
Efficient discovery of potent α-glucosidase inhibitors from Paeoniae lactiflora using enzyme–MOF nanocomposites and competitive indicators†
Xinlin
Chen
 a,
Ying
Wu
a,
Yucheng
Gu
b,
Jianguang
Luo
*a and
Lingyi
Kong
*a
a,
Ying
Wu
a,
Yucheng
Gu
b,
Jianguang
Luo
*a and
Lingyi
Kong
*a
aState Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Bioactive Natural Product Research, School of Traditional Chinese Pharmacy, China Pharmaceutical University, 24 Tong Jia Xiang, Nanjing 210009, PR China. E-mail: luojg@cpu.edu.cn; cpu_lykong@126.com; Tel: +8625 8327 1402 Tel: +86 25 8327 1405
bSyngenta, Jealott's Hill International Research Centre, Bracknell, Berkshire RG42 6EY, UK
First published on 28th November 2022
Abstract
A great deal of attention has been paid to the seeds of Paeoniae lactiflora pall., an underutilized food resource, since its extract exhibits excellent α-glucosidase (GAA) inhibitory activity. In the present study, to gain further insight into this plant and find out potent GAA inhibitors, we established a novel ligand fishing strategy by introducing a competitive inhibitor as an indicator. After the successful establishment of this approach was verified by a series of methods, including kinetic assay, fluorescence determination, and HPLC, the newly developed ligand fishing method was applied to acquire potent GAA inhibitors from P. lactiflora seeds. Nine bioactive compounds were captured, and seven of them were identified as suffruticosol A, suffruticosol B, resveratrol, vitisin E, luteolin, trans-δ-viniferin, and ampelopsin E. The data of their GAA inhibitory activity demonstrated that these constituents were vigorously active against GAA with IC50 values of 1.67–30.47 μM, while such value of 1-DNJ was 228.77 μM. Among them, vitisin E and ampelopsin E were reported to show such inhibitory activity for the first time. Collectively, our findings provide valuable clues for the further utilization of P. lactiflora seeds as a functional food, and offer a new avenue for acquiring potent inhibitors from natural resources.
1. Introduction
Diabetes mellitus is one of the most prevalent epidemic diseases worldwide.1 Fortunately, type 2 diabetes is considered a preventable condition and can be managed by changes in diet or medicines that help control blood glucose levels by inhibiting the activity of α-glucosidase (GAA).2,3 Additionally, patients are also willing to eat functional foods with hypoglycemic effects to help reduce postprandial blood glucose.4 In China, such functional foods can be seen on the market, such as mango leaf tea,5 lotus leaf tea,6 and vine tea,7 which show GAA inhibitory activity and good effects on diabetic patients. These functional foods come from some underused materials of commercial fruits or plants. There are many unutilized by-products globally, and they are worthy of further potential applications as ingredients in the food industrial and pharmaceutical fields.Increasing research studies on plants of Paeoniaceae for its nutritive and medicinal value have been reported because of the presence of adequate unsaturated fatty acids, monoterpene glycosides, phenolic compounds, and stilbene oligomers in it.8Paeoniae lactiflora pall., a member of the Paeoniaceae family, is a kind of desirable ornamental plant.9 As a kind of traditional Chinese medicine, the roots of this plant are used as analgesic, sedative, anti-inflammatory, and pyretic drugs.10 Nowadays, investigation and application put more emphasis on the nutritive and medicinal value of its seed oil.11 Besides, our experiments illustrated that the extract of P. lactiflora seeds (PS) showed potent inhibitory activity against GAA. Despite having found some GAA inhibitors in PS,12 scientific evidence discloses that this underutilized by-product of PS still has enormous potential as a functional food for further analysis due to its extraordinary nutritive value and pharmaceutical potency.13
The screening of bioactive compounds has been one of the challenges due to their presence in complex chemical systems or low concentrations.14 The conventional methods, including systematic separation and bioactivity-guided separation, are tedious and time-consuming.15 The affinity-based ligand fishing screening assay has been established to simplify the process, which depends on the principle of protein target-ligand binding.16 The target protein can be immobilized on materials to directly obtain potential inhibitors/activators from mixed samples.17 Moreover, the overall performance of this analytical method improves with the development of enzymatic support materials.18 For example, compared with magnetic beads, using nanoparticles like metal–organic frameworks (MOFs) can enhance the protein loading capacity and catalytic activity of immobilized enzymes due to their high ratio of surface area and biocompatibility.19–21 However, despite the advantages of ligand fishing in terms of efficiency and selectivity in screening active compounds, ligands can be “fished out” once they bind to the target protein regardless of whether they show the corresponding activity.22 Inevitably, screening once again via activity assay in vitro is needed to find potent inhibitors from potential ligands. Thus, although the efficiency of ligand fishing is better than that of the traditional way for discovering bioactive constituents from natural resources, it still needs improvement.
As a unique and innovative approach for molecular sensing, indicator displacement assay is widely used to detect complex analytes or discriminate unknown sample mixtures.23 It relies on the principle of competitive displacement between labeled and unlabeled analytes for host molecules.24 In recent years, this principle has also been applied to identify enzyme inhibitors.25 This screening method based on a competitive displacement assay shows greater efficiency since it can afford potent inhibitors with superior activity compared with indicators.26 Inspired by this principle, we assumed that probes or inhibitors with moderate inhibitory activity could be added as the indicator and they bound to the active site of GAA competitively to obtain potent active compounds from the extract of PS. Simultaneously, combined with immobilized enzymes, potent inhibitors were harvested directly from the mixture through simple two-dimensional separation. Under an ideal situation, the indicator–enzyme complex could acquire inhibitors with better activity than the indicator by competitive substitution from the plant extract.
Hence, to further explore potent GAA inhibitors from PS efficiently and provide another idea for the method of ligand fishing, we established the method based on a competitive indicator and GAA@MOF nanoparticles in the present study. Using MOF-based immobilized GAA (GAA@UiO-66-NH2) nanoparticles as a vehicle and a GAA competitive inhibitor as an indicator, the established strategy had been applied successfully to acquire potent GAA inhibitors from PS. Nine compounds in PS were discovered efficiently as potent GAA inhibitors via this approach. Seven of these potent inhibitors were identified with the aid of HPLC-Q-TOF-MS/MS and NMR. Besides, their powerful activities against GAA were proved by GAA inhibitory assay in vitro as proof to further verify the feasibility of this established approach. Meanwhile, these data mentioned above indicated which constituents of PS had GAA inhibitory activities. This study could supply scientific clues for the further development of PS as a functional food.
2. Materials and methods
2.1. Materials and instruments
All chemicals and reagents were of analytical grade and used without any further purification. ZrCl4, aminoterephthalic acid (ATA), gallic acid, p-nitrophenyl-α-D-glucopyranoside (pNPG), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDCI), N-hydroxysuccinimide (NHS), 1-deoxynojirimycin (1-DNJ), p-toluene sulfochloride, 4-pentyn-1-ol, 2H-1-benzopyran-2-one, and 7-hydroxy-4-trifluoromethylcoumarin were obtained from Aladdin Reagent Co., Ltd (Shanghai, China). α-Glucosidase (EC.3.2.1.20, from Saccharomyces cerevisiae) and 3-azido-1-propanol were supplied by Sigma-Aldrich (MA, USA). Naringenin and resveratrol were obtained from Shanghai Yuanye Bio-Technology Co., Ltd (Shanghai, China). N,N-Dimethyl formamide (DMF), acetic acid (HAc), HCl, methanol, dimethyl sulfoxide (DMSO), ethanol and acetonitrile were provided by CINC High Purity Solvents Co., Ltd (Shanghai, China).The samples were subjected to HPLC-Q-TOF-MS analysis on an Agilent 1290 LC instrument coupled with a 6520 Quadrupole-Time-of-Flight Mass Spectrometer via an ESI source. Data acquisition and analysis were conducted using Agilent Masshunter software Ver. B. 04. 00. The HPLC analysis was carried out using an Agilent 1260 series HPLC instrument. Preparative HPLC was performed on a Shimadzu LC-6A equipped with a Shim-pack RP-C18 column (20 × 200 mm, 10 μm). The flow rate was set at 10.0 mL min−1, the column temperature was maintained at 25 °C, and the eluents were detected at a wavelength of 254 nm using a binary-channel UV detector. The 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were recorded on a Bruker ACF-500 II spectrometer, and TMS was employed as an internal standard. A Bruker Tensor 27 spectrometer was employed to record IR (KBr-disc) spectra. Ni-filtered Cu Kα radiation (k1 = 1.54 Å) generated at a voltage of 40 keV and a current of 40 mA was used for powder X-Ray diffraction (XRD) analysis with a Bruker D8 Advance X-Ray diffractometer. Field emission scanning electron microscopy (FESEM) of MOFs was carried out on a Hitachi FESEM SU8200 operated at 15.0 kV.
2.2. Synthesis of the indicator
The compound 5 was synthesized according to previously reported literature with some modifications.27 Briefly, the N-alkylation of 1-DNJ through nucleophilic displacement of 1-pentyn-4-tosylate with K2CO3 alone afforded the desired N-alkynyl DNJ derivative (compound 2). Besides, compound 4 was produced by coupling 7-hydroxy-4-trifluoromethylcoumarin to 3-azido-1-propanol tosylate in the presence of K2CO3 in DMF. Then, the click azide–alkyne cycloaddition was carried out between 2 and 4 in the presence of catalytic copper sulfate, producing the target compound 5. The detailed procedures are listed in the ESI.†2.3. Characterization of the indicator
Kinetic assay used pNPG as the substrate, which was hydrolyzed by GAA to release p-nitrophenol. The enzyme kinetic analysis was performed according to the above reaction using Lineweaver–Burk plots 1/v versus 1/[S]. The quantity of GAA was maintained at 0.5 U mL−1, and inhibitors of different concentrations (0, 125, 250, 500, and 625 μM) were measured in various concentrations of pNPG (0.3125, 0.625, 1.25, 2.5, and 5 mM).For fluorescence assay, compound 5 was dissolved in PBS and the concentration was brought to 1 mM. The excitation and emission spectra were recorded to find the maximum emission of compound 5. Then, 90 μL PBS solution of compound 5 (1 mM) was incubated with 10 μL GAA solutions of different concentrations (0.078 mg mL−1 to 1.25 mg mL−1) at 37 °C for 30 min to assay the changes of fluorescence intensity. Besides, to test the displacement ability of the potent inhibitor, 10 μL GAA solution (0.5 U mL−1) was incubated with 89 μL compound 5 PBS solution (0.1 mM) and 1 μL naringenin (NG) solutions of different concentrations (0 μg mL−1 to 20 μg mL−1) at 37 °C for 30 min. The fluorescence spectra were recorded at an emission wavelength range from 300 to 550 nm with an excitation wavelength of 270 nm using a Shimadzu FR-6000 luminescence spectrometer.
2.4. Synthesis of the immobilized α-glucosidase (GAA@UiO-66-NH2)
The synthesis of the MOFs was carried out using our previously reported method.21 The immobilized GAA was synthesized through covalent linkages. Precisely, 5 mg UiO-66-NH2 was mixed with 100 μL GAA solution (10 mg mL−1 in 50 mM PBS, pH 6.8), and then 100 μL NHS (5 mg mL−1 in 50 mM PBS, pH 6.8) and 100 μL EDCI solution (5 mg mL−1 in 50 mM PBS, pH 6.8) were added. The resulting mixture was agitated at 4 °C for 90 min. Subsequently, the suspension was centrifuged (8000 rpm, 5 min) and washed three times, meanwhile, the supernatant was collected for calculating the capacity of protein loading. The quantity of immobilized GAA on MOFs and the relative activity recovery were determined following a previously described method.28 The immobilized GAA activity and free GAA activity were tested in 0.5 mL centrifuge tubes. 1 μL enzyme solution was diluted with 59 μL PBS (50 mM, pH 6.8) and mixed with 40 μL 2.5 mM pNPG solution to start reacting for 10 min at 37 °C. Then, 80 μL Na2CO3 was injected to stop the reaction. The GAA activity was detected at 405 nm and a calibration curve was constructed from the generation of p-nitrophenol (pNP) per minute at 37 °C. The quantity of enzyme needed to catalyze the release of 1 μM pNP per min at pH 6.8 and 37 °C was defined as one unit (U) of enzyme activity (initial reaction rate).2.5. Measurements and characterization of GAA@UiO-66-NH2
The introduction of functional groups obtained from the reaction was explored by the FT-IR technique. Moreover, qualitative analysis was conducted in a parallel analysis:20 (1) free GAA and (2) GAA@UiO-66-NH2. In brief, 10 μL of the above-mentioned sample solutions was injected into 1.5 mL centrifuge tubes. Subsequently, 1 μL NG of six different concentrations (0, 31.25, 62.5, 125, 250, and 500 μM) was added into each tube and diluted with 49 μL PBS. The mixed solution was incubated at 37 °C for 10 min. Then, 40 μL of 2.5 mM pNPG was added, and the mixture was reacted at 37 °C for 10 min. After the reaction, 80 μL Na2CO3 was injected to stop the reaction. Moreover, the supernatant was collected and recorded at an absorbance of 405 nm.2.6. Performances of ligand fishing based on the competitive indicator and GAA@UiO-66-NH2
The compound 5 was designated as an indicator. For verifying the displacement effect between the potent inhibitor and indicator, the NG was chosen as a potent inhibitor. In detail, 5 mg GAA@UiO-66-NH2 was suspended and incubated with 500 μL indicator solution (2 mg indicator in 50 mM PBS solution, pH 6.8) at 37 °C for 30 min, and then 1 μL NG (40 mg mL−1 in DMSO) was added and incubated at 37 °C for 90 min. After that, the mixture was centrifuged and washed with 50 mM PBS (pH 6.8) three times. Finally, 95% (v/v) methanol solution was injected to release ligands from GAA, and the supernatant was collected and named E1A. The same procedures were performed for immobilized GAA only with the indicator to get the sample named E1B. UiO-66-NH2 was tested in the same way to get a blank control. Chromatographic separation was performed on a Shim-pack: Agilent ZORBAX SB-C18 column (4.6 × 250 mm, 5 μm), with the mobile phase consisting of 0.2% (v/v) formic acid aqueous solution (A) and methanol (B) according to the following gradient program: 5%–100% B at 0–50 min. The UV detection wavelength was set at 230 nm, and the column temperature was maintained at 25 °C.The self-made model mixed sample of E2 (E2M) consisted of 100 μg NG, 100 μg gallic acid and 100 μg (+)-catechin. 5 mg GAA@UiO-66-NH2 was suspended in and incubated with 500 μL indicator solution (2 mg indicator in 50 mM PBS solution, pH 6.8) at 37 °C for 30 min, and then the model mixed sample was added. The mixed solution was incubated at 37 °C for 90 min, followed by centrifugation. Then the precipitate was rinsed with 50 mM PBS (pH 6.8) three times to remove non-specific compounds. Finally, 1 mL of 95% (v/v) methanol solution was added to inactivate the GAA and release the potential potent inhibitors, condensing and yielding the eluent named E2A. The same incubation of GAA@UiO-66-NH2 without an indicator was carried out and the sample named E2B was obtained. Besides, the same incubation of UiO-66-NH2 with E2M was performed as a blank control. Chromatographic separation was performed as mentioned above. To observe whether other compounds with similar characteristics of compound 5 could also be regarded as indicators or not, experiments were carried out in the same way as for E2 except for replacing compound 5 with 500 μg NG, and choosing resveratrol as a potent inhibitor. Chromatographic separation was performed on a Shim-pack: Agilent ZORBAX SB-C18 column (4.6 × 250 mm, 5 μm), with the mobile phase consisting of 0.2% (v/v) formic acid aqueous solution (A) and methanol (B) according to the following gradient program: 5%–100% B at 0–50 min. The UV detection wavelength was set at 230 nm, and the column temperature was maintained at 25 °C.
2.7. Plant material and sample preparation
P. lactiflora seeds were obtained from Anhui Province, China, and identified by Prof. Mian Zhang of the research department of pharmacognosy, China Pharmaceutical University. The P. lactiflora seeds (500 g) were extracted by ultrasonication with 70% (v/v) ethanol solution three times (each time for 3 h). The solvent was removed under reduced pressure, and the seed extract was obtained. Moreover, ampelopsin E, suffruticosol A, suffruticosol B, vitisin E, resveratrol and luteolin were isolated from PS via kinds of chromatography methods, and detailed procedures are listed in the ESI.†2.8. Application of ligand fishing in the extract of PS
5 mg GAA@UiO-66-NH2 was mixed with 2 mg indicator (dissolved in 5 μL DMSO) and diluted with 495 μL PBS solution, followed by incubation at 37 °C for 30 min. Subsequently, as the target sample, 1 mg extract of PS (dissolved in 5 μL DMSO) was added in the mixed solution mentioned above and incubated at 37 °C for 90 min. Afterwards, the suspension was centrifuged, and the precipitate was harvested and then rinsed with 500 μL of 50 mM PBS (pH 6.8) three times to remove non-specific constituents. Finally, 1 mL of 95% (v/v) methanol solution was injected to inactivate GAA and release potential ligands. After centrifugation, the supernatant was obtained and named PS-S for further analysis. The same incubation procedures for GAA@UiO-66-NH2 and UiO-66-NH2 were performed and they were named PS-S′ and blank control, respectively.2.9. Identification of compounds by HPLC-Q-TOF-MS/MS
An Agilent ZORBAX SB-C18 column (4.6 × 250 mm, 5 μm) was employed to perform chromatographic separation with a flow rate of 0.2 mL min−1 and an injection volume of 5 μL. The temperature of the column was set at 30 °C. A quadrupole-time-of-flight mass spectrometer was used in negative ionization mode. The major parameters were set as follows: gas temperature, 320 °C; drying gas (nitrogen) flow rate, 10 L min−1; nebulizer, 45 psig; capillary voltage, 4.0 kV; fragmentor, 170 V; and skimmer, 65 V. Nitrogen was used as the collision gas for MS/MS scan. The mobile phase consisted of 0.1% (v/v) formic acid aqueous solution (A) and methanol (B). The elution program was as follows: 5%–40% B at 0–30 min, 40%–50% B at 30–35 min, 50%–50% B at 35–45 min, 50%–80% B 45–70 min, and 80%–100% B at 70–80 min.2.10. α-Glucosidase inhibitory activity assay
The inhibitory assay was performed according to the previously described method with some modifications.29 The assay was performed in a 96-well microplate. GAA was dissolved in 50 mM PBS (pH 6.8) to a concentration of 0.5 U mL−1. 10 μL GAA solution was added in a microplate and mixed with 50 μL sample solution (diluting with PBS). The mixture was preincubated with an inhibitor at 37 °C for 10 min, and then 40 μL pNPG (2.5 mM) was added and incubated for 10 min at 37 °C. Subsequently, 80 μL Na2CO3 was injected to stop the reaction. Using a microplate reader, the amount of released 4-nitrophenyl was measured in terms of the absorbance at 405 nm. The percent inhibition [%] was calculated using the following formula: inhibition [%] = [1 − (AS − ASB)/(AC − ACB)] × 100, where AC, ACB, AS, and ASB were the absorbance of the control, control blank, sample and sample blank, respectively. The IC50 value was calculated from three independent assays performed in triplicate. 1-DNJ was used as a positive control.2.11. Data presentation and statistical analysis
All the experiments were repeated at least three times, and were presented as the mean ± standard deviation (SD). The calculation of the 50% inhibitory concentration (IC50) in the enzyme activity was performed by nonlinear regression. The data of kinetics of GAA inhibition were analyzed using Origin 2018 64Bit software.3. Results and discussion
3.1. Design of ligand fishing based on the competitive indicator and GAA@UiO-66-NH2
To find out potent GAA inhibitors efficiently from the extract of PS, meanwhile, enriching methods of ligand fishing, we designed the indicator-based ligand fishing approach and drew a schematic diagram (Fig. 1). In this strategy, the indicator played the role of a “goalkeeper”, which occupied the active region of GAA and prevented inhibitors with weaker inhibitory activity to enter. Ideally, the indicator held the active sites of catalytic areas first. Then, the potent inhibitors of PS were added to displace the indicator via a competitive strategy. Consequently, lower-affinity GAA inhibitors would be excluded by the immobilized enzyme-integrated indicator to get the potent inhibitors efficiently. Undoubtedly, the active site-directed competitive indicator and immobilized enzyme were used as practical tools to profile the proposed method of ligand fishing. Before implementing the established strategy, the amounts, kinds and GAA inhibitory activity levels of compounds in plants were unknown. Thus, a competitive GAA inhibitor with moderate activity was chosen to act as the indicator to avoid missing out all potent GAA inhibitors as far as possible. For immobilized enzymes, the relative recovery activity and protein loading capacity were essential for this newly established approach.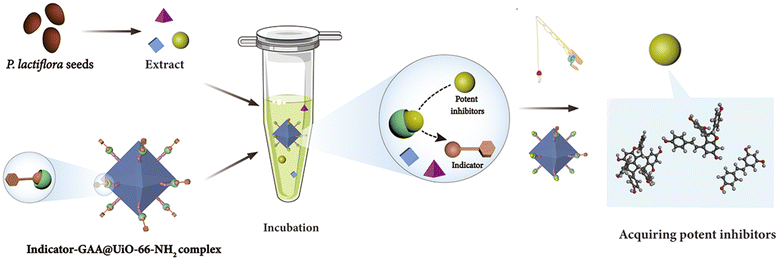 | ||
| Fig. 1 Schematic illustration of the ligand fishing based on the competitive indicator and GAA@UiO-66-NH2. | ||
3.2. Characterization of GAA@UiO-66-NH2
Previous studies have demonstrated that the MOFs are a type of nano-scale supporter of immobilized enzymes with lots of merits.30,31 The typical MOFs, UiO-66-NH2, show excellent water stability and large surface areas.32 The nano-scale UiO-66-NH2 was acquired successfully through the method from our previous work21 and proved by XRD and SEM (Fig. S1†). This MOF was selected as a supporter to immobilize GAA through covalent linkages. Therefore, GAA was supposed to be anchored onto the surface of UiO-66-NH2 by forming the amide bond between the amino group of MOFs and the carboxyl group of protein. FT-IR was used as a valuable method to disclose changes in the functional groups to verify such a hypothesis. Fig. 2E describes the positions and differences of band areas for GAA@UiO-66-NH2 compared with controls. The bands at 1607 cm−1 and 3418 cm−1 of UiO-66-NH2 were assigned to the scissor bending and stretching vibrations of the amino group, respectively.33 However, in the spectrum of immobilized GAA, the characteristic vibration bands can be seen at 1626 cm−1 (amide I), 1575 cm−1 (amide II), and 1440 cm−1 (amide III),34 indicating the presence of the amide group.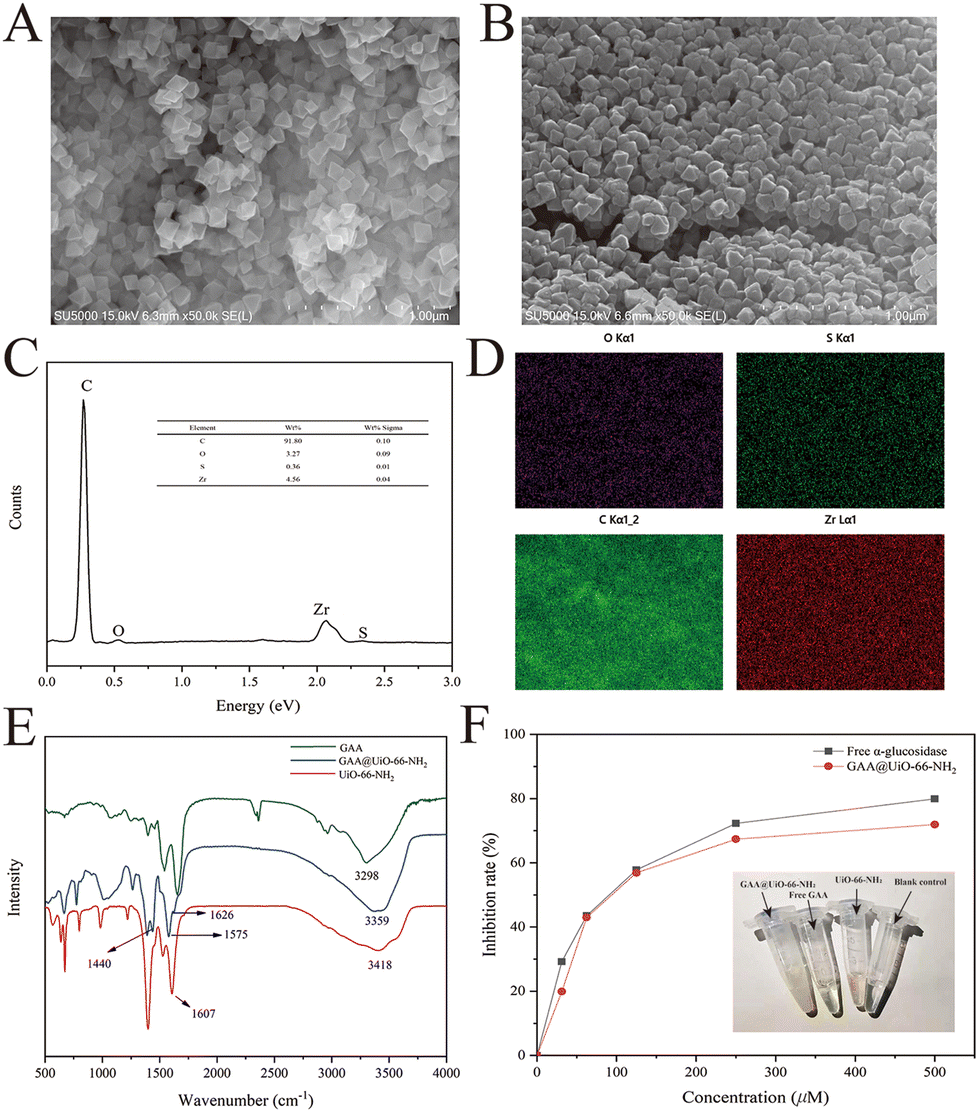 | ||
| Fig. 2 Characterization of GAA@UiO-66-NH2via SEM, EDS, FT-IR and parallel experiments. FESEM images of (A) UiO-66-NH2 and (B) GAA@UiO-66-NH2. EDS analysis (C) and mapping (D) of GAA@UiO-66-NH2. (E) FT-IR analysis of GAA, UiO-66-NH2 and GAA@UiO-66-NH2. (F) Qualitative analysis of the free GAA and GAA@UiO-66-NH2 with different concentrations of NG. Inset: the macroscopic appearances of GAA@UiO-66-NH2 and free GAA and their blank controls. | ||
Additionally, Fig. 2B shows that the surface of immobilized GAA was rougher than that of UiO-66-NH2, further indicating the successful immobilization of GAA. Besides, the EDS mapping data testified that GAA was successfully immobilized on MOFs, evidenced by the presence of the element sulfur (Fig. 2C and D). The element sulfur is a component of the disulfide bond in protein,35 and it was only from GAA in this research. As a result, data of FT-IR, SEM, and EDS proved the successful immobilization of GAA via covalent linking with MOFs. In addition, to observe whether the site of the active area was exposed or not, the activity of GAA@UiO-66-NH2 was assayed by measuring the capacity for hydrolyzing the glycosylic bond of pNPG. According to the above-mentioned approaches, the relative activity recovery of GAA@UiO-66-NH2 was 46.28%, and the protein loading capacity achieved 72.07 mg g−1. Meanwhile, the catalytic activity of GAA@UiO-66-NH2 was also tested through parallel assay testing with free GAA. The activity of immobilized GAA was gradually decreased when the inhibitor concentration was increased, which was consistent with the trend of free GAA (Fig. 2F). Next, the optimal reaction temperature and pH were studied. Fig. S2† depicts the effects of pH and temperature on the activity of immobilized enzymes. The optimal pH of GAA@UiO-66-NH2 was 6.8, the optimal reaction temperature was 37 °C, and these values were identical to those of free GAA. Altogether, our experimental data demonstrated that the GAA was successfully immobilized onto UiO-66-NH2, and the activity of GAA was maintained during the whole synthetic process.
3.3 Synthesis and characterization of the indicator
1-DNJ, a typical phytochemical GAA inhibitor with moderate inhibitory activity, has been reported to bind with GAA via competitive inhibition.36,37 To verify the inhibitory characteristics of 1-DNJ, the kinetic assay was performed using Lineweaver–Burk plots 1/v versus 1/[S] (Fig. 3A). The Lineweaver–Burk plot of 1-DNJ generated straight lines that had intersections on the Y-axis, demonstrating that it was exactly a competitive inhibitor of GAA. However, it is difficult to monitor this inhibitor through HPLC-DAD because of no ultraviolet absorption.38 Fortunately, 1-DNJ can be modified with a chromophore. To get a suitable indicator, 1-DNJ was modified according to a previously described approach with some modifications.27 The secondary amine of 1-DNJ was nucleophilic and displaced by 4-pentyn-1-tosylate, and compound 2 was acquired with a yield of 44%. Secondly, the 7-hydroxy-4-trifluoromethyl was selected as a chromophore and reacted with 1-azidopropyl-3-tosylate to obtain compound 4 with a yield of 84%. Finally, compound 2 was linked with compound 4via click chemistry to get the target compound 5 with a yield of 33%. All synthetic compounds were characterized by 1H and 13C NMR, as well as ESI-MS (Fig. S8–20†).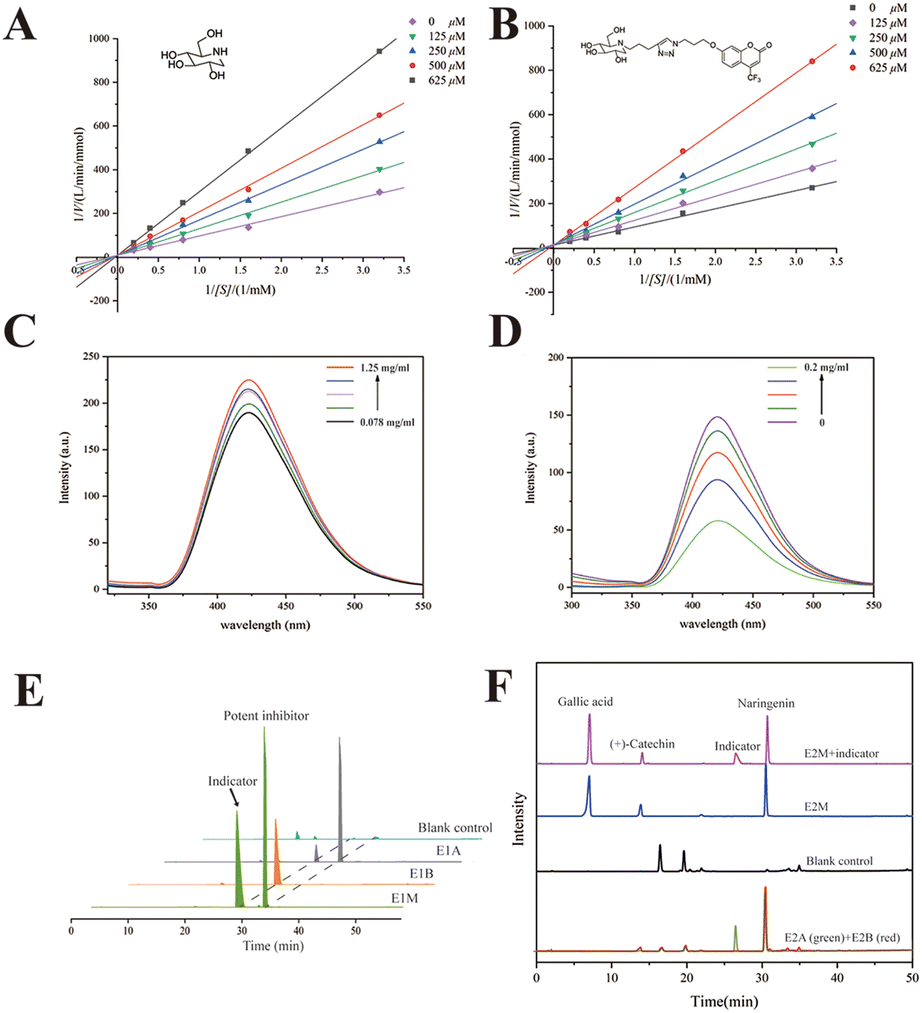 | ||
| Fig. 3 Characterization of the indicator. Lineweaver–Burk double reciprocal plots of 1-DNJ (A) and the indicator (B); c(GAA) = 0.5 U mL−1 and c(pNPG) = 0.3125, 0.625, 1.25, 2.5, and 5 mM. (C) Fluorescence spectra of the indicator in the presence of GAA with different concentrations (pH 6.8, T = 25 °C, λex = 270 nm). c(indicator) = 1 mM, and c(GAA) = 0.078, 0.156, 0.312, 0.625 and 1.25 mg mL−1. (D) Fluorescence spectra of GAA and the indicator in the presence of NG with different concentrations (pH 6.8, T = 25 °C, λex = 270 nm). c(indicator) = 1 mM, c(GAA) = 0.05 mg mL−1, and c(NG) = 0, 0.025, 0.05, 0.1 and 0.2 mg mL−1. (E) The HPLC chromatograms obtained from GAA@UiO-66-NH2 incubation with the indicator and NG (E1A), GAA@UiO-66-NH2 only incubation with the indicator (E1B), and UiO-66-NH2 incubation with mixed solution (contained NG and indicator, E1M) as the blank control. (F) HPLC chromatograms of a mixed sample of E2 (E2M, containing the indicator, potent inhibitor NG, weak inhibitor (+)-catechin and no active compound gallic acid). HPLC chromatograms obtained from GAA@UiO-66-NH2 incubation with the indicator and E2M (E2A), GAA@UiO-66-NH2 only incubation with E2M (E2B) and UiO-66-NH2 incubation with E2M (blank control). | ||
Then, the 1-DNJ derivative (5) was assessed to determine whether it was eligible to be an indicator. Compared with 1-DNJ, the GAA inhibitory activity of compound 5 was slightly decreased, with an IC50 value of 309.10 ± 1.97 μM (1-DNJ, IC50 = 228.77 ± 3.78 μM), indicating that it also could bind with the catalytically hydrolyzed site of GAA and had a moderate GAA inhibitory activity. Besides, the Lineweaver–Burk double reciprocal plots were further used to study the behaviour of 1-DNJ and compound 5 against GAA. Fig. 3 shows the GAA inhibitory activities in the presence of pNPG and test samples (1-DNJ and compound 5) at various concentrations. All of the lines intersected at a certain point on the Y-axis, and the slope increased with the increase of inhibitor concentration. These results demonstrated that the GAA inhibitory actions of 1-DNJ and compound 5 were similar, and were all based on competition.
To verify whether a potent inhibitor could displace the indicator from the active site of GAA, compound 5 was dissolved in 50 mM PBS (pH 6.8) to measure fluorescence characteristics. Spectral analysis displayed a fluorescence peak at 410 nm, which belonged to 7-hydroxy-4-trifluoromethylcoumarin (Fig. S3†). Subsequently, compound 5 at a constant concentration was incubated with GAA at a range of concentrations. It was found that the fluorescence peak intensity of compound 5 could be enhanced when it bound with GAA (Fig. 3C). Utilizing this increase in fluorescence peak intensity, it could be tested whether a potent GAA inhibitor could displace the indicator. Theoretically, the fluorescence enhancement effect would be impaired when a potent inhibitor was added because the potent inhibitor destroyed the binding between GAA and the indicator. Therefore, the fluorescence changes were observed before and after the addition of a potent inhibitor. The NG was chosen as a potent inhibitor because of its better GAA inhibitory activity with an IC50 value of 83.51 ± 3.34 μM. Fig. 3D shows that after NG of different concentrations was added, the fluorescence intensity decreased with the increase of NG concentration. In conclusion, compound 5 was a suitable vehicle for follow-up study, and a potent inhibitor could displace the indicator from GAA.
3.4. Performances of ligand fishing based on the competitive indicator and GAA@UiO-66-NH2
In solution, it has been verified that the indicator can be displaced by a potent indicator from the active site of free GAA. Then, the indicator should be proved to have the same effect in ligand fishing. Relevant comparative experiments were divided into two groups, named E1 and E2. The model mixed solution of E1 only contained one potent inhibitor to certify whether the indicator could be displaced by a potent inhibitor from the active site of immobilized GAA. Changes in the indicator's peak intensity were monitored in a sample in the presence of a potent inhibitor (E1A) or in the absence of a potent inhibitor (E1B). The NG was chosen as the potent inhibitor. Fig. 3E shows that the immobilized GAA could capture the indicator and NG from the model solution (E1M). Besides, comparing the difference in the indicator's peak intensity in E1A and E1B, it was easy to find that the concentration of indicator became smaller after NG was added. In detail, comparing the change of the peak area, the peak of the indicator in E1A was decreased by 81.1% compared with that in E1B. This revealed that NG could displace the indicator from the catalytic area of GAA@UiO-66-NH2, even though the number of indicators was quite in excess.To test the specificity and feasibility of ligand fishing based on the indicator and GAA@UiO-66-NH2, the model mixed solution of E2 (E2M) contained one potent inhibitor, one weak inhibitor, and one compound that was not active against GAA. The NG was chosen as a potent inhibitor, and gallic acid with no GAA inhibitory activity was chosen as a negative control. (+)-Catechin was chosen as a weak inhibitor because of its weaker activity against GAA with an IC50 value of 357.53 ± 1.24 μM, and its activity was slightly weaker than that of the indicator as well. To test whether weak inhibitors could be ruled out in this way, the peak intensity of weak inhibitors was monitored in the samples with the indicator (E2A), without the indicator (E2B), and blank control containing only MOFs incubated with the model mixed solution to exclude the false-positive control. These experiments were carried out parallelly. Fig. 3F shows that the potent inhibitor and weak inhibitor were recognized by GAA@UiO-66-NH2 in the sample of E2B. Besides, gallic acid was not detected in all samples, suggesting that GAA@UiO-66-NH2 could recognize GAA inhibitors accurately. Significantly, only the potent inhibitor was recognized after introducing the indicator, evidenced by the drastically decreased peak intensity of the weak inhibitor in E2A. This demonstrated that the weak inhibitor failed to displace the indicator from GAA@UiO-66-NH2, and only the potent inhibitor was acquired by ligand fishing based on the competitive indicator and GAA@UiO-66-NH2. Meanwhile, it was proved that the indicator helped impede low-affinity ligands from coming into the active site of GAA, and an inhibitor with higher affinity was capable of being acquired directly through the competitive way. As we successfully verified the accuracy and feasibility of this newly established method, we assumed that other compounds with similar characteristics to compound 5 could also be regarded as the indicator. The kinetic assay of NG was tested and is displayed in Fig. S3.† NG was proved to be a reversible inhibitor with a mixed inhibition mechanism, indicating that it could also get into the active site of GAA and meet the requirements of the indicator. Therefore, the same procedures of E2 were repeated, except that the potent indicator was replaced by resveratrol (stronger activity against GAA than against NG). The sample obtained by adding an indicator group was named E2A′, while the group with no indicator was named E2B′. As predicted, NG as an indicator was also a “goalkeeper” to help screen potent inhibitors (Fig. S5†). Overall, all the above data illustrated the accuracy and feasibility of ligand fishing based on the competitive indicator and immobilized GAA. Subsequently, this newly established method of ligand fishing would be tested in the PS extract and identify potent GAA inhibitors efficiently at the same time.
3.5. Ligand fishing and HPLC-Q-TOF-MS/MS analysis of the extract of PS
The bioassay data displayed that the extract of PS exhibited strong GAA inhibitory activity with an IC50 value of 8.84 ± 0.23 μg mL−1. The HPLC chromatogram also indicated complicated constituents of this plant (Fig. S6†). To test our hypothesis, the extract was incubated with GAA@UiO-66-NH2 and an indicator (PS-S). Meanwhile, GAA@UiO-66-NH2 (PS-S′) and UiO-66-NH2 (blank control) were respectively used following the same steps to make a comparison and exclude non-specific compounds. Fig. 4 shows that 11 potential ligands, peaks 2, 4, 5, 6, 7, 8, 9, 10, 11, 12 and 13, were captured by GAA@UiO-66-NH2. After the indicator was added, only peaks 5, 6, 7, 8, 9, 10, 11, 12 and 13 were obtained, indicating that peaks 2 and 4 were not potent GAA inhibitors. Peak 7 was covered by the peak of the indicator, while its molecular ion peak was found in the data of PS-S via HPLC-Q-TOF-MS/MS. To gain an insight into what constituents existed in PS and contributed to GAA inhibition, all samples were analysed with the help of HPLC-Q-TOF-MS/MS. All the compounds shown in the sample of PS-E were figured out according to the literature,39 and are listed in Table S3.† Interestingly, it was easily found that the constituents acquired by the indicator–enzyme complex were almost stilbene oligomers. Monoterpene glycosides showed weak GAA inhibitory activity and even no activity against GAA. These results illustrated that stilbene oligomers of PS were a critical component for GAA inhibition. However, it was difficult to distinguish them merely using mass spectra because of plentiful isomerides. Therefore, peaks 5, 6, 7, 10, 11, and 13 were isolated from PS through traditional extraction and separation methods, and they were identified by NMR (Fig. S21–40†) as suffruticosol A (peak 5), suffruticosol B (peak 6), resveratrol (peak 7), vitisin E (peak 10), luteolin (peak 11), and ampelopsin E (peak 13). Peaks 8, 9 and 12 were not obtained via traditional extraction and separation methods. Peak 4 was also isolated and identified as paeoniflorin to prove the validity of the established method. Generally speaking, nine potent inhibitors in the extract of PS were captured via our established strategy of ligand fishing, and six compounds were acquired through the traditional separation method for further GAA inhibitory activity assay.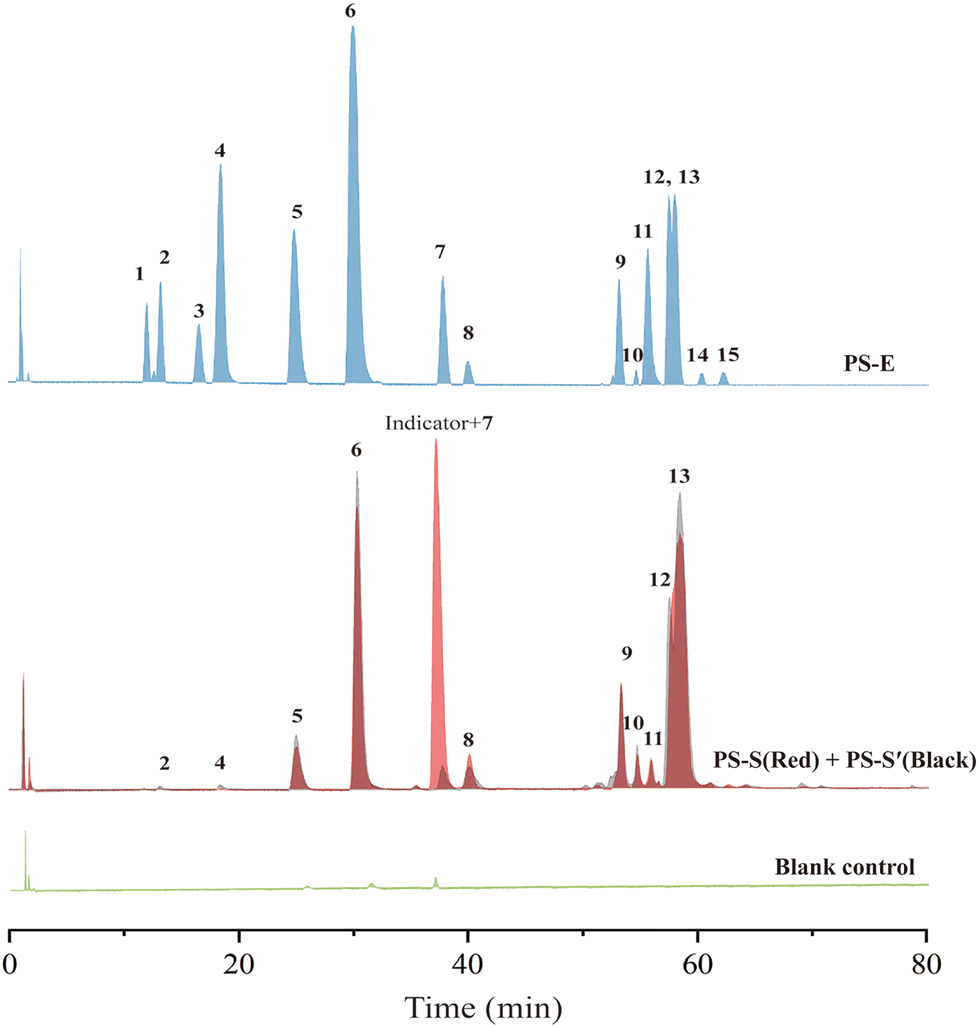 | ||
| Fig. 4 HPLC-Q-TOF-MS chromatograms of the extract of PS (PS-E) and the ligand fishing results obtained from the extract of PS with the indicator and GAA@UiO-66-NH2 (PS-S), GAA@UiO-66-NH2 (PS-S′) and UiO-66-NH2 (blank control). | ||
3.6. Validation of results obtained from the established method of ligand fishing
For further verification, suffruticosol A, suffruticosol B, resveratrol, vitisin E, luteolin, and ampelopsin E were tested for their GAA inhibitory activity in vitro and the results are listed in Table 1. As expected, peak 4 indicated a weak GAA inhibitor, which could suppress GAA with a large IC50 value of 4.18 mM. Table 1 shows that all six compounds exhibited excellent activities, with IC50 values ranging from 1.67 to 30.47 μM compared with 1-DNJ, which was regarded as a positive control with an IC50 value of 228.77 ± 3.78 μM. Among these GAA inhibitors, previous studies on vitisin E and ampelopsin E had reported their antioxidant, anti-cancer, and tyrosinase inhibitory activities.40 However, the two compounds were reported to have strong GAA inhibitory activity for the first time. To promote further understanding on the inhibitory characteristics of these potent GAA inhibitors, the kinetic assay was carried out using Lineweaver–Burk plots 1/v versus 1/[S] (Fig. 5). The Lineweaver–Burk plots of these inhibitors demonstrated that suffruticosol A, vitisin E, and ampelopsin E inhibited GAA in a competitive manner, while suffruticosol B, resveratrol, and luteolin inhibited GAA in a mixed manner. Therefore, according to the results in the forepart, the feasibility of our established method used for obtaining potent GAA inhibitors from PS was proved. To sum up, the information mentioned above elucidated that the nine potent GAA inhibitors were obtained from PS using the established method. Besides, this underutilized by-product of P. lactiflora could be selected as a considerable potential source for reducing postprandial blood glucose since it contained abundant stilbene oligomers with excellent GAA inhibitory activity.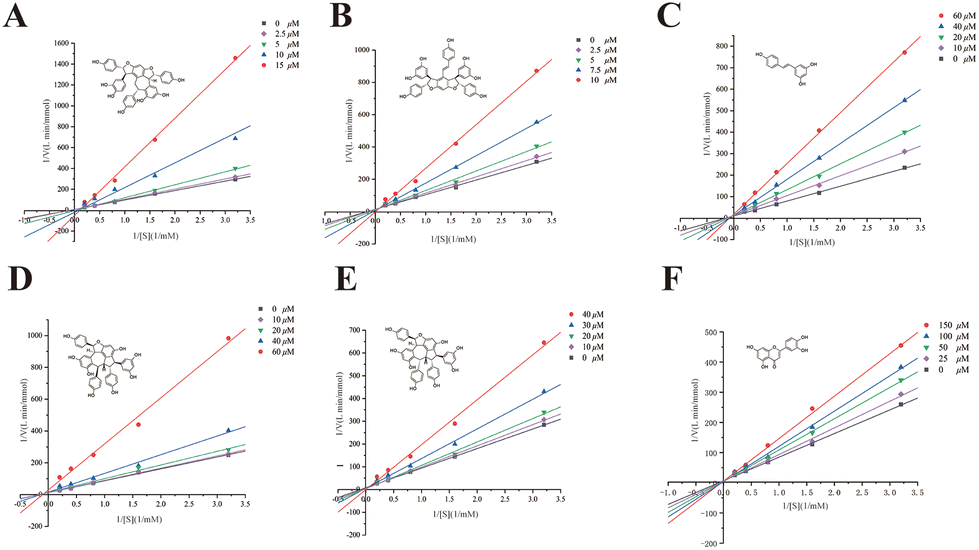 | ||
| Fig. 5 Lineweaver–Burk double reciprocal plots of vitisin E (A), ampelopsin E (B), resveratrol (C), suffruticosol B (D), suffruticosol A (E) and luteolin (F), c(GAA) = 0.5 U mL−1, c(pNPG) = 0.3125, 0.625, 1.25, 2.5, and 5 mM. | ||
| Peak no. | Compounds | IC50 (μM) | Inhibitory type |
|---|---|---|---|
| 5 | Suffruticosol A | 17.79 ± 0.86 | Competitive |
| 6 | Suffruticosol B | 10.21 ± 0.4 | Mixed |
| 7 | Resveratrol | 12.86 ± 1.02 | Mixed |
| 10 | Vitisin E | 4.15 ± 0.19 | Competitive |
| 11 | Luteolin | 30.47 ± 0.48 | Mixed |
| 13 | Ampelopsin E | 1.67 ± 0.13 | Competitive |
| Positive control | 1-DNJ | 228.77 ± 3.78 | Competitive |
4. Conclusions
In the present study, we successfully established a novel ligand fishing strategy by introducing competitive inhibitors as the indicator for efficiently screening potent GAA inhibitors from PS. Nine potent GAA inhibitors were acquired, and the GAA inhibitory activity of six of them was verified via activity assay in vitro. The results indicated that these constituents were vigorously active against GAA. Among these potent inhibitors, two compounds, ampelopsin E and vitisin E, have been reported to show such inhibitory activity for the first time. Besides, our findings revealed that abundant stilbene oligomers with potent GAA inhibitory activity in PS made this “scrap” a candidate functional food for reducing postprandial blood glucose. Meanwhile, overall research demonstrated that integrating competitive indicators with immobilized enzymes based on MOFs improved the efficiency of ligand fishing, to a certain extent. The established competitive method of ligand fishing was expected to rapidly acquire potent bioactive components from complex mixtures, and be widely used in the food industry.Author contributions
Xinlin Chen: conceptualization, methodology, formal analysis, validation, software, and writing – original draft; Ying Wu: software, data curation, and resources; Jianguang Luo: writing – reviewing & editing and project administration; Lingyi Kong: funding acquisition. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the 111 Project from the Ministry of Education of China and the State Administration of Foreign Export Affairs of China (B18056) and a Syngenta Ph. D. fellowship awarded to X. L. C.References
- R. C. Turner, R. R. Holman, C. A. Cull and I. M. Stratton, et al., Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33), Lancet, 1998, 9131, 837–853 Search PubMed.
- M. L. Barrett and J. K. Udani, A proprietary alpha-amylase inhibitor from white bean (Phaseolus vulgaris): A review of clinical studies on weight loss and glycemic control, Nutr. J., 2011, 10, 24–34 CrossRef PubMed.
- H. E. Lebovitz, Alpha-glucosidase inhibitors as agents in the treatment of diabetes, Diabetes Rev., 1998, 6, 132–145 Search PubMed.
- P. Mirmiran, Z. Bahadoran and F. Azizi, Functional foods-based diet as a novel dietary approach for management of type 2 diabetes and its complications: A review, World J. Diabetes, 2014, 3, 267–281 CrossRef.
- F. Y. Shi, L. W. Xie, Q. L. Lin, C. Y. Tong, Q. C. Fu, J. J. Xu, J. B. Xiao and S. Y. Shi, Profiling of tyrosinase inhibitors in mango leaves for a sustainable agroindustry, Food Chem., 2020, 312, 126042–126053 CrossRef CAS PubMed.
- G. Uddin, A. Rauf, B. S. Siddiqui, M. Arfan, I. U. Rahman and I. Khan, Proximate chemical composition and antimicrobial activities of fixed oils from Diospyros lotus L., Med. Chem., 2013, 3, 282–285 Search PubMed.
- R. C. V. Carneiro, L. Y. Ye, N. Baek, G. H. A. Teixeira and S. F. O'Keefe, Vine tea (Ampelopsis grossedentata): A review of chemical composition, functional properties, and potential food applications, J. Funct. Foods, 2021, 76, 104317–104326 CrossRef CAS.
- S. H. Wu, D. G. Wu and Y. W. Chen, Chemical constituents and bioactivities of plants from the genus Paeonia, Chem. Biodivers., 2010, 7, 90–104 CrossRef CAS.
- J. J. Li, Tree peony of China, Encyclopedia of China Publishing House, Beijing, China, 2011, pp. 1–20 Search PubMed.
- R. Nie, Y. R. Zhang, H. Zhang, Q. Z. Jin, G. C. Wu and X. G. Wang, Effect of different processing methods on physicochemical properties, chemical compositions and in vitro antioxidant activities of Paeonia lactiflora Pall seed oils, Food Chem., 2020, 332, 127408–127415 CrossRef CAS PubMed.
- J. S. Meng, Y. H. Tang, J. Sun, D. Q. Zhao, K. L. Zhang and J. Tao, Identification of genes associated with the biosynthesis of unsaturated fatty acid and oil accumulation in herbaceous peony ‘Hangshao’ (Paeonia lactiflora ‘Hangshao’) seeds based on transcriptome analysis, BMC Genomics, 2021, 22, 94–115 CrossRef CAS PubMed.
- C. C. Zhang, C. A. Geng, X. Y. Huang, X. M. Zhang and J. J. Chen, Antidiabetic stilbenes from peony seeds with PTP1B, α-glucosidase, and DPPIV inhibitory activities, J. Agric. Food Chem., 2019, 24, 6765–6772 CrossRef PubMed.
- R. X. Deng, J. Y. Gao, J. P. Yi and P. Liu, Peony seeds oil by-products: Chemistry and bioactivity, Ind. Crops Prod., 2022, 187, 115333–115359 CrossRef CAS.
- D. J. Newman and G. M. Cragg, Natural products as sources of new drugs from 1981 to 2014, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- B. Wang, J. Deng, Y. Gao, L. Zhu, H. Rui and Y. Xu, The screening toolbox of bioactive substances from natural products: A review, Fitoterapia, 2011, 82, 1141–11517 CrossRef CAS PubMed.
- M. W. Wang, X. Hao and K. Chen, Biological screening of natural products and drug innovation in China, Philos. Trans. R. Soc. B, 2007, 362, 1093–1105 CrossRef CAS PubMed.
- C. Łukasz and R. Moaddel, Comparison of analytical techniques for the identification of bioactive compounds from natural products, Nat. Prod. Rep., 2016, 33, 1131–1145 RSC.
- Y. F. Ping, L. M. Zhang, X. Wang and A. Van Schepdael, Off-line and on-line liquid chromatography-mass spectrometry methods with immobilized bio-macromolecules for drug screening from natural sources, J. Chromatogr. A, 2022, 1683, 463538–463547 CrossRef CAS PubMed.
- Y. P. Shen, M. Wang, J. W. Zhou, Y. F. Chen, M. R. Wu, Z. Z. Yang, C. Y. Yang, G. H. Xia, J. P. Tam, C. S. Zhou, H. Yang and X. B. Jia, Construction of Fe3O4@alpha-glucosidase magnetic nanoparticles for ligand fishing of alpha-glucosidase inhibitors from a natural tonic Epimedii Folium, Int. J. Biol. Macromol., 2020, 165, 1361–1372 CrossRef CAS PubMed.
- Z. Wang, X. Q. Li, M. H. Chen, F. Y. Liu, C. Han, L. Y. Kong and J. G. Luo, A strategy for screening of α-glucosidase inhibitors from Morus alba root bark based on the ligand fishing combined with high-performance liquid chromatography mass spectrometer and molecular docking, Talanta, 2018, 180, 337–345 CrossRef CAS PubMed.
- X. L. Chen, S. Xue, Y. L. Lin, J. G. Luo and L. Y. Kong, Immobilization of porcine pancreatic lipase onto a metal-organic framework, PPL@MOF: A new platform for efficient ligand discovery from natural herbs, Anal. Chim. Acta, 2020, 1099, 94–102 CrossRef CAS PubMed.
- R. J. Zhuo, L. Hao, N. N. Liu and Y. Wang, Ligand fishing: A remarkable strategy for discovering bioactive compounds from complex mixture of natural products, Molecules, 2016, 21, 1516–1527 CrossRef PubMed.
- B. T. Nguyen and E. V. Anslyn, Indicator-displacement assays, Coord. Chem. Rev., 2006, 250, 3118–3127 CrossRef CAS.
- A. C. Sedgwicj, J. T. Brewster, T. H. Wu, X. Feng, S. D. Bull, X. H. Qian, J. L. Sessler, T. D. James, E. V. Anslyn and X. L. Sun, Indicator displacement assays (IDas): the past, present and future, Chem. Soc. Rev., 2021, 50, 9–38 RSC.
- Z. P. Xiao, D. Chen, S. S. Song, R. Van der vlag, P. E. Van der Wouden, R. Van Merkerk, R. H. Cool, A. K. H. Hirsch, B. N. Melgert, W. J. Quax, G. J. Poelarends and F. J. Dekker, 7-Hydroxycoumarins are affinity-based fluorescent probes for competitive binding studies of macrophage migration inhibitory factor, J. Med. Chem., 2020, 20, 11920–11933 CrossRef PubMed.
- P. Koutnik, E. G. Shcherbakova, S. Gozem, M. G. Caglayan, T. Minami and P. Anzenbacher, Fluorescence-based assay for carbonic anhydrase inhibitors, Chem, 2017, 2, 271–282 CAS.
- A. Hatano, Y. Kanno, Y. Kondo, Y. Sunaga, H. Umezawa, M. Okada, H. Yamada, R. Lwaki, A. Kato and K. Fukui, Synthesis and characterization of novel, conjugated, fluorescent DNJ derivatives for α-glucosidase recognition, Bioorg. Med. Chem., 2016, 25, 773–778 CrossRef PubMed.
- S. L. Cao, D. M. Yue, X. H. Li, T. J. Smith, N. Li, M. H. Zong, H. Wu, Y. Z. Ma and W. Y. Lou, Novel nano/micro-biocatalyst: soybean epoxide hydrolase immobilized on UiO-66-NH2 MOF for efficient biosynthesis of enantipure (R)-1, 2-octanediol in deep eutectic solvents, ACS Sustainable Chem. Eng., 2016, 4, 3586–3595 CrossRef CAS.
- X. Q. Wu, H. Xu, H. Yue, K. Q. Liu and X. Y. Wang, Inhibition kinetics and the aggregation of α-glucosidase by different denaturants, Protein J., 2009, 28, 448–456 CrossRef CAS PubMed.
- W. J. Duan, Z. F. Zhao, H. D. An, Z. J. Zhang, P. Chen, Y. Chen and H. Huang, State-of-the-art and prospects of biomolecules: Incorporation in functional metal-organic frameworks, Top. Curr. Chem., 2019, 377, 34–65 CrossRef PubMed.
- Z. Y. Gu, J. Park, A. Raiff, Z. W. Wei and H. C. Zhou, Metal-organic frameworks as biomimetic catalysts, ChemCatChem, 2014, 6, 1–9 CrossRef.
- P. M. Schoenecker, C. G. Carson, H. Jasuja, C. J. J. Flemming and K. S. Walton, Effect of water adsorption on retention of structure and surface area of metal-organic frameworks, Ind. Eng. Chem. Res., 2012, 51, 6513–6519 CrossRef CAS.
- J. Yang, Y. Dai, X. Y. Zhu, Z. Wang, Y. S. Li, Q. X. Zhuang, J. L. Shi and J. L. Gu, Metal-organic frameworks with inherent recognition sites for selective phosphate sensing through their coordination-induced fluorescence enhancement effect, J. Mater. Chem. A, 2015, 3, 7445–7452 RSC.
- B. Jovi, M. Pani, N. Radnovi, K. Zivojevic, M. Mladenovic, V. Crnojevic and N. Knezevic, Investigation of the surface interactions of selected amides with mesoporous silica using FTIR spectroscopy and hyperspectral imaging, J. Mol. Struct., 2020, 1219, 128562–128569 CrossRef.
- M. Depuydt, J. Messens and J. F. Collet, How proteins form disulfide bonds, Antioxid. Redox Signaling, 2011, 15, 49–66 CrossRef CAS PubMed.
- H. J. Wang, Y. Shen, Y. L. Zhao and Y. F. Ye, 1-Deoxnojirinicin and its derivatives: A mini review of the literature, Curr. Med. Chem., 2021, 28, 628–643 CAS.
- Z. H. Liu, Y. Yang, W. J. Dong, Q. Liu, R. Y. Wang, J. M. Pang, X. J. Xia, X. Y. Zhu, S. N. Liu, Z. F. Shen, Z. Y. Xiao and Y. L. Liu, Investigation on the enzymatic profile of mulberry alkaloids by enzymatic study and molecular docking, Molecules, 2019, 24, 1776–1791 CrossRef CAS PubMed.
- B. Saunier, R. D. Kilker, J. S. Tkacz, A. Quaroni and A. Herscovics, Inhibition of N-linked complex oligosaccharide formation by 1-deoxynojirimycin, an inhibitor of processing glucosidases, J. Biol. Chem., 1982, 257, 14155–14161 CrossRef CAS PubMed.
- R. Nie, Y. R. Zhang, Q. Z. Jin, S. Zhang, G. C. Wu, L. Chen, H. Zhang and X. G. Wang, Identification and characterization of bioactive compounds from the seed kernels and hulls of Paeonia lactiflora, Pall by UPLC-QTOF-MS, Food Res. Int., 2021, 139, 109916–109929 CrossRef CAS PubMed.
- Y. Q. Xue, J. M. Di, Y. Luo, K. J. Cheng, X. Wei and Z. Shi, Resveratrol oligomers for the prevention and treatment of cancers, Oxid. Med. Cell. Longevity, 2014, 2014, 765832–765841 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2fo02783f |
| This journal is © The Royal Society of Chemistry 2023 |