
Critical factors for levulinic acid production from starch-rich food waste: solvent effects, reaction pressure, and phase separation†
Shanta
Dutta
 a,
Iris K. M.
Yu
ab,
Jiajun
Fan
c,
James H.
Clark
c and
Daniel C. W.
Tsang
*a
a,
Iris K. M.
Yu
ab,
Jiajun
Fan
c,
James H.
Clark
c and
Daniel C. W.
Tsang
*a
aDepartment of Civil and Environmental Engineering, The Hong Kong Polytechnic University, 181 Chatham Road South, Hung Hom, Kowloon, Hong Kong, China. E-mail: dan.tsang@polyu.edu.hk
bDepartment of Chemistry and Catalysis Research Institute, TU München, Lichtenbergstrasse 4, 85748 Garching, Germany
cGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, York, YO10 5DD, UK
First published on 30th September 2021
Abstract
A considerable amount of food waste generated globally could be upcycled to synthesise platform chemicals to enhance environmental sustainability and realise a circular economy. This study investigates the catalytic production of the vital platform molecule levulinic acid (LA) from bread waste, a typical stream of starch-rich food waste generated worldwide. Gamma-valerolactone (GVL), isopropanol (IPA), and propylene carbonate (PC) were evaluated as bio-derived and CO2-derived green co-solvents for LA synthesis. In-vessel pressure generated in PC/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent was conducive to rapid LA production from bread waste compared to GVL/H2O and IPA/H2O. In PC/H2O, 72 mol% total soluble product yield was observed quickly within 1 min in moderate reaction conditions (130 °C, 0.5 M H2SO4), whereas ∼15–20 mol% of LA could be obtained when the reaction was prolonged for 10–20 min at 130 °C. The yield of LA could be significantly enhanced in GVL/H2O through phase separation using NaCl (30 wt%(aq)). LA yield increased up to a maximum of ∼2.5 times in the biphasic system (28 mol%, 150 °C, 15 min) (representing a theoretical yield of 66%) in GVL/H2O (1:1) compared to the monophasic system (∼11 mol%) under the same reaction conditions. The partition coefficient for LA achieved was 4.2 in the GVL/H2O (1:1) biphasic medium, indicating that the system was efficient for simultaneous production and extraction of LA. Biphasic GVL/H2O facilitated selective LA production, which could be optimised by tuning the reaction conditions. These new insights can foster the development of high-performance LA production and sustainable biorefinery.
:1) solvent was conducive to rapid LA production from bread waste compared to GVL/H2O and IPA/H2O. In PC/H2O, 72 mol% total soluble product yield was observed quickly within 1 min in moderate reaction conditions (130 °C, 0.5 M H2SO4), whereas ∼15–20 mol% of LA could be obtained when the reaction was prolonged for 10–20 min at 130 °C. The yield of LA could be significantly enhanced in GVL/H2O through phase separation using NaCl (30 wt%(aq)). LA yield increased up to a maximum of ∼2.5 times in the biphasic system (28 mol%, 150 °C, 15 min) (representing a theoretical yield of 66%) in GVL/H2O (1:1) compared to the monophasic system (∼11 mol%) under the same reaction conditions. The partition coefficient for LA achieved was 4.2 in the GVL/H2O (1:1) biphasic medium, indicating that the system was efficient for simultaneous production and extraction of LA. Biphasic GVL/H2O facilitated selective LA production, which could be optimised by tuning the reaction conditions. These new insights can foster the development of high-performance LA production and sustainable biorefinery.
Introduction
Appropriate and sustainable management of the growing amount of food waste is a big issue in modern society. Approximately 931 million tonnes of food waste were globally generated in the year of 2019 at the retail and consumer level (households and commercial food services), excluding food loss during post-harvest to distribution in the food supply chain.1 Such an enormous amount of food waste requires proper handling to minimise pollution risks and be diverted from landfill disposal.2 Transforming food waste into platform chemicals offers a sustainable opportunity to utilise the vast waste stream for biorefinery applications and pave the way forward to realising a circular bioeconomy.3Levulinic acid (LA) is one of the top bio-derived platform chemicals with various applications, e.g., pharmaceuticals, plasticisers, solvents, fuels, and personal care products.4,5 Besides, LA serves as a building block for the production of various value-added derivatives such as gamma-valerolactone (GVL),6 succinic acid, diphenolic acid,7,8 alkyl levulinates, 2-methyl-tetrahydrofuran, etc.5 Considering the low cost and high catalytic activity for tandem hydrolysis and dehydration reactions, mineral acids such as H2SO4 and HCl are generally used for LA production from diverse biomass feedstock including food waste, paper waste, etc.9,10 Starch-rich foods such as rice, bread, and potatoes are commonly consumed and constitute a significant proportion of global food waste,11 which can serve as a potential feedstock for biorefineries. Recent studies demonstrated that catalytic production of sugars and platform chemicals could be an effective technique for recycling/valorising a considerable amount of starch-rich food waste generated globally.12,13 In this study, bread waste serves as the representative starch-rich feedstock for the catalytic production of LA.
Appropriate reaction conditions and solvent selection are critical for selective and cost-effective LA production.14 As the greenest and environmentally benign solvent, water is preferred as a reaction medium for LA synthesis. However, harsh reaction conditions, i.e., high temperature and acidity, are required when using water as the reaction medium resulting in an energy-intensive process and high byproduct (humin) formation.15 The selection of a suitable solvent is necessary, as it not only serves as a reaction medium, but also influences the catalytic process through solvent-solute interactions, adjusting the reactivity of proton, suppressing the byproduct formation, etc.13,16 Owing to the need for selective and high-efficiency chemical synthesis, various organic solvents as reaction medium have been widely investigated, such as dimethylsulfoxide (DMSO),9 tetrahydrofuran (THF), dimethylformamide (DMF),7 GVL, acetone, etc.10,17 However, conventional organic solvents DMSO, DMF, THF, etc., are often considered “non-green” due to their direct or indirect detrimental effects on the natural environment and human health and safety. Therefore, the use of alternative “green” solvents, and especially those which are renewable and bio-derived (GVL, alcohols, etc.) or CO2-derived (propylene carbonate (PC), dimethyl carbonate (DMC), etc.), are recommended for future applications.18,19 Furthermore, enhanced catalytic conversion of biowaste using these bio-derived and CO2-derived solvents was reported in recent studies,10,13,20 yet their influences should be evaluated in comparable conditions to validate their efficiency and elucidate the critical factors for catalytic LA production.
Apart from using environmentally friendly and safe reaction systems, process intensification is indispensable to improve the final concentration of LA for scaling up at the industrial level.14 Efficient separation and purification of LA are necessary for the possible recovery of mineral acid catalyst and further conversion of LA to valuable derivatives such as GVL, which can be negatively affected by the presence of mineral acid.17,21 A viable strategy could be using the biphasic solvent system comprising two immiscible layers. The aqueous layer contains the acid catalyst and serves as the reactive phase, while the organic layer acts as the extractive phase, facilitating simultaneous production and extraction of LA, which might enhance LA yield and selectivity.21,22 For instance, a recent study on the conversion of remnant algal biomass reported a significant enhancement of hydroxymethylfurfural (HMF) and LA yield using acidic ZSM-5 zeolite as a heterogeneous catalyst when utilising a biphasic solvent system composed of methyl isobutyl ketone (MIBK) and H2O with NaCl as the phase modifier.23 Another study achieved a ∼4 fold increase in LA yield from rice straw using a biphasic reaction medium containing dichloromethane (DCM) and H2O (DCM/H2O, 1:1), in which the enhanced performance was attributed to the good extraction capability of DCM solvent.24 However, there is insufficient experimental evidence and limited understanding of critical impacts of the biphasic system on LA production using the recommended green solvents.
This study examines LA production from bread waste using PC, GVL, and isopropanol (IPA) as green co-solvent systems, i.e., PC/H2O, GVL/H2O, IPA/H2O, catalysed by dilute H2SO4 to investigate how and why an efficient yield of LA could be achieved at moderate reaction conditions using these bio-derived and CO2-derived green co-solvents. Water (100%) as the greenest solvent was also evaluated as the baseline for comparison. Furthermore, this study explores the application and tuning of the binary biphasic reaction medium to intensify the concentration of the target product LA considering the potential advantages of phase separation for reactive LA extraction.
Materials and methods
Bread waste and chemicals
Bread waste was collected from catering outlets in the Hong Kong International Airport (HKIA) and dried, ground, sieved through a 0.3 mm mesh, and stored in an airtight container for further experimental use. The bread waste contains 72.6% available carbohydrates, 4.2% total dietary fibre, 14.8% protein, 6.1% total fat, 2.3% ash, and 41.5% total organic carbon (TOC) on a dry mass basis.13 Solvents were purchased as follows, PC (99%, Aladdin), IPA (99%, Honeywell), GVL (99%, Sigma-Aldrich), and propylene glycol (PG) (99.8%, J&K Scientific). The standard chemical/compounds for calibration of the analytical equipment and catalytic reaction included glucose (99%, UNI-chem, China); fructose (≥99%) and maltose monohydrate (98%) (WAKO); levoglucosan (LG) (Fluorochem); LA (98%), and formic acid (FA) (98%) (Alfa Aesar); furfural (99%) and HMF (≥99%, Sigma Aldrich); and H2SO4 (98%, Honeywell Fluka). All standard chemicals were used as received.Catalytic conversion
The catalytic conversion of bread waste substrate was performed under microwave heating in Ethos Up Microwave Reactor (Milestone, maximum power of 1.9 kW) following the method reported in our recent studies.13,20 To perform catalytic tests using a monophasic system, 0.5 g bread waste (5 wt/v%), a mixture of H2O and organic solvent (PC/GVL/IPA) (total volume 10 ml; solvent ratio 1:1 for monophasic reaction), and 0.5 M H2SO4 were loaded in teflon vessel and sealed, then heated to reach the desired reaction temperature (110–150 °C) following a constant ramp rate (32 °C min−1), followed by a holding stage for 1–20 min, and cooling down for 20–30 min using mechanical ventilation. Each experimental run was conducted in duplicates to ensure reliable analysis. The reaction conditions were selected based on the latest studies on starch-rich food waste conversion11,13 and adjusted if necessary based on the experimental observations. To substantiate the experimental results obtained in PC/H2O solvent, additional tests under the selected conditions were conducted using PG and H2O (PG/H2O, 1:1) for comparison.
After analysing the results obtained from different monophasic solvent systems, GVL/H2O and IPA/H2O solvents were investigated for biphasic reactions. As GVL and IPA are completely miscible in water, 30 wt%(aq) NaCl was applied as a phase modifier to prepare the biphasic system,21,25 and catalytic tests were conducted following the conditions mentioned above. It is noted that the concentration of H2SO4 refers to the whole solvent system (i.e., consistent for all experimental runs with different solvents, both monophasic and biphasic), whereas the concentration of NaCl refers to the reaction phase (H2O) only, which is denoted as NaCl(aq). To analyse the influence of the reaction phase and extraction phase on catalytic performance in the biphasic system, different ratios of H2O and organic solvent (1:1, 1:3, 3:1) were applied for the conversion. Control runs without bread waste substrate were performed for solvent systems under the selected conditions. Standard thermocouple and pressure data-logger were used during catalytic reactions to monitor and record the in-vessel reaction temperature and autogenous pressure, respectively.
Analysis of samples
Soluble samples were obtained from each replicate, diluted with deionised water (DIW), and filtered through a membrane filter (0.45 μm) before analysis. For biphasic reactions, samples were obtained from each layer and prepared separately for analysis after dilution in DIW. High-performance liquid chromatography (HPLC) consisting of a Chromaster instrument equipped with Aminex HPX-87H column (Bio-Rad) and a refractive index detector (Hitachi, Japan) was used to analyse the soluble products; 0.01 M H2SO4 was used as the mobile phase at 0.5 ml min−1 flow rate at 50 °C.10,26 To ensure reliable analysis, spiked samples with known concentrations of standard compounds were injected before analysing the experimental samples. The yield of products was calculated based on the carbon content (eqn (1)) of the bread waste substrate.13 | (1) |
 | (2) |
Post-reaction solid residues were collected through centrifugation and decantation; washed with DIW three times and oven-dried for 48 h at 60 °C and then ground as a powder for further analysis using 13C nuclear magnetic resonance (NMR). Solid 13C NMR were recorded with bulk powder samples on a Jeol JNM-ECZ500R MHz spectrometer operating at a resonance frequency of 125 MHz. A commercial 3.2 mm magic-angle spinning (MAS) NMR probe was used with a standard cross-polarisation MAS (CPMAS) pulse sequence. The MAS frequency was 10 kHz with relaxation delay, scan times, and contact time at 5 s, 1200–2000, and 2 ms, respectively. Liquid samples subjected to the selected reaction conditions were analysed by 13C NMR (liquid-state) using D2O solution. Chemical shifts (δ) were given in ppm and measured relative to tetramethylsilane (TMS) as the internal standard. The solid residues were also characterised by X-ray diffraction (XRD) (Rigaku Smatlab, 5° to 50° 2θ, rate: 10° min−1 at 45 kV and 200 mA) and the crystallinity index (CrI) was calculated from XRD patterns following the method reported in the literature.27
Results and discussion
Catalytic conversion of bread waste under bio-derived and CO2-derived solvents
Facile conversion of bread waste was achieved under moderate reaction conditions, obtaining 45–72 mol% total product yield at 130 °C under bio-derived and CO2-derived co-solvent systems (Fig. 1a). For H2O, PC/H2O, and GVL/H2O solvents, the maximum total sugars released from bread waste were 64–70 mol% in a short reaction duration of 1 min (Fig. S1a†). In contrast, using IPA/H2O, the total sugar yield after 1 min of reaction was 43.5 mol%, increasing up to 48.8 mol% when the reaction was prolonged for 20 min. Given the total sugar yield generated at 110 °C (Fig. 1b), hydrolysis of bread waste was faster in GVL/H2O (48.1 mol%) and PC/H2O (53.7 mol%) compared to H2O (∼30 mol%) and IPA/H2O (9.4 mol%). With increasing reaction duration up to 20 min, sugar yield in H2O and IPA/H2O remained steady and generated an insignificant LA yield ∼1.5 mol% after 20 min of reaction. In contrast, sugars were consumed, and LA yield increased with increasing reaction duration gradually in GVL/H2O (up to 4.8 mol%) and sharply in PC/H2O (up to 19.6 mol%) (Fig. 1a). 13C NMR spectra of reacted solutions (1 and 20 min reaction) (Fig. 2a) provided further evidence for the disparity in catalytic conversion under different solvent systems. 13C NMR chemical shift (δ) at 95.8 ppm, attributed to glucose,28 was observed for all reactions. The highest intensity of glucose peak was observed for 1 min reacted solution in PC/H2O, which agrees with the maximum sugar yield measured by HPLC. In contrast, the lowest intensity of glucose peak and emerged LA peak (at 27.7, 37.6, 177 pm) along with an FA peak (at 165.45 ppm)29 for 20 min reaction in PC/H2O further prove the efficient tandem catalysis in PC/H2O. The characteristic peaks for LA and FA were not detectable in IPA/H2O and H2O, while low-intensity peaks could be observed for GVL/H2O. Besides, representative solvent peaks29 were detected in the reacted solutions indicated in Fig. 2a. | ||
| Fig. 1 Total sugar yield and LA yield during the catalytic conversion of bread waste in different solvents (a) at 130 °C, 1–20 min, (b) at 110 °C, 1 min (reaction conditions: 5 wt% substrate loading, 0.5 M H2SO4, 1:1 solvent ratio). | ||
 | ||
| Fig. 2 (a) 13C NMR spectra of soluble samples obtained after bread waste conversion in different solvents (reaction condition: 5 wt% substrate loading, 0.5 M H2SO4, 130 °C, 1 min & 20 min, 1:1 solvent ratio), (b) 13C solid-state NMR spectra of untreated bread waste and solid residues collected after reaction under different solvent systems (reaction conditions: 5 wt% substrate loading, 0.5 M H2SO4, 130 °C, 20 min, 1:1 solvent ratio). | ||
The higher soluble product yields in PC/H2O and GVL/H2O (Fig. 1b) could be related to the high reactivity of the Brønsted acid catalyst due to the availability of highly active proton in the presence of aprotic co-solvents in the reaction system.13,30 The extent of proton stabilisation influences the acid dissociation constant, and in water lowers the proton reactivity, which subsequently elevates the required energy level for acid-catalysed biomass conversion reactions such as hydrolysis and dehydration.30 Therefore, reaction kinetics could be slower in H2O (100%) compared to a solvent system consisting of a polar aprotic solvent such as GVL and PC, which might enhance the reaction rates owing to reactive proton and facile glycosidic bond cleavage.20,31 For instance, a recent study reported a ten-fold increase in reaction rate for acid-catalysed conversion of HMF to LA using GVL with 10% H2O compared to 100% H2O as solvent.32 Slower conversion of bread waste was observable in the case of IPA/H2O solvent (total sugar ∼10 mol%, at 110 °C, 1 min) (Fig. 1b), where both co-solvents are protic and probably hamper the proton reactivity during conversion.
The starch contained in the bread waste substrate represents a complex structure combining linear amylose chains and highly branched amylopectin comprising α(1 → 4) and α(1 → 6) glycosidic bonds. 13C NMR spectra of solid residues (Fig. 2b) subjected to reaction under different solvent systems (20 min) showed differences in chemical shifts than the untreated bread waste, possibly related to the changes in starch structures.33 The chemical shifts observed at resonance values of 93–103, 74–85, 64–73, 55–60 ppm can be attributed to C1, C4, C2,3,5, and C6 of glucose units, respectively.11,34 Resonance values attributed to C1 and C4 can be helpful to understand the changes in the amorphous and highly ordered/crystalline state of starch. The C1 resonance for post-reaction solid residues shifted toward 98–103 ppm, presenting comparatively sharp peaks compared to the broad peak observed at 93–100 ppm for untreated bread waste. The broad shoulder observed around 95 ppm for untreated bread waste is characteristic of the amorphous domain of C1. In contrast, a sharp peak observed around 100 ppm (C1) for solid residues might indicate a decrease in amorphous content and a subsequent rise in relative crystallinity, which was previously reported for acid-modified starch.34,35 In contrast, the resonance for C4 around 82 ppm was observed for solid residues, characteristic of an amorphous state.36 Different changes in chemical shifts in different carbon regions can reflect varied transformations between amylose and amylopectin within the starch structures. Amylose is mainly amorphous and more susceptible to acid hydrolysis than amylopectin, which comprises highly ordered/crystalline domain building double-helical structures. However, initially during acid hydrolysis, amorphous amylose could be partially transformed into double helices resistant to acid hydrolysis, and consequently, crystalline content could be enhanced relative to amorphous content.37,38 The observed changes in solid residues compared to untreated bread waste was substantiated by XRD analysis. The XRD pattern of untreated bread waste (Fig. S2†) showed a broad crystalline peak at ∼2θ = 20°, and the calculated CrI value was 0.51, whereas solid residues in different solvent systems provided CrI values ranging 0.73–0.78. This also suggested the rapid decomposition of amorphous region of starch granules during acid hydrolysis resulting in an increased relative crystallinity.27
LA yield enhanced by in-vessel high pressure generated in PC/H2O
Comparing different co-solvent systems, PC/H2O was found to be the best for LA production, generating 19.6 mol% after 20 min reaction at 130 °C (Fig. 1a), which is promising considering mild reaction conditions used in this study and the reported literature results under comparable experimental conditions.9,39 Under GVL/H2O solvent, sugars depleted gradually during 3–20 min of reaction and generated 2–4.8 mol% LA. In contrast, in PC/H2O solvent, sugars started to deplete rapidly during 3–20 min of reaction, and 19.6 mol% LA yield was achieved after 20 min. A significant in-vessel pressure build-up was recorded during reaction in PC/H2O, reaching a maximum ∼27 bar at 130 °C, 20 min compared to reactions in other solvents (Fig. 3a). Compared to the autogenous in-vessel pressure (∼13 bar) generated in PC/H2O during 1 min reaction, the reaction pressure level almost doubled (∼23–27 bar) during 3–20 min reaction (Fig. 3b), which was analogous to rapid sugar consumption and a sharp increase in LA yield (4–19.6 mol%) during 3–20 min. In acidic conditions, PC solvent could decompose into PG and CO2 as reported in the literature.13,40 Noticeably, ∼5–15% solvent loss was recorded for reactions in PC/H2O (1–20 min), indicating that a fraction of the PC solvent was decomposed during the reaction. Besides, 13C NMR spectra (Fig. 2a) for the reacted solutions under PC/H2O (1 and 20 min) showed peaks at 66.4 and 67.7 ppm representing PG.29 The PG peaks had comparatively higher intensity after 20 min reaction than 1 min, suggesting that a higher fraction of PC was decomposed to PG, which was consistent with higher CO2 pressure recorded for more prolonged reactions in PC/H2O (Fig. 3b). | ||
| Fig. 3 (a) Pressure profile during the catalytic conversion of bread waste in different solvents for 20 min reaction, (b) pressure profile in PC/H2O for different reaction durations (1–20 min), (c) sugar and LA yield in PC/H2O after 20 min and 5 + 15 min (pressure release after 5 min) reaction, (d) sugar and LA yield in PG/H2O after 5 and 20 min reaction (reaction conditions: 5 wt% substrate loading, 0.5 M H2SO4, 130 °C, 1:1 solvent ratio). | ||
Apart from the aprotic nature of the PC solvent, significant pressure build-up resulting from CO2 generation during catalytic conversion in PC/H2O might enhance the LA production rate. Another influential factor could be the increased acidity due to carbonic acid derived from partial dissolution of CO2 during the reaction, which might enhance sugar dehydration.41 Solution acidity can arise through in situ formation and dissociation of carbonic acid, as given in eqn (3).42
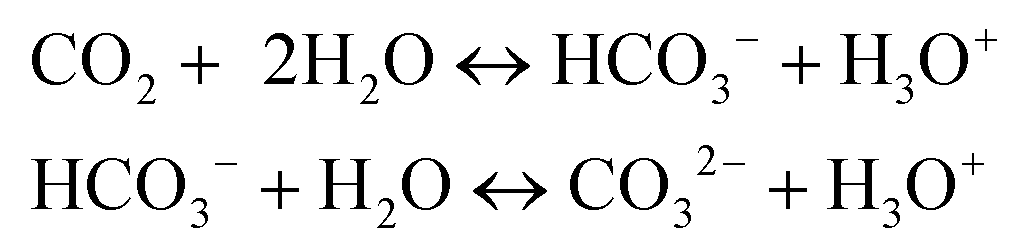 | (3) |
The acidity (pH) associated with CO2 generated during 3–20 min reaction in PC/H2O was estimated, following the scheme reported in the literature.43,44 The estimated CO2-derived pH value for specified reaction conditions was ∼3.5, i.e., considerably higher than the H2SO4 derived pH (∼0.5) in this study. Therefore, CO2-derived pH on its own might not be sufficient to catalyse tandem reactions and significantly enhance LA yield. In other words, LA yield might be improved by PC/H2O generated CO2 pressure more than CO2-derived acidity during the catalytic conversion of bread waste. To testify this postulation, an additional test using PC/H2O was conducted, in which autogenous pressure developed due to PC degradation (∼22 bar) was released after 5 min reaction, and then the solution underwent reaction for another 15 min (Fig. 3c). Compared to continuous 20 min reaction in PC/H2O (LA yield 19.6 mol%), only about half of LA was produced (LA yield 11.2 mol%) when the pressure was released prematurely during the reaction. After the pressure release, the autogenous pressure was ∼4.2 bar from the beginning till the end point of additional 15 min reaction. Moreover, supplementary tests using PG/H2O only generated ∼3 mol% LA yield (130 °C, 20 min) (Fig. 3d), which was insignificant compared to LA yield obtained in PC/H2O, therefore disproving any favourable influences by PG solvent (decomposed from PC) on the catalytic performance. These experimental evidences confirm the crucial role of reaction pressure in enhancing catalytic LA production.
Intensification of LA yield through phase separation
Among co-solvent systems, PC/H2O generated the highest LA yield 19.6 mol% at 130 °C, and ∼9 mol% soluble sugar remained after a 20 min reaction. In contrast, ∼47–49 mol% soluble sugar was available after 20 min reaction in GVL/H2O and IPA/H2O (Fig. 1a), which could be further converted to LA or other sugar derivatives. To improve the catalytic performance in GVL/H2O and IPA/H2O, conversion of bread waste was conducted at 150 °C for 1–20 min reaction duration (Fig. 4a and b). As higher temperature provided more energy for the reaction, LA yield increased gradually up to 16.4 mol% in GVL/H2O (150 °C, 20 min) (Fig. 4b), which was more than three times higher than the maximum LA yield achieved at 130 °C (4.8 mol%, 20 min). The maximum LA yield achieved in IPA/H2O was only 6.6 mol% (150 °C, 20 min) (Fig. 4a), notably lower than GVL/H2O system, corroborating the positive role of aprotic GVL solvent in enhancing proton reactivity for less energy-intensive tandem catalysis.10,30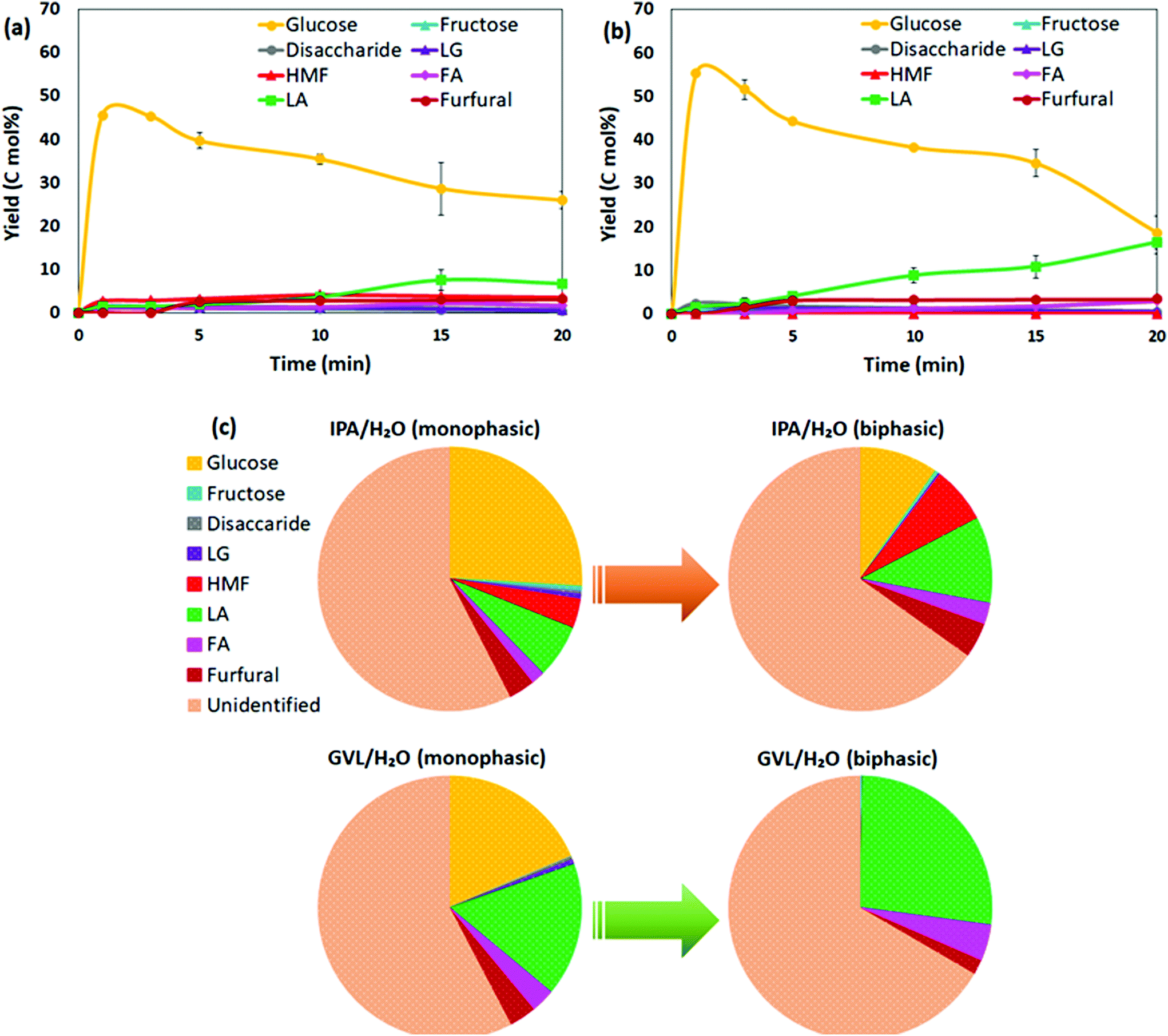 | ||
| Fig. 4 Product yields during the catalytic conversion of bread waste at 150 °C and 1–20 min reaction duration in (a) IPA/H2O (monophasic) and (b) GVL/H2O (monophasic); (c) comparative product yields in IPA/H2O and GVL/H2O monophasic and biphasic medium (30 wt% NaCl(aq)) at 150 °C, 20 min (reaction conditions: 5 wt% substrate loading, 0.5 M H2SO4, 1:1 solvent ratio). | ||
The biphasic solvent systems (prepared using 30 wt% NaCl(aq)) were further investigated as an intensification scheme to improve the concentration of the target product LA in this study. Promisingly, the concentration of LA at the end of the reaction (150 °C, 20 min) increased ∼1.6 times in both biphasic GVL/H2O and IPA/H2O systems (Fig. 4c) compared to analogous monophasic systems. Though total soluble product yields were comparable in the two biphasic systems (∼33–35 mol%) (Fig. S1b†), the distributions of soluble products were distinctive between GVL/H2O and IPA/H2O. While biphasic GVL/H2O selectively generated LA as the main product 26.8 mol% yield and 4.4 mol% FA as co-product, IPA/H2O biphasic contained 10.6 mol% LA, 2.6 mol% FA, ∼7 mol% HMF, and 10.2 mol% remaining sugars (glucose and fructose) (Fig. 4c). A considerable fraction of HMF present in both monophasic and biphasic IPA/H2O indicated that the solvent could hinder the HMF rehydration to LA. In comparison to DMSO, which is a widely recommended solvent for HMF production, IPA can create a shielding effect around HMF that helps to prevent its rehydration and suppress further conversion to byproducts such as insoluble humins.45,46 A recent study revealed that the rate constant for fructose to HMF was considerably higher than that of HMF to degradation products when IPA was present in the solvent system,46 echoing the experimental observations in this study where low LA selectivity was obtained in IPA/H2O solvent system. In other words, IPA/H2O could be considered as a potential solvent for HMF production in future investigations. By contrast, in the presence of GVL solvent, H2SO4 catalysed HMF production could be rapid and readily converted to LA at high acid strength.47 Considerable LA yield achieved in biphasic GVL/H2O in this study (26.8 mol%, at 150 °C, 20 min) proves the high efficiency of H2SO4/GVL/H2O system for LA production.
As GVL/H2O biphasic solvent was the most efficient for intensifying LA yield at moderate conditions, a comprehensive investigation was conducted for bread waste conversion in the co-solvent system following varied reaction conditions (150 °C, 10–20 min). In biphasic GVL/H2O, LA yield increased with increasing reaction duration (∼19–28 mol% in 10–15 min) (GVL/H2O, 1:1) (Fig. 5a). However, a slight decrease in LA yield was observed after 20 min, suggesting that LA started to degrade with prolonged reaction. The major product in monophasic GVL/H2O (1:1, 15 min) was glucose (34.6 mol%), while the biphasic system selectively generated LA (28 mol%) as the major product. The LA yield increased up to ∼2.5 times in the biphasic system compared to the monophasic one (∼11 mol%) (150 °C, 15 min), implying that phase separation could be beneficial for intensive LA production and upscaling to industrial scale.
 | ||
| Fig. 5 (a) Product yields during the catalytic conversion of bread waste in GVL/H2O monophasic and biphasic medium comprising different solvent ratios; (b) LA and (c) FA distribution in the organic and aqueous phase and partition coefficients obtained for the catalytic conversion of bread waste in GVL/H2O biphasic medium comprising different solvent ratios (reaction conditions: 5 wt% substrate loading, 0.5 M H2SO4, 30 wt% NaCl(aq) 150 °C, 10–20 min). | ||
Essentially, to develop an efficient biphasic reaction medium, it is necessary to understand the contribution and influences of the reaction phase (H2O) and the extraction phase (GVL) to optimise the yield of the target product. To tune the reaction and extraction phases in GVL/H2O biphasic system, catalytic tests were conducted with various ratios of GVL and H2O (1:1, 1:3, and 3:1 of GVL/H2O). The partition coefficient for LA (RLA) in the GVL/H2O biphasic solvent was calculated to evaluate the effect of the extraction phase (GVL) in the system. The RLA achieved for 10–15 min reaction was 4.2 when 1:1 GVL/H2O was applied for reaction (Fig. 5b), indicating a higher distribution of LA in the organic phase compared to the aqueous phase. Therefore, GVL solvent can be considered highly effective for reactive LA extraction. The partitioning of LA obtained in this study (RLA 4.2) is comparable to a previous study that reported RLA ranging 3.7–4 using cellulose as feedstock.21
The total yield of LA increased (27.4–32 mol%) when a higher fraction of H2O (1:3, GVL/H2O) was applied during catalytic conversion, whereas the total LA yield decreased (14.5–17.1 mol%) when a lower fraction of H2O (3:1, GVL/H2O) was applied (Fig. 5b). Though the total LA yield increased with a higher fraction of H2O, the extraction of LA was found to be inefficient. The partition coefficient achieved was only 1.1 (1:3, GVL/H2O), indicating nearly even distribution of produced LA in the reaction and extraction phases due to the lower fraction of GVL applied. In comparison, ∼97% of the total LA produced was extracted into the GVL phase when a higher fraction of GVL (3:1, GVL/H2O) was applied for reaction. However, a considerable decrease in total LA yield was observed, possibly due to the lack of sufficient aqueous phase available for reaction. These observations suggested the indispensable contribution of both reaction and extraction phases for an efficient biphasic system for LA production. Considering LA yield (28 mol%) and RLA (4.2), the 1:1 GVL/H2O biphasic system (150 °C, 15 min) provided the best performance for intensive LA production.
To reflect further on the mechanism and roles of catalytic species in NaCl modified GVL/H2O biphasic system, additional tests were performed under the selected reaction conditions. In the case of reaction conducted with only NaCl (30 wt%) without acid catalyst, only marginal product yield was observed (Fig. 6a). When NaCl was applied together with H2SO4 for bread waste conversion, an increase in LA yield was observed compared to the reaction in the presence of H2SO4 without salt addition. These observations indicate that NaCl alone could not catalyse the desired reaction yet it may act synergistically with H2SO4 enhancing the LA production in the aqueous phase during biphasic conversion of bread waste. Product distributions in the aqueous and GVL phases in the specified reaction conditions are provided in Fig. S3.† The synergistic catalysis by NaCl and H2SO4 together with simultaneous efficient extraction of LA into GVL could account for the high LA yield obtained in the GVL/H2O biphasic system.
 | ||
| Fig. 6 (a) LA yield and total product yield under different reaction conditions (reaction conditions: 5 wt% substrate loading, 150 °C), (b) LA yield and other product yields (sugar + HMF + furfural) in consecutive runs in GVL/H2O biphasic system (reaction conditions: 5 wt% substrate loading, 0.5 M H2SO4, 30 wt% NaCl(aq) 150 °C, 15 min, solvent ratio 1:1). | ||
Moreover, the recyclability of the aqueous phase containing H2SO4 was investigated. After the catalytic run (reaction and cooling), the reacted organic phase was removed and fresh GVL and bread waste substrate were applied for another cycle of catalytic reaction (run-2) again while retaining the aqueous phase from the previous run without adding extra H2SO4. Similarly, the third cycle of catalytic reaction (run-3) was conducted. In the 2nd and 3rd run (Fig. 6b), 15 mol% and 13 mol% LA was obtained, respectively, compared to the LA yield (28 mol%) obtained in the initial run, which suggested that the acid catalyst could be retained in the aqueous phase and recycled for further conversion, though a certain degree of decrease in LA yield was observed. Future studies would be required for evaluating the biphasic system in a continuous reaction mode, which may be more efficient compared to the batch reaction mode.
Apart from LA as the target product, FA yield was analysed for bread waste conversion in monophasic and biphasic GVL/H2O, as FA is co-produced during HMF rehydration to LA.48 Similar to LA, the FA yield was enhanced through phase separation with the yield from ∼2–3 mol% in the monophasic system to a maximum 5.4 mol% FA yield in biphasic GVL/H2O (150 °C, 15 min, 1:3 solvent ratio) (Fig. 5c). Using 1:1 GVL/H2O, FA partition coefficient (RFA) obtained was ∼2–3 (150 °C, 10–20 min, solvent ratio 1:1) (Fig. 5c), indicating that FA could also be simultaneously extracted into the GVL layer during bread waste conversion. Besides, partition coefficients for LA, FA, and HMF were evaluated in IPA/H2O solvent (150 °C, 20 min, 1:1 solvent ratio), and the values were 6.6, 5.4, and 5.7, respectively (Fig. S4†), corroborating an effective extraction of these compounds into the organic phase. Considering the low LA selectivity and total LA yield (10.6 mol% at 150 °C, 20 min), further investigation was discontinued for IPA/H2O.
Perspectives on LA production and solvent recycling
To evaluate the efficiency of the catalytic system for LA production, obtained LA yield relative to the theoretical LA yield from bread waste substrate (considering total available carbohydrate and possible maximum theoretical yield (64.5%)) was calculated and compared with the reported achievements with commercially available starch substrates in the existing literature (Table 1). For PC/H2O (130 °C, 20 min) and GVL/H2O biphasic solvents (150 °C, 15 min), 45.7% and 66% LA yield was achieved relative to theoretical yield, respectively. This is considered promising in comparison to the reported LA yield (45.6–66.4%) from commercially available starch substrates in previous studies49,50 where the conversion was conducted using comparatively harsh reaction conditions. For other recent studies where no direct comparison is possible due to the lack of data regarding relative LA yield,17,51 it should be noted that much longer reaction duration was required to obtain the reported LA yield.| Substrate | Reaction conditions | Catalyst | LA yield based on total organic carbon (mol%) | LA yield based on weight of substrate (%) | LA yield relative to theoretical yield (%) | Ref. |
|---|---|---|---|---|---|---|
| Bread waste | 130 °C, 20 min, H2O | 0.5 M H2SO4 | 1.5 | 1.2 | 3.6 | This study |
| 130 °C, 20 min, IPA/H2O | 1.0 | 0.8 | 2.4 | |||
| 130 °C, 20 min, GVL/H2O | 4.8 | 3.8 | 11.3 | |||
| 130 °C, 20 min, PC/H2O | 19.6 | 15.5 | 45.7 | |||
| 150 °C, 20 min, IPA/H2O (mono) | 6.6 | 5.3 | 15.6 | |||
| 150 °C, 20 min, IPA/H2O (bi) | 10.6 | 8.5 | 25.1 | |||
| 150 °C, 15 min, GVL/H2O (mono) | 10.9 | 8.8 | 25.8 | |||
| 150 °C, 15 min, GVL/H2O (bi) | 28 | 22.4 | 66.0 | |||
| 150 °C, 20 min, GVL/H2O (mono) | 16.4 | 13.2 | 38.8 | |||
| 150 °C, 20 min, GVL/H2O (bi) | 26.8 | 21.6 | 63.4 | |||
| Bread | 130 °C, 8 h | 1.5 ml 5 M H2SO4 and 10 ml GVL | — | 30.2 | — | 17 |
| Starch | 165 °C, 5 h | Sulfonated hyperbranched poly (arylene oxindole)s | — | 31.1 | — | 51 |
| Starch | 200 °C, 60 min | 4% H2SO4 | — | 47.5 | 66.4 | 50 |
| Sorghum grain | 200 °C, 40 min | 8% H2SO4 | 32.6 | 45.6 | 49 |
The recyclability of GVL/H2O biphasic solvent was investigated and discussed in the previous section. For PC/H2O solvent, despite partial decomposition of PC during acid catalysis, it offers various advantages as a solvent because a high product yield can be obtained with a moderate temperature and a short duration, which play a critical role in reducing the energy consumption and reactor size. The production of PC from CO2 is beneficial for process economy and environmental sustainability in terms of CO2 utilisation. In the application of PC solvent, the derived CO2 after reaction can be captured and recycled for PC production or other CO2 based biorefineries, which are conducive to fostering the circular economy. The spent organic solvents can be recycled after separation and purification following commonly used techniques such as fractional distillation. For instance, a comprehensive life cycle assessment (LCA) on solvent waste recovery52 demonstrated that implementing a solvent recovery system can reduce the environmental footprint of the total solvent manufacturing, usage, and disposal process. The energy requirements and emissions associated with the solvent recovery process have been recognised to be trivial in comparison to the emissions due to virgin solvent production and spent solvent disposal by incineration.
Conclusions
This study provides comprehensive insights into the critical factors for intensive LA production, which can be conducive to the technology scale-up. The bio-derived and CO2-derived co-solvents in this study, i.e., PC/H2O, GVL/H2O, IPA/H2O, could facilitate H2SO4 catalysed tandem reactions during bread waste conversion but demonstrated varied catalytic efficiency. PC/H2O solvent was the most efficient for LA production using moderate reaction conditions, which could be attributed to the CO2 pressure generated in situ through solvent (PC) degradation. Besides, catalytic performance could be significantly enhanced in GVL/H2O and IPA/H2O using the biphasic system, facilitating reactive extraction of LA in the organic layer that intensified the final concentration of LA. GVL/H2O biphasic solvent seems promising considering LA yield (28 mol%) and LA partitioning (RLA 4.2) achieved in the study. The bio-derived and CO2-derived solvents applied in this study can be considered as emerging solvents and potential alternatives to common industrial solvents in future biorefinery applications.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors appreciate the financial support from the Hong Kong International Airport Environmental Fund (Phase 2) and Hong Kong Research Grants Council (PolyU 15222020). The authors also acknowledge the support of the University Research Facility on Chemical and Environmental Analysis (UCEA) of PolyU.References
- UNEP. Food Waste Index Report 2021 http://www.unep.org/resources/report/unep-food-waste-index-report-2021 (accessed Apr 14, 2021).
- X. Xiong, I. K. M. Yu, D. C. W. Tsang, N. S. Bolan, Y. S. Ok, A. D. Igalavithana, M. B. Kirkham, K.-H. Kim and K. Vikrant, Value-Added Chemicals from Food Supply Chain Wastes: State-of-the-Art Review and Future Prospects, Chem. Eng. J., 2019, 375, 121983, DOI:10.1016/j.cej.2019.121983.
- T. M. W. Mak, X. Xiong, D. C. W. Tsang, I. K. M. Yu and C. S. Poon, Sustainable Food Waste Management towards Circular Bioeconomy: Policy Review, Limitations and Opportunities, Bioresour. Technol., 2020, 297, 122497, DOI:10.1016/j.biortech.2019.122497.
- S. S. Chen, T. Maneerung, D. C. W. Tsang, Y. S. Ok and C.-H. Wang, Valorization of Biomass to Hydroxymethylfurfural, Levulinic Acid, and Fatty Acid Methyl Ester by Heterogeneous Catalysts, Chem. Eng. J., 2017, 328, 246–273, DOI:10.1016/j.cej.2017.07.020.
- G. C. Hayes and C. R. Becer, Levulinic Acid: A Sustainable Platform Chemical for Novel Polymer Architectures, Polym. Chem., 2020, 11, 4068–4077, 10.1039/D0PY00705F.
- S. Dutta, I. K. M. Yu, D. C. W. Tsang, Y. H. Ng, Y. S. Ok, J. Sherwood and J. H. Clark, Green Synthesis of Gamma-Valerolactone (GVL) through Hydrogenation of Biomass-Derived Levulinic Acid Using Non-Noble Metal Catalysts: A Critical Review, Chem. Eng. J., 2019, 372, 992–1006, DOI:10.1016/j.cej.2019.04.199.
- C. Antonetti, D. Licursi, S. Fulignati, G. Valentini and A. M. Raspolli Galletti, New, Frontiers in the Catalytic Synthesis of Levulinic Acid: From Sugars to Raw and Waste Biomass as Starting Feedstock, Catalysts, 2016, 6, 196, DOI:10.3390/catal6120196.
- F. D. Pileidis and M.-M. Titirici, Levulinic Acid Biorefineries: New Challenges for Efficient Utilization of Biomass, ChemSusChem, 2016, 9, 562–582, DOI:10.1002/cssc.201501405.
- S. S. Chen, I. K. M. Yu, D. C. W. Tsang, A. C. K. Yip, E. Khan, L. Wang, Y. S. Ok and C. S. Poon, Valorization of Cellulosic Food Waste into Levulinic Acid synthesized by Heterogeneous Brønsted Acids: Temperature and Solvent Effects, Chem. Eng. J., 2017, 327, 328–335, DOI:10.1016/j.cej.2017.06.108.
- S. Dutta, I. K. M. Yu, D. C. W. Tsang, Z. Su, C. Hu, K. C. W. Wu, A. C. K. Yip, Y. S. Ok and C. S. Poon, Influence of Green Solvent on Levulinic Acid Production from Lignocellulosic Paper Waste, Bioresour. Technol., 2020, 298, 122544, DOI:10.1016/j.biortech.2019.122544.
- I. K. M. Yu, J. Fan, V. L. Budarin, F. P. Bouxin, J. H. Clark and D. C. W. Tsang, NaCl-Promoted Phase Transition and Glycosidic Bond Cleavage under Microwave Heating for Energy-Efficient Biorefinery of Rice Starch, Green Chem., 2020, 22, 7355–7365, 10.1039/D0GC01761B.
- I. K. M. Yu, D. C. W. Tsang, S. S. Chen, L. Wang, A. J. Hunt, J. Sherwood, K. De Oliveira Vigier, F. Jérôme, Y. S. Ok and C. S. Poon, Polar Aprotic Solvent-Water Mixture as the Medium for Catalytic Production of Hydroxymethylfurfural (HMF) from Bread Waste, Bioresour. Technol., 2017, 245, 456–462, DOI:10.1016/j.biortech.2017.08.170.
- I. K. M. Yu, D. C. W. Tsang, A. C. K. Yip, A. J. Hunt, J. Sherwood, J. Shang, H. Song, Y. S. Ok and C. S. Poon, Propylene Carbonate and γ-Valerolactone as Green Solvents Enhance Sn(IV)-Catalysed Hydroxymethylfurfural (HMF) Production from Bread Waste, Green Chem., 2018, 20, 2064–2074, 10.1039/C8GC00358K.
- C. Antonetti, D. Licursi and A. M. Raspolli Galletti, New, Intensification Strategies for the Direct Conversion of Real Biomass into Platform and Fine Chemicals: What Are the Main Improvable Key Aspects?, Catalysts, 2020, 10, 961, DOI:10.3390/catal10090961.
- S. S. Chen, L. Wang, I. K. M. Yu, D. C. W. Tsang, A. J. Hunt, F. Jérôme, S. Zhang, Y. S. Ok and C. S. Poon, Valorization of Lignocellulosic Fibres of Paper Waste into Levulinic Acid Using Solid and Aqueous Brønsted Acid, Bioresour. Technol., 2018, 247, 387–394, DOI:10.1016/j.biortech.2017.09.110.
- P. J. Dyson and P. G. Jessop, Solvent Effects in Catalysis: Rational Improvements of Catalysts via Manipulation of Solvent Interactions, Catal. Sci. Technol., 2016, 6, 3302–3316, 10.1039/C5CY02197A.
- M. Y. Lui, C. Y. Y. Wong, A. W.-T. Choi, Y. F. Mui, L. Qi and I. T. Horváth, Valorization of Carbohydrates of Agricultural Residues and Food Wastes: A Key Strategy for Carbon Conservation, ACS Sustainable Chem. Eng., 2019, 7, 17799–17807, DOI:10.1021/acssuschemeng.9b04242.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, CHEM21 Selection Guide of Classical- and Less Classical-Solvents, Green Chem., 2016, 18, 288–296, 10.1039/C5GC01008J.
- F. Gao, R. Bai, F. Ferlin, L. Vaccaro, M. Li and Y. Gu, Replacement Strategies for Non-Green Dipolar Aprotic Solvents, Green Chem., 2020, 22, 6240–6257, 10.1039/D0GC02149K.
- S. Dutta, I. K. M. Yu, D. C. W. Tsang, J. Fan, J. H. Clark, Z. Jiang, Z. Su, C. Hu and C. S. Poon, Efficient Depolymerization of Cellulosic Paper Towel Waste Using Organic Carbonate Solvents, ACS Sustainable Chem. Eng., 2020, 8, 13100–13110, DOI:10.1021/acssuschemeng.0c04102.
- S. G. Wettstein, D. M. Alonso, Y. Chong and J. A. Dumesic, Production of Levulinic Acid and Gamma-Valerolactone (GVL) from Cellulose Using GVL as a Solvent in Biphasic Systems, Energy Environ. Sci., 2012, 5, 8199, 10.1039/c2ee22111j.
- J. Wang, H. Cui, Y. Wang, R. Zhao, Y. Xie, M. Wang and W. Yi, Efficient Catalytic Conversion of Cellulose to Levulinic Acid in the Biphasic System of Molten Salt Hydrate and Methyl Isobutyl Ketone, Green Chem., 2020, 22, 4240–4251, 10.1039/D0GC00897D.
- L. K. Rihko-Struckmann, O. Oluyinka, A. Sahni, K. McBride, M. Fachet, K. Ludwig and K. Sundmacher, Transformation of Remnant Algal Biomass to 5-HMF and Levulinic Acid: Influence of a Biphasic Solvent System, RSC Adv., 2020, 10, 24753–24763, 10.1039/D0RA02784G.
- S. Kumar, V. Ahluwalia, P. Kundu, R. S. Sangwan, S. K. Kansal, T. M. Runge and S. Elumalai, Improved Levulinic Acid Production from Agri-Residue Biomass in Biphasic Solvent System through Synergistic Catalytic Effect of Acid and Products, Bioresour. Technol., 2018, 251, 143–150, DOI:10.1016/j.biortech.2017.12.033.
- Z. Jiang, J. Remón, T. Li, V. L. Budarin, J. Fan, C. Hu and J. H. Clark, A One-Pot Microwave-Assisted NaCl–H2O/GVL Solvent System for Cellulose Conversion to 5-Hydroxymethylfurfural and Saccharides with in Situ Separation of the Products, Cellulose, 2019, 26, 8383–8400, DOI:10.1007/s10570-019-02362-8.
- I. K. M. Yu, X. Xiong, D. C. W. Tsang, Y. H. Ng, J. H. Clark, J. Fan, S. Zhang, C. Hu and Y. S. Ok, Graphite Oxide- and Graphene Oxide-Supported Catalysts for Microwave-Assisted Glucose Isomerisation in Water, Green Chem., 2019, 21, 4341–4353, 10.1039/C9GC00734B.
- M. L. Foresti, M. Williams, P. del, R. Martínez-García and A. Vázquez, Analysis of a Preferential Action of α-Amylase from B. Licheniformis towards Amorphous Regions of Waxy Maize Starch, Carbohydr. Polym., 2014, 102, 80–87, DOI:10.1016/j.carbpol.2013.11.013.
- L. Qi, R. Alamillo, W. A. Elliott, A. Andersen, D. W. Hoyt, E. D. Walter, K. S. Han, N. M. Washton, R. M. Rioux, J. A. Dumesic and S. L. Scott, Operando Solid-State NMR Observation of Solvent-Mediated Adsorption-Reaction of Carbohydrates in Zeolites, ACS Catal., 2017, 7, 3489–3500, DOI:10.1021/acscatal.7b01045.
- AIST. Spectral Database for Organic Compounds (SDBS). 2018, https://sdbs.db.aist.go.jp/sdbs/cgi-bin/cre_index.cgi (accessed May 10, 2021) Search PubMed.
- M. A. Mellmer, C. Sener, J. M. R. Gallo, J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Solvent Effects in Acid-Catalyzed Biomass Conversion Reactions, Angew. Chem., Int. Ed., 2014, 53, 11872–11875, DOI:10.1002/anie.201408359.
- M. A. Mellmer, D. M. Alonso, J. S. Luterbacher, J. M. R. Gallo and J. A. Dumesic, Effects of γ-Valerolactone in Hydrolysis of Lignocellulosic Biomass to Monosaccharides, Green Chem., 2014, 16, 4659–4662, 10.1039/C4GC01768D.
- M. A. Mellmer, C. Sanpitakseree, B. Demir, P. Bai, K. Ma, M. Neurock and J. A. Dumesic, Solvent-Enabled Control of Reactivity for Liquid-Phase Reactions of Biomass-Derived Compounds, Natl. Catal., 2018, 1(3), 199–207, DOI:10.1038/s41929-018-0027-3.
- B. M. Flanagan, M. J. Gidley and F. J. Warren, Rapid Quantification of Starch Molecular Order through Multivariate Modelling of 13C CP/MAS NMR Spectra, Chem. Commun., 2015, 51, 14856–14858, 10.1039/C5CC06144J.
- J. Cai, C. Cai, J. Man, Y. Yang, F. Zhang and C. Wei, Crystalline and Structural Properties of Acid-Modified Lotus Rhizome C-Type Starch, Carbohydr. Polym., 2014, 102, 799–807, DOI:10.1016/j.carbpol.2013.10.088.
- N. Atichokudomchai, S. Varavinit and P. Chinachoti, A Study of Ordered Structure in Acid-Modified Tapioca Starch by 13C CP/MAS Solid-State NMR, Carbohydr. Polym., 2004, 58, 383–389, DOI:10.1016/j.carbpol.2004.07.017.
- C. Mutungi, L. Passauer, C. Onyango, D. Jaros and H. Rohm, Debranched Cassava Starch Crystallinity Determination by Raman Spectroscopy: Correlation of Features in Raman Spectra with X-Ray Diffraction and 13C CP/MAS NMR Spectroscopy, Carbohydr. Polym., 2012, 87, 598–606, DOI:10.1016/j.carbpol.2011.08.032.
- W. R. Morrison, R. F. Tester, M. J. Gidley and J. Karkalas, Resistance to Acid Hydrolysis of Lipid-Complexed Amylose and Lipid-Free Amylose in Lintnerised Waxy and Non-Waxy Barley Starches, Carbohydr. Res., 1993, 245, 289–302, DOI:10.1016/0008-6215(93)80078-S.
- R. Hoover, Acid-treated Starches, Food Rev. Int., 2000, 16, 369–392, DOI:10.1081/FRI-100100292.
- K. Lappalainen, N. Vogeler, J. Kärkkäinen, Y. Dong, M. Niemelä, A. Rusanen, A. L. Ruotsalainen, P. Wäli, A. Markkola and U. Lassi, Microwave-Assisted Conversion of Novel Biomass Materials into Levulinic Acid, Biomass Convers. Biorefin., 2018, 8, 965–970, DOI:10.1007/s13399-018-0334-6.
- Z. Zhang, I. M. O'Hara, D. W. Rackemann and W. O. S. Doherty, Low Temperature Pretreatment of Sugarcane Bagasse at Atmospheric Pressure Using Mixtures of Ethylene Carbonate and Ethylene Glycol, Green Chem., 2013, 15, 255–264, 10.1039/C2GC36323B.
- A. R. C. Morais, M. D. D. J. Matuchaki, J. Andreaus and R. Bogel-Lukasik, A Green and Efficient Approach to Selective Conversion of Xylose and Biomass Hemicellulose into Furfural in Aqueous Media Using High-Pressure CO2 as a Sustainable Catalyst, Green Chem., 2016, 18, 2985–2994, 10.1039/C6GC00043F.
- S. Jing, X. Cao, L. Zhong, X. Peng, X. Zhang, S. Wang and R. Sun, In Situ Carbonic Acid from CO2: A Green Acid for Highly Effective Conversion of Cellulose in the Presence of Lewis Acid, ACS Sustainable Chem. Eng., 2016, 4, 4146–4155, DOI:10.1021/acssuschemeng.6b00623.
- G. P. Van Walsum, Severity Function Describing the Hydrolysis of Xylan Using Carbonic Acid. in Twenty-Second Symposium on Biotechnology for Fuels and Chemicals, ed. B. H. Davison, J. McMillan and M. Finkelstein, ABAB Symposium; Humana Press, Totowa, NJ, 2001, DOI:10.1007/978-1-4612-0217-2_27.
- A. Toscan, A. R. C. Morais, S. M. Paixão, L. Alves, J. Andreaus, M. Camassola, A. J. P. Dillon and R. M. Lukasik, High-Pressure Carbon Dioxide/Water Pre-Treatment of Sugarcane Bagasse and Elephant Grass: Assessment of the Effect of Biomass Composition on Process Efficiency, Bioresour. Technol., 2017, 224, 639–647, DOI:10.1016/j.biortech.2016.11.101.
- S. H. Mushrif, S. Caratzoulas and D. G. Vlachos, Understanding Solvent Effects in the Selective Conversion of Fructose to 5-Hydroxymethyl-Furfural: A Molecular Dynamics Investigation, Phys. Chem. Chem. Phys., 2012, 14, 2637, 10.1039/c2cp22694d.
- J. K. C. N. Agutaya, R. Inoue, S. S. V. Tsie, A. T. Quitain, J. de la Peña-García, H. Pérez-Sánchez, M. Sasaki and T. Kida, Metal-Free Synthesis of HMF from Glucose Using the Supercritical CO2 –Subcritical H2O–Isopropanol System, Ind. Eng. Chem. Res., 2020, 59, 16527–16538, DOI:10.1021/acs.iecr.0c03551.
- L. Qi, Y. F. Mui, S. W. Lo, M. Y. Lui, G. R. Akien and I. T. Horváth, Catalytic Conversion of Fructose, Glucose, and Sucrose to 5-(Hydroxymethyl)Furfural and Levulinic and Formic Acids in γ-Valerolactone As a Green Solvent, ACS Catal., 2014, 4, 1470–1477, DOI:10.1021/cs401160y.
- T. Flannelly, M. Lopes, L. Kupiainen, S. Dooley and J. J. Leahy, Non-Stoichiometric Formation of Formic and Levulinic Acids from the Hydrolysis of Biomass Derived Hexose Carbohydrates, RSC Adv., 2016, 6, 5797–5804, 10.1039/C5RA25172A.
- Q. Fang and M. A. Hanna, Experimental Studies for Levulinic Acid Production from Whole Kernel Grain Sorghum, Bioresour. Technol., 2002, 81, 187–192, DOI:10.1016/S0960-8524(01)00144-4.
- J. Y. Cha and M. A. Hanna, Levulinic Acid Production Based on Extrusion and Pressurized Batch Reaction, Ind. Crops Prod., 2002, 16, 109–118, DOI:10.1016/S0926-6690(02)00033-X.
- S. V. de Vyver, J. Thomas, J. Geboers, S. Keyzer, M. Smet, W. Dehaen, P. A. Jacobs and B. F. Sels, Catalytic Production of Levulinic Acid from Cellulose and Other Biomass-Derived Carbohydrates with Sulfonated Hyperbranched Poly(Arylene Oxindole)s, Energy Environ. Sci., 2011, 4, 3601–3610, 10.1039/C1EE01418H.
- M. J. Raymond, C. S. Slater and M. J. Savelski, LCA Approach to the Analysis of Solvent Waste Issues in the Pharmaceutical Industry, Green Chem., 2010, 12, 1826–1834, 10.1039/C003666H.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc01948a |
| This journal is © The Royal Society of Chemistry 2022 |