
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Debondable adhesives and their use in recycling
Kira R.
Mulcahy
 ab,
Alexander F. R.
Kilpatrick
a,
Gavin D. J.
Harper
c,
Allan
Walton
c and
Andrew P.
Abbott
*a
ab,
Alexander F. R.
Kilpatrick
a,
Gavin D. J.
Harper
c,
Allan
Walton
c and
Andrew P.
Abbott
*a
aSchool of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: apa1@le.ac.uk
bSchool of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK
cSchool of Metallurgy and Materials, University of Birmingham, Birmingham, B15 2TT, UK
First published on 22nd November 2021
Abstract
Structural adhesives are commonly used to join dissimilar materials and are of particular interest in complex technological devices used in automotive, telecommunications, photonic devices, aerospace and sustainable power production and storage. Their strength, ease of application and stability are of significant help in the manufacture and service of the device, however, they can present significant issues at end of life, particularly when technology critical metals need to be recovered. In this paper, we highlight some of the issues, and discuss an alternative approach: the use of adhesives that can be debonded upon application of an external stimulus. The aim of this critical review is to consider the polymer systems which have been investigated and suggest some alternative strategies. It also aims to demonstrate the different stimuli that can be used to debond and comment on the applicability of these with a number of case studies. Finally, the service conditions and process economics are considered together with a discussion of the Green benefits of this type of methodology.
1. Introduction
Most technological devices consist of organic, inorganic, and metallic components, joined into complex architectures using organic adhesives; many of which play a multifaceted role, and impart structural integrity, in addition to binding joints. At present, removal of such adhesives is time-consuming and inefficient, requiring solvents, pyrolysis, or comminution (grinding/shredding). The considerable challenges posed by these adhesives, to disassembly and recycling, have recently been reviewed with respect to lithium-ion batteries (LiBs);1 however, these issues extend to a wide range of devices and components, including mobile electronics, motors and photonic devices.This is of particular concern, with respect to the recovery of technology critical metals (TCMs), which are diffusely distributed within components but essential to function.2 Maintaining the structural integrity of the adhesive prevents the preferred green alternatives of repair, remanufacture, reuse and recycle. In addition, due to current non-selective hydrometallurgical techniques, maintaining value in recycled material is difficult, often rendering reuse within the same context unviable. Given the essential nature of TCMs in most digital and clean energy technologies, their demand is growing. However, there remain significant bottlenecks in the development of mining and processing capacity for the production of new primary TCMs. Therefore, recovery of these elements will become an essential component in the supply chain of raw materials for technologies that aid decarbonisation. In addition, due to limited resources, and ethical and environmental issues associated with extraction, a move towards a more circular economy is vital.
The solution to these issues lies with adhesive design, as easy debonding or degradation would facilitate disassembly and allow higher value outputs from recycling. At present, the structural adhesives market is dominated by epoxy and polyurethane chemistries, which form thermoset resins on cross-linking of their polymer networks. Due to their high strength and fast setting times, the epoxide market alone had an estimated worth of $5.9 billion in 2019, with projected growth to $10.3 billion by 2027.3 However, these adhesives are not without their issues. Formed from the reactions of di- or polyols, such as bisphenol A (BPA), with epichlorohydrin (ECH), these ubiquitous polymers do not biodegrade and emit toxic gases, when incinerated. Recent research has considered replacing monomers or cross-linkers with bio-based components, or using novel adhesives based on biopolymers such as polysaccharides.4 An alternative approach proposes the use of polymers which can debond on application of an external stimulus. While this technology is not widely available at present, this critical review will discuss the approaches used in the design of debondable adhesives, and the limitations of systems already reported in the literature.
1.1. Service requirements
For an adhesive to function effectively, the polymer molecules need to stick to each other (cohesive forces) as well as the surfaces they are sticking together (adhesive forces). This adhesion is a complex mixture of chemical and mechanical interactions, and may have components of absorption, chemisorption, diffusion and physical ‘lock and key’ interactions. Typical bond strengths, for structural adhesives, are in the range of 10 to 35 MPa, but are highly dependent upon substrate type, pre-treatment, surface porosity and surface cleanliness.5 Precise strengths required, depend on service applications but usually adhesives are designed to fail at typically five times the required stress to provide the relevant safety factors. It is also essential for adhesives to function under all operating conditions experienced during device lifetime. Temperature is a crucial consideration here: adhesives lose cohesive strength at elevated temperatures, and only function effectively below their glass transition temperature, meaning few are suitable above 150 °C. In addition, for the adhesive to remain effective throughout device use, product and bond lifetimes must be considered. This is highly variable with adhesive use ranging from a few days, for biological applications such as wound dressings, to hundreds of years, such as with construction. The wide range of applications and service requirements will probably necessitate a variety of approaches to produce debondable adhesives for all possible scenarios.2. Debonding stimuli
For the purpose of this review, adhesive debonding will be considered to occur via one of two basic mechanisms, shown in Scheme 1. The first is cohesive failure, where there is loss of mechanical strength in the bulk adhesive for example, on depolymerisation or melting. The second is adhesive failure, where the interactions between the adhesive and substrate are influenced, for example on formation of gas bubbles at the interface. Fractures in the substrate are not relevant to this review and will not be discussed.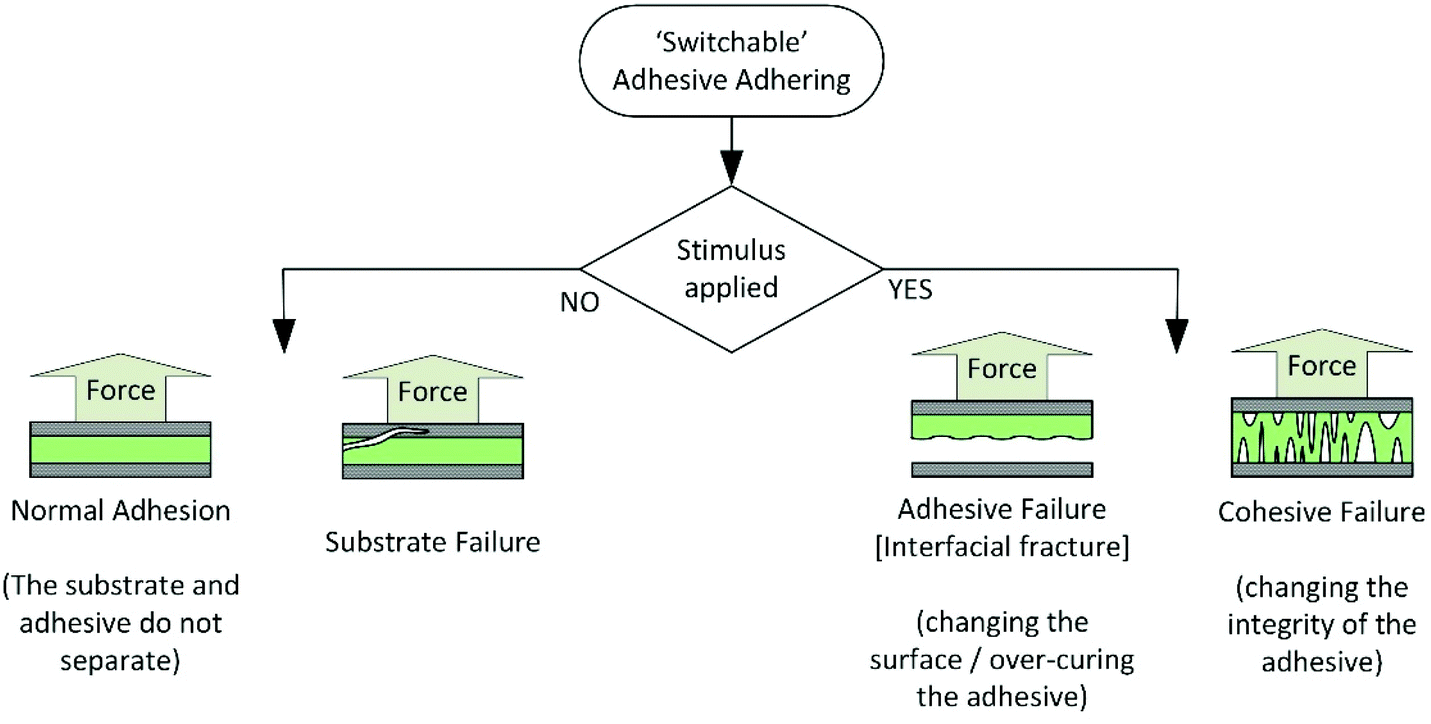 | ||
| Scheme 1 Alternative methods of debonding adhesives. | ||
The choice of stimulus for debonding is a key design consideration, as it constrains the potential applications and lifetime of the final adhesive. Stimuli may be physical or chemical, but must allow on-demand debonding, within a commercially viable timeframe (typically 1 to 100 minutes), without damaging the adhered substrates. In addition, it is ideal for stimuli to be highly specific, to reduce the likelihood of exposure during product lifetime, where unwanted debonding could lead to catastrophic failures. Preferred debonding stimuli are those not ordinarily experienced during adhesive service, such as ultrasound or microwaves.
Aside from mechanical separating joints, currently ultraviolet (UV) radiation and elevated temperatures are the most utilised debonding stimuli. However, these face many limitations with respect to the accessibility of bonded regions and high probability of unintended exposure. Other debonding mechanisms utilise electrical, magnetic, ultrasonic, or chemical stimuli, which each pose their own challenges. This review will cover each debonding stimulus in turn, presenting the current chemistries utilised for debondable adhesion. The debonding process is shown schematically in Scheme 2.
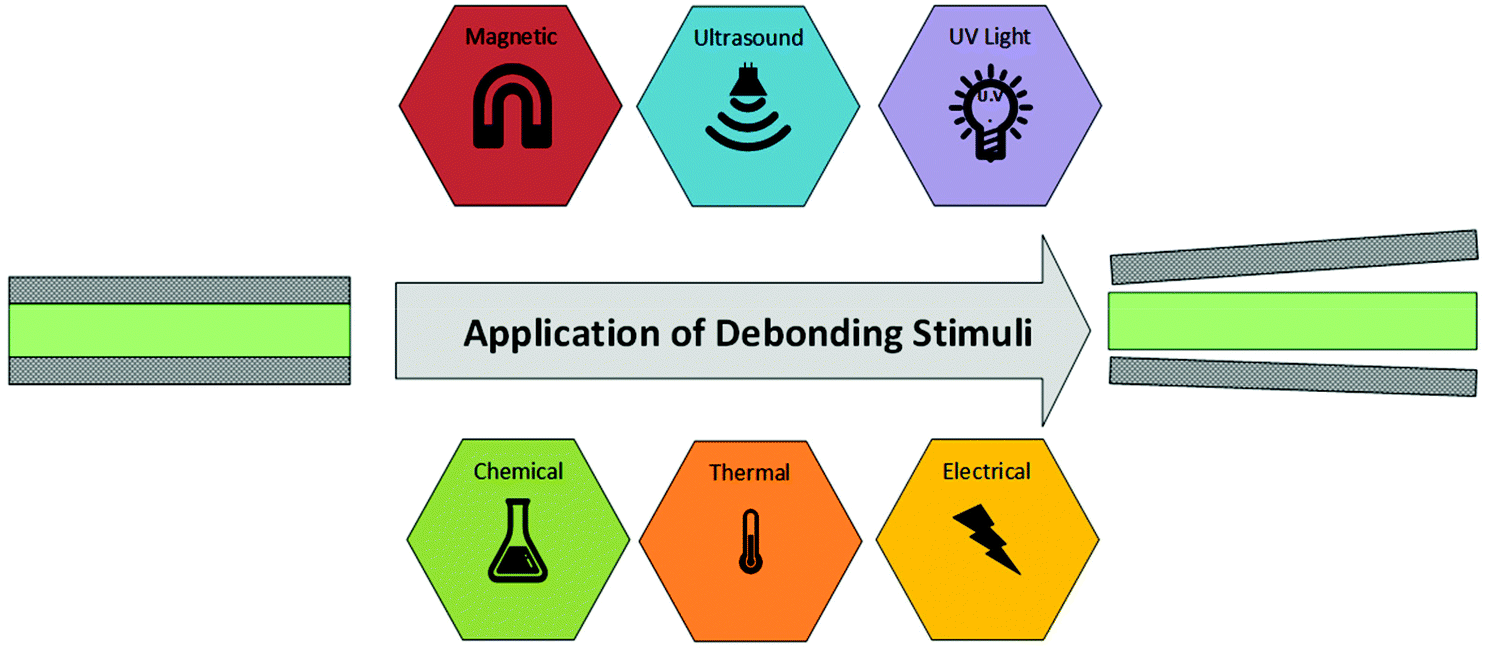 | ||
| Scheme 2 Schematic representation of the debonding process using different stimuli. | ||
2.1. Photo-debondable adhesives
One method that has been widely researched in recent years is the incorporation of photo responsive moieties into traditional commercial adhesives. This has been extensively reviewed in a recent publication by Hohl and Weder.6 Most developments in this area fall into two distinct categories: photoinduced overcuring and photodegradation, although other strategies utilising isomerisation and dimerisation have also been investigated.6,7 These mechanisms allow for rapid on-demand debonding; however, substrates must be transparent at the appropriate wavelengths. Use is also limited to joints where little or no exposure to the debonding wavelength will occur during product lifetime.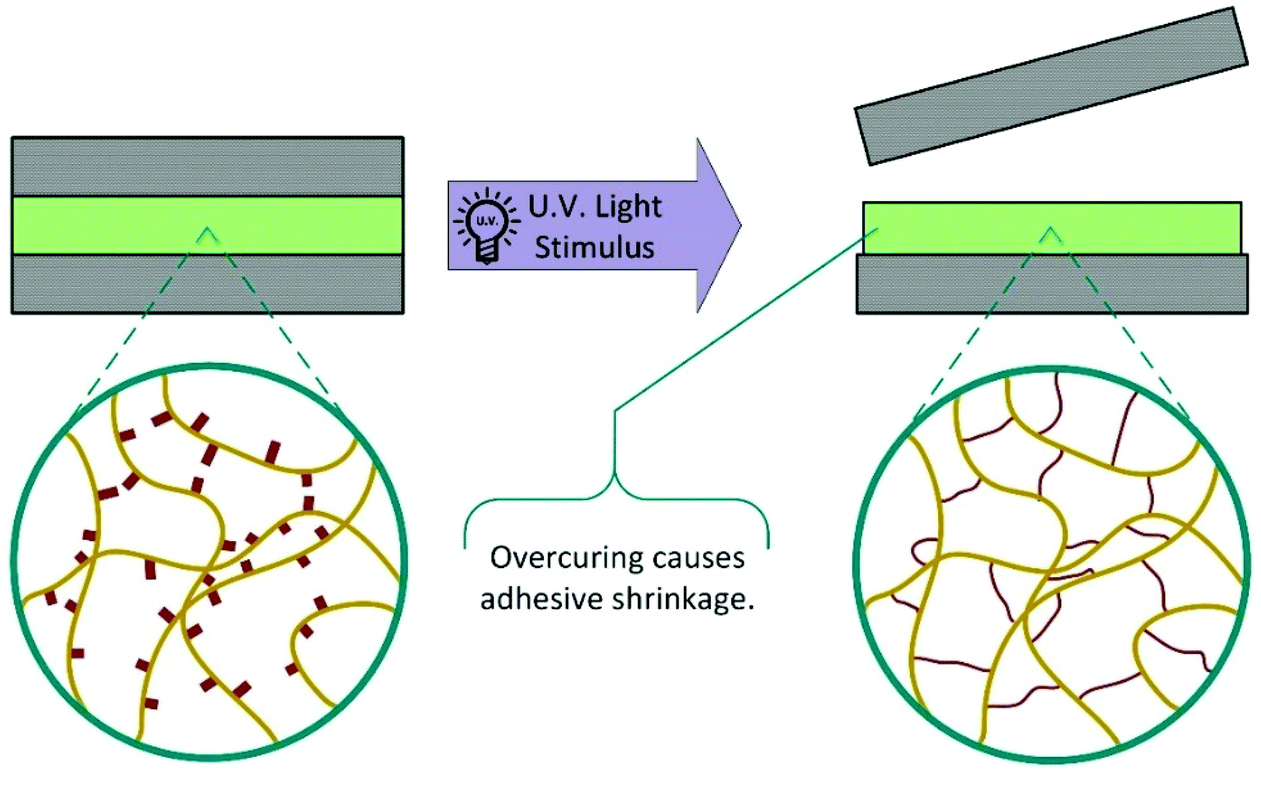 | ||
| Scheme 3 Schematic representation of photoinduced overcuring causing adhesive failure. | ||
In 1981, the 3M company developed UV debondable adhesives, utilising the ring opening polymerisation of epoxies.8 Oxirane ring-containing monomers were copolymerised into conventional pressure sensitive adhesives with overcuring causing a reduction in adhesive strength. Since then, other strategies have been developed using unsaturated multifunctional monomers or oligomers,9–13 with functional groups including acrylates,10,11,14–20 itaconic acid,21 alcohols,16 thiols,22,23 aziridines24 and combinations thereof.6 The various cross-linkable entities may be incorporated into the adhesive by chemical integration (Scheme 4), copolymerisation, or physical blending and additional cross-linking agents may be added to increase efficiency.
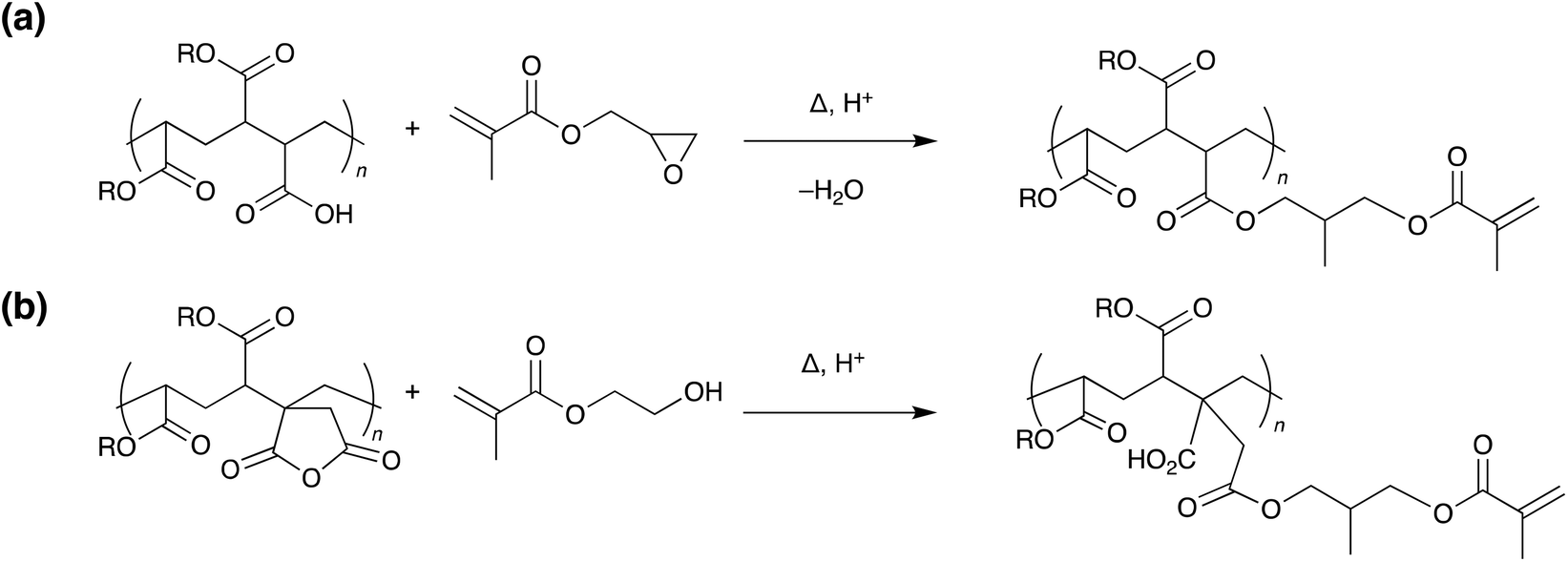 | ||
| Scheme 4 Representative examples of side chain functionalisation of acrylic copolymers with methyacrylate residues.9,15 | ||
There are few restrictions in the choice of photo initiator, and recent work has shown the use of a wide-range of molecules including 1-hydroxycyclohexyl phenyl ketone,6 triaryl sulfonium salts,8 acyl phosphine oxide,22 benzophenone,23 C60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25,26 and other singlet oxygen generators such as Rose Bengal.27 Adding the curable monomer and photo initiator by stirring them into a commercial adhesive reduces the synthetic steps required, however, free photo initiators may cause issues during the adhesive lifetime including bleeding, degradation of gloss or skin irritation, when used in a biological setting. Copolymerisation of the photo initiator into the polymer backbone, may reduce these issues and improve both adhesive lifetime and efficiency of debonding.28 These strategies are accessible to a wide range of adhesives including acrylates, epoxies, polyesters, and vinyl block copolymers allowing a wide range of adhesive properties. In addition, the debonding efficiency and level of residue can be tuned by controlling the ratios of cross-linker to photo initiator. These adhesives share the limitations of all UV debonding classes, with the requirement for transparent substrates, however, there is also an issue with the incorporation of photo initiators: these may affect the mechanical properties of the adhesive and cause unwanted debonding or side reactions during the product lifetime.
25,26 and other singlet oxygen generators such as Rose Bengal.27 Adding the curable monomer and photo initiator by stirring them into a commercial adhesive reduces the synthetic steps required, however, free photo initiators may cause issues during the adhesive lifetime including bleeding, degradation of gloss or skin irritation, when used in a biological setting. Copolymerisation of the photo initiator into the polymer backbone, may reduce these issues and improve both adhesive lifetime and efficiency of debonding.28 These strategies are accessible to a wide range of adhesives including acrylates, epoxies, polyesters, and vinyl block copolymers allowing a wide range of adhesive properties. In addition, the debonding efficiency and level of residue can be tuned by controlling the ratios of cross-linker to photo initiator. These adhesives share the limitations of all UV debonding classes, with the requirement for transparent substrates, however, there is also an issue with the incorporation of photo initiators: these may affect the mechanical properties of the adhesive and cause unwanted debonding or side reactions during the product lifetime.
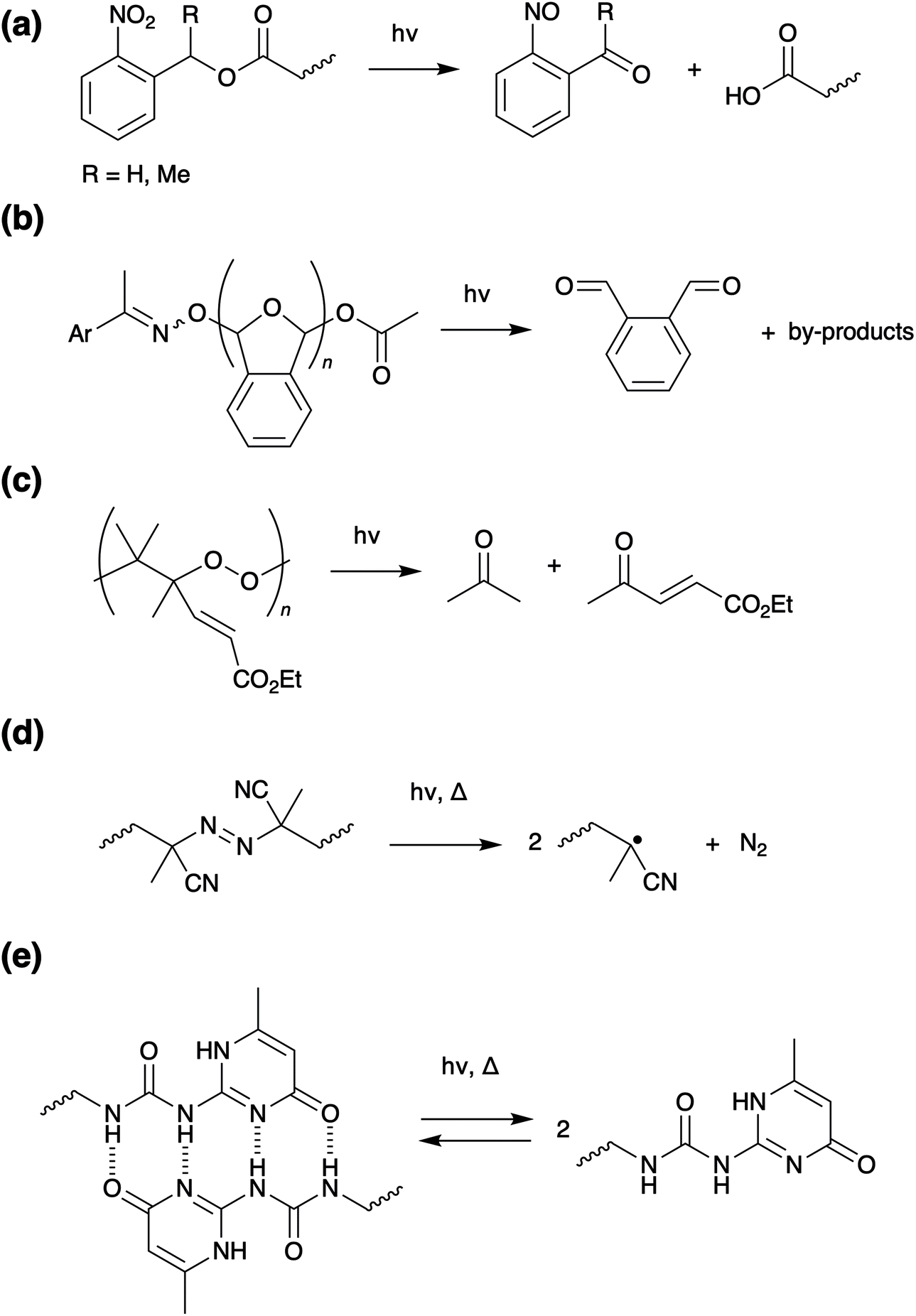 | ||
| Scheme 5 Photo-induced dissociation of (a) ortho-nitro-benzyl-esters, (b) aromatic acyl oximes, (c) poly peroxides (d) azo-compounds and (e) a 2-ureido 4-pyrimidinone (UPy) hydrogen bonding interaction. | ||
Applying the same strategy to supramolecular polymers, UV can be used to induce degradation of non-covalent cross-linkages. The 2-ureido 4-pyrimidinone (UPy) hydrogen bonding motif, can be incorporated into a wide range of backbones, including polyacrylates,46–48 polyacrylamides,49 polyvinyls,50 and polyureas,51 to form polymers with excellent adhesive properties.52 On application of UV irradiation, reversible dissociation of the hydrogen bonds occurs (Scheme 5e), converting the adhesive into a low viscosity liquid and allowing debonding on demand within minutes.
Although the individual cross-linkers are weaker, hydrogen bond networks are more stable than the covalent systems and will not degrade under ambient conditions. Most networks fail to absorb sufficient light for photodissociation and require UV sensitisers, which can compromise adhesive properties. Likewise, at ambient temperatures the adhesive is stable, but with temperature elevation thermal decomposition is observed.
For stronger supramolecular interactions, metal ligand motifs have been utilised for UV induced photodissociation and photochemical liquefaction. Weng and co-workers developed a self-healing gel utilising pyridine-zinc complexation, which had good adhesion to metal surfaces.53 Also using zinc-pyridine complexation, Heinzmann et al. reported a poly(ethylene-co-butylene) based adhesive where the metal–ligand motif absorbed UV irradiation, dissociating without the need for a photosensitiser; decomplexation occurred within 80 seconds due to photo liquefaction.50 Gao et al. developed a poly(acrylic acid) based adhesive with FeIII–carboxyl coordination complexes as cross linkers.54 Application of UV light reduced FeIII to FeII triggering dissociation and gel–sol transition allowing debonding on demand. Histidine-NiII and catechol-FeIII complexes have also been utilised for debondable adhesives; however, these require additional photoacid generators and photo initiators, respectively.6,55,56
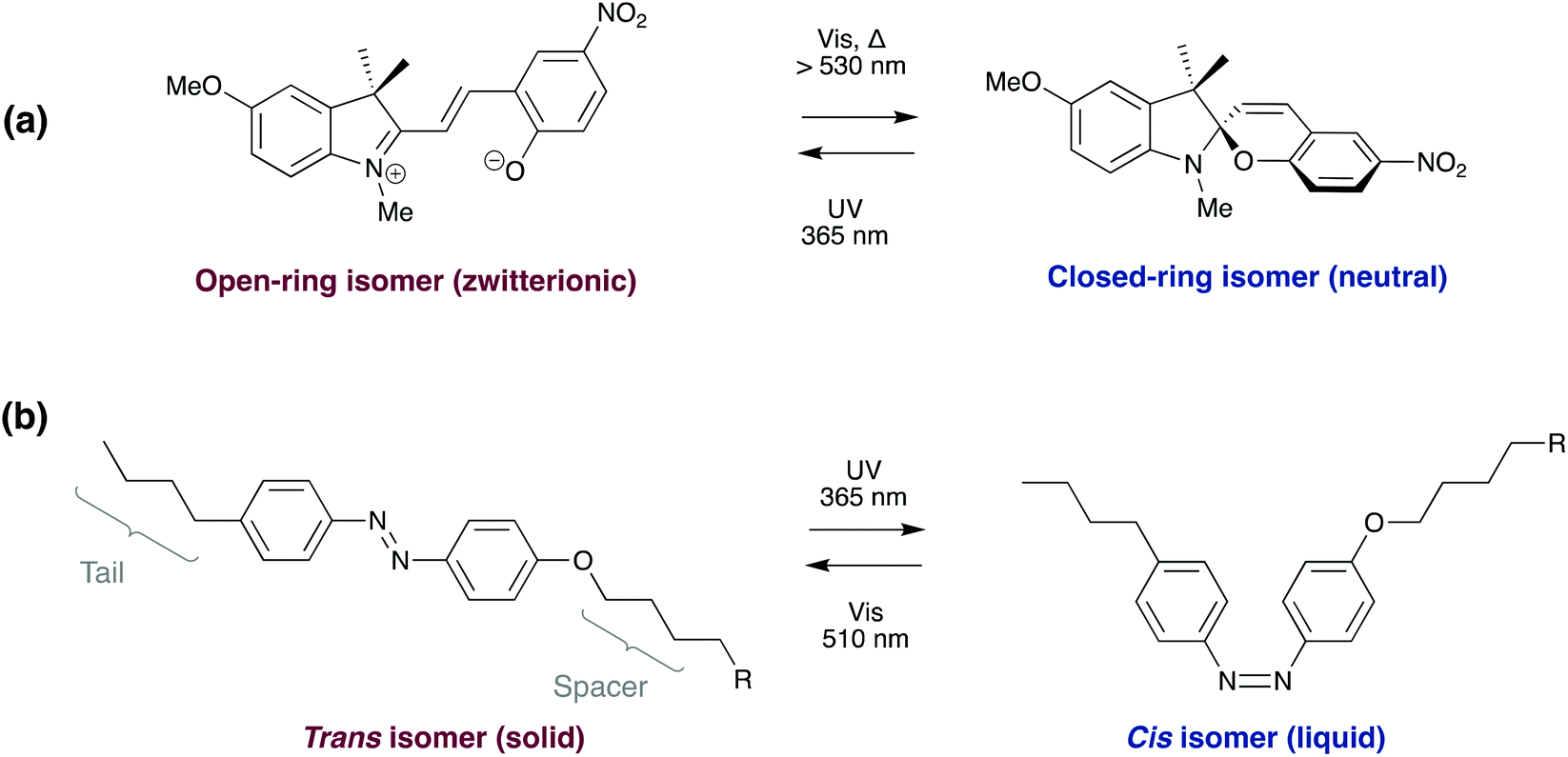 | ||
| Scheme 6 Photo-induced isomerisation of (a) spiropyran and (b) azobenzene derivatives, utilised in reversibly bonding adhesives. | ||
Photoinduced [4πs + 4πs] cycloaddition reaction of polymers functionalised with terminal anthracene groups can result in cross-linking of individual polymer chains (bonding). The reverse reaction (leading to debonding) can be triggered either via thermal dissociation of the dimers,87,88 physically grinding the polymer sample, or alternatively by an additional light treatment with a wavelength less than 300 nm (Scheme 7a). The latter photo-reversibility method has been recently demonstrated by Schlögl and co-workers in hydrogenated carboxylated nitrile butadiene rubbers,87 and by Zhang and co-workers in epoxy resins.89 Saito and co-workers recently reported a π-stacked liquid crystal carbon framework with rigid anthracene units fused with a flexible cyclooctatetraene ring, which upon UV irradiation undergoes photodimerisation, causing the columnar liquid crystal to melt. Heating the melted mixture at 160 °C induces a thermal back reaction of the photodimer into the monomer, which recovered the columnar liquid crystal phase.90
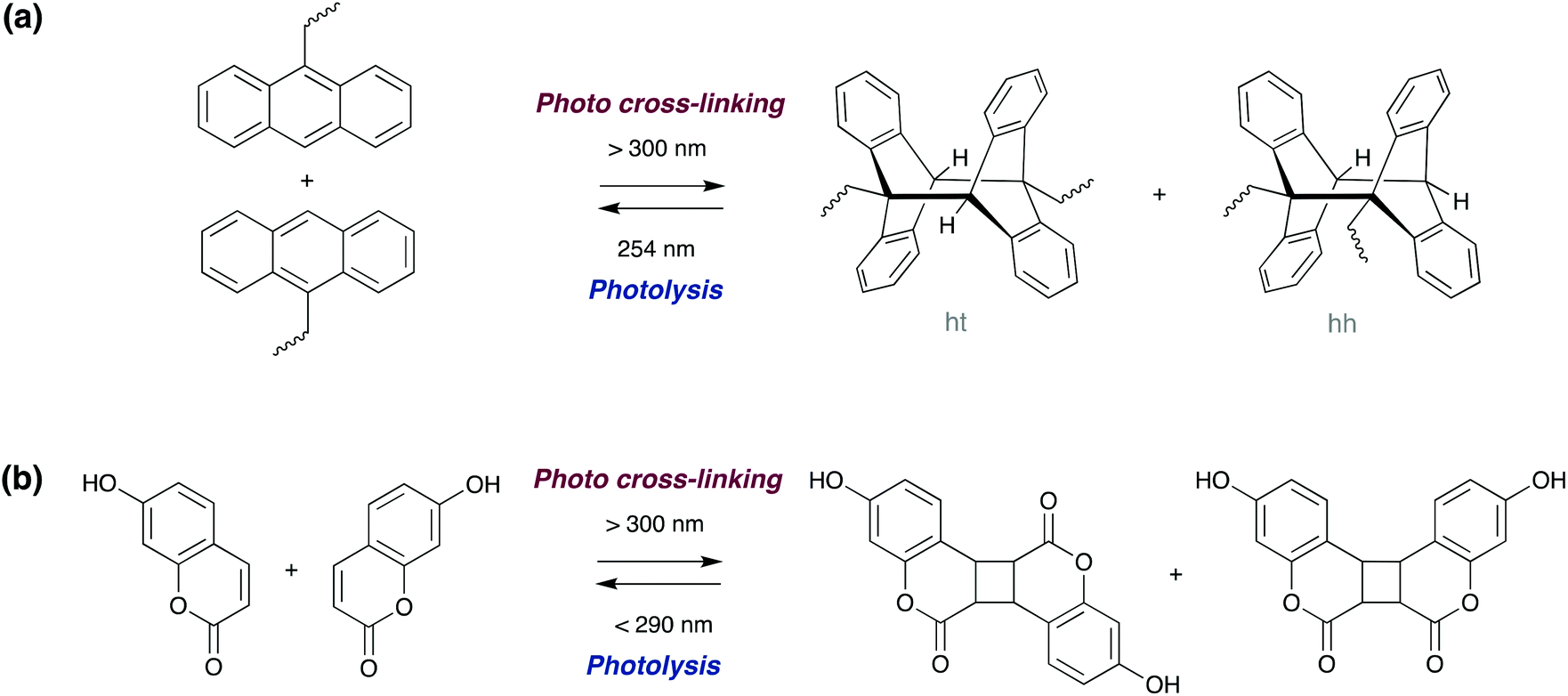 | ||
| Scheme 7 Photoreversible dimerization of (a) anthracene and (b) hydroxy-coumarin derivatives via [4π + 4π] and [2π + 2π] cycloadditions, respectively. | ||
The photoreversible [2π + 2π] dimerisation of 7-hydroxy-coumarin derivatives (Scheme 7b) also allows for reversible cross-linking in polyacrylates91 and polyurethane92 adhesives. Although less studied, other motifs which undergo cycloadditions such as cinnamates,93 maleimides and chitosan, can also be incorporated into photoreversible adhesives.6
2.2. Thermally debondable adhesives
Currently, thermal treatment is the most widely studied mechanism for debondable adhesion. It could be claimed that all polymeric adhesives can be thermally debonded as all go through a melting or decomposition at a readily available activation temperature. Within adhesive technology, hot-melt adhesives utilise the same properties for debonding on demand. Unfortunately, however, many strong structural adhesives are thermosets and decompose at extreme temperatures before melting. For a commercial application, temperatures above 250 °C are undesirable due to expense and damage to substrates, and some caution should be used when considering some of the adhesives described. The main approaches for thermal debonding within a realistic temperature range have included thermally cleavable cross-linkers, thermally activated additives and exploiting specific physical and chemical changes.Thermal debonding allows for the selection of substrates without concern over transparency, however they must be sufficiently conductive and stable up to the debonding temperature. There are also additional limitations associated with specific heating methods such as light-heat conversion or microwaves which limit substrate choice.
One recent strategy involves the thermal degradation of a tert-butoxy-carbonyl group within an acrylate copolymer (Scheme 8a). At 200 °C, the protecting group decomposes, releasing isobutene and carbon dioxide, and causing a loss of polymer molecular weight and interfacial failure of the adhesive.101 Another similar approach involves the deblocking and degradation of blocked isocyanate crosslinkers (Scheme 8b).102,103 These polymers contain weak bonds from the reaction of an isocyanate with a hydrogen active compound such as an acetate, amine or alcohol. Above the glass transition temperature, the blocking equilibrium shifts, regenerating the free isocyanate which decomposes releasing carbon dioxide. Introduction of porosity and loss of mechanical strength allows on-demand debonding.
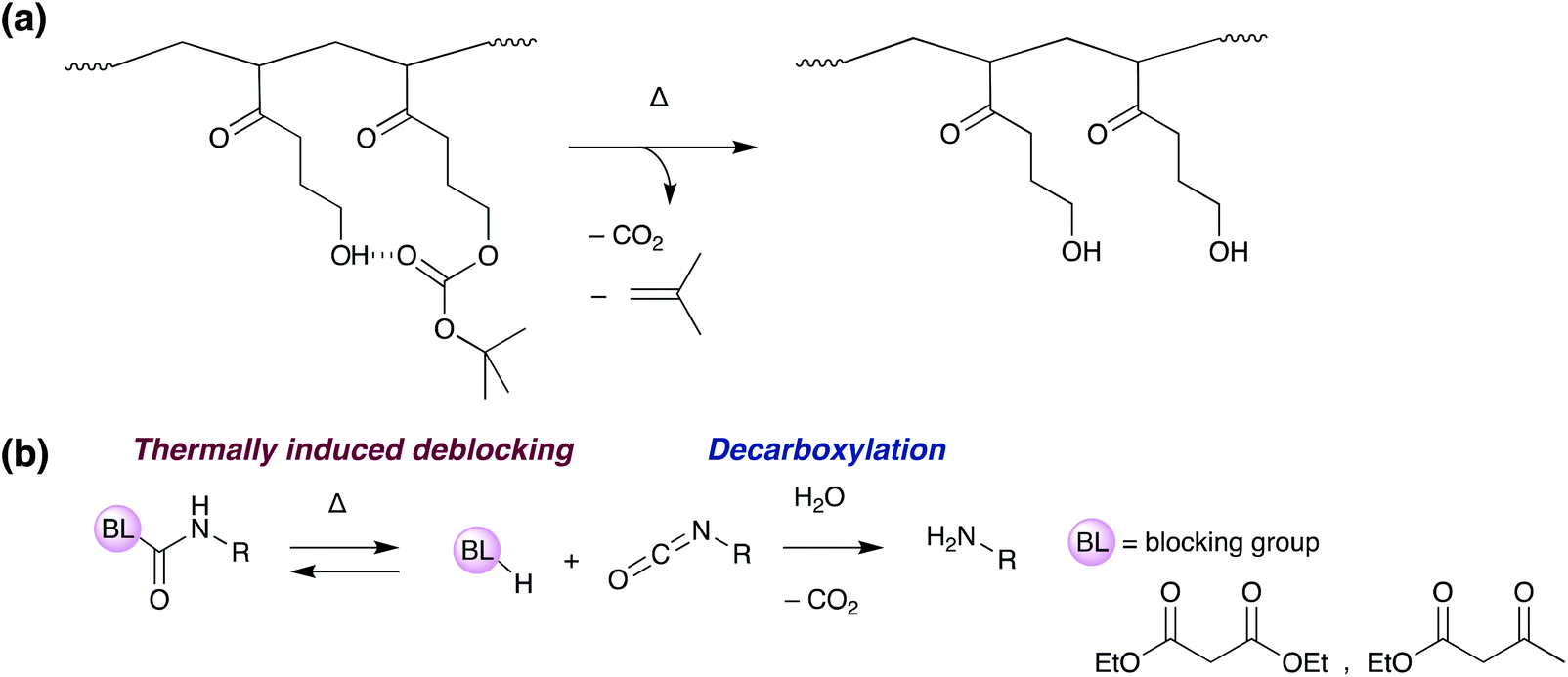 | ||
| Scheme 8 Mechanism of thermally induced “debonding-on-demand” by the use of (a) thermal degradation of a tert-butoxy-carbonyl,101 (b) deblocking and degradation of blocked isocyanate crosslinkers.102,103 | ||
Furthermore, there are several hydrogen bonding motifs which allow thermal debonding, in addition to the UV debondable UPy discussed earlier.46–52 Balkenende et al. combined a 5-aminoisophthalic acid, bipyridines and UPy motifs, to generate a supramolecular glass, where the hydrogen bonds dissembled on heating allowing application as a hot-melt adhesive.52 A similar strategy was used by Ferahian et al. who incorporated isophthalic acid and bipyridines into telechelic poly(ethylene-co-butylene) (PEB)104 and Weder and co-workers who chemically incorporated isophthalic acid into a triglyceride backbone from soybean oil.105 Another approach utilises the reversible interactions between nucleobases.106,107 For example, a polyacrylate adhesive with adenine-thymine cross linkages, had degradation of hydrogen bonds above 60 °C, allowing thermally induced debonding. In yet another method for thermal debonding, the lower critical solution temperatures (LCST) of supramolecular networks were employed.108–112 For poly(N-isopropylacrylamide) or poly(N-vinyl caprolactam) with poly(ethylene glycol) linkages, the extensive hydrogen bond network is responsible for the adhesive properties. When the temperature is increased above the LCST, the polymer is no longer fully miscible and phase separation occurs, causing degradation of the hydrogen bond network. This combined with the subsequent surface hydrophobisation, causes interfacial failure. The strength of the adhesion for this adhesive type is reliant on the precise ratio of oligomer linkages to polymer chain and the LCST temperature can be tuned by adjusting water content and substrate hydrophilicity.
The intrinsic glass and melt transitions of polymers can also be utilised for debonding on demand. Asua and co-workers developed a semi-crystalline polymer adhesive, where heating above the melt transition causes migration of the crystalline domain to the interface.114 With recrystallisation on cooling, the interface becomes hard and non-tacky allowing for easy debonding. Luo et al. used similar chemistry in an epoxy composite with polycaprolactone (PCL).115 When heated above 60 °C, the PCL liquefies and migrates to the interface, acting as a hot-melt adhesive, with high adhesive strength on cooling. On re-heating, the PCL layer at the interface melts, allowing easy debonding. Another related approach was utilised for debonding on cooling. At operating temperature, the polymer was fully amorphous with good adhesive properties, but when cooled below the crystallisation temperature, surface contact between the adhesive and substrate was reduced by the formation of crystalline, non-tacky domains.7,116–118 Each of these strategies, relies on cycles of heating and cooling, so debonding cannot be achieved instantaneously on application of a stimulus. They are also highly constrained by the specific melt and glass transitions of each polymer, and debonding temperatures can be highly variable depending on the chemical composition. Commercially, this technology has been utilised by the Nitta corporation with the Landec Intelimer™, technology for electronic component manufacturing, and with Johnson and Johnson for medical adhesives operable at body temperature.
Direct thermal expansion of the adhesive and substrate can also be utilised.119,120 For example, a laser applied to the interface of a poly(dimethylsiloxane) adhesive allows rapid on-demand debonding for use in silicon chip manufacturing. Another strategy for thermally induced debonding is structural rearrangement.121 Higashihara et al. developed a poly(cyanurate) adhesive, with a low number of amide units in the main chain. On exposure to high air temperatures (260 °C) structural rearrangement to isocyanurates occurred (Scheme 9), accompanied by a reduction in molecular weight and cohesive strength. This allowed thermally induced debonding within an hour.
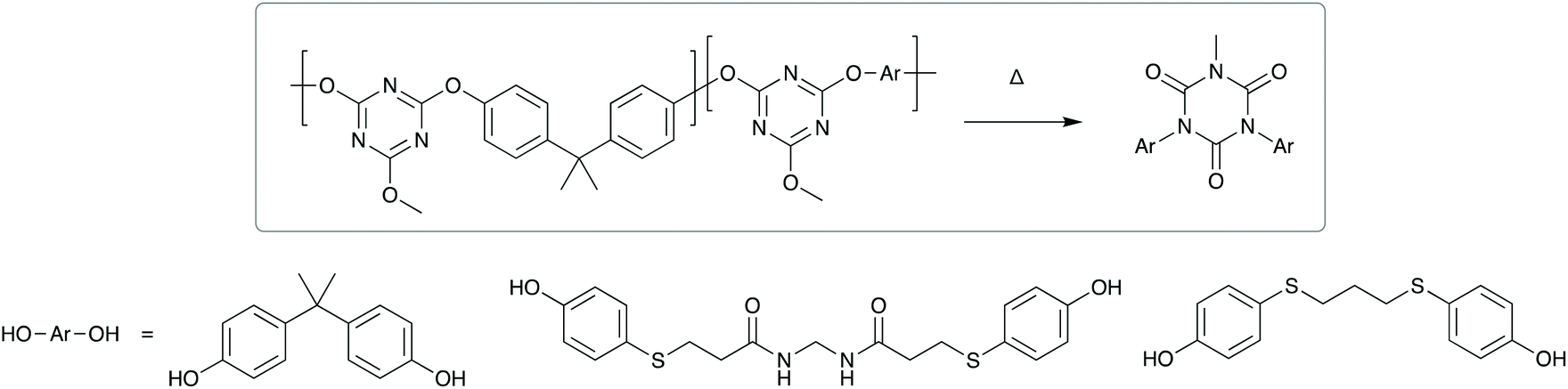 | ||
| Scheme 9 Thermal degradation of random polycyanurates via thermal rearrangement of a 2,4-bis(4-aryloxy)-6-methoxy-1,3,5-triazine unit. | ||
Most of the current technology involves incorporation of thermally responsive functional groups or copolymers into conventional adhesives. However, recent studies have suggested a novel approach using sublimable molecular solids could be applicable. Mirica and co-workers have developed polycrystalline films of molecular solids where debonding occurs due to sublimation, without the need for solvent or mechanical force.122,123 Various compounds were trialled including several poly aromatic hydrocarbons, iodine hexachlorobenzene, octa-cyclic sulfur, (+)-camphor, dimethyl sulfone, (−)-menthol and (S)-ibuprofen. Rapid bonding to glass, metal and plastic was achieved via melt adhesion, with maximum cohesive strength of 2.1 MPa. Controlled debonding on demand was possible in closed systems without exposure to the external environment, allowing capture of the toxic gas produced. Lack of control over crystallinity and polymorphism and the requirement for melt-bonding currently present challenges; however, tuning of molecular and intermolecular interactions, and crystal optimisation, could help overcome these challenges.
Thermally expandable additives (microparticles). Another approach for thermal debonding with additives, is the incorporation of expandable microparticles into commercial adhesive formulations.7,124–132 A wide range of chemical or physical foaming agents can be incorporated into polymeric shells, such that elevated temperatures triggers considerable expansion of the microcapsule. This generates internal stress with propagation of cracks through the adhesive, leading to a loss of modulus and consequently, cohesive failure, (Scheme 10). This effect is exacerbated by the softening of the matrix and change in the interface at higher temperatures. Various methods of thermal treatment can be used including microwave radiation and light-heat conversion, and nanoparticles such as ZnO or graphite can be used to accelerate debonding. The physical properties of the adhesive are dependent on the polymer while the debonding time is reliant on the type and concentration of expandable particles. Unfortunately, this strategy is not as simple as matching high performance commercial adhesives with thermally expandable capsules, as there can be compatibility issues and side reactions between adhesives and additives. Another major issue with incorporating these microparticles into commercial adhesives is the associated reduction in maximum adhesive strength, which decreases with microparticle concentration. In addition, the presence of these additives causes the adhesive to become opaque, limiting the range of useful applications. Furthermore, microcapsule expansion is irreversible, limiting the adhesive to one bonding–debonding application.
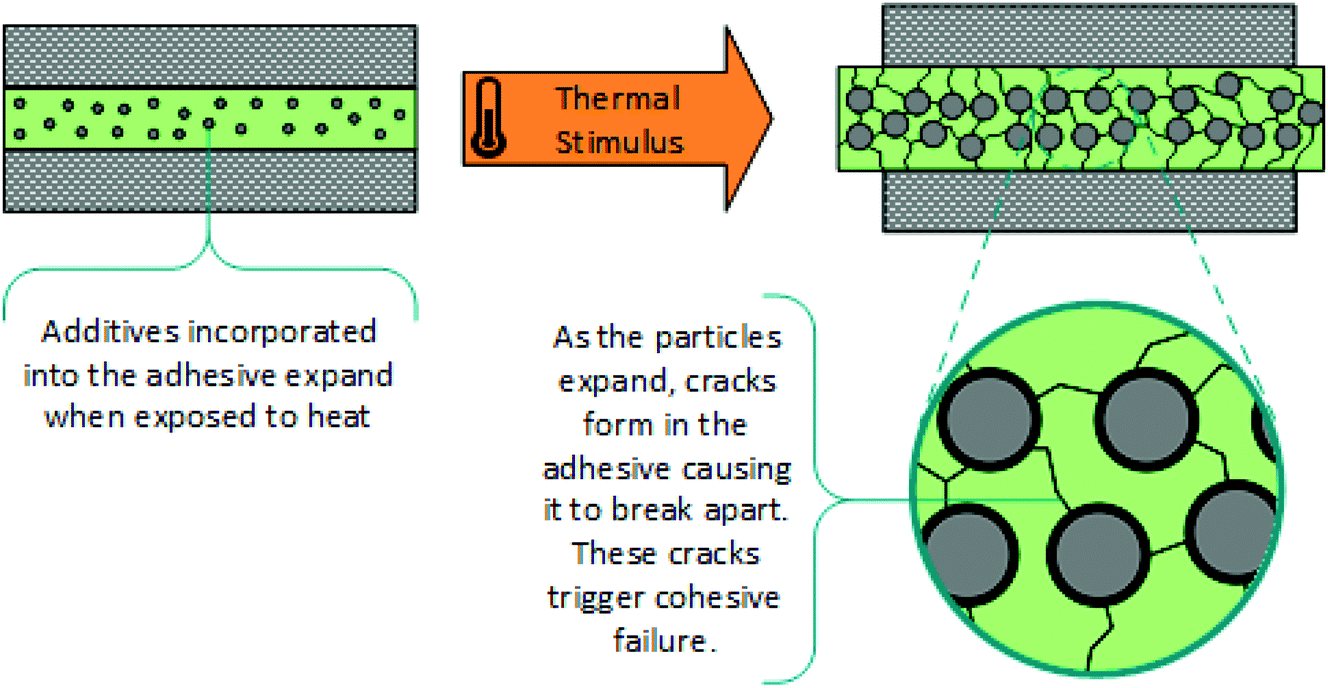 | ||
| Scheme 10 Schematic representation showing thermal debonding, via cohesive failure, due to the expansion of additive microparticles.124 | ||
Evaporable and decomposable additives. In an alternative method, organic additives are utilised which evaporate or decompose on application of thermal stimulus, as shown in Scheme 11.124,125,133–136 The gas produced migrates to the interfaces, causing volume expansion of the polymer matrix, and in some cases foaming.
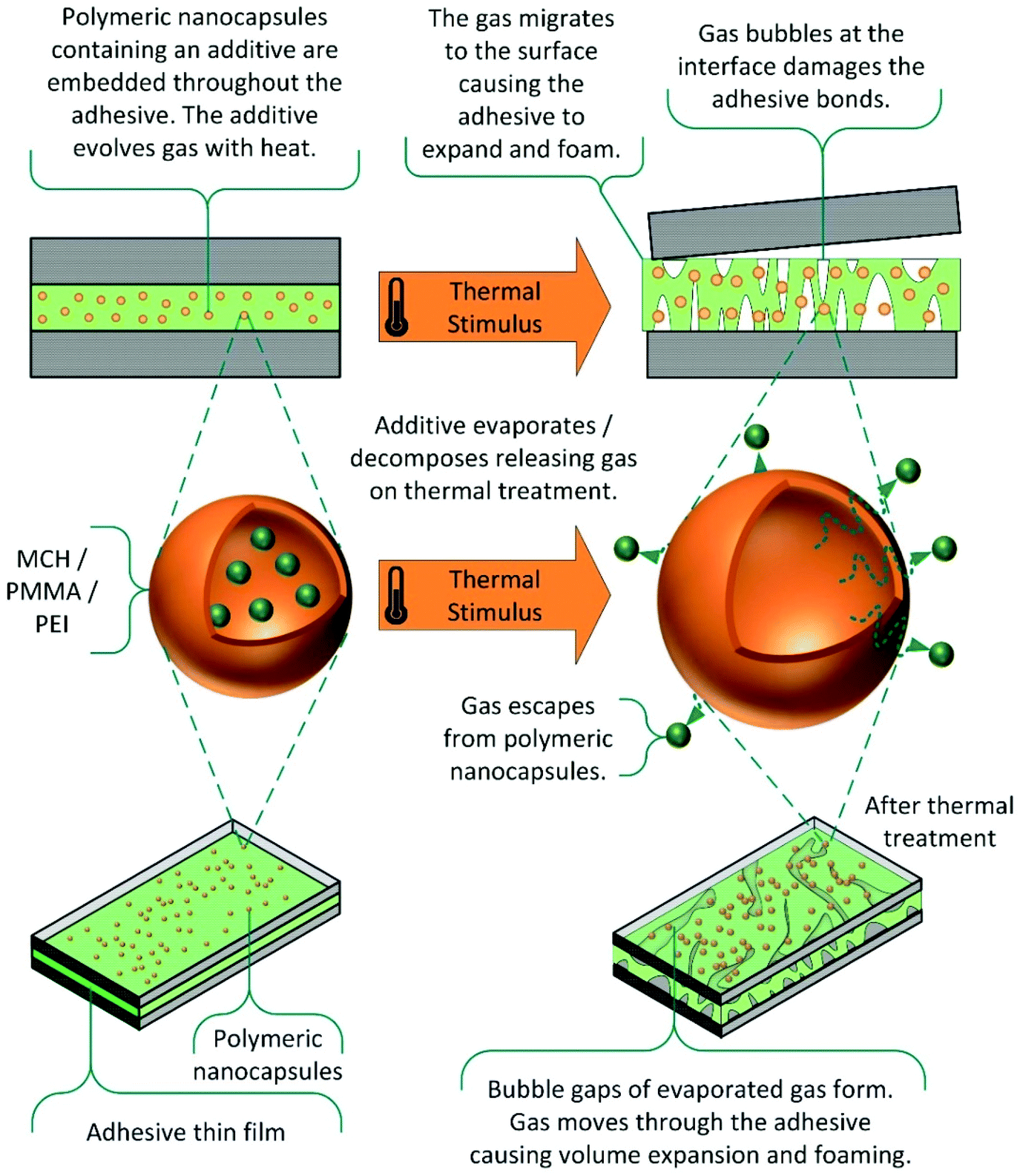 | ||
| Scheme 11 Thermally induced debonding via evaporable and decomposable additives.124 | ||
The debonding mechanism is not purely cohesive, as the presence of gas bubbles at the interface facilitates adhesive failure, by disrupting interactions with the substrate. This technology can be applied to a wide range of commercial adhesives, such as epoxies, urethanes, acrylates and silicone matrixes, and numerous additives, including carboxylic acids, azodicarbonamides and nano-capsules of methylcyclohexane. Debonding on demand is generally achieved within 10 minutes, using microwave radiation or high temperatures. As these additives are generally on the nanoscale, the effect on mechanical strength is considerably reduced, relative to microparticle alternatives. Commercially, this debonding technology is available in the form of the INDAR technology patented by Rescoll, and has applications in the aerospace, aeronautics, and automotive industries.7,134,135 More recently, a patent by the Nike company has also utilised this technology, applying it to the removal of trainer soles for recycling.136
Shape memory additives. In another approach, shape memory additives (SMA) are utilised. These are deformed into flat geometries and incorporated into commercial adhesives, as polymer layers or randomly distributed fibres.137–142 On application of thermal stimulus, the SMA releases the internal strain from deformation and returns to its ‘remembered’ geometry. Transition of the SMA from flat to curved or corrugated, causes fractures in the adhesive matrix allowing debonding. Generally, using shape memory materials, such as Nitinol, is the better approach, as these can actively improve the mechanical performance of the adhesive instead of compromising modulus and strength, as observed for copolymer layers.140 This mechanism for debonding is shown in Scheme 12.
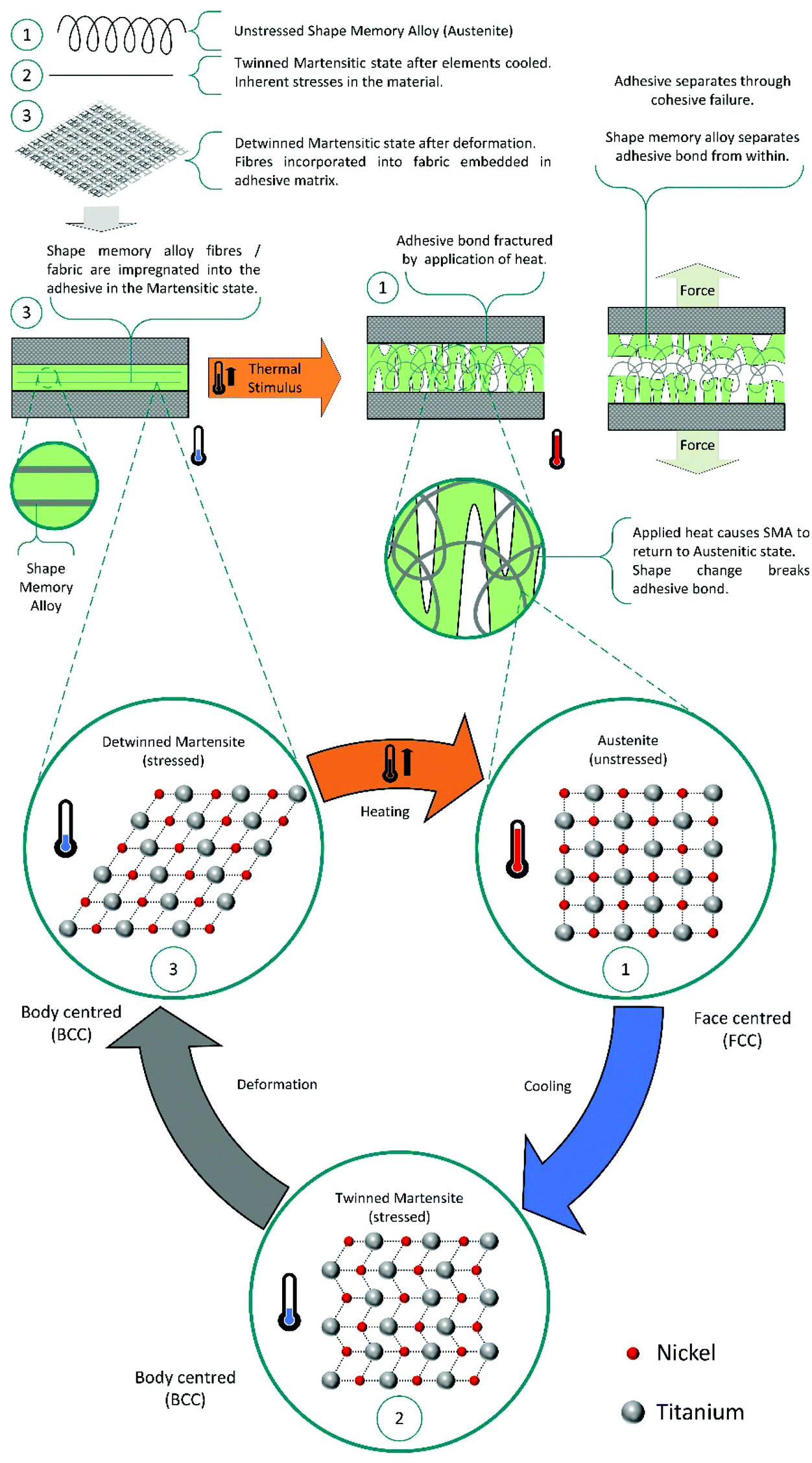 | ||
| Scheme 12 Schematic representation of the debonding mechanism using shape memory additives. | ||
Other additives. Throughout the literature, other additive properties have also been exploited to induce thermal debonding, with varying levels of success. One such example, is the use of amphiphilic or crystalline nanoparticles, which, when heated above the glass transition temperature, migrate to the interface and coalesce.143–146 The sintering of these nanoparticles causes an irreversible change in the network and interface, changing the modulus and tackiness of the adhesive. Debonding can be achieved within 30 seconds using intense infrared radiation, or 30 minutes with a convection oven. In an entirely different strategy, an epoxy resin was loaded with imidazolium ionic liquid, dramatically increasing the amount of microwave radiation absorbed, and facilitating thermal degradation of the adhesive.7 Another approach utilised the LCST of microgel beads, forming an adhesive where a small reduction in strength was observed after 12 hours immersion in solution.147 Harris and co-workers incorporated cellulose nanocrystals into an epoxy thermoset, increasing the shear strength by 30% and reducing the thermal degradation temperature to 220 °C.148
Thermal stimuli: Diels–Alder chemistry. In addition to the photo-controlled versions discussed previously, cycloaddition reactions can also be utilised for thermally induced debonding on demand. The most common of these is the Diels–Alder [4 + 2] reaction, as shown in Scheme 13a, where retro-cycloaddition is induced at elevated temperatures (approx. 80–90 °C), causing significant decrease in modulus.7 Diels–Alder moieties can be incorporated into a wide range of polymers including acrylates,149,150 epoxies,149–153 urethanes,149,150 vinyls154,155 and polycarbonates,156 where choice of cross-linkers and binders allows tuning of adhesive properties. In general, good adhesion, mechanical properties and solvent resistance are achieved, however, the compatibility of the diene/dieneophile with the polymer backbone must be considered to avoid unwanted side reactions and degradation. There is also considerable potential for manipulation of the Diels–Alder system. Branda and co-workers developed a linear diarylethene adhesive, where a photoinduced ring-closing reaction removes the cyclohexene created during the [4 + 2] reaction, the system is “locked” into this form and the retro Diels–Alder reaction is prevented. Exposing the system to visible light reverses the photoreaction, regenerates the ring-open isomer and “unlocks” the system to the retro Diels–Alder reaction, and the adhesive could then be easily debonded using thermal treatment.154 In another interesting development, Schenzel et al. developed a vinyl adhesive where debonding was self-reporting due to the production of a red di-thioester species on degradation.155 Other stimuli have also been explored for retro-Diels–Alder, including ultrasound152 and hysteresis heating157 – these will be discussed further in later sections.
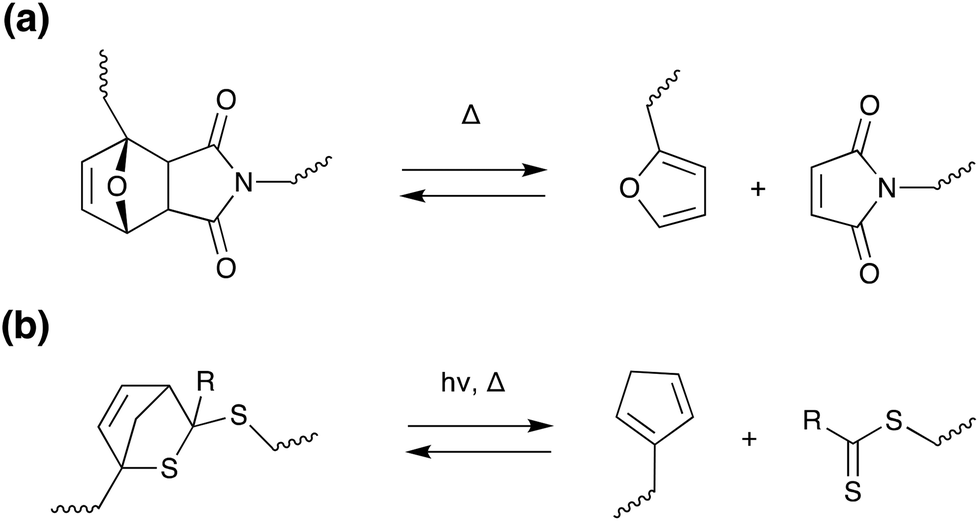 | ||
| Scheme 13 Retro (a) Diels–Alder (b) hetero Diels–Alder [4 + 2] reactions employed in debonding on-demand.155,156,158,159 | ||
Oehlenschlaeger et al. reported a low temperature reversible system utilising a cyanodithioester (CDTE) compound as a dienophile in hetero Diels–Alder reactions with cyclopentadiene (Cp), as shown in Scheme 13b.158 This system does not require a catalyst and shows hysteresis-free repetitive cyclability between 40–120 °C in a very rapid fashion (less than 5 min).158 This CDTE/Cp hetero Diels–Alder pair was subsequently utilised to design and characterize a novel self-healing material.159
These adhesives share the weaknesses of all dynamic covalent networks, with creep deformation and unwanted reverse reactions. Additionally, on continued exposure to high temperatures, over an extended period, side reactions occur leading to a loss of reversibility.
2.3. Magnetically debondable adhesives
Some adhesive formulations, utilise magnetic additives to activate degradation under an oscillating magnetic field.157,160–163 Addition of iron oxide (Fe3O4) nanoparticles to commercial adhesives, allows magnetic stimulation of hysteresis heating, generating local heat. This heat may melt the adhesive, directly causing debonding, or activate other debonding mechanisms such as the degradation of cross-links or activation of blowing agents. The debonding time is highly dependent on the loading of Fe3O4 and can be tuned further by the addition of other nanoparticles such as graphene, which increase melt efficiency. Unlike many other additives, which compromise adhesive properties, the addition of these nanoparticles can increase the strength and modulus of the adhesive formulation, allowing strong bonding to a range of surfaces.162 Other significant advantages of this approach include: its low cost, application to existing adhesives and the low likelihood of unintended exposure to oscillating magnetic fields. However, it is important to note that this debonding stimulus is only appropriate for binding non-ferromagnetic substrates, which could limit its applicability. The magnetic fields required are also quite strong and would require specialist devices.2.4. Electrically debondable adhesives
One niche debonding mechanism, is the application of a low voltage (10–50 V) to cause electrical debonding.164–168 These adhesives contain inorganic or organic salts additives, often in the form of ionic liquids which, when voltage is applied, migrate to an electrode interface affecting the interfacial interaction and enabling debonding (Scheme 14). Bonding of the adhesive to a metal, on at least one side, is essential, and both substrates must be sufficiently conductive to act as electrodes during debonding. To avoid this limitation, for use on non-conducting surfaces, the substrates may be connected via an intermediate consisting of adhesive laminated aluminium sheets. Commercially, this technology is dominated by EIC laboratories, with their ElectRelease™ adhesives. These are primarily amine cured epoxy resins; however, a variety of different adhesive strengths and formulations are available for different applications.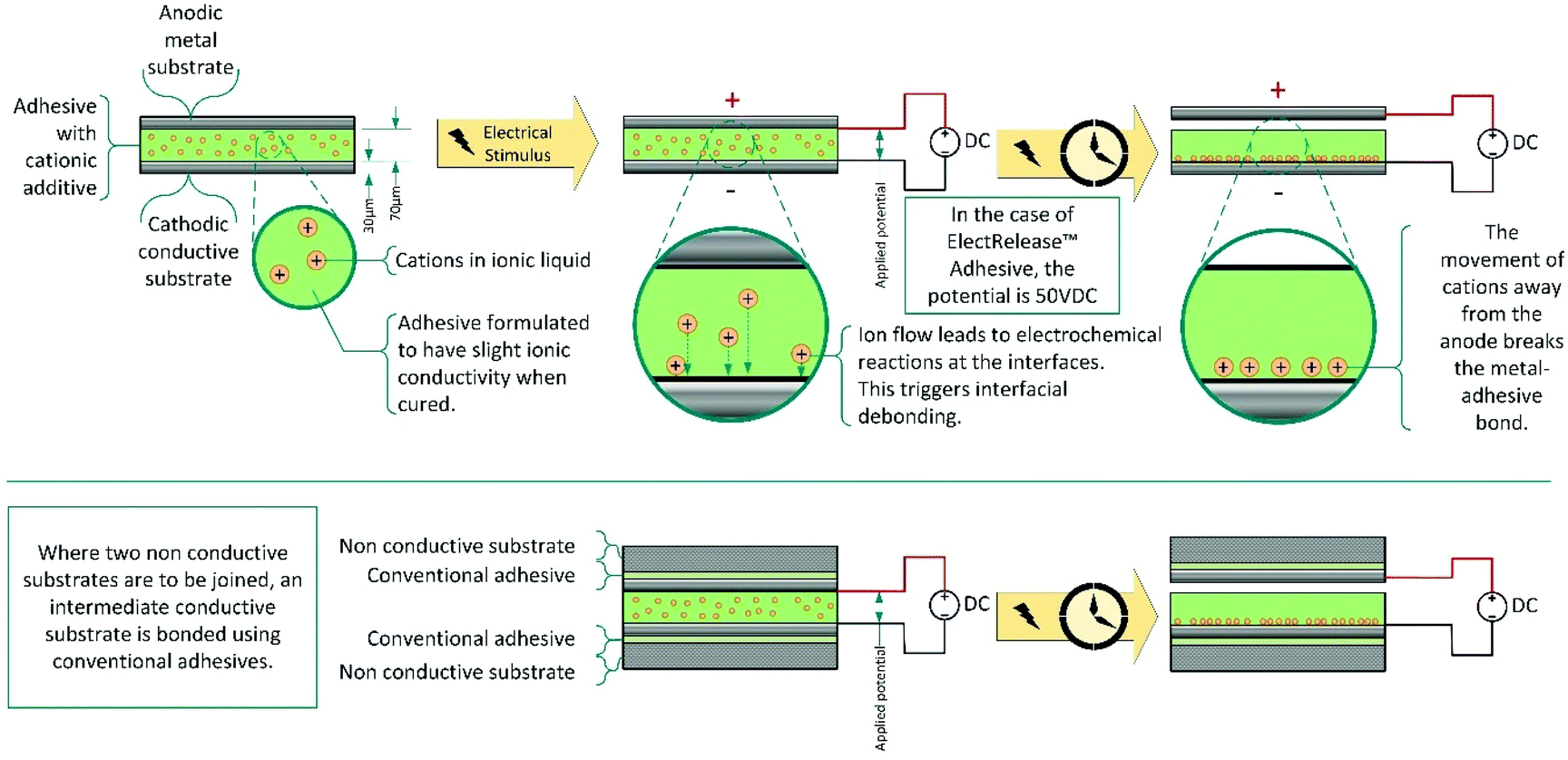 | ||
| Scheme 14 Schematic representation of electrically debondable adhesives. | ||
2.5. Ultrasound debondable adhesives
Ultrasound has been widely used as a non-destructive imaging technique for the analysis of adhesive degradation.169–173 Despite this however, there is some (albeit limited) potential for ultrasound as a stimulus for adhesive debonding. In 2017, Tachi and Suyama reported an acid generating microcapsule, which was activated using 28 kHz ultrasonic irradiation, for 20 minutes at 80 °C.174 When incorporated into a polyurethane, this generated a pressure sensitive adhesive, which underwent debonding on demand via acid catalysed degradation. Ultrasound has also been used for cleavage of Diels–Alder bonds in an epoxy-amine thermoset.152 Zhang and co-workers utilised a range of irradiation times (25–125 min) and ultrasonic power (30–50% intensity: 195–325 W), to produce polymers of different molecular weights and solubilities. Although the thermoset in question was not an adhesive, this technique could potentially be applied to other epoxies. Mechanochemical scission has also been used for other polymers and supramolecular polymers. Craig and co-workers developed perfluorocyclobutane polymers which degraded on demand when subject to pulsed ultrasound (120 min, 30% amplitude 8.7 W cm−2), but were mendable above 150 °C.175 Groote et al. conducted computational analysis on a supramolecular polymer, of poly(tetrahydofuran) backbone with silver-N-heterocyclic carbene cross-linkers, which were cleaved using ultrasound.1762.6. Chemically and fluidically debondable adhesives
Chemical stimuli present an interesting alternative to the aforementioned physical debonding methods. For many polymeric systems, there will be a chemical method to break the polymeric structure or react with a component of a composite. One issue that does need to be addressed in technological devices is that the amount of adhesive used is often minimal and access of a chemical debonding agent of often hindered which may affect the rate at which debonding can occur.Babra et al. have developed a polyurethane adhesive, where debonding on demand is possible using fluoride ions as the chemical stimulus.177,178 On application of a fluoride source, such as tetra-n-butylammonium fluoride, there is rapid degradation of a silyl protected phenol unit, leading to a swift decrease in molecular weight and cohesive failure; while, in the absence of fluoride, the polyurethanes, or copolymers thereof, behave as effective thermoplastic adhesives for metal, wood, plastic, and glass substrates.
Robertson and co-workers reported an acrylic adhesive, with glycerol sebacate oligomers, where debonding was achieved within 20 seconds by spraying with isopropanol.179 This worked by disruption of the intramolecular hydrogen bond network and swelling of the oligo(glycerol sebacate), which reduced the adhesive modulus.
Various approaches have utilised physical stimuli to generate acidic or basic catalysts for adhesive degradation. A simple alternative, however, is the direct application of an acidic or basic solution for chemical debonding. Many adhesives contain backbones or cross-linkages which are solvent sensitive or susceptible to hydrolysis, facilitating efficient breakdown.
One example of this, is the addition of a basic solution to a composite containing a hydrogen bonded block.180,181 Without the cross-linking interactions, between the fatty acids and ureido-diethylene-triamine, heating to 80 °C causes melting, allowing debonding via an adhesive failure mechanism. Malik and Clarson also developed a debonding strategy utilising elevated temperatures and a basic species, however their adhesive degraded via cleavage of sterically hindered urea linkages, rather than disruption of hydrogen bonds.182 Acidic or basic solutions could also be utilised to activate decomposition of additives, allowing degradation via the same mechanism as the thermally activated alternatives. One example of this, is the addition of a basic solution to a composite containing a hydrogen bonded block.7,181 Without the cross-linking interactions, between the fatty acids and ureido-diethylene-triamine, heating to 80 °C causes melting, allowing debonding via an adhesive failure mechanism. Malik and Clarson also developed a debonding strategy utilising elevated temperatures and a basic species, however their adhesive degraded via cleavage of sterically hindered urea linkages, rather than disruption of hydrogen bonds.182 Acidic or basic solutions could also be utilised to activate decomposition of additives, allowing degradation via the same mechanism as the thermally activated alternatives.
Another strategy for chemically induced debonding is through the application of an oxidising or reducing agent. Oguri et al. utilised sodium hypochlorite, for the oxidation of diacyl hydrazine moieties in an epoxy resin.183 Once oxidised, the hydrazine moieties degraded to carboxylic acids and nitrogen gas, with the subsequent drop in polymer molecular weight, allowing debonding on demand. In the absence of a specific oxidant, the epoxy was a strong adhesive for glass and metal, and stable in a wide range of chemical and thermal conditions. Oxidising agents can also be used for degradation of supramolecular interactions. Harada and co-workers utilised high host–guest complementarity, to adhere, polyacrylamide hydrogels with β-cyclodextrin moieties to ferrocene functionalised surfaces.81 On addition of iron trichloride, as an oxidant, there was dissociation of the host–guest complex, due to the low affinity of β-cyclodextrin for cationic ferrocene. This allowed on-demand debonding on application of a chemical agent. Similarly, the high complementarity of cucurbit [7] uril hosts, to amino-methyl ferrocene guests, was used for the ‘underwater-velcro’ developed by Ahn et al.184 Upon oxidation, using a redox reagent, binding affinity was weakened allowing on-demand debonding between the functionalised surfaces. In addition, chemical reagents can also be used in place of UV radiation, to oxidise or reduce metal centres, leading to cleavage of supramolecular cross linkages.54
Metal ions can also be added as chemical debonding agents. For example, injection of a multivalent metal ion solution, into a medical hydrocolloid adhesive, alters viscoelastic properties allowing debonding.7,185 Metal ions also control adhesion between two hydrogels in a complex system developed by Nakamura et al.186 In the absence of a transition metal (MII where M = Mn, Fe, Co, Ni, Cu, Zn), all β-cyclodextrin (βCD) moieties are bonded to 2,2′-bipyridyl (bpy) groups, within the same hydrogel and there is no adhesion to the guest gel. With addition of MII, bpy coordinates to the metal centre, dissociating from βCD and generating free βCD moieties; these form host–guest links with guests on a second hydrogel, generating supramolecular cross-linkages and adhering the two gels together. Varying the choice of bipyridine, metal ion and guest gel, allows access to a wide range of gel properties and differing adhesive interactions.
Many traditional, bio-based adhesives have high water solubility allowing easy debonding. Casein, for example, is widely utilised for attaching labels to glass bottles and jars, where removal using hot water is desirable for glass recycling.4 Another common example is wallpaper paste, which is made from modified starch, cellulose or clay, and allows facile removal of old paper for redecorating. However, due to the likelihood of moisture exposure in product lifetimes, these adhesives have generally been superseded with synthetic alternatives, where better strength and higher water resistance is achieved.
2.7. Reversible adhesion: dynamic covalent networks and vitrimers
One recent development in the field of debondable adhesives, is the use of dynamic covalent bonds and vitrimers. Covalent equilibria generally have slower manipulation, but higher mechanical strength, than their supramolecular equivalents. In the absence of a stimulus, bond exchange is slow, and networks are fixed into rigid structures with strong adhesive bonds. On application of a stimulus, usually heat, there is rapid exchange of the covalent bonds, leading to a drop in modulus and viscosity. This allows the adhesive to flow, acting as a viscoelastic fluid, and allowing debonding and reprocessing. Vitrimers are distinguishable from general dynamic covalent chemistry, in that bond exchange occurs through an associative, rather than dissociative, mechanism, such that the number of cross-links remains constant. Consequently, there is higher solvent resistance and better stability for these networks, and adhesive systems can be optimised to have beneficial properties of both thermosets and thermoplastics.Several recent reviews have considered the development of this new class of materials, and their potential applications in a wide variety of fields.187–191 One of the most common approaches, used for vitrimer based adhesives, is to incorporate motifs for transesterification into an epoxy resin.188,191–194 Choice of hardener, catalyst, epoxy prepolymer, and regulation of epoxy vs. ester motif stoichiometry allows tuning of adhesive and mechanical properties. At ambient temperature, these resins act as elastomers/adhesives with strength around 5 MPa, but elevated temperatures or application of a catalyst, such as [Zn(acac)2], allows debonding on demand. One interesting example is the fully bio-based adhesive developed by Zhang et al. employing ozone treated kraft lignin cured with sebic acid, which provides a lap strength of 6.5 MPa on aluminium sheers, and easy detachment of substrates at elevated temperature via a cohesive failure mode.194 Adhesives utilising trans-esterification are not limited to epoxies. Meyer et al. developed an aromatic thermosetting co-polyester, for use in future space structures, which used vitrimer properties resulting from transesterification.195
Disulfide bond exchange, is also commonly exploited for debonding, with adhesives based on a variety of networks including, epoxy resins and poly(benzoxazines).196–203 These systems can be responsive to a wide range of stimuli, in addition to temperature, and may be debonded on demand utilising: light, heat, pH, nucleophiles or redox reagents, depending on the system. Adhesion is tuneable depending on pre-treatment and the mechanical properties can be controlled by the cross-linking density, number of branches per crosslink and the presence of additives. This was demonstrated by Rowan and co-workers, who added cellulose nanocrystals to a di-sulfide based adhesive, achieving strengths of 23 MPa for binding to metals and up to 50 MPa with other substrates.202 As with ester linkages, natural products can also be utilised in the synthesis of the adhesive network: with Verge and co-workers using cardanol, a non-edible waste product from the cashew industry, in a poly(benzoxazine) adhesive containing disulfide bonds.203 Another natural product used for debondable adhesives is vanillin.204,205 Poly(imines), based on vanillin, have shear strengths up to 6 MPa, and may also display other properties such as antifungal activity or conductivity. Degradation of these adhesives can be achieved rapidly using 0.1 M HCl, or dynamic imine exchange at elevated temperatures. Huang and co-workers reported self-healable adhesive based on poly(1,2,3-triazolium) vitrimers, that has extremely high adhesive strength as high as 23.7 MPa (Scheme 15).
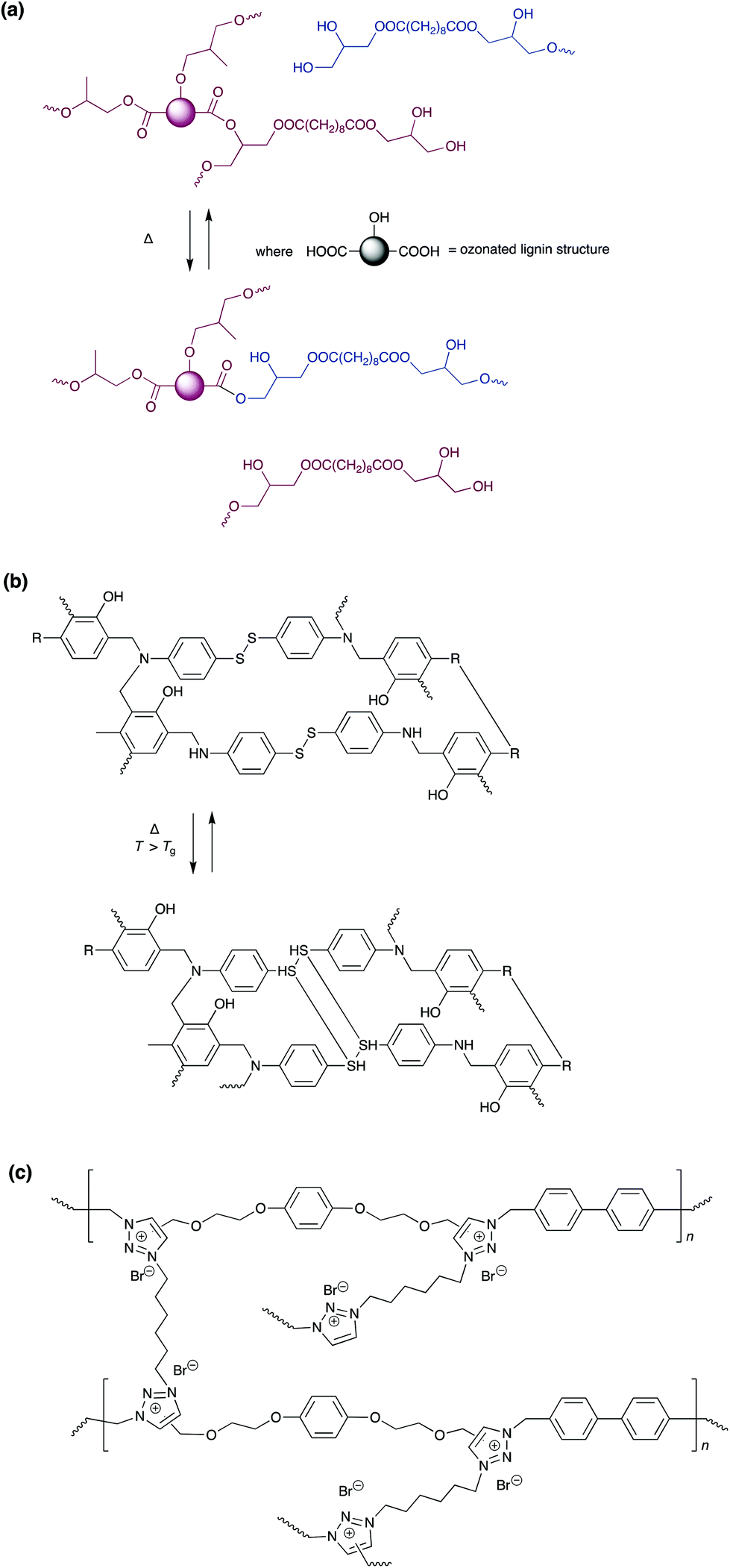 | ||
| Scheme 15 Simplified cross-linked network and mechanism of chemical bond re-arrangement upon heating in (a) lignin-based vitrimer containing β-hydroxylester linkages;194 (b) poly(benzoxazine) vitrimer containing disulfide bonds,203 and (c) poly(1,2,3-triazolium) vitrimers which display reversible adhesion.206 | ||
Although uncommon at present, there are many other lesser investigated dynamic networks with potential for debondable adhesives. These include but are not limited to: carbonate exchange, the thiol-Michael reaction, trans-amination,196,207trans-carbamoylation,208trans-alkylation,209,210 and olefin, dioxaborolane(boronic ester),211,212 or silyl ether metathesis.191
Hayashi et al. developed a polyacrylate elastomer, with unique solubility, which flowed at elevated temperatures due to trans-N-alkylation.209 Wang et al. synthesised a polyurethane, with combined benefits of a thermoplastic and thermoset, utilising thermally reversible oxime–carbamate bonds.208 Yang et al. reported self-healable polyurethanes, exploiting catechol derived boronic esters, for moderate adhesion of 0.5–1.0 MPa.211
There is great potential for the use of biologically derived materials and waste products, for vitrimer and dynamic adhesives. Various natural products including vegetable oils,210,213 rosin,210 and natural rubber,214 and can be utilised, in addition to those mentioned previously.191 This provides an opportunity to step beyond the current commercial adhesives, such as epoxy resins, with their associated sustainability and environmental issues. Unfortunately, some major weaknesses still remain in this area of chemistry. Many vitrimers require catalysts for malleability and these can be unstable, toxic and prone to leaching. Furthermore, due to issues with creep deformation and stress-relaxation, particularly at high pressure, the strength of vitrimer adhesives is limited relative to non-dynamic systems. Nevertheless, this is a new and emerging field which possesses considerable potential for the future of debondable adhesives.
2.8. Other debondable adhesives
Although not currently applicable to recycling, it is worth mentioning an emerging field related to debondable adhesion. Dry adhesives use non-covalent interactions to adhere to surfaces and are predominantly based on systems observed in nature, such as the fibrillar footpads of lizards and geckos. For these adhesives, high surface area is essential for bonding and changing surface topography allows smart adhesion. Topographical changes can be achieved using similar strategies to those mentioned previously including: utilisation of magnetic additives or structures,215–220 UV induced isomerisation,6,221,222 and thermal treatment of shape memory polymers.137,223–225 Presently, this technology is of interest for a range of applications including climbing robots and biosensors, but complex surface pre-treatment and intricate fibrillar structures make this too expensive for general use in recycling.2.9. Sustainability and natural products
All adhesives prior to the 20th century were bio-based and formulated from wood and animal products. These were typically debondable using water, which due to the common nature of this stimulus, posed a high risk of unintended adhesive failure. Recently however, there is a demand for greater sustainability and a movement to return to more bio-based adhesives, where advantages extend beyond renewability. This may involve using naturally derived monomers, or polymer backbones, and has been covered extensively in a recent review by Heinrich.4 One of the most widely researched bio-adhesives is starch, where a wide range of adhesive properties can be accessed via modification. Modification techniques include but are not limited to: acylation, alchoholysis, alkylation, carboxymethylation, cross-linking, dextrinization, oxidation, reduction, phosphorylation, succinylation, acid or base hydrolysis, substitution and enzyme conversions.226–230 Cross-linking is one of the major methods for improving properties, particularly with respect to cohesive strength and water resistance. The traditional cross-linking agents adipate, epicholorhydrin, and phosphorous oxychloride, are highly efficient, but have associated issues in relation to toxicity and environmental hazards.226,230 Considerable research has been done into alternative cross-linking reagents, with lower cost, lower toxicity or less environmental damage, including: sodium trimetaphosphate;226,230 epoxy silanes;231,232 citric acid;233 glutaraldehyde, glyoxal and glycidyl esters;227 and hexmethoxymethylamine.4In addition to starch, many other natural products can be used for adhesives. Gao et al. developed a wood-adhesive utilising whey protein, a waste product from the dairy industry, with isocyanate cross-linkers.234 Berger et al. utilised chitosan, sourced from the shells of crustaceans, to make adhesive hydrogels using a wide range of reversible and irreversible crosslinkers.235 Altuna et al. generated self-healing polymer networks, using epoxidized soybean oil and citric acid solution.236 Zheng et al. reported a defatted soy-flour based adhesive with self-crosslinking by hydrolysation237 and in yet another example Lee et al. used the ring opening polymerisation of ε-decalactone and L-lactide.238
Introducing novel cross-linking in biopolymers, provides a potential opportunity in the field of debondable adhesives. Cross-linking allows for improved mechanical properties, which could then be removed by degradation of the cross-linker, allowing debonding on demand. Alternatively, at the end of product lifetime the adhesive could be degraded by hydrolysis, redox or enzymatic methods, allowing recycling of components. Therefore, starch and other bio-based adhesives, and their composites, could potentially provide the debondable adhesives of the future. The fact that they are already used on a large scale for low strength adhesives in applications such as paper and cardboard shows that supply to the smaller application field of structural adhesives should not unduly affect ethical issues such as competition with food supplies.
3. Process kinetics
A major consideration, for the application of debondable adhesives to recycling, is the debonding timeframe. Ideally, the debonding process should occur in a controlled manner within 30 minutes, to allow for an economically viable production line: however, longer processes may be viable for specific situations. Reports in the literature have not accurately or consistently defined debonding timescales, nevertheless rough ranges and specific examples can be used to compare between the different mechanisms and stimuli. In general, debonding time is limited by the accessibility of the joint and the application of the stimulus to the entire adhesive.For UV induced overcuring, debonding is rapid and usually occurs under 5 minutes. For example, the vinyl block copolymers, with thiol cross-linkers, debond within 3 minutes.22,23 Other methods using UV stimuli typically have slower debonding, in the range of minutes to several hours, depending on the bonds and/or additives in question. With thermal stimuli there is also considerable variation in bonding times from milliseconds, using laser thermal expansion, to several hours for acid degradation of esters. However, most optimised thermal methods allow debonding within an hour.
Rapid debonding is observed for electrical and magnetic stimuli with debonding times, ranging from 30 seconds to several minutes.157,160–168 Ultrasound induced debonding, currently, lacks evidence, but existing methods require irradiation for around 20 minutes.174 For mechanisms, utilising additives, debonding is faster with higher weight percentages, but these can compromise mechanical properties. Consequently, longer debonding times may be preferred so as not to limit adhesive applications.
Chemical debonding is controlled by the rate of diffusion and transport of the chemical agent to the adhesive–substrate interface, in addition to the rate of the debonding reaction. Therefore, debonding times are highly variable within this class. For example, the acrylate developed by Robertson et al. contains perforations, designed to accelerate the transport of isopropanol through the matrix; facilitating a reduction in adhesive strength within 20 seconds.179 While, at the opposite end of the spectrum, debonding using 5% sodium hypochlorite solution, required soaking for 4–30 hours before spontaneous debonding occurred.183 However, due to the ease and low energy requirements of this debonding method, it may be commercially viable to recover high value substrates by soaking for extended periods; particularly if this allows cheaper and/or less toxic debonding agents to be used.
4. Applications of debondable adhesives
Debondable adhesives will be most applicable where adhesive mass is low and product value is high, encouraging recycling. This includes sectors with significant usage of TCMs including aerospace, transport, construction, engineering, manufacturing, mining and renewable energy. However, there is also potential for debondable adhesives in healthcare and the life sciences, where reversible adhesion for dressing and prosthetics would be revolutionary. A variety of debondable adhesives are already commercially available and some are listed in Table 1.| Company | Product | Method of debonding | Notes |
|---|---|---|---|
| Recoll | INDAR® | Additives generate gas on heating | Aerospace, automotive and aeronautics |
| EIC labs Inc. | ElectRelease™ | Electrically debondable | Bond survey equipment to light aircraft |
| Fielco Adhesives | BondAway (two part epoxy-based) and BondAway UV (1 part epoxy) | Heat debondable adhesive systems. Disassembled by immersion in a 95 °C water bath | UV curable and fast setting. Processing or machining of glass, ceramics, or metals |
| Henkel | Loctite® 3214 and Loctite® 3215 | Thermally (200–350 °C) debondable adhesives | Designed for adhesion to glass. UV-cure |
| Henkel | Loctite® 3382 (epoxy) | Water debondable | Bonds silicon to glass and metal |
| Nitto Denko | REVALPHA product range | Thermally debondable: range from 90 to 170 °C | Adhesive tape for electronics manufacturing |
| Nitto Denko | ELEP HOLDER™ | UV-switchable debondable adhesives | Dicing tape for electronic components |
| Adwill | D Series | UV switchable tape | Electronic component manufacturing |
| Nitta corporation | Intelimer™ (semi-crystalline graft copolymers) | Cool-off type (debonds T < 55 °C), warm-off type (debonds T > 60 °C) | Temperature activated adhesives for various uses e.g. electronic components |
| Brewer Science | BrewerBOND® and WaferBOND® | Debonding using lasers, thermal slide or low mechanical force | Electrical component manufacturing and coatings |
| 3M | Various | Heat debondable adhesives. Some require laser debonding | Tape for electronic component manufacture |
| Lumina Adhesives | Adhelight™ | Light mediated (UV or visible) switchable adhesives | Medical dressings |
| Evonik | AdNano® MagSilica and MagSilica® | Magnetically debondable (and curable) | Commercially available additives for addition to the adhesive |
| Stanelco RF technology | Currently in commercialisation | Magnetically debondable | Use in technological devices to aid recycling |
In addition, reversible adhesion would also have considerable impact in the repair market where materials have set lifetimes: for applications such as mobile phone screen replacement, on-demand detachment would be highly beneficial. Repair extends product lifetimes, reducing the need for replacement materials, and is the Greenest approach for dealing with waste material. This is especially important with respect to electronic waste, which is one of the fastest growing waste streams (estimated 53.6 Mt of e-waste was produced globally in 2019) and has a significant environmental impact.239,240 The use of debondable adhesives has already been proposed for e-waste applications and could make a substantial contribution towards repair and recycling, in the future. One key example of this, is the magnetically debondable adhesive currently being commercialised by Stanelco RF Technology.160
A recent review discussed the importance of design for the facile recycling of three key decarbonisation technologies: LiBs, photovoltaic cells and wind turbine blades.241 These three devices, are discussed below, as case studies, illustrating the potential use of debondable adhesives for increasing recycling efficiency, and highlighting some of the issues which may be faced in their application.
5. Case-studies
5.1. Lithium-ion batteries for electric vehicles
At present, LiBs for electric vehicles are not designed with disassembly in mind, and recycling is limited to pyrometallurgy or comminution, followed by hydrometallurgy.1 These techniques are inadequate and lead to low value product streams, or high-cost recovery with low product fractions. Unfortunately, the complex structure of battery packs and the extensive use of adhesives, provides a major barrier to disassembly, preventing more efficient recycling.Typically, battery packs have a hierarchical structure and are composed of modules, which in turn are built up from connected cells.242 For example, in the Nissan Leaf Mk 1 22 kW battery pack, there are 192 pouch cells, divided between 24 modules.1,243,244 Modules are assembled by stacking cells together using an adhesive and adhering them into a metal case. With adhesives in addition to fixings, this complicates disassembly using smart robots making it currently unviable and even highly trained professionals may take several hours to mechanically disassemble a single module. These issues are not restricted to the Nissan Leaf. Tesla battery packs, contain rows of cylindrical cells, adhered to neighbours and cooling plates using a polyurethane adhesive.1,245 Adhesives may also be used for a range of other purposes including sealing cells, modules and/or battery packs; fixing modules into place and absorbing crash energy (Scheme 16).246
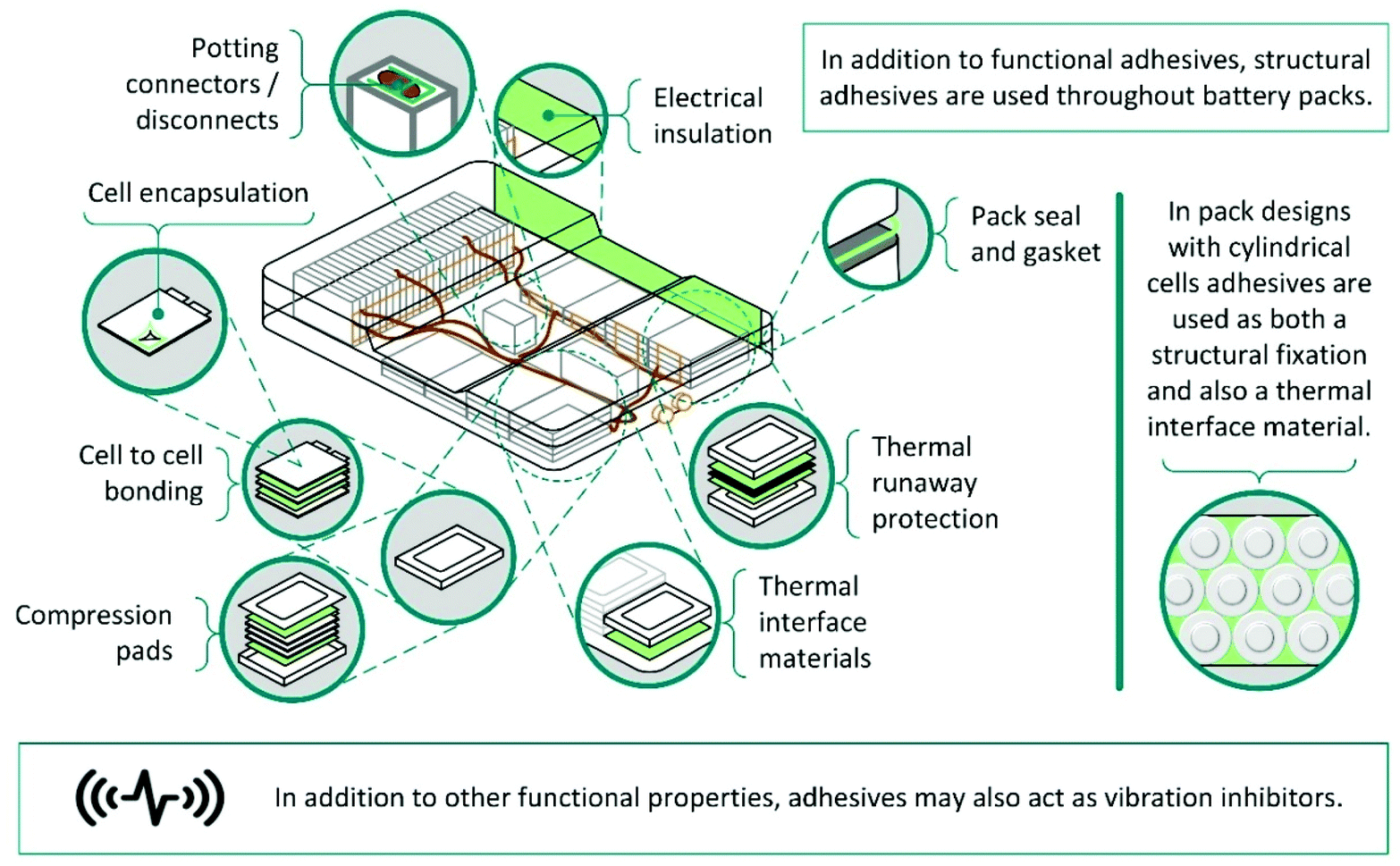 | ||
| Scheme 16 Schematic depiction showing the location of adhesives in a lithium-ion battery. | ||
Unfortunately, there are numerous issues with debondable adhesives to applying to LiBs at pack and module level due mostly to the service conditions and the materials used. UV is an inappropriate stimulus, as the bonded substrates are not transparent at the appropriate wavelengths and the possibility of exposure to UV radiation during product lifetime is high. Within modules, thermally conductive adhesives are generally required to facilitate cooling, rendering thermal debonding highly problematic. In addition, a battery pack that falls apart on exposure to high or low temperatures would be catastrophic during vehicle lifetime, particularly in extreme weather events or during vehicle fires. Use of electrically debonding adhesives would also be highly unsuitable, as the conductive additives would cause short circuits and fires (electrically insulating adhesives are required to prevent this). Magnetic debonding also poses issues, due to the presence of magnetic components within the battery pack. Furthermore, chemical debonding is also limited as many reagents are incompatible with the battery pack. All chemical redox reagents must be avoided for debonding due to risk of metal corrosion and water is too prevalent in the environment to be a safe debonding stimulus. At the battery pack level, this leaves limited debonding options, where issues such as adhesive strength, expense, debonding time and human/environmental toxicity still require solving. It is possible debondable adhesives could have some use in battery packs, however a more sensible strategy would focus on design that minimise adhesive use.1
At the cellular level, however, debondable adhesives are more promising. Recent work has shown that adhesives currently used within cells (for binding the electrode current collectors to the active materials) can be debonded, in milliseconds, using focussed, high-powered ultrasound as the stimulus.247 This process has several significant benefits: ultrasound is not generally experienced during material lifetime, reducing the risk of accidental debonding; recycling metrics are substantially better than for other techniques;248 and while already efficient, there is potential for future improvement with modification of the adhesive (binder) used. Therefore, on a cellular level, debonding of adhesives could be highly beneficial for recycling.
5.2. Photovoltaic devices
The decarbonisation of energy will necessitate a significant production of photovoltaic (PV) devices. While much of this will be crystalline silicon, other technologies such as thin film, hybrid and dye sensitised solar cells will also be used. It is estimated that photovoltaic waste, will cumulatively reach 80 Mt by 2050.249 Use of recycled silicon in PV modules, has the potential to save 250 kW h per module, a greater than 50% energy reduction.249 There are significant research and development gaps in the valorisation of PV modules at the end-of-life through recycling and reuse.250 In a similar manner to LIBs, photovoltaic devices use adhesives and sealants in a variety of roles to create structure in the device, affix the device to a variety of surfaces and seal the device contents from the elements.251Functional electrically conductive adhesives (ECAs) have been proposed.252 These pose a quandary as whilst they impose a challenge at end-of-life from a materials separation perspective, they also impart useful functional properties, and can contribute to efficient photovoltaic module design. Using ECA's can reduce the need for lead solders,250 which reduces the handling of toxins in manufacturing and at the end of life and aid compliance with Restriction of Hazardous Substances (RoHS) legislation. They also allow assembly processes to occur at low-temperatures, which can be important for some high-efficiency photovoltaic cells.250,253 The materials used are diverse and include bituminous asphalts, silicones, epoxy resins, methacrylates and polyurethanes. Similarly, present glues and adhesives frustrate the recycling process of PV panels.
PV panels are encapsulated and adhered to substrates, and this step presents “the most difficult step in separating the module's components” at the end of life to remove the cells cleanly.254 Mechanical separation of crushed PV panels produces a problematic intermediate fraction of dissimilar materials joined by glue.255 Even after successive mechanical separation steps, the adhesive properties of EVA used in cell construction result in a residual fraction that is difficult to recycle.256 Alternatives exist to polymer encapsulation, including for example, the inclusion of silicon sheets to form a seal rather than an adhesive which is problematic at the end-of-life.250 Clearly photolytic and thermal debonding would be problematic but chemical, magnetic or ultrasound debonding could be viable.
In addition to new debondable adhesives, design for recycle initiatives could negate the use of some adhesives. Bifacial solar cells.257 use the active material sandwiched between two transparent sheets to enable energy to be generated from light hitting the cell in either direction. Hybrid solar cells comprise a number of different photovoltaic materials with different properties stacked on top of each other. The complimentary bandgaps of the different semiconductors used respond to different wavelengths increasing the efficiency of the hybrid construction and the design of these devices could potentially decrease some adhesive use.258
Many current approaches to photovoltaic recycling utilise thermal decomposition methods to deal with organics and separate cells from the substrate.254 Processes have been developed where cells, glued to EVA and Tedlar substrates are removed through heating in order to debond them to facilitate their reuse. However, this process using current adhesives, results in the cells retaining 80–100% of their efficiency.255 There is significant scope to improve the adhesives used such that they debond below the current temperatures of 500 °C but above the maximum normal operating temperatures of ca. 100 °C.
5.3. Wind turbines
In addition to solar energy a significant proportion of renewable energy will be harvested using wind turbines which have a design life of around 25 years.259 The logistics associated with end-of-life wind turbines are significant. The effort to decommission a turbine and ship components for end-of-life treatment is at least comparable to initial commissioning.259 It has also been estimated that 12 bn t of material will be needed to create wind turbine blades. Turbine blades are mostly made from carbon fibre or fibreglass constructs. These materials are notoriously hard to recycle because of their composition and the use of glues and epoxies.249 To put the magnitude of the issue into perspective some turbines have blade diameters in excess of 100 m and blade masses of >20 t. Current end of life solutions involve either energy recovery or use as fillers.260 Carbon fibre has a particularly high entrapped energy associated with its manufacture so these solutions are not particularly viable from a circularity perspective. Unlike PV cells and LIBs, the simple composition of turbine blades means that many of the constraints listed in Table 2 are not applicable so epoxy resin for example could be debonded by almost any stimulus with the possible exception of photolytic. The large size of the blades would make chemical debonding less likely unless it was a catalytic process. It is also evident that given the design for recycle criteria blades which could be dismantled for transportation and processing would clearly be advantageous.| Stimulus |
|
|
|
|
|
|
|---|---|---|---|---|---|---|
| Potential uses | Thin transparent materials | Inorganic and metallic components | Bonding metals and conductive substrates | Reactive composite adhesives with stable substrates | Biological/electronic applications | Better for weak adhesive bonds |
| Limitations | Opaque additives | Equipment operating at high temp. | Non-metals | Needs to not etch the substrate | Magnetic substrates | Needs to be wet recycle process |
| Non-thermally stable/conductive substrates. Metals when using microwave radiation | Non-conductive substrates (these must be bonded with an intermediate patch) | |||||
| Notes | Harder with complex geometries | Potentially applicable to most adhesives | Better for thin samples | Diffusion can be slow | Low cost, easy to develop | Surface wetting important |



Additionally, for some designs of wind-turbine generator, particularly offshore wind turbines, rare earth magnets, based upon neodymium, iron and boron (Nd–Fe–B), are used in the generators. These materials are particularly important with the drive to larger turbines, placed offshore, and there is a suggestion that the share of turbines in the market employing this type of generator will increase.261 The use of rare earth magnets allows turbine designers to dispense with the intermediate gearbox, which improves reliability, reduces maintenance costs and extends service life. This is particularly important in inaccessible, difficult to service locations such as the marine environment.
Wind turbine magnets are typically encapsulated into stainless steel cans which are hermetically sealed to prevent salt spray getting onto the surface of the magnets. They are often coated with an epoxy resin and then glued into place with multiple magnets making up each module with around 5 kg of Nd–Fe–B magnet per module. The magnets are typically in a bread loaf shape and made in fully dense form by sintering of an Nd–Fe–B alloy powder.
Re-use of magnets is difficult as there are many different magnet grades and compositions which have shifted historically as the design of applications has changed. Also the magnets themselves are very brittle and therefore they are difficult to handle without breaking the material. However there is a large incentive to remove the epoxy/binder material as if this can be achieved the sintered Nd–Fe–B can be “short loop” recycled by breaking the magnet down in hydrogen and milling, blending and re-sintering the alloy powder back into a sintered magnet at a “tailored grade”.262 If the epoxy binder is not removed it would produce carbon contamination in the sintered magnet which would significantly deteriorate the magnetic properties of the recycled sintered product. In some instances hydrogen can be used to break down the embedded magnet in order to produce a demagnetised alloy powder and the epoxy can be removed by mechanical means (HPMS process – Hydrogen Processing of Magnet Scrap).263 However this is very dependent on the choice of polymer.
Some authors have suggested that in order to reuse magnets from large machines such as those employed in wind turbines then standardised shapes would be required.264 One approach that has been suggested to improve the reusability of magnets in large machines is to use a modular “Lego™” like concept to create the “bread loaf” design as shown in Scheme 17.264 This has been successfully trialled using an adhesive but it was noted that it would present some challenges at the end of life for the reuse of these modular magnetic components.265 The thermal treatments needed at the end-of-life degrade the properties of the magnet.264 Again, here a debondable adhesive could aid in end of life treatment and while magnetic debonding is clearly not feasible, almost any other type of debonding could be used. Thermal demagnetisation to enable safe disassembly requires in excess of 320 °C to take the magnets past their Curie temperature. Many adhesives are likely to fail before this temperature has been reached, so adhesive failure may occur before modular magnets are demagnetised. Furthermore, the process of thermal treatment results in a sacrificial layer being formed on the surface of the magnet, which cannot be usefully recycled and must therefore be discarded. The much greater surface area presented by a great number of modular magnets may result in additional magnetic material being lost at the recycling stage.
 | ||
| Scheme 17 (a) A solid piece of bread loaf magnet. (b) Segmented bread loaf magnet. (c) Combined rectangular shape magnet. (d) Magnet pole with standard segments for reusing Redrawn from ref. 264. | ||
Other concepts include the use of magnetic powder in a thermoplastic binder.266 This results in some significant compromises in terms of the magnets strength, and as such, polymer bonded magnets are currently not used in wind turbines commercially.
6. Future perspectives
As this review has shown, design for debondable adhesion has received considerable attention with some commercial products now available. From a different perspective however, it could be argued that all adhesives are debondable, as all polymers will eventually decompose or lose adhesion on heating. The question then becomes whether the substrate and device can cope with the applied temperature.Most studies have focused on thermal and light-induced debonding, despite being the two stimuli which are most likely to be encountered naturally in service, and while numerous other stimuli exist, there are relatively few case studies for them. Each debonding stimulus will have specific mechanisms, applications, and limitations of use and these are summarised in Table 2.
Research has generally focused on the adaption of current adhesive technologies using modified monomers, additives, copolymers or composite phases. These require little modification, affect the adhesion only minimally and are usually economically viable. While polymer systems based on new monomers have been developed, these are less likely to be used for anything other than small volume, niche applications.
The economics of a debondable adhesive system must be comparable to commercial structural adhesives. Using an epoxide system as an example, the key monomers ECH and BPA retail for $500–2500 per tonne and $800–2400 per tonne respectively, with exact costs dependent on purity. Competing debondable systems should be less than $5000 per tonne, ideally considerably less. This excludes many of the more exotic approaches discussed above as even relatively common components, such as cyclodextrins have costs in the region of $3000–$100000 per tonne. Consequently, debondable adhesives based on novel monomers will be unlikely for anything other than niche, low volume applications. Simple additives, such as graphite, zinc, or iron oxide, are more affordable with prices in the range of $200–800 per tonne, in relatively pure states. Therefore, from a practical and economic perspective, the addition of small quantities of additives, to known structural adhesives is more likely. These additives are often found in the waste products of other processes, such as primary battery recycling, where purification is rarely economically viable; however, as an inert black mass, the waste product could be added to adhesives directly. This would catalyse debonding at a lower cost and the use of a waste product would significantly increase the Green metrics of the process. Other low-cost fillers could also be used as debonding agents, for example, carbonates and bicarbonates react with mild acid, producing gaseous products which aid physical debonding. In addition, composites, involving biopolymers, such as starch or alginates, could enable low cost-fillers which are water soluble (low molecular weight starches) or digestible by enzymes.
Design for recycle is central to creating a circular economy and debondable adhesives will provide opportunities for more efficient recycling processes-essential for TCMs. Future debondable adhesives, are likely to be modifications of current formulations, utilising renewables, or waste products as cheaper, less hazardous components. There is also significant scope for the use of traditional bio-based adhesives, with additional cross-linking allowing access to structural adhesive properties. As this review has demonstrated, regardless of monomers and stimuli utilised, debondable adhesives have an important role to play in the design of recyclable and repairable products. Some limitations in the systems and stimuli remain, for specific applications and architectures, but this is clearly a research area that will grow in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Faraday Institution (Faraday Institution grant codes FIRG005 and FIRG006, project website https://relib.org.uk), the UKRI Interdisciplinary Circular Economy Centre for Technology Metals, Met4Tech project (EP/V011855/1) for funding this work. Jade Lewin and Sally Heavey from Structural Adhesives Ltd are also acknowledged for useful discussions.References
- D. L. Thompson, J. M. Hartley, S. M. Lambert, M. Shiref, G. D. J. Harper, E. Kendrick, P. Anderson, K. S. Ryder, L. Gaines and A. P. Abbott, Green Chem., 2020, 22, 7585–7603 RSC.
- University of Birmingham, Securing Technology-Critical Metals for Britain, 2021 Search PubMed.
- A. Choudhary and E. Prasad, Epoxy Resin Market, https://www.alliedmarketresearch.com/epoxy-resins-market CAS, (accessed 22 August 2021).
- L. A. Heinrich, Green Chem., 2019, 21, 1866–1888 RSC.
- J. F. Lancaster, in Metallurgy of Welding, ed. J. F. Lancaster, Woodhead Publishing, 6th edn, 1999, pp. 54–84 Search PubMed.
- D. K. Hohl and C. Weder, Adv. Opt. Mater., 2019, 7, 1900230 CrossRef.
- N. Schüwer and R. Vendamme, in Green Chemistry for Surface Coatings, Inks and Adhesives: Sustainable Applications, ed. R. Höfer, A. S. Matharu and Z. Zhang, Royal Society of Chemistry, 2019, pp. 310–338 CAS.
- R. E. Bennett and M. A. Hittner, US Pat, US4286047, 1981 Search PubMed.
- H. S. Do, J. H. Park and H. J. Kim, Eur. Polym. J., 2008, 44, 3871–3882 CrossRef CAS.
- K. Ebe, H. Seno and K. Horigome, J. Appl. Polym. Sci., 2003, 90, 436–441 CrossRef CAS.
- C. M. Ryu, B. L. Pang, J. H. Han and H. Il Kim, J. Photopolym. Sci. Technol., 2012, 25, 705–712 CrossRef CAS.
- C. M. Ryu, B. Pang, H. Il Kim, H. J. Kim, J. W. Park, S. W. Lee and K. M. Kim, Int. J. Adhes. Adhes., 2013, 40, 197–201 CrossRef.
- Z. Czech, A. Kowalczyk, J. Kabatc, L. Shao, Y. Bai and J. Świderska, Polym. Bull., 2013, 70, 479–488 CrossRef CAS.
- T. Ozawa, S. Ishiwata, Y. Kano and T. Kasemura, J. Adhes., 2000, 72, 1–16 CrossRef.
- R. A. Chivers, Int. J. Adhes. Adhes., 2001, 21, 381–388 CrossRef CAS.
- H. H. Chu, C. K. Wang, C. K. Sein and C. Y. Chang, J. Polym. Res., 2014, 21, 1–7 CAS.
- J. M. Boyne, E. J. Millan and I. Webster, Int. J. Adhes. Adhes., 2001, 21, 49–53 CrossRef CAS.
- S. W. Lee, J. W. Park, Y. H. Lee, H. J. Kim, M. Rafailovich and J. Sokolov, J. Adhes. Sci. Technol., 2012, 26, 1629–1643 CrossRef CAS.
- M. Tunius, US Pat, US20110224593A1, 2008 Search PubMed.
- M. Tunius, US Pat, US20130017246A1, 2010 Search PubMed.
- N. Schüwer and R. Vendamme, Eur. Pat, EP2957611A1, 2014 Search PubMed.
- C. Decker and T. Nguyen Thi Viet, J. Appl. Polym. Sci., 2000, 77, 1902–1912 CrossRef CAS.
- J. K. Kim, W. H. Kim and D. H. Lee, Polymer, 2002, 43, 5005–5010 CrossRef CAS.
- J. Han, Y. Zhou, G. Bai, W. Wei, X. Liu and X. Li, J. Adhes. Sci. Technol., 2021, 5, 1–13 CrossRef.
- J. P. Phillips, X. Deng, R. R. Stephen, E. L. Fortenberry, M. L. Todd, D. M. McClusky, S. Stevenson, R. Misra, S. Morgan and T. E. Long, Polymer, 2007, 48, 6773–6781 CrossRef CAS.
- J. P. Phillips, X. Deng, M. L. Todd, D. T. Heaps, S. Stevenson, H. Zhou and C. E. Hoyle, J. Appl. Polym. Sci., 2008, 109, 2895–2904 CrossRef CAS.
- D. M. McCluskey, T. N. Smith, P. K. Madasu, C. E. Coumbe, M. A. MacKey, P. A. Fulmer, J. H. Wynne, S. Stevenson and J. P. Phillips, ACS Appl. Mater. Interfaces, 2009, 1, 882–887 CrossRef CAS PubMed.
- Z. Czech, A. Kowalczyk, J. Kabatc, J. Świderska, L. Shao and Y. Bai, Polym. Bull., 2012, 68, 441–452 CrossRef CAS.
- S. M. June, T. Suga, W. H. Heath, Q. Lin, R. Puligadda, L. Yan, D. Dillard and T. E. Long, J. Adhes., 2013, 89, 548–558 CrossRef CAS.
- Y. Z. Wang, L. Li, F. S. Du and Z. C. Li, Polymer, 2015, 68, 270–278 CrossRef CAS.
- M. Kim and H. Chung, Polym. Chem., 2017, 8, 6300–6308 RSC.
- Z. Shafiq, J. Cui, L. Pastor-Pérez, V. San Miguel, R. A. Gropeanu, C. Serrano and A. Del Campo, Angew. Chem., Int. Ed., 2012, 51, 4332–4335 CrossRef CAS PubMed.
- A. Romano, I. Roppolo, E. Rossegger, S. Schlögl and M. Sangermano, Materials, 2020, 13, 2777 CrossRef CAS.
- H. Hayashi, H. Tachi and K. Suyama, J. Photopolym. Sci. Technol., 2020, 33, 269–278 CrossRef CAS.
- K. Suyama and H. Tachi, J. Photopolym. Sci. Technol., 2015, 28, 45–48 CrossRef CAS.
- K. Suyama and H. Tachi, Prog. Org. Coat., 2016, 100, 94–99 CrossRef CAS.
- K. Suyama and H. Tachi, J. Photopolym. Sci. Technol., 2017, 30, 247–252 CrossRef CAS.
- K. Suyama and H. Tachi, J. Photopolym. Sci. Technol., 2018, 31, 517–522 CrossRef CAS.
- E. Sato, C. Omori, T. Nishiyama and H. Horibe, J. Photopolym. Sci. Technol., 2018, 31, 511–515 CrossRef CAS.
- E. Sato, M. Yuri, S. Fujii, T. Nishiyama, Y. Nakamura and H. Horibe, RSC Adv., 2016, 6, 56475–56481 RSC.
- E. Sato, T. Hagihara and A. Matsumoto, ACS Appl. Mater. Interfaces, 2012, 4, 2057–2064 CrossRef CAS PubMed.
- E. Sato, H. Tamura and A. Matsumoto, ACS Appl. Mater. Interfaces, 2010, 2, 2594–2601 CrossRef CAS.
- A. Matsumoto and S. Taketani, J. Am. Chem. Soc., 2006, 128, 4566–4567 CrossRef CAS.
- M. A. Ayer, S. Schrettl, S. Balog, Y. C. Simon and C. Weder, Soft Matter, 2017, 13, 4017–4023 RSC.
- M. A. Ayer, Y. C. Simon and C. Weder, Macromolecules, 2016, 49, 2917–2927 CrossRef CAS.
- K. Yamauchi, J. R. Lizotte and T. E. Long, Macromolecules, 2003, 36, 1083–1088 CrossRef CAS.
- C. Heinzmann, U. Salz, N. Moszner, G. L. Fiore and C. Weder, ACS Appl. Mater. Interfaces, 2015, 7, 13395–13404 CrossRef CAS PubMed.
- A. Faghihnejad, K. E. Feldman, J. Yu, M. V. Tirrell, J. N. Israelachvili, C. J. Hawker, E. J. Kramer and H. Zeng, Adv. Funct. Mater., 2014, 24, 2322–2333 CrossRef CAS.
- C. Heinzmann, I. Lamparth, K. Rist, N. Moszner, G. L. Fiore and C. Weder, Macromolecules, 2015, 48, 8128–8136 CrossRef CAS.
- C. Heinzmann, S. Coulibaly, A. Roulin, G. L. Fiore and C. Weder, ACS Appl. Mater. Interfaces, 2014, 6, 4713–4719 CrossRef CAS PubMed.
- B. Qin, S. Zhang, P. Sun, B. Tang, Z. Yin, X. Cao, Q. Chen, J. F. Xu and X. Zhang, Adv. Mater., 2020, 32, 2000096 CrossRef CAS.
- D. W. R. Balkenende, C. A. Monnier, G. L. Fiore and C. Weder, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- J. Yuan, X. Fang, L. Zhang, G. Hong, Y. Lin, Q. Zheng, Y. Xu, Y. Ruan, W. Weng, H. Xia and G. Chen, J. Mater. Chem., 2012, 22, 11515–11522 RSC.
- Y. Gao, K. Wu and Z. Suo, Adv. Mater., 2019, 31, 1806948 CrossRef.
- S. C. Grindy and N. Holten-Andersen, Soft Matter, 2017, 13, 4057–4065 RSC.
- E. M. White, J. E. Seppala, P. M. Rushworth, B. W. Ritchie, S. Sharma and J. Locklin, Macromolecules, 2013, 46, 8882–8887 CrossRef CAS.
- G. L. Eian and J. E. Trend, US Pat, US4980096A, 1990 Search PubMed.
- H. Sugita, K. Itou, Y. Itou, N. Wada, T. U. Shin-ya Kurita, Y. Hirose, K. Hatase, H. Matsumoto and D. Ichinohe, Int. J. Adhes. Adhes., 2021, 104, 102758 CrossRef CAS.
- H. Sugita, Y. Miyashita, H. Suzuki, S. Takasugi, N. Matsumura and M. Yoshizawa, J. Appl. Polym. Sci., 2018, 135, 45871 CrossRef.
- M. Shirai, Polym. J., 2014, 46, 859–865 CrossRef CAS.
- H. Okamura and M. Shirai, J. Photopolym. Sci. Technol., 2011, 24, 561–564 CrossRef CAS.
- B. Cardineau, P. Garczynski, W. Earley and R. L. Brainard, J. Photopolym. Sci. Technol., 2013, 26, 665–671 CrossRef CAS.
- T. Inui, E. Sato and A. Matsumoto, ACS Appl. Mater. Interfaces, 2012, 4, 2124–2132 CrossRef CAS PubMed.
- K. Yamanishi, E. Sato and A. Matsumoto, J. Photopolym. Sci. Technol., 2013, 26, 239–244 CrossRef CAS.
- T. Inui, K. Yamanishi, E. Sato and A. Matsumoto, Macromolecules, 2013, 46, 8111–8120 CrossRef CAS.
- T. Inui, E. Sato and A. Matsumoto, RSC Adv., 2014, 4, 24719–24728 RSC.
- E. Sato, K. Taniguchi, T. Inui, K. Yamanishi, H. Horibe and A. Matsumoto, J. Photopolym. Sci. Technol., 2014, 27, 531–534 CrossRef.
- E. Sato, K. Yamanishi, T. Inui, H. Horibe and A. Matsumoto, Polymer, 2015, 64, 260–267 CrossRef CAS.
- E. Sato, S. Iki, K. Yamanishi, H. Horibe and A. Matsumoto, J. Adhes., 2017, 93, 811–822 CrossRef CAS.
- Y. Fukamoto, E. Sato, H. Okamura, H. Horibe and A. Matsumoto, Appl. Adhes. Sci., 2017, 5, 1–11 CrossRef.
- T. Sasaki, S. Hashimoto, N. Nogami, Y. Sugiyama, M. Mori, Y. Naka and K. V. Le, ACS Appl. Mater. Interfaces, 2016, 8, 5580–5585 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- P. Tannouri, K. M. Arafeh, J. M. Krahn, S. L. Beaupré, C. Menon and N. R. Branda, Chem. Mater., 2014, 26, 4330–4333 CrossRef CAS.
- H. Zhou, C. Xue, P. Weis, Y. Suzuki, S. Huang, K. Koynov, G. K. Auernhammer, R. Berger, H. J. Butt and S. Wu, Nat. Chem., 2017, 9, 145–151 CrossRef CAS.
- H. Akiyama, T. Fukata, A. Yamashita, M. Yoshida and H. Kihara, J. Adhes., 2017, 93, 823–830 CrossRef CAS.
- S. Ito, A. Yamashita, H. Akiyama, H. Kihara and M. Yoshida, Macromolecules, 2018, 51, 3243–3253 CrossRef CAS.
- S. Ito, H. Akiyama, R. Sekizawa, M. Mori, M. Yoshida and H. Kihara, ACS Appl. Mater. Interfaces, 2018, 10, 32649–32658 CrossRef CAS.
- S. Ito, H. Akiyama, M. Mori, M. Yoshida and H. Kihara, Macromol. Chem. Phys., 2019, 220, 1900105 CrossRef.
- I. Tomatsu, A. Hashidzume and A. Harada, J. Am. Chem. Soc., 2006, 128, 2226–2227 CrossRef CAS PubMed.
- H. Yamaguchi, Y. Kobayashi, R. Kobayashi, Y. Takashima, A. Hashidzume and A. Harada, Nat. Commun., 2012, 3, 1–5 Search PubMed.
- Y. Takashima, T. Sahara, T. Sekine, T. Kakuta, M. Nakahata, M. Otsubo, Y. Kobayashi and A. Harada, Macromol. Rapid Commun., 2014, 35, 1646–1652 CrossRef CAS.
- O. Roling, L. Stricker, J. Voskuhl, S. Lamping and B. J. Ravoo, Chem. Commun., 2016, 52, 1964–1966 RSC.
- F. Van De Manakker, T. Vermonden, C. F. Van Nostrum and W. E. Hennink, Biomacromolecules, 2009, 10, 3157–3175 CrossRef CAS.
- P. E. Williams, Z. Walsh-Korb, S. T. Jones, Y. Lan and O. A. Scherman, Langmuir, 2018, 34, 13104–13109 CrossRef CAS PubMed.
- C. S. Y. Tan, J. Del Barrio, J. Liu and O. A. Scherman, Polym. Chem., 2015, 6, 7652–7657 RSC.
- J. Liu, C. S. Y. Tan and O. A. Scherman, Angew. Chem., Int. Ed., 2018, 57, 8854–8858 CrossRef CAS.
- S. Kaiser, S. V. Radl, J. Manhart, S. Ayalur-Karunakaran, T. Griesser, A. Moser, C. Ganser, C. Teichert, W. Kern and S. Schlögl, Soft Matter, 2018, 14, 2547–2559 RSC.
- H. Akiyama, Y. Okuyama, T. Fukata and H. Kihara, J. Adhes., 2018, 94, 799–813 CrossRef CAS.
- Z. Liu, J. Cheng and J. Zhang, Macromol. Chem. Phys., 2021, 222, 27–32 Search PubMed.
- S. Saito, S. Nobusue, E. Tsuzaka, C. Yuan, C. Mori, M. Hara, T. Seki, C. Camacho, S. Irle and S. Yamaguchi, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- S. R. Trenor, T. E. Long and B. J. Love, J. Adhes., 2005, 81, 213–229 CrossRef CAS.
- J. Ling, M. Z. Rong and M. Q. Zhang, Polymer, 2012, 53, 2691–2698 CrossRef CAS.
- N. Schüwer and G. De Visscher, Eur. Pat, EP2957610A1, 2014 Search PubMed.
- L. Wang and C. P. Wong, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2991–3001 CrossRef CAS.
- L. Wang, H. Li and C. P. Wong, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3771–3782 CrossRef CAS.
- X. Zhang, G. C. Chen, A. Collins, S. Jacobson, P. Morganelli, Y. L. Dar and O. M. Musa, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1073–1084 CrossRef CAS.
- O. M. Musa, X. Zhang, G. C. Chen, A. Collins, S. Jacobson, P. Morganelli and Y. L. Dar, in NATO Science for Peace and Security Series A: Chemistry and Biology, Springer, 2009, pp. 361–374 Search PubMed.
- H. Li, L. Wang, K. Jacob and C. P. Wong, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1796–1807 CrossRef CAS.
- E. Themistou, A. Kanari and C. S. Patrickios, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5811–5823 CrossRef CAS.
- S. Ma and D. C. Webster, Prog. Polym. Sci., 2018, 76, 65–110 CrossRef CAS.
- M. Iseki, Y. Suzuki, H. Tachi and A. Matsumoto, ACS Omega, 2018, 3, 16357–16368 CrossRef CAS.
- C. Gorsche, C. Schnoell, T. Koch, N. Moszner and R. Liska, Macromolecules, 2018, 51, 660–669 CrossRef CAS.
- E. Delebecq, J. P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS.
- A. C. Ferahian, D. K. Hohl, C. Weder and L. Montero de Espinosa, Macromol. Mater. Eng., 2019, 304, 1900161 CrossRef.
- A. del Prado, D. K. Hohl, S. Balog, L. M. de Espinosa and C. Weder, ACS Appl. Polym. Mater., 2019, 1, 1399–1409 CrossRef CAS.
- S. Cheng, M. Zhang, N. Dixit, R. B. Moore and T. E. Long, Macromolecules, 2012, 45, 805–812 CrossRef CAS.
- N. Ishikawa, M. Furutani and K. Arimitsu, ACS Macro Lett., 2015, 4, 741–744 CrossRef CAS.
- K. A. Bovaldinova, M. M. Feldstein, N. E. Sherstneva, A. P. Moscalets and A. R. Khokhlov, Polymer, 2017, 125, 10–20 CrossRef CAS.
- M. M. Feldstein, K. A. Bovaldinova, E. V. Bermesheva, A. P. Moscalets, E. E. Dormidontova, V. Y. Grinberg and A. R. Khokhlov, Macromolecules, 2014, 47, 5759–5767 CrossRef CAS.
- K. A. Boval'dinova, N. E. Sherstneva, M. M. Fel'dshtein, A. P. Moskalets and A. R. Khokhlov, Polym. Sci., Ser. B, 2019, 61, 458–470 CrossRef.
- M. M. Feldstein, WO2015088368A1, 2015.
- N. A. Cortez-Lemus and A. Licea-Claverie, Prog. Polym. Sci., 2016, 53, 1–51 CrossRef CAS.
- W. Fu and B. Zhao, Polym. Chem., 2016, 7, 6980–6991 RSC.
- E. Mehravar, M. A. Gross, G. P. Leal, B. Reck, J. R. Leiza and J. M. Asua, Polymer, 2018, 10, 975 Search PubMed.
- X. Luo, K. E. Lauber and P. T. Mather, Polymer, 2010, 51, 1169–1175 CrossRef CAS.
- R. F. Stewart and E. E. Schmitt, Eur. Pat, EP0471767B1, 1996 Search PubMed.
- C. H. Beede, US Pat, US3635754A, 1972 Search PubMed.
- G. De Crevoisier, P. Fabre, J. M. Corpart and L. Leibler, Science, 1999, 285, 1246–1249 CrossRef CAS PubMed.
- A. B. Croll, N. Hosseini and M. D. Bartlett, Adv. Mater. Technol., 2019, 4, 1900193 CrossRef.
- R. Saeidpourazar, R. Li, Y. Li, M. D. Sangid, C. Lu, Y. Huang, J. A. Rogers and P. M. Ferreira, J. Microelectromech. Syst., 2012, 21, 1049–1058 CAS.
- T. Higashihara, M. C. Fu, T. Uno and M. Ueda, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1153–1158 CrossRef CAS.
- N. D. Blelloch, H. T. Mitchell, C. C. Tymm, D. W. Van Citters and K. A. Mirica, Chem. Mater., 2020, 32, 9882–9896 CrossRef CAS.
- H. T. Mitchell, M. K. Smith, N. D. Blelloch, D. W. Van Citters and K. A. Mirica, Chem. Mater., 2017, 29, 2788–2793 CrossRef CAS.
- M. D. Banea, Rev. Adhes. Adhes., 2019, 7, 33–50 CrossRef CAS.
- B. Lee, I. Son, J. H. Kim, C. Kim, J. Y. Yoo, B. W. Ahn, J. Hwang, J. Lee and J. H. Lee, J. Appl. Polym. Sci., 2018, 135, 46586 CrossRef.
- Y. Nishiyama, N. Uto, C. Sato and H. Sakurai, Int. J. Adhes. Adhes., 2003, 23, 377–382 CrossRef CAS.
- R. H. McCurdy, A. R. Hutchinson and P. H. Winfield, Int. J. Adhes. Adhes., 2013, 46, 100–113 CrossRef CAS.
- M. D. Banea, L. F. M. da Silva, R. J. C. Carbas and S. de Barros, J. Adhes., 2017, 93, 756–770 CrossRef CAS.
- J. Bonaldo, M. D. Banea, R. J. C. Carbas, L. F. M. Da Silva and S. De Barros, J. Adhes., 2019, 95, 995–1014 CrossRef CAS.
- M. D. Banea, L. F. M. Da Silva and R. J. C. Carbas, Int. J. Adhes. Adhes., 2015, 59, 14–20 CrossRef CAS.
- D. Kim, G. Kim, G. Song and I. Chung, Polymer, 2016, 40, 216–224 Search PubMed.
- Y. Lu, J. Broughton and P. Winfield, Int. J. Adhes. Adhes., 2014, 50, 119–127 CrossRef CAS.
- M. Bauer, A. Concord, E. Langkabel, H. Luinge and G. Wachinger, US Pat, US20130196143A1, 2013 Search PubMed.
- M-P. Foulc, T. Bergara and M. Olive, WO2011080477A1, 2011.
- M-P. Foulc, T. Bergara and M. Olive, Pat, WO2011080478A1, 2011 Search PubMed.
- E. A. G. De Los Santo, K. Chittibabu, D. M. Martino, S. Trakhtenberg and J. C. Warner, Eur. Pat, EP3568447B3, 2018 Search PubMed.
- X. Tao and X. Xingcheng, Chem. Mater., 2008, 20, 2866–2868 CrossRef.
- J. D. Rule, R. E. Behling, N. A. Lee and L. M. Lebow, Pat, WO2013012973, 2013 Search PubMed.
- M. Frensemeier, J. S. Kaiser, C. P. Frick, A. S. Schneider, E. Arzt, R. S. Fertig and E. Kroner, Adv. Funct. Mater., 2015, 25, 3013–3021 CrossRef CAS PubMed.
- H. Y. Hwang, Funct. Compos. Struct., 2020, 2, 15003 CrossRef CAS.
- J. D. Rule, K. M. Lewandowski and M. D. Determan, US Pat, US8592034B2, 2013 Search PubMed.
- J. D. Rule, R. E. Behling, N. A. Lee and L. M. LeBow, US Pat, US9821529B2, 2012 Search PubMed.
- R. S. Gurney, D. Dupin, J. S. Nunes, K. Ouzineb, E. Siband, J. M. Asua, S. P. Armes and J. L. Keddie, ACS Appl. Mater. Interfaces, 2012, 4, 5442–5452 CrossRef CAS PubMed.
- S. Umemoto and M. Fujita, US Pat, US20110008552A1, 2008 Search PubMed.
- E. E. Schmitt, R. Clarke, A. W. Larson, S. P. Bitler, R. S. Tsugita and D. A. Schultz, US Pat, US5412035A, 1992 Search PubMed.
- R. R. Burch, US Pat, US20120021215A1, 2012 Search PubMed.
- S. Y. Lin, K. S. Chen and L. Run-Chu, Biomaterials, 2001, 22, 2999–3004 CrossRef CAS.
- J. S. Kang, A. J. Myles and K. D. Harris, ACS Appl. Polym. Mater., 2020, 2, 4626–4631 CrossRef CAS.
- M. Wouters, M. Burghoorn, B. Ingenhut, K. Timmer, C. Rentrop, T. Bots, G. Oosterhuis and H. Fischer, Prog. Org. Coat., 2011, 72, 152–158 CrossRef CAS.
- B. C. Mac Murray, T. H. Tong and R. D. Hreha, US Pat, US9260640, 2016 Search PubMed.
- J. H. Aubert, J. Adhes., 2003, 79, 609–616 CrossRef CAS.
- Y. Min, S. Huang, Y. Wang, Z. Zhang, B. Du, X. Zhang and Z. Fan, Macromolecules, 2015, 48, 316–322 CrossRef CAS.
- K. Luo, T. Xie and J. Rzayev, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4992–4997 CrossRef CAS.
- A. M. Asadirad, S. Boutault, Z. Erno and N. R. Branda, J. Am. Chem. Soc., 2014, 136, 3024–3027 CrossRef CAS PubMed.
- A. M. Schenzel, C. O. Klein, K. Rist, N. Moszner and C. Barner-Kowollik, Adv. Sci., 2016, 3, 1500361 CrossRef PubMed.
- A. M. Schenzel, N. Moszner and C. Barner-Kowollik, Polym. Chem., 2017, 8, 414–420 RSC.
- B. J. Adzima, C. J. Kloxin and C. N. Bowman, Adv. Mater., 2010, 22, 2784–2787 CrossRef CAS PubMed.
- K. K. Oehlenschlaeger, N. K. Guimard, J. Brandt, J. O. Mueller, C. Y. Lin, S. Hilf, A. Lederer, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Polym. Chem., 2013, 4, 4348–4355 RSC.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed.
- S. Salimi, T. S. Babra, G. S. Dines, S. W. Baskerville, W. Hayes and B. W. Greenland, Eur. Polym. J., 2019, 121, 109264 CrossRef.
- R. Ciardiello, G. Belingardi, B. Martorana and V. Brunella, J. Adhes., 2020, 96, 1003–1026 CrossRef CAS.
- R. Ciardiello, G. Belingardi, F. Litterio and V. Brunella, Int. J. Adhes. Adhes., 2021, 107, 102850 CrossRef CAS.
- J. Kolbe, T. Kowalik, M. Popp, M. Sebald, O. Schorsch, S. Heberer, M. Pridohl, G. Zimmermann, A. Hartwig and E. Born, US Pat, US20040249037A1, 2009 Search PubMed.
- K. Chino and M. Matsumura, Eur. Pat, EP1914285A1, 2008 Search PubMed.
- R. Heucher, S. Kopannia, C. McArdle, M. Stuve and J. Kolbe, US Pat, US20140374032A1, 2014 Search PubMed.
- M. D. Gilbert, US Pat, US6620308B2, 2003 Search PubMed.
- D. W. Haydon, Assem. Autom., 2002, 22, 326–329 CrossRef.
- S. Leijonmarck, A. Cornell, C. O. Danielsson, T. Åkermark, B. D. Brandner and G. Lindbergh, Int. J. Adhes. Adhes., 2012, 32, 39–45 CrossRef CAS.
- G. Scarselli and F. Nicassio, in Health Monitoring of Structural and Biological Systems 2017, SPIE, Portland, Oregon, 2017, vol. 10170, p. 1017020 Search PubMed.
- Y. Okabe, J. Kuwahara, K. Natori, N. Takeda, T. Ogisu, S. Kojima and S. Komatsuzaki, Smart Mater. Struct., 2007, 16, 1370–1378 CrossRef.
- B. Yan and B. Tang, in 13th International Conference on Ubiquitous Robots and Ambient Intelligence, Institute of Electrical and Electronics Engineers Inc., Xian, 2016, pp. 26–30 Search PubMed.
- D. Palumbo, R. Tamborrino, U. Galietti, P. Aversa, A. Tatì and V. A. M. Luprano, NDT&E Int., 2016, 78, 1–9 CrossRef.
- D. Ginzburg, F. Ciampa, G. Scarselli and M. Meo, Smart Mater. Struct., 2017, 26, 105018 CrossRef.
- H. Tachi and K. Suyama, J. Photopolym. Sci. Technol., 2017, 30, 253–257 CrossRef CAS.
- H. M. Klukovich, Z. S. Kean, S. T. Iacono and S. L. Craig, J. Am. Chem. Soc., 2011, 133, 17882–17888 CrossRef CAS PubMed.
- R. Groote, B. M. Szyja, E. A. Pidko, E. J. M. Hensen and R. P. Sijbesma, Macromolecules, 2011, 44, 9187–9195 CrossRef CAS.
- T. S. Babra, M. Wood, J. S. Godleman, S. Salimi, C. Warriner, N. Bazin, C. R. Siviour, I. W. Hamley, W. Hayes and B. W. Greenland, Eur. Polym. J., 2019, 119, 260–271 CrossRef CAS.
- T. S. Babra, A. Trivedi, C. N. Warriner, N. Bazin, D. Castiglione, C. Sivour, W. Hayes and B. W. Greenland, Polym. Chem., 2017, 8, 7207–7216 RSC.
- F. Robertson, Y. Wang and H. Rosing, Int. J. Adhes. Adhes., 2014, 55, 64–68 CrossRef CAS.
- N. Schüwer and R. Vendamme, in RSC Green Chemistry, Royal Society of Chemistry, 2019, vol. 2019-Janua, pp. 310–338 Search PubMed.
- D. Goubard and N. Sajot, US Pat, US20110281045A1, 2011 Search PubMed.
- J. Malik and S. J. Clarson, Int. J. Adhes. Adhes., 2002, 22, 283–289 CrossRef CAS.
- T. Oguri, A. Kawahara and N. Kihara, Polymer, 2016, 99, 83–89 CrossRef CAS.
- Y. Ahn, Y. Jang, N. Selvapalam, G. Yun and K. Kim, Angew. Chem., Int. Ed., 2013, 52, 3140–3144 CrossRef CAS PubMed.
- C. Soulie-Ziakovic and L. Leibler, WO2003061720A1, 2003.
- T. Nakamura, Y. Takashima, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- T. Liu, B. Zhao and J. Zhang, Polymer, 2020, 194, 122392 CrossRef CAS.
- B. Krishnakumar, R. V. S. P. Sanka, W. H. Binder, V. Parthasarthy, S. Rana and N. Karak, Chem. Eng. J., 2020, 385, 123820 CrossRef CAS.
- W. Alabiso and S. Schlögl, Polymer, 2020, 12, 1660 CAS.
- Y. Yang, Y. Xu, Y. Ji and Y. Wei, Prog. Mater. Sci., 2021, 120, 100710 CrossRef CAS.
- S. Zhao and M. M. Abu-Omar, Macromolecules, 2019, 52, 3946–3654 Search PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- S. Zhang, T. Liu, C. Hao, L. Wang, J. Han, H. Liu and J. Zhang, Green Chem., 2018, 20, 2995–3000 RSC.
- J. L. Meyer, P. Lan, S. Pang, K. Chui, J. Economy and I. Jasiuk, J. Adhes. Sci. Technol., 2021, 1–18 Search PubMed.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- U. Lafont, H. Van Zeijl and S. Van Der Zwaag, ACS Appl. Mater. Interfaces, 2012, 4, 6280–6288 CrossRef CAS PubMed.
- Y. Z. Meng, S. C. Tjong and A. S. Hay, Polymer, 2001, 42, 5215–5224 CrossRef CAS.
- M. Furutani and K. Arimitsu, J. Adhes. Soc. Jpn., 2018, 54, 302–309 CrossRef CAS.
- M. Furutani, A. Kakinuma and K. Arimitsu, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 237–241 CrossRef CAS.
- F. Ji, X. Liu, D. Sheng and Y. Yang, Polymer, 2020, 197, 122514 CrossRef CAS.
- E. Cudjoe, K. M. Herbert and S. J. Rowan, ACS Appl. Mater. Interfaces, 2018, 10, 30723–30731 CrossRef CAS PubMed.
- A. Trejo-Machin, L. Puchot and P. Verge, Polym. Chem., 2020, 11, 7026–7034 RSC.
- Z. Zhou, X. Su, J. Liu and R. Liu, ACS Appl. Polym. Mater., 2020, 2, 5716–5725 CrossRef CAS.
- S. Gao, Z. Cheng, X. Zhou, Y. Liu, J. Wang, C. Wang, F. Chu, F. Xu and D. Zhang, Chem. Eng. J., 2020, 394, 124896 CrossRef CAS.
- J. Tang, L. Wan, Y. Zhou, H. Pan and F. Huang, J. Mater. Chem. A, 2017, 5, 21169–21177 RSC.
- F. Hajiali, S. Tajbakhsh and M. Marić, Polymer, 2021, 212, 123126 CrossRef CAS.
- S. Wang, Z. Liu, L. Zhang, Y. Guo, J. Song, J. Lou, Q. Guan, C. He and Z. You, Mater. Chem. Front., 2019, 3, 1833–1839 RSC.
- M. Hayashi, Y. Oba, T. Kimura and A. Takasu, Polym. J., 2021, 53, 835–840 CrossRef CAS.
- E. E. L. Maassen, J. P. A. Heuts and R. P. Sijbesma, Polym. Chem., 2021, 12, 3640–3649 RSC.
- Y. Yang, F. S. Du and Z. C. Li, ACS Appl. Polym. Mater., 2020, 2, 5630–5640 CrossRef CAS.
- Y. Lei, A. Zhang and Y. Lin, Polymer, 2020, 209, 123037 CrossRef CAS.
- Y. Zhu, F. Gao, J. Zhong, L. Shen and Y. Lin, Eur. Polym. J., 2020, 135, 109865 CrossRef CAS.
- J. Wang, S. Chen, T. Lin, J. Ke, T. Chen, X. Wu and C. Lin, RSC Adv., 2020, 10, 39271–39276 RSC.
- J. Krahn, E. Bovero and C. Menon, ACS Appl. Mater. Interfaces, 2015, 7, 2214–2222 CrossRef CAS PubMed.
- J. Risan, A. B. Croll and F. Azarmi, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 48–57 CrossRef CAS.
- W. Wang, J. V. I. Timonen, A. Carlson, D. M. Drotlef, C. T. Zhang, S. Kolle, A. Grinthal, T. S. Wong, B. Hatton, S. H. Kang, S. Kennedy, J. Chi, R. T. Blough, M. Sitti, L. Mahadevan and J. Aizenberg, Nature, 2018, 559, 77–82 CrossRef CAS PubMed.
- A. G. Gillies, J. Kwak and R. S. Fearing, Adv. Funct. Mater., 2013, 23, 3256–3261 CrossRef CAS.
- D. M. Drotlef, P. Blümler and A. Del Campo, Adv. Mater., 2014, 26, 775–779 CrossRef CAS PubMed.
- M. T. Northen, C. Greiner, E. Arzt and K. L. Turner, Adv. Mater., 2008, 20, 3905–3909 CrossRef CAS.
- D. Liu, C. W. M. Bastiaansen, J. M. J. Den Toonder and D. J. Broer, Macromolecules, 2012, 45, 8005–8012 CrossRef CAS.
- E. Kizilkan, J. Strueben, A. Staubitz and S. N. Gorb, Sci. Robot., 2017, 2, eaak9454 CrossRef PubMed.
- H. Zhang, H. Lai, Z. Cheng, D. Zhang, W. Wang, P. Liu, X. Yu, Z. Xie and Y. Liu, Chem. Eng. J., 2021, 420, 129862 CrossRef CAS.
- J. D. Eisenhaure, T. Xie, S. Varghese and S. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 7714–7717 CrossRef CAS PubMed.
- S. Reddy, E. Arzt and A. Del Campo, Adv. Mater., 2007, 19, 3833–3837 CrossRef CAS.
- M. C. R. Franssen and C. G. Boeriu, in Starch Polymers: From Genetic Engineering to Green Applications, ed. P. Halley and L. R. Avérous, Elsevier B.V., 2014, pp. 145–184 Search PubMed.
- P. Tomasik and C. H. Schilling, Adv. Carbohydr. Chem. Biochem., 2004, 59, 175–403 CrossRef CAS.
- U. Funke and M. G. Lindhauer, Starch/Staerke, 2001, 53, 547–554 CrossRef CAS.
- N. Shah, R. K. Mewada and T. Mehta, Rev. Chem. Eng., 2016, 32, 265–270 CAS.
- M. P. Guarás, L. N. Ludueña and V. A. Alvarez, in Starch-Based Materials in Food Packaging: Processing, Characterization and Applications, ed. M. A. Villar, S. E. Barbosa, M. A. García, L. A. Castillo and O. V. López, Elsevier Inc., 2017, pp. 77–124 Search PubMed.
- R. V. Gadhave, P. A. Mahanwar and P. T. Gadekar, BioResources, 2019, 14, 3833–3843 CAS.
- L. Chen, Y. Wang, Z. Din, P. Fei, W. Jin, H. Xiong and Z. Wang, Int. J. Biol. Macromol., 2017, 104, 137–144 CrossRef CAS PubMed.
- N. Reddy and Y. Yang, Food Chem., 2010, 118, 702–711 CrossRef CAS.
- Z. Gao, W. Wang, Z. Zhao and M. Guo, J. Appl. Polym. Sci., 2011, 120, 220–225 CrossRef CAS.
- J. Berger, M. Reist, J. M. Mayer, O. Felt, N. A. Peppas and R. Gurny, Eur. J. Pharm. Biopharm., 2004, 57, 19–34 CrossRef CAS PubMed.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Green Chem., 2013, 15, 3360–3366 RSC.
- P. Zheng, Y. Li, F. Li, Y. Ou, Q. Lin and N. Chen, Polymer, 2017, 9, 153–165 Search PubMed.
- S. Lee, K. Lee, Y. W. Kim and J. Shin, ACS Sustainable Chem. Eng., 2015, 3, 2309–2320 CrossRef CAS.
- V. K. Soo and M. Doolan, Procedia CIRP, 2014, 15, 263–271 CrossRef.
- Electronic waste generated worldwide from 2010 to 2019.
- A. Norgren, A. Carpenter and G. Heath, J. Sustain. Metall., 2020, 6, 761–774 CrossRef.
- A. Das, D. Li, D. Williams and D. Greenwood, World Electr. Veh. J., 2018, 9, 22 CrossRef.
- Nissan LEAF Teardown: Lithium-ion battery pack structure, https://www.marklines.com/en/report_all/rep1786_201811, (accessed 22 August 2021).
- 2011 Nissan LEAF Battery-Deep Dive, https://youtu.be/vYQJatWpBXY, (accessed 22 August 2021).
- Model Y E35: Individual Battery Cell Removal and Comparing the MY-M3 Battery Modules, https://youtu.be/NoD4jzdReNo, (accessed 22 August 2021).
- V. Oehl, Adhes. Adhes. + SEALANTS, 2019, 16, 16–19 CrossRef.
- C. Lei, I. Aldous, J. M. Hartley, D. L. Thompson, S. Scott, R. Hanson, P. A. Anderson, E. Kendrick, R. Sommerville, K. S. Ryder and A. P. Abbott, Green Chem., 2021, 23, 4710–4715 RSC.
- D. Thompson, C. Hyde, J. M. Hartley, A. P. Abbott, P. A. Anderson and G. D. J. Harper, Resour., Conserv. Recycl., 2021, 175, 105741 CrossRef.
- D. Mulvaney, R. M. Richards, M. D. Bazilian, E. Hensley, G. Clough and S. Sridhar, Renewable Sustainable Energy Rev., 2021, 137, 110604 CrossRef.
- J. A. Tsanakas, A. van der Heide, T. Radavičius, J. Denafas, E. Lemaire, K. Wang, J. Poortmans and E. Voroshazi, Prog. Photovolt.: Res. Appl., 2020, 28, 454–464 CrossRef.
- B. Ekiss, PV junction box mounting and sealing advances, https://www.renewableenergyworld.com/solar/pv-junction-box-mounting-and-sealing-advances/, (accessed 22 August 2021).
- L. Theunissen, B. Willems, J. Burke, D. Tonini, M. Galiazzo and A. Henckens, AIP Conf. Proc., 2018, 1999, 80003 CrossRef.
- W. Mühleisen, L. Neumaier, J. Scheurer, B. Stoesser, W. Pranger, A. Schütz, F. Vollmaier, T. Fischer, R. Lorenz, M. Schwark, R. Ebner and C. Hirschl, in 35th European Photovoltaic Solar Energy Conference and Exhibition, Brussels, 2018, pp. 75–78 Search PubMed.
- P. Dias, S. Javimczik, M. Benevit and H. Veit, Waste Manag., 2017, 60, 716–722 CrossRef CAS.
- F. C. S. M. Padoan, P. Altimari and F. Pagnanelli, Sol. Energy, 2019, 177, 746–761 CrossRef.
- A. Rubino, P. G. Schiavi, P. Altimari and F. Pagnanelli, Waste Manag., 2021, 122, 89–99 CrossRef CAS PubMed.
- X. Jia, C. Zhou, Y. Tang and W. Wang, Sol. Energy Mater. Sol. Cells, 2021, 227, 111112 CrossRef CAS.
- I. Mathews, D. O'Mahony, K. Thomas, E. Pelucchi, B. Corbett and A. P. Morrison, Prog. Photovolt.: Res. Appl., 2015, 23, 1080–1090 CrossRef CAS.
- A. Kolios and M. Martínez-Luengo, Renew. Energy Focus, 2016, 17, 109–111 CrossRef.
- R. Cherrington, V. Goodship, J. Meredith, B. M. Wood, S. R. Coles, A. Vuillaume, A. Feito-Boirac, F. Spee and K. Kirwan, Energy Policy, 2012, 47, 13–21 CrossRef.
- P. D. Jensen, P. Purnell and A. P. M. Velenturf, Sustain. Prod. Consum., 2020, 24, 266–280 CrossRef.
- M. Zakotnik, I. R. Harris and A. J. Williams, J. Alloys Compd., 2009, 469, 314–321 CrossRef CAS.
- A. Walton, H. Yi, N. A. Rowson, J. D. Speight, V. S. J. Mann, R. S. Sheridan, A. Bradshaw, I. R. Harris and A. J. Williams, J. Clean. Prod., 2015, 104, 236–241 CrossRef CAS.
- Z. Li, A. K. Lebouc, J.-M. Dubus, L. Garbuio and S. Personnez, in Symposium de Génie Electrique, Nancy, 2018 Search PubMed.
- S. Högberg, T. S. Pedersen, F. B. Bendixen, N. Mijatovic, B. B. Jensen and J. Holboll, in XXII International Conference on Electrical Machines (ICEM), Lausanne, 2016, pp. 1625–1629 Search PubMed.
- Z. Li, A. Kedous-Lebouc, J.-M. Dubus, J. Legranger and R. Fratila, in International Joint MMM-Intermag Conference, Washington, DC, 2019 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |