 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImage processing tools for the validation of CryoEM maps†
C. O. S.
Sorzano
 *a,
J. L.
Vilas
a,
E.
Ramírez-Aportela
a,
J.
Krieger
a,
D.
del Hoyo
a,
D.
Herreros
a,
E.
Fernandez-Giménez
a,
D.
Marchán
a,
J. R.
Macías
a,
I.
Sánchez
a,
L.
del Caño
a,
Y.
Fonseca-Reyna
a,
P.
Conesa
a,
A.
García-Mena
a,
J.
Burguet
b,
J.
García Condado
c,
J.
Méndez García
d,
M.
Martínez
a,
A.
Muñoz-Barrutia
e,
R.
Marabini
f,
J.
Vargas
b and
J. M.
Carazo
a
*a,
J. L.
Vilas
a,
E.
Ramírez-Aportela
a,
J.
Krieger
a,
D.
del Hoyo
a,
D.
Herreros
a,
E.
Fernandez-Giménez
a,
D.
Marchán
a,
J. R.
Macías
a,
I.
Sánchez
a,
L.
del Caño
a,
Y.
Fonseca-Reyna
a,
P.
Conesa
a,
A.
García-Mena
a,
J.
Burguet
b,
J.
García Condado
c,
J.
Méndez García
d,
M.
Martínez
a,
A.
Muñoz-Barrutia
e,
R.
Marabini
f,
J.
Vargas
b and
J. M.
Carazo
a
aNatl. Center of Biotechnology, CSIC, c/Darwin, 3, 28049, Madrid, Spain. E-mail: coss@cnb.csic.es; Fax: +34 91 585 4506; Tel: +34 91 585 45 10
bDepto. de Óptica, Univ. Complutense de Madrid, Pl. Ciencias, 1, 28040, Madrid, Spain
cBiocruces Bizkaia Instituto Investigación Sanitaria, Cruces Plaza, 48903, Barakaldo, Bizkaia, Spain
dUniv. Tecnológica de Pereira, 660003, Pereira, Colombia
eUniv. Carlos III de Madrid, Avda. de la Universidad 30, 28911, Leganés, Madrid, Spain
fEscuela Politécnica Superior, Univ. Autónoma de Madrid, CSIC, C. Francisco Tomás y Valiente, 11, 28049, Madrid, Spain
First published on 4th April 2022
Abstract
The number of maps deposited in public databases (Electron Microscopy Data Bank, EMDB) determined by cryo-electron microscopy has quickly grown in recent years. With this rapid growth, it is critical to guarantee their quality. So far, map validation has primarily focused on the agreement between maps and models. From the image processing perspective, the validation has been mostly restricted to using two half-maps and the measurement of their internal consistency. In this article, we suggest that map validation can be taken much further from the point of view of image processing if 2D classes, particles, angles, coordinates, defoci, and micrographs are also provided. We present a progressive validation scheme that qualifies a result validation status from 0 to 5 and offers three optional qualifiers (A, W, and O) that can be added. The simplest validation state is 0, while the most complete would be 5AWO. This scheme has been implemented in a website https://biocomp.cnb.csic.es/EMValidationService/ to which reconstructed maps and their ESI can be uploaded.
Cryo-electron microscopy is currently one of the most active techniques in structural biology. The number of maps deposited at the Electron Microscopy Data Bank is rapidly growing every year1,2 and keeping the quality of the submitted maps is essential to maintain the scientific quality of the field. Additionally, all scientific domains have recognized open science as a way to accelerate research.3 In this way, disclosing sufficient information in order to understand the limitations and strengths of a CryoEM map is crucial for a better use of the map. Ideally, reproducibility of the results should be achieved, and the possibility of depositing the raw data at the EMPIAR (Electron Microscopy Public Image Archive, Iudin et al.4) has certainly been a huge step forward. However, as a community we are still far from having generally adopted this reproducibility goal. For instance, from the 950 EMDB entries labelled as SARS-CoV-2, only 23 (less than 2.5%) are deposited at EMPIAR (as of March 7th, 2022). The availability of the raw data could be complemented with the availability of the image processing workflow and decisions taken to go from the raw acquisition to the final map. Despite the fact that scientific articles are generally trusted by other scientists and the general public, severe concerns about a reproducibility crisis in science have been raised.5
The ultimate quality measure is the consistency of the map and an atomic model.6 However, this is only possible for high-resolution maps. Alternatively, the standard map validation practice has mostly been restricted to the internal consistency of two half-maps calculated from independent halves of the whole dataset.7 This internal consistency is essential, and it is a good measure of the presence of random fluctuations. Its main drawback is that it is not immune to systematic biases,8 that is, systematic mistakes committed in both halves would be rewarded in terms of the Fourier shell correlation.
Over the years, there have been many suggestions about validation measures of CryoEM maps.9 Unfortunately, most of these measures are not currently used due to their spread across multiple software tools and the associated difficulty accessing them. To alleviate this problem, we present a validation grading system and its public availability through a web server that qualifies the CryoEM map depending on the information available to assess it. This system grades a structure map at six different levels. In this way, a map could be validated at level 0 (the deposited map), 1 (two half maps), 2 (2D classes), 3 (particles), 4 (…+angular assignment), 5 (…+micrographs and coordinates). For each of these levels, we list algorithms that can be employed. In addition to this grading system, we have three optional qualifiers: A (…+atomic model), W (…+image processing workflow), and O (…+other techniques). Those depositions wanting to achieve the highest level of validation should deposit a relatively small number of particles (in the order of 10k randomly chosen from the final set of particles) along with their micrographs and all alignment parameters. They should also include a detailed description of the image processing workflow so that the final result can be fully understood (and ideally reproduced) and some extra validation by other experimental data. This high-quality standard is ideal and will not be accomplished shortly for all deposited structures. Nevertheless, it is essential to have it as a compass to direct our community efforts.
The web server is publicly available at https://biocomp.cnb.csic.es/EMValidationService/. It returns a PDF report that evaluates the correctness of the submitted map from multiple figures of merit. Obviously, map submission to a public database cannot require a compulsory deposition of all these elements. Still, a validation grading system could be adopted in which, if they are given, the consistency of the map with the different aspects that give rise to it can be assessed.
The server is aimed at structures determined by single particle analysis (SPA). Maps determined by subtomogram averaging (STA) in cryo-electron tomography can share the levels 0, 1, A, and W with the maps coming from SPA. However, more specific analyses could be developed for STA maps.
We hope that the validation server proposed in this paper will help improve the understanding of CryoEM maps and reduce the reproducibility crisis, especially if the image processing workflow is also disclosed.
1 Validation methods
The following sections describe the different methods at one’s disposal to validate a CryoEM map. The availability of these methods usually depends on the accessibility of extra information, like the set of particles supporting it, their angular assignment, etc. The level of map validation is defined as the highest consecutive number up to which there is information available. The three extra qualifiers can be added to any of the levels. The highest degree of validation would be 5AWO. Still, we could have, for instance, a map whose validation is 4W, meaning that it has reached level 4 and a detailed description of the image processing workflow is available. At present, the highest validation that a typical map from EMDB can achieve is 1A (map + half maps + atomic model). Interestingly, since February 2022, the deposition of half maps at the EMDB has been compulsory, meaning that we have moved from validation level 0 to level 1.1.0 Level 0: map
The first level of validation is performed when just the reconstructed map is available along with a visualization threshold. At this point, several methods can evaluate the local resolution of the map and its hand.Although not yet implemented, it would be possible to detect preferential orientations or artifacts by analyzing the macromolecule’s local texture and noise.
1.1 Level 1: …+half maps
If independent half maps are available, then we can further assess the local resolution with different means:1.2 Level 2: …+2D classes
If 2D classes of the particles used for the reconstruction are available, then the following method can be applied:1.3 Level 3: …+particles
If a random subset of the particles is provided, the following actions can be performed:1.4 Level 4: …+angular assignment
If the angular assignment of the particles in level 3 is available, then the following methods can be applied:1.5 Level 5: …+micrographs and coordinates
If a random subset of micrographs and their corresponding coordinates are available, then the following measure can be taken:1.6 Level A: …+atomic model
If a fitted atomic model is available, then we may apply the following validation methods:We are aware that the report generated at the submission to EMDB/PDB is relatively rich in this area, including methods such as Molprobity36 or TEMPy2.37 Our goal is to complement this analysis with alternative tools. Some of these analyses (0.a, 0.b, 0.c, 2.a, 3.b, 3.c, 4.a, 4.d, 4.e, and 4.i) have been newly developed for this server.
1.7 Qualifier W: …+workflow
Another level of validation purely based on image processing is a detailed description of each of the image processing steps used to produce the final map so that an external user can understand the results of each step and has all the information to reproduce the whole pipeline. This information is produced, for example, by Scipion38 in the form of a JSON that can be submitted to EMPIAR1 or Scipion’s workflow repository (https://nolan.cnb.csic.es/cryoemworkflowviewer) and visualized with a JavaScript viewer integrated into these databases. Alternative methods would also be valid, specifically tailored to each one of the different image processing packages.1.8 Qualifier O: …+other techniques
Finally, if extra experimental work is available, then we may apply the following techniques.1.9 Availability
We have created a web server located at https://biocomp.cnb.csic.es/EMValidationService/. The server has a web interface that guides the user through the different steps. We have not made the levels to be compulsorily progressive. For instance, one could have validation levels 0 and 2 without having the information for level 1. Although possible, this option is discouraged. Method A.c is sensible, but this method takes much time to execute due to the molecular dynamics underneath (about 6 hours per constructed model). For this reason, we recommend not to run it regularly to not saturate the server.Once a job is submitted to the server, the execution time varies from 20 minutes to 16 hours if A.c is not executed or 3 days if it is. The execution time depends on the number of validations to perform, the size of the reconstructed map, and the number of images provided for validation. During its execution, the user gets a token so that they can check whether the job has already been finished.
The server asks for a URL where the image processing workflow can be visualized for validation. If Scipion has created the workflow, it is included in the report. However, the validation information truly lies on the given URL. For submitting the Scipion workflow, the user may use the scipion-em-datamanager plugin that submits the current project to the workflow repository at https://nolan.cnb.csic.es/cryoemworkflowviewer/entries. An example of such a workflow can be seen in Fig. 1.
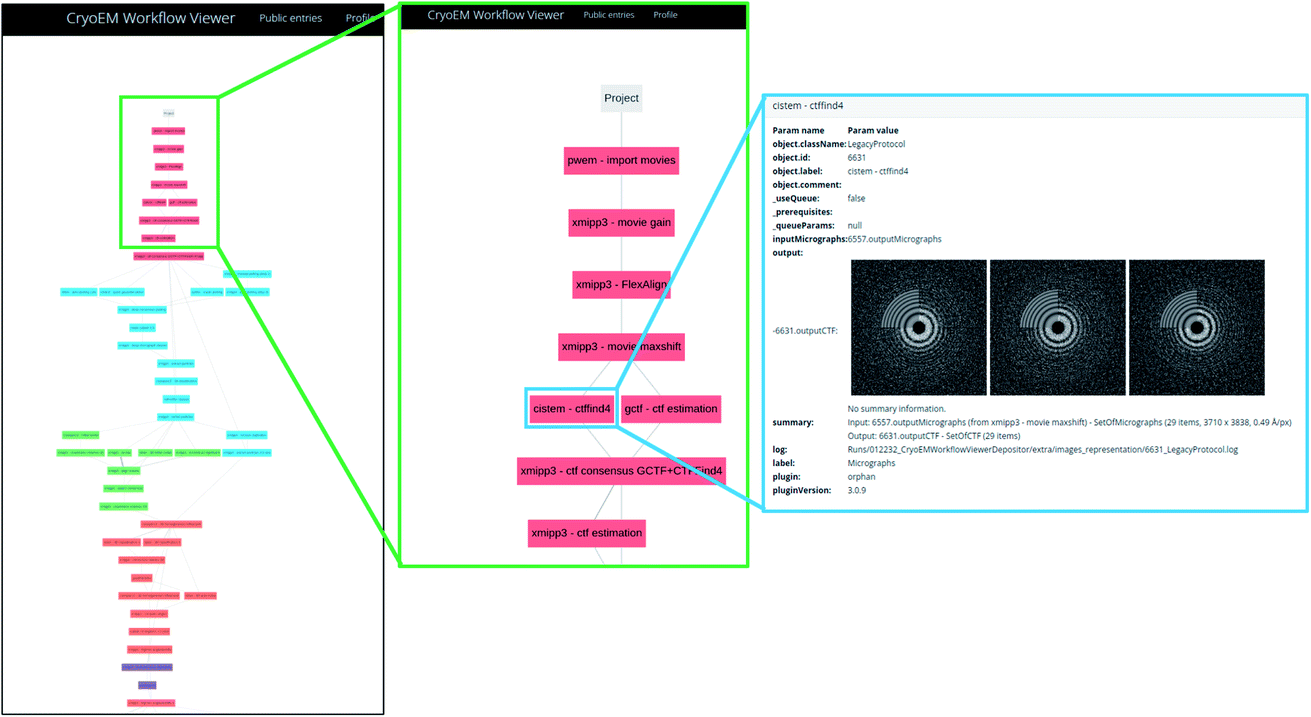 | ||
| Fig. 1 An example of workflow visualization at Scipion’s workflow repository. Each of the colored boxes represent a protocol executed within Scipion. The last panel on the right shows how the input parameters, data and results can be inspected using a web browser. | ||
The server automatically constructs a Scipion workflow38 based on the input data. Depending on the amount of data available (levels 0, 1, 2, …), the appropriate protocols are instanced and interconnected. The source code of the server is available at https://github.com/I2PC/scipion-em-validation. The program creates the Scipion workflow, automatically analyzes the results, and constructs a Latex document which is later compiled into a PDF. This report is handed to the user.
2 Results
To show the usefulness of the validation reports, we have applied this methodology to multiple datasets. The first one comes from the tutorial of Scipion,38 while all others are examples from the EMDB. In the first one, we have information for evaluating all validation levels (5AWO), while for the EMDB, the validation reports are of levels 0A or 1A.2.1 Full report
ESI 1† shows the validation report for Scipion’s tutorial. The data corresponds to 30 micrographs of the apoferritin EMPIAR 10248 dataset.42 The tutorial used 1457 experimental projections to construct a map whose nominal resolution is 2.6 Å. Although generally, the map is good (see Fig. 2) and reasonably agrees with its atomic model by visual inspection, the validation report shows that it has several problems:(1) The background outside the map does not have zero mean, and it contains a significant amount of intensity outliers.
(2) The reported resolution seems to be overestimated according to several local resolution algorithms (DeepRes, Resmap, and MonoDir) and Phenix’s comparison to the atomic model. The average resolution seems to be more around 4.4 Å, rather than 2.6 Å.
(3) Images are difficult to align, as reported by the alignability smoothness, CryoSparc alignment, and the comparison between CryoSparc and Relion alignments. This uncertainty in the alignment was also shown in MonoDir radial plots.
(4) Relion could not reliably determine the scaling factor of the images.
(5) The resolvability of the side chains is not good, as reported by EMRinger and DAQ.
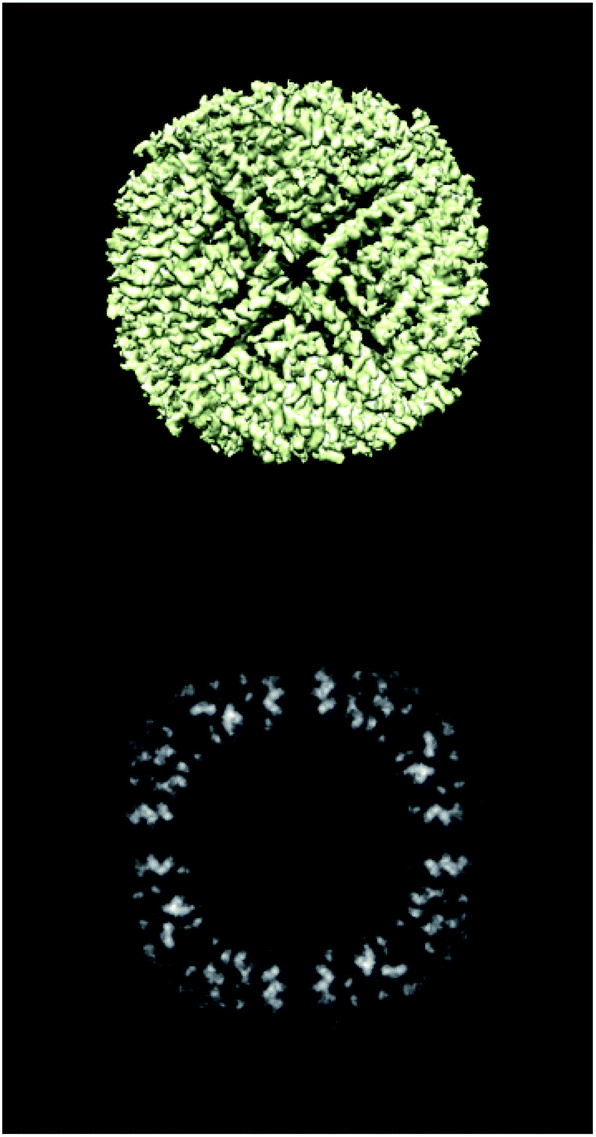 | ||
| Fig. 2 Isosurface and central slice of the Scipion’s tutorial dataset, apoferritin. | ||
None of these errors is terrible, and it is the expected result for a tutorial reconstruction from only 30 micrographs. However, this contrasts with the resolution reported by the FSC, which gives the false impression of having achieved a better map than the one obtained.
Fig. 3–5 show the summary of the report generated for this dataset. In a single glimpse, the main problems can be easily identified.
 | ||
| Fig. 3 Example of the abstract of the full report generated for Scipion’s tutorial data. | ||
 | ||
| Fig. 4 Example of the index of the full report generated for Scipion’s tutorial data. | ||
 | ||
| Fig. 5 Example of the warnings abstract of the full report generated for Scipion’s tutorial data. | ||
2.2 A comparison of three SARS-CoV 2 spike structures
With the Covid-19 pandemic, many structural studies have addressed all the proteins amenable to CryoEM. We have chosen three reconstructions of the spike (EMDB entries 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 337,43 22301,44 and 2283845) which are supposed to be reconstructed at similar resolutions (3.3, 3.7 and 3.84 Å, respectively; see Fig. 6). The first entry could be validated at level 1A, while the other two could only be validated at level 0A. The validation reports for these structures can be seen in ESI 2.†
337,43 22301,44 and 2283845) which are supposed to be reconstructed at similar resolutions (3.3, 3.7 and 3.84 Å, respectively; see Fig. 6). The first entry could be validated at level 1A, while the other two could only be validated at level 0A. The validation reports for these structures can be seen in ESI 2.†
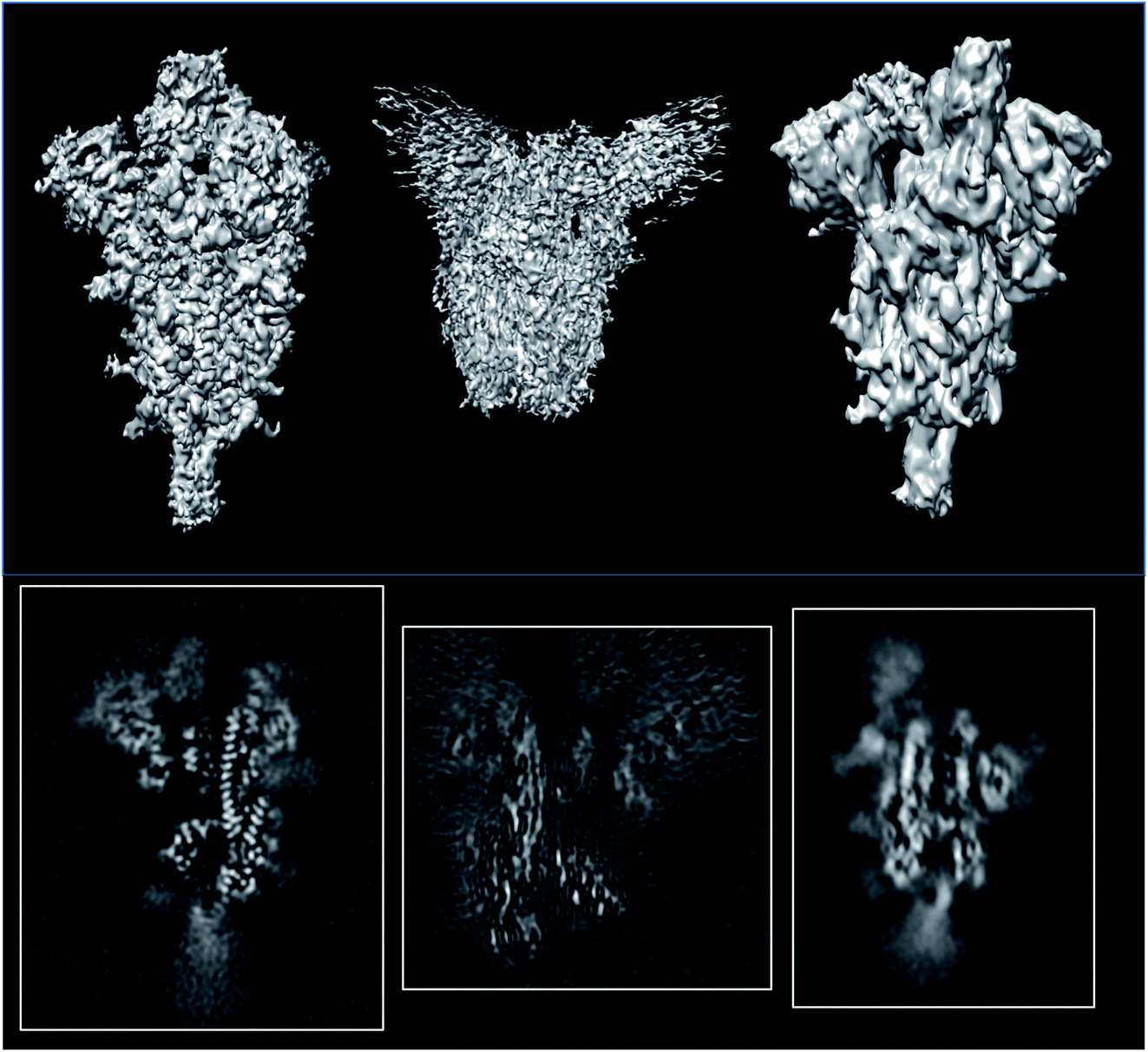 | ||
| Fig. 6 Isosurfaces and central slices of EMDB 11337 (left), EMDB 22301 (middle), and EMDB 22838 (right), all of which are SARS-CoV2 spikes. | ||
The average resolutions measured by several methods for the different structures were 4.9, 8.2, and 3.8 Å, respectively. This points out that the internal resolution variability is much higher than the one reported by the FSC. We have empirically verified that very often, the resolution based on the FSC is at the lower extreme of the distribution of resolutions reported by many local resolution methods.11 For this reason, this FSC resolution must be understood as “there is a region in the map whose local resolution is this number”.
In this analysis, we can identify the following problems:
(1) The backgrounds of the three structures have problems as they are not equal to 0 and contain significant outliers.
(2) Two of the maps, EMDB11337 and EMDB22838, have a reported resolution that is particularly good compared to the one computed by DeepRes (level 0) or other local resolution methods (level 1).
(3) EMDB22838 and EMDB22301 have either too low a resolution or a severe problem of anisotropic resolution, probably caused by the attraction of particles towards specific directions. Both issues are translated into the uncertainty of DeepHand to determine the handedness of the structure.
(4) EMDB11337 has problems fitting to the atomic map according to FSC-Q and Phenix. EMDB22301 has issues in this fitting according to MapQ, Phenix, and DAQ. Finally, EMDB 22838 has problems according to MapQ, EMRinger, and DAQ.
2.3 An analysis of a 1.15 Å resolution apoferritin
EMDB11668 is currently the map with the best resolution in the EMDB reconstructed by single particle analysis (see Fig. 7). It is the reconstruction of human apoferritin in a Titan Krios with a second-generation spherical aberration correction.46 An analysis of the deposited structure reveals that the recommended threshold, 0.15, is too high and causes many mass and mask problems (see ESI 3 Report 11668_015†). In the following, we will analyze the map at a threshold of 0.05 (see ESI 3 Report 11668_005†).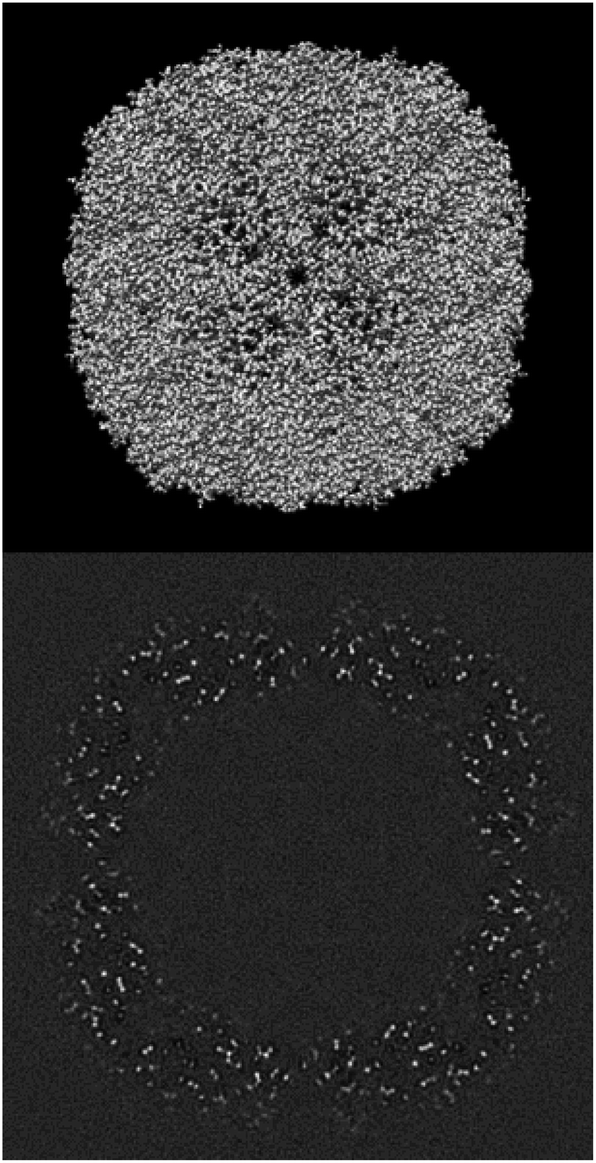 | ||
| Fig. 7 Isosurface and central slice of EMDB 11668, apoferritin. | ||
In this analysis, we can identify the following problems:
(1) The mask of the map is very fragmented, with 478289 connected components. The largest component only takes 42% of the mass. This is due to the boosting of the high frequencies caused by the B-factor correction.
(2) The mean of the background is not 0, and there is a significant amount of outlier values.
(3) The map has been B-factor corrected, resulting in a boost of noise at high frequency and a disagreement between the falloffs of the map and the model.
(4) There is a significant disagreement between the map and its model according to MapQ, Phenix, and DAQ in some regions. In Fig. 8, we show a region of the map and model in Coot. Although the model is excellent in many regions, there are portions in which the map and model do not match, as highlighted in the figure.
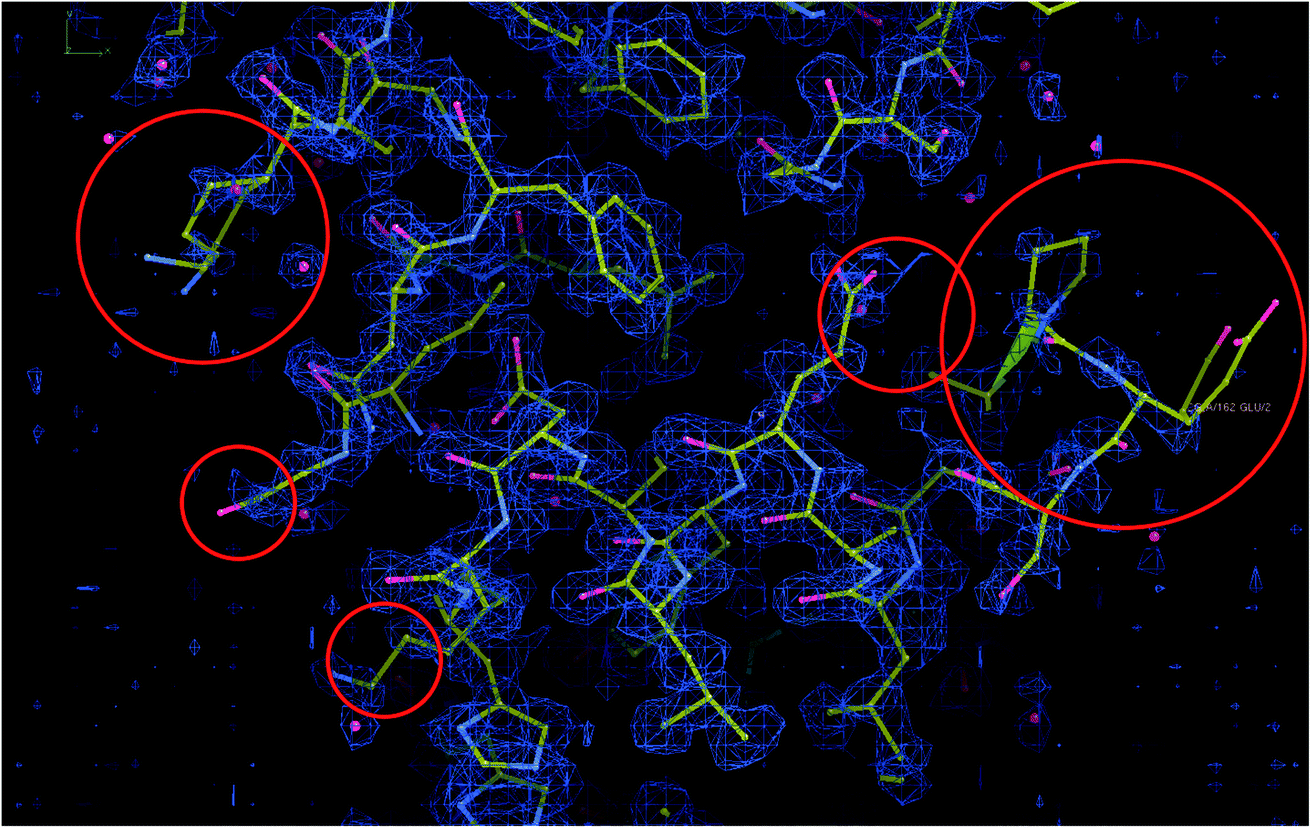 | ||
| Fig. 8 Map and model representations of apoferritin with some mismatching areas highlighted. | ||
3 Discussion
The determination of a three-dimensional map compatible with the CryoEM measurements of a macromolecule is full of decisions along the image processing pipeline (whether to keep or not this micrograph according to its contrast transfer function or beam-induced alignment, whether a region of that micrograph represents a centered projection of the macromolecule, which is the orientation of that projection with respect to a reference map and which is the exact conformation of that macromolecule, etc.). All these decisions involve parameter estimates. Some of these parameters are continuous, such as the micrograph defocus or the in-plane shifts of a given projection with respect to a reference map. Some others are discrete, such as whether a given projection is in one conformation or another. From this point of view, the reconstructed map is a signal (another parameter) to be estimated in a N3-dimensional space, with N3 being the number of map voxels.All algorithms that have to estimate a parameter can commit an error (false positives or negatives if the parameter is binary, or a residual if the parameter is continuous). Our final map is a mixture of our good and bad decisions for all the particles involved in the reconstruction. To simplify the analysis, let us consider that we perfectly estimate all parameters of a given projection with probability p, and we do not, with probability 1 − p. The fraction of well-estimated projections will provide us with a perfect reconstruction, while the fraction of incorrectly estimated parameters will result in an imperfect reconstruction:
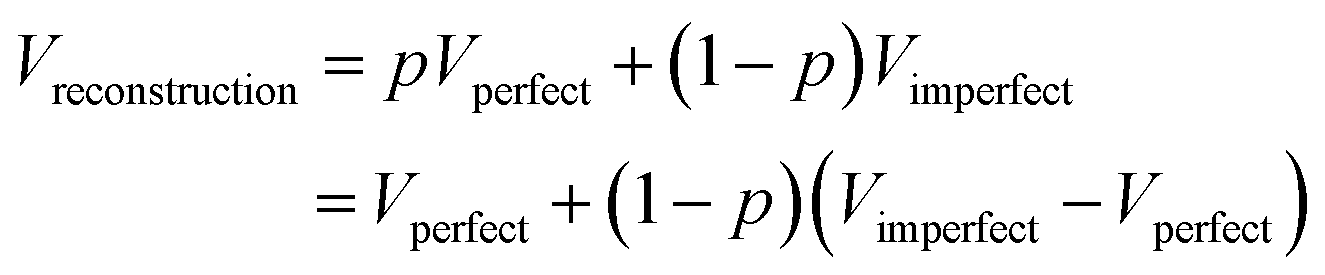
In this way, we see that our reconstruction is a perfect map plus bias that depends on the fraction of incorrectly estimated parameters, 1 − p, and the difference between the imperfect and perfect reconstructions (if we make a mistake in the estimation of the projection direction of an experimental projection, but the map from the wrong direction is very similar to the map from the correct direction, the mistake will have a negligible effect). It is this bias that we call overfitting in our field. Hence, overfitting is directly linked to incorrect parameter estimates. In Sorzano et al.,8 we discuss the different sources of map bias and how we can design image processing workflows that try to identify the incorrectly estimated parameters. Ultimately, this identification requires multiple estimates of the same parameter by, ideally, different algorithms. In this way, we can determine the reliability of any particular value.
The use of multiple algorithms to discard incorrectly estimated parameters or average those that agree more is seldom seen in our applied papers. However, this practice of estimating parameters only once leaves us in a position in which we cannot know whether the estimated parameter is stable or not.
Map and model validations have become a relevant research line in the field, responding to the need to assess the reliability of the reconstructed maps. Despite the multiple methods available to perform this validation, the most common is the reconstruction of two half-maps and their subsequent comparison through the Fourier shell correlation. This practice is known in the field as the gold standard. It is a cheap substitute for the multiple estimations of the same parameter: we use the estimate of the N3-dimensional signal as a proxy of the various estimations at a cost that involves estimating the parameters only once for each of the experimental images. If the parameter estimation errors are independent, zero-mean processes, this is a good practice. However, it does not protect us if the estimation errors are dependent or do not have a zero mean. For instance, a wrong estimate of the defocus of a micrograph will systematically affect the CTF correction of many particles. Suppose we have an attraction problem8 (a projection direction has a larger signal-to-noise ratio, SNR, than its surrounding or a 3D class has a larger SNR than the alternatives). In that case, the parameters would also be systematically misestimated.
In Sorzano et al.,8 we show that splitting the data into two halves is not a sufficient nor necessary condition for avoiding overfitting. It is not sufficient because we can make systematic mistakes in estimating the parameters in both halves. It is not necessary because not splitting the data into two halves does not necessarily lead to an incorrect estimation of the underlying parameters. The most common measure to assess the correction of a reconstructed map is the Fourier shell correlation between the two half maps. This measure is excellent when the difference between the two maps is independent and randomly distributed with zero mean. However, it is misled by systematic errors appearing in both halves.8
Despite the plethora of alternative validation methods, these are seldom used due to the difficulty of accessing them conveniently and smoothly. Scipion38 is an appropriate platform for this evaluation as it integrates all the methods described in this paper, a total of 37, and provides effective ways of allowing them to interoperate. The server is open to new approaches, and any new validation tool is welcome to be incorporated into the validation report.
4 Conclusions
In this work, we have presented a validation server that assesses CryoEM maps’ correction from multiple perspectives. The validation can be more profound as more data is available about the structure, the experimental images supporting it, and the image processing workflow followed to achieve it. In this way, we provide a progressive validation level that evaluates the map from different perspectives: (0) the map itself, (1) its two-halves, (2) the 2D classes of the particles, (3) the particles themselves, (4) the angular assignment of those particles, (5) the coordinates of those particles in the micrographs, (A) an atomic model of the structure, (W) the image processing workflow leading to the final result, and (O) validation through alternative experiments.We believe that disclosing as much information as possible about the supporting experimental evidence leading to a CryoEM map will help the user of that structure better understand the strengths and weaknesses of the map at hand. And finally, it will ultimately contribute to more open and reproducible science.
Author contributions
COSS conceptualized the project, implemented its calculation engine, coordinated the integration of the different methods and performed the experiments. COSS and JMC analyzed the application and wrote the article. IS and JRM implemented the webserver. All others helped to make the methods available through Scipion plugins. All authors have revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to Philip Baldwin, Dmitry Lyumkis, and Gabriel Lander for making their validation methods available (4.h and A.c). Javier Vargas and Jordi Burguet would like to thank the Spanish Ministry of Science and Innovation for financial support through the call 2019 Proyectos de I+D+i – RTI Tipo A (PID2019-108850RA-I00) and Arrate Muñoz Barrutia, PID2019-109820RB-I00, MCIN/AEI/10.13039/501100011033/, cofinanced by European Regional Development Fund (ERDF), “A way of making Europe.”. The authors acknowledge the economic support from MICIN of the Instruct Image Processing Center (I2PC) as part of the Spanish participation in Instruct-ERIC, the European Strategic Infrastructure Project (ESFRI) in the area of Structural Biology, Grant PID2019-104757RB-I00 funded by MCIN/AEI/10.13039/501100011033/ and “ERDF A way of making Europe”, by the European Union and Grant PRE2020 - 093527 funded by MCIN/AEI/ 10.13039/501100011033 and by “ESF Investing in your future”. We also acknowledge support from “Comunidad Autónoma de Madrid” through Grant: S2017/BMD-3817, Instituto de Salud Carlos III (project IMPaCT-Data, exp. IMP/00019), co-funded by the European Union, European Regional Development Fund (ERDF, “A way to make Europe”), and European Union (EU) and Horizon 2020 through grants: EOSC Life (INFRAEOSC-04-2018, Proposal: 824087), HighResCells (ERC – 2018 – SyG, Proposal: 810057), IMpaCT (WIDESPREAD-03-2018 – Proposal: 857203), EOSC – Synergy (EINFRA-EOSC-5, Proposal: 857647), iNEXT-Discovery (Proposal: 871037), EnLaCES (H2020-MSCA-IF-2020, Proposal: 101024130).Notes and references
- A. Patwardhan, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 503–508 CrossRef CAS.
- C. O. S. Sorzano and J.M. Carazo, Cryo-Electron Microscopy: the field of 1000+ methods, J. Struct. Biol., 2022, 214, 107861 CrossRef CAS PubMed.
- M. Woelfle, P. Olliaro and M. H. Todd, Nat. Chem., 2011, 3, 745–748 CrossRef CAS PubMed.
- A. Iudin, P. K. Korir, J. Salavert-Torres, G. J. Kleywegt and A. Patwardhan, Nat. Methods, 2016, 13, 387–388 CrossRef CAS PubMed.
- M. Baker, Nature, 2016, 533, 452–454 CrossRef CAS PubMed.
- C. L. Lawson, A. Kryshtafovych, P. D. Adams, P. V. Afonine, M. L. Baker, B. A. Barad, P. Bond, T. Burnley, R. Cao and J. Cheng, et al. , Nat. Methods, 2021, 18, 156–164 CrossRef CAS.
- R. Henderson, A. Sali, M. L. Baker, B. Carragher, B. Devkota, K. H. Downing, E. H. Egelman, Z. Feng, J. Frank, N. Grigorieff, W. Jiang, S. J. Ludtke, O. Medalia, P. A. Penczek, P. B. Rosenthal, M. G. Rossmann, M. F. Schmid, G. F. Schröder, A. C. Steven, D. L. Stokes, J. D. Westbrook, W. Wriggers, H. Yang, J. Young, H. M. Berman, W. Chiu, G. J. Kleywegt and C. L. Lawson, Structure, 2012, 20, 205–214 CrossRef CAS.
- C. O. S. Sorzano, A. Jimenez-Moreno, D. Maluenda, M. Martinez, E. Ramirez-Aportela, R. Melero, A. Cuervo, J. Conesa, J. Filipovic, P. Conesa, L. del Caño, Y. C. Fonseca, J. Jiménez-de la Morena, P. Losana, R. Sáchez-García, D. Strelak, E. Fernandez-Gimenez, F. de Isidro, D. Herreros, J. L. Vilas, R. Marabini and J. M. Carazo, On bias, variance, overfitting, gold standard and consensus in Single Particle Analysis by Cryo-electron microscopy, Acta Crystallogr., Sect. D: Struct. Biol., 2022, D78, 410–423 Search PubMed.
- J. L. Vilas, N. Tabassum, J. Mota, D. Maluenda, A. Jiménez-Moreno, T. Majtner, J. M. Carazo, S. T. Acton and C. O. S. Sorzano, Curr. Opin. Struct. Biol., 2018, 52, 127–145 CrossRef CAS PubMed.
- P. B. Rosenthal and R. Henderson, J. Mol. Biol., 2003, 333, 721–745 CrossRef CAS PubMed.
- E. Ramírez-Aportela, J. Mota, P. Conesa, J. M. Carazo and C. O. S. Sorzano, IUCrJ, 2019, 6, 1054–1063 CrossRef.
- S. Kaur, J. Gomez-Blanco, A. A. Khalifa, S. Adinarayanan, R. Sanchez-Garcia, D. Wrapp, J. S. McLellan, K. H. Bui and J. Vargas, Nat. Commun., 2021, 12, 1 CrossRef PubMed.
- J. García Condado, A. Muñoz-Barrutia and C. O. S. Sorzano, bioRxiv, 2022 DOI:10.1101/2022.03.01.482513.
- C. O. S. Sorzano, J. Vargas, J. Oton, V. Abrishami, J. M. de la Rosa-Trevin, J. Gomez-Blanco, J. L. Vilas, R. Marabini and J. M. Carazo, Prog. Biophys. Mol. Biol., 2017, 124, 1–30 CrossRef CAS PubMed.
- M. Beckers and C. Sachse, J. Struct. Biol., 2020, 212, 107579 CrossRef CAS PubMed.
- G. Cardone, J. B. Heymann and A. C. Steven, J. Struct. Biol., 2013, 184, 226–236 CrossRef PubMed.
- A. Kucukelbir, F. J. Sigworth and H. D. Tagare, Nat. Methods, 2014, 11, 63–65 CrossRef CAS PubMed.
- J. L. Vilas, J. Gómez-Blanco, P. Conesa, R. Melero, J. M. de la Rosa Trevín, J. Otón, J. Cuenca, R. Marabini, J. M. Carazo, J. Vargas and C. O. S. Sorzano, Structure, 2018, 26, 337–344 CrossRef CAS PubMed.
- J. L. Vilas, H. D. Tagare, J. Vargas, J. M. Carazo and C. O. S. Sorzano, Nat. Commun., 2020, 11, 55 CrossRef CAS PubMed.
- Y. Z. Tan, P. R. Baldwin, J. H. Davis, J. R. Williamson, C. S. Potter, B. Carragher and D. Lyumkis, Nat. Methods, 2017, 14, 793–796 CrossRef CAS PubMed.
- C. O. S. Sorzano, J. Vargas, J. M. de la Rosa-Trevín, A. Zaldívar-Peraza, J. Otón, V. Abrishami, I. Foche, R. Marabini, G. Caffarena and J. M. Carazo, Proc. Intl. Work-Conference on Bioinformatics and Biomedical Engineering, IWBBIO, 2014, p. 950 Search PubMed.
- J. Méndez, E. Garduño, J. M. Carazo and C. O. S. Sorzano, J. Struct. Biol., 2021, 213, 107771 CrossRef PubMed.
- J. Vargas, J. Otón, R. Marabini, J. M. Carazo and C. O. S. Sorzano, Sci. Rep., 2016, 6, 21626 CrossRef CAS PubMed.
- J. Vargas, R. Melero, J. Gómez-Blanco, J. M. Carazo and C. O. S. Sorzano, Sci. Rep., 2017, 7, 6307 CrossRef CAS PubMed.
- B. Heymann, AIMS Biophys., 2015, 2, 21–35 Search PubMed.
- K. Naydenova and C. J. Russo, Nat. Commun., 2017, 8, 629 CrossRef.
- P. R. Baldwin and D. Lyumkis, Prog. Biophys. Mol. Biol., 2020, 150, 160–183 CrossRef CAS.
- R. Sanchez-Garcia, J. Segura, D. Maluenda, C. O. S. Sorzano and J. M. Carazo, J. Struct. Biol., 2020, 210, 107498 CrossRef PubMed.
- G. Pintilie and W. Chiu, Acta Crystallogr., Sect. D: Struct. Biol., 2021, 77, 1142–1152 CrossRef CAS PubMed.
- E. Ramírez-Aportela, D. Maluenda, Y. C. Fonseca, P. Conesa, R. Marabini, J. B. Heymann, J. M. Carazo and C. O. S. Sorzano, Nat. Commun., 2021, 12, 42 CrossRef PubMed.
- M. A. Herzik, J. S. Fraser and G. C. Lander, Structure, 2019, 27, 344–358 CrossRef CAS PubMed.
- J. L. Vilas, J. Vargas, M. Martínez, E. Ramírez-Aportela, R. Melero, A. Jiménez-Moreno, E. Garduño, P. Conesa, R. Marabini, D. Maluenda, J. M. Carazo and C. O. S. Sorzano, J. Struct. Biol., 2020, 209, 107447 CrossRef CAS PubMed.
- P. V. Afonine, B. P. Klaholz, N. W. Moriarty, B. K. Poon, O. V. Sobolev, T. C. Terwilliger, P. D. Adams and A. Urzhumtsev, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 814–840 CrossRef CAS.
- B. A. Barad, N. Echols, R. Y.-R. Wang, Y. Cheng, F. DiMaio, P. D. Adams and J. S. Fraser, Nat. Methods, 2015, 12, 943–946 CrossRef CAS PubMed.
- G. Terashi, X. Wang, S. R. M. V. Subramaniya, J. J. G. Tesmer and D. Kihara, 2022, submitted, https://colab.research.google.com/drive/1Q-Dj42QjVO8TCOLXMQBJlvm1zInxPkOu?usp=sharing.
- C. J. Williams, J. J. Headd, N. W. Moriarty, M. G. Prisant, L. L. Videau, L. N. Deis, V. Verma, D. A. Keedy, B. J. Hintze, V. B. Chen, S. Jain, S. M. Lewis, W. B. Arendall, J. Snoeyink, P. D. Adams, S. C. Lovell, J. S. Richardson and D. C. Richardson, Protein Sci., 2018, 27, 293–315 CrossRef CAS.
- T. Cragnolini, H. Sahota, A. P. Joseph, A. Sweeney, S. Malhotra, D. Vasishtan and M. Topf, Acta Crystallogr., Sect. D: Struct. Biol., 2021, 77, 41–47 CrossRef CAS PubMed.
- J. M. de la Rosa-Trevín, A. Quintana, L. Del Cano, A. Zaldívar, I. Foche, J. Gutiérrez, J. Gómez-Blanco, J. Burguet-Castell, J. Cuenca-Alba, V. Abrishami, J. Vargas, J. Otón, G. Sharov, J. L. Vilas, J. Navas, P. Conesa, M. Kazemi, R. Marabini, C. O. S. Sorzano and J. M. Carazo, J. Struct. Biol., 2016, 195, 93–99 CrossRef PubMed.
- M. Sinnott, S. Malhotra, M. S. Madhusudhan, K. Thalassinos and M. Topf, Structure, 2020, 28, 1061–1070 CrossRef CAS PubMed.
- A. Jiménez, S. Jonic, T. Majtner, J. Oón, J. L. Vilas, D. Maluenda, J. Mota, E. Ramírez-Aportela, M. Martínez, Y. Rancel, J. Segura, R. Sánchez-García, R. Melero, L. Del Caño, P. Conesa, L. Skjaerven, R. Marabini, J. M. Carazo and C. O. S. Sorzano, Bioinformatics, 2019, 35, 2427–2433 CrossRef PubMed.
- R. Henderson, S. Chen, J. Z. Chen, N. Grigorieff, L. A. Passmore, L. Ciccarelli, J. L. Rubinstein, R. A. Crowther, P. L. Stewart and P. B. Rosenthal, J. Mol. Biol., 2011, 413, 1028–1046 CrossRef CAS PubMed.
- T. Kato, F. Makino, T. Nakane, N. Terahara, T. Kaneko, Y. Shimizu, S. Motoki, I. Ishikawa, K. Yonekura and K. Namba, Microsc. Microanal., 2019, 25, 998–999 CrossRef.
- R. Melero, C. O. S. Sorzano, B. Foster, J. L. Vilas, M. Martínez, R. Marabini, E. Ramírez-Aportela, R. Sanchez-Garcia, D. Herreros, L. Del Caño, P. Losana, Y. Fonseca-Reyna, P. Conesa, D. Wrapp, P. Chacón, J. S. McLellan, H. D. Tagare and J. M. Carazo, IUCrJ, 2020, 7, 1059–1069 CrossRef CAS PubMed.
- L. Yurkovetskiy, X. Wang, K. E. Pascal, C. Tomkins-Tinch, T. P. Nyalile, Y. Wang, A. Baum, W. E. Diehl, A. Dauphin, C. Carbone, K. Veinotte, S. B. Egri, S. F. Schaffner, J. E. Lemieux, J. B. Munro, A. Rafique, A. Barve, P. C. Sabeti, C. A. Kyratsous, N. V. Dudkina, K. Shen and J. Luban, Cell, 2020, 183, 739–751 CrossRef CAS PubMed.
- S. M.-C. Gobeil, K. Janowska, S. McDowell, K. Mansouri, R. Parks, K. Manne, V. Stalls, M. F. Kopp, R. Henderson, R. J. Edwards, B. F. Haynes and P. Acharya, Cell Rep., 2021, 34, 108630 CrossRef CAS PubMed.
- K. M. Yip, N. Fischer, E. Paknia, A. Chari and H. Stark, Nature, 2020, 587, 157–161 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2fd00059h |
| This journal is © The Royal Society of Chemistry 2022 |