 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spiers Memorial Lecture: prospects for photoelectron spectroscopy
Wendy R.
Flavell
 *
*
Department of Physics and Astronomy, Photon Science Institute and Henry Royce Institute, School of Natural Sciences, The University of Manchester, Manchester, M13 9PL, UK. E-mail: wendy.flavell@manchester.ac.uk
First published on 20th May 2022
Abstract
An overview is presented of recent advances in photoelectron spectroscopy, focussing on advances in in situ and time-resolved measurements, and in extending the sampling depth of the technique. The future prospects for each are considered. Progress in the last few years has been remarkable, aided by substantial advances in photon sources, environmental cells and in analyser and detection technology. It is now possible to use the technique at ambient pressures and above, and to routinely probe interfaces buried many tens of nanometres below the surface in a non-destructive way. Our capabilities in time-resolved photoemission of the solid state have extended so far as to enable exploration of the fundamental timescale of the photoemission process itself.
1. Introduction and some basic principles
Einstein famously proposed a direct link between the kinetic energy (EKE) of a photoelectron emitted from a solid and the energy of the photon absorbed (hν) in 1905,1 an achievement for which he was awarded the Nobel Prize in 1921:| EKE = hν − EBE − Φ, | (1) |
The simple relationship between the measured kinetic energy and the binding energy in the solid (eqn (1)) immediately defines photoelectron spectroscopy as a technique sensitive to chemical state. The demonstration that the binding energy of an electron in a solid depends on its chemical state was made by Siegbahn (who received the Nobel Prize in 1981);3 his work led to the acronym ‘ESCA’ (electron spectroscopy for chemical analysis), although a chemical shift (in X-ray absorption) was first measured by Robinson in 1930.4 In nearly a century since, X-ray photoelectron spectroscopy (XPS) has become ubiquitously used as a ‘go-to’ technique for chemical analysis and is implemented in many thousands of labs around the world. The development of photoemission excited by UV-sources (ultraviolet photoelectron spectroscopy or UPS), primarily as a technique for measuring the energy- and momentum-dependent filled bandstructure of solids, using angle-resolved photoelectron spectroscopy (ARPES, Fig. 1),5 proceeded in parallel by Spicer and others.6 More recently, the gap between the two photon energy regimes has been plugged by synchrotron sources, and photoelectron spectroscopy, implemented in multiple ways, is now one of the most important and direct techniques for measuring the chemical and electronic structure of solids.
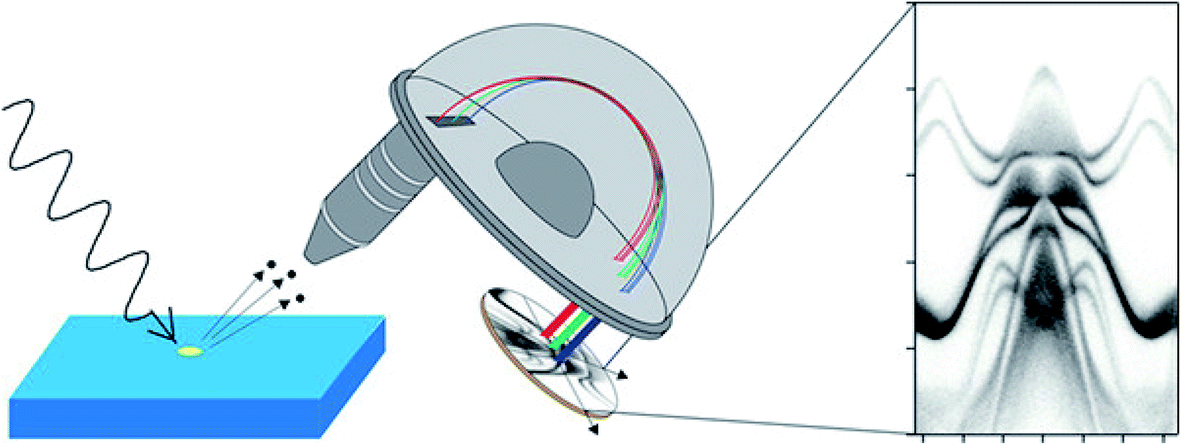 | ||
| Fig. 1 Schematic diagram of angle-resolved photoelectron spectroscopy (ARPES) using a conventional hemispherical electron energy analyser. By measuring the intensity of the photoelectrons as a function of their kinetic energy and emission angle, ARPES gives access to the electron-removal spectral function in the crystal momentum space, allowing band dispersions to be obtained (right). Reprinted with permission from ref. 5. Copyright (2021) American Chemical Society. | ||
A large number of excellent reviews has been written5,7 so here we focus on recent developments in the technique that promise to be of key importance to the future. A characteristic of the technique is that the exiting photoelectrons are of relatively low energy and are charged. This has historically led to interlinked difficulties (described below) that have imposed some limits on the applications of photoemission. A hallmark of the last decade is that significant progress has been made in tackling these issues.
Firstly, when a large group of photoelectrons leaves a solid at the same time (for example if excited with a short and intense pulse from a free-electron laser (FEL)), they interact with one another, causing space charge effects in the vacuum above the solid. This, until the last few years, slowed the progress in developing ultrafast time-resolved photoemission from solids. This roadblock has been largely removed very recently with the advent of new time-of-flight detectors and higher repetition-rate sources. The rapid recent progress in this field is reviewed in Section 4.
A related issue is that the low energy and charge of the photoelectrons means that they interact strongly with any medium they pass through. Photoelectron spectroscopy measurements have historically been carried out in ultrahigh vacuum (UHV), partly to allow the photoemitted electrons to reach the electron energy analyser without attenuation by an intervening gas. This leaves the technique open to the criticism that results cannot be obtained under realistic pressures (for example in studies of catalysts or thin film growth) or in a liquid (for example in studies of electrocatalysts). Here again, astonishing progress has been made in stretching the capabilities of near-ambient pressure XPS (NAP-XPS) so that in the last few years, several measurements have been reported at ambient pressure, and substantial progress has been made in our capability to carry out real-time measurements of cells and devices under working conditions. This progress in NAP-XPS is summarised in Section 2.
A further consequence of the strong interaction of charged low-energy photoelectrons with matter is that they are very strongly attenuated by inelastic scattering as they leave the solid. This leads to the hallmark surface sensitivity of photoelectron spectroscopy, which in many ways is a boon, but can also be a limitation, as results from it are often taken to apply to the bulk of the material. The initial peak intensity, I0, of a photoelectron signal is attenuated with depth, d, as
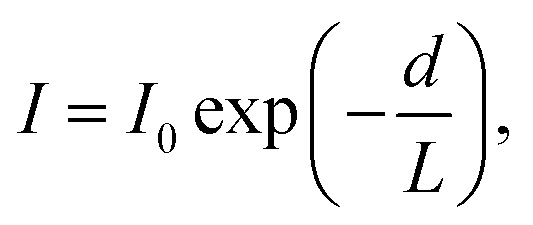 | (2) |
dS = 3λ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ, cosθ, | (3) |
The sampling depth as defined in eqn (3) applies to the use of primary photoelectron peaks, i.e., those electrons which have escaped the surface to detection without inelastic scattering. These ‘elastic’ peaks are used for chemical state determination (as chemical state changes lead to chemical shifts in core level electron binding energies), and for the measurement of the distribution of elements near the surface. Probing beyond the elastic limit can be achieved by modelling the inelastic (secondary electron) background associated with each core level excitation at kinetic energies (KEs) lower than the primary peak.8–12 The shape of this background is dependent on the depth distribution below the surface,13 and this background signal remains significant even after the signals from the core level peaks have been attenuated by overlayers. This may be used to yield information from depths much larger than 3λ.8 The mean energy loss is approximately linear with the distance the electron has travelled, meaning that extracting a depth distribution requires measurement of the inelastic background over several hundred eV.13 This technique can be applied to surface-sensitive XPS with photon energies of around 1.5 keV. However, in conventional XPS, the inelastic background from a single core level is often obscured by other core level peaks and Auger lines present in the spectrum, meaning that the energy range over which the inelastic background is measured may be limited. The information depth has been found to be ∼ 8λ (perhaps some tens of nm) when the background is measured over a range of ∼100 eV to the low kinetic energy of the primary peak, as might be typical in XPS.14 There are a great many applications (for example to device heterostructures) where this is simply too short to be useful, and there is therefore a strong incentive to extend the sampling depth of XPS. Recent progress in extending the sampling depth via hard X-ray photoelectron spectroscopy (HAXPES) and X-ray standing wave techniques has been remarkable, and has also aided the realisation of ambient-pressure XPS. This progress is reviewed in Section 3.
This paper serves as an introduction to the 2022 Faraday Discussion meeting ‘Photoelectron spectroscopy and the future of surface analysis’. I have framed it around three of the key themes of the meeting, developments in in situ techniques (based on NAP-XPS), in measurements of buried interfaces by non-destructive techniques, and in time-resolved photoemission. I present a personal view, so the paper is not exhaustive and cannot fully reflect the diversity and wealth of discussions that took place during the meeting. Far from being impacted by the pandemic, there has been quite astonishing progress in all three areas in the last few years alone, which is reflected in the papers presented at this meeting.
2. In situ methods: developments in near-ambient pressure XPS
2.1. Background
Without doubt, one of the most important developments in photoemission in the last decade has been the significant increase in the availability of near-ambient pressure XPS instruments, and their widespread use. The important applications are well-represented by the submissions to this meeting, and include heterogeneous catalysis (https://doi.org/10.1039/D1FD00120E), including water splitting15 electrochemistry (https://doi.org/10.1039/D1FD00114K),16 thin film growth, for example by atomic-layer deposition,17 gas sensing18 and corrosion and degradation.19 A large number of excellent reviews has been published.20–23The low IMFP of low-energy photoelectrons, which gives XPS its surface sensitivity, also of course limits the IMFP of electrons in gases (in the case of 100 eV electrons travelling through 1 mbar of water vapour, to about 1 mm).20 This is short compared to the typical distance between the sample and the electron energy analyser in XPS, so in order to carry out XPS under non-vacuum conditions, typically differential pumping stages are used that reduce the pathlength of the electrons through the gas phase, together with scattering of the electrons and signal attenuation. Near-ambient pressure- (NAP-)XPS (also called ambient-pressure- (AP-)XPS) was developed initially by the Siegbahn group in Uppsala as long ago as the 1970’s,24,25 but suffered from low count rates. The technique did not start to become deployed in a widespread way until the early 2000’s when NAP-XPS instruments were developed at high flux synchrotron sources and were combined with a new generation of high transmission analysers (the first of these at the Lawrence Berkeley National Laboratory (LBNL) in the late 1990’s).26 Further technical developments, in particular by SPECS Surface Nano Analysis GmbH (with LBNL and the Fritz Haber Institute in Berlin), based on the SPECS Phoibos 150 NAP analyser, have seen an explosion of activity in the last decade, and NAP-XPS is now widely available at both synchrotron sources and in commercial lab-based instruments.20,23
Most NAP-XPS instruments utilise an electron energy analyser with multiple (typically four) pumping stages that reduce the pressure from (typically) a few mbar in the sample environment to <10−7 mbar in the hemispherical analyser, with the aperture nozzle of the first pumping stage held typically around 1 mm from the sample. Different NAP-XPS systems realise the sample environment in different ways (Fig. 2),20 and in the last few years, this has led to specialised commercial instruments with rather different applications. In the simplest experiments, the whole of the vacuum chamber that isolates the sample from the laboratory environment is exposed to the gas atmosphere during data taking (Fig. 2(a)). This method is in use, for example at the TEMPO beamline at SOLEIL, Paris and at beamlines 9.3.2 and 11.0.2 at the Advanced Light Source (ALS), Berkeley. The main disadvantage of this approach is the time taken to regain ultra-high vacuum (UHV) if UHV XPS measurements are required (which normally involves bake-out), and also the possibility of cross-contamination between experiments. However, these disadvantages may be turned to spectacular advantage if the instrument can be dedicated to one purpose (e.g. wet environments), and if UHV experiments are not required, as discussed further in Section 2.6.
 | ||
| Fig. 2 Schematic diagram of the main arrangements of in situ measurement cells used in NAP-XPS systems. (a) The analysis chamber/in situ cell is part of a vacuum system (often with load lock and preparation chamber). The whole chamber is exposed to gases during APXPS experiments. (b) The in situ cell is located inside a larger vacuum chamber. Only the in situ cell is exposed to gases during APXPS measurements. (c) Exchangeable in situ cells, tailored to a particular experiment, are attached to the analyser. Reproduced from ref. 20. Copyright © 2013 Author(s). | ||
In instruments where other sample analyses (including UHV XPS) and complex sample preparation are needed, it is more typical to use an environmental or in situ cell, which is placed inside the analyser chamber and sealed from it during the exposure of the sample to gases (Fig. 2(b)). This allows UHV to be regained more rapidly between experiments (a particular consideration for synchrotron experiments), but adds to the complexities of sample manipulation, as the sample must be manoeuvred in and out of the cell. In a refinement of this approach, called the ‘Lund’ or cell-in-cell concept (developed by Lund University and SPECS Surface Nano Analysis GmbH), the nozzle that is normally attached to the analyser aperture is incorporated into the cell, so that the analyser collection angle is not constrained by it during UHV measurements.27 The ‘cell-in-cell’ concept has been implemented at the SPECIES and HIPPIE beamlines at MAX IV.28,29 It is also possible to make use of a set of interchangeable sample cells, each of which can be devoted to one purpose (e.g. corrosive environments, liquid samples, electrochemical cells etc., Fig. 2(c)). This again adds to experimental complexity, but improves flexibility. This type of arrangement is available, for example, at the VERSOX beamline at Diamond Light Source and at HIPPIE and SPECIES (MAX IV).
2.2. Applications in heterogeneous catalysis
A number of excellent reviews of NAP-XPS have noted the preponderance of applications to heterogeneous gas-phase catalysis,20,23 and these applications are well-represented at this meeting. Even though new applications of NAP-XPS are emerging rapidly, in 2020, surface science and catalysis still accounted for around 50% of all NAP-XPS publications. In heterogeneous catalysis, studies of model reactions, such as the CO oxidation reaction, accounted for 45% of NAP-XPS research at this point.23 In these studies, it has become clear that under NAP-XPS conditions, the reaction is typically mass-transfer limited (for example in CO oxidation, all CO that reaches the surface is instantaneously oxidised to CO2, and there is a CO depletion layer at the surface that extends for some mm above the surface).23 Under these conditions, the total conversion rate is determined by the rate of gas diffusion through the depletion layer.23 Planar laser-induced fluorescence has been used to explore the significant differences between the CO2 concentration measured above the sample and that measured by mass spectrometry at the gas outlet.30 Although there is now an increasing number of studies of oxide catalysts (https://doi.org/10.1039/D1FD00120E),31 there is a preponderance of studies of metal single crystal surfaces (including curved crystals). These have been well-reviewed,20,23 and have been used to elucidate the differing catalytic activity of different metals (e.g. Pt(111) vs. Ir(111) in CO oxidation,32 and in the preferential and partial oxidation of CO/H2 mixtures23) and of different metal surfaces, highlighting the catalytic activity of stepped and faceted surfaces (e.g. in studies of a Pt curved crystal in the same reaction33).The study of the reactions of larger molecules, particularly carbon-containing ones, is of huge importance in the production of fine chemicals and pharmaceuticals. However, the study of these reactions by NAP-XPS remains challenging because of issues of rapid surface contamination, complexity of the mechanism, and the low vapour pressure of the reactants. In addition, the binding energies of the gas and surface-phase components often overlap, necessitating parallel UHV studies. As an example, a recent study of the Sonogashira cross-coupling reaction (a C–C bond-formation reaction between iodo- or chlorobenzene and phenylacetylene to form diphenylacetylene) confirmed earlier UHV results that Au(111) catalyses the reaction at moderate temperature and pressure.34 However, rapid inactivation due to the formation of a carbonaceous species was observed at higher temperature and pressure,34 illustrating the importance of NAP-XPS in providing more industrially relevant conditions. Although, as we discuss in Section 2.7, it has recently become possible to carry out NAP-XPS at pressures of 1 bar and above of a few mbar, and there remain significant challenges to solve in studying real catalytic reactions of large molecules.
2.3. Electrochemistry and electrocatalysis
NAP-XPS now contributes powerfully to the study of electrocatalysis and electrochemical devices, such as batteries, fuel cells and supercapacitors. Although such studies only began around 2008,36 the capacity of NAP-XPS for in situ and operando measurements has led to a rapid expansion in these studies, which by 2020 accounted for about 6% of NAP-XPS publications.23 The first devices studied in this way were solid oxide electrochemical cells (SOCs), which typically operate in an atmosphere of gaseous fuel at temperatures > 700 °C, precluding the application of most surface analytical tools.36–39 The first experiments were carried out at the ALS, and used a specially designed SOC in which a Au–ceria working electrode and a Pt counter electrode were deposited on a single crystal yttria-stabilised zirconia (YSZ) electrolyte disc.36,37 This was designed with all the components located on the same side of the electrolyte disc so that all the cell components were exposed to the gas atmosphere, and were accessible to NAP-XPS. As well as providing chemically specific information about each element of the cell under in situ conditions (at ca. 750 °C, in a pressure of 1.3 mbar (1:1H2/H2O)) while the cell was working, this study showed that local electrical potential changes at the surface can be determined from the changes in the KE of the core level photoelectron peaks.36,37 The authors were also able to show that the active electrochemical region on ceria extends up to 150 mm away from the current collector, with significant changes in the equilibrium Ce3+/Ce4+ ratio observed throughout this region.37 The first measurement of individual overpotentials across an interface was made using a Ni/YSZ/Pt SOC;38 in the water splitting reaction, and it was used to show that H2O splitting is faster than H2 oxidation on Ni, while the converse is true on Pt.38
Clearly a particular target for NAP-XPS is to understand the oxidation and reduction half-reactions in lithium-ion and lithium–air batteries. These pose particular challenges, not least the chemical complexity, leading to parasitic side reactions, but also the use of a liquid electrolyte in many conventional cells. Currently most NAP-XPS uses soft X-rays (with energies <2 keV), which limits the probing depth to less than about 5 nm below the sample surface. This in general allows access to gas/solid or liquid/solid interfaces but buried interfaces (liquid/solid or solid/solid) are more problematic. It means that the liquid electrolyte surrounding the immersed electrode must be stable (to evaporation) and less than a few nm thick.40 As a result, some of the first NAP-XPS studies were carried out instead on solid state Li-ion/O2 cells.41 These allowed the Li–O2 reactions to be probed in the absence of binder and liquid electrolyte, and for the first time revealed reversible Li+ intercalation into a LixV2O5 electrode in situ under applied potential in ca. 0.5 mbar O2.41 This general approach has been extended to Na–O2 model batteries, where it showed the formation of Na2CO3 as the main discharge product of the cell.42
In the years following the initial measurements, there has been a strong drive to towards developing methods to access buried interfaces (particularly the liquid/solid interface) with NAP-XPS, so that more realistic devices with liquid electrolytes may be studied under real working (operando) conditions with a temporal resolution high enough to detect metastable states.43 Two main threads have been pursued: the development of liquid cells for XPS,16,44–52 and the use of harder X-rays with energies of a few keV (often called ‘tender’ X-rays) producing photoelectrons with a higher IMPF, and thus a larger sampling depth (Section 3).53,54 The latter also facilitates the use of standing wave techniques (Section 3.4) combined with NAP-XPS in a technique known as standing-wave ambient-pressure photoemission spectroscopy (SWAPPS).53 The use of tender X-rays has also made possible the study of electrode/electrolyte interfaces created via the meniscus or ‘dip and pull’ approach.54 This has been recently implemented, for example, at the HIPPIE beamline at MAX IV (Sweden)28 and at the SpAnTeX beamline at BESSY II.58
In the last decade, there has been significant development of cells that use ultrathin membranes of graphene, graphene oxide or silicon,16,44–51,59,60 which separate the sample volume from the vacuum (Fig. 3). If these are sufficiently thin, the emerging photoelectrons can be emitted through the membrane into the electron energy analyser. The approach has been implemented in flow cell geometries with both liquid and vapour flows, and used, for example, to monitor the electrodeposition of an oxidised cobalt layer from a CoSO4 solution onto the graphene membrane, anchored by Co(CO)x species.16In situ electrochemical cells using proton exchange membranes (PEMs) have been implemented in both 2-electrode and 3-electrode (flow) manifestations to probe the oxygen evolution reaction (OER) during water electrolysis over Pt- and Ir-based electrodes, revealing the working oxidation states of both anions and cations.48,52,56
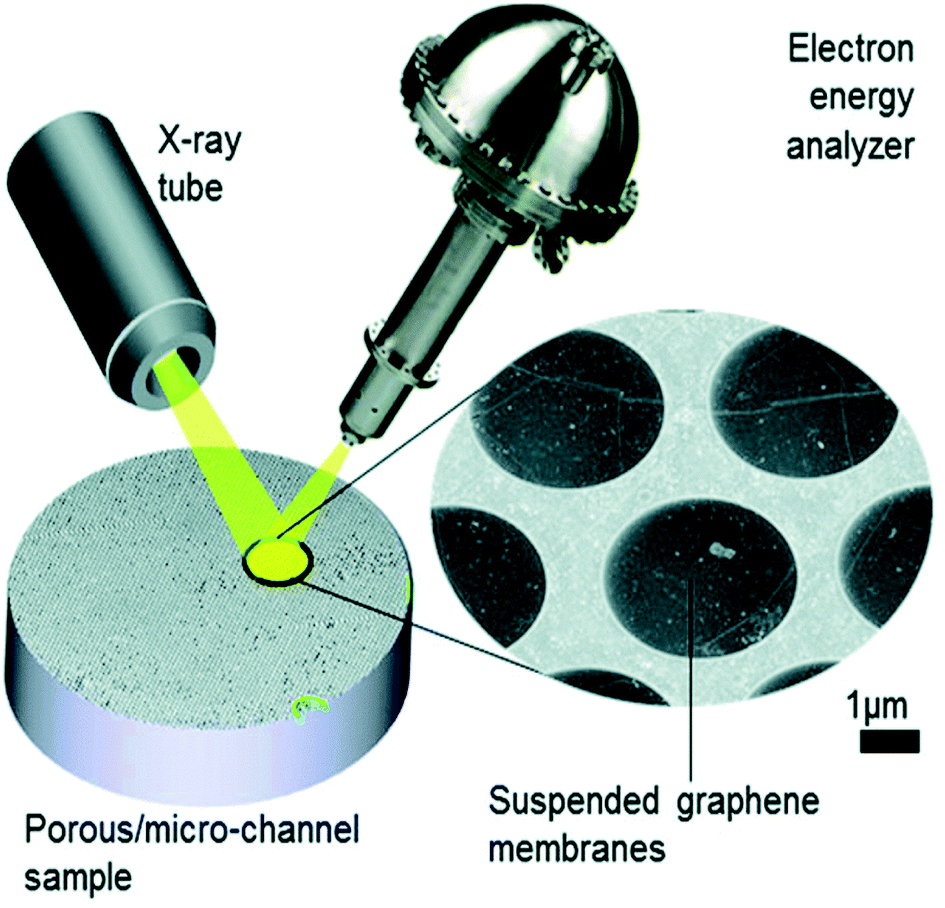 | ||
| Fig. 3 Schematic illustration of the collection of XPS through graphene; in this example, a microporous substrate that contains a high density of individual micro-volumes is separated from the vacuum by electron-transparent graphene membranes. The sample is impregnated with the analyte. The concept is implemented at the ESCA Microscopy beamline at Elettra (Trieste).61 Reproduced from ref. 51 with permission from the Royal Society of Chemistry. | ||
Current research on electrochemical systems is clearly benefitting from a number of simultaneous technical developments such as those described above; particularly important future directions are the development of liquid-jet sources22 and the rapid development of capability to perform XPS at pressures of 1 bar and above.35,62–64 These developments are discussed further in Section 2.7.
