
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile formation of black titania films using an atmospheric-pressure plasma jet†
M. Emre
Sener
a,
Raul
Quesada-Cabrera
 ab,
Ivan P.
Parkin
a and
Daren J.
Caruana
*a
ab,
Ivan P.
Parkin
a and
Daren J.
Caruana
*a
aDepartment of Chemistry, Christopher Ingold Laboratories, University College London, 20 Gordon St., London WC1H 0AJ, UK. E-mail: D.J.Caruana@ucl.ac.uk
bDepartment of Chemistry, Photoelectrocatalysis for Environmental Applications (FEAM), Institute of Environmental Studies and Natural Resources (i-UNAT), Universidad de Las Palmas de Gran Canaria, Campus de Tafira 35017, Spain
First published on 23rd February 2022
Abstract
A rapid atmospheric-pressure pulsed helium/hydrogen plasma jet method for conversion of TiO2 films into defective, black TiO2 is demonstrated. This method can reduce nanoporous anatase films with fine spatial control, allowing precise 2D patterning of pre-deposited films under ambient conditions. The resulting modified TiO2 films are stable in air and show enhanced photocatalytic activity with respect to the as-deposited films. From photoelectrochemical current measurements, the plasma-treated anatase TiO2 films supported on FTO-glass showed a six-fold increase in photocurrent density under both sub- and super-bandgap illumination compared to untreated anatase films. The changes in optical absorption are mainly due to introduced mid-gap states, with negligible overall band gap narrowing. Thorough characterisation of these black TiO2 materials using X-ray photoelectron, Raman and UV-VIS spectroscopy showed strong evidence for the presence of Ti3+ states in the reduced films. There is a clear relationship between the Ti3+ content of the plasma-treated TiO2 and the enhanced photocurrent, although increased Ti3+ content leads to a decrease in photocurrent, which we ascribe to an increase in the number of electron–hole recombination centres.
Introduction
A disorder-engineered, hydrogenated titania (TiO2) material, commonly known as black TiO2,1,2 has found broad applications across diverse fields such as catalysis and fuel cells, through to dye sensitised solar cells and environmental pollution removal.3–7 This interest is mainly fuelled by black TiO2's ability to harvest low-energy photons across the visible light region of the solar spectrum, suggesting that its inherent photocatalytic activity may be improved.However, the practical use of this material is currently limited due to the harsh conditions needed to form black TiO2. Methods relying on the treatment of pristine TiO2 under high pressure and/or temperature H2 flow conditions over periods ranging from 2 hours to 5 days.8,9 Comparatively, atmospheric pressure plasma jet (APPJ) work at low temperature and present an energy efficient method for conversion of TiO2 films into oxygen deficient black TiO2. This study uses APPJ at low power, <10 W, very low concentration of hydrogen (<5%) and a carrier gas which is not consumed and may potentially be recycled. This opens the route for a very sustainable approach to preparing oxygen deficient black TiO2.
The origin of the broad visible light absorption of black TiO2 ranging from the ultraviolet (UV) to the near-infrared region is object of current active research.2,7,10,11 The peculiar optical properties of black TiO2 have been attributed to disorder-induced band tail states, forming a continuum that extends to and even overlaps with the conduction band edge.2,12 Discussion on the factors governing light absorption and photocatalytic performance of black TiO2 and other reduced versions of TiO2 containing Ti3+, have been extensively described.10,11 The role of hydrogen in the mechanism of defect formation and diffusion is unclear.10 Not all hydrogenation procedures result in black TiO2.13 The presence of Ti3+ species, oxygen vacancies, Ti–OH and Ti–H groups seems arbitrary.1 For almost all hydrogenation approaches, disordered structural features were observed near the surface of black TiO2, with a general lattice contraction of the disordered layer, although expansion of the shell has also been reported.2 The role of the disordered layer is still controversial and it is not clear how it participates in the photocatalytic properties of the material.14,15 The structure–property landscape of black TiO2 remains largely incomplete and it will undoubtedly be subject of further active research throughout this new decade.
The need for rapid, accessible and controllable avenues to black TiO2 formation is required. Plasma processing can be a viable alternative to reduced pressure hydrogenation approaches as a promising synthesis route towards black TiO2. Islam et al.16 demonstrated the reduction of TiO2 using a 400 W H2 plasma at 10 Torr. Similarly, Yan et al.17 reported on the synthesis of black TiO2 using a 3000 W inductively coupled H2 plasma at 25 Torr. Mohammadizadeh et al.18 synthesised black TiO2via a hollow cathode plasma process. In all these reports, it was found that the reduced TiO2 versions showed enhanced photocatalytic activity compared to pristine TiO2 samples. It must be noted that the majority of previous studies have relied on powder TiO2 forms and processes based on vacuum apparatus that limits the practicality of the reduction process.
Despite progress, to our knowledge, no sustainable route to black TiO2 films have ever been reported at atmospheric pressure and convenient substrate temperatures (<100 °C), other than chemical routes. In the current work, a rapid atmospheric pressure plasma method is described for the reduction of TiO2 films into black TiO2 utilising a pulsed helium atmospheric pressure plasma jet containing a minimal concertation of hydrogen. We have previously demonstrated the reducing capability of atmospheric pressure plasma jets on various metal oxides.19,20 In this work, we show that it is possible to reduce nanoporous anatase films with fine spatial control, allowing large area processing of pre-deposited TiO2 films at ambient conditions.
Experimental
Plasma jet treatment of TiO2 photoelectrodes
Pristine and modified electrodes were screen-printed from nano-titania paste (Solaronix Inc.) onto fluorine doped tin oxide (FTO)-coated glass substrates (Osilla Ltd) and calcined at 550 °C for 1 hour in air. The film thickness was estimated as 2.5 µm via cross-sectional scanning electron microscopy (see ESI†). The particle size within individual TiO2 grains was estimated as 20–30 nm after calcination (ESI, Fig. S1†).The pulsed plasma jet used in all experiments (Fig. 1a and b) was generated at a tungsten pin electrode inserted coaxially into a glass capillary or an alumina tube, as described elsewhere.20 Briefly, the electrode is driven by a radio frequency generator operating at 13.56 MHz and 10 W, the driving signal is pulse-width modulated at 30 kHz, 15% duty cycle to minimise the thermal load on the samples. The driving signal is passed through a matching network tuned to match the high impedance plasma load to the 50 Ω output of the generator. The working gas mixture of helium and hydrogen (5% v/v) is then ignited between the electrode and the TiO2 surface at a total flow rate of 200 ml min−1. To prepare the black TiO2 photoanodes, surfaces of identically prepared electrodes were scanned with the plasma jet (foot print 1 mm2 on pristine TiO2) over a 1 cm × 1 cm area at constant input power. The rate of travel was fixed at 1 mm s−1 and 20 traverse passes were required for full coverage of the treatment area with 0.5 mm stepover. We refer to the residence time per unit area in the samples produced, with a single pass over the surface corresponding to a residence time of 2 s mm−2.
 | ||
| Fig. 1 (a) Photograph of the plasma jet treatment on a titania film showing a sample area of black TiO2. (b) Diagrammatic view of the reduction process. | ||
Surface, structural and optical characterization
A Thermo K-Alpha photoelectron spectroscopy system equipped with a monochromatic Al-Kα (1486.6 eV) X-ray source was used for X-ray photoelectron spectra acquisition. Spectra were collected from a spot of 50 μm diameter on the sample surface, at a pass energy of 50 eV. All samples were washed with deionised water prior to XPS spectra collection, and no ion beam sputtering was used to further clean the surfaces to avoid damage to the film surface. Curve fitting was used for deconvolution of high-resolution spectra using Voigt line shapes and Shirley background functions. An Ocean Optics HR4000 spectrometer with a 0.5 nm (FWHM) resolution was used for all UV/Vis measurements. Renishaw inVia confocal Raman microscope with an unpolarized 532 nm argon ion laser excitation source was used for the characterization of the films. Raman spectra were obtained from an area of approximately 10 μm diameter with a 100× objective. The spectra were recorded in the 140 cm−1 to 1000 cm−1 Raman shift range at ambient temperature. Scanning electron micrographs were obtained with a JEOL JSM 6701F SEM at an accelerating voltage of 10 kV with samples placed perpendicularly to the electron gun.Functional characterisation
For photoelectrochemical current measurements, all photoanodes were masked to expose a circular area of 28 mm2 with opaque masking tape. All measurements were done in 0.5 M Na2SO4 electrolyte with a saturated Ag/AgCl reference electrode (BAS, Instruments) and high surface area platinum counter electrode. Potentiostatic control was employed via a potentiostat (AutolabII) to keep the photoanodes biased at +0.3 V vs. Ag/AgCl. For UV illumination, a 365 nm UV LED was used with power density of 30 mW cm−2 measured at the target. For visible illumination, a 420 nm blue LED was used. The light was diffused with a ground quartz window to ensure even illumination across different experiments. All photoelectrodes were front-lit.Photocurrent response simulation
Simulated photocurrents were generated by solving (differential equation) in Mathematica software using the Range–Kutta method. This approach is based on the work of de Araujo et al.21 Details can be found in ESI.†Results and discussion
Surface-reduced TiO2 films were prepared upon scanning of the plasma jet, henceforth labelled as H:TiO2-n, with n indicating the total processing time of the surface with the plasma jet. As the plasma jet was open to ambient air, atmospheric nitrogen was entrained into the plume during the reduction process. The temperature of the plasma plume was directly monitored by fitting synthetic spectra to the measured optical emission from the second positive system of N2. During the on-period of the pulsed plasma jet, the gas plume temperature was determined to be 240 ± 60 °C (ESI, Fig. S2†). The substrate temperature, as measured with a thermocouple, did not rise above 90 °C during the experiments. Visually, the reduced films showed significant darkening upon plasma exposure, turning from white to deep blue/black, as shown in Fig. 2a. The colour change was concomitant with increasing broadband absorption across the visible range upon reduction of the films (Fig. 2b). The plasma treatment was most effective on the surface of relatively thick TiO2 films and thus the effect of the reduction in optical properties is likely underestimated using bulk methods such as UV-Vis spectroscopy. It should be noted that the optical band gap, estimated from Tauc plot analysis (Fig. 2c, see ESI† for full details of analysis) did not shift significantly (<0.05 eV) compared to the pristine sample (Ebg = 3.34 eV), in contrast to multiple literature reports on the electronic properties of black TiO2.22–24 Nevertheless, bulk resistivity analysis suggested that the impact of the plasma treatment was not insignificant in the bulk, as discussed below (vide infra). | ||
| Fig. 2 (a) Photographs of reference sample after consecutive plasma jet passes (from top to bottom, pristine TiO2 and H:TiO2-n with n = 0, 4, 12, 24, 36 s mm−2). (b) Diffuse reflectance spectrum of pristine (TiO2) and plasma-treated (H:TiO2-n) films upon increasing treatment time with the plasma jet and (c) corresponding Tauc plots of the transformed Kubelka–Munk functions. | ||
XPS analysis confirmed the reduction of surface Ti4+ species upon increasing plasma jet exposure residence time on TiO2 films. Fig. 3a shows the evolution of the Ti 2p XP spectra as a function of number of passes of the plasma jet. Decomposition of XPS spectra in the Ti 2p region of pristine TiO2 samples showed two prominent peaks (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio) due to Ti 2p1/2 and 2p2/3 spin orbit split states assigned to Ti4+, shown in Fig. 3b. Plasma-treated samples showed the evolution of two additional broad peaks and the associated spin orbit split components (Fig. 3c). Based on previous studies of Ti2O3 and doped d1 titanium oxides, the multiple peaks were assigned to final state effects of a nominal Ti3+ state.25 Valence band spectra obtained from the highly reduced H:TiO2-36 sample showed differences in band edge position and band tail with respect to pristine TiO2. Valence band maxima of −2.1 and −2.3 eV were estimated from extrapolation of the linear portion of the corresponding band edges for pristine and H:TiO2-36 samples, respectively. The defect states in the plasma-treated samples formed an exponential tail extending towards the Fermi level. In nominally Ti3+ compounds, a distinct mid-gap Ti(a1g) state would be expected just below the Fermi level.26 In our samples, this peak was not detected, which may be attributed to re-oxidation of surface defects via ambient oxygen. For samples with the highest exposure to the plasma jet extent of reduction, repeated XPS experiments showed that the film composition was stable even after storage in air for 6 months with no significant re-oxidation. This stability suggests that the defects are not only at the surface, which would reoxidise rapidly, but deeper in the lattice which would be more stable at low temperature. We were able to entirely re-oxidise the coatings by annealing in air at 400 °C for 10 minutes.
:2 ratio) due to Ti 2p1/2 and 2p2/3 spin orbit split states assigned to Ti4+, shown in Fig. 3b. Plasma-treated samples showed the evolution of two additional broad peaks and the associated spin orbit split components (Fig. 3c). Based on previous studies of Ti2O3 and doped d1 titanium oxides, the multiple peaks were assigned to final state effects of a nominal Ti3+ state.25 Valence band spectra obtained from the highly reduced H:TiO2-36 sample showed differences in band edge position and band tail with respect to pristine TiO2. Valence band maxima of −2.1 and −2.3 eV were estimated from extrapolation of the linear portion of the corresponding band edges for pristine and H:TiO2-36 samples, respectively. The defect states in the plasma-treated samples formed an exponential tail extending towards the Fermi level. In nominally Ti3+ compounds, a distinct mid-gap Ti(a1g) state would be expected just below the Fermi level.26 In our samples, this peak was not detected, which may be attributed to re-oxidation of surface defects via ambient oxygen. For samples with the highest exposure to the plasma jet extent of reduction, repeated XPS experiments showed that the film composition was stable even after storage in air for 6 months with no significant re-oxidation. This stability suggests that the defects are not only at the surface, which would reoxidise rapidly, but deeper in the lattice which would be more stable at low temperature. We were able to entirely re-oxidise the coatings by annealing in air at 400 °C for 10 minutes.
 | ||
| Fig. 3 (a) High resolution Ti 2p XP spectra from pristine and increasingly plasma-treated reduced samples. Peak decomposition analysis of Ti 2p XP spectra is highlighted for pristine (b) and H:TiO2-36 (c) samples. (d) Corresponding valence bands of pristine and H:TiO2-36 samples. | ||
To further understand the changes induced by plasma exposure, Raman spectroscopy was used to probe the local structure of the treated TiO2 films. Raman spectra of the samples showed that, at moderate reduction levels, the only bands visible could be attributed to anatase TiO2 with Raman active modes at 142, 197, 399, 515 and 640 cm−1, as shown in Fig. 4. Reduction leads to an overall decrease in Raman scattering intensity, which suggests an induced short-range disorder upon plasma treatment, together with slight blue shift of the Eg mode at 142 cm−1 (Fig. 4a, inset). This blue shift has been previously attributed to an increase in disorder within the anatase lattice leading to phonon confinement effects.27 No anatase to rutile phase transformation could be detected in the Raman spectra, owing to the low temperatures encountered during the plasma treatment. Prolonged plasma treatment was followed by the appearance of new broad bands at 247 and 341 cm−1 in films with XPS determined Ti3+/Ti ratio above 50% (Fig. 4b). These additional bands have been attributed to the emergence of a brookite phase during amorphization of the highly defective anatase surface.25 These observations are consistent with reported analysis of black TiO2.1,25
 | ||
| Fig. 4 (a) Raman spectra of pristine and reduced TiO2 samples. Inset: normalised Eg band region. (b) Normalised spectra of a highly reduced sample compared to pristine TiO2. | ||
The absolute increase in photocurrent efficiency was up to six-fold compared to that of pristine TiO2. The photocurrent density of plasma-treated samples increased upon illumination under UV (λ = 365 nm) and visible (λ = 420 nm) light is shown in Fig. 5a and b. The photocurrent efficiency did not follow a linear trend, however, and prolonged plasma treatment (beyond a residence time of 25 s mm−2) resulted in a relative decrease of photocurrent density (Fig. 5b). The surface Ti3+/Ti ratio at optimum current density was around 45%, as determined by XPS. This value represents an upper bound on the optimal defect density since the XPS sampling depth is very small compared to the thickness of the film and the diffusion of the reduced species is most probably not fast enough so as to induce uniform reduction across the entire bulk. No immediate change in either photocatalytic performance or a change in optical properties were observed under on–off illumination for up to 6 hours during our experiments for any of the films.
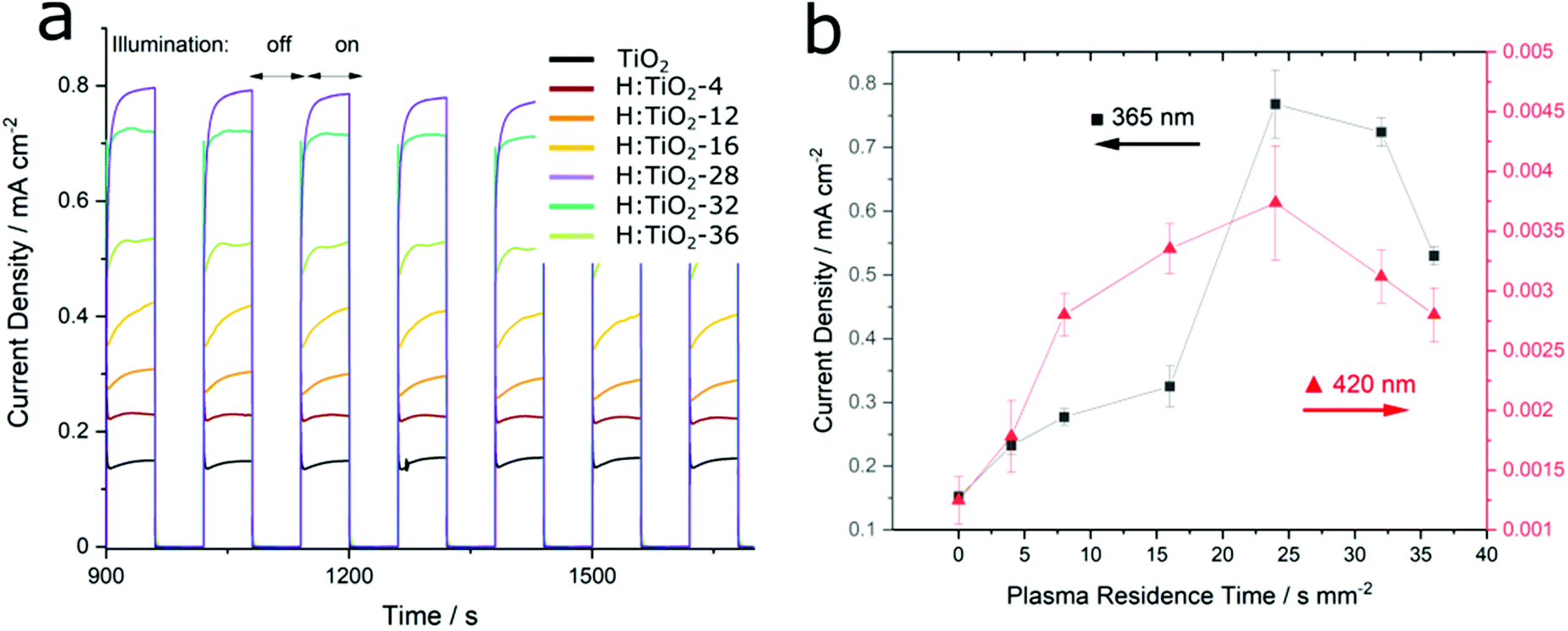 | ||
| Fig. 5 (a) Photocurrent density of prepared TiO2 photoanodes under chopped illumination at 365 nm, (photocurrent density transients under 420 nm in ESI†). (b) Peak steady-state current obtained during plasma treatment, expressed in terms of residence time (units, s mm−2). | ||
It is worth noting that highly reduced samples showing significant disruption of the anatase phase, with additional brookite Raman bands (at 247 and 341 cm−1), resulted in relatively low photocurrent densities, as expected from high distribution of structural defects acting as charge recombination centres. This correlation between high defect density and low photocatalytic efficiency has been previously described in the literature.21
In contrast to some previous reports,28,29 we found only a weak enhancement of visible light activity. If the band gap of the film were significantly reduced, we would expect to see a significant increase in photocurrent density obtained under sub-band gap illumination. Despite this, even under 2.95 eV (420 nm) light which is close to the determined band gap energy, we found that the photocurrent enhancement did not differ significantly from values obtained from super-band gap illumination experiments (ESI, Fig. S3†). This observation agreed with the optical band-gap measurements and valence band XP spectra obtained from the samples. The band positions in plasma reduced TiO2 were not shifted to a significant extent. The visible light absorption was attributed to tail states above the valence band maximum that are formed due to self-doping of TiO2 with Ti3+, yet holes stemming from these tail states do not have a large influence on the overall photocurrent efficiency. A possible explanation for this phenomenon has been brought forward by Wheeler et al.30 who observed that electron–hole pairs produced under visible light excitation are liable to rapidly recombine in oxygen vacancy trap states that lie below the conduction band.
Considering the transient response of photocurrent in chopped light experiments, we observed that the simulated response profiles (Fig. 6a) fitted to a simple model (see ESI†) simulating photocurrent transients with increasing electron mobilities. The apparent change in transient shape could be qualitatively explained by the two modes of induced photocurrent in the photoanode. The initial transient spike observed in the low mobility case seen in Fig. 5a, corresponds to the photocapacitive behaviour of the film as electron hole pairs are generated and electrons close to the FTO/TiO2 interface rapidly diffuse across the junction. This is followed by a second transient corresponding to the catalytic process driving hole depletion in reactions and concurrent electron diffusion. In this case, where electron mobility is significantly increased, more photogenerated electrons are extracted from deep regions within the bulk of the film. This in-turn leads to a decrease in the overall recombination rate and therefore increases the overall photocatalytic reaction rate. As the electron extraction is not limited to just a shallow layer, there is an absence of the initial photocapacitive peak. For the case were electron mobilities are low, the initial photocapacitive peak is dominant, as shown in Fig. 6a. Instead, disregarding space-charge effects, photocurrents in the optimally doped samples increase continuously until they reach a plateau determined by the incident light intensity and photoconversion efficiency.21 In an n-type semiconductor such as TiO2, the conductivity of the film is proportional to electron mobility and electron density. In reduced films, a drastic drop in bulk resistivity is expected due to increased carrier donor (Ti3+) density and increased electron mobility, as discussed. The resistivity of the highly reduced TiO2 films was around 3 orders of magnitude lower than that of pristine TiO2, as shown in Fig. 6b.
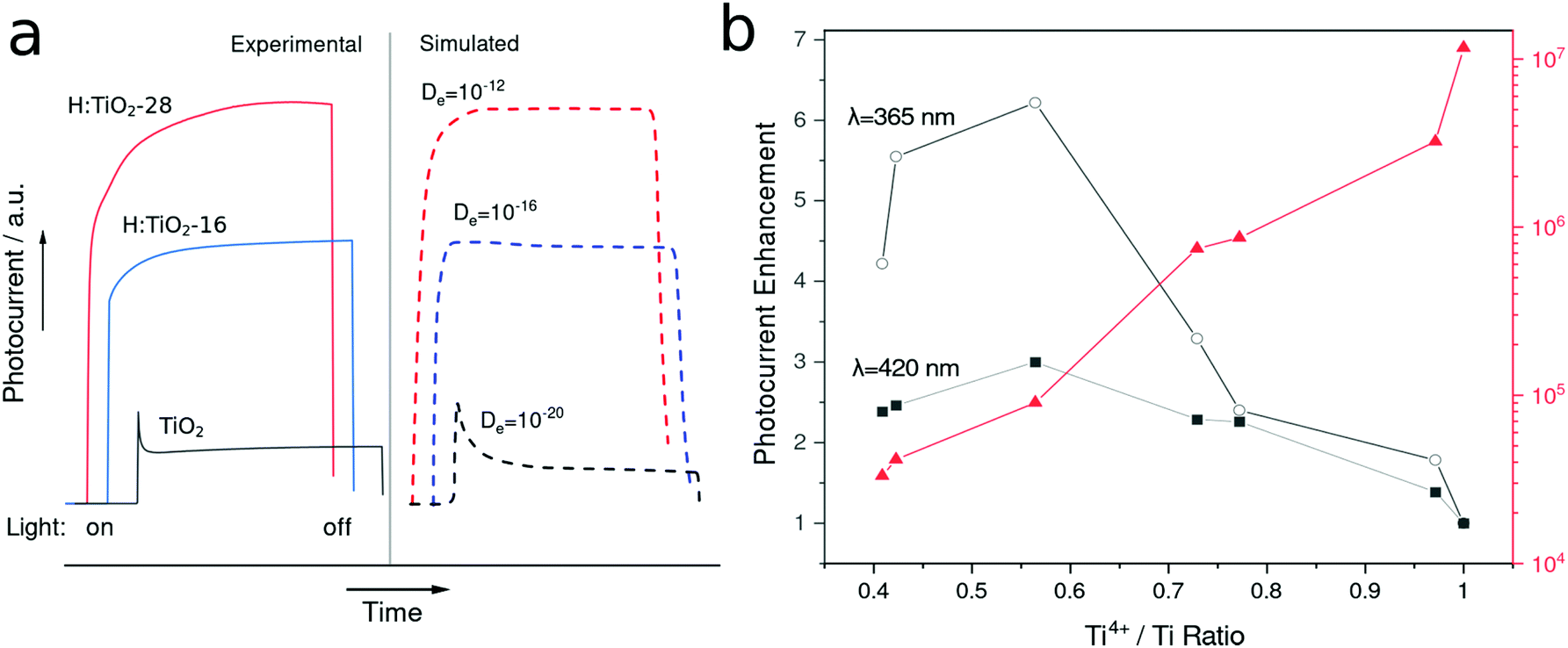 | ||
| Fig. 6 (a) Experimental and simulated photocurrent transients in pristine and selected reduced TiO2 samples. (b) Dependence of photocurrent enhancement ratio and conductivity as a function of defect density, given as Ti4+/total Ti ratio. | ||
Conclusions
Atmospheric-pressure plasma is shown to rapidly reduce TiO2 films providing a highly accessible approach to the synthesis of black titania-based photoelectrodes. The proposed reduction process overcomes the need for high pressure and high temperature treatment methods while at the same time avoiding costly vacuum equipment and strict safety measures. This rapid increase in rate has been attributed to the generation of atomic hydrogen in the plasma plume, evidenced by optical emission spectroscopy (ESI, Fig. S2a†). Photocurrent density improved by a factor of up to 6 under both sub and super-bandgap illumination, which was attributed to an improved conductivity due to the formation of surface Ti3+ defects. A saturation point was also found, where significant Ti3+ content induced a relative decrease in photocurrent density. This was ascribed to an increase in the number of electron–hole recombination centres. Contrary to literature reports of visible light activity and band gap narrowing in black TiO2, we showed that the changes in optical absorption are mainly due to introduced tail states while the overall band gap narrowing is negligible. Similarly, we found that plasma reduced TiO2 showed negligible visible light activity despite intense broadband light absorption. This is supported by the weak photocurrent response obtained under visible light illumination (ESI Fig. S3†).Computational studies of hydrogenation of TiO2 by atomic hydrogen have shown to be thermodynamically favourable at low temperatures both on the surface and subsurface regions.21 The properties of the reduced TiO2 reported here are comparable to those based on black TiO2 generated using high temperature and vacuum methods even when conventional deposition techniques and substrates are used. Furthermore, using a low power plasma jet permits the potential to pattern a single surface coating to provide various levels of Ti4+/Ti3+ ratios at a low temperature, avoiding anatase to rutile phase transformation and damage to catalyst support materials. This is an important step towards the promotion of black titania in commercial settings as photoanodes or environmental catalysts.
Author contributions
This work was conceptualised by Daren J. Caruana and M. Emre Sener, and conducted by M. Emre Sener. The manuscript was written by Daren Caruana and M. Emre Sener with contributions from Raul Quesada-Cabrera and Ivan Parkin.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by EPSRC (EP/T024836/1). MES thanks the EPSRC for a PhD studentship. RQC thanks the Beatriz Galindo Programme (BG20/00072) from the Ministry of Education and Vocational Training (MEFP), Spain, for funding. Prof. Rob Palgrave is thanked for assistance with XPS analysis. Mike Parkes is thanked for assistance with the UV/Vis studies and Dr Steve Firth for his assistance with SEM analysis.References
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- X. B. Chen, L. Liu and F. Q. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- Q. Y. Bi, X. Y. Huang, Y. C. Dong and F. Q. Huang, Catal. Lett., 2020, 150, 1346–1354 CrossRef CAS.
- D. El-Gendy, N. A. A. Ghany and N. K. Allam, RSC Adv., 2019, 9, 12555–12566 RSC.
- J. L. Xu, X. S. Qi, C. Z. Luo, J. Qiao, R. Xie, Y. Sun, W. Zhong, Q. Fu and C. X. Pan, Nanotechnology, 2017, 28, 12 Search PubMed.
- S. G. Ullattil, A. V. Thelappurath, S. N. Tadka, J. Kavil, B. K. Vijayan and P. Periyat, Sol. Energy, 2017, 155, 490–495 CrossRef CAS.
- B. Wang, S. H. Shen and S. S. Mao, J. Materiomics, 2017, 3, 96–111 CrossRef.
- H. Q. Lu, B. B. Zhao, R. L. Pan, J. F. Yao, J. H. Qiu, L. Luo and Y. C. Liu, RSC Adv., 2014, 4, 1128–1132 RSC.
- G. M. Wang, H. Y. Wang, Y. C. Ling, Y. C. Tang, X. Y. Yang, R. C. Fitzmorris, C. C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- N. Rahimi, R. Pax and E. M. Gray, Prog. Solid State Chem., 2019, 55, 1–19 CrossRef CAS.
- K. Zhang and J. H. Park, J. Phys. Chem. Lett., 2017, 8, 199–207 CrossRef CAS PubMed.
- X. B. Chen, L. Liu, Z. Liu, M. A. Marcus, W. C. Wang, N. A. Oyler, M. E. Grass, B. H. Mao, P. A. Glans, P. Y. Yu, J. H. Guo and S. S. Mao, Sci. Rep., 2013, 3, 1510 CrossRef PubMed.
- S. G. Ullattil, S. B. Narendranath, S. C. Pillai and P. Periyat, Chem. Eng. J., 2018, 343, 708–736 CrossRef CAS.
- T. S. Rajaraman, S. P. Parikh and V. G. Gandhi, Chem. Eng. J., 2020, 389, 29 CrossRef.
- U. Diebold, Nat. Chem., 2011, 3, 271–272 CrossRef CAS PubMed.
- S. Z. Islam, A. Reed, S. Nagpure, N. Wanninayake, J. F. Browning, J. Strzalka, D. Y. Kim and S. E. Rankin, Microporous Mesoporous Mater., 2018, 261, 35–43 CrossRef CAS.
- Y. Yan, M. Y. Han, A. Konkin, T. Koppe, D. Wang, T. Andreu, G. Chen, U. Vetter, J. R. Morante and P. Schaaf, J. Mater. Chem. A, 2014, 2, 12708–12716 RSC.
- M. R. Mohammadizadeh, M. Bagheri, S. Aghabagheri and Y. Abdi, Appl. Surf. Sci., 2015, 350, 43–49 CrossRef CAS.
- M. E. Sener and D. J. Caruana, Electrochem. Commun., 2018, 95, 38–42 CrossRef.
- M. E. Sener, S. Sathasivam, R. Palgrave, R. Q. Cabrera and D. J. Caruana, Green Chem., 2020, 22, 1406–1413 RSC.
- M. A. de Araujo, M. F. Gromboni, F. Marken, S. C. Parker, L. M. Peter, J. Turner, H. C. Aspinall, K. Black and L. H. Mascaro, Appl. Catal., B, 2018, 237, 339–352 CrossRef CAS.
- A. Naldoni, M. Allieta, S. Santangelo, M. Marelli, F. Fabbri, S. Cappelli, C. L. Bianchi, R. Psaro and V. Dal Santo, J. Am. Chem. Soc., 2012, 134, 7600–7603 CrossRef CAS PubMed.
- X. Liu, S. M. Gao, H. Xu, Z. Z. Lou, W. J. Wang, B. B. Huang and Y. Dai, Nanoscale, 2013, 5, 1870–1875 RSC.
- A. Sinhamahapatra, J. P. Jeon and J. S. Yu, Energy Environ. Sci., 2015, 8, 3539–3544 RSC.
- R. L. Kurtz and V. E. Henrich, Surf. Sci. Spectra, 1998, 5, 179–181 CrossRef CAS.
- R. L. Kurtz and V. E. Henrich, Surf. Sci. Spectra, 1998, 5, 182–185 CrossRef CAS.
- V. E. Henrich, Surf. Sci. Spectra, 1998, 5, 176–178 CrossRef CAS.
- Z. X. Pei, L. Y. Ding, H. Lin, S. X. Weng, Z. Y. Zheng, Y. D. Hou and P. Liu, J. Mater. Chem. A, 2013, 1, 10099–10102 RSC.
- X. X. Zou, J. K. Liu, J. Su, F. Zuo, J. S. Chen and P. Y. Feng, Chem. – Eur. J., 2013, 19, 2866–2873 CrossRef CAS PubMed.
- D. A. Wheeler, Y. C. Ling, R. J. Dillon, R. C. Fitzmorris, C. G. Dudzik, L. Zavodivker, T. Rajh, N. M. Dimitrijevic, G. Millhauser, C. Bardeen, Y. Li and J. Z. Zhang, J. Phys. Chem. C, 2013, 117, 26821–26830 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03646g |
| This journal is © The Royal Society of Chemistry 2022 |