and X. Zhao, J. Energy Chem., 2021, 57, 247–280 CrossRef.
- Y. Liao, S. F. Koelewijn, G. van den Bossche, J. van Aelst, S. van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- Y.-Y. Wang, C. E. Wyman, C. M. Cai and A. J. Ragauskas, ACS Appl. Polym. Mater., 2019, 1, 1672–1679 CrossRef CAS.
- W. Zhang, Y. Ma, C. Wang, S. Li, M. Zhang and F. Chu, Ind. Crops Prod., 2013, 43, 326–333 CrossRef CAS.
- S. Nikafshar, J. Wang, K. Dunne, P. Sangthonganotai and M. Nejad, ChemSusChem, 2021, 14, 1184–1195 CrossRef CAS PubMed.
- K. Van Aelst, E. Van Sinay, T. Vangeel, Y. Zhang, T. Renders, S. den Bosch, J. Van Aelst and B. Sels, Chem. Commun., 2021, 57, 5642–5645 RSC.
- S. Van Den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- M. V. Galkin, A. T. Smit, E. Subbotina, K. A. Artemenko, J. Bergquist, W. J. J. Huijgen and J. S. M. Samec, ChemSusChem, 2016, 9, 3280–3287 CrossRef CAS.
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, G. T. Beckham and Y. Román-Leshkov, Joule, 2017, 1, 613–622 CrossRef CAS.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttämaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC.
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS PubMed.
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS.
- T. Renders, S. den Bosch, S.-F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- T. Renders, S. Van Den Bosch, T. Vangeel, T. Ennaert, S. F. Koelewijn, G. Van Den Bossche, C. M. Courtin, W. Schutyser and B. F. Sels, ACS Sustainable Chem. Eng., 2016, 4, 6894–6904 CrossRef CAS.
- K. Van Aelst, E. Van Sinay, T. Vangeel, E. Cooreman, G. Van den Bossche, T. Renders, J. Van Aelst, S. Van den Bosch and B. Sels, Chem. Sci., 2020, 11, 11498–11508 RSC.
- H. Li and G. Song, ACS Catal., 2019, 9, 4054–4064 CrossRef CAS.
- Z. Sun, J. Cheng, D. Wang, T.-Q. Yuan, G. Song and K. Barta, ChemSusChem, 2020, 1–15 Search PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2020, 14, 262–292 RSC.
- Z. Sun, J. Cheng, D. Wang, T.-Q. Yuan, G. Song and K. Barta, ChemSusChem, 2020, 13(19), 5199–5212 CrossRef CAS PubMed.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS PubMed.
- E. Cooreman, T. Vangeel, K. Van Aelst, J. Van Aelst, J. Lauwaert, J. W. Thybaut, S. Van den Bosch and B. F. Sels, Ind. Eng. Chem. Res., 2020, 59, 17035–17045 CrossRef CAS.
- T. I. Korányi, B. Fridrich, A. Pineda and K. Barta, Molecules, 2020, 25(12), 2815 CrossRef.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- X. Jiang, D. Savithri, X. Du, S. Pawar, H. Jameel, H. Chang and X. Zhou, ACS Sustainable Chem. Eng., 2017, 5, 835–842 CrossRef CAS.
- M. Gigli and C. Crestini, Green Chem., 2020, 22, 4722–4746 RSC.
- R. Ebrahimi Majdar, A. Ghasemian, H. Resalati, A. Saraeian, C. Crestini and H. Lange, ACS Sustainable Chem. Eng., 2020, 8, 16803–16813 CrossRef.
- C. Allegretti, O. Boumezgane, L. Rossato, A. Strini, J. Troquet, S. Turri, G. Griffini and P. D'Arrigo, Molecules, 2020, 25(12), 2893 CrossRef CAS.
- F. J. Calvo-Flores, G. G. Dobado, J. A. Isac-Garcia and J. Martin-Martinez, Lignin and Lignans as renewable raw materials, John Wiley & Sons, Ltd, 2015 Search PubMed.
- H. Lange, P. Giannì and C. Crestini, in Lignin Valorization: Emerging Approaches, The Royal Society of Chemistry, 2018, pp. 413–476 Search PubMed.
- J. S. Lupoi, S. Singh, R. Parthasarathi, B. A. Simmons and R. J. Henry, Renewable Sustainable Energy Rev., 2015, 49, 871–906 CrossRef CAS.
- H. Ma, H. Li, W. Zhao, L. Li, S. Liu, J. Long and X. Li, Green Chem., 2019, 21, 658–668 RSC.
- C. S. Lancefield, S. Constant, P. de Peinder and P. C. A. Bruijnincx, ChemSusChem, 2019, 12, 1139–1146 CrossRef CAS PubMed.
- X. Bai, K. H. Kim, R. C. Brown, E. Dalluge, C. Hutchinson, Y. J. Lee and D. Dalluge, Fuel, 2014, 128, 170–179 CrossRef CAS.
- T. Renders, W. Schutyser, S. Van den Bosch, S.-F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, ACS Catal., 2016, 6, 2055–2066 CrossRef CAS.
- Y. S. Choi, P. A. Johnston, R. C. Brown, B. H. Shanks and K.-H. Lee, J. Anal. Appl. Pyrolysis, 2014, 110, 147–154 CrossRef CAS.
- M. B. Figueirêdo, R. H. Venderbosch, H. J. Heeres and P. J. Deuss, J. Anal. Appl. Pyrolysis, 2020, 149, 104837 CrossRef.
- M. D. M. Arruda, S. da Paz Leôncio Alves, I. J. da Cruz Filho, G. F. de Sousa, G. A. de Souza Silva, D. K. D. do Nascimento Santos, M. do Carmo Alves de Lima, G. J. de Moraes Rocha, I. A. de Souza and C. M. L. de Melo, Int. J. Biol. Macromol., 2021, 180, 286–298 CrossRef CAS PubMed.
- F. Leng, Y. Wang, J. Chen, S. Wang, J. Zhou and Z. Luo, Chin. J. Chem. Eng., 2017, 25, 324–329 CrossRef CAS.
- H. N. Lyckeskog, C. Mattsson, L. Olausson, S.-I. Andersson, L. Vamling and H. Theliander, Biomass Convers. Biorefin., 2017, 7, 401–414 CrossRef CAS.
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, W. Muchero, K. J. Ramirez, G. T. Beckham and Y. Román-Leshkov, Nat. Commun., 2019, 10, 2033 CrossRef PubMed.
- D. C. Dayton and T. D. Foust, in Emerging Issues in Analytical Chemistry, Elsevier, 2020, pp. 75–88 Search PubMed.
- M. Kinne, M. Poraj-Kobielska, R. Ullrich, P. Nousiainen, J. Sipilä, K. Scheibner, K. E. Hammel and M. Hofrichter, Holzforschung (HF), 2011, 65, 673–679 CAS.
- E. Kiyota, P. Mazzafera and A. C. H. F. Sawaya, Anal. Chem., 2012, 84, 7015–7020 CrossRef CAS.
- B. C. Owen, L. J. Haupert, T. M. Jarrell, C. L. Marcum, T. H. Parsell, M. M. Abu-Omar, J. J. Bozell, S. K. Black and H. I. Kenttämaa, Anal. Chem., 2012, 84, 6000–6007 CrossRef CAS.
- D. Tomasini, F. Cacciola, F. Rigano, D. Sciarrone, P. Donato, M. Beccaria, E. B. Caramão, P. Dugo and L. Mondello, Anal. Chem., 2014, 86, 11255–11262 CrossRef CAS PubMed.
- H. Sheng, W. Tang, J. Gao, J. S. Riedeman, G. Li, T. M. Jarrell, M. R. Hurt, L. Yang, P. Murria, X. Ma, J. J. Nash and H. I. Kenttämaa, Anal. Chem., 2017, 89, 13089–13096 CrossRef CAS.
- S. O. Asare, K. R. Dean and B. C. Lynn, Anal. Bioanal. Chem., 2021, 413, 4037–4048 CrossRef CAS PubMed.
- J. Zhang, Y. Jiang, A. Astner, H. Zhu, J. J. Bozell and H. I. Kenttämaa, Green Chem., 2021, 23, 4024–4033 RSC.
- W. Schutyser, S. Van Den Bosch, T. Renders, T. De Boe, S.-F. F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Green Chem., 2015, 17, 5035–5045 RSC.
- S. Van Den Bosch, W. Schutyser, S.-F. F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC.
- Y. Wang, R. Yin, M. Chai, Nishu, C. Li, M. Sarker and R. Liu, J. Energy Inst., 2020, 93, 2163–2175 CrossRef CAS.
- Z. Ji-lu, J. Anal. Appl. Pyrolysis, 2007, 80, 30–35 CrossRef.
- K. G. Kalogiannis, S. D. Stefanidis, C. M. Michailof, A. A. Lappas and E. Sjöholm, J. Anal. Appl. Pyrolysis, 2015, 115, 410–418 CrossRef CAS.
- M. R. Djokic, T. Dijkmans, G. Yildiz, W. Prins and K. M. Van Geem, J. Chromatogr. A, 2012, 1257, 131–140 CrossRef CAS PubMed.
- C. Michailof, T. Sfetsas, S. Stefanidis, K. Kalogiannis, G. Theodoridis and A. Lappas, J. Chromatogr. A, 2014, 1369, 147–160 CrossRef CAS.
- T. Sfetsas, C. Michailof, A. Lappas, Q. Li and B. Kneale, J. Chromatogr. A, 2011, 1218, 3317–3325 CrossRef CAS.
- F. Mao, H. Fan and J. Wang, J. Anal. Appl. Pyrolysis, 2019, 139, 213–223 CrossRef CAS.
- N. V. Hung, C. Mohabeer, M. Vaccaro, S. Marcotte, V. Agasse-Peulon, L. Abdelouahed, B. Taouk and P. Cardinael, J. Mass Spectrom., 2020, 55, e4495 CrossRef PubMed.
- V. O. Nunes, R. V. S. Silva, G. A. Romeiro and D. A. Azevedo, Microchem. J., 2020, 153, 104514 CrossRef CAS.
- R. V. S. Silva, N. S. Tessarolo, V. B. Pereira, V. L. Ximenes, F. L. Mendes, M. B. B. de Almeida and D. A. Azevedo, Talanta, 2017, 164, 626–635 CrossRef CAS PubMed.
- C. R. Kumar, N. Anand, A. Kloekhorst, C. Cannilla, G. Bonura, F. Frusteri, K. Barta and H. J. Heeres, Green Chem., 2015, 17, 4921–4930 RSC.
- L. Negahdar, A. Gonzalez-Quiroga, D. Otyuskaya, H. E. Toraman, L. Liu, J. T. B. H. Jastrzebski, K. M. Van Geem, G. B. Marin, J. W. Thybaut and B. M. Weckhuysen, ACS Sustainable Chem. Eng., 2016, 4, 4974–4985 CrossRef CAS PubMed.
- C. M. Michailof, K. G. Kalogiannis, T. Sfetsas, D. T. Patiaka and A. A. Lappas, Wiley Interdiscip. Rev.: Energy Environ., 2016, 5, 614–639 CAS.
- R. Vendamme, J. Behaghel de Bueren, J. Gracia-Vitoria, F. Isnard, M. M. Mulunda, P. Ortiz, M. Wadekar, K. Vanbroekhoven, C. Wegmann, R. Buser, F. Héroguel, J. S. Luterbacher and W. Eevers, Biomacromolecules, 2020, 21(10), 4135–4148 CrossRef CAS PubMed.
- J. E. Q. Quinsaat, E. Feghali, D. J. van de Pas, R. Vendamme and K. M. Torr, ACS Appl. Polym. Mater., 2021, 3, 5845–5856 CrossRef CAS.
- E. Feghali, D. J. van de Pas and K. M. Torr, Biomacromolecules, 2020, 21, 1548–1559 CrossRef CAS PubMed.
- J. S. Mahajan, R. M. O'Dea, J. B. Norris, L. T. J. Korley and T. H. Epps, ACS Sustainable Chem. Eng., 2020, 8, 15072–15096 CrossRef CAS.
- A. Moreno and M. H. Sipponen, Mater. Horiz., 2020, 7, 2237–2257 RSC.
- M. N. Collins, M. Nechifor, F. Tanasă, M. Zănoagă, A. McLoughlin, M. A. Stróżyk, M. Culebras and C.-A. Teacă, Int. J. Biol. Macromol., 2019, 131, 828–849 CrossRef CAS PubMed.
- I. Zaborniak, P. Chmielarz and K. Matyjaszewski, Eur. Polym. J., 2019, 120, 109253 CrossRef CAS.
- M. A. Jedrzejczyk, S. Van den Bosch, J. Van Aelst, K. Van Aelst, P. D. Kouris, M. Moalin, G. R. M. Haenen, M. D. Boot, E. J. M. Hensen, B. Lagrain, B. F. Sels and K. V. Bernaerts, ACS Sustainable Chem. Eng., 2021, 9, 12548–12559 CrossRef CAS.
- E. O. Ebikade, N. Samulewicz, S. Xuan, J. D. Sheehan, C. Wu and D. G. Vlachos, Green Chem., 2020, 22, 7435–7447 RSC.
- A. Parodi, E. Diguilio, S. Renzini and I. Magario, Carbohydr. Res., 2020, 487, 107885 CrossRef CAS PubMed.
- J.-Y. de Saint Laumer, S. Leocata, E. Tissot, L. Baroux, D. M. Kampf, P. Merle, A. Boschung, M. Seyfried and A. Chaintreau, J. Sep. Sci., 2015, 38, 3209–3217 CrossRef CAS PubMed.
- J.-Y. de Saint Laumer, E. Cicchetti, P. Merle, J. Egger and A. Chaintreau, Anal. Chem., 2010, 82, 6457–6462 CrossRef CAS PubMed.
- S. Y. Park, J.-Y. Kim, H. J. Youn and J. W. Choi, Int. J. Biol. Macromol., 2018, 106, 793–802 CrossRef CAS PubMed.
- C. S. Lancefield, H. L. J. Wienk, R. Boelens, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Sci., 2018, 9, 6348–6360 RSC.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem.,– Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- H. Li and G. Song, ACS Catal., 2020, 10(20), 12229–12238 CrossRef CAS.
- F. Yue, F. Lu, S. Ralph and J. Ralph, Biomacromolecules, 2016, 17, 1909–1920 CrossRef CAS PubMed.
- Y. Li, T. Akiyama, T. Yokoyama and Y. Matsumoto, Biomacromolecules, 2016, 17, 1921–1929 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03822b |
| ‡ Both authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2022 |