2.4. Thin film growth
In contrast to many processes in gas-phase heterogeneous catalysis, the surface chemistry and mechanisms of thin film growth remain largely unknown.65 In the last decade, the potential of NAP-XPS to contribute to the understanding of commercially important thin film growth processes has been realised, and although there remain rather few published studies, investments are being made to support these challenging experiments at a number of synchrotron sources, including at the SPECIES and HIPPIE beamlines at MAX-IV.66,67 The pressure used in atomic layer deposition (ALD) (and the useful range in some chemical vapour deposition (CVD) processes) is typically in the 10−3 to 20 mbar range, which is well-matched to NAP-XPS.23 However, there are also formidable challenges. ALD makes use of dedicated reactor geometries for self-limiting film growth, using short vapour pulses of each precursor in alternation, with purging of the sample volume with an inert gas between every precursor pulse – so NAP-XPS instruments of the types shown in Fig. 2(b) and (c) are needed, with a small cell volume. Because the precursors are designed to stick well to surfaces and decompose easily, there are serious issues with the contamination of succeeding experiments, and X-rays can induce surface reactions that dominate the experiment.23 It is nevertheless possible to obtain completely new insights. An example is the use of NAP-XPS to probe the growth of TiO2 on RuO2(110) by ALD using tetrakis(dimethylamido) titanium (TDMAT) and water in alternate half-cycles of the reaction.17 NAP-XPS was recorded in real time during repeated dosing of the surface with first TDMAT and then water at 60 °C and pressures up to 0.1 mbar (Fig. 4).23 | ||
| Fig. 4 NAP-XPS study of TiO2 ALD from TDMAT and H2O on RuO2(110): Ti 2p, N 1s and O 1s spectra acquired during the initial cycles of ALD. The sample temperature was 110 °C, and a photon energy of 580 eV was used in the recording of all spectra. Reprinted with permission from ref. 17 and ref. 23. Copyright (2016) American Chemical Society. | ||
Prior to these experiments, the proposed reaction mechanism was a simple Ti ligand displacement by surface hydroxyl to bind the TDMAT to the surface, followed by hydrolysis of the remaining ligands by water, involving no change the oxidation state of Ti(IV).68 However, a much more complex picture was revealed by NAP-XPS. In the first half cycle, evidence of Ti(II), (III) and (IV) is observed, together with a much more complex N 1s chemistry than expected. In addition to the expected surface –N(CH3)2 ligands from the precursor, dimethylamine (HN(CH3)2) and methylene imine (CH3N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) are observed (Fig. 4).17,23 These species persist during the water half cycle, while the –N(CH3)2 ligands react with water, and dimethylammonium ions are formed. It has been proposed that the reaction mechanism in the initial TDMAT half-cycle is dominated by a β-hydride elimination reaction involving the reduction of Ti(IV).17,23 The expected ligand exchange mechanism appears to take over in subsequent cycles; the imine peak is removed and a hydroxyl peak appears in the O 1s spectrum during the first water half-cycle (Fig. 4).17,23
CH2) are observed (Fig. 4).17,23 These species persist during the water half cycle, while the –N(CH3)2 ligands react with water, and dimethylammonium ions are formed. It has been proposed that the reaction mechanism in the initial TDMAT half-cycle is dominated by a β-hydride elimination reaction involving the reduction of Ti(IV).17,23 The expected ligand exchange mechanism appears to take over in subsequent cycles; the imine peak is removed and a hydroxyl peak appears in the O 1s spectrum during the first water half-cycle (Fig. 4).17,23
Although in the example discussed above, spectra were recorded in real time during ALD, they were not recorded during typical reactor operating conditions. As the typical pulse durations in ALD are on the ms to s timescale,69 it is clear that this will usually not allow enough time to accumulate NAP-XPS with sufficient statistics. It is necessary then to develop a ‘snapshot’ mode capability, where data from many repeated cycles may be summed at the same temporal delay between pulse and spectrum (akin to those developed for some forms of time-resolved pump–probe photoelectron spectroscopy, discussed in Section 4.4).66 This can then be used to study ‘steady-state’ ALD, when the chemistry is identical for a large number of successive pulses. This capability was realised for a test reaction (CO oxidation on Pd(100)) in 2021.67 Because the mass flow controllers that switch the gas pulses in the cell are generally distant from the surface, gas switching may not be a suitable signal for triggering the averaging. In this demonstration, the work-function-induced binding energy shift of the gas-phase O 1s peak as the gas environment around the sample changes from CO2 (at the start of a CO pulse) to O2 (at the end of the pulse) was used, and recognised using image recognition software.67 It was possible to record data at frame rates of 6–17 Hz, giving a time resolution of the order of 150–50 ms, limited by the software and frame rate of the detector camera.67 Clearly, with a delay-line or similar detector, the time resolution can be increased substantially if there is a sufficiently large trigger signal. Indeed, a demonstration of sub-ms time resolution in gas-pulsing experiments was published in 2021.70 This utilised gated detection based on a fast camera, combined with short gas pulses generated using a fast valve that generates pulses with a rising edge of a few 100 μs, and internal pressures in the mbar range.70 The fast camera shutter was triggered by the valve operation. The development of time-resolved NAP-XPS measurements is an exciting area of current development that is discussed further in Section 2.7.
2.5. Material degradation in humid environments
There still remains a ‘pressure-gap’ in NAP-XPS between the conditions of most experiments (a few mbar) and atmospheric pressure and the pressures used in real heterogeneous catalysis. This is rapidly being closed, as we describe in Section 2.7. Nevertheless, there is a number of fields where the ‘few mbar’ pressure range that is already readily covered in NAP-XPS is ideal – an example is the thin film growth studies described above. A further important application is to problems of material degradation in wet environments,15 and the surface passivation necessary to avoid it. A pressure of 9 mbar of water vapour (easily achieved in NAP-XPS) is equivalent to an atmospheric relative humidity (RH) of ca. 30% at a standard temperature of 25 °C, a fairly typical ambient atmosphere in temperate climates. This means that realistic water pressures can be achieved within the controlled environment of the spectrometer.Organic–inorganic hybrid perovskites, exemplified by the ‘MAPI’ (methylammonium lead iodide, CH3NH3PbI3) perovskite, have shown immense promise as cheap light harvesters for a new generation of solar cells, with device efficiencies rising in the last 9 years to more than 25% – rivalling that of the conventional material, silicon.71,72 However, the exploitation of these materials is currently prevented by their instability when exposed to water and air, which leads to rapid degradation. Understanding the mechanism of this reaction may allow researchers to identify passivation routes. The prototypical perovskite MAPI is particularly unstable.73,74 The key difficulties in understanding the degradation mechanism of MAPI are then the difficulty in determining the surface chemistry of the first few atomic layers of the pristine material without any exposure to ambient atmospheric conditions, and, conversely, the difficulty in measuring this same surface chemistry in real time under exposure to realistic environments. Both problems were addressed simultaneously by utilising NAP-XPS to investigate the real-time degradation of an in situ-deposited MAPI film, exposed to 9 mbar water vapour, which provides a realistic model of the partial pressure of water vapour in the ambient environment (Fig. 5).19 A thin film of MAPI was grown onto a SrTiO3(100) substrate in situ in the spectrometer using vapour deposition.75 The use of a film thickness of only a few nm meant that peak intensities could be reliably quantified by referencing to the attenuation of signals from the underlying substrate. When exposed to water vapour (Fig. 5), complete decomposition of the methylammonium cation of the perovskite was observed, evidenced by the complete loss of the N 1s signal, and the evolution of C:N stoichiometry (Fig. 5).19 This occurred concomitantly with the collapse of the lattice of PbI6 octahedra of the perovskite structure to form PbI2, evidenced by changes in the Pb 4f and I 3d binding energies and a change in the Pb:I stoichiometry (Fig. 5).19 The data indicate a degradation pathway by reaction with water, which involves the complete loss of nitrogen from the film without the initial formation of methylammonium iodide, consistent with a degradation reaction of the type:
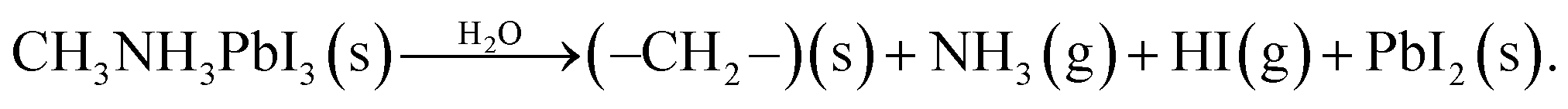 | (4) |
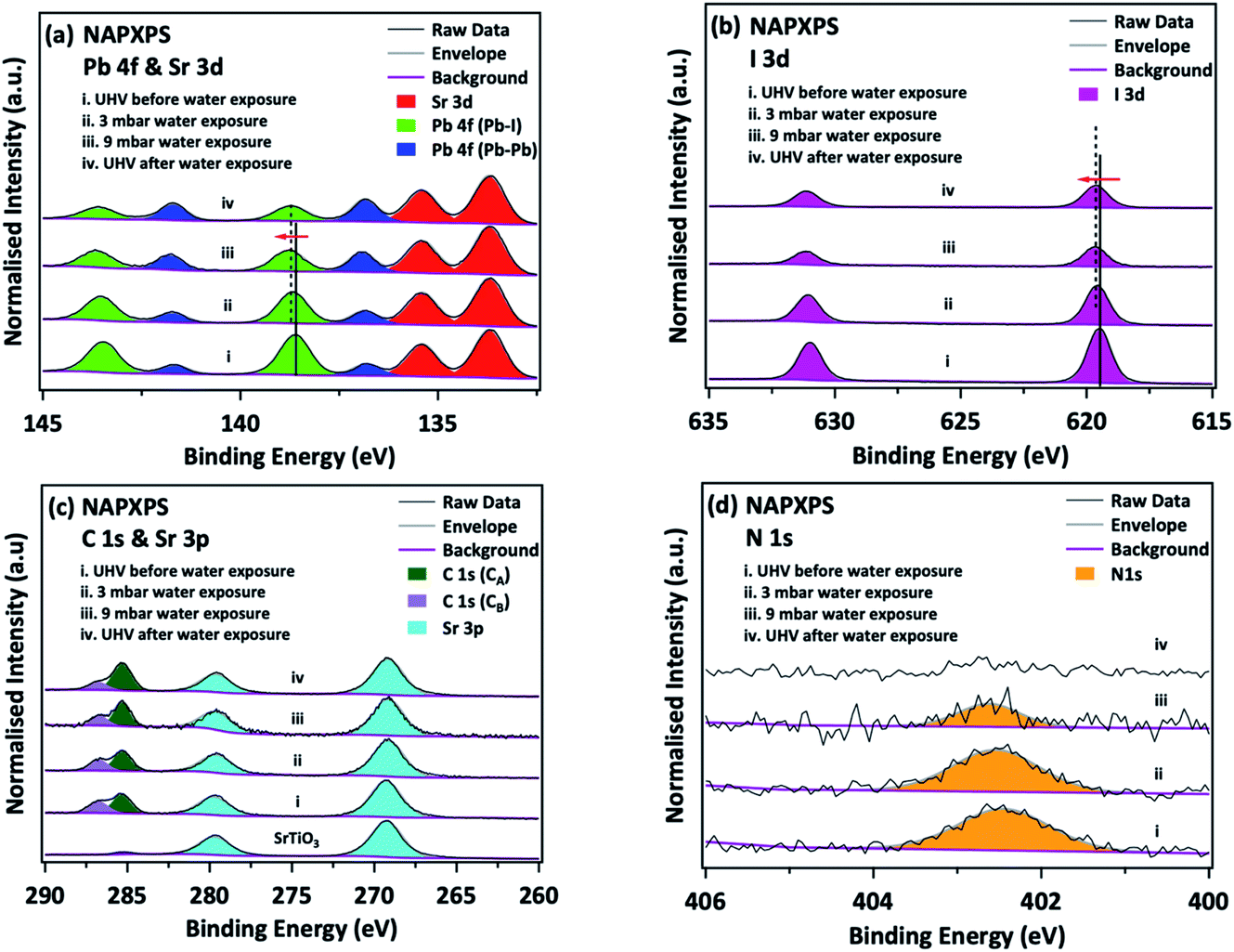 | ||
| Fig. 5 Degradation of in situ-grown MAPI monitored by NAP-XPS: (a) Pb 4f & Sr 3d, (b) I 3d, (c) C 1s & Sr 3p, and (d) N 1s core levels before/during/after water exposure. C 1s (CA) and C 1s (CB) refer to adventitious carbon and to carbon associated with the perovskite methylammonium ion/CO/C–OH, respectively. All spectra are normalised to the corresponding integrated area of the Sr 3d5/2 peak. Reproduced from ref. 20. Copyright © 2017 Author(s). | ||
2.6. Natural and biological samples
A simple NAP-XPS spectrometer of the type shown in Fig. 2(a) has some serendipitous advantages in the study of materials with little vacuum compatibility. Typically, these instruments operate at a significantly higher working pressure than conventional XPS, which can lead to much reduced sample pump-down times, and can remove the need for a load lock as the sample may be directly introduced into the analytical chamber of the instrument. This reduces the time needed for sample loading, and increases the capability for high throughput.76 A perhaps more significant advantage is the in-built charge compensation that NAP-XPS provides, because the incident X-rays ionize the gas molecules around the sample, creating cations (together with photoelectrons, secondary and Auger electrons). These efficiently compensate the surface charging of insulating molecules.77–79 The amount of charge compensation can be tuned by changing the X-ray flux or the pressure and type of gas in the analysis chamber.78 In the last 5 years, these advantages have been turned to good use in a new generation of high-throughput, simple NAP-XPS instruments exemplified by the SPECS EnviroESCA.78 These instruments are suitable for the routine and fast measurement of a wide range of materials that are insulating and/or outgas in UHV, such as zeolites and polymers, biological and other liquid and wet samples (including nanoparticle suspensions) and consumer goods, such as foods and cosmetics.76,80 This approach has made XPS easy and accessible to new communities. It has been possible, for example, to use NAP-XPS to study Escherichia coli in the hydrated state,81 and to show that freeze drying (which has been commonly used to study biological systems by XPS82) alters the composition of carbon species at the surface.81 In later studies of the bacterium Pseudomonas fluorescens, it has been shown that this difference can be minimised by using fast freezing in cryo-XPS.83 Recently, the first NAP-XPS studies of radiation damage in fully hydrated double-strand DNA have been published.84 DNA damage by ionising radiation is of fundamental importance to cancer therapy, but is difficult to study by XPS in realistic environments. XPS in vacuum, nitrogen and water was used to deconvolute direct DNA damage, caused by low energy electrons, from the indirect effects caused by the ionisation of water molecules to form reactive oxygen species, such as OH˙ and H2O+. The latter initiate indirect damage processes which lead to deoxyribose and nucleobase modification and base release. In the presence of water, the authors show that these processes predominate over the strand breakage, which is the dominant type of damage in dry DNA, although damage of all types increases when water is introduced.84 Thus, it is clear that NAP-XPS is beginning to make powerful contributions to resolving long-standing controversies in biological systems.2.7. Future directions for NAP-XPS
As noted above, there is currently a strong drive to close the pressure gap completely in NAP-XPS, and to make possible studies at atmospheric pressure and above35,62–64 (finally allowing the ‘N’ in the acronym to be dropped without any shade of exaggeration!) This is coupled with a strong drive to use harder X-rays, and to implement the technique in the regime of hard X-ray photoelectron spectroscopy (HAXPES). HAXPES is discussed in more detail in Section 3, but here we note that these two drivers are coupled; the use of higher kinetic energies reduces the scattering of photoelectrons in the gas phase, allowing higher pressures to be used. The use of X-rays in the few keV energy range, coupled with a very small analyser aperture placed very close to the sample, can then allow AP-XPS at atmospheric pressure, for example as implemented on BL36XU at SPring-8, using a 30 μm aperture at a working distance of 60 μm from the sample.85 Of course, the use of higher kinetic energies also increases the inelastic mean free path in the solid. Surface sensitivity can then be enhanced by using grazing angles of incidence of ca. 1–5° relative to the surface (close to the condition for total external reflection).58,62,85 In this geometry, the footprint of the X-ray beam on the surface becomes significantly elongated in the plane of incidence. In the POLARIS high-pressure HAXPES instrument recently developed for beamline P22 at PETRA III, this has led to the development of an analyser aperture made up of a linear array of evenly spaced μm-sized holes, matching the beam footprint on the sample.63,64 In this system, the sample-to-aperture distance is a few tens of μm, and gas dosing directed at the sample is incorporated into the aperture front cone. This leads to a very localised high-pressure pocket at the sample, allowing for pressures exceeding 1 bar to be achieved locally at the sample, while maintaining the main chamber of the spectrometer at a pressure of a few mbar.62 This has been used to measure CO oxidation on Pd(100) in the ‘light-off’ regime (when CO2 begins to be produced) in pressures of up to 1 bar for the first time.35 The measurements indicate that under the conditions of the experiment, the system is not in the mass-transfer-limited regime immediately after light-off, allowing the measurement to be performed in the kinetic regime. The variation with temperature of the intensity of the C 1s and O 1s signals from the working catalyst allowed the extraction of an apparent activation energy.35 In this arrangement, there is no separation between the sample and the main chamber, but effectively a ‘virtual’ cell is achieved by the localised high-pressure pocket. An alternative approach to closing the pressure gap is to further develop the in situ cells discussed in Section 2.3. In a development of the graphene membrane technology for electrochemical cells discussed earlier, AP-XPS at 1 atm has been achieved by separating the vacuum and high pressure environments by a silicon nitride grid with an array of 1 μm-diameter holes coated by bilayer graphene.64A further strong area of development in the last decade has been in studies of the liquid/vapour interface – partly driven by the work in electrocatalysis described above (Section 2.3), but also by studies of water and heterogeneous reactions at liquid–vapour surfaces, such as those of atmospheric aerosols.86 One driver here is climate change, as about one third of anthropogenically generated CO2 is sequestered at the air/water interface.87 With the exception of ionic liquids (which have sufficiently low vapour pressures at room temperature that they can be studied in UHV), these studies pose significant challenges, which have been recently reviewed,22,88 but include issues of charge referencing, vapour pressure, contamination and beam damage. The development of in situ cells with thin membranes has allowed some significant advances, for example the study of dispersed nanoparticles in solution.89 However, in the main the approaches to preparing a liquid surface fall into two categories: fast flowing jet or droplet sources suitable for short (ms) reaction times, and the use of a bulk reservoir of liquid inside the vacuum chamber generated, for example, via a wet wire, Langmuir trough,22 deliquesced salt,90,91 supported droplet (or offset droplet for studies of the liquid/solid interface92) or dip-and-pull approach.22,54 The latter requires the experiment to be done in the presence of an equilibrium vapour pressure (to avoid evaporation), while fast moving jets can be used in the vacuum environment as the jet or droplet train moves through the chamber at a speed of many tens of m s−1, so the interaction time is brief.22 Jet and droplet sources can also be used in fast mixing modes, for example where two identical laminar jets collide just inside the measurement chamber, forming a stable liquid sheet known as a ‘flatjet’, and so far implemented primarily in X-ray absorption spectroscopy.93,94 The solutions can also be fast-mixed just prior to entering the analysis chamber, an approach used in studies of the aqueous carbonate system to mix HCl and NaHCO3, generating carbonic acid (H2CO3) in situ.86 Depth-profiling the mixed jets by varying the photon energy was used to reveal surface partitioning of CO32− over HCO32−.86 The effects of organic surfactants may also be probed, as in recent studies of octanoic acid at the water–gas interface.95 This experiment exploited the geometry of liquid-jet experiments to extract photoelectron angular distributions as a means of characterising the orientation and relative position of the surfactant molecules at the interface.95 These experiments are not without their difficulties and a number of studies have outlined the care that must be taken in interpreting these measurements;95,96 an example is the distortion of the spectra by the quasi-elastic scattering of low kinetic energy electrons recently highlighted in measurements from liquid water made using the liquid jet sources at the PLEIADES and DESIRS beamlines at SOLEIL.96 However, as liquid jet sources have now been implemented in a significant number of labs, this area is burgeoning. Although many studies have been made of water, the approach has been extended to other liquids. A particularly elegant example from BESSY II is studies of solvated electrons in liquid ammonia microjets.97 Here, increasing amounts of Li, Na or K were introduced into the jets, and the transition from a blue, dilute electrolyte solution of paired electrons to a delocalised bronze metallic solution with a strong Fermi edge was tracked.97
As noted in Section 2.4, there is currently significant work devoted to improving the time-resolution of NAP-XPS. A number of spectrometers incorporating delay-line detectors have been developed in the last decade, implemented for example at beamline 11.0.2 at the ALS98 and at the SpAnTeX beamline at BESSY II.58 These detectors in principle have a precision of ca. 150 ps, as the detector records the arrival time and position of every electron at the exit plane of the analyser. However, the ultimate time resolution in such systems is limited by the spread in the times of flight of the photoelectrons in the analyser to values on the order of 1 ns.98 The spectra are typically collected by repeated scans of a narrow binding energy window, so in reality the time resolution is limited by the signal level and the duty cycle of the repeated process. However, 30 ns snapshot capability with viable signal has been demonstrated.58 Methods for accessing faster timescales, including using free-electron laser (FEL) and high-harmonic generation (HHG) light sources, have been recently reviewed.99 Time-resolved photoemission is discussed further in Section 4.
In the next few years, we are likely to see the very rapid development of multi-modal experiments incorporating NAP-XPS.100 The grazing incidence geometry used with tender X-rays allows for the incorporation of a number of additional techniques that use a reflection geometry. As an example, a schematic layout of an instrument under construction at the Fritz Haber Institute in Berlin is shown in Fig. 6.22 This is designed to probe surfactant chemistry at the aqueous solution vapour interface. The samples will be prepared in a Langmuir trough, located inside the spectrometer. A micro-focussed X-ray source at grazing incidence will provide 3 different photon energies, allowing depth-profiling in NAP-XPS, and is combined with 2 other techniques in reflection geometry – reflection absorption infrared spectroscopy, RAIRS (providing optical characterisation) and grazing incidence small angle X-ray scattering, GISAXS (providing information on the morphology and ordering of the surfactant layer, supplemented by Brewster angle microscopy). X-ray fluorescence, measured with a fluorescence yield (FY) detector, will be used to measure the bulk composition, and an in-vacuum force sensor will be used to measure the surface tension as a function of the compression of the surfactant layer.22
 | ||
| Fig. 6 Schematic layout of a combined AP-XPS–Langmuir trough instrument under construction at the Fritz Haber Institute in Berlin. Published in ref. 22. Copyright © 2021 Author(s). | ||
In general, there is a strong push to combine NAP-XPS with structural techniques and/or lateral resolution. In addition to grazing incidence X-ray scattering,101 examples include its combination with SWAPPS53,55–57 (Section 2.7), where there are now prospects to extend the technique to studies at elevated temperature,57 and near-edge X-ray absorption fine structure (NEXAFS).102 Combination with scanning photoelectron microscopy (SPEM) adds lateral imaging capability to the picture, and is implemented with NAP-XPS, for example, at the ESCA Microscopy beamline at Elettra (Trieste), offering sub-μm resolution.61,103,104 The use of a 3D delay-line detector with a lateral resolution lens at the SpAnTeX beamline at BESSY II58 has opened up the possibility of combined time- and lateral resolution in NAP-XPS with tender X-rays.
3. Buried interfaces
3.1. Developments in HAXPES
Over the last decades, XPS has become embedded and ubiquitously used as a core technique for materials characterisation. However, as the emerging photoelectrons have a photoelectron inelastic mean free pathlength, λ, of only a few nm, there are some dangers in probing only the surface. Surface contamination, degradation, and segregation effects are very significant over these probing depths. Indeed, as a consequence of the absence of the crystal potential at the surface, the equilibrium electronic structure and composition of the surface is expected to differ from that of the bulk. Thus, although conventional XPS is very commonly applied to materials, the information obtained about the bulk is at best an inference and at worst meaningless. There is now a large number of advanced functional materials (such as the MAPI perovskite photovoltaic) where the surface is sufficiently unstable as to render conventional XPS useless, and an increasing number of 2D layered materials (such as van der Waals heterostructures) where it is necessary to determine the layer-by-layer composition in order to tune the electronic properties. There are numerous technological applications (for example to device heterostructures and passivating coatings) where there is a need to study interfaces buried some tens of nm below the surface. For some decades, there has thus been a recognition of the desirability of extending the probing depth of XPS.105At electron kinetic energies larger than around 100 eV, the electron mean free pathlength, λ, scales roughly with E1/2 (where E is the photoelectron kinetic energy),106 so one obvious way to achieve this is to use a higher X-ray energy source to increase the energy of the photoelectrons leaving the sample. Hard X-ray photoelectron spectroscopy (HAXPES), which typically uses X-rays with energies ca. 3–10 keV, has developed from the first experiments in 2001–2002 to be an important area of research, as illustrated by the rapid increase in HAXPES publications and citations to them.105 One significant drawback that has slowed development is the rapid drop in photoelectron cross sections as the photon energy is increased (Fig. 7).107,108 Until recently, any form of lab-based HAXPES has utilised conventional low-intensity, unfocussed X-ray sources (typically of the twin-anode type using 5.41 keV Cr Kα or 2.9 keV Ag Lα sources), making many HAXPES experiments inviable in reality as unrealistic counting times are needed. The need for a brilliant photon source, however, led to rapid development of the technique at synchrotron facilities in the last 20 years (such as the GALAXIES beamline at SOLEIL, I09 at Diamond, 9.3.1 at ALS and no fewer than 12 beamlines at SPring-8), which in general became rapidly oversubscribed. In the last few years, there has been a significant effort by manufacturers to alleviate this situation by providing higher flux HAXPES sources with high transmission analysers in order to make high-throughput lab-based HAXPES feasible. The capability to perform such spectroscopy in the laboratory environment with sufficient flux and sensitivity to be able to make rapid measurements is a step change in photoemission spectroscopy. The widespread deployment of these instruments could see HAXPES become as ubiquitously used in the next 20 years as XPS is now.
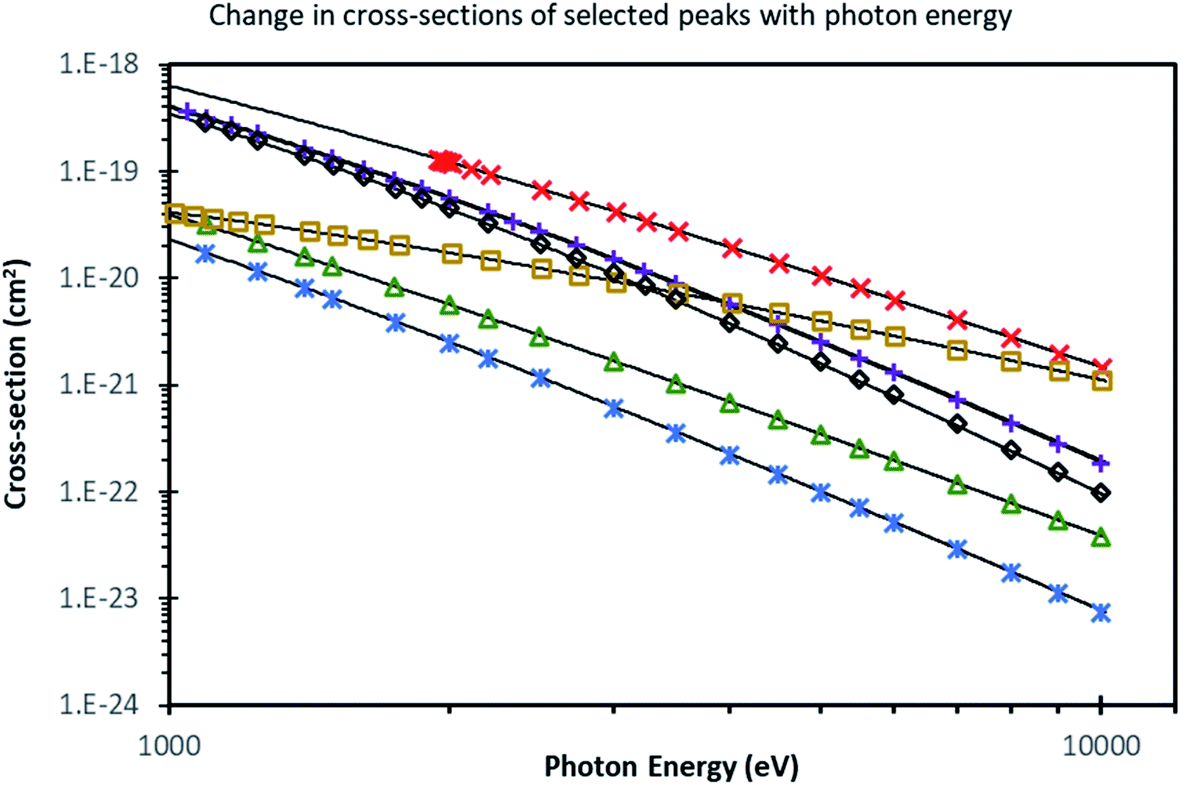 | ||
Fig. 7 Cross section values from Sabbatucci and Salvat (ref. 107) for some selected subshells, along with the associated model (lines), depicted on a log–log plot. Values from the database are shown as points, with descriptive curves from ref. 108 shown as lines. Markers correspond to subshells as follows: ( – C 1s); ( – C 1s); ( – Si 1s); ( – Si 1s); ( – Si 2p3/2); ( – Si 2p3/2); ( – Ag 3d5/2); ( – Ag 3d5/2); ( – Au 4s); ( – Au 4s); ( – Au 4f7/2). Reproduced from ref. 108. – Au 4f7/2). Reproduced from ref. 108. | ||
The new generation of high flux lab-based HAXPES instruments is primarily based on Cr Kα and Ga Kα sources. A high flux, monochromated and focussed Cr Kα source was first combined with a high transmission electron energy analyser around 2010.109,110 This has now been implemented in a number of commercially available sources, including the Physical Electronics Quantes (ULVAC-PHI) system.111 More recently, Scienta Omicron GmbH launched the HAXPES-lab instrument.112 This system uses a 9.25 keV metal jet X-ray source (Excillum)113,114 coupled with a bespoke monochromator and a high-transmission EW-4000 electron energy analyser. The focussed X-ray source (with 50 μm spot size) has an intensity three orders of magnitude greater than that of traditional twin-anode X-ray sources, sufficient to obviate the core-level sensitivity issue. The result is that HAXPES measurements can be made on similar timescales to conventional Al Kα XPS (https://doi.org/10.1039/D2FD00021K). Lab-based HAXPES measurements of light elements such as lithium, boron, carbon and nitrogen are made more feasible (https://doi.org/10.1039/D2FD00021K).
Applications of HAXPES are diverse, ranging from the identification of buried oxygen in Cu catalysts for CO2 reduction115 to characterising the likely outcomes of nuclear accidents.116 In particular, HAXPES has proved strikingly powerful in locating and characterising layers and buried interfaces,117 for example in devices (such as prototype metal-oxide-semiconductor field effect transistor devices, https://doi.org/10.1039/D1FD00110H118 and spintronic materials119) and interphases, for example the solid electrolyte interphase in battery materials.120–122 In both cases, there are significant advantages in making measurements in a more realistic environment in a working device or cell, moving towards in operando conditions. Although much of this work has been carried out with synchrotron sources, the new generation of lab-based sources make this work viable in the laboratory. As an example, in this meeting, lab-based HAXPES of a prototypical oxide memory device taken during resistive switching is presented. The chemical modifications under bias and the potential distribution at the interface between a Ti electrode and a film of La0.8Sr0.2MnO3 is investigated (https://doi.org/10.1039/D1FD00110H). In the case of battery materials, the difficulties in approaching operando conditions are perhaps more significant, and much of the initial work was carried out with ex situ electrochemical cycling, with the material subsequently recovered from the cells for examination.120,121 More recently, however, it has become possible to obtain HAXPES from prototype cells during charging and discharging in situ inside the UHV chamber. An example, from BL28XU at SPring-8, is the observation of reversible changes in the Co 2p and O 1s spectra during in situ cycling of a LiCoO2 thin film cell.122
3.2. Depth-profiling using the inelastic background
Probing beyond the elastic limit by modelling the inelastic background associated with each core level excitation is proving very productive in HAXPES experiments.8–13,123–125 This is because, compared with XPS, rather few Auger lines are typically present in the regions of interest, so often the inelastic background can be measured over a very wide kinetic energy range. This is especially true for the deep core levels of heavier atoms, such as in the example shown in Fig. 8,126 where the Ir 3d core level at ca. 2000 eV binding energy is used. This shows 9.25 keV HAXPES from a thin (18 nm-thick) layer of metal–organic complex (Ir(ppy)2(acac), C27H23IrN2O2) buried at various depths of up to 200 nm in organic material (Irganox1010 (C73H108O12)).126 The core level peaks from Ir are strongly attenuated by the 50 nm overlayer, and are completely lost for the 100 and 200 nm layers. However, the inelastic background shows a striking depth-dependent oscillation to lower kinetic energy (higher binding energy) from each core level peak. These features shift as the overlayer thickness is increased, and it can be seen that the onset of the inelastic background from each Ir core level shifts to lower kinetic energy as the Ir photoelectrons travel through more overlayer material, and therefore lose more energy to inelastic scattering, before escaping the surface to detection. Modelling of this background, done here using the QUASES-Generate software package,127 generates overlayer thicknesses within an error of ±5% for the 50 and 100 nm overlayers, and ±10% for the thicker 200 nm overlayer.126 Use of the inelastic background has been demonstrated to enable information to be extracted from an inelastic sampling depth of up to 20λ (potentially corresponding to more than hundreds of nm).126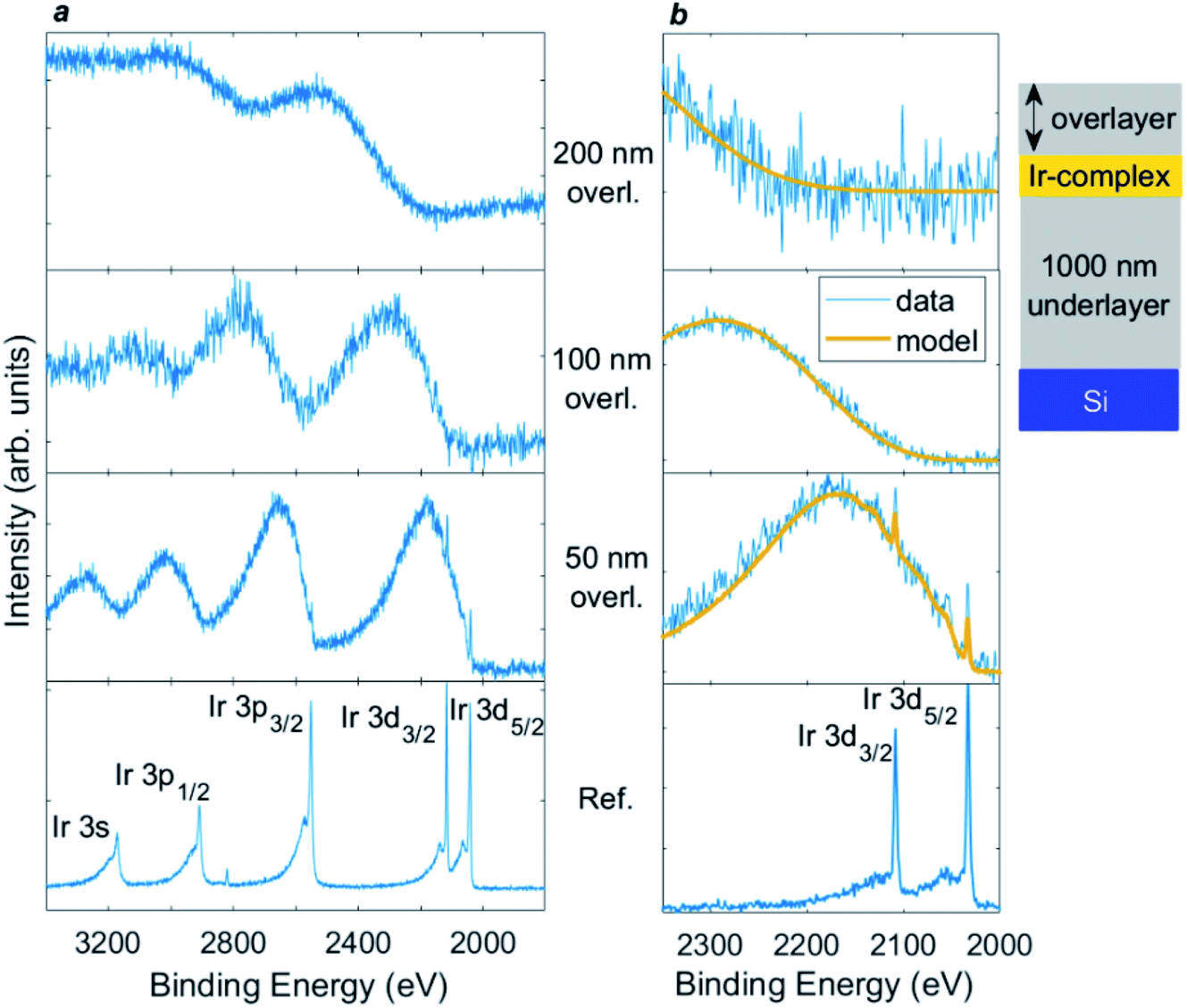 | ||
| Fig. 8 9.25 keV HAXPES for (a) Ir 3s/3p/3d region, and (b) Ir 3d with inelastic background modelling for (from bottom upwards, as labelled) an Ir(ppy)2(acac) layer without an overlayer, and with overlayers of 50, 100, and 200 nm Irganox1010. The layers were deposited on a 1000 nm Irganox1010 underlayer on top of a silicon substrate. Yellow lines show inelastic background modelling using the QUASES-Generate software package (ref. 127) used to obtain the overlayer thickness. The ordering of the layers is illustrated in the inset diagram (not to scale). Reproduced from ref. 126. Copyright © 2021 Author(s). | ||
3.3. Future directions for HAXPES
A further metrological aspect currently receiving attention is the extent to which species identification via the Auger parameter can be extended into the HAXPES regime. As was first recognised for XPS by Wagner, the sum of the binding energies of a strong core level photoelectron line and the kinetic energy of a sharp core–core Auger transition from the same element is a characteristic quantity which can be used to diagnose the local chemical state of the atom.133 The advent of commercial HAXPES instruments which combine traditional low energy XPS sources (such as Al Kα) with a higher energy source (such as Cr Kα or Ga Kα) means that it is now possible to determine the Auger parameter for some very common elements that have high energy KLL transitions (such as Si, Al and Ti) without the use of a synchrotron.134 If the photoelectron and Auger lines are chosen carefully, it is possible to choose pairs with similar probing depth, adding to the accuracy of the analysis.134
In general, the other rapidly developing areas in HAXPES are strikingly similar to those commented on above for NAP-XPS, and have been recently reviewed.105 There is a strong push to combine HAXPES with structural techniques and/or lateral and time resolution. Time-resolved experiments are discussed in detail in Section 4, but it is worth noting here that sub-100 ps HAXPES has been achieved at both synchrotron135 and free-electron laser136 sources. As we shall see below, developments in time-resolved spectroscopy have occurred simultaneously with advances in full-field imaging momentum microscopes operating using time-of-flight.137–139 Although developed primarily for its valence band mapping capability, the momentum microscope can be used directly for its diffraction capability in the hard X-ray regime,140 leading to the rapid development of hard X-ray photoelectron diffraction combined with HAXPES.105,141 The use of harder X-rays in HAXPES also allows for the possibility of extracting structural information using the X-ray standing wave that is set up above and below the surface as the incoming and outgoing beam interference, discussed below.
