 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gently does it!: in situ preparation of alkali metal–solid electrolyte interfaces for photoelectron spectroscopy†
Joshua S.
Gibson
 ab,
Sudarshan
Narayanan
ab,
Jack E. N.
Swallow
ab,
Pardeep
Kumar-Thakur
c,
Mauro
Pasta
ab,
Tien-Lin
Lee
c and
Robert S.
Weatherup
*ab
ab,
Sudarshan
Narayanan
ab,
Jack E. N.
Swallow
ab,
Pardeep
Kumar-Thakur
c,
Mauro
Pasta
ab,
Tien-Lin
Lee
c and
Robert S.
Weatherup
*ab
aDepartment of Materials, University of Oxford, Parks Road, Oxford, OX1 3PH, UK. E-mail: robert.weatherup@materials.ox.ac.uk
bThe Faraday Institution, Quad One, Harwell Science and Innovation Campus, Didcot, OX11 0RA, UK
cDiamond Light Source, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
First published on 26th January 2022
Abstract
The key charge transfer processes in electrochemical energy storage devices occur at electrode–electrolyte interfaces, which are typically buried, making it challenging to access their interfacial chemistry. In the case of Li-ion batteries, metallic Li electrodes hold promise for increasing energy and power densities and, when used in conjunction with solid electrolytes, the adverse safety implications associated with dendrite formation in organic liquid electrolytes can potentially be overcome. To better understand the stability of solid electrolytes when in contact with alkali metals and the reactions that occur, here we consider the deposition of thin (∼10 nm) alkali metal films onto solid electrolyte surfaces, where the metal is thin enough that X-ray photoelectron spectroscopy can probe the buried electrode–electrolyte interface. We highlight the importance of in situ alkali metal deposition by assessing the contaminant species that are present after glovebox handling and the use of ‘inert’ transfer devices. Consequently, we compare and contrast three available methods for in situ alkali-metal deposition; Li sputter deposition, Li evaporation, and Li plating induced by e− flood-gun irradiation. Studies on both a sulphide solid electrolyte (Li6PS5Cl), and a single-layer graphene probe surface reveal that the more energetic Li deposition methods, such as sputtering, can induce surface damage and interfacial mixing that are not seen with thermal evaporation. This indicates that the appropriate selection of the Li deposition method for in situ studies is required to observe representative behaviour, and the results of previous studies involving energetic deposition may warrant further evaluation.
Introduction
Batteries with improved energy densities, power densities, and cycling lifetimes are needed to increase the range and fast-charging capabilities of electric vehicles, and lower their lifetime cost.1,2 Alkali metal anodes offer very high theoretical specific capacities (3860 mA h g−1 for Li),1,3–5 however the propensity for filament/dendrite formation during cyclic metal stripping/plating renders them unsafe when used with the flammable liquid electrolytes found in conventional Li-ion batteries.6,7 The use of a solid electrolyte (SE) as the ion (e.g. Li+, Na+, Mg2+etc.) conducting medium avoids flammable organic electrolytes and can mechanically suppress dendrite formation, making the commercial realisation of solid-state batteries (SSBs) a promising prospect.1,8 Amongst the alkali-metal SSB systems, Li metal anodes have the highest theoretical capacity and lowest electrode potential,5 and the improvement in energy density this offers has made them the focus of many SSB studies to date.1–3,9 Given the highly reducing character of Li metal, its reaction with the battery electrolyte to form a stable interfacial layer, or a solid electrolyte interphase (SEI), is of vital importance to the extended battery cycling performance. Many studies have thus focussed on attempting to understand the nature of the reactions that proceed, to inform future battery development.6,10–15 The exact nature of the interaction layers formed, and therefore the reaction mechanisms, are dependent upon the electrolyte used. For example, LLZO (Li7La3Zr2O12) garnets have been suggested to form oxygen deficient layers,4,16 sulphide glasses to form lithium sulphides and phosphides,17,18 while lithium phosphorus oxynitride (LiPON) can react to form phosphides and nitrides.19,20 Argyrodite-type SEs, in particular the chloride variety Li6PS5Cl, have attracted much attention given their potentially high Li-ion conductivities (10−3 to 10−2 S cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21), and relative ease of preparation, e.g. through the cold-pressing of commercially available powders. Despite high Li-ion conductivities and successful cycling having been experimentally observed,22,23 degradation of the SE when in contact with Li metal leads to poor long-term performance.24,25 Elucidating the chemical processes occurring at such interfaces and the nature of the SEI formed remains a significant challenge, with the SEI referred to as the “most important and least understood” element within Li-ion batteries.26
21), and relative ease of preparation, e.g. through the cold-pressing of commercially available powders. Despite high Li-ion conductivities and successful cycling having been experimentally observed,22,23 degradation of the SE when in contact with Li metal leads to poor long-term performance.24,25 Elucidating the chemical processes occurring at such interfaces and the nature of the SEI formed remains a significant challenge, with the SEI referred to as the “most important and least understood” element within Li-ion batteries.26
SSB performance, and the formation of the associated SEI, requires good physical contact between Li and the SE. Methods previously employed to achieve this include pressing Li metal into the solid electrolyte surface27 and dropping it on as a molten liquid.28 To obtain chemically specific information about these buried interfaces with surface sensitive techniques, such as X-ray photoelectron spectroscopy (XPS),18,20,29 post-mortem studies are usually performed, where the Li electrode must subsequently be peeled off. However, this risks mechanical damage to the interfacial layers and can allow undesired side reactions to occur by exposure to trace contaminants within glovebox atmospheres.27,30,31 A recent systematic study suggested that Li metal from different suppliers may have different passivation layers, also affecting the interfacial chemistry.32 Alternatively, thin Li films can be deposited onto the solid electrolyte surface through evaporation,20,33,34 sputtering11,35 or even plating, driven by electron beam irradiation10,34 to ensure good physical contact. For characterisation, these can then either be controllably etched away14 or, if thin-enough, non-destructive probing directly through them may be possible.10,35 Given the high reactivity of the species involved and variety in Li preparation methods available, it is important to establish what impact these deposition processes may have on the reaction species observed.
Thin films can be deposited in situ, or in external deposition systems, prior to surface analysis. External deposition systems generally allow greater convenience, without the necessity for compatibility with the analysis system or the challenges of manipulation under vacuum conditions (at least for glovebox-based systems). The main disadvantage is the fact that even in a glovebox there is the possibility that surfaces react with contaminant species. A glovebox operating at 1 bar with sensors reading <0.1 ppm for water or oxygen could still present a partial pressure up to 10−4 mbar, and other contaminants such as CO2 are often not controlled for. Transfer from the glovebox to the ultra-high vacuum (UHV) conditions typical of surface analysis systems is then required, often with an O-ring sealed transfer vessel, which over time will permit some leakage of air, leading to unwanted surface reactions. Such issues were investigated recently when looking at the surface passivation of Li metal,32 concluding that the storage and processing history of the Li metal surface is very important for the interpretation of the surface analysis.
A number of approaches have been reported for in situ Li deposition in commercial UHV surface analysis systems. Sputtering of a Li metal chip with an Ar+ beam, leading to deposition of the sputtered material onto an adjacent material surface11,35 can be achieved relatively easily with a simple modification to the sample holder and using an existing sputter gun used for surface cleaning or depth-profiling. There is, however, the necessity for inert transfer, and consequent surface contamination of the Li metal target that will be sputtered onto the surface. A low energy e− beam (the e− charge neutraliser or flood-gun) has also been used to drive surface Li plating, fed from a reservoir of Li metal in contact with another region of the SE.10,34 However, it has been observed for carbon nanotube and graphene surfaces that the irradiation of sample surfaces with an electron beam leads to increased deposition of amorphous carbon,36,37 which could affect the observed surface chemistry. In situ evaporation often requires more extensive modification of the analysis system. In the case of alkali metals, an adjacent deposition chamber may be preferable to prevent contamination of the analysis chamber with these reasonably high vapour pressure metals38 during evaporation. The benefit of such a system, however, is that transfer of the sample from the deposition chamber to the analysis chamber occurs under UHV (≈10−8 mbar), without significant exposure to contaminants.
In this work, we use lab- and synchrotron-based XPS to compare the interfacial chemistry of Li deposited onto Li6PS5Cl by thermal evaporation, sputtering, and e− flood-gun driven Li plating. This reveals that in situ deposition is critical to avoiding the conversion of the Li film to hydroxide and carbonate species by reaction with environmental contaminants present in gloveboxes and ‘inert’ transfer devices. We discuss the different methods for Li deposition and assess the benefits and disadvantages that each may have on the interfacial chemistry seen during SEI formation. In particular, we use Raman spectroscopy to reveal the surface damage induced by each deposition method. This reveals that the use of more energetic deposition methods, such as sputtering, can lead to significant surface damage and interfacial mixing that is not seen with thermal evaporation, and is not expected to be representative of the SEI formed through the mechanical contacting of Li to the SE. These findings will inform improved experimental procedures for alkali metal deposition and handling when performing surface analysis, contributing to an enhanced understanding of the interfacial stability of solid electrolytes and alkali metals. Furthermore, these results indicate that previous studies of SE systems where Li deposition by sputtering or charge neutralisation using Ar+ ions may require revisiting.
Experimental
Argyrodite (Li6PS5Cl) solid electrolyte pellets were prepared by cold pressing commercial powders (∼10 μm grain size, purchased from Ampcera™, USA) into 8 mm disks of 700–750 μm thickness by the application of 500 MPa of pressure using a hydraulic press. The handling of Li6PS5Cl powders and pellets was performed in an Ar-filled glovebox with H2O and O2 contamination below the detectable limits (both <0.1 ppm, partial pressure <1 × 10−4 mbar). Pellets were used as pressed, with no further surface treatment. Single-layer graphene (SLG), used as a model surface for evaluating the surface damage related to different lithium deposition methods, was grown on copper foil substrates by chemical vapour deposition,39 and transferred to 300 nm SiO2 on Si(100) substrates (Pi-Kem Ltd) using the well-established wet transfer method.40,41 In brief, the SLG on copper was supported by polymethylmethacrylate (PMMA), after which the sample was floated on top of a sodium persulphate etchant solution (0.21 M aqueous Na2S2O8, Acros Organics >98%) for a minimum of 5 hours. The etched samples were transferred to distilled water to rinse away residual etchant, and then the PMMA coated SLG was picked up onto SiO2(300 nm)/Si substrates and dried. The PMMA support was then removed by soaking in acetone (>99%, Sigma-Aldrich) for 1 hour, followed by rinsing in isopropyl alcohol (>99.5%, Sigma-Aldrich).For in situ Li deposition, either by sputtering or driven by the e− flood-gun, bare Li6PS5Cl pellets were loaded into an ULVAC Phi Versaprobe III XPS system from glovebox environments using an inert transfer device (ULVAC Phi GmbH). In situ sputter deposition of Li has already been described in detail elsewhere.11,35 Briefly, metallic Li foil (750 μm thick, purchased from Sigma-Aldrich®), was mechanically cleaned by scraping in a glovebox, affixed vertically to an L-shaped in situ metal deposition stage (ULVAC Phi GmbH) and inertly transferred to the XPS system. The Li is then bombarded with an Ar+ ion beam (4 keV, 3 × 3 mm2 raster, 2.8 μA beam current or 2 keV, 3 × 3 mm2 raster, 1 μA beam current), to sputter Li onto samples placed horizontally beneath the Li foil (see the schematic in Fig. 1). The Li deposition rate is determined based on the attenuation of Cu 3p peaks during calibration experiments on bare Cu foil samples, assuming a continuous layer and using the Beer–Lambert law with calculated values for the inelastic mean free path.42,43 The attenuation of the Cu 3s and Cu 2p peaks corroborated the deposition rate obtained using the Cu 3p peaks. Li-deposition using a “virtual electrode” created by an e− flood-gun has been previously demonstrated10,34 and involves irradiating the surface of the Li6PS5Cl pellet with low-energy e−, such as those generated by an e− flood-gun within the XPS system. The e−-flooded electrolyte surface draws Li+ ions from a Li metal reservoir on the underside of the pellet, leading to plating of Li metal on the irradiated surface. During in situ Li deposition, surfaces are studied by X-ray photoelectron spectroscopy (XPS) using a water-cooled monochromatic Al Kα source (hν = 1486.6 eV, 15 kV anode voltage, 25 W beam power). Charge compensation with the in-built e− flood-gun or low-energy Ar+ ion source was avoided for all samples, except the Li sputtered Li6PS5Cl surface, or cases where the use of the charge neutraliser was being studied (such as the “virtual electrode” measurements where only the e− flood-gun is used, or the evaluation of SLG damage). In these cases the electron neutraliser was set to 1.35 V bias to accelerate thelow energy electrons, while the argon ion neutraliser used a beam voltage of 110 V and a−100 V float to provide ions with energies of approximately 10 eV.71 Core level spectra were collected with a pass energy of 55 eV and a step size of 0.05 eV/step (giving a Ag 3d5/2 peak with a fwhm of 0.69 eV). The analysis chamber pressure was maintained below 5 × 10−9 mbar during measurement. Data analysis was performed using the CasaXPS software package.44
For in situ Li evaporation, a purpose built UHV chamber was attached to the I09 end station at the Diamond Light Source synchrotron facility (Didcot, UK), where the bare Li6PS5Cl pellets could be loaded from a glovebox environment using an inert transfer device sealed under Ar. The inert transfer device was constructed entirely from UHV compatible components and sealed with a gate-valve with a Viton® seal. Li was deposited by thermal evaporation from a boron nitride crucible heated by a tantalum wire basket. The deposition rate was calibrated for the chamber geometry to be 4 nm min−1, with a chamber pressure below 1 × 10−6 mbar during evaporation. The samples could then be transferred to the analysis chamber whilst maintaining a base pressure below 5 × 10−10 mbar. Once in the analysis chamber, the surfaces were probed with hard X-ray photoelectron spectroscopy (HAXPES), using photon energies of 2.2 keV and 6.6 keV, and soft X-ray photoelectron spectroscopy (SOXPES), such that the kinetic energy of core level photoelectrons was ∼315 eV (i.e. photon energies of 845 eV, 600 eV, 510 eV, 475 eV, 450 eV and 370 eV for O 1s, C 1s, Cl 2p, S 2p, P 2p and Li 1s, respectively). The use of different photon energies provides depth-resolved data for the studied surfaces with photoelectrons having inelastic mean free paths through Li metal of approximately 1.1 nm, 6.0–7.5 nm and 18.5–19.8 nm for the SOXPES, 2.2 keV and 6.6 keV photons, respectively.43 To minimise beam damage (as monitored using the S 2p core level) the undulator was detuned to provide an X-ray intensity of ∼I0/10 for HAXPES and ∼I0/100 for SOXPES, where I0 is the full beam intensity. The defocussed X-ray beam gave dimensions of ∼300 (vertical) × 300 (horizontal) μm and impinged upon the sample surface at an angle of 15° to enhance the photoelectron signal and yield a ∼300 × 1200 μm illuminated area. The energy distribution curves (EDCs) of the photoelectrons leaving the surface were measured with a concentric hemispherical analyser (VG Scienta EW4000 10 keV, lens acceptance angle ±28°). The hemispherical analyser was operated with a pass energy of 200 eV for HAXPES and 100 eV for SOXPES.
Raman spectra of the SLG samples were collected using a Renishaw inVia confocal Raman microscope, with a 532 nm wavelength laser having a maximum power of 50 mW. Following Li deposition samples were transferred to air to allow Raman spectra to be collected. Raman data was collected over the range 1000 to 3000 cm−1 using 1% of the maximum power to avoid laser-induced damage to the SLG. Data was recorded using a 1800 mm−1 grating, with 30 acquisitions per scan and a 10 s exposure time. Data was analysed and fitted using Lorentzian line shapes in the Igor Pro software package.45
Results and discussion
Fig. 1 shows XPS core level spectra for the C 1s, O 1s, S 2p and Li 1s regions for different stages of Li deposition on Li6PS5Cl solid electrolytes, with the peak area of each region normalised to unity (except S 2p (iii), due to its lack of discernible peaks). The top row of spectra, Fig. 1(i), show measurements of the as-loaded bare Li6PS5Cl. The O 1s region contains a broad peak at ∼531.6 eV, consistent with the presence of Li2CO3 and LiOH.10 The C 1s region shows a contribution at ∼288.6 eV, again attributable to Li2CO3,4,46 as well as adventitious C at ∼284.8 eV. Changes in the relative intensities of these components are apparent with increasing photon energy, which corresponds to increased information depth, as faster photoelectrons can escape from deeper below the surface. Between SOXPES and 2.2 keV HAXPES, the ratio of Li2CO3 to adventitious C peaks in the C 1s region is seen to increase, suggesting that the top surface is covered in adventitious C (e.g. hydrocarbons), with a carbonate layer beneath it. For the 6.6 keV HAXPES, the signal to noise ratio decreases significantly due to both the lower photoionisation cross section at higher energies and the increased probing depth of the faster photoelectrons, resulting in the peak attributable to Li2CO3 no longer being well-resolved. These variations in peak intensity with depth are thus consistent with a Li2CO3 layer on the outermost surface of the pellet that is covered with adventitious C. | ||
| Fig. 1 Synchrotron HAXPES (hν = 2.2 keV, black and 6.6 keV, green) and SOXPES (kinetic energy = 315 eV, red) data for (i) as-loaded bare Li6PS5Cl argyrodite, (ii) 20 nm Li evaporated on Li6PS5Cl in situ and (iii) 20 nm of Li on Li6PS5Cl after having been transferred in an inert Ar atmosphere. Data has been normalised so that the total peak area is the same, with the exception of the S 2p data in (iii), and the binding energy is calibrated such that the Cl 2p3/2 sits at 198.5 eV (see ESI, Fig. S1†). | ||
The S 2p core level of the as-loaded Li6PS5Cl shows a strong doublet with a S 2p3/2 component at ∼161.4 eV, attributable to Li6PS5Cl, whilst a much weaker doublet with S 2p3/2 at ∼160.1 eV is assigned to S2− or Li2S.12 The S 2p shows a slightly lower contribution of Li2S relative to Li6PS5Cl in the SOXPES measurements, which indicates some depletion of S, perhaps related to the formation of Li2CO3 at the surface. Nevertheless the fact that the Li6PS5Cl doublet is well-resolved even in the SOXPES data indicates that the carbonate layer is relatively thin, with a uniform carbonate layer of >3 nm thickness expected to fully attenuate any Li6PS5Cl signal at this energy.42 The Li 1s core level shows only a single peak for the bare pellet at ∼55.4 eV, which is consistent with Li6PS5Cl, however likely also includes contributions from Li2CO3, LiHCO3 and LiOH, which are expected to have overlapping peaks at ∼55 eV for the charge referencing used here.4,46 The Cl 2p and the P 2p spectra (Fig. S1†) do not reveal any significant variations in peak shape with photon energy. These results thus confirm the presence of surface-contaminants on the Li6PS5Cl, particularly Li2CO3 and adventitious C, which likely arise from glovebox storage and processing. Since the powders are stored in a glovebox prior to room-temperature pellet pressing, it is likely that each of the micron-sized grains is coated with these contaminants. Although the presence of Li2CO3 has not been rigorously assigned in Li6PS5Cl previously, we note that it has been observed on many other lithium containing solid electrolytes and has been implicated in increasing interfacial resistance (e.g. LLZO garnets,4 (LiPON)20 and Li2S–P2S5 sulphides47).
Cl 2p was chosen as an internal reference for charge correction (Cl 2p3/2 = 198.5 eV) as prior reports have indicated that Cl 2p peak distortion is not observed with Li metal deposition.12 Recoil effects in photoelectron emission, where the outgoing photoelectron transfers momentum to the photoemitting nucleus, has been shown to be well approximated by the use of the momentum equation ΔE = EKEm/M where ΔE is the recoil shift, EKE is the photoelectron kinetic energy, m is the photoelectron mass and M the nuclear mass. Recoil effects become significant for high photoelectron kinetic energies, or for lighter elements, causing an increase in the experimentally observed binding energy.48,49 It follows that for the photoelectron energies produced in this work, the Cl 2p is expected to experience recoil shifts of 5 meV, 31 meV and 99 meV, whilst the Li 1s would experience shifts of 25 meV, 168 meV and 513 meV for the SOXPES, 2.2 keV and 6.6 keV photon energies, respectively. Recoil effects are not significant enough (below 0.1 eV) to affect most of the photoelectron peaks nor the choice of Cl 2p for alignment. They will, however, cause significant BE increases in the Li 1s, C 1s and O 1s of ∼0.5 eV, 0.3 eV and 0.2 eV for the 6.6 keV (green) data, respectively.
The spectra in the second row, Fig. 1(ii), were acquired from an Li6PS5Cl pellet following in situ evaporation of ∼20 nm of Li onto the surface. The O 1s region shows an oxygen peak at ∼528.4 eV, assigned to Li2O from the reaction of the deposited Li with trace oxygen,10,11 in addition to the carbonate/hydroxide peak at ∼531.2 eV that is already present in the as-loaded bare pellet. The C 1s data show a single peak due to adsorbed hydrocarbons at ∼284.8 eV, with the lack of a significant contribution from any carbonate species suggesting that the high BE oxygen peak is primarily attributable to lithium hydroxide, presumably due to reaction with background water. The enhanced hydroxide to oxide peak ratio in the SOXPES data compared to the 2.2 keV and 6.6 keV HAXPES data indicates that the hydroxide species are at the outermost surface of the deposited Li, while the formation of Li2O by reaction with residual oxygen in the UHV system is more uniform throughout. The S 2p spectra show a change in the ratio of Li6PS5Cl to Li2S, compared to the as-loaded bare pellet, with Li2S now being dominant following Li evaporation. The Li 1s spectra show a large component at ∼53.7 eV, indicative of interfacial products, such as Li2S, Li2P and Li2O,10 A metallic Li component is observed at ∼52.2 eV, once recoil effects have been accounted for, and is more prominent at greater probing depths (higher photon energies).
The deposition of Li significantly attenuates the Li6PS5Cl species observed in the Cl 2p, S 2p, P 2p and Li 1s core levels, as well as the Li2CO3 and adventitious C seen at the surface of the bare pellet in the C 1s region. The depth resolution available from the different photon energies shows that the topmost surface layers (SOXPES data, red) appear to contain a greater proportion of Li2S, following from the reaction of mobile S2− species with metallic Li.12 With increasing probing depth, the ratio of Li6PS5Cl to Li2S can be seen to increase, starting from complete attenuation of the Li6PS5Cl component in the SOXPES data, and increasing when moving from 2.2 keV to 6.6 keV photons (see Fig. 1(ii), S 2p). This behaviour is consistent with a continuous film deposition, suggesting that the Li metal has adequately wet the pellet surface.
The third row of Fig. 1(iii) shows EDCs collected from a surface with 20 nm Li evaporated onto Li6PS5Cl, transferred inertly via a vacuum suitcase. Samples were sealed under an Ar glovebox environment and spent <6 hours in this environment prior to transfer into UHV for measurement. The O 1s spectra show a broad peak at ∼531.3 eV, attributable to Li2CO3, LiHCO3 and/or LiOH.10 The C 1s data show the presence of the adsorbed hydrocarbons at ∼284.8 eV, as well as two higher binding energy peaks at ∼290.3 eV and ∼288.9 eV assigned to LiHCO3 and Li2CO3, respectively.4 With increasing probing depth (increasing photon energy) the high binding energy peak in the O 1s can be observed to decrease in binding energy from ∼531.6 eV to 531.2 eV, which is not attributable to recoil effects, and a peak corresponding to Li2O at 528.4 eV is seen to appear and grow in intensity. This is consistent with the outermost surface being composed predominantly of the higher binding energy carbonate species34 on top of LiOH and Li2O. These results support the idea of gaseous CO2, presumably present in trace amounts in the “inert” glovebox atmosphere, reacting with the surface LiOH that is present following UHV deposition.14 Additionally, the C 1s shows adventitious hydrocarbon adsorption on the outer surface and an increasing ratio of LiHCO3 to Li2CO3 when the probing depth increases. The reaction of CO2 and LiOH to form Li2CO3 releases water;14 we thus tentatively suggest that at the buried LiOH/Li2CO3 interface this results in LiHCO3 formation, whilst closer to the surface we hypothesise that H2O diffuses out into the low water content “glovebox” atmosphere used for inert transfer, leading to more Li2CO3 towards the outer surface.
The S 2p spectra show complete attenuation of the Li2S and Li6PS5Cl species in the SOXPES data, indicating that the carbonate passivation layer has a thickness of >3 nm, as discussed earlier. The Li 1s data show a high binding energy peak at ∼54.9 eV, consistent with the presence of Li2CO3, LiHCO3 and LiOH. The Li 1s HAXPES data show a shift towards the lower BE side, when we consider the respective ∼0.15 eV and ∼0.5 eV recoil shifts for 2.2 and 6.6 keV data, indicating the presence of other reacted Li species (e.g. Li2O and Li2S seen in Fig. 1(ii)) beneath the carbonate layer. However, there is a lack of any metallic Li peak for all probing depths. The 2.2 keV S 2p HAXPES shows the emergence of Li2S at 160.0 eV, closest to the carbonate layer, with pristine Li6PS5Cl observed beneath this (162.4 eV), as shown by the increased Li6PS5Cl to Li2S ratio at greater probing depths. The absence of Li2S at the outermost surface could be due to either the fact that the Li2CO3 is more thermodynamically stable50 or that Li2S hydrolyses to H2S(g), highlighting the importance of ensuring CO2-free and H2O-free processing of materials used in alkali-metal batteries. We note that the observed behaviour may not be solely attributable to glovebox exposure but may also arise from contaminants present during ