
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEco-friendly processes for the synthesis of amorphous calcium carbonate nanoparticles in ethanol and their stabilisation in aqueous media†
Lauriane
Chuzeville
ab,
Frank
Boury
 c,
David
Duday
a,
Resmi
Anand
a,
Enzo
Moretto
ab and
Jean-Sébastien
Thomann
*a
c,
David
Duday
a,
Resmi
Anand
a,
Enzo
Moretto
ab and
Jean-Sébastien
Thomann
*a
aMaterials Research and Technology, Luxembourg Institute of Science and Technology, 5 Avenue des Hauts Fourneaux, Esch/Alzette L-4362, Luxembourg. E-mail: jean-sebastien.thomann@list.lu
bUniversity of Luxembourg, Department of Physics & Materials Science, 162a Avenue de la faiencerie, 1511 Luxembourg city, Luxembourg
cCRCINA-GLIAD Team 17, INSERM, UMRS 1232, Université de Nantes, Université d'Angers, 49933 Angers, France
First published on 13th January 2022
Abstract
Amorphous calcium carbonate nanoparticles (ACC NPs) are promising multifunctional materials for healthcare applications. Due to their instability in aqueous media, pure ACC NPs for the biomedical field are increasingly synthesised in absolute ethanol, using the ammonia diffusion method (ADM). Although this method presents the advantage of providing stable ACC NPs without additives, it requires the use of pure ethanol as solvent. New insights into the formation mechanisms of ACC NPs in ethanol using gas diffusion are presented in this article. The optimisation of the process according to these findings can increase the mass concentration of ACC NPs by a factor of 3.5. As a result, the amount of ethanol required to produce a target mass of particles is significantly decreased, reducing the ecological impact of the process. The stabilisation of the resulting ACC NPs in aqueous media is achieved by a short-time process using phospholipids based on the ethanol injection method. By using the natural electrostatic affinity of negatively charged materials for the positive surface of ACC NPs in ethanol, we reduced the process time from 24 h to 2 minutes, compared with the closest state of the art, decreasing the operating time and corresponding energy consumption. The process does not require the use of synthetic PEGylated lipids for steric stabilisation. In addition, a natural egg-sourced phospholipid was identified as an efficient stabiliser for the first time. The upscaling of our process was successfully demonstrated using a 50 L reactor for bulk synthesis, as well as a continuous flow reactor for industrial continuous flow production.
Introduction
In recent years, there has been increasing interest in calcium carbonate nanoparticles (CaCO3 NPs) from various industrial fields.1–5 Besides their excellent biocompatibility and biodegradability, they present numerous advantages for cosmetic formulations and biomedical applications.6–12 For instance, they are intensively investigated for drug delivery, diagnostics and tumour therapy due to their pH responsive and buffering properties.6–12 In both fields, nanosize can be of extreme importance, for long-lasting effect and increased stability in cosmetic formulations, or for targeting tumoral and inflamed tissues in biomedical applications for instance.13–17CaCO3 exists in three major crystalline polymorphs (vaterite, aragonite and calcite), and in an amorphous phase.18,19 The later presents great advantages for the healthcare and biomedical fields, including a higher porosity and the ease of obtaining highly monodisperse NPs via facile processes.20–22 The high porosity of the amorphous form (100–200 m2 g−1) compared to the other polymorphs (<30 m2 g−1) leads to higher capacity of active molecule loading, as well as a higher sensitivity to water. The latter is a key advantage for enhancing burst dissolution at target sites.20,22–25 Amorphous CaCO3 nanoparticles (ACC NPs) are also employed for their highly homogeneous spherical shape, including for the synthesis of core–shell multifunctional nanomaterials.26,27
Due to its transient nature, ACC is the most unstable CaCO3 form when in contact with water, spontaneously dissolving and crystalising into other CaCO3 polymorphs, vaterite, calcite and/or aragonite.28,29 Despite being an advantage for enhancing burst dissolution, the instability in aqueous media remains a major hindrance to the use of ACC NPs in the fields where the phase and size conservation is critical in aqueous media.30,31 This is particularly the case for applications requiring intravenous injection for instance.6,20,26
The synthesis and stabilisation of CaCO3 colloids at nanoscale is widely described in the literature using one-pot strategies in aqueous media. This includes the synthesis of CaCO3 with stabilising molecules such as synthetic polymers, lipids or a combination of therapeutic molecules and synthetic polymers.5,9,32–40 The phase of the particles is generally not amorphous, with a mean diameter often higher than 300 nm.5,9,32,33,35,36 Vidallon et al. recently reported the synthesis of a hybrid polydopamine/bovine serum albumin/CaCO3 NPs stable in aqueous media for contrast-enhanced ultrasound imaging.5 The NPs obtained are a polycrystalline mixture of vaterite and calcite, with a mean diameter above 500 nm.5 Furthermore, these methods provide hybrid nanoparticles, with a CaCO3 content lower than pure ACC NPs materials.5,9,32–36 Another aqueous strategy consists of the synthesis of CaCO3 nanoparticles inside liposomes, to control the final size of the particles.41–44 These methods use toxic organic solvents to synthesise the liposomes.41–43 The size homogeneity and phase control of the CaCO3 particles obtained is uncontrolled with these methods.41–44
Pure ACC NPs with a size under 150 nm can be easily obtained without synthetic additives via a low cost and facile vacuum-assisted process in absolute ethanol developed over the last ten years.6,11,20,21,23,26,45–50 This process is the ammonia diffusion method (ADM), usually known for the synthesis of CaCO3 NPs in water or in a mix water/ethanol.32,33,51 In this variation, absolute ethanol is used as unique solvent to limit the amount of water and prevent the crystallisation of ACC NPs into vaterite or calcite without the need for synthetic additives.6,20,21 The final nanoparticles are dispersed in ethanol and are stable, with a highly homogeneous size under 150 nm, and a spherical shape.6,20 Although this method is becoming increasingly popular in the biomedical field, there are only few studies that briefly discuss the process parameters and the ACC NPs formation mechanism.21,49 Therefore, several variations of the methods are found in the literature, highlighting the need for a deeper understanding of ethanol ADM mechanisms for the synthesis of ACC NPs. Indeed, this process uses ethanol as solvent and should be carefully optimised to minimise the quantity of solvent required to synthesise a certain amount of particles.
For applications requiring the dispersion of ACC NPs in aqueous media, the use of an albumin solution or of lipidic additives have been reported to achieve the stabilisation of the particles in presence of water.6,11,20,23,26,47 However, stabilisation without the use of synthetic additives and/or harmful solvents remains a challenge.7,11,20,37,38,40,52 Hybrid lipid-ACC NPs (LCC NPs) are of particular interest as they combine the advantages of ACC and lipid nanomaterials.11,37,38 The major drawback of lipid-based stabilisation is the need for hazardous organic solvents like cyclohexane or chloroform to dissolve the lipids,37,38 and/or the use of PEGylated lipids.20,39 The work of Wang et al. for example uses a mix of ethanol and chloroform to dissolve the lipid mix used as stabilisers.11 Recently, Wang et al. successfully developed a method without toxic organic solvent to stabilise ACC NPs in water using phospholipids.20 However, their method requires a 24 h continuous mixing of the ACC NPs/lipid mixture at 37 °C prior to the injection in water, representing a long operating time and energy consumption.20 Furthermore, the lipid mix reported includes PEGylated phospholipids as key stabiliser.20 One of the recent challenges faced by the pharmaceutical and cosmetic industries is the development of more sustainable products and technologies.53–56 Therefore, there is a need for the development of PEG-free materials and as PEGs are synthetic and nonbiodegradable materials.53,54,56 This need is also motivated by the emergence of anti-PEG immune responses due to the extensive use of PEGylated additives in the food, cosmetic and pharmaceutical industries.57–62 In the biomedical field, the apparition of anti-PEG IgG and IgM is responsible for efficacy loss and hypersensitivity reactions.59,63
In this work, we report on valuable new insights into the formation mechanisms of ACC NPs by ADM in absolute ethanol by investigating several influential parameters (diffusion of water and ammonia in the ethanol solution of CaCl2, size of the gas–ethanol interphase, presence or absence of a diffusion barrier) and in situ monitoring of the pH in ethanol (pHe) and conductivity during the reaction. These new insights lead to a better understanding of the influence of several reaction parameters, allowing the optimisation of the process efficiency to significantly reduce the amount of ethanol required to synthesise a certain mass of ACC NPs.
Subsequently, this work reports new advances for the stabilisation of the resulting ACC NPs in aqueous media using phospholipids. Similarly to the work of Wand et al., the developed process is based on the ethanol injection method and is not using toxic organic solvents.20 We studied the influence of the phospholipid polar head group on the stabilisation efficiency, and rationally selected the lipids according to their natural electrostatic affinity for calcium ions and ACC NPs. As a result, we were able to significantly reduce the process duration from 24 h to 2 min, decreasing the operating time and energy consumption of the process. The selected lipids are not PEGylated, increasing the sustainability of the process. Lastly, a bio-sourced phospholipid has been identified as suitable stabiliser. CryoTEM imaging is used to investigate the particular structuration behaviour of these lipids around the ACC NPs. This work also presents two different strategies to adapt the process to industrial needs, using a 50 L large scale reactor and a continuous flow reactor.
Experimental
Materials
Calcium chloride hexahydrate (CaCl2·6H2O), ammonium bicarbonate (NH4HCO3), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), 1,2-distearoyl-sn-glycero-3-phospho-L-serine (DSPS), 1,2-dihexadecanoyl-sn-glycero-3-phospho-L-serine (DPPS), 1,2-dimyristoyl-sn-glycero-3-phospho-L-serine (DMPS), 1,2-didodecanoyl-sn-glycero-3-phospho-L-serine (DLPS), and 1,2-dipalmitoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DPPG) were purchased from Sigma Aldrich (Overijse, and Diegem, Belgium). N-(Carbonyl-methoxypolyethylene glycol-2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-PEG 2000) was purchased from Avanti (Alabaster, Alabama, Unites States). Egg phosphatidyl glycerol (1,2-diacyl-sn-glycero-3-phospho-rac-glycerol, sodium salt) (E PG) was purchased from Lipoid (Ludwigshafen, Germany). Absolute Ethanol was purchased from VWR International (Fontenay-sous-Bois, France). The 3 Å molecular sieve was purchased from Sigma Aldrich (Gillingham, United Kingdoms).ACC NPs preparation
CaCl2·6H2O was dissolved in absolute ethanol in a glass container, at a mass concentration of 4.4 mg mL−1. Optionally, the container can be covered by parafilm punctured with 12 holes of 1 mm diameter. The container is then placed in a desiccator at room temperature, along with excess NH4HCO3 distributed in 4 vials of (∼10 g per vial). The system is placed under vacuum for 2–96 h. The vacuum is set at 200 mbars, then the system is sealed, and the pumping is stopped. At the end of the reaction, the particles were centrifuged at 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min. The supernatant is discarded, the particles are resuspended in the proper amount of ethanol and dispersed with an ultrasonic bath for few minutes. The resulting solution of ACC NPs in ethanol is stored at 4 °C. ACC NPs growth was monitored with and without 3 Å molecular sieve in the desiccator, to evaluate the influence of ammonia and water on ACC NPs growth.
000g for 10 min. The supernatant is discarded, the particles are resuspended in the proper amount of ethanol and dispersed with an ultrasonic bath for few minutes. The resulting solution of ACC NPs in ethanol is stored at 4 °C. ACC NPs growth was monitored with and without 3 Å molecular sieve in the desiccator, to evaluate the influence of ammonia and water on ACC NPs growth.
Stabilisation of ACC NPs in water
The stabilisers (i.e. DPPC, DOPS, DSPS, DPPS, DMPS, DLPS DOTAP, E-PG, DPPG, DSPE-PEG2000) were prepared in ethanol at a mass concentration of 0.25 mg mL−1 using an ultrasonic bath.Then, ACC NPs suspended in ethanol are mixed with the stabiliser at a mass ratio ACC:stabiliser of 1. After a one-minute of homogenisation in an ultrasonic bath, the obtained solution was mixed with deionised water by a one-pot injection at a ratio of ethanol:deionised water 1:9. The injection was followed by a 15-second stirring by vortex (Fig. 1).
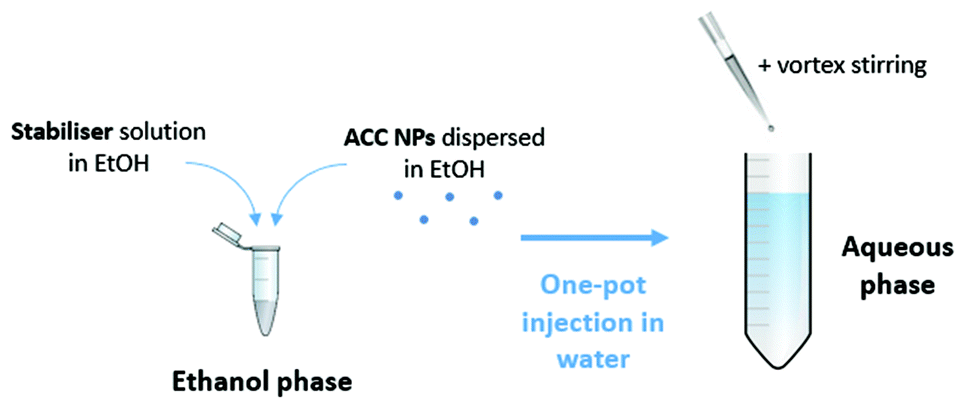 | ||
| Fig. 1 Process for the stabilisation of ACC NPs in aqueous media. | ||
Surface charge modulation
Lipid-coated ACC (LCC) using DOPS as stabilisers dispersed in deionised water were mixed with a stock solution of DOTAP in ethanol, molar ratio DOTAP/DOPS between 1–10. The stock solution of DOTAP was mixed with deionised water by a one-pot injection at a ratio of ethanol:deionised water 1:9. The injection was followed by a 15-second stirring by vortex.
Characterisation
The particle size distribution and zeta potential were measured in ethanol64 or in deionized water at pH 7, with a Zetasizer (Nano-ZS90, Malvern Instruments Ltd, UK) based on dynamic light scattering (DLS) or micro-electrophoretic light scattering technique respectively. The nanoparticles suspensions were neither diluted nor filtered prior to the analysis. A minimum of three measurements are performed per sample. The refractive indexes used are 1.681 for pure ACC and LCC NPs, with an absorbance of 0.The mass concentration of ACC NPs after synthesising was determined by centrifuging 20 mL of solution at 30000g for 10 min, discarding the supernatant, and drying the pellet under vacuum for 15 minutes. Afterward, the tube was weighed (mass tube). The mass of the empty tube is referred to as “mass tare”.
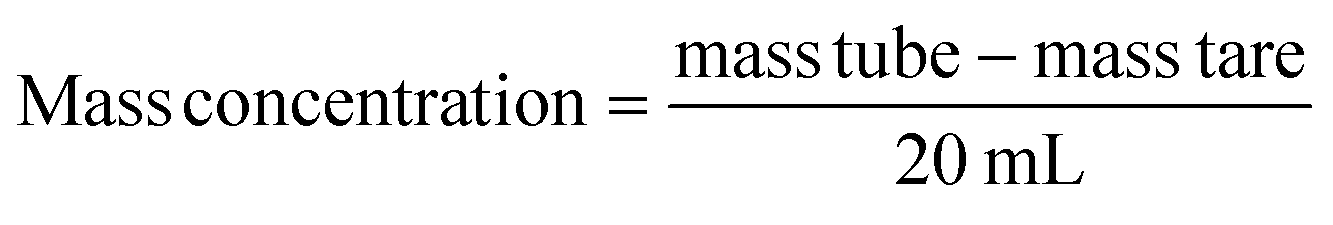
The size and surface morphology of the NPs were characterized by Scanning Electron Microscopy (SEM). A droplet of solution was deposited on to copper tape and air-dried. The ACC NPs non coated by lipids were imaged immediately after drying to avoid degradation due to air moisture contamination. SEM images were performed with a Helios Nanolab, 650 Scanning Electron Microscope (FEI Company, Hillsboro, Oregon, United States), operating at a voltage of 2–10 kV and current of 25–100 pA.
The concentration of the nanoparticle suspensions was determined by Nanoparticles Tracking Analysis (NTA), using the Nanosight® NS500, with the temperature control set at 25 °C. The samples were diluted at a ratio 1:1000 in deionised water at pH 7 prior to analysis. A minimum of five measurements were performed by sample.
The XRD measurements were performed on an XRD X'Pert Pro in reflection mode with a Cu K-alpha tube (45 kV, 40 mA). The primary optics contain a programmable divergence slit whereas the secondary optics consist of a PIXcel detector run in 1D mode with a programmable antiscatter slit. Before measuring the sample, the solutions containing the nanoparticles were deposited on to a zero-background holder and covered with a thin Kapton film in order to prevent the solvent from evaporating. During the measurement, the zero-background holder was spun.
Cryogenic transmission electron microscopy (CryoTEM) was performed as follows. Four microliters of sample were deposited on a holey carbon film (Quantifoil type R1.2/1.3), rendered hydrophilic by either a low-power plasma cleaning treatment (Model 1070 NanoClean, Fishione) for the LCC NPs sample or by a negative glow discharge in air (PELCO easiGlow™) for the ACC NPs sample. For the ACC NPs samples, blotting was performed for 2 s with a blot force of 8, and for LCC NPs, 0.5 seconds with a blot force of 5. Then, vitrification in liquid ethane was performed using an automated plunger (Vitrobot, FEI Compagny). Blotting was carried out at 10 °C and 90–95% humidity. Images were recorded using the SerialEM software on a FEI Tecnai F30 microscope (FEI Company, Hillsboro, Oregon, United States) operating at a voltage of 100 kV and equipped with a Falcon camera (FEI company).
The helium ion microscope (HIM) images were recorded with an Orion Nanofab Instrument (Zeiss, Peabody, US). Helium ion microscopy was based on the detection, with a conventional Everhart–Thornley (ETD) detector, of the secondary electrons emitted from the sample surface scanned with a focused energetic He+ or Ne+ ion beam.65 With a helium beam resolution of 0.3 nm and a Neon beam resolution of 1.5 nm, this microscope offers Secondary Electron (SE) images with a high contrast for imaging insulator. Compared to the latest generation of Scanning Electron Microscope (SEM), the HIM allows specimens to be probed with a better surface sensitivity, a higher depth of field and the ability to analyse uncoated insulator samples.65 In this study, the images were acquired with a 25 keV He+ beam of 0.2 pA for a matrix of 1024 × 1024 pixels and a counting time of 40 μs per pixel.
ACC particle size monitoring
Five desiccators containing all the reagents (see ACC NPs preparation) were connected to the vacuum manifold of a Schlenk line (ESI Fig. S1†). The system was placed under vacuum. The 5 desiccators were exposed to the same vacuum conditions. At several time intervals, a desiccator was isolated from the Schlenk line by a valve and opened to measure the nanoparticle size by DLS (see Characterisation).The particle size after 2 h of reaction time was characterised by SEM, and also by Helium Ion Microscopy (HIM) (see Characterisation) to corroborate the SEM measurements.
pHe and conductivity monitoring during ACC NPs synthesis
A pH probe and a conductivity probe were inserted into a modified plastic desiccator (ESI Fig. S2†), and dipped into the CaCl2·6H2O solution in ethanol. The reaction was conducted by following the ACC NPs preparation protocol.The variation of electrical conductivity (EC) was followed by a USP compliant EC, TDS, NaCl, Resistivity, Temperature Meter from Hanna Instruments (Temse, Belgium), HI98192, with a four-ring conductivity resistivity electrode from Hanna Instruments, HI763123.
The pHe variation was followed by a pH/temperature Bench Meter from Hanna Instruments (Temse, Belgium), HI2002, with a digital, glass body, double junction, conical tip, pH/temperature electrode from Hanna Instruments, HI10530.
Process upscaling for the stabilisation of ACC NPs in water and the surface charge modulation for volumes >50 mL
Stock solutions of stabilisers and charge modulation agent in absolute ethanol were prepared at a mass concentration of 0.25 mg mL−1, using an ultrasonic bath.The solution of ACC NPs in ethanol was mixed with the stock solution of stabiliser using an ultrasonic bath. The mass ratio of ACC:stabiliser is ranging between 1 and 2. After one minute of mixing, the ethanol solution was added to a large volume of deionised water under magnetic stirring (>500 rpm), at a ratio of ethanol:deionised water 1:9. For an Erlenmeyer of 2 L with a bottom diameter of 15 cm, a magnetic bar of 5 cm was used.
The magnetic stirring (>500 rpm) replaced the one-pot injection using a pipette (see Stabilisation of ACC NPs in water), allowing larger volumes to be handled.
Process upscaling for the stabilisation of ACC NPs in water adapted to industrial volumes
50 L reactor: the upscaling was performed using the reactor from Orb Pilot 50 L (Syrris, Royston, United Kingdom), equipped with a mechanical stirrer, on a 40 L final volume. 36 L of water were put under stirring with a stirring speed of 300 rpm. Then, 4 L of an ethanolic solution of EPG and suspended ACC NPs (mass ratio ACC NPs:EPG of 1) were poured in 13 seconds inside the water phase.
Continuous flow reactor: for the upscaling of the stable ACC NPs in water, we performed a facile continuous flow mixing method using Coflore® Agitated Cell Reactor (ACR) (AM Technology, Cheshire, England). The Coflore ACR is a mechanically agitated plug flow reactor, in which efficient mixing is performed using multiple interlinked cell reactors. The cell reactor was connected to a syringe pump and a peristaltic pump for the continuous supply of reactants. The stabiliser DOPS was prepared in ethanol at a mass concentration of 0.25 mg mL−1 and ACC NPs suspension was mixed with the stabiliser at the mass ratio of 1, followed by homogenization in an ultrasonic bath. Then, the ACC NPs:stabilizer colloidal solution was transferred into a 50 mL syringe connected to a syringe pump. A controlled flow of deionised water to the cell reactor was established using a peristaltic pump. Both reactants were introduced to the Coflore mixing cell at the following conditions: flow rate of ACC NPs:stabilizer 1 mL min−1, flow rate of water 9 mL min−1, agitation of 9 Hz at room temperature.
Results and discussion
ACC NPs formation by ethanol ADM
ACC NPs were prepared by a vacuum-assisted ammonia diffusion method (ADM) in absolute ethanol inspired by the work of S–F. Chen et al.21 and A. Som et al.6 (Fig. 2). CaCl2·6H2O serves as Ca2+ precursor, ammonium bicarbonate as CO32− precursor, and absolute ethanol as solvent. The synthesis in ethanol limits the presence of water, preventing the spontaneous aggregation and crystallisation of CaCO3 NPs without the need for synthetic additives.6,11,20,21,23,26,45–50 This method leads to the obtention of pure ACC nanoparticles with a size under 150 nm.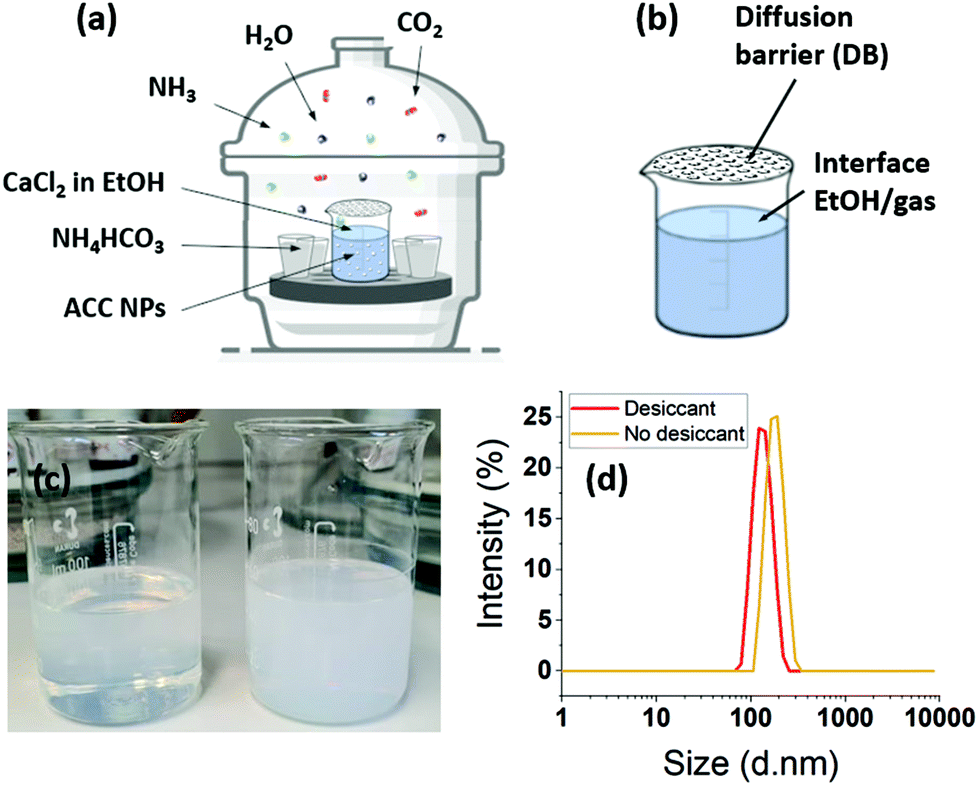 | ||
| Fig. 2 (a) Schematic representation of the reaction setup. (b) Schematic representation of the diffusion barrier (DB) and the gas–ethanol interface. (c) Final ACC NPs solution obtained with desiccant (left) and without desiccant (right). (d) Size distribution measured by DLS of final ACC NPs obtained with and without desiccant. | ||
This method is increasingly employed in the biomedical field to produce highly regular pure ACC NPs in terms of size and shape.6,11,20,23,26,46–50 However, the investigation of ACC NPs formation mechanisms in absolute ethanol using ADM is at its beginning.21,49 Therefore, several variations in the process parameters are present in the literature.6,11,20,21,23,26,45–50 The following work intends to bring new data and understanding on the ethanol ADM formation of ACC NPs, as a base for process efficiency optimisation. As ethanol is used as only solvent, improving the process efficiency reduces the amount of ethanol needed to synthesise a specific mass of particles, decreasing the ecological impact of the process.
In aqueous ADM, the CaCO3 formation mechanisms are well known and are described by eqn (1)–(6):51
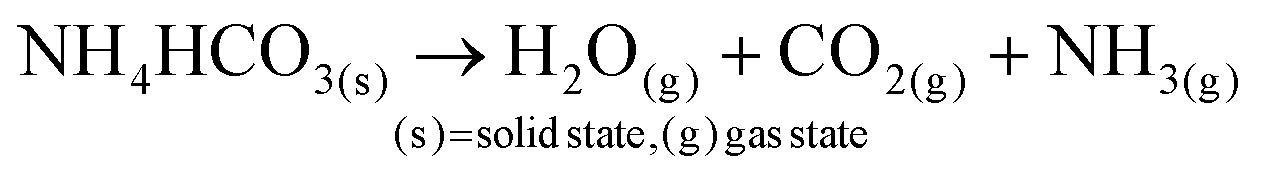 | (1) |
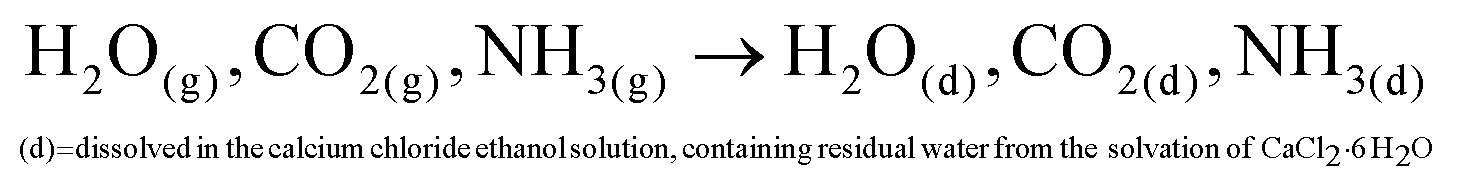 | (2) |
| CO2(d) + H2O(d) ↔ H2CO3(d) | (3) |
| H2CO3(d) ↔ HCO−3(d) + H+ | (4) |
| HCO−3(d) ↔ CO2−3(d) + H+ | (5) |
| CO2−3(d) + Ca2+(d) → CaCO3(s)↓ | (6) |
Ammonium carbonate or bicarbonate are used as the CO2 source, and placed in a sealed container with an aqueous solution of calcium chloride. Under vacuum, the decomposition of ammonium carbonate or bicarbonate is spontaneous66 and leads to a progressive saturation of the gas phase with CO2, NH3 and H2O (eqn (1)).51 These gases diffuse into the calcium chloride solution through the gas–liquid interface (eqn (2)). Once in solution, water molecules react with the dissolved CO2 to form H2CO3 (eqn (3)), in acid–base equilibria with HCO3− and CO32− (eqn (4) and (5)). The diffusion of ammonia in the solution increases the pH, generating a basic environment where the formation rate of CO32− is increased.51 The final reaction between Ca2+ and CO32− produces CaCO3 (eqn (6)).51,67 In water, without additives, the amorphous form of CaCO3 at nano-size is not stable.22,28,29
In absolute ethanol ADM, the generation of H2O from the decomposition of ammonium carbonate or bicarbonate, is an essential step for the formation of the precursor ions NH4+ and CO32−.46,48,67,68 In our process, water molecules also come from the solvation of CaCl2·6H2O in ethanol. We verified the influence of the water and ammonia vapours coming from the decomposition of ammonium bicarbonate by placing a 3 Å molecular sieve in the desiccator. A 3 Å molecular sieve has the ability of trapping of water and ammonia without trapping the CO2 (Fig. 2). The particle size distribution monitored by DLS shows a size of 132 nm with a PDI of 0.008 with the sieve. The size increases to 177 nm without molecular sieve, with a PDI of 0.025. The size dispersion remains narrow with a particle size under 200 nm. These results seems to indicate that the water coming from the decomposition of ammonium bicarbonate do not lead to uncontrolled crystallisation or aggregation. The final mass concentration is 0.6 mg mL−1 with sieve, compared to a final mass without thieve of 2.1 mg mL−1. As a consequence, the solution turbidity (Fig. 2c) appears more important without molecular sieve. These results evidence that the initial water content coming from the solvation of CaCl2·6H2O in absolute ethanol is sufficient to obtain monodisperse ACC NPs. However, the results without sieve show that the diffusion of ammonia and water in ethanol catalyses the formation of ACC NPs, leading to an increase in the final mass concentration by a factor of 3.5. In a similar way to aqueous ADM, the role of water is to generate the aqueous forms of CO2, i.e. H2CO3, HCO3−, CO32− (eqn (3)). The role of ammonia is to alkalinise the ethanol solution, promoting the precipitation of CaCO3 NPs by displacing the equilibrium in favour of CO32− (eqn (3)–(5)).46,48,67,68 The final mass concentration increase by a factor of 3.5 without molecular sieve demonstrates the importance of the diffusion of water and ammonia in the process efficiency. In several studies, drierite is employed to trap the water coming from the decomposition of ammonium bicarbonate.6,48 Our results strongly suggest a better process efficiency without trapping these water molecules. Indeed, the increase in final mass concentration observed without drierite could divide by 3.5 the volume of ethanol required to produce the same mass of particles.
We investigated the growth mechanisms of ACC NPs in ethanol ADM. The size of ACC NPs was monitored by DLS at different reaction times (Fig. 3, ESI Fig. S8†). The particle size after 2 h reaction is below the limit of DLS sensitivity and was successfully determined by SEM imaging. Clusters with a mean diameter of 10 nm were observed. The results were confirmed by helium ion microscopy imaging to exclude a possible contamination or artefact from the SEM measurement. Thanks to this observation, we successfully determined that the growth of ACC NPs followed a classical power law between 2 h and 49 h reaction time in ethanol ADM (Fig. 3c). The nature of the power law variables (temperature, pressure) were not determined in this work, and would require further modelling. The ACC NPs PDI after 49 h reaction time was below 0.05. Such a narrow size distribution suggests a growth mechanism with the nucleation and growth steps occurring separately. A simultaneous nucleation and growth mechanism would lead to polydisperse size dispersions.69 The Lamer model was considered, as it is the most common model of the classical nucleation theory describing a burst nucleation followed by a diffusion driven growth.69 However, the CryoTEM characterisation of the final ACC NPs revealed an irregular shape of NPs (Fig. 3e). The NPs appeared to be constituted from an aggregation of nuclei, forming an irregular granular structure (Fig. 3e). The SEM and helium beam characterisation at 2 h confirms the granular structure, revealing small clusters with a similar particle constitution of several nuclei (Fig. 3a and b). These observations exclude both diffusion-driven growth and the Lamer model, which would lead to a more homogeneous structure and shape.69 Growth by nucleus aggregation was also excluded by the PDI values <0.05 at 49 h reaction time. These observations led us to consider the hypothesis of a homogeneous nucleation followed by an autocatalytic heterogeneous nucleation & growth on the surface irregularities of the NPs. This hypothesis is consistent with the PDI < 0.05 after 49 h reaction time, and with the irregular granular structure of the nanoparticle revealed by CryoTEM.
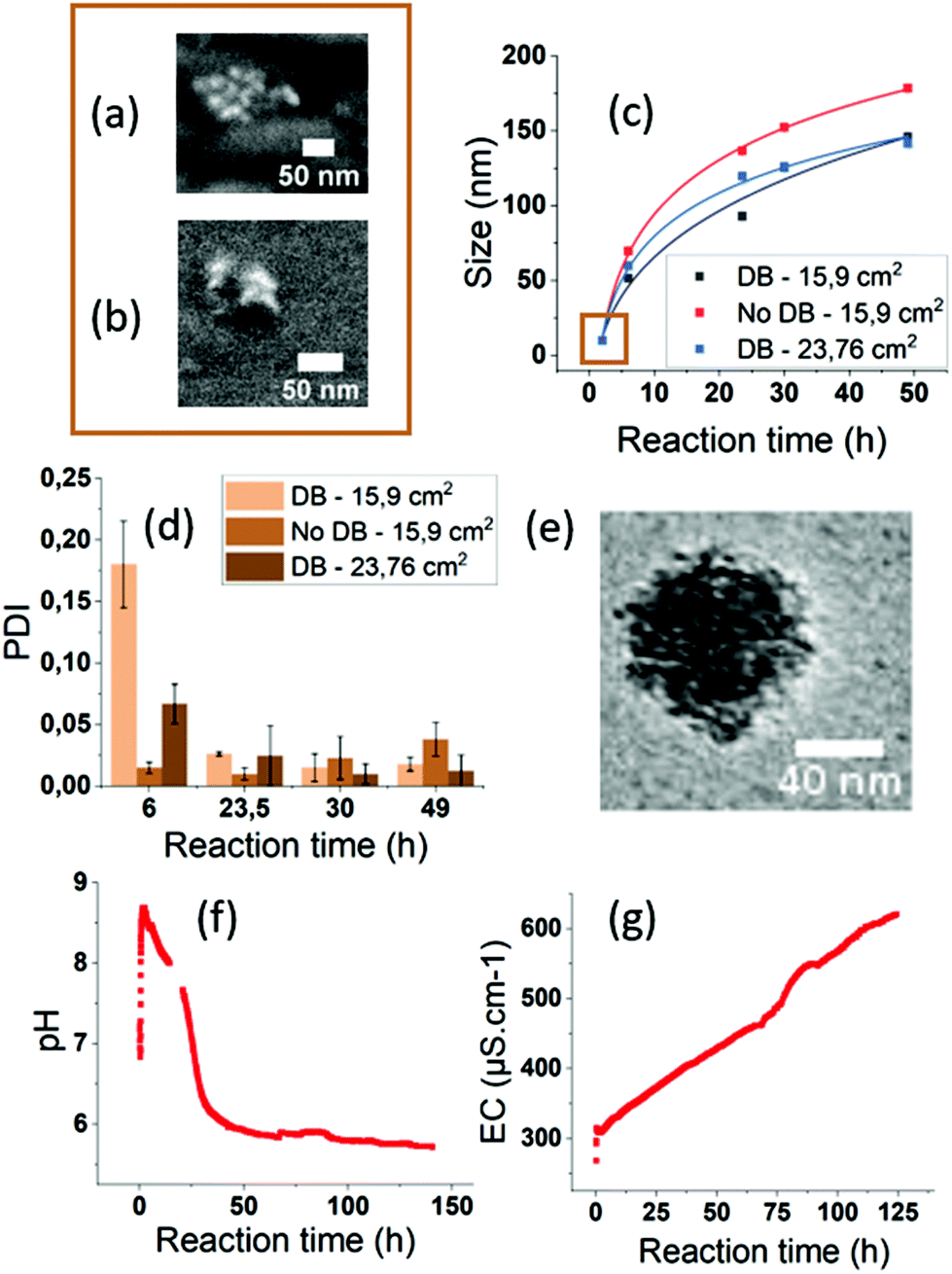 | ||
| Fig. 3 ACC NPs growth in function of the reaction time. (a) SEM imaging after 2 h reaction. (b) Helium ion beam imaging after 2 h. (c) ACC NPs growth curves, with or without DB, for gas–ethanol interfaces of 15.9 cm2 and 23.8 cm2. The fitting curves are following a power law y = a + b × xc. The r2 are of 0.981 (black), 0.997 (blue) and 0.992 (red). (d) PDI values. (e) CryoTEM imaging of an ACC NP after synthesis. (f) pHe monitoring for 140 h. (g) Electric conductivity (EC) monitoring for 140 h. | ||
The influence of the gas–ethanol interface area (Fig. 3, ESI Fig. S7†) was also investigated. An increase in the gas–ethanol interface area generates a higher gas diffusion rate in the CaCl2 solution according to the Fick Law.70 In our experiment, for a gas–ethanol interface of 15.9 cm2, the final mass concentration reached 1.1 mg mL−1, compared to 2.1 mg mL−1 with an interface of 23.8 cm2. It represents a final mass concentration increase by a factor of 1.9. No PDI alteration was observed. The results of the present study show an increase in size and mass concentration when the gas–ethanol interface area increases. Hence, process efficiency in terms of end product quantity can be significantly improved by increasing the gas–ethanol interface area.
For the synthesis of ACC NPs by ADM in water or ethanol, a diffusion barrier (DB) constituted of parafilm punctured with small holes is generally placed on the CaCl2 solution container (Fig. 2b). These DB are known to act as reaction regulators.51 We investigated the influence of and need for such a DB barrier in a process in absolute ethanol. Indeed, the literature suggests that performing the reaction in absolute ethanol is sufficient to limit the amount of water and prevent the spontaneous aggregation and crystallisation of CaCO3 NPs.49,71,72 We compared the size, dispersion and mass concentration between an uncovered container, and a container covered by a parafilm tape punctured with small holes. A difference in size and mass was observed between the two samples (Fig. 3, ESI Fig. S6†). The final mass concentration without a DB reached 1.2 mg mL−1, compared to 0.5 mg mL−1 with a DB, which represents a final mass increase by a factor of 2.4. The PDI of both samples were under 0.1 after 49 h reaction time (Fig. 3). Without a DB, the diffusion rate of CO2, NH3 and H2O in the CaCl2 solution increases. Hence, this may generate a faster concentration increase of the precursor ion CO32− in solution, and therefore lead to higher particle concentration without alteration of the particle size dispersion. These results strongly suggest that a regulation with DBs is not required in ethanol ADM to generate monodisperse ACC NPs. Furthermore, they demonstrate that the removal of the DB lead to a significantly higher efficiency in terms of final mass concentration of ACC NPs.
The conductivity and pHe were monitored during the synthesis for 96 h inside the chamber to get a better understanding of the formation mechanisms of ACC NPs in ethanol ADM. The trends observed were confirmed by a second monitoring up to 140 h of synthesis (Fig. 3f, ESI Fig. S9†). The size measured by DLS after 96 h reaction was 150.8 nm with a PDI of 0.066. These values were similar to the sizes obtained after 49 h reaction (Fig. 3c). Hence, the size of ACC NPs seems to reach a maximum close to 150 nm. After 140 h reaction, white aggregates were visually observed in the solution, with a polydisperse size distribution characterised by DLS (ESI Fig. S10†).
In ADM, pH is a critical parameter known to govern the balance between the different aqueous carbonate species.51 The pHe monitoring of our reaction showed an initial burst up to a pHe of 8.75 after 5 minutes, followed by a progressive decrease and a stationary phase at pH 5.7 (Fig. 3f). The pHe burst is probably a consequence of ammonia and water diffusion in the ethanol solution, following the decomposition of ammonium bicarbonate. Indeed, in aqueous or ethanol ADM, ammonia is known to cause a pH increase favouring the existence of CO32−.46,48,67,68 However, the pHe evolution we observed in ethanol was surprisingly different from pH studies conducted in aqueous ADM.51 With water as the solvent, the pH increases during 5 to 30 minutes before reaching a maximum value between 9.8 and 10.5.51 Afterwards, in water, the pH mildly decreased to reach a stationary phase at pH 9.8.51 The differences in the pH and pHe variation rate and amplitude observed between aqueous and ethanol ADM could originate from the differences in ionic solubility and the solvation limit between the two solvents. The presence of CO2 in ethanol or in water is known to decrease the pHe or the pH.51,67,73 CO2 has a higher solubility in ethanol compared to water.67 On the other hand, ammonia is less soluble in ethanol compared to water.74,75 These two differences could explain the amplitude of the pHe decrease in ethanol compared to water ADM. As the pHe decreases, the equilibrium between the different forms of hydrated CO2 will progressively be displaced in disfavour of the formation of CO32−. As a result, the formation of ACC NPs will progressively slow down (Fig. 3c). A stationary phase is reached at pHe 5.7 in our solution. At such pHe, CO32− becomes a minority. This stationary phase is reached close to 48 h, when the growth of ACC NPs becomes negligible, with the maximum size of 150 nm observed by DLS at 50 h and 96 h reaction time.
The conductivity was also monitored to follow the ionic and water content of the CaCl2 ethanol solution. In absolute ethanol, the conductivity is negligible (0.06 μS cm−1). The initial conductivity measured in the CaCl2·6H2O ethanol solution is significantly higher, with values reaching 290 μS cm−1. This increase evidences the presence of Ca2+, Cl− and water coming from the solvation of CaCl2·6H2O. The follow up of conductivity shows a burst when the vacuum is set, followed by a steady and linear increase along the reaction. This increase is linear up to the end of the 140 h monitoring. The initial burst when the vacuum is set might result from the significant bubbling occurring in the solution during the vacuum pumping, as the conductivity electrode is very sensitive to air bubbles. However, it might also be a consequence of the gas diffusion in the ethanol solution following the decomposition of ammonium bicarbonate. The steady increase testifies to a regular generation of ionic species in the solution. Despite Ca2+ and CO32− being involved in the formation of ACC NPs, NH4+ and Cl− also exist in the solution and contribute to the final ionic content. In contrast with pHe and size evolution, no maximum or stationary phase was observed. Besides the ionic content evolution, the linear increase of conductivity strongly suggests a continuous diffusion of water inside the ethanol solution. An increase of water content would certainly explain the aggregation of ACC NPs occurring between 96 h and 140 h reaction, through dissolution/crystallisation catalysed by the slightly acidic environment.28,29
The XRD spectrum of the final ACC NPs shows two wide peaks centred on 2θ values of 25° and 40° respectively, characteristic of amorphous CaCO3 NPs (Fig. 4a).76 The typical porosity and granular structure of ACC are visible by CryoTEM (Fig. 4b). The size dispersion characterized by DLS evidences monodisperse spherical NPs with a polydispersity index (PDI) under 0.05 (Fig. 4c). SEM imaging corroborate monodisperse nanoparticles with spherical and porous structure, characteristic of non-crystalline CaCO3 NPs with an average diameter of 70 nm (Fig. 4d). The size dispersion in DLS is centred on 127 nm, a higher value compared to SEM measurement, due to the hydrodynamic diameter of ACC NPs in ethanol.6
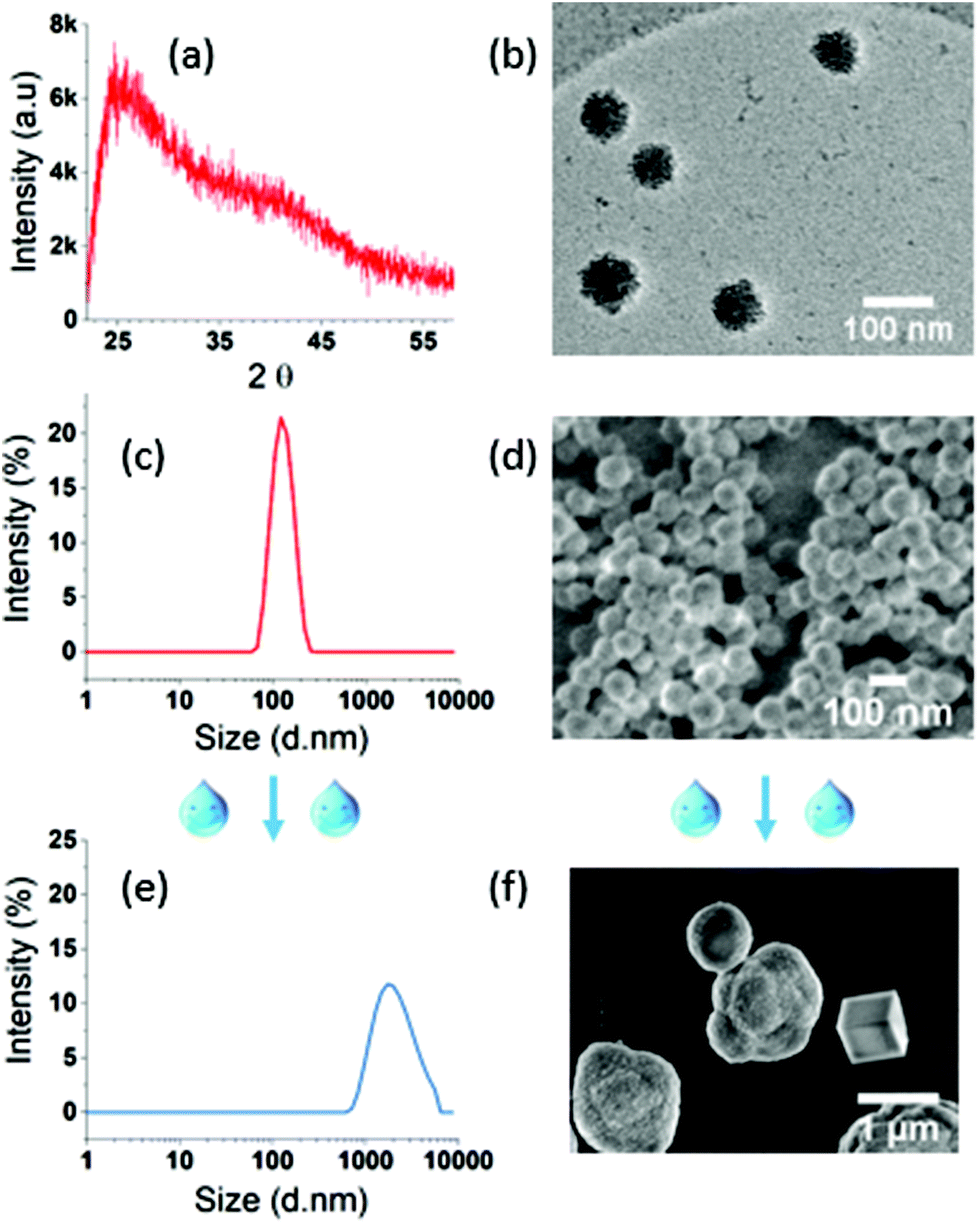 | ||
| Fig. 4 ACC NPs characterisation in ethanol and water. In ethanol: (a) XRD spectra. (b) CryoTEM imaging. (c) Size distribution characterized by DLS. (d) SEM characterisation. (e) Particle size distribution of ACC NPs in presence of water. (f) SEM images of ACC NPs after water exposition. | ||
ACC NPs size and shape in the presence of water have been characterised by DLS and SEM to evaluate the influence of water on the size and crystallinity of the particles (Fig. 4e and f). Both analyses clearly evidenced the instability of ACC NPs in aqueous media. The DLS size distribution is larger with PDI above 0.25 and centred on micrometre scale values >1 μm. SEM characterisation confirms the presence of large microparticles, with characteristic shapes of crystalline polymorphs, typically vaterite aggregates and trigonal rhombohedral calcite.77 These results testify to the instability of ACC NPs in the presence of water, and strongly suggest a crystalline transition to calcite through vaterite intermediates. This behaviour is inherent to the transient nature of non-stabilised ACC NPs in the presence of water.78
The following section presents a facile green process to preserve the amorphous structure and nanosize of ACC NPs in aqueous media. This process is exclusively using a water–ethanol mix and non-PEGylated phospholipids. The process was optimised by rationalising the selection of phospholipids according to their polar head structure.
PEG-free lipid stabilisation of ACC NPs in water
Up to this day, the greenest strategy for the stabilisation in aqueous media of ACC NPs synthesised using ethanol ADM has been reported by Wang et al.20 The process is based on solvent exchange using ethanol and water, a well-known method used for the synthesis of liposomes and more commonly known as the ethanol injection method.20,79,80 A mix of PEGylated lipids and phosphatidylcholine (PC) phospholipids is used for the stabilisation.20 The lipids are put in contact with ACC NPs for 24 h under continuous magnetic stirring before injection in a large volume of water. Lipids self-organise around the ACC NPs when they come into contact with water.20Our work aimed to lower the contact time between the lipids and the ACC NPs to lower the among of energy and operating time required for the process. The objective is also to employ more sustainable stabilisers and provide an effective stabilisation without using PEGylated lipids. To achieve these goals, we rationalised the phospholipid selection according to the polar head structure. Indeed, the polar head group affinity for Ca2+ ions is likely to influence the stabilisation efficiency, as ACC NPs have a positive surface charge in ethanol64 (Fig. 5d). 1,2-Dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), a negatively charged phospholipid was selected for the proof of concept. Phosphatidylserine (PS) phospholipids are known to interact strongly with calcium by non-covalent electrostatic interactions, resulting in the formation of dehydrated complexes.81–83 According to some studies, PS polar head groups are more rigid than PC head groups, and have a stronger ability to interact with each other by electrostatic interactions or hydrogen bonds.84,85 As a result, membranes composed of PS phospholipids are more condensed in the presence of Ca2+ ions compared to other phospholipids membranes.84–86 PS lipids, due to their negative net charge are likely to self-organise more rapidly and efficiently around ACC NPs. They should provide an effective protective layer against water without the need for PEGylated lipids, due to their condensed structural arrangement in the presence of Ca2+ ions. In our process, ACC NPs are put into contact with DOPS in ethanol for 1–2 minutes, followed by a one-pot injection into water with 15 seconds vortex at a ratio ethanol:water 1:9. Compared to the state of the art,20 the contact time between ACC NPs and the lipids is reduced from 24 h to 1–2 min before injection into water. SEM images of the resulting LCC NPs reveal monodisperse particles with an average size of 90 nm. An average size increase of 20 nm compared to absolute ACC NPs is observed (Fig. 4d and 5a), testifying from the structuration of DOPS around ACC NPs.
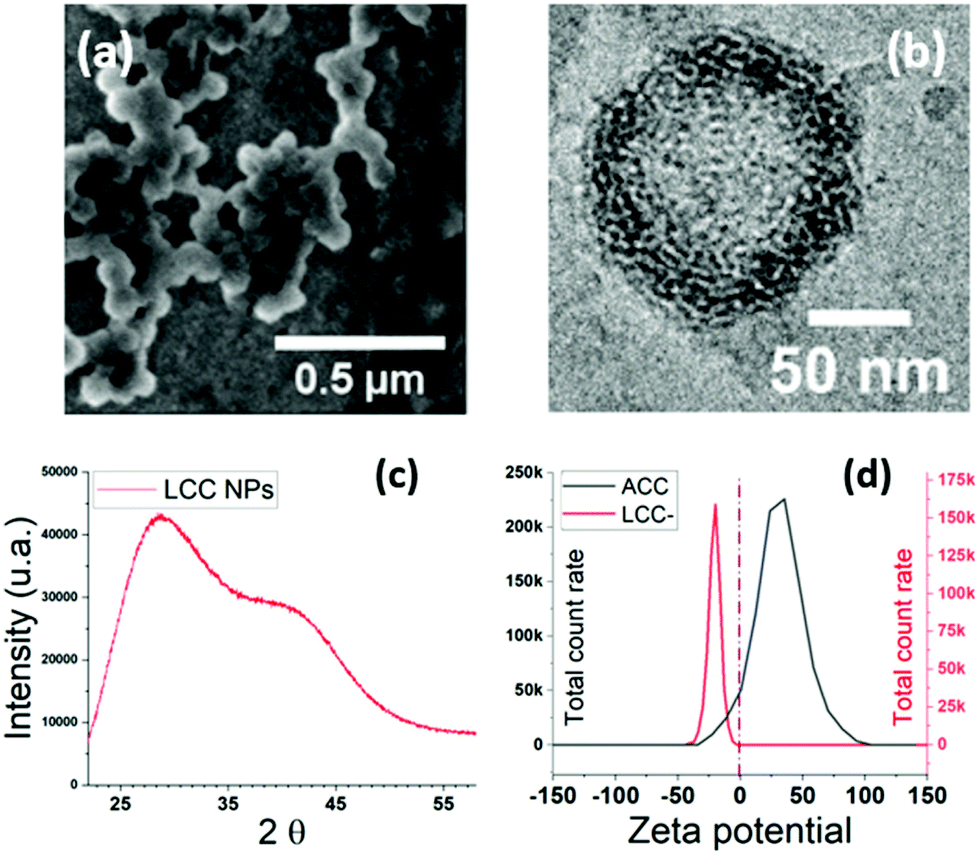 | ||
| Fig. 5 Characterisation of DOPS LCC NPs. (a) SEM imaging of LCC NPs. (b) CryoTEM imaging of LCC NPs in water. (c) XRD spectra of LCC NPs in water. (d) Zeta potential of ACC NPs in ethanol (black) and LCC NPs in water. | ||
A monitoring of zeta potential at pH 7 before and after stabilisation shows a switch from positive to negative values, corroborating the presence of DOPS at the surface of the NPs87 (Fig. 5d). ACC NPs are not stable in aqueous media. Therefore, the zeta potential measurement is performed in ethanol.64 CryoTEM imaging (Fig. 5b) reveals a singular structuration of the lipids around the LCC core, with interconnected lipid multi-assemblies. Studies using “one-pot” injection method for the stabilisation of ACC NPs using phospholipids suggest a lipid structuration in a supported lipid bilayer around the ACC NPs.20,39 Our study suggests that the structuration of PS phospholipids around ACC NPs does not lead to a supported lipid bilayer but to several lipid layers that are interconnected probably through hemi-fusion mechanisms88 (Fig. 5b). Indeed, on the CryoTEM image, the multilayers are not concentric and are not separated by a water layer.89 The fusion or hemi-fusion of PS membrane in the presence of Ca2+ ions is a well-known phenomenon.90,91 It seems that some layers are infiltrated into the first few nanometres of the porous structure of ACC NPs. The XRD spectrum confirmed the conservation of the amorphous structure of the LCC NPs core (Fig. 5c). Two wide peaks centred on 2θ values of 27° and 42° are observed, characteristics from amorphous CaCO3.76
The increase of the DOPS/CaCO3 molar ratio leads to a size increase in the resulting LCC NPs (Fig. 6a and b). The size and PDI of each sample were measured by DLS six times in a 15 min period, to ensure that the particles were stable. The minor standard deviations testify to the stability of the LCC NPs after injection in water. The PDI values around 0.1 evidence monodisperse size dispersions, even at low molar ratios DOPS/CaCO3 (Fig. 6b). With DOPS as stabiliser, LCC NPs remain stable in water with significant variations in temperature, up to 60 °C (Fig. 6c). The stability in time is up to several months in water, which is really promising in terms of in vivo circulation time, storage conditions, handling and transport resistance.
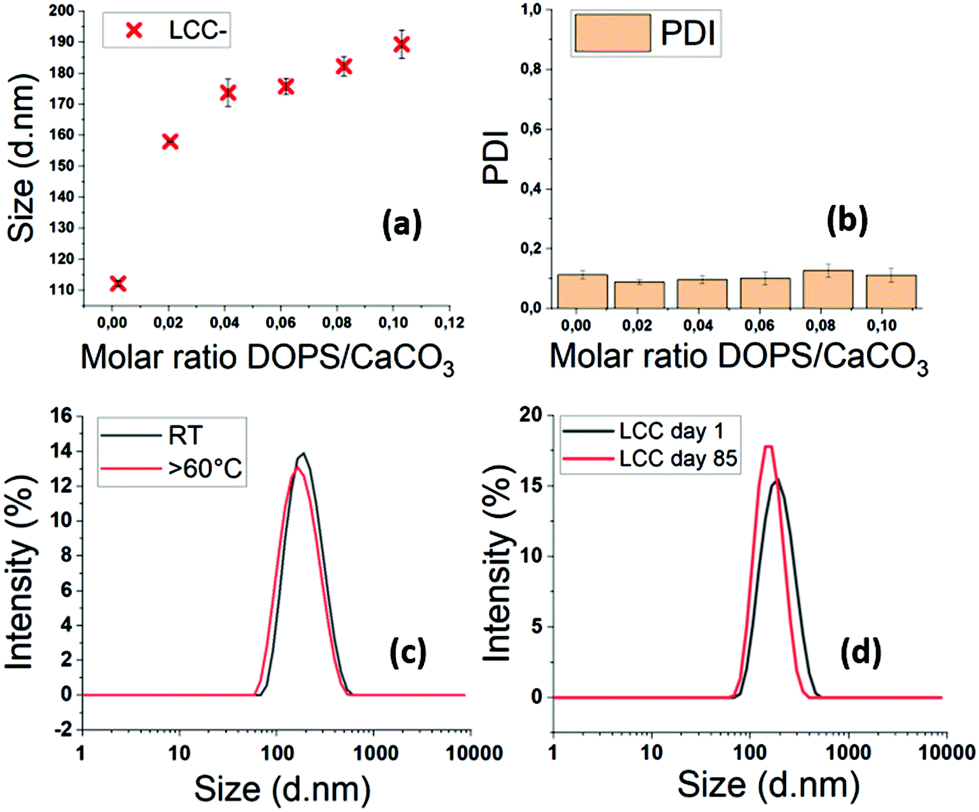 | ||
| Fig. 6 (a) LCC size evolution according to the molar ratio DOPS/CaCO3 characterized by DLS. 6 measurements were performed by sample. (b) Corresponding PDI values. (c) LCC size evolution after exposure to a temperature of 60–80 °C for 1 h, characterized by DLS. (d) LCC size dispersion at day 1 and day 85 characterized by DLS. | ||
The influence of the lipid polar head structure on stabilisation efficiency was investigated. Several lipids with cationic (trimethylammonium-propane (TAP)), zwitterionic (PC), or anionic head group phosphatidylglycerol (PG) were selected (Table 1). The particle size and PDI measured by DLS, as well as the concentration monitored by Nanoparticle Tracking Analysis (NTA) of the resulting LCC NPs, were compared to the values obtained with PS phospholipids, which were used as a reference. Samples were synthesised at a constant mass of lipid and ACC NPs.
| Stabiliser | Size (nm) | PDI | Concentration (particles per mL) |
|---|---|---|---|
| a Egg-PH composition: 95% mixed C16–C18 saturated and unsaturated (∼50–50). | |||
| 18:1 PS (DOPS) |
162.5 | 0.18 | 1.46 × 1011 |
| 18:1 TAP |
104.3 | 0.246 | 6.38 × 1010 |
| 16:0 PS |
139.3 | 0.102 | 2.31 × 1011 |
| 16:0 PC |
326.4 | 0.322 | 1.25 × 1011 |
| 16:0 PC + DSPE-PEG (80/20 w/w) |
Out of quality range | 5.71 × 1011 | |
| 16:0 PG |
142.7 | 0.017 | 2.07 × 1011 |
| E-PGa | 148.7 | 0.022 | 1.88 × 1011 |
An effective stabilisation was also observed using negatively charged lipids with a PG polar head. The LCC NPs sizes and concentrations using PG phospholipids were comparable with the values obtained with PS phospholipids of a similar aliphatic chain length (Table 1). PDIs were inferior to 0.1, evidencing monodisperse size populations.
On the contrary, the comparison between 18:1 TAP and 18:1 PS lipids showed a PDI increase from 0.18 to 0.246, and a concentration drop from 1.46 × 1011 NPs per mL to 6.38 × 1010 NPs per mL when using TAP lipids as stabilisers. This evidenced a lack of stabilisation, leading to polydisperse samples and concentration drop.
Using 16:0 PS as a reference, the comparison between the PC and PS head groups showed a PDI increase from 0.102 to 0.322, and a size increase from 139.3 nm to 326.4 nm when using PC lipids as stabilisers. This evidences a lack of stabilisation leading to polydisperse samples, with larger particles which may result from aggregation and/or crystallisation.28,29
A mix of PC and DSPE-PEG phospholipids was also tested with proportions similar to the work of C. Wang et al., to investigate the influence of the addition of PEGylated lipids on the NPs stabilisation with a reduced contact time between ACC NPs and lipids.20 PEGylated lipids could restore the stabilisation by providing steric stabilisation to the ACC NPs.92 However, with our process, this lipid mix did not lead to efficient stabilisation. The size and PDI could not be monitored as the values were out of the quality range of the DLS, due to excessive polydispersity of the sample (Table 1).
To summarise, these results evidence an efficient stabilisation with negatively charged lipids, and a deficient stabilisation when using zwitterionic and cationic lipids. The addition of PEGylated lipids did not provide efficient steric stabilisation with our process when using a mix of PC + DSPE-PEG. These results demonstrated the correlation between the stabilisation efficiency and the phospholipid head structure. The lipid affinity for calcium ions is a determinant parameter for a short process using a one-pot ethanol injection. A reduction of the process time from 24 h to 2 min has been achieved compared with the closest state of the art using the ethanol injection method.20 This achievement could lead to significant optimisations in the future in terms of process efficiency, operating time reduction and corresponding energy saving for the stabilisation of ACC NPs in aqueous media.
This affinity of PG lipids for ACC NPs led us to consider egg PG (E-PG), a bio-sourced phospholipid, as a stabiliser. E-PG is a mix of PG with different aliphatic chain lengths and unsaturation degrees. The size dispersion of E-PG LCC NPs characterized by DLS is narrow, with a PDI of 0.022 for a mean diameter of 148.7 nm, evidencing an efficient stabilisation (Table 1). Using E-PG as exclusive stabiliser, we achieved in this work the stabilisation of ACC NPs in water with a size under 150 nm using only a bio-sourced phospholipid. Replacing the use of synthetic or hemi-synthetic phospholipids by E-PG could lead to the development of a more stainable process compared with the closest state of the art, using a PEGylated phospholipid in the stabilising mix.20 Furthermore, the cost of bio-sourced highly purified natural phospholipids from soybeans or egg yolk suitable for injectable applications is becoming comparable with the price of PEGylated phospholipids. Hence, the use of egg-sourced PG represents a promising green alternative in terms of process cost to the use of synthetic stabilisers for calcium carbonate nanoparticles in aqueous media. A comparative table of the different existing methods to stabilize CaCO3 NPs in aqueous media is presented in the ESI ( Fig. S12†) to illustrate the benefits of our process in terms of sustainability and stabilisation efficiency.
A test was performed in phosphate buffer solution 0.1 M with DOPS, to evaluate the stability of LCC NPs in a media suitable for biological applications (ESI Fig. S13†). The size dispersion of the DOPS-LCC NPs in PBS was characterised by DLS, showing a PDI of 0.21 for a mean diameter of 187.3 nm. The control in water presented a PDI of 0.15 for a mean diameter of 168.5 nm. Despite an increase in mean diameter and polydispersity, particles stabilise in PBS present a narrow size distribution and a mean diameter below 200 nm. The difference observed in PBS might come from the ions existing in PBS, interfering with the interaction phospholipid/ACC NPs. For possible biological application, the 10% ethanol remaining in the solution can be evaporated by rotavapor if necessary.
LCC NPS surface charge modulation
The stabilisation of ACC NPs using cationic lipids was not efficient (Table 1). However, the surface charge modulation of the LCC NPs could be achieved by the post-insertion of cationic lipids in a solution of LCC NPs. Hence, there is an electrostatic affinity between the negatively charged phospholipids used for the stabilisation of ACC NPs in water and positively charges lipids.An ethanolic solution of 18:1 TAP cationic lipids is injected at room temperature in an aqueous solution of LCC NPs stabilised by DOPS (Fig. 7a). The injection is followed by high intensity mixing using a vortex. The zeta potential shifted from negative values to positive values, testifying from the structuration of the cationic lipids around the LCC NPs (Fig. 7b).
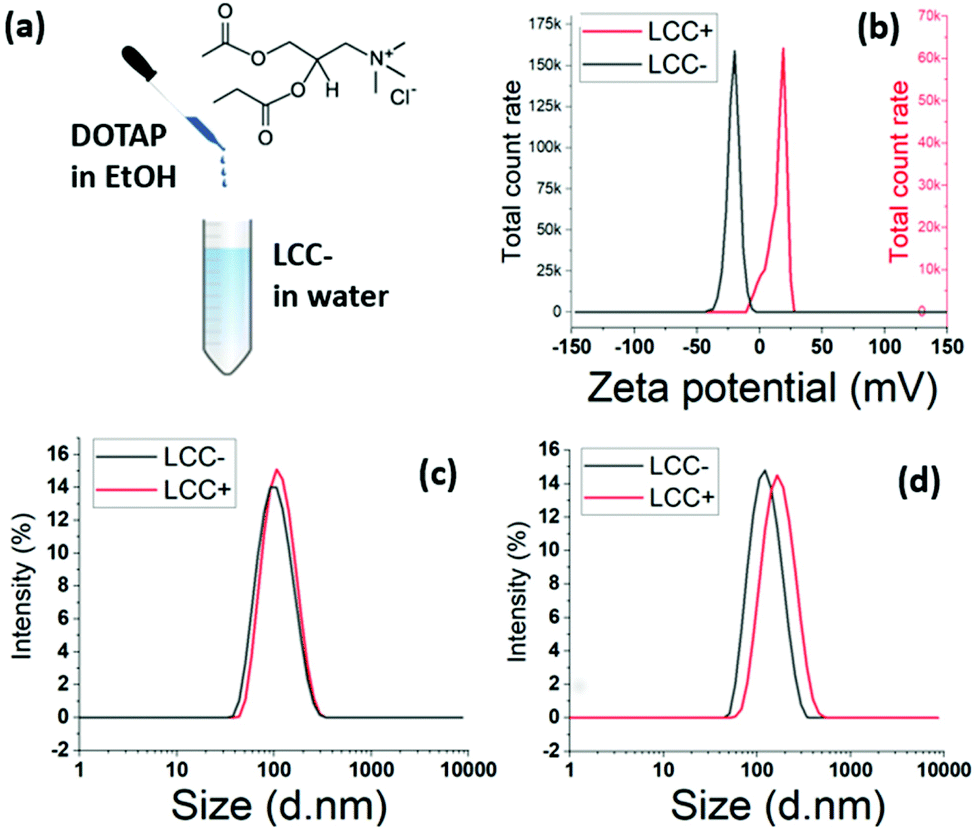 | ||
| Fig. 7 Post-insertion of DOTAP to modulate the charge of LCC NPS. (a) Schematic synthesis process. (b) Zeta potential of DOPS LCC NPs (black) and DOPS/DOTAP LCC NPs (red). (c) DLS characterisation of DOPS LCC NPs before (black)and after (red) the injection of DOTAP. Molar ratio DOTAP/DOPS = 1. (d) DLS characterisation of DOPS LCC NPs before (black)and after (red) the injection of DOTAP. Molar ratio DOTAP/DOPS = 10. | ||
The DLS characterisation showed a narrow size distribution, with a mean diameter shifting from 95.4 nm to 105.3 nm after the injection of 18:1 TAP lipids at a ratio DOPS:TAP of 1 (Fig. 7c). When the DOPS:TAP ratio is increased to 10, the mean diameter shift is larger, with a shift from 115.4 nm to 152.8 nm following the injection of 18:1 TAP lipids (Fig. 7d). The variation is mean diameter shift in accordance with the 18:1 TAP concentration increase also evidences the structuration of the cationic lipids around the already formed LCC NPs stabilised by DOPS.
These results proves the possibility of post-inserting cationic lipids after the stabilisation of ACC NPs in water using DOPS in water. This post-insertion technique allows to effectively tune the surface charge of the final LCC NPs and could pave the way for the modulation of other surface properties.
Upscaling
The ratio ethanol/water (1/9) used for the stabilisation of ACC NPs in water with the ethanol injection technique20 plays an important role in the final concentration of LCC NPs. This final concentration of LCC NPs after injection is limited. The synthesis of larger amounts of LCC NPs leads to larger volumes of the water phase and of the ethanol phase. According to our experiments, the pipette injection method is efficient up to a volume of 40 mL of LCC NPs, using a 10 mL pipette for the injection of a 4 mL ethanol phase. Above this volume, we observed that the injection strength became weaker, which strongly affected the lipid structuration around the particles. We tested on a 2 L volume the replacement of the injection method by a magnetic stirring of the water phase above 500 rpm. The size dispersion of the particles remains narrow, with PDI < 0.1, and a comparable particle stability is obtained compared with the pipette injection method (ESI Fig. S14†).These results showed injection can be replaced by strong stirring of the water phase. We tested two different upscaling techniques suitable for industrial transfer. The first one is a 50 L reactor with mechanical stirring, and the second one is a continuous mixing flow process allowing continuous flow production of the stabilised nanoparticles (Fig. 8a and b).
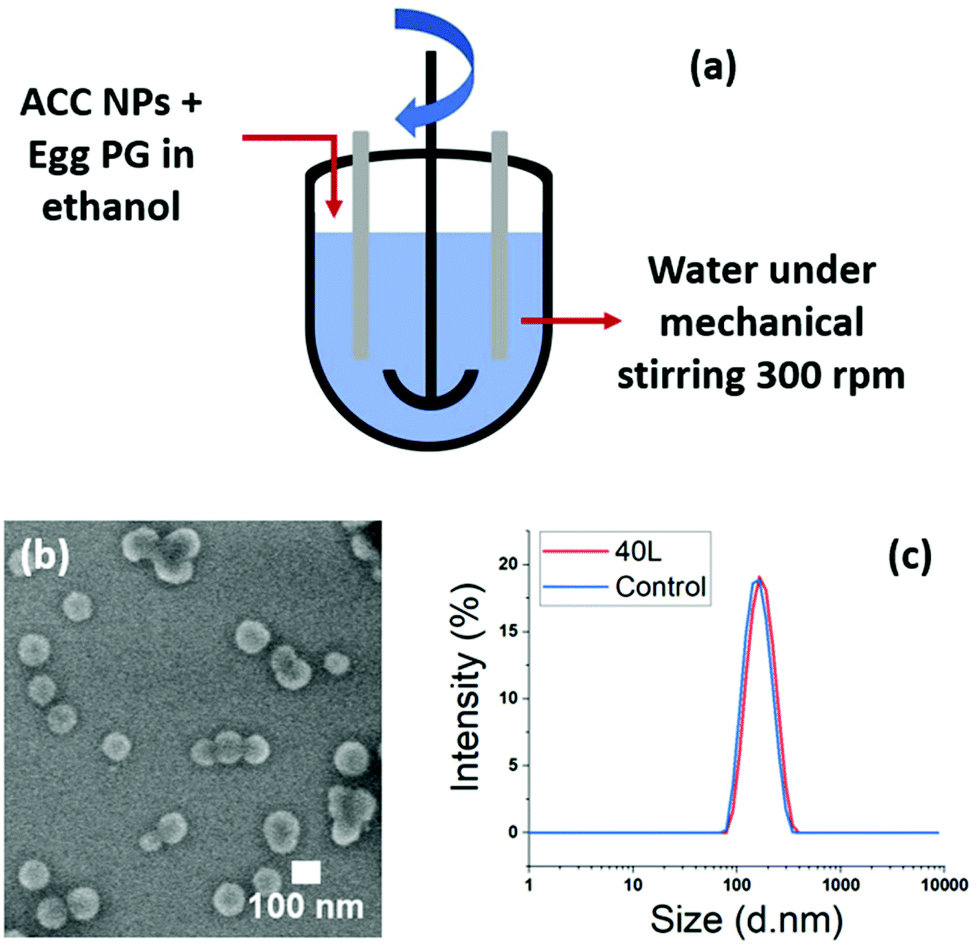 | ||
| Fig. 8 Upscaling stabilisation ACC NPs by egg PG in aqueous media using a 50 L chemical reactor, for a final volume of 40 L LCC NPs. (a) Schematic representation of the reactor. (b) SEM imaging of the resulting LCC NPs in aqueous media. (c) DLS characterisation of the resulting LCC NPs size dispersion with a control performed by ethanol injection in a volume of 5 mL. | ||
In the 50 L reactor we performed a synthesis on a 40 L final volume, using egg PG as stabiliser. 4 L of an ethanolic solution containing ACC NPs and egg PG was poured into 36 L of water under strong mechanical mixing. A 5 mL control was also performed using ethanol injection method. The DLS and SEM results show highly homogeneous nanoparticles, stable in water. The size increase between the control and the upscaled sample is neglectable, under 10 nm in mean diameter (Fig. 8b and c). This experiment demonstrates the ability to upscale our process for industrial needs using large scale mechanical stirring reactors. The concern with this method was the one-pot addition of the ethanolic phase containing the ACC NPs and the stabiliser. Indeed, the addition time with larger volumes can increase. However, despite the increase of addition time from 3 s (5 mL control) to 13 s (40 L volume), the nanoparticles obtained were stable and homogeneous in size and shape.
To overcome the potential limitation of the one-pot addition of large volumes, we investigated the possibility of industrial flow production with our process. We developed a facile continuous flow mixing method using Coflore® Agitated Cell Reactor (ACR). This continuous flow reactor performs efficient mechanical agitation using multiple interlinked cell reactors (Fig. 9a). The upscaling proof of concept was performed on a final volume of 250 mL using DOPS as stabiliser. The fluxes of the ethanol phase containing ACC NPs and DOPS, and of the water phase were determined according to the ratio water:ethanol 9:1 used for the injection method. The flux of the ethanol phase is 1 mL min−1, and the flux of water of 9 mL min−1, in order to obtain a ratio 9:1 of water:ethanol at the mixing points. The final nanoparticles are stable in aqueous media and highly homogeneous in size and shape, with a mean size of 70 nm measured on the SEM picture (Fig. 9b and c). A control was performed on 5 mL using the ethanol injection method, showing no significant difference in size and dispersity. These results demonstrate the feasibility of switching from bulk injection and stirring to a continuous flow production using agitated cell mixing. This method has a great potential for industrial upscaling as a continuous flow process, allowing the continuous production of stabilised LCC NPs with a high control and reproducibility of the mixing conditions. High volumes can be produced with this technique. With these fluxes, 25 min are required to synthesise 250 mL.
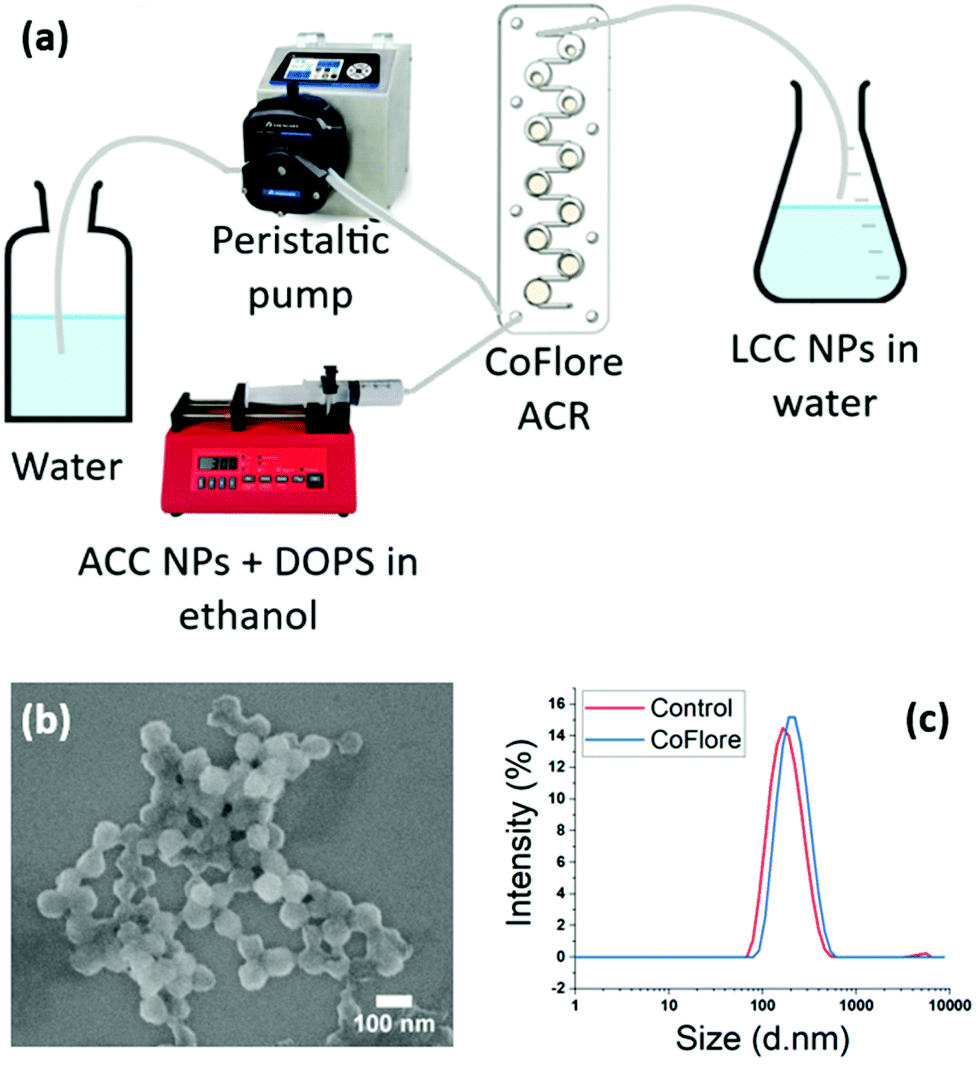 | ||
| Fig. 9 Upscaling stabilisation ACC NPs by DOPS in aqueous media using the CoFlore continuous flow reactor. (a) Schematic representation of the reactor and of the process. (b) SEM imaging of the resulting LCC NPs in aqueous media. (c) DLS characterisation of the resulting LCC NPs size dispersion with a control performed by ethanol injection in a volume of 5 mL. | ||
Conclusions
ACC NPs synthesised using ethanol as solvent are monodispersed with a mean diameter under 150 nm without the need for synthetic additives. The particle size growth follows a power law between 2 h and 49 h reaction, maintaining a narrow size dispersion with PDI values under 0.05. Combined with the porous granular structure of ACC NPs, these results suggest a formation of ACC NPs by a first homogeneous nucleation event, followed by a growth by autocatalytic heterogeneous nucleation happening on the ACC NPs surface irregularities. We demonstrated the importance of the diffusion of water and ammonia to catalyse the formation of ACC NPs in ethanol. Consequently, we observed that the removal of the DB and of drierite increased the process efficiency by a factor of 3.5. Therefore, the volume of ethanol required to produce a determined mass of ACC NPs can be divided by 3.5, reducing the ecological impact of the process.Subsequently, our works focussed on the stabilisation in aqueous media of the resulting ACC NPs for biomedical and healthcare applications. The adaptation of the ethanol injection process using phospholipids represents to this date the greenest process for the stabilisation of ACC NPs in water. We rationalised the selection of the phospholipids according to their natural electrostatic affinity for ACC NPs and were able to reduce the process time from 24 h to 2 min. The use of PS and PG phospholipids provides an efficient electrostatic stabilisation of the ACC NPs, without the need for additional PEGylated lipids. Compared with the state of the art, the absence of PEGylated lipids increases the sustainability of the process and the biotolerance of the final particles. The reduction of the process duration consequently decreases its operating time and energy consumption. The obtained LCC NPs were highly monodisperse, with an excellent stability in temperature (up to 60 °C) and time (up to three months) variations. The efficiency of PG lipids led to the identification of an egg-sourced phospholipid as a suitable bio-sourced stabiliser. Our process was demonstrated suitable for industrial large-scale production in one pot using a 50 L chemical reactor with mechanical stirring, and in continuous flow production using the Coflore® Agitated Cell Reactor.
This work aims to contribute to the transition toward sustainable processes in the development of ACC NPs technologies suitable for biomedical and cosmetic applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Fonds National de la Recherche (FNR) of Luxembourg for supporting this work through the projects MASSENA PRIDE/15/10935404. The authors also acknowledge the support and the use of resources of the French Infrastructure for Integrated Structural Biology FRISBI ANR-10-INBS-05 and of Instruct-ERIC. They particularly thank Corinne Crucifix for her help with CryoTEM sample preparation and images collection. From the Luxembourg Institute of Science and Technology, the authors acknowledge J. N. Audinot for insightful discussions and the acquisition of the HIM data, Y. Fleming from the Characterization Platform for performing the XRD measurements and his valuable help with the analysis of the results, and S. Krishnamoorthy for his valuable insights on the writing. The authors acknowledge the programs Nanofar (EMJD Europ) and Nanofar+ (Pays de la Loire, France) for their help and support in the first author training.Notes and references
- A. Syafiq, et al., Facile synthesize of transparent hydrophobic nano-CaCO3 based coatings for self-cleaning and anti-fogging, Mater. Chem. Phys., 2020, 239, 121913 CrossRef.
- N. Farghal and H. Elkafrawy, The effects of activated charcoal and calcium carbonate based toothpastes on surface properties of composite resin restorative materials, Egypt. Dent. J., 2020, 66, 2431–2438 CrossRef.
- E. T. Hwang, et al., Highly Stable and Fine-Textured Hybrid Microspheres for Entrapment of Cosmetic Active Ingredients, ACS Omega, 2020, 5, 29577–29584 CrossRef CAS PubMed.
- A. D. Woldetsadik, S. K. Sharma, S. Khapli, R. Jagannathan and M. Magzoub, Hierarchically porous calcium carbonate scaffolds for bone tissue engineering, ACS Biomater. Sci. Eng., 2017, 3, 2457–2469 CrossRef CAS PubMed.
- M. L. P. Vidallon, et al., Gas–Generating, pH–Responsive Calcium Carbonate Hybrid Particles with Biomimetic Coating for Contrast–Enhanced Ultrasound Imaging, Part. Part. Syst. Charact., 2020, 37, 1900471 CrossRef CAS.
- A. Som, et al., Monodispersed calcium carbonate nanoparticles modulate local pH and inhibit tumor growth in vivo, Nanoscale, 2016, 8, 12639–12647 RSC.
- P. Zhao, et al., Enhancing anti-tumor efficiency in hepatocellular carcinoma through the autophagy inhibition by miR-375/sorafenib in lipid-coated calcium carbonate nanoparticles, Acta Biomater., 2018, 72, 248–255 CrossRef CAS PubMed.
- M. d'Amora, F. Liendo, F. A. Deorsola, S. Bensaid and S. Giordani, Toxicological profile of calcium carbonate nanoparticles for industrial applications, Colloids Surf., B, 2020, 190, 110947 CrossRef PubMed.
- N. M. Elbaz, A. Owen, S. Rannard and T. O. McDonald, Controlled synthesis of calcium carbonate nanoparticles and stimuli-responsive multi-layered nanocapsules for oral drug delivery, Int. J. Pharm., 2020, 574, 118866 CrossRef CAS PubMed.
- N. I. Hammadi, et al., Formulation of a sustained release docetaxel loaded cockle shell-derived calcium carbonate nanoparticles against breast cancer, Pharm. Res., 2017, 34, 1193–1203 CrossRef CAS PubMed.
- C. Wang, et al., Mitoxantrone-preloaded water-responsive phospholipid-amorphous calcium carbonate hybrid nanoparticles for targeted and effective cancer therapy, Int. J. Nanomed., 2019, 14, 1503 CrossRef CAS PubMed.
- R. K. Thapa, et al., Progressive slowdown/prevention of cellular senescence by CD9-targeted delivery of rapamycin using lactose-wrapped calcium carbonate nanoparticles, Sci. Rep., 2017, 7, 1–11 CrossRef PubMed.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Exploiting the enhanced permeability and retention effect for tumor targeting, Drug Discovery Today, 2006, 11, 812–818 CrossRef CAS PubMed.
- H. Nehoff, N. N. Parayath, L. Domanovitch, S. Taurin and K. Greish, Nanomedicine for drug targeting: strategies beyond the enhanced permeability and retention effect, Int. J. Nanomed., 2014, 9, 2539 Search PubMed.
- G. Fytianos, A. Rahdar and G. Z. Kyzas, Nanomaterials in cosmetics: Recent updates, Nanomaterials, 2020, 10, 979 CrossRef CAS PubMed.
- F. Carrouel, S. Viennot, L. Ottolenghi, C. Gaillard and D. Bourgeois, Nanoparticles as anti-microbial, anti-inflammatory, and remineralizing agents in oral care cosmetics: a review of the current situation, Nanomaterials, 2020, 10, 140 CrossRef CAS PubMed.
- J. Marto, et al., Pickering Emulsions Stabilized by Calcium Carbonate Particles: A New Topical Formulation, Cosmetics, 2020, 7, 62 CrossRef CAS.
- F. A. Andersen and L. Brecevic, Infrared spectra of amorphous and crystalline calcium carbonate, Acta Chem. Scand., 1991, 45, 1018–1024 CrossRef CAS.
- N. H. de Leeuw and S. C. Parker, Surface structure and morphology of calcium carbonate polymorphs calcite, aragonite, and vaterite: an atomistic approach, J. Phys. Chem. B, 1998, 102, 2914–2922 CrossRef CAS.
- C. Wang, et al., Facile preparation of phospholipid–amorphous calcium carbonate hybrid nanoparticles: toward controllable burst drug release and enhanced tumor penetration, Chem. Commun., 2018, 54, 13080–13083 RSC.
- S.-F. Chen, H. Cölfen, M. Antonietti and S.-H. Yu, Ethanol assisted synthesis of pure and stable amorphous calcium carbonate nanoparticles, Chem. Commun., 2013, 49, 9564–9566 RSC.
- C. Wang, S. Chen, Q. Yu, F. Hu and H. Yuan, Taking advantage of the disadvantage: employing the high aqueous instability of amorphous calcium carbonate to realize burst drug release within cancer cells, J. Mater. Chem. B, 2017, 5, 2068–2073 RSC.
- C. Wang, et al., Lipase–triggered water–responsive “Pandora's Box” for cancer therapy: Toward induced neighboring effect and enhanced drug penetration, Adv. Mater., 2018, 30, 1706407 CrossRef PubMed.
- D. Gebauer, X. Liu, B. Aziz, N. Hedin and Z. Zhao, Porous tablets of crystalline calcium carbonate via sintering of amorphous nanoparticles, CrystEngComm, 2013, 15, 1257–1263 RSC.
- R. Sun, et al., Amorphous calcium carbonate constructed from nanoparticle aggregates with unprecedented surface area and mesoporosity, ACS Appl. Mater. Interfaces, 2018, 10, 21556–21564 CrossRef CAS PubMed.
- Y. Zhao, et al., A preloaded amorphous calcium carbonate/doxorubicin@ silica nanoreactor for pH–responsive delivery of an anticancer drug, Angew. Chem., Int. Ed., 2015, 54, 919–922 CrossRef CAS PubMed.
- Y. Nakashima, C. Takai, H. Razavi-Khosroshahi, W. Suthabanditpong and M. Fuji, Synthesis of ultra-small hollow silica nanoparticles using the prepared amorphous calcium carbonate in one-pot process, Adv. Powder Technol., 2018, 29, 904–908 CrossRef CAS.
- J. Xiao, Z. Wang, Y. Tang and S. Yang, Biomimetic mineralization of CaCO3 on a phospholipid monolayer: from an amorphous calcium carbonate precursor to calcite via vaterite, Langmuir, 2010, 26, 4977–4983 CrossRef CAS PubMed.
- Q. Shen, et al., Properties of amorphous calcium carbonate and the template action of vaterite spheres, J. Phys. Chem. B, 2006, 110, 2994–3000 CrossRef CAS PubMed.
- G.-B. Cai, G.-X. Zhao, X.-K. Wang and S.-H. Yu, Synthesis of polyacrylic acid stabilized amorphous calcium carbonate nanoparticles and their application for removal of toxic heavy metal ions in water, J. Phys. Chem. C, 2010, 114, 12948–12954 CrossRef CAS.
- A. Barhoum, et al., Effect of cationic and anionic surfactants on the application of calcium carbonate nanoparticles in paper coating, ACS Appl. Mater. Interfaces, 2014, 6, 2734–2744 CrossRef CAS PubMed.
- C.-F. Dai, et al., Methotrexate intercalated calcium carbonate nanostructures: synthesis, phase transformation and bioassay study, Mater. Sci. Eng. C, 2016, 69, 577–583 CrossRef CAS PubMed.
- C.-F. Dai, W.-Y. Wang, S.-P. Li and X.-D. Li, Synthesis of nanostructured calcium carbonate/methotrexate@ silica and its application in cancer therapy, RSC Adv., 2016, 6, 68335–68340 RSC.
- C. Xu, et al., Biodegradable Nanoparticles of Polyacrylic Acid–Stabilized Amorphous CaCO3 for Tunable pH–Responsive Drug Delivery and Enhanced Tumor Inhibition, Adv. Funct. Mater., 2019, 29, 1808146 CrossRef.
- K. H. Min, et al., pH-controlled gas-generating mineralized nanoparticles: a theranostic agent for ultrasound imaging and therapy of cancers, ACS Nano, 2015, 9, 134–145 CrossRef CAS PubMed.
- J. Lee, et al., Theranostic gas-generating nanoparticles for targeted ultrasound imaging and treatment of neuroblastoma, J. Controlled Release, 2016, 223, 197–206 CrossRef CAS PubMed.
- S. K. Kim, M. B. Foote and L. Huang, Targeted delivery of EV peptide to tumor cell cytoplasm using lipid coated calcium carbonate nanoparticles, Cancer Lett., 2013, 334, 311–318 CrossRef CAS PubMed.
- P. Zhao, et al., MiR-375 delivered by lipid-coated doxorubicin-calcium carbonate nanoparticles overcomes chemoresistance in hepatocellular carcinoma, Nanomedicine, 2017, 13, 2507–2516 CrossRef CAS PubMed.
- J. Q. Peng, et al., Targeted co-delivery of protein and drug to a tumor in vivo by sophisticated RGD-modified lipid-calcium carbonate nanoparticles, J. Controlled Release, 2019, 302, 42–53 CrossRef CAS PubMed.
- J. Peng, et al., One-step formation of lipid-polyacrylic acid-calcium carbonate nanoparticles for co-delivery of doxorubicin and curcumin, J. Drug Targeting, 2017, 25, 704–714 CrossRef CAS PubMed.
- C. C. Tester, et al., In vitro synthesis and stabilization of amorphous calcium carbonate (ACC) nanoparticles within liposomes, CrystEngComm, 2011, 13, 3975–3978 RSC.
- C. C. Tester, C.-H. Wu, S. Weigand and D. Joester, Precipitation of ACC in liposomes—a model for biomineralization in confined volumes, Faraday Discuss., 2012, 159, 345–356 RSC.
- A. Szcześ, Effects of DPPC/Cholesterol liposomes on the properties of freshly precipitated calcium carbonate, Colloids Surf., B, 2013, 101, 44–48 CrossRef PubMed.
- A. Szcześ and D. Sternik, Properties of calcium carbonate precipitated in the presence of DPPC liposomes modified with the phospholipase A 2, J. Therm. Anal. Calorim., 2016, 123, 2357–2365 CrossRef.
- H. S. Lee, T. H. Ha and K. Kim, Fabrication of unusually stable amorphous calcium carbonate in an ethanol medium, Mater. Chem. Phys., 2005, 93, 376–382 CrossRef CAS.
- H. Zhou, et al., Co-precipitation of calcium carbonate and curcumin in an ethanol medium as a novel approach for curcumin dissolution enhancement, J. Drug Delivery Sci. Technol., 2019, 51, 397–402 CrossRef CAS.
- Z. Dong, et al., CaCO3 nanoparticles as an ultra-sensitive tumor-pH-responsive nanoplatform enabling real-time drug release monitoring and cancer combination therapy, Biomaterials, 2016, 110, 60–70 CrossRef CAS PubMed.
- A. Som, et al., Calcium carbonate nanoparticles stimulate tumor metabolic reprogramming and modulate tumor metastasis, Nanomedicine, 2019, 14, 169–182 CrossRef CAS PubMed.
- C. Rao, et al., In vitro preparation and characterization of amorphous calcium carbonate nanoparticles for applications in curcumin delivery, J. Mater. Sci., 2019, 54, 11243–11253 CrossRef CAS.
- S. F. Lam, K. W. Bishop, R. Mintz, L. Fang and S. Achilefu, Calcium carbonate nanoparticles stimulate cancer cell reprogramming to suppress tumor growth and invasion in an organ-on-a-chip system, Sci. Rep., 2021, 11, 1–12 CrossRef PubMed.
- J. Ihli, P. Bots, A. Kulak, L. G. Benning and F. C. Meldrum, Elucidating Mechanisms of Diffusion–Based Calcium Carbonate Synthesis Leads to Controlled Mesocrystal Formation, Adv. Funct. Mater., 2013, 23, 1965–1973 CrossRef CAS.
- M. Mantilaka, R. Rajapakse, D. Karunaratne and H. Pitawala, Preparation of amorphous calcium carbonate nanoparticles from impure dolomitic marble with the aid of poly (acrylic acid) as a stabilizer, Adv. Powder Technol., 2014, 25, 591–598 CrossRef CAS.
- S. Bom, et al., Replacing Synthetic Ingredients by Sustainable Natural Alternatives: A Case Study Using Topical O/W Emulsions, Molecules, 2020, 25, 4887 CrossRef CAS PubMed.
- N. Hadjesfandiari and A. Parambath, in Engineering of Biomaterials for Drug Delivery Systems, ed. A. Parambath, Woodhead Publishing, 2018, pp. 345–361 Search PubMed.
- L. Simon, et al., Polyoxazolines based mixed micelles as PEG free formulations for an effective quercetin antioxidant topical delivery, Int. J. Pharm., 2019, 570, 118516, DOI:10.1016/j.ijpharm.2019.118516.
- I. M. Banat, Q. Carboué, G. Saucedo-Castañeda and d. J. Cázares-Marinero, J. Biosurfactants: The green generation of speciality chemicals and potential production using Solid-State fermentation (SSF) technology, Bioresour. Technol., 2021, 320, 124222, DOI:10.1016/j.biortech.2020.124222.
- P. Zhang, F. Sun, S. Liu and S. Jiang, Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation, J. Controlled Release, 2016, 244, 184–193 CrossRef CAS PubMed.
- Q. Yang and S. K. Lai, Anti–PEG immunity: emergence, characteristics, and unaddressed questions, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 655–677 CAS.
- G. Kozma, T. Shimizu, T. Ishida and J. Szebeni, Anti-PEG antibodies: Properties, formation and role in adverse immune reactions to PEGylated nano-biopharmaceuticals, Adv. Drug Delivery Rev., 2020, 154–155, 163–175 CrossRef CAS PubMed.
- T. T. Hoang Thi, et al., The Importance of Poly(ethylene glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation, Polymers, 2020, 12, 298 CrossRef PubMed.
- P. Sellaturay, S. Nasser and P. Ewan, Polyethylene Glycol–Induced Systemic Allergic Reactions (Anaphylaxis), J. Allergy Clin. Immunol., 2021, 9, 670–675, DOI:10.1016/j.jaip.2020.09.029.
- F. Cox, K. Khalib and N. Conlon, PEG That Reaction: A Case Series of Allergy to Polyethylene Glycol, J. Clin. Pharmacol., 2021, 61, 832–835, DOI:10.1002/jcph.1824.
- R. S. Lane, F. M. Haller, A. A. Chavaroche, A. Almond and P. L. DeAngelis, Heparosan-coated liposomes for drug delivery, Glycobiology, 2017, 27, 1062–1074 CrossRef CAS PubMed.
- M. E. Labib and R. Williams, The use of zeta-potential measurements in organic solvents to determine the donor—acceptor properties of solid surfaces, J. Colloid Interface Sci., 1984, 97, 356–366 CrossRef CAS.
- T. Wirtz, O. De Castro, J.-N. Audinot and P. Philipp, Imaging and analytics on the helium ion microscope, Annu. Rev. Anal. Chem., 2019, 12, 523–543 CrossRef CAS PubMed.
- J. Zernike and Q. J. Ahmed, Three–phase curve of ammonium bicarbonate, Recl. Trav. Chim. Pays-Bas, 1954, 73, 95–101 CrossRef CAS.
- K.-S. Seo, C. Han, J.-H. Wee, J.-K. Park and J.-W. Ahn, Synthesis of calcium carbonate in a pure ethanol and aqueous ethanol solution as the solvent, J. Cryst. Growth, 2005, 276, 680–687 CrossRef CAS.
- E. Peh, A. Taubert and K. Tauer, Ammonium carbonate gas diffusion crystallization in a continuous organic medium leads to dendritic calcium carbonate, Matters, 2017, 3, e201706000012 Search PubMed.
- N. T. Thanh, N. Maclean and S. Mahiddine, Mechanisms of nucleation and growth of nanoparticles in solution, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- D. Jaynes and A. Rogowski, Applicability of Fick's law to gas diffusion, Soil Sci. Soc. Am. J., 1983, 47, 425–430 CrossRef CAS.
- N. Koga, Y. Yamane and T. Kimura, Thermally induced transformations of calcium carbonate polymorphs precipitated selectively in ethanol/water solutions, Thermochim. Acta, 2011, 512, 13–21 CrossRef CAS.
- M. Farhadi-Khouzani, D. M. Chevrier, P. Zhang, N. Hedin and D. Gebauer, Water as the key to proto–aragonite amorphous CaCO3, Angew. Chem., Int. Ed., 2016, 55, 8117–8120 CrossRef CAS PubMed.
- S. R. Golisz, J. S. Yang and R. D. Johnson, Understanding the effect of CO2 on the pHe of fuel ethanol, Fuel, 2017, 199, 1–3 CrossRef CAS.
- T. K. Sherwood, Solubilities of sulfur dioxide and ammonia in water, Ind. Eng. Chem., 1925, 17, 745–747 CrossRef CAS.
- L.-J. Huang, W.-L. Xue and Z.-X. Zeng, The Solubility of ammonia in ethanol between 277.35 K and 328.15 K, Fluid Phase Equilib., 2011, 303, 80–84 CrossRef CAS.
- A. Radha, T. Z. Forbes, C. E. Killian, P. Gilbert and A. Navrotsky, Transformation and crystallization energetics of synthetic and biogenic amorphous calcium carbonate, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16438–16443 CrossRef CAS PubMed.
- G.-T. Zhou, Q.-Z. Yao, S.-Q. Fu and Y.-B. Guan, Controlled crystallization of unstable vaterite with distinct morphologies and their polymorphic transition to stable calcite, Eur. J. Mineral., 2010, 22, 259–269 CrossRef CAS.
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Sea urchin spine calcite forms via a transient amorphous calcium carbonate phase, Science, 2004, 306, 1161–1164 CrossRef CAS PubMed.
- Z. Yaari, et al., Theranostic barcoded nanoparticles for personalized cancer medicine, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- M. Pons, M. Foradada and J. Estelrich, Liposomes obtained by the ethanol injection method, Int. J. Pharm., 1993, 95, 51–56 CrossRef CAS.
- H. Hauser, E. Finer and A. Darke, Crystalline anhydrous Ca-phosphatidylserine bilayers, Biochem. Biophys. Res. Commun., 1977, 76, 267–274 CrossRef CAS.
- R. Dluhy, D. G. Cameron, H. H. Mantsch and R. Mendelsohn, Fourier transform infrared spectroscopic studies of the effect of calcium ions on phosphatidylserine, Biochemistry, 1983, 22, 6318–6325 CrossRef CAS.
- M. Roux and M. Bloom, Calcium binding by phosphatidylserine headgroups. Deuterium NMR study, Biophys. J., 1991, 60, 38–44 CrossRef CAS PubMed.
- J. L. Browning and J. Seelig, Bilayers of phosphatidylserine: a deuterium and phosphorus nuclear magnetic resonance study, Biochemistry, 1980, 19, 1262–1270 CrossRef CAS PubMed.
- G. Büldt and R. Wohlgemuth, The headgroup conformation of phospholipids in membranes, J. Membr. Biol., 1981, 58, 81–100 CrossRef PubMed.
- A. Melcrová, et al., The complex nature of calcium cation interactions with phospholipid bilayers, Sci. Rep., 2016, 6, 38035 CrossRef PubMed.
- M. H. Derkani, et al., Mechanisms of surface charge modification of carbonates in aqueous electrolyte solutions, Colloids Interfaces, 2019, 3, 62 CrossRef CAS.
- X. Banquy, K. Kristiansen, D. W. Lee and J. N. Israelachvili, Adhesion and hemifusion of cytoplasmic myelin lipid membranes are highly dependent on the lipid composition, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 402–410 CrossRef CAS PubMed.
- H. L. Scott, et al., On the mechanism of bilayer separation by extrusion, or why your LUVs are not really unilamellar, Biophys. J., 2019, 117, 1381–1386 CrossRef CAS PubMed.
- N. Düzgüne, et al., Calcium-and magnesium-induced fusion of mixed phosphatidylserine/phosphatidylcholine vesicles: effect of ion binding, J. Membr. Biol., 1981, 59, 115–125 CrossRef PubMed.
- Z. D. Schultz, I. M. Pazos, F. K. McNeil-Watson, E. N. Lewis and I. W. Levin, Magnesium-induced lipid bilayer microdomain reorganizations: implications for membrane fusion, J. Phys. Chem. B, 2009, 113, 9932–9941 CrossRef CAS PubMed.
- J. Zhai, et al., Lipid–PEG conjugates sterically stabilize and reduce the toxicity of phytantriol-based lyotropic liquid crystalline nanoparticles, Langmuir, 2015, 31, 10871–10880 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03396d |
| This journal is © The Royal Society of Chemistry 2022 |