3.4. Probing buried interfaces using X-ray standing waves (XSW)
The X-ray standing wave (XSW) or SW-XPS (standing wave X-ray photoelectron spectroscopy) technique uses the interference between the incident and a reflected X-ray beam to add depth resolution to XPS, allowing buried interfaces (for example in superlattices) to be probed non-destructively.143 An XSW is formed in the region of spatial overlap between the waves, which results in a periodic modulation of the X-ray intensity perpendicular to the reflecting planes (normally vertically through the sample). Inelastic scattering from the atoms within the XSW acts as the probe, and is measured by X-ray fluorescence (XRF) or photoelectron yield (typically for light atoms). Varying the period of the XSW then allows successive atoms to be brought into regions of high and low XSW electric field amplitude, causing periodic modulation in the XRF or photoelectron yield. The experiment has been implemented in two main modes; in the standard rocking-curve method, the X-rays are incident at the Bragg angle of the surface plane, giving a strong reflected wave at the Bragg condition for the superlattice:| λX-ray = 2dSLsinθB | (5) |
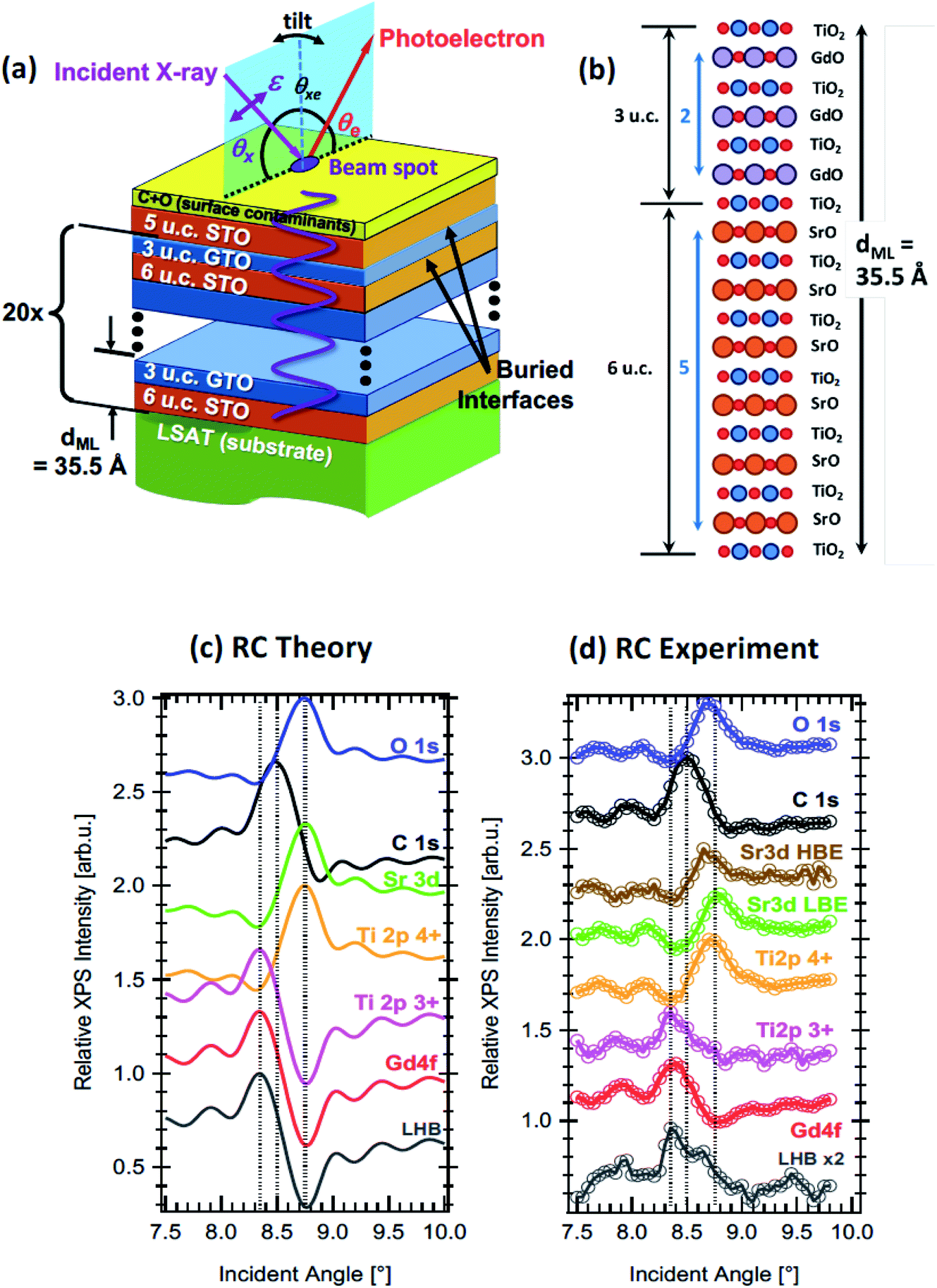 | ||
| Fig. 9 (a) The experimental geometry of the XSW rocking curve method applied to a heterostructure of SrTiO3 and GdTiO3 layers, with various key parameters defined. The angle of incidence, θx is tuned in the plane of reflection through the Bragg angle, changing the period of the XSW shown in purple. θxe is fixed. Tilt in the perpendicular plane can be used to obtain angle-resolved photoemission data. (b) A layer-by-layer drawing of a single GdTiO3/SrTiO3 bilayer in the sample, with 3 unit cells (u.c.) of GdTiO3 (GTO) and 6 u.c. of SrTiO3 (STO). (c) Calculated RCs for various core levels and the lower Hubbard band. (d) Experimental RCs, taken with excitation at 1181 eV, chosen just below the Gd M5 resonance, for the same core levels and the lower Hubbard band. The low binding energy and high binding energy Sr 3d signals are shown separately. Experimental data are from the ALS. Adapted with permission from ref. 142. Copyright 2016 the American Physical Society. | ||
XSW measurements have been typically implemented at synchrotron sources, where the energy tunability can be used to match the XSW to the superlattice/multilayer period. The rocking curve method allows the signal from the buried interface to dominate over the surface signal when the surface is in a region of low electric field amplitude of the XSW. Nevertheless, the photoelectron IMFP still poses a limitation which may be alleviated to some extent by implementing the approach combined with HAXPES at soft-X-ray beamlines, allowing deeply buried layers to be probed. In the last ten years, this approach has been very productively implemented at a number of synchrotron beamlines, including the Galaxies beamline at SOLEIL (and at lower energies, Cassiopee),147 beamline BL15XU at SPring-8,142,148 BLP09 at PETRA III142,148 and beamline 9.3.1 at the ALS.142,146,148 In parallel with these developments, the theoretical background that allows the rocking curves to be simulated in order to fit experiment has been developed and made available,143,149 and measurements have often been combined with resonant photoemission or complemented by real-space imaging, such as by high angular dark-field scanning tunnelling electron microscopy.144 The result is that the chemical structure, oxidation state, interdiffusion or roughness at interfaces,144 and even the built-in potential,147 can now be measured with few-ångstrom resolution. This has been particularly powerful in studies of various types of oxide superlattices, for example in characterising the polarisation-induced charge density accumulation at the interface between a ferroelectric and a Mott insulator,146 in characterising multilayer magnetic tunnel junctions,144 and (by isolating the binding energy shifts due to individual multilayers) in determining the form of the built-in potential in a LaCrO3/SrTiO3 superlattice.147Fig. 9 shows an example where standing wave techniques in the hard and soft and X-ray regime are used with resonant photoemission and electronic structure calculation to characterise the two-dimensional electron gas at the interface between two oxides, SrTiO3 and GdTiO3.142
The structural sensitivity and accuracy of the XSW technique has also been demonstrated using a fs pulsed source of photons from a FEL, FLASH at DESY in Hamburg.145 Photoelectron yield was measured from Si/Mo multilayers terminated with surface SiO2 layers of various thicknesses, allowing determination of the oxide layer thickness to an accuracy of a few percent.145 This opens up the prospect of combining the fs temporal resolution of a FEL with a high level of structural accuracy, in order to better understand the structural dynamics of surfaces. Because XSW does not require lateral long-range order (unlike, for example, low energy electron diffraction), the technique could provide a route to measuring the structural evolution occurring during chemical reactions at surfaces, significantly enhancing our understanding of heterogeneous catalysis. Recent developments in time-resolved photoemission at FEL and other light sources are reviewed in the next section.
4. Time-resolved surface analysis
4.1. Introduction
The study of the ‘real-time’ dynamics of charge carriers at material interfaces is becoming ever more important as next generation technologies increasingly access nanoscale dimensions. An example is solar harvesting technologies, where a clear understanding of the dynamics of photoexcited carriers is of key fundamental importance. Similarly, in catalysis, there is a need to underpin our understanding of reaction pathways by acquiring data on the timescales of bond making and bond breaking, so that we may improve our understanding of the transition state and the way it is modified by the catalyst. The last 20 years in particular has been characterised by a drive to complement our often detailed understanding of static structure and chemical composition by an improved understanding of dynamics of various types. Surface scientists have been in the vanguard of this campaign, driven by the fundamental importance of the surface to problems in passivation, catalysis and device design.In fact, the primary processes of interest to physical scientists occur over a wide range of timescales (Table 1), and so require a range of different experimental arrangements for their measurement. One common feature of many such experiments is that they operate on the ‘pump–probe’ principle, where a pulse of (typically) electromagnetic radiation induces the process of interest, and a second pulse, with a known but variable time delay from the first, probes the dynamics of the system. Clearly, the shortest dynamics that can be measured are then limited by the pulse length of the pump pulse, so typical synchrotron pulses (of some tens of ps or longer) may only be used to probe the longer timescales listed in Table 1, such as exciton lifetimes. Studies of molecular vibrations and bond breaking (in the 10–100 fs range) have been addressed primarily using high power pulsed table-top lasers or FELs, which can attain pulse lengths of a few fs routinely. Time-resolved photoemission has emerged as an important technique in all these ranges, using lab-based high-harmonic generation (HHG) sources,137 at FELs such as FLASH,138 the linac coherent light source (LCLS) at Stanford150 SACLA at SPring-8151and FERMI at Elettra,152 and at synchrotrons including SOLEIL (https://doi.org/10.1039/D1FD00107H), ALS153 and SPring-8.135
| Timescale | Process | Providing access to |
|---|---|---|
| as | Fundamental electron motion | Wave packet dynamics |
| 10s as | Photoemission from valence and low-lying core levels | Correlation dynamics and relative photoemission delays |
| 100s as to few fs | Core hole lifetime | Core-hole clock spectroscopy |
| Few fs | Nuclear motion | Nuclear dynamics |
| <10 fs | Some charge transfers | Charge transfer into some semiconductors |
| 10–100s fs | Vibrations, bond-breaking | Reaction dynamics |
| 100s fs | Photoexcited quasiparticle relaxation | Electron dynamics |
| 100s fs | Spin and lattice changes | Spin dynamics |
| 1 ps | Electron-phonon thermalisation | Charge-density-wave melting |
| Few ps | Cooper-pair formation in superconductors | Superconducting dynamics |
| >50 ps | Spin-lattice equilibration | Magnetisation dynamics |
| 10s ps to ms | Exciton lifetimes in semiconductors | Charge dynamics |
| 10s ns | Domain wall motion | Magnetisation switching |
| 100s ns to s | Formation of reaction products | Reaction kinetics |
4.2. Attosecond and few-femtosecond dynamics probed using the ‘core-hole clock’ method
At the shortest ultrafast times, attosecond (as) pulse lengths can now be achieved using lab-based tabletop lasers, but this is still far from routine, and studies have so far focussed on gas-phase studies where Auger decay and valence electron motion have been observed in real time (for reviews, see e.g. ref. 154–157) and wavepacket dynamics are probed on timescales where the nuclei are not effectively frozen, as assumed by the Franck–Condon principle. In the solid state, the space charge effect in ultrafast photoelectron generation has proved a huge challenge, and, as we shall see in Sections 4.3 and 4.5, significant progress enabling solid-state attosecond (or even fs) photoemission has only been made very recently. By contrast, research using all-optical ‘photon-in, photon-out’ techniques (such as fs and attosecond XAS) has progressed more rapidly.155,156 To date, an alternative photoemission-based approach, the ‘core-hole clock’ method, has been used more widely to probe ultrafast dynamics in solid state systems, particularly for molecules adsorbed at surfaces.The core-hole clock method uses the core hole created by photoelectron emission as an internal clock to detect changes in electronic configuration that occur before it decays. This is typically implemented at soft X-ray (often called ‘tender X-ray’) synchrotron beamlines, using photon energies in the range of 2–10 keV. The core holes created by this radiation typically have a lifetime on the order of a few fs, and sometimes even hundreds of attoseconds, allowing (in particular) the detection of some ultrafast charge transfer processes that occur faster than this (or on similar timescales).158,159 The technique, as applied to surface adsorbates, is typically implemented by a careful comparison of X-ray absorption (XAS) and resonant photoemission (or resonant photoelectron spectroscopy, RPES) spectra, aided by the high resolution now available at soft X-ray synchrotron beamlines. RPES monitors decay events of the core excited state that involve the photoexcited electron (Fig. 10). If a transfer of the photoexcited electron (say out of a surface-adsorbed molecule) takes place on a timescale that competes with the normal decay of the core hole, this modifies the RPES intensities (Fig. 10). The RPES intensities can then be compared with XAS intensities to estimate the electron transfer time.158
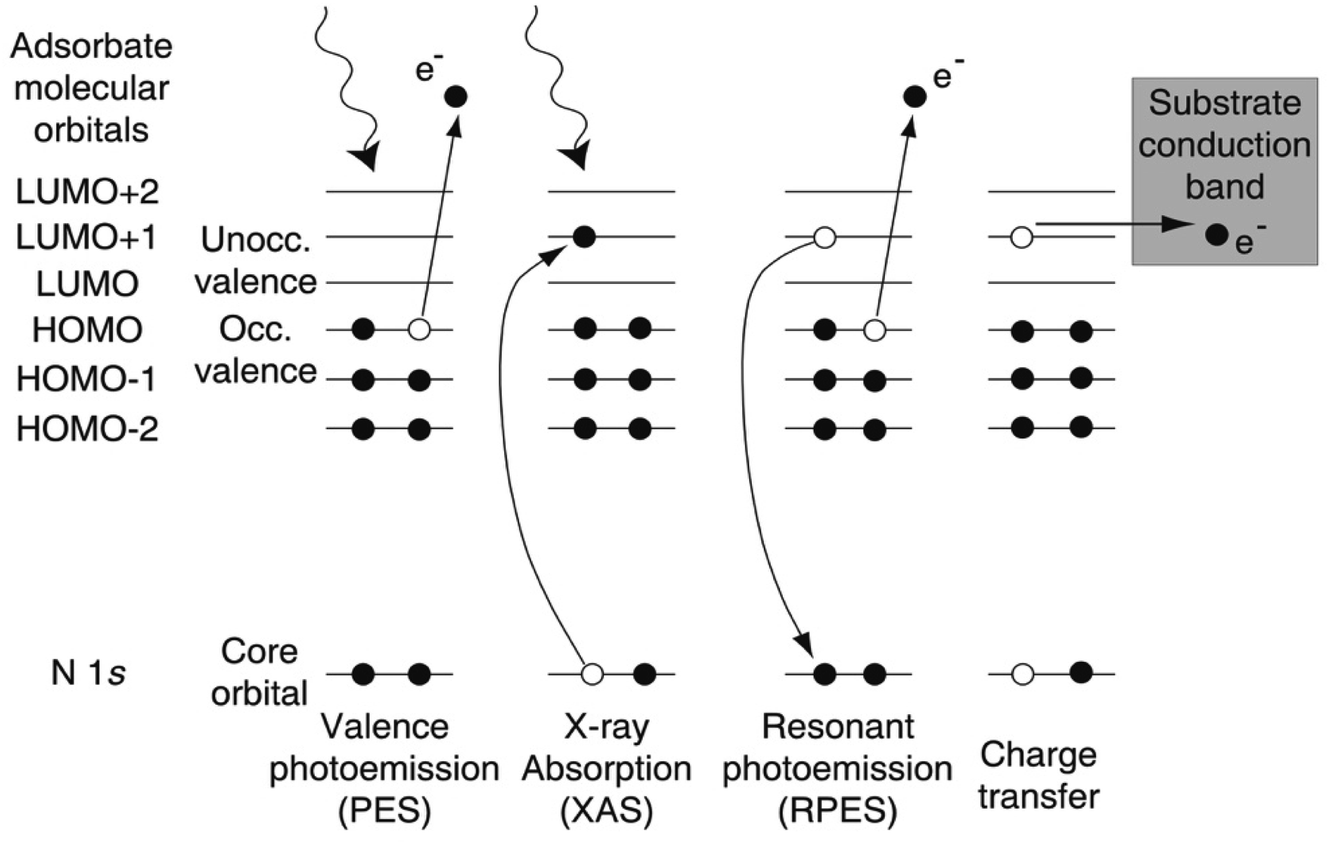 | ||
| Fig. 10 Left to right: photoelectron spectroscopy (PES), X-ray absorption spectroscopy (XAS) and resonant photoelectron spectroscopy (RPES) applied to a nitrogen-containing molecule. The electronic states are indicated by lines. PES measures the occupied and XAS the local unoccupied density-of-states. RPES monitors the decay of the excited state. Far right: If the molecular orbital couples to the substrate density-of-states, an electron transfer may be possible that competes with RPES. HOMO, highest occupied molecular orbital; LUMO, lowest unoccupied molecular orbital. Here, an electron transfer is energetically possible from LUMO+1 and LUMO+2, but not from the LUMO. If the depicted electron charge transfer occurs, the RPES is reduced in spectral intensity. Copyright Joachim Schnadt, 2022, private communication, reproduced with permission. | ||
A seminal early demonstration of this technique was its application to dye-sensitised solar cells, where measurements at the nitrogen K-edge were used to show that the transfer of a photoexcited electron from a surface-adsorbed bi-isonicotinic acid molecule (a model for the ligands of the dye160) into a TiO2 substrate occurs on an estimated timescale of <3 fs.161 At the time of this experiment, the available time-resolved laser techniques had shown only that the upper time limit was of the order of 20–100 fs.162 This was the first study to demonstrate that the electron transfer from an aromatic adsorbate to a semiconductor can occur in only a few fs, and so competes effectively with loss processes, such as the intramolecular thermalisation of excited states.161 In 2008, this approach was extended to the full so-called ‘N3’ dye molecule (cis-bis(isothiocyanato)bis(2,2′-bipyridyl-4,4′-dicarboxylato)-ruthenium(II)), deposited intact onto TiO2 by electrospray deposition.163 This showed that electron injection from the N3 dye into the substrate takes place in <16 fs.163 By focussing on the creation of short-lived holes, where the initial and final states are in the same electronic shell, the resolution has been improved into the attosecond regime. Measurements on the c(4 × 2)–S/Ru(0001) surface (using the S LLM decay) showed that electron transfer from an adsorbed S atom to the Ru surface takes place in around 320 as.164 The approach has now been implemented at tender X-ray HAXPES/XAS beamlines (for example at the GALAXIES beamline at SOLEIL), allowing the comparison between resonant Auger emission and resonant inelastic X-ray scattering (RIXS).159 Tender X-ray beamlines provide access to a wider range of K-edges of important elements, including those of S and P. Recently studies at the S K-edge have been used to probe charge transfer in organic heterojunctions,165 in MoS2)166 and SnS2, using polarised light to probe the effects of in-plane and out-of-plane excitation.167 Charge transfer times as short as tens of attoseconds have been determined.166
4.3. Towards reaction dynamics and molecular movies – femtosecond pump–probe measurements
On slightly slower, fs timescales, the concept of making a ‘molecular movie’ showing atoms and molecules moving and reacting in real time has been a long-held goal, as this would revolutionise our understanding of reaction dynamics. The ability to probe species on surfaces at these timescales took a major leap forward with the development in the last 15 years of FELs that can deliver ultrashort (few fs) soft X-ray pulses, combined with atom-specific spectroscopic tools that can monitor the evolution of the electronic structure.139,151,168,169 The implementation of these measurements in solid state photoemission has not been without its difficulties (associated with space charge issues, discussed further below), and in general, until very recently most progress had been made with ‘photon-in-photon-out’ approaches that do not change the electron count of the sample.In fact, the first direct time-resolved measurement of the transition state (TS) region during a surface reaction made using an X-ray FEL (XFEL) employed X-ray emission spectroscopy (XES) and XAS applied to the oxidation of CO on a Ru(0001) substrate.170 Here, an optical laser pump pulse was used to excite the substrate electrons, with energy transfer leading to a rapid increase in adsorbate-substrate vibrational excitation (in several hundred fs), sufficient to drive the CO oxidation. The LCLS was used to provide synchronised X-ray pulses probing the O K edge. Within about 800 fs, new electronic states were detected due to the TS region en route to the formation of CO2. Remarkably, about 10% of the CO was observed to populate the TS region within around 1 ps, the majority of which dissociated back to O and CO.170 This ability to probe transient species close to the TS gives completely new insights into the electronic states of reacting molecules at surfaces and provides essential input to theory. Further experiments in the last few years have allowed the timescales of electron excitation, phonon thermalization, and the excitation of various adsorbate modes to be distinguished.171,172 An example is recent studies using the seeded FEL facility at FERMI at Elettra, using variably polarised soft X-ray pulses to excite the C K-edge to probe CO dynamics at the Ru surface (Fig. 11).173 As the C K-edge is subject to less vibrational broadening than the O K-edge, this allows the evolution of the spectral features to be distinguished more clearly. This has revealed the surface diffusion of excited CO molecules to highly coordinated sites in the first ps, before a fraction enter the precursor state. In this state, the molecules are free to rotate isotropically for several ps before being desorbed or readsorbed, illustrated in Fig. 11.173
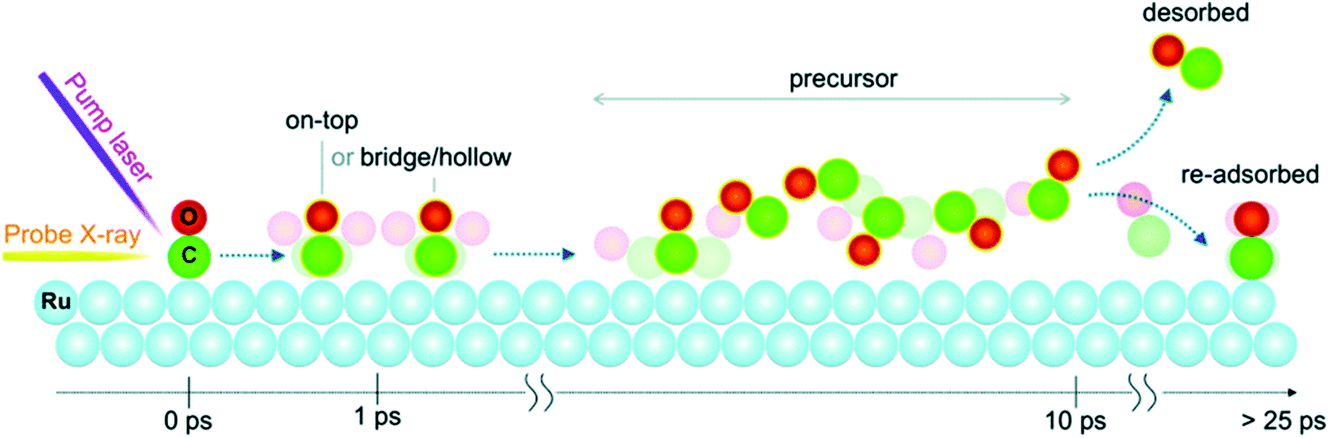 | ||
| Fig. 11 Dynamic evolution as function of pump–probe delay for CO chemisorbed on Ru(0001) following optical laser excitation. After laser irradiation of Ru(0001), CO molecules are excited and diffuse from the on-top sites towards more highly coordinated sites, followed by isotropically rotating in a precursor state. A fraction of CO in the precursor state desorbs while the majority re-adsorb back onto the surface. Reproduced from ref. 173 with permission from the PCCP Owner Societies. | ||
Although these experiments are undoubtedly challenging, the future looks exciting for studies of surfaces and solid-state systems via time-resolved XAS. While FELs typically deliver few-fs pulses, the delivery of attosecond pulses with good coherence and sufficiently high repetition rate has been demonstrated.174 Meanwhile, the development of lab-based HHG sources has continued apace, and isolated soft X-ray attosecond pulses with a broad bandwidth suitable for XAS measurements have become available. This has allowed the sub-cycle optoelectronic response in a range of quantum solids to be probed.155 A recent example is studies of the transition-metal dichalcogenide TiS2, where attosecond Ti L2,3 XANES is used to probe the 3d-state dynamics, in the presence of a near-IR optical control field (bridging the bandgap). This shows that localization of electron density around the Ti atoms occurs within a fraction of the cycle of the optical control field.155
Turning to photoemission, in the last few decades, fs time-resolved photoemission excited with optical lasers has become well-established (typically using as a pump an infrared (1.5 eV) output from a Ti:sapphire laser with a pulse length of a few tens of fs). For one-photon photoemission, the probe pulse energy must at least exceed the sample work function, requiring a probe pulse energy of typically ≥6 eV. This can be readily provided by cascaded frequency upconversion in non-linear crystals (as in e.g. ref. 175–177), although it does mean that only low binding energy states may be probed, and the low tunability of the source can mean that only states around the Brillouin zone centre can be probed in most materials. This approach has been used to probe a variety of phenomena in condensed matter physics and chemistry, such as charge-density wave melting,176 the momentum dependence of Cooper pair dynamics in high temperature superconductors,175,178 insulator–metal transitions in Mott insulators,179 the non-thermal carrier dynamics in graphite,180 and the ultrafast electron dynamics at the surface of the topological insulator Bi2Se3.181
These laser-excited photoemission studies are limited by the lack of chemical specificity that arises as the core levels cannot be accessed. There has been a strong drive in recent years to provide extreme ultraviolet (XUV) or soft X-ray pulses suitable for photoemission measurements from solids. Rapid developments have occurred along two main fronts, the development of HHG sources that provide sufficient flux in the few tens of eV energy range,182 and FEL sources with sufficiently reproducible pulse trains.139 However, until very recently, the great potential for time-resolved photoemission from solids using these sources has been hampered by vacuum space charge effects, which arise when too many photoelectrons are emitted from the same ultrashort high intensity photon pulse. This causes broadening and shifting of the spectral features. The effect was noted, in the case of FEL radiation, in studies of W(110) using FLASH (Hamburg) as early as 2008,169 and, by controlling the fluence, some measurements of CDW melting in 1T-TaS2 were possible.168,183 However, the developmental work needed to find ways of bringing vacuum space charge effects under control139,168,184–188 has delayed the widespread deployment of both FEL and HHG sources in time-resolved photoemission from the solid state. In order to control these effects, it is necessary to control the number of emitted photoelectrons per pulse,139 which is obviously counterproductive to the spectral signal-to-noise. It is thus necessary to use the most efficient detection schemes in combination with high repetition-rate sources. In the last few years, significant progress has been made in the development of both FEL139 and HHG189,190 pulse trains with higher repetition rate. As noted in Section 3.3.2, these developments have occurred simultaneously with advances in full-field imaging momentum microscopes operating using time-of-flight.137–139 This has occurred alongside an improved theoretical understanding of the problem,151,187 and experimental developments such as a retarding the electrostatic front lens, which suppresses the pump-induced slow photoelectrons.186 These developments have transformed the prospects for solid state photoemission using FEL and HHG sources, and made possible solid-state time- and angle-resolved photoemission with combined time-resolved momentum microscopy (tr-ARPES/tr-MM, Fig. 12).139 As full-field photoelectron diffraction is now demonstrated in static experiments, there is a realistic prospect that tr-ARPES/tr-MM can be used with fs core-level excitation to simultaneously probe how the both the electronic states and the coherent lattice motion evolve after a pump pulse, by exploiting photoelectron diffraction and X-ray standing waves.138,145,191
 | ||
| Fig. 12 Time-resolved momentum microscopy of graphene. (a) Schematic diagram of the experiment. The time-resolved photoemission intensity is measured from the graphene C 1s core level and valence band by combining an infrared pump pulse with a soft X-ray probe pulse from FLASH. The electrons are detected over a wide angular range using a momentum microscope. (b) C 1s photoemission intensity as a function of the pump–probe delay and binding energy. (c) Same as in (b) but with the average spectrum before excitation subtracted. Red and blue indicate an increase and decrease in electron counts, respectively. (d) Selected spectra (cuts at specific time delays) fitted with a model described in ref. 138 (blue line). The temperature-dependent asymmetry kernel for each fit is shown in green, showing the way in which the low temperature asymmetry due to electron–hole pair excitations around the Fermi energy is reduced as energy gains become possible at high electronic temperature. The electronic temperatures resulting from the fit are noted close to the spectra. Adapted with permission from ref. 138. Copyright 2021 by the American Physical Society. | ||
These recent developments are enabling a new generation of time-resolved photoemission experiments, giving unprecedented insight into ultrafast dynamics in the excited state, ranging from the nonequilibrium band structure of WSe2 (ref. 139) to momentum-space imaging of transiently excited unoccupied molecular orbitals in surface-adsorbed organic molecules.137 An example is the use of an IR pump pulse and soft X-ray probe pulse at FLASH to measure the ultrafast linewidth broadening of the C 1s core level of graphene, illustrated in Fig. 12.139 This is used to show that the broadening is caused by an exchange of energy and momentum between the outgoing photoelectron and the hot electron gas, rather than by vibrational excitation. The momentum change of the outgoing photoelectrons leads to a small change in the time-resolved photoelectron diffraction pattern measured using a momentum microscope.
4.4. Slower dynamics; probing carrier recombination dynamics and reaction kinetics
As illustrated in Table 1, a great many dynamic processes, particularly those of interest to chemists, occur on timescales of the order of some tens of ps or longer, and do not require the use of an ultrashort pulsed source for their investigation. Here, the highly reproducible, lower intensity and high repetition rate source provided by a synchrotron is highly suitable, as it largely avoids the space charge issues encountered in photoemission with ultrashort pulses from FEL and HHG sources. In the last decade or so, various types of ‘hybrid’ or ‘low α-’ mode operation (where both continuous and few-ps pulsed electron bunch trains are available in the same machine fill) have been developed at several synchrotron sources (including SOLEIL, SPring-8 and BESSY II) with the aim of providing access to dynamic measurements.192–194 Photoemission measurements using these sources have focussed on probing carrier recombination dynamics at surfaces.Carrier dynamics at the surfaces of semiconductors may be probed by pump–probe spectroscopy using time-resolved surface photovoltage (SPV) spectroscopy.195–197 The space charge layer at the surface of semiconductors where the Fermi level is pinned by surface states within the band gap leads to band bending in this region. For an n-type semiconductor with a depletion layer at the surface, the bands bend upwards, and downwards in a p-type semiconductor. A laser is used to promote carriers across the band gap, and, in the depletion region of an n-type semiconductor, electrons and holes migrate away from and towards the surface, respectively (and vice versa in a p-type semiconductor, Fig. 13).198 This additional electric field reduces the band bending, creating a shift in the surface photovoltage which shifts the energy of the valence band maximum in the space charge region (in n-type semiconductors to a higher BE and to a lower BE in p-type).197 The SPV shift relaxes once all carriers have recombined and the surface potential is returned to its original value, on timescales that vary from typically a few μs to seconds or longer in the case of persistent photoconductivity.195 In the period before this occurs, the SPV shift can be measured with photoemission as a function of time, either as a change in the position of the valence band maximum (VBM), or a change in the binding energy position of a core level peak.197,199,200 In the last 30 years, the technique has been implemented in various ways, including with both continuous (multibunch) and pulsed (single bunch or hybrid mode) synchrotron X-ray sources as the probe beam.193,196,197,201,202 In the latter case, the X-ray probe pulses are synchronised to the initial photoexcitation, and used to excite core or valence level photoelectrons (using XPS). By varying the time delay between the laser pump and XPS probe, the recombination dynamics are monitored over time as the carriers recombine and the band bending and binding energies return to equilibrium.196,197,201,202 This allows the recombination dynamics at the surface to be inferred (https://doi.org/10.1039/D1FD00107H).196,197,201,202 Such measurements have been implemented at a number of world synchrotron sources, one of the earliest being the TEMPO beamline at SOLEIL.192,194,203 As well as measurements of recombination at semiconductor surfaces (primarily Si) (https://doi.org/10.1039/D1FD00107H),196,197,201,202 charge injection across interfaces where there is a surface depletion layer has also been monitored, for example from gold nanoparticles into TiO2 153 or from light-absorbing quantum dots into ZnO.204 In combination with time-resolved magneto-optical Kerr effect measurements, pump–probe photoemission of this type has also been used to study magnetisation dynamics, such as the antiferromagnetic–ferromagnetic transition in FeRh films, which occurs within 100 ps205 and electron relaxation dynamics over several hundreds of ps in the half metallic La1−xSrxMnO3.135
 | ||
| Fig. 13 Schematic band diagrams of the surface photovoltage effect in (top) a p-type semiconductor material with a hole depletion layer at the surface and (bottom) an n-type semiconductor material with an electron depletion layer at the surface. The change in the band bending is indicated by the SPV shift (V0), and the positions of the core levels relative to their equilibrium positions are indicated. Reproduced from ref. 198. Copyright © 2021 Author(s). | ||
Laser-pump X-ray-probe SPV experiments using pulsed lasers, where the pump laser is synchronized to individual X-ray pulses from a synchrotron, are limited by the time window available, determined by the synchrotron pulse length and repetition rate. For synchrotron excitation, this limits the time resolution to (typically) some tens of ps and the maximum time window to some hundreds of ns, often less. This means that for processes occurring on slower timescales, the observed SPV transient is influenced by the residual SPV induced by preceding laser pump pulses.197 If separate experiments can be carried out using as a probe both the normal ‘multibunch’ train from the synchrotron and the isolated pulse of hybrid mode, then it becomes possible to start to deconvolute processes occurring on ps timescales from longer-lived effects. An example is work carried out at the ALOISA beamline at Elettra, where a lattice distortion occurring in 2H-MoTe2 on sub-ns timescales is distinguished from a SPV shift persisting for μs (https://doi.org/10.1039/D1FD00105A). The use of angle-resolved time-of-flight (ARToF) analysers with two-dimensional delay-line detectors (such as at BESSY II206 and at the BL07LSU beamline at SPring-8193) has allowed synchronisation of the pump laser pulse with hybrid mode pulses in multiple synchrotron periods, enabling SPV measurements extending to μs timescales, for example in studies of Si(111),207 ZnO(0001),208 TiO2(110)209 and phthalocyanine/fullerene layers adsorbed on TiO2.210
A different approach is necessary to enable SPV measurements over much longer timescales (up to ms or longer), which may be required where the electron dynamics are intrinsically very slow, for example due to persistent photoconductivity caused by carrier trapping.197,204 This has been enabled at the TEMPO beamline at SOLEIL, where pump–probe measurements are now possible over timescales ranging from 50 ps to ms or longer. Fast (ps to ns) measurements use a pulsed fs laser in combination with hybrid mode synchrotron radiation.192 Dynamics with slower characteristic times are conveniently monitored using a CW, rather than a pulsed laser. This is modulated using the output of a signal generator, where the period and duty cycle are easily controlled over wide ranges, and a fast XPS detection system (with a two-dimensional delay-line detector) is used to monitor XPS spectra at small time intervals over this modulation period.197,211,212 The access that this provides to a wide range of timescales is very convenient in systems where small changes in the chemistry may result in a change of orders of magnitude in the carrier decay rates.
One such example is in colloidal quantum dot (CQD) solids, made up of an ordered arrays of quantum dots passivated with ligands. These solids take on bulk material properties, as the wave functions of adjacent CQDs overlap,213 coupling them together.214 This makes them fascinating subjects for the study of the transition from localised to itinerant properties. It also provides potential for controlling bulk properties, including the semiconductor doping level, carrier concentration, mobility, and lifetimes, through control of the CQD material, size, shape and surface chemistry.214 They have a range of optoelectronic devices ranging from solar cells215,216 to photodetectors.217–219 Tailoring the band alignment and band bending at the junctions between CQD solids and oxides, metal cathodes or other CQD solids has become important for device design.220,221 For example, in CQD photovoltaics, this has led to more efficient carrier separation and extraction.222 It is thus important to develop a clear understanding of how band bending occurs in these solids. Time-resolved SPV spectroscopy has been used to show that band bending is intrinsic to CQD solids and occurs at the CQD solid–vacuum interface (Fig. 14).198,223 It is observed in the presence and in the absence of detectable surface oxidation, and for a number of ligand types and synthesis routes. The timescale of the photoexcited SPV dynamics is found to depend dramatically on the surface chemistry of the film. If large amounts of oxygen-containing contaminants are present in the film (as in the example in Fig. 14(b)), the rates of carrier recombination decrease by 6 orders of magnitude compared to those of a film with no observable contamination (Fig. 14(c)).198 This is consistent with the hypothesis that dynamic deep traps are formed when photoexcited carriers react with contaminants, decreasing the mobility of the photoexcited carriers.224,225
 | ||
| Fig. 14 (a) Time-resolved SPV shift of the Pb 4f core level in a PbS CQD film with the CW laser ‘on–off’ cycle superimposed; (b) a film treated with 3-MPA ligands under illumination by a λ = 375 nm CW laser modulated on a 50% duty cycle with a period of 40 s; (c) a film with a quasi-epitaxial MAI/PbI2 ligand shell, with the CW laser modulated on a 50% duty cycle with a period of 100 μs. The X-ray photon energy was 475 eV, corresponding to a sampling depth of 1.4 nm.223 The rise and decay of the pump-induced SPV shift are fitted using double exponential functions (yellow and blue lines, respectively). Reproduced from ref. 198. Copyright © 2021 Author(s). | ||
Finally in this section, we note that the advent of high transmission electron energy analysers (often with two- or three-dimensional delay-line detectors) means they may conveniently be used in ‘snapshot’ mode to record in real time the evolution of core level peaks over the comparatively long timescales of 103 s or longer that may be needed for kinetic analysis. Some examples are measurements of the adsorption kinetics of water on the Si(001)-2 × 1 surface,226 of ammonia on the hydroxylated Si(001)-2 × 1 surface227 and the role of adsorbed atomic oxygen in the insertion of Pd ions into the 2H-TPP porphyrin.228
4.5. Future directions in time-resolved experiments
As we have noted in previous sections, there is in general a very strong push towards multi-modal analysis, and to improve the time resolution of both NAP-XPS (discussed in Section 2.7) and HAXPES (discussed in Section 3.3.2). It is also clear that the advent of time-resolved momentum microscopy, together with higher repetition rate pulse trains, is transforming the prospects for fs time-resolved solid-state photoemission in greatly alleviating the problems associated with sample charging in the solid state. The drive for shorter and shorter pulses, allowing the fastest dynamics to be probed, will continue. However, measurements in the attosecond regime remain challenging. One fundamental issue affecting attosecond photoemission is that it has proved very hard to attain the few meV resolution that is routinely attainable in static photoemission, as the use of an attosecond pulse intrinsically implies a broad bandwidth of up to a few tens of eV.229 Since 2000, two major experimental advances in attosecond metrology have made such photoemission possible; attosecond streaking, using isolated attosecond pulses and RABBIT (reconstruction of attosecond harmonic beating by interference of two-photon transitions), using attosecond pulse trains. A detailed description can be found in a number of reviews,154,156,157 but both exploit the sub-cycle temporal structure of HHG radiation, and its synchronisation with the driving field provided by the intense visible/near IR laser pulses used to create it, to analyse the evolution of the photoelectron spectra with the varying time delay between the two optical fields. Both techniques have been used extensively to measure attosecond photoemission delays from gases, providing insight into electron dynamics within the atom.154,156,157,230 By contrast, solid-state attosecond photoemission has proved much more challenging, largely because the space charge effects we discussed in the context of fs spectroscopy are significantly more problematic in the case of shorter attosecond pulses.231 As in fs photoemission, this has accelerated the drive to produce pulse trains with high repetition rates which allow for a high average photoelectron flux while releasing only a relatively low number of photoelectrons per pulse. Recently, high flux MHz HHG sources have become available,156,232 enabling measurements from solids. In the photoemission process, the outgoing photoelectron wavepacket forms in a few tens of attoseconds, so these studies tell us a great deal about the photoemission process itself,233 requiring us to revise our understanding of the process. For example, studies of WSe2 have shown that electrons with low angular momentum, l, are emitted first, while those with high l are delayed.234 This effect, due to electron correlation, is well-established in studies of atoms,230,235 but is neglected in models of solid-state photoemission, which historically have treated photoemission as an instantaneous single-particle phenomenon.230,234 More recently, a high flux 18.4 MHz HHG source has been used to enable a study of attosecond intra-valence band dynamics in W(110), showing that sp-band photoemission is delayed by almost 40 as compared with emission from the d-band.231 It seems clear that in the immediate future we are set to learn more than we ever thought possible about the photoemission process itself.5. Outlook and conclusions
With the obvious pun fully intended, the future is bright for surface analysis by photoelectron spectroscopy. In the last decade, we have seen the development of a number of truly disruptive technologies. These have included the development of lab-based, high intensity tender X-ray sources, and high repetition rate HHG sources. The deployment of three-dimensional delay-line detectors in full-field imaging momentum microscopes has transformed the prospects for ultrafast time- and angle-resolved photoemission from solids. In some cases, an advance in one area has aided another – for example, the rapid uptake of HAXPES has made it easier to achieve ambient pressure XPS measurements, and the geometries of these experiments lend themselves to multimodal approaches with the incorporation of other spectroscopies. The clever design of sample environments has meant that we are just beginning to be able to make genuinely operando measurements in real time on working systems. There is a strong drive to harness these capabilities together and to provide all-encompassing measurements with lateral resolution and time resolution (and also spin resolution, a topic which I have not discussed, but where there is also huge progress5,236). Advances in our theoretical understanding have supported these developments – for example in developing our understanding of the vacuum space charge effect in time-resolved photoemission and of the inelastic background in HAXPES. In turn, experimental advances, for example the observation of photoemission delay in the solid state, are a salutary demonstration to us that our understanding of photoelectron emission from surfaces is not complete. It is not an over-statement to say that we will learn more in the next decade than we currently know about how real systems behave in real time. Certainly, it seems clear that today’s postgraduate students inherit an exciting and burgeoning field (which will be worth the investment needed to learn an exponentially rising collection of acronyms!).Data availability
This review brought together existing research data obtained upon request and subject to licence restrictions from a number of different sources. No data were created or analysed in this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I am grateful to all my co-workers for the studies reported in this paper that come from our own work. I also thank a number of colleagues, including Joachim Schnadt, Slavomir Nemšák, Davide Curcio, Ben Spencer, Pip Clark, Andrew Thomas and Chun-Ren Ke, for providing or editing original figures. Aspects of the research described in this paper have been supported by the EPSRC, including the Henry Royce Institute for Advanced Materials (funded through the EPSRC grants EP/R00661X/1, EP/P025021/1 and EP/P025498/1) and EP/K009710. I acknowledge SOLEIL for providing access to synchrotron radiation facilities. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence (where permitted by UKRI, ‘Open Government Licence’ or ‘Creative Commons Attribution No-derivatives (CC BY-ND) licence may be stated instead) to any Author Accepted Manuscript version arising.References
- A. Einstein, Ann. Phys., 1905, 322, 132–148 CrossRef.
- C. N. Berglund and W. E. Spicer, Phys. Rev., 1964, 136, A1030–A1044 CrossRef.
- S. Hagström, C. Nordling and K. Siegbahn, Z. Phys., 1964, 178, 439–444 CrossRef.
- H. R. Robinson and C. L. Young, London, Edinburgh Dublin Philos. Mag. J. Sci., 1930, 10, 71–75 CrossRef CAS.
- P. D. C. King, S. Picozzi, R. G. Egdell and G. Panaccione, Chem. Rev., 2021, 121, 2816–2856 CrossRef CAS PubMed.
- W. E. Spicer, CRC Crit. Rev. Solid State Sci., 1976, 6, 317–336 CrossRef CAS.
- S. Hüfner, Photoelectron Spectroscopy: Principles and Applications, Springer Series in Solid State Sciences, Springer, Berlin, 3rd edn, 2003, vol. 82 Search PubMed.
- Y.-T. Cui, S. Tougaard, H. Oji, J.-Y. Son, Y. Sakamoto, T. Matsumoto, A. Yang, O. Sakata, H. Song and I. Hirosawa, J. Appl. Phys., 2017, 121, 225307 CrossRef.
- C. Zborowski, O. Renault, A. Torres, Y. Yamashita, G. Grenet and S. Tougaard, Appl. Surf. Sci., 2018, 432, 60–70 CrossRef CAS.
- P. Risterucci, O. Renault, C. Zborowski, D. Bertrand, A. Torres, J.-P. Rueff, D. Ceolin, G. Grenet and S. Tougaard, Appl. Surf. Sci., 2017, 402, 78–85 CrossRef CAS.
- C. Zborowski, O. Renault, A. Torres, C. Guedj, Y. Yamashita, S. Ueda, G. Grenet and S. Tougaard, J. Appl. Phys., 2018, 124, 085115 CrossRef.
- C. Zborowski and S. Tougaard, Surf. Interface Anal., 2019, 51, 857–873 CrossRef CAS.
- S. Tougaard, J. Surf. Anal., 2017, 24, 107–114 CrossRef CAS.
- S. Tougaard, Surf. Interface Anal., 1998, 26, 249–269 CrossRef CAS.
- P.-M. Deleuze, H. Magnan, A. Barbier, M. Silly, B. Domenichini and C. Dupont, J. Phys. Chem. Lett., 2021, 12, 11520–11527 CrossRef CAS PubMed.
- J. J. Velasco-Velez, V. Pfeifer, M. Hävecker, R. S. Weatherup, R. Arrigo, C.-H. Chuang, E. Stotz, G. Weinberg, M. Salmeron, R. Schlögl and A. Knop-Gericke, Angew. Chem., Int. Ed., 2015, 54, 14554–14558 CrossRef CAS PubMed.
- A. R. Head, S. Chaudhary, G. Olivieri, F. Bournel, J. N. Andersen, F. Rochet, J.-J. Gallet and J. Schnadt, J. Phys. Chem. C, 2016, 120, 243–251 CrossRef CAS.
- M. Vorokhta, I. Khalakhan, M. Vondráček, D. Tomeček, M. Vorokhta, E. Marešová, J. Nováková, J. Vlček, P. Fitl, M. Novotný, P. Hozák, J. Lančok, M. Vrňata, I. Matolínová and V. Matolín, Surf. Sci., 2018, 677, 284–290 CrossRef CAS.
- J. C.-R. Ke, A. S. Walton, D. J. Lewis, A. Tedstone, P. O’Brien, A. G. Thomas and W. R. Flavell, Chem. Commun., 2017, 53, 5231–5234 RSC.
- D. E. Starr, Z. Liu, M. Hävecker, A. Knop-Gericke and H. Bluhm, Chem. Soc. Rev., 2013, 42, 5833–5857 RSC.
- Ambient Pressure Spectroscopy in Complex Chemical Environments, ed. A. R. Head, S. Nemšák and B. Eren, American Chemical Society, 2021, vol. 1396, ACS Symposium Series Search PubMed.
- R. Dupuy, C. Richter, B. Winter, G. Meijer, R. Schlögl and H. Bluhm, J. Chem. Phys., 2021, 154, 060901 CrossRef CAS PubMed.
- J. Schnadt, J. Knudsen and N. Johansson, J. Phys.: Condens. Matter, 2020, 32, 413003 CrossRef CAS PubMed.
- H. Siegbahn and K. Siegbahn, J. Electron Spectrosc. Relat. Phenom., 1973, 2, 319–325 CrossRef CAS.
- H. Siegbahn, J. Phys. Chem., 1985, 89, 897–909 CrossRef CAS.
- D. F. Ogletree, H. Bluhm, G. Lebedev, C. S. Fadley, Z. Hussain and M. Salmeron, Rev. Sci. Instrum., 2002, 73, 3872–3877 CrossRef CAS.
- J. Knudsen, J. N. Andersen and J. Schnadt, Surf. Sci., 2016, 646, 160–169 CrossRef CAS.
- S. Zhu, M. Scardamaglia, J. Kundsen, R. Sankari, H. Tarawneh, R. Temperton, L. Pickworth, F. Cavalca, C. Wang, H. Tissot, J. Weissenrieder, B. Hagman, J. Gustafson, S. Kaya, F. Lindgren, I. Källquist, J. Maibach, M. Hahlin, V. Boix, T. Gallo, F. Rehman, G. D’Acunto, J. Schnadt and A. Shavorskiy, J. Synchrotron Radiat., 2021, 28, 624–636 CrossRef CAS PubMed.
- E. Kokkonen, F. Lopes da Silva, M.-H. Mikkelã, N. Johansson, S.-W. Huang, J.-M. Lee, M. Andersson, A. Bartalesi, B. N. Reinecke, K. Handrup, H. Tarawneh, R. Sankari, J. Knudsen, J. Schnadt, C. Såthe and S. Urpelainen, J. Synchrotron Radiat., 2021, 28, 588–601 CrossRef CAS PubMed.
- J. Zhou, S. Blomberg, J. Gustafson, E. Lundgren and J. Zetterberg, Catalysts, 2017, 7, 29 CrossRef.
- X. Garcia, L. Soler, N. J. Divins, X. Vendrell, I. Serrano, I. Lucentini, J. Prat, E. Solano, M. Tallarida, C. Escudero and J. Llorca, Catalysts, 2020, 10, 286 CrossRef CAS.
- N. Johansson, M. Andersen, Y. Monya, J. N. Andersen, H. Kondoh, J. Schnadt and J. Knudsen, J. Phys.: Condens. Matter, 2017, 29, 444002 CrossRef CAS PubMed.
- F. Schiller, M. Ilyn, V. Pérez-Dieste, C. Escudero, C. Huck-Iriart, N. Ruiz del Arbol, B. Hagman, L. R. Merte, F. Bertram, M. Shipilin, S. Blomberg, J. Gustafson, E. Lundgren and J. E. Ortega, J. Am. Chem. Soc., 2018, 140, 16245–16252 CrossRef CAS PubMed.
- N. Johansson, S. Sisodiya, P. Shayesteh, S. Chaudhary, J. N. Andersen, J. Knudsen, O. F. Wendt and J. Schnadt, J. Phys.: Condens. Matter, 2017, 29, 444005 CrossRef CAS PubMed.
- S. Blomberg, U. Hejral, M. Shipilin, S. Albertin, H. Karlsson, C. Hulteberg, P. Lömker, C. Goodwin, D. Degerman, J. Gustafson, C. Schlueter, A. Nilsson, E. Lundgren and P. Amann, ACS Catal., 2021, 11, 9128–9135 CrossRef CAS PubMed.
- S. C. DeCaluwe, G. S. Jackson, R. Farrow, A. McDaniel, F. E. Gabaly, K. McCarty, S. Nie, M. Linne, H. Bluhm, J. Newberg, Z. Liu and Z. Hussain, ECS Trans., 2009, 16, 253 CrossRef CAS.
- C. Zhang, M. E. Grass, A. H. McDaniel, S. C. DeCaluwe, F. E. Gabaly, Z. Liu, K. F. McCarty, R. L. Farrow, M. A. Linne, Z. Hussain, G. S. Jackson, H. Bluhm and B. W. Eichhorn, Nat. Mater., 2010, 9, 944–949 CrossRef CAS PubMed.
- F. E. Gabaly, M. Grass, A. H. McDaniel, R. L. Farrow, M. A. Linne, Z. Hussain, H. Bluhm, Z. Liu and K. F. McCarty, Phys. Chem. Chem. Phys., 2010, 12, 12138–12145 RSC.
- C. Zhang, M. E. Grass, Y. Yu, K. J. Gaskell, S. C. DeCaluwe, R. Chang, G. S. Jackson, Z. Hussain, H. Bluhm, B. W. Eichhorn and Z. Liu, ACS Catal., 2012, 2, 2297–2304 CrossRef CAS.
- E. J. Crumlin, H. Bluhm and Z. Liu, J. Electron Spectrosc. Relat. Phenom., 2013, 190, 84–92 CrossRef CAS.
- Y.-C. Lu, E. J. Crumlin, G. M. Veith, J. R. Harding, E. Mutoro, L. Baggetto, N. J. Dudney, Z. Liu and Y. Shao-Horn, Sci. Rep., 2012, 2, 715 CrossRef PubMed.
- B. Mao, Y. Dai, J. Cai, Q. Li, C. Jiang, Y. Li, J. Xie and Z. Liu, Top. Catal., 2018, 61, 2123–2128 CrossRef CAS.
- S. Narayanan, J. S. Gibson, J. Aspinall, R. S. Weatherup and M. Pasta, Curr. Opin. Solid State Mater. Sci., 2022, 26, 100978 CrossRef CAS.
- C. H. Wu, R. S. Weatherup and M. B. Salmeron, Phys. Chem. Chem. Phys., 2015, 17, 30229–30239 RSC.
- A. Kolmakov, L. Gregoratti, M. Kiskinova and S. Günther, Top. Catal., 2016, 59, 448–468 CrossRef CAS PubMed.
- R. S. Weatherup, Top. Catal., 2018, 61, 2085–2102 CrossRef CAS.
- R. S. Weatherup, B. Eren, Y. Hao, H. Bluhm and M. B. Salmeron, J. Phys. Chem. Lett., 2016, 7, 1622–1627 CrossRef CAS PubMed.
- V. Streibel, M. Hävecker, Y. Yi, J. J. Velasco Vélez, K. Skorupska, E. Stotz, A. Knop-Gericke, R. Schlögl and R. Arrigo, Top. Catal., 2018, 61, 2064–2084 CrossRef CAS.
- A. Kolmakov, D. A. Dikin, L. J. Cote, J. Huang, M. K. Abyaneh, M. Amati, L. Gregoratti, S. Günther and M. Kiskinova, Nat. Nanotechnol., 2011, 6, 651–657 CrossRef CAS PubMed.
- T. Masuda, H. Yoshikawa, H. Noguchi, T. Kawasaki, M. Kobata, K. Kobayashi and K. Uosaki, Appl. Phys. Lett., 2013, 103, 111605 CrossRef.
- J. Kraus, R. Reichelt, S. Günther, L. Gregoratti, M. Amati, M. Kiskinova, A. Yulaev, I. Vlassiouk and A. Kolmakov, Nanoscale, 2014, 6, 14394–14403 RSC.
- R. Arrigo, M. Hävecker, M. E. Schuster, C. Ranjan, E. Stotz, A. Knop-Gericke and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52, 11660–11664 CrossRef CAS PubMed.
- O. Karslıoğlu, S. Nemšák, I. Zegkinoglou, A. Shavorskiy, M. Hartl, F. Salmassi, E. M. Gullikson, M. L. Ng, C. Rameshan, B. Rude, D. Bianculli, A. A. Cordones, S. Axnanda, E. J. Crumlin, P. N. Ross, C. M. Schneider, Z. Hussain, Z. Liu, C. S. Fadley and H. Bluhm, Faraday Discuss., 2015, 180, 35–53 RSC.
- S. Axnanda, E. J. Crumlin, B. Mao, S. Rani, R. Chang, P. G. Karlsson, M. O. M. Edwards, M. Lundqvist, R. Moberg, P. Ross, Z. Hussain and Z. Liu, Sci. Rep., 2015, 5, 9788 CrossRef CAS PubMed.
- S. Nemšák, A. Shavorskiy, O. Karslioglu, I. Zegkinoglou, A. Rattanachata, C. S. Conlon, A. Keqi, P. K. Greene, E. C. Burks, F. Salmassi, E. M. Gullikson, S.-H. Yang, K. Liu, H. Bluhm and C. S. Fadley, Nat. Commun., 2014, 5, 5441 CrossRef PubMed.
- Y. Takagi, H. Wang, Y. Uemura, T. Nakamura, L. Yu, O. Sekizawa, T. Uruga, M. Tada, G. Samjeské, Y. Iwasawa and T. Yokoyama, Phys. Chem. Chem. Phys., 2017, 19, 6013–6021 RSC.
- O. Karslıoğlu, L. Trotochaud, F. Salmassi, E. M. Gullikson, A. Shavorskiy, S. Nemšák and H. Bluhm, J. Vac. Sci. Technol., A, 2022, 40, 013207 CrossRef.
- M. Favaro, P. C. J. Clark, M. J. Sear, M. Johansson, S. Maehl, R. van de Krol and D. E. Starr, Surf. Sci., 2021, 713, 121903 CrossRef CAS.
- D. M. Itkis, J. J. Velasco-Velez, A. Knop-Gericke, A. Vyalikh, M. V. Avdeev and L. V. Yashina, ChemElectroChem, 2015, 2, 1427–1445 CrossRef CAS.
- E. S. Jones, J. E. N. Swallow and R. S. Weatherup, in Ambient Pressure Spectroscopy in Complex Chemical Environments, American Chemical Society, 2021, vol. 1396, pp. 175–218 Search PubMed.
- M. Amati, V. Bonanni, L. Braglia, F. Genuzio, L. Gregoratti, M. Kiskinova, A. Kolmakov, A. Locatelli, E. Magnano, A. A. Matruglio, T. O. Menteş, S. Nappini, P. Torelli and P. Zeller, J. Electron Spectrosc. Relat. Phenom., 2019, 146902 CrossRef.
- P. Amann, D. Degerman, M.-T. Lee, J. D. Alexander, M. Shipilin, H.-Y. Wang, F. Cavalca, M. Weston, J. Gladh, M. Blom, M. Björkhage, P. Löfgren, C. Schlueter, P. Loemker, K. Ederer, W. Drube, H. Noei, J. Zehetner, H. Wentzel, J. Åhlund and A. Nilsson, Rev. Sci. Instrum., 2019, 90, 103102 CrossRef.
- C. Schlueter, A. Gloskovskii, K. Ederer, I. Schostak, S. Piec, I. Sarkar, Yu. Matveyev, P. Lömker, M. Sing, R. Claessen, C. Wiemann, C. M. Schneider, K. Medjanik, G. Schönhense, P. Amann, A. Nilsson and W. Drube, AIP Conf. Proc., 2019, 2054, 040010 CrossRef.
- J. J. Velasco-Vélez, V. Pfeifer, M. Hävecker, R. Wang, A. Centeno, A. Zurutuza, G. Algara-Siller, E. Stotz, K. Skorupska, D. Teschner, P. Kube, P. Braeuninger-Weimer, S. Hofmann, R. Schlögl and A. Knop-Gericke, Rev. Sci. Instrum., 2016, 87, 053121 CrossRef PubMed.
- F. Zaera, Coord. Chem. Rev., 2013, 257, 3177–3191 CrossRef CAS.
- E. A. Redekop, N. Johansson, E. Kokkonen, S. Urpelainen, F. Lopes da Silva, M. Kaipio, H.-E. Nieminen, F. Rehman, V. Miikkulainen, M. Ritala and U. Olsbye, Rev. Sci. Instrum., 2021, 92, 044101 CrossRef CAS PubMed.
- J. Knudsen, T. Gallo, V. Boix, M. D. Strømsheim, G. D’Acunto, C. Goodwin, H. Wallander, S. Zhu, M. Soldemo, P. Lömker, F. Cavalca, M. Scardamaglia, D. Degerman, A. Nilsson, P. Amann, A. Shavorskiy and J. Schnadt, Nat. Commun., 2021, 12, 6117 CrossRef CAS PubMed.
- Q. Xie, Y.-L. Jiang, C. Detavernier, D. Deduytsche, R. L. Van Meirhaeghe, G.-P. Ru, B.-Z. Li and X.-P. Qu, J. Appl. Phys., 2007, 102, 083521 CrossRef.
- V. Miikkulainen, M. Leskelä, M. Ritala and R. L. Puurunen, J. Appl. Phys., 2013, 113, 021301 CrossRef.
- A. Shavorskiy, G. D’Acunto, V. Boix de la Cruz, M. Scardamaglia, S. Zhu, R. H. Temperton, J. Schnadt and J. Knudsen, ACS Appl. Mater. Interfaces, 2021, 13, 47629–47641 CrossRef CAS PubMed.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS PubMed.
- H. Min, D. Y. Lee, J. Kim, G. Kim, K. S. Lee, J. Kim, M. J. Paik, Y. K. Kim, K. S. Kim, M. G. Kim, T. J. Shin and S. Il Seok, Nature, 2021, 598, 444–450 CrossRef CAS PubMed.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D’Haen, L. D’Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. D. Angelis and H.-G. Boyen, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- R. K. Misra, S. Aharon, B. Li, D. Mogilyansky, I. Visoly-Fisher, L. Etgar and E. A. Katz, J. Phys. Chem. Lett., 2015, 6, 326–330 CrossRef CAS PubMed.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed.
- D. I. Patel, T. Roychowdhury, V. Jain, D. Shah, T. G. Avval, S. Chatterjee, S. Bahr, P. Dietrich, M. Meyer, A. Thißen and M. R. Linford, Surf. Sci. Spectra, 2019, 26, 016801 CrossRef.
- M. Salmeron and R. Schlögl, Surf. Sci. Rep., 2008, 63, 169–199 CrossRef CAS.
- P. M. Dietrich, S. Bahr, T. Yamamoto, M. Meyer and A. Thissen, J. Electron Spectrosc. Relat. Phenom., 2019, 231, 118–126 CrossRef CAS.
- H. Bluhm, J. Electron Spectrosc. Relat. Phenom., 2010, 177, 71–84 CrossRef CAS.
- M. Kjærvik, A. Hermanns, P. Dietrich, A. Thissen, S. Bahr, B. Ritter, E. Kemnitz and W. E. S. Unger, J. Phys.: Condens. Matter, 2017, 29, 474002 CrossRef PubMed.
- M. Kjærvik, K. Schwibbert, P. Dietrich, A. Thissen and W. E. S. Unger, Surf. Interface Anal., 2018, 50, 996–1000 CrossRef.
- P. G. Rouxhet and M. J. Genet, Surf. Interface Anal., 2011, 43, 1453–1470 CrossRef CAS.
- M. Kjærvik, M. Ramstedt, K. Schwibbert, P. M. Dietrich and W. E. S. Unger, Front. Chem., 2021, 9, 666161 CrossRef PubMed.
- M. B. Hahn, P. M. Dietrich and J. Radnik, Commun. Chem., 2021, 4, 1–8 CrossRef.
- Y. Takagi, T. Nakamura, L. Yu, S. Chaveanghong, O. Sekizawa, T. Sakata, T. Uruga, M. Tada, Y. Iwasawa and T. Yokoyama, Appl. Phys. Express, 2017, 10, 076603 CrossRef.
- R. K. Lam, J. W. Smith, A. M. Rizzuto, O. Karslıoğlu, H. Bluhm and R. J. Saykally, J. Chem. Phys., 2017, 146, 094703 CrossRef.
- C. L. Sabine, R. A. Feely, N. Gruber, R. M. Key, K. Lee, J. L. Bullister, R. Wanninkhof, C. S. Wong, D. W. R. Wallace, B. Tilbrook, F. J. Millero, T.-H. Peng, A. Kozyr, T. Ono and A. F. Rios, Science, 2004, 305, 367–371 CrossRef CAS PubMed.
- C. Richter, R. Dupuy and H. Bluhm, in Ambient Pressure Spectroscopy in Complex Chemical Environments, American Chemical Society, 2021, vol. 1396, pp. 39–66 Search PubMed.
- L. Nguyen, P. P. Tao, H. Liu, M. Al-Hada, M. Amati, H. Sezen, L. Gregoratti, Y. Tang, S. D. House and F. F. Tao, Langmuir, 2018, 34, 9606–9616 CrossRef CAS PubMed.
- H. Tissot, G. Olivieri, J.-J. Gallet, F. Bournel, M. G. Silly, F. Sirotti and F. Rochet, J. Phys. Chem. C, 2015, 119, 9253–9259 CrossRef CAS.
- H. Tissot, J.-J. Gallet, F. Bournel, G. Olivieri, M. G. Silly, F. Sirotti, A. Boucly and F. Rochet, Top. Catal., 2016, 59, 605–620 CrossRef CAS.
- S. G. Booth, A. M. Tripathi, I. Strashnov, R. A. W. Dryfe and A. S. Walton, J. Phys.: Condens. Matter, 2017, 29, 454001 CrossRef CAS PubMed.
- M. Ekimova, W. Quevedo, M. Faubel, P. Wernet and E. T. J. Nibbering, Struct. Dyn., 2015, 2, 054301 CrossRef PubMed.
- M. Kubin, M. Guo, M. Ekimova, M. L. Baker, T. Kroll, E. Källman, J. Kern, V. K. Yachandra, J. Yano, E. T. J. Nibbering, M. Lundberg and P. Wernet, Inorg. Chem., 2018, 57, 5449–5462 CrossRef CAS PubMed.
- R. Dupuy, J. Filser, C. Richter, R. Seidel, F. Trinter, T. Buttersack, C. Nicolas, J. Bozek, U. Hergenhahn, H. Oberhofer, B. Winter, K. Reuter and H. Bluhm, Phys. Chem. Chem. Phys., 2022, 24, 4796–4808 RSC.
- S. Malerz, F. Trinter, U. Hergenhahn, A. Ghrist, H. Ali, C. Nicolas, C.-M. Saak, C. Richter, S. Hartweg, L. Nahon, C. Lee, C. Goy, D. M. Neumark, G. Meijer, I. Wilkinson, B. Winter and S. Thürmer, Phys. Chem. Chem. Phys., 2021, 23, 8246–8260 RSC.
- T. Buttersack, P. E. Mason, R. S. McMullen, H. C. Schewe, T. Martinek, K. Brezina, M. Crhan, A. Gomez, D. Hein, G. Wartner, R. Seidel, H. Ali, S. Thürmer, O. Marsalek, B. Winter, S. E. Bradforth and P. Jungwirth, Science, 2020, 368, 1086–1091 CrossRef CAS PubMed.
- A. Shavorskiy, S. Neppl, D. S. Slaughter, J. P. Cryan, K. R. Siefermann, F. Weise, M.-F. Lin, C. Bacellar, M. P. Ziemkiewicz, I. Zegkinoglou, M. W. Fraund, C. Khurmi, M. P. Hertlein, T. W. Wright, N. Huse, R. W. Schoenlein, T. Tyliszczak, G. Coslovich, J. Robinson, R. A. Kaindl, B. S. Rude, A. Ölsner, S. Mähl, H. Bluhm and O. Gessner, Rev. Sci. Instrum., 2014, 85, 093102 CrossRef PubMed.
- A. Shavorskiy, J. Schnadt and J. Knudsen, in Ambient Pressure Spectroscopy in Complex Chemical Environments, American Chemical Society, 2021, vol. 1396, pp. 219–248 Search PubMed.
- H. Kersell, L. Falling, A. Shavorskiy and S. Nemšák, in Ambient Pressure Spectroscopy in Complex Chemical Environments, American Chemical Society, 2021, vol. 1396, pp. 333–358 Search PubMed.
- H. Kersell, P. Chen, H. Martins, Q. Lu, F. Brausse, B.-H. Liu, M. Blum, S. Roy, B. Rude, A. Kilcoyne, H. Bluhm and S. Nemšák, Rev. Sci. Instrum., 2021, 92, 044102 CrossRef CAS PubMed.
- J. Cole, Z. Henderson, A. G. Thomas, C. L. Compeán-González, A. J. Greer, C. Hardacre, F. Venturini, W. Q. Garzon, P. Ferrer, D. C. Grinter, G. Held and K. L. Syres, J. Phys. Chem. C, 2021, 125, 22778–22785 CrossRef CAS.
- M. Scardamaglia, C. Struzzi, A. Zakharov, N. Reckinger, P. Zeller, M. Amati and L. Gregoratti, ACS Appl. Mater. Interfaces, 2019, 11, 29448–29457 CrossRef CAS PubMed.
- M. Amati, L. Gregoratti, P. Zeller, M. Greiner, M. Scardamaglia, B. Junker, T. Ruß, U. Weimar, N. Barsan, M. Favaro, A. Alharbi, I. J. T. Jensen, A. Ali and B. D. Belle, J. Phys. D: Appl. Phys., 2021, 54, 204004 CrossRef CAS.
- C. Kalha, N. K. Fernando, P. Bhatt, F. O. L. Johansson, A. Lindblad, H. Rensmo, L. Z. Medina, R. Lindblad, S. Siol, L. P. H. Jeurgens, C. Cancellieri, K. Rossnagel, K. Medjanik, G. Schönhense, M. Simon, A. X. Gray, S. Nemšák, P. Lömker, C. Schlueter and A. Regoutz, J. Phys.: Condens. Matter, 2021, 33, 233001 CrossRef CAS PubMed.
- M. P. Seah and W. A. Dench, Surf. Interface Anal., 1979, 1, 2–11 CrossRef CAS.
- L. Sabbatucci and F. Salvat, Radiat. Phys. Chem., 2016, 121, 122–140 CrossRef CAS.
- D. J. H. Cant, B. F. Spencer, W. R. Flavell and A. G. Shard, Surf. Interface Anal., 2022, 54(4), 442–454 CrossRef CAS.
- K. Kobayashi, M. Kobata and H. Iwai, J. Electron Spectrosc. Relat. Phenom., 2013, 190, 210–221 CrossRef CAS.
- M. Kobata, I. Píš, H. Iwai, H. Yamazui, H. Takahashi, M. Suzuki, H. Matsuda, H. Daimon and K. Kobayashi, Anal. Sci., 2010, 26, 227–232 CrossRef CAS PubMed.
- O. Renault, E. Martinez, C. Zborowski, J. Mann, R. Inoue, J. Newman and K. Watanabe, Surf. Interface Anal., 2018, 50, 1158–1162 CrossRef CAS.
- A. Regoutz, M. Mascheck, T. Wiell, S. K. Eriksson, C. Liljenberg, K. Tetzner, B. A. D. Williamson, D. O. Scanlon and P. Palmgren, Rev. Sci. Instrum., 2018, 89, 73105 CrossRef PubMed.
- M. Otendal, T. Tuohimaa, U. Vogt and H. M. Hertz, Rev. Sci. Instrum., 2008, 79, 016102 CrossRef CAS PubMed.
- M. Wansleben, C. Zech, C. Streeck, J. Weser, C. Genzel, B. Beckhoff and R. Mainz, J. Anal. At. Spectrom., 2019, 34, 1497–1502 RSC.
- H.-Y. Wang, M. Soldemo, D. Degerman, P. Lömker, C. Schlueter, A. Nilsson and P. Amann, Angew. Chem., Int. Ed., 2022, 61, e202111021 CAS.
- T. Okane, M. Kobata, I. Sato, K. Kobayashi, M. Osaka and H. Yamagami, Nucl. Eng. Des., 2016, 297, 251–256 CrossRef CAS.
- H.-Q. Wang, J. Xu, X. Lin, Y. Li, J. Kang and J.-C. Zheng, Light: Sci. Appl., 2021, 10, 153 CrossRef CAS PubMed.
- G. Panaccione and K. Kobayashi, Surf. Sci., 2012, 606, 125–129 CrossRef CAS.
- T. Pincelli, V. Lollobrigida, F. Borgatti, A. Regoutz, B. Gobaut, C. Schlueter, T.-L. Lee, D. J. Payne, M. Oura, K. Tamasaku, A. Y. Petrov, P. Graziosi, F. M. Granozio, M. Cavallini, G. Vinai, R. Ciprian, C. H. Back, G. Rossi, M. Taguchi, H. Daimon, G. van der Laan and G. Panaccione, Nat. Commun., 2017, 8, 16051 CrossRef CAS PubMed.
- G. Assat, D. Foix, C. Delacourt, A. Iadecola, R. Dedryvère and J.-M. Tarascon, Nat. Commun., 2017, 8, 2219 CrossRef PubMed.
- R. Dubey, J. Sastre, C. Cancellieri, F. Okur, A. Forster, L. Pompizii, A. Priebe, Y. E. Romanyuk, L. P. H. Jeurgens, M. V. Kovalenko and K. V. Kravchyk, Adv. Energy Mater., 2021, 11, 2102086 CrossRef CAS.
- H. Kiuchi, K. Hikima, K. Shimizu, R. Kanno, F. Toshiharu and E. Matsubara, Electrochem. Commun., 2020, 118, 106790 CrossRef CAS.
- P. Risterucci, O. Renault, E. Martinez, B. Detlefs, V. Delaye, J. Zegenhagen, C. Gaumer, G. Grenet and S. Tougaard, Appl. Phys. Lett., 2014, 104, 051608 CrossRef.
- C. Zborowski and S. Tougaard, Surf. Interface Anal., 2022, 54, 433–441 CrossRef CAS.
- S. Tougaard, J. Vac. Sci. Technol., A, 2021, 39, 011201 CrossRef CAS.
- B. F. Spencer, S. Maniyarasu, B. P. Reed, D. J. H. Cant, R. Ahumada-Lazo, A. G. Thomas, C. A. Muryn, M. Maschek, S. K. Eriksson, T. Wiell, T.-L. Lee, S. Tougaard, A. G. Shard and W. R. Flavell, Appl. Surf. Sci., 2021, 541, 148635 CrossRef CAS.
- Quases-Tougaard - QUASES, https://www.quases.com/products/quases-tougaard/, accessed March 11, 2022.
- J. H. Scofield, Theoretical Photoionization Cross Sections from 1 to 1500 keV, U.S. Atomic Energy Commission, 1973 Search PubMed.
- M. B. Trzhaskovskaya and V. G. Yarzhemsky, At. Data Nucl. Data Tables, 2018, 119, 99–174 CrossRef CAS.
- M. B. Trzhaskovskaya and V. G. Yarzhemsky, At. Data Nucl. Data Tables, 2019, 129–130, 101280 CrossRef CAS.
- A. G. Shard, J. D. P. Counsell, D. J. H. Cant, E. F. Smith, P. Navabpour, X. Zhang and C. J. Blomfield, Surf. Interface Anal., 2019, 51, 763–773 CrossRef CAS.
- D. J. H. Cant, B. P. Reed, B. F. Spencer, W. R. Flavell and A. G. Shard, unpublished work.
- C. D. Wagner, J. Electron Spectrosc. Relat. Phenom., 1977, 10, 305–315 CrossRef CAS.
- S. Siol, J. Mann, J. Newman, T. Miyayama, K. Watanabe, P. Schmutz, C. Cancellieri and L. P. H. Jeurgens, Surf. Interface Anal., 2020, 52, 802–810 CrossRef CAS.
- T. Pincelli, R. Cucini, A. Verna, F. Borgatti, M. Oura, K. Tamasaku, H. Osawa, T.-L. Lee, C. Schlueter, S. Günther, C. H. Back, M. Dell’Angela, R. Ciprian, P. Orgiani, A. Petrov, F. Sirotti, V. A. Dediu, I. Bergenti, P. Graziosi, F. Miletto Granozio, Y. Tanaka, M. Taguchi, H. Daimon, J. Fujii, G. Rossi and G. Panaccione, Phys. Rev. B, 2019, 100, 045118 CrossRef CAS.
- L.-P. Oloff, M. Oura, K. Rossnagel, A. Chainani, M. Matsunami, R. Eguchi, T. Kiss, Y. Nakatani, T. Yamaguchi, J. Miyawaki, M. Taguchi, K. Yamagami, T. Togashi, T. Katayama, K. Ogawa, M. Yabashi and T. Ishikawa, New J. Phys., 2014, 16, 123045 CrossRef.
- R. Wallauer, M. Raths, K. Stallberg, L. Münster, D. Brandstetter, X. Yang, J. Güdde, P. Puschnig, S. Soubatch, C. Kumpf, F. C. Bocquet, F. S. Tautz and U. Höfer, Science, 2021, 371, 1056–1059 CrossRef CAS PubMed.
- D. Curcio, S. Pakdel, K. Volckaert, J. A. Miwa, S. Ulstrup, N. Lanatà, M. Bianchi, D. Kutnyakhov, F. Pressacco, G. Brenner, S. Dziarzhytski, H. Redlin, S. Y. Agustsson, K. Medjanik, D. Vasilyev, H.-J. Elmers, G. Schönhense, C. Tusche, Y.-J. Chen, F. Speck, T. Seyller, K. Bühlmann, R. Gort, F. Diekmann, K. Rossnagel, Y. Acremann, J. Demsar, W. Wurth, D. Lizzit, L. Bignardi, P. Lacovig, S. Lizzit, C. E. Sanders and P. Hofmann, Phys. Rev. B, 2021, 104, L161104 CrossRef CAS.
- D. Kutnyakhov, R. P. Xian, M. Dendzik, M. Heber, F. Pressacco, S. Y. Agustsson, L. Wenthaus, H. Meyer, S. Gieschen, G. Mercurio, A. Benz, K. Bühlman, S. Däster, R. Gort, D. Curcio, K. Volckaert, M. Bianchi, Ch. Sanders, J. A. Miwa, S. Ulstrup, A. Oelsner, C. Tusche, Y.-J. Chen, D. Vasilyev, K. Medjanik, G. Brenner, S. Dziarzhytski, H. Redlin, B. Manschwetus, S. Dong, J. Hauer, L. Rettig, F. Diekmann, K. Rossnagel, J. Demsar, H.-J. Elmers, Ph. Hofmann, R. Ernstorfer, G. Schönhense, Y. Acremann and W. Wurth, Rev. Sci. Instrum., 2020, 91, 013109 CrossRef CAS PubMed.
- O. Fedchenko, A. Winkelmann, K. Medjanik, S. Babenkov, D. Vasilyev, S. Chernov, C. Schlueter, A. Gloskovskii, Y. Matveyev, W. Drube, B. Schönhense, H. J. Elmers and G. Schönhense, New J. Phys., 2019, 21, 113031 CrossRef CAS.
- K. Medjanik, S. V. Babenkov, S. Chernov, D. Vasilyev, B. Schönhense, C. Schlueter, A. Gloskovskii, Y. Matveyev, W. Drube, H. J. Elmers and G. Schoenhense, J. Synchrotron Radiat., 2019, 26, 1996–2012 CrossRef CAS PubMed.
- S. Nemšák, G. Conti, A. X. Gray, G. K. Palsson, C. Conlon, D. Eiteneer, A. Keqi, A. Rattanachata, A. Y. Saw, A. Bostwick, L. Moreschini, E. Rotenberg, V. N. Strocov, M. Kobayashi, T. Schmitt, W. Stolte, S. Ueda, K. Kobayashi, A. Gloskovskii, W. Drube, C. A. Jackson, P. Moetakef, A. Janotti, L. Bjaalie, B. Himmetoglu, C. G. Van de Walle, S. Borek, J. Minar, J. Braun, H. Ebert, L. Plucinski, J. B. Kortright, C. M. Schneider, L. Balents, F. M. F. de Groot, S. Stemmer and C. S. Fadley, Phys. Rev. B, 2016, 93, 245103 CrossRef.
- S.-H. Yang, A. X. Gray, A. M. Kaiser, B. S. Mun, B. C. Sell, J. B. Kortright and C. S. Fadley, J. Appl. Phys., 2013, 113, 073513 CrossRef.
- C. S. Conlon, G. Conti, S. Nemšák, G. Palsson, R. Moubah, C.-T. Kuo, M. Gehlmann, J. Ciston, J. Rault, J.-P. Rueff, F. Salmassi, W. Stolte, A. Rattanachata, S.-C. Lin, A. Keqi, A. Saw, B. Hjörvarsson and C. S. Fadley, J. Appl. Phys., 2019, 126, 075305 CrossRef.
- G. Mercurio, I. A. Makhotkin, I. Milov, Y. Y. Kim, I. A. Zaluzhnyy, S. Dziarzhytski, L. Wenthaus, I. A. Vartanyants and W. Wurth, New J. Phys., 2019, 21, 033031 CrossRef CAS.
- M. Marinova, J. E. Rault, A. Gloter, S. Nemsak, G. K. Palsson, J.-P. Rueff, C. S. Fadley, C. Carrétéro, H. Yamada, K. March, V. Garcia, S. Fusil, A. Barthélémy, O. Stéphan, C. Colliex and M. Bibes, Nano Lett., 2015, 15, 2533–2541 CrossRef CAS PubMed.
- S.-C. Lin, C.-T. Kuo, R. B. Comes, J. E. Rault, J.-P. Rueff, S. Nemšák, A. Taleb, J. B. Kortright, J. Meyer-Ilse, E. Gullikson, P. V. Sushko, S. R. Spurgeon, M. Gehlmann, M. E. Bowden, L. Plucinski, S. A. Chambers and C. S. Fadley, Phys. Rev. B, 2018, 98, 165124 CrossRef CAS.
- G. Conti, A. M. Kaiser, A. X. Gray, S. Nemšák, G. K. Pálsson, J. Son, P. Moetakef, A. Janotti, L. Bjaalie, C. S. Conlon, D. Eiteneer, A. A. Greer, A. Keqi, A. Rattanachata, A. Y. Saw, A. Bostwick, W. C. Stolte, A. Gloskovskii, W. Drube, S. Ueda, M. Kobata, K. Kobayashi, C. G. Van de Walle, S. Stemmer, C. M. Schneider and C. S. Fadley, J. Appl. Phys., 2013, 113, 143704 CrossRef.
- O. Karslıoğlu, M. Gehlmann, J. Müller, S. Nemšák, J. A. Sethian, A. Kaduwela, H. Bluhm and C. Fadley, J. Electron Spectrosc. Relat. Phenom., 2019, 230, 10–20 CrossRef.
- K. R. Siefermann, C. D. Pemmaraju, S. Neppl, A. Shavorskiy, A. A. Cordones, J. Vura-Weis, D. S. Slaughter, F. P. Sturm, F. Weise, H. Bluhm, M. L. Strader, H. Cho, M.-F. Lin, C. Bacellar, C. Khurmi, J. Guo, G. Coslovich, J. S. Robinson, R. A. Kaindl, R. W. Schoenlein, A. Belkacem, D. M. Neumark, S. R. Leone, D. Nordlund, H. Ogasawara, O. Krupin, J. J. Turner, W. F. Schlotter, M. R. Holmes, M. Messerschmidt, M. P. Minitti, S. Gul, J. Z. Zhang, N. Huse, D. Prendergast and O. Gessner, J. Phys. Chem. Lett., 2014, 5, 2753–2759 CrossRef CAS PubMed.
- L.-P. Oloff, A. Chainani, M. Matsunami, K. Takahashi, T. Togashi, H. Osawa, K. Hanff, A. Quer, R. Matsushita, R. Shiraishi, M. Nagashima, A. Kimura, K. Matsuishi, M. Yabashi, Y. Tanaka, G. Rossi, T. Ishikawa, K. Rossnagel and M. Oura, Sci. Rep., 2016, 6, 35087 CrossRef CAS PubMed.
- M. Fushitani, S. T. Pratt, D. You, S. Saito, Y. Luo, K. Ueda, H. Fujise, A. Hishikawa, H. Ibrahim, F. Légaré, P. Johnsson, J. Peschel, E. R. Simpson, A. Olofsson, J. Mauritsson, P. A. Carpeggiani, P. K. Maroju, M. Moioli, D. Ertel, R. Shah, G. Sansone, T. Csizmadia, M. Dumergue, N. G. Harshitha, S. Kühn, C. Callegari, O. Plekan, M. Di Fraia, M. B. Danailov, A. Demidovich, L. Giannessi, L. Raimondi, M. Zangrando, G. De Ninno, P. R. Ribič and K. C. Prince, J. Chem. Phys., 2021, 154, 144305 CrossRef CAS PubMed.
- M. Borgwardt, J. Mahl, F. Roth, L. Wenthaus, F. Brauße, M. Blum, K. Schwarzburg, G. Liu, F. M. Toma and O. Gessner, J. Phys. Chem. Lett., 2020, 11, 5476–5481 CrossRef CAS PubMed.
- M. Nisoli, P. Decleva, F. Calegari, A. Palacios and F. Martín, Chem. Rev., 2017, 117, 10760–10825 CrossRef CAS PubMed.
- B. Buades, A. Picón, E. Berger, I. León, N. Di Palo, S. L. Cousin, C. Cocchi, E. Pellegrin, J. H. Martin, S. Mañas-Valero, E. Coronado, T. Danz, C. Draxl, M. Uemoto, K. Yabana, M. Schultze, S. Wall, M. Zürch and J. Biegert, Appl. Phys. Rev., 2021, 8, 011408 CAS.
- J. Li, J. Lu, A. Chew, S. Han, J. Li, Y. Wu, H. Wang, S. Ghimire and Z. Chang, Nat. Commun., 2020, 11, 2748 CrossRef CAS PubMed.
- F. Krausz and M. I. Stockman, Nat. Photonics, 2014, 8, 205–213 CrossRef CAS.
- P. A. Brühwiler, O. Karis and N. Mårtensson, Rev. Mod. Phys., 2002, 74, 703–740 CrossRef.
- M. N. Piancastelli, G. Goldsztejn, T. Marchenko, R. Guillemin, R. K. Kushawaha, L. Journel, S. Carniato, J.-P. Rueff, D. Céolin and M. Simon, J. Phys. B: At., Mol. Opt. Phys., 2014, 47, 124031 CrossRef.
- A. G. Thomas, W. R. Flavell, C. Chatwin, S. Rayner, D. Tsoutsou, A. R. Kumarasinghe, D. Brete, T. K. Johal, S. Patel and J. Purton, Surf. Sci., 2005, 592, 159–168 CrossRef CAS.
- J. Schnadt, P. A. Brühwiler, L. Patthey, J. N. O’Shea, S. Södergren, M. Odelius, R. Ahuja, O. Karis, M. Bässler, P. Persson, H. Siegbahn, S. Lunell and N. Mårtensson, Nature, 2002, 418, 620–623 CrossRef CAS PubMed.
- T. A. Heimer, E. J. Heilweil, C. A. Bignozzi and G. J. Meyer, J. Phys. Chem. A, 2000, 104, 4256–4262 CrossRef CAS.
- L. C. Mayor, J. Ben Taylor, G. Magnano, A. Rienzo, C. J. Satterley, J. N. O’Shea and J. Schnadt, J. Chem. Phys., 2008, 129, 114701 CrossRef PubMed.
- A. Föhlisch, P. Feulner, F. Hennies, A. Fink, D. Menzel, D. Sanchez-Portal, P. M. Echenique and W. Wurth, Nature, 2005, 436, 373–376 CrossRef PubMed.
- F. O. L. Johansson, M. Ivanović, S. Svanström, U. B. Cappel, H. Peisert, T. Chassé and A. Lindblad, J. Phys. Chem. C, 2018, 122, 12605–12614 CrossRef CAS.
- F. O. L. Johansson, U. B. Cappel, M. Fondell, Y. Han, M. Gorgoi, K. Leifer and A. Lindblad, Phys. Chem. Chem. Phys., 2020, 22, 10335–10342 RSC.
- F. O. L. Johansson, X. Chen, O. Eriksson, B. Sanyal and A. Lindblad, Phys. Rev. B, 2020, 102, 035165 CrossRef CAS.
- S. Hellmann, C. Sohrt, M. Beye, T. Rohwer, F. Sorgenfrei, M. Marczynski-Bühlow, M. Kalläne, H. Redlin, F. Hennies, M. Bauer, A. Föhlisch, L. Kipp, W. Wurth and K. Rossnagel, New J. Phys., 2012, 14, 013062 CrossRef.
- A. Pietzsch, A. Föhlisch, M. Beye, M. Deppe, F. Hennies, M. Nagasono, E. Suljoti, W. Wurth, C. Gahl, K. Döbrich and A. Melnikov, New J. Phys., 2008, 10, 033004 CrossRef.
- H. Öström, H. Öberg, H. Xin, J. LaRue, M. Beye, M. Dell’Angela, J. Gladh, M. L. Ng, J. A. Sellberg, S. Kaya, G. Mercurio, D. Nordlund, M. Hantschmann, F. Hieke, D. Kühn, W. F. Schlotter, G. L. Dakovski, J. J. Turner, M. P. Minitti, A. Mitra, S. P. Moeller, A. Föhlisch, M. Wolf, W. Wurth, M. Persson, J. K. Nørskov, F. Abild-Pedersen, H. Ogasawara, L. G. M. Pettersson and A. Nilsson, Science, 2015, 347, 978–982 CrossRef PubMed.
- M. Beye, H. Öberg, H. Xin, G. L. Dakovski, M. Dell’Angela, A. Föhlisch, J. Gladh, M. Hantschmann, F. Hieke, S. Kaya, D. Kühn, J. LaRue, G. Mercurio, M. P. Minitti, A. Mitra, S. P. Moeller, M. L. Ng, A. Nilsson, D. Nordlund, J. Nørskov, H. Öström, H. Ogasawara, M. Persson, W. F. Schlotter, J. A. Sellberg, M. Wolf, F. Abild-Pedersen, L. G. M. Pettersson and W. Wurth, J. Phys. Chem. Lett., 2016, 7, 3647–3651 CrossRef CAS PubMed.
- E. Diesen, H.-Y. Wang, S. Schreck, M. Weston, H. Ogasawara, J. LaRue, F. Perakis, M. Dell’Angela, F. Capotondi, L. Giannessi, E. Pedersoli, D. Naumenko, I. Nikolov, L. Raimondi, C. Spezzani, M. Beye, F. Cavalca, B. Liu, J. Gladh, S. Koroidov, P. S. Miedema, R. Costantini, T. F. Heinz, F. Abild-Pedersen, J. Voss, A. C. Luntz and A. Nilsson, Phys. Rev. Lett., 2021, 127, 016802 CrossRef CAS PubMed.
- H.-Y. Wang, S. Schreck, M. Weston, C. Liu, H. Ogasawara, J. LaRue, F. Perakis, M. Dell’Angela, F. Capotondi, L. Giannessi, E. Pedersoli, D. Naumenko, I. Nikolov, L. Raimondi, C. Spezzani, M. Beye, F. Cavalca, B. Liu, J. Gladh, S. Koroidov, P. S. Miedema, R. Costantini, L. G. M. Pettersson and A. Nilsson, Phys. Chem. Chem. Phys., 2020, 22, 2677–2684 RSC.
- J. Duris, S. Li, T. Driver, E. G. Champenois, J. P. MacArthur, A. A. Lutman, Z. Zhang, P. Rosenberger, J. W. Aldrich, R. Coffee, G. Coslovich, F.-J. Decker, J. M. Glownia, G. Hartmann, W. Helml, A. Kamalov, J. Knurr, J. Krzywinski, M.-F. Lin, J. P. Marangos, M. Nantel, A. Natan, J. T. O’Neal, N. Shivaram, P. Walter, A. L. Wang, J. J. Welch, T. J. A. Wolf, J. Z. Xu, M. F. Kling, P. H. Bucksbaum, A. Zholents, Z. Huang, J. P. Cryan and A. Marinelli, Nat. Photonics, 2020, 14, 30–36 CrossRef CAS.
- C. L. Smallwood, J. P. Hinton, C. Jozwiak, W. Zhang, J. D. Koralek, H. Eisaki, D.-H. Lee, J. Orenstein and A. Lanzara, Science, 2012, 336, 1137–1139 CrossRef CAS PubMed.
- F. Schmitt, P. S. Kirchmann, U. Bovensiepen, R. G. Moore, L. Rettig, M. Krenz, J.-H. Chu, N. Ru, L. Perfetti, D. H. Lu, M. Wolf, I. R. Fisher and Z.-X. Shen, Science, 2008, 321, 1649–1652 CrossRef CAS PubMed.
- A. Gauthier, J. A. Sobota, N. Gauthier, K.-J. Xu, H. Pfau, C. R. Rotundu, Z.-X. Shen and P. S. Kirchmann, J. Appl. Phys., 2020, 128, 093101 CrossRef CAS.
- J. Graf, C. Jozwiak, C. L. Smallwood, H. Eisaki, R. A. Kaindl, D.-H. Lee and A. Lanzara, Nat. Phys., 2011, 7, 805–809 Search PubMed.
- L. Perfetti, P. A. Loukakos, M. Lisowski, U. Bovensiepen, H. Berger, S. Biermann, P. S. Cornaglia, A. Georges and M. Wolf, Phys. Rev. Lett., 2006, 97, 067402 CrossRef CAS PubMed.
- G. Rohde, A. Stange, A. Müller, M. Behrendt, L.-P. Oloff, K. Hanff, T. J. Albert, P. Hein, K. Rossnagel and M. Bauer, Phys. Rev. Lett., 2018, 121, 256401 CrossRef CAS PubMed.
- Y. H. Wang, D. Hsieh, D. Pilon, L. Fu, D. R. Gardner, Y. S. Lee and N. Gedik, Phys. Rev. Lett., 2011, 107, 207602 CrossRef CAS PubMed.
- E. J. Sie, T. Rohwer, C. Lee and N. Gedik, Nat. Commun., 2019, 10, 3535 CrossRef PubMed.
- S. Hellmann, M. Beye, C. Sohrt, T. Rohwer, F. Sorgenfrei, H. Redlin, M. Kalläne, M. Marczynski-Bühlow, F. Hennies, M. Bauer, A. Föhlisch, L. Kipp, W. Wurth and K. Rossnagel, Phys. Rev. Lett., 2010, 105, 187401 CrossRef CAS PubMed.
- S. Hellmann, K. Rossnagel, M. Marczynski-Bühlow and L. Kipp, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 035402 CrossRef.
- J. Graf, S. Hellmann, C. Jozwiak, C. L. Smallwood, Z. Hussain, R. A. Kaindl, L. Kipp, K. Rossnagel and A. Lanzara, J. Appl. Phys., 2010, 107, 014912 CrossRef.
- G. Schönhense, D. Kutnyakhov, F. Pressacco, M. Heber, N. Wind, S. Y. Agustsson, S. Babenkov, D. Vasilyev, O. Fedchenko, S. Chernov, L. Rettig, B. Schönhense, L. Wenthaus, G. Brenner, S. Dziarzhytski, S. Palutke, S. K. Mahatha, N. Schirmel, H. Redlin, B. Manschwetus, I. Hartl, Yu. Matveyev, A. Gloskovskii, C. Schlueter, V. Shokeen, H. Duerr, T. K. Allison, M. Beye, K. Rossnagel, H. J. Elmers and K. Medjanik, Rev. Sci. Instrum., 2021, 92, 053703 CrossRef PubMed.
- L.-P. Oloff, K. Hanff, A. Stange, G. Rohde, F. Diekmann, M. Bauer and K. Rossnagel, J. Appl. Phys., 2016, 119, 225106 CrossRef.
- M. Dell’Angela, T. Anniyev, M. Beye, R. Coffee, A. Föhlisch, J. Gladh, S. Kaya, T. Katayama, O. Krupin, A. Nilsson, D. Nordlund, W. F. Schlotter, J. A. Sellberg, F. Sorgenfrei, J. J. Turner, H. Öström, H. Ogasawara, M. Wolf and W. Wurth, Struct. Dyn., 2015, 2, 025101 CrossRef PubMed.
- C. W. Nicholson, A. Lücke, W. G. Schmidt, M. Puppin, L. Rettig, R. Ernstorfer and M. Wolf, Science, 2018, 362, 821–825 CrossRef CAS PubMed.
- A. K. Mills, S. Zhdanovich, M. X. Na, F. Boschini, E. Razzoli, M. Michiardi, A. Sheyerman, M. Schneider, T. J. Hammond, V. Süss, C. Felser, A. Damascelli and D. J. Jones, Rev. Sci. Instrum., 2019, 90, 083001 CrossRef CAS PubMed.
- S. Gerber, S.-L. Yang, D. Zhu, H. Soifer, J. A. Sobota, S. Rebec, J. J. Lee, T. Jia, B. Moritz, C. Jia, A. Gauthier, Y. Li, D. Leuenberger, Y. Zhang, L. Chaix, W. Li, H. Jang, J.-S. Lee, M. Yi, G. L. Dakovski, S. Song, J. M. Glownia, S. Nelson, K. W. Kim, Y.-D. Chuang, Z. Hussain, R. G. Moore, T. P. Devereaux, W.-S. Lee, P. S. Kirchmann and Z.-X. Shen, Science, 2017, 357, 71–75 CrossRef CAS PubMed.
- N. Bergeard, M. G. Silly, D. Krizmancic, C. Chauvet, M. Guzzo, J. P. Ricaud, M. Izquierdo, L. Stebel, P. Pittana, R. Sergo, G. Cautero, G. Dufour, F. Rochet and F. Sirotti, J. Synchrotron Radiat., 2011, 18, 245–250 CrossRef CAS PubMed.
- M. Ogawa, S. Yamamoto, Y. Kousa, F. Nakamura, R. Yukawa, A. Fukushima, A. Harasawa, H. Kondoh, Y. Tanaka, A. Kakizaki and I. Matsuda, Rev. Sci. Instrum., 2012, 83, 023109 CrossRef PubMed.
- M. G. Silly, T. Ferté, M. A. Tordeux, D. Pierucci, N. Beaulieu, C. Chauvet, F. Pressacco, F. Sirotti, H. Popescu, V. Lopez-Flores, M. Tortarolo, M. Sacchi, N. Jaouen, P. Hollander, J. P. Ricaud, N. Bergeard, C. Boeglin, B. Tudu, R. Delaunay, J. Luning, G. Malinowski, M. Hehn, C. Baumier, F. Fortuna, D. Krizmancic, L. Stebel, R. Sergo and G. Cautero, J. Synchrotron Radiat., 2017, 24, 886–897 CrossRef CAS PubMed.
- L. Kronik and Y. Shapira, Surf. Sci. Rep., 1999, 37, 1–206 CrossRef CAS.
- J. P. Long, H. R. Sadeghi, J. C. Rife and M. N. Kabler, Phys. Rev. Lett., 1990, 64, 1158–1161 CrossRef CAS PubMed.
- B. F. Spencer, D. M. Graham, S. J. O. Hardman, E. A. Seddon, M. J. Cliffe, K. L. Syres, A. G. Thomas, S. K. Stubbs, F. Sirotti, M. G. Silly, P. F. Kirkham, A. R. Kumarasinghe, G. J. Hirst, A. J. Moss, S. F. Hill, D. A. Shaw, S. Chattopadhyay and W. R. Flavell, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 195301 CrossRef.
- P. C. J. Clark, N. K. Lewis, J. C.-R. Ke, R. Ahumada-Lazo, Q. Chen, D. C. J. Neo, E. A. Gaulding, G. F. Pach, I. Pis, M. G. Silly and W. R. Flavell, Nanoscale, 2021, 13, 17793–17806 RSC.
- B. F. Spencer, M. J. Cliffe, D. M. Graham, S. J. O. Hardman, E. a Seddon, K. L. Syres, A. G. Thomas, F. Sirotti, M. G. Silly, J. Akhtar, P. O’Brien, S. M. Fairclough, J. M. Smith, S. Chattopadhyay and W. R. Flavell, Faraday Discuss., 2014, 171, 275–298 RSC.
- S. Tanaka, S. D. Moré, K. Takahashi and M. Kamada, J. Phys. Soc. Jpn., 2003, 72, 659–663 CrossRef CAS.
- M. Marsi, R. Belkhou, C. Grupp, G. Panaccione, A. Taleb-Ibrahimi, L. Nahon, D. Garzella, D. Nutarelli, E. Renault, R. Roux, M. E. Couprie and M. Billardon, Phys. Rev. B: Condens. Matter, 2000, 61, R5070–R5073 CrossRef CAS.
- W. Widdra, D. Bröcker, T. Gießel, I. V. Hertel, W. Krüger, A. Liero, F. Noack, V. Petrov, D. Pop, P. M. Schmidt, R. Weber, I. Will and B. Winter, Surf. Sci., 2003, 543, 87–94 CrossRef CAS.
- F. Polack, M. Silly, C. Chauvet, B. Lagarde, N. Bergeard, M. Izquierdo, O. Chubar, D. Krizmancic, M. Ribbens, J. -P. Duval, C. Basset, S. Kubsky and F. Sirotti, AIP Conf. Proc., 2010, 1234, 185–188 CrossRef CAS.
- B. F. Spencer, M. A. Leontiadou, P. C. J. Clark, A. I. Williamson, M. G. Silly, F. Sirotti, S. M. Fairclough, S. C. E. Tsang, D. C. J. Neo, H. E. Assender, A. A. R. Watt and W. R. Flavell, Appl. Phys. Lett., 2016, 108, 091603 CrossRef.
- F. Pressacco, V. Uhlíř, M. Gatti, A. Nicolaou, A. Bendounan, J. A. Arregi, S. K. K. Patel, E. E. Fullerton, D. Krizmancic and F. Sirotti, Struct. Dyn., 2018, 5, 034501 CrossRef PubMed.
- A. Vollmer, R. Ovsyannikov, M. Gorgoi, S. Krause, M. Oehzelt, A. Lindblad, N. Mårtensson, S. Svensson, P. Karlsson, M. Lundvuist, T. Schmeiler, J. Pflaum and N. Koch, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 55–60 CrossRef CAS.
- M. Ogawa, S. Yamamoto, R. Yukawa, R. Hobara, C.-H. Lin, R.-Y. Liu, S.-J. Tang and I. Matsuda, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 235308 CrossRef.
- R. Yukawa, S. Yamamoto, K. Ozawa, M. Emori, M. Ogawa, Sh. Yamamoto, K. Fujikawa, R. Hobara, S. Kitagawa, H. Daimon, H. Sakama and I. Matsuda, Appl. Phys. Lett., 2014, 105, 151602 CrossRef.
- K. Ozawa, M. Emori, S. Yamamoto, R. Yukawa, S. Yamamoto, R. Hobara, K. Fujikawa, H. Sakama and I. Matsuda, J. Phys. Chem. Lett., 2014, 5, 1953–1957 CrossRef CAS PubMed.
- K. Ozawa, S. Yamamoto, M. D’angelo, Y. Natsui, N. Terashima, K. Mase and I. Matsuda, J. Phys. Chem.C, 2019, 123, 4388–4395 CrossRef CAS.
- B. F. Spencer, M. J. Cliffe, D. M. Graham, S. J. O. Hardman, E. A. Seddon, K. L. Syres, A. G. Thomas, F. Sirotti, M. G. Silly, J. Akhtar, P. O’Brien, S. M. Fairclough, J. M. Smith, S. Chattopadhyay and W. R. Flavell, Surf. Sci., 2015, 641, 320–325 CrossRef CAS.
- H. Magnan, P. M. Deleuze, J. Brehin, T. Plays, D. Stanescu, W. R. Flavell, M. G. Silly, B. Domenichini and A. Barbier, J. Phys. Chem. C, 2020, 124, 10315–10323 CrossRef CAS.
- S. Koley, J. Cui, Y. E. Panfil and U. Banin, Acc. Chem. Res., 2021, 54, 1178–1188 CrossRef CAS PubMed.
- C. R. Kagan and C. B. Murray, Nat. Nanotechnol., 2015, 10, 1013–1026 CrossRef CAS PubMed.
- E. M. Sanehira, A. R. Marshall, J. A. Christians, S. P. Harvey, P. N. Ciesielski, L. M. Wheeler, P. Schulz, L. Y. Lin, M. C. Beard and J. M. Luther, Sci. Adv., 2017, 3, eaao4204 CrossRef PubMed.
- M. Hao, Y. Bai, S. Zeiske, L. Ren, J. Liu, Y. Yuan, N. Zarrabi, N. Cheng, M. Ghasemi, P. Chen, M. Lyu, D. He, J. H. Yun, Y. Du, Y. Wang, S. Ding, A. Armin, P. Meredith, G. Liu, H. M. Cheng and L. Wang, Nat. Energy, 2020, 5, 79–88 CrossRef CAS.
- A. Maulu, P. J. Rodríguez Cantó, J. Navarro Arenas, R. Abargues, J. F. Sanchez-Royo, R. García Calzada and J. P. Martínez-Pastor, RSC Adv., 2016, 6, 80201–80212 RSC.
- A. Yousefiamin, N. A. Killilea, M. Sytnyk, P. Maisch, K. C. Tam, H. J. Egelhaaf, S. Langner, T. Stubhan, C. J. Brabec, T. Rejek, M. Halik, K. Poulsen, J. Niehaus, A. Köck and W. Heiss, ACS Nano, 2019, 13, 2389–2397 CAS.
- I. Ramiro, O. Özdemir, S. Christodoulou, S. Gupta, M. Dalmases, I. Torre and G. Konstantatos, Nano Lett., 2020, 20, 1003–1008 CrossRef CAS PubMed.
- A. G. Pattantyus-Abraham, I. J. Kramer, A. R. Barkhouse, X. Wang, G. Konstantatos, R. Debnath, L. Levina, I. Raabe, M. K. Nazeeruddin, M. Grätzel and E. H. Sargent, ACS Nano, 2010, 4, 3374–3380 CrossRef CAS PubMed.
- N. Oh, B. H. Kim, S.-Y. Cho, S. Nam, S. P. Rogers, Y. Jiang, J. C. Flanagan, Y. Zhai, J.-H. Kim, J. Lee, Y. Yu, Y. K. Cho, G. Hur, J. Zhang, P. Trefonas, J. A. Rogers and M. Shim, Science, 2017, 355, 616–619 CrossRef CAS PubMed.
- C.-H. M. Chuang, P. R. Brown, V. Bulović and M. G. Bawendi, Nat. Mater., 2014, 13, 796–801 CrossRef CAS PubMed.
- P. C. J. Clark, H. Radtke, A. Pengpad, A. I. Williamson, B. F. Spencer, J. O. Hardman, M. A. Leontiadou, D. C. J. Neo, S. M. Fairclough, A. A. R. Watt, I. Pis, S. Nappini, F. Bondino, E. Magnano, K. Schulte, M. Silly, F. Sirotti and W. R. Flavell, Nanoscale, 2017, 9, 6056–6067 RSC.
- Y. Zhang, Q. Chen, A. P. Alivisatos and M. Salmeron, Nano Lett., 2015, 15, 4647–4663 Search PubMed.
- L. Hu, S. Huang, R. Patterson and J. E. Halpert, J. Mater. Chem. C, 2019, 7, 4497–4502 RSC.
- D. Pierucci, J.-J. Gallet, F. Bournel, F. Sirotti, M. G. Silly, H. Tissot, A. Naitabdi and F. Rochet, J. Phys. Chem. C, 2016, 120, 21631–21641 CrossRef CAS.
- L. Pérez Ramírez, J.-J. Gallet, F. Bournel, F. Lim, S. Carniato, F. Rochet, O. V. Yazyev, A. Pasquarello, E. Magnano and F. Bondino, J. Phys. Chem. A, 2020, 124, 5378–5388 CrossRef PubMed.
- F. Armillotta, E. D’Incecco, M. Corva, M. Stredansky, J.-J. Gallet, F. Bournel, A. Goldoni, A. Morgante, E. Vesselli and A. Verdini, Angew. Chem., Int. Ed., 2021, 60, 25988–25993 CrossRef CAS PubMed.
- E. Goulielmakis, M. Schultze, M. Hofstetter, V. S. Yakovlev, J. Gagnon, M. Uiberacker, A. L. Aquila, E. M. Gullikson, D. T. Attwood, R. Kienberger, F. Krausz and U. Kleineberg, Science, 2008, 320, 1614–1617 CrossRef CAS PubMed.
- M. Ossiander, F. Siegrist, V. Shirvanyan, R. Pazourek, A. Sommer, T. Latka, A. Guggenmos, S. Nagele, J. Feist, J. Burgdörfer, R. Kienberger and M. Schultze, Nat. Phys., 2017, 13, 280–285 Search PubMed.
- S. Heinrich, T. Saule, M. Högner, Y. Cui, V. S. Yakovlev, I. Pupeza and U. Kleineberg, Nat. Commun., 2021, 12, 3404 Search PubMed.
- T. Saule, S. Heinrich, J. Schötz, N. Lilienfein, M. Högner, O. deVries, M. Plötner, J. Weitenberg, D. Esser, J. Schulte, P. Russbueldt, J. Limpert, M. F. Kling, U. Kleineberg and I. Pupeza, Nat. Commun., 2019, 10, 458 Search PubMed.
- V. S. Yakovlev and N. Karpowicz, Science, 2017, 357, 1239–1240 CrossRef CAS PubMed.
- F. Siek, S. Neb, P. Bartz, M. Hensen, C. Strüber, S. Fiechter, M. Torrent-Sucarrat, V. M. Silkin, E. E. Krasovskii, N. M. Kabachnik, S. Fritzsche, R. D. Muiño, P. M. Echenique, A. K. Kazansky, N. Müller, W. Pfeiffer and U. Heinzmann, Science, 2017, 357, 1274–1277 CrossRef CAS PubMed.
- M. Schultze, M. Fieß, N. Karpowicz, J. Gagnon, M. Korbman, M. Hofstetter, S. Neppl, A. L. Cavalieri, Y. Komninos, Th. Mercouris, C. A. Nicolaides, R. Pazourek, S. Nagele, J. Feist, J. Burgdörfer, A. M. Azzeer, R. Ernstorfer, R. Kienberger, U. Kleineberg, E. Goulielmakis, F. Krausz and V. S. Yakovlev, Science, 2010, 328, 1658–1662 CrossRef CAS PubMed.
- Y.-J. Chen, M. Hoffmann, B. Zimmermann, G. Bihlmayer, S. Blügel, C. M. Schneider and C. Tusche, Commun. Phys., 2021, 4, 1–6 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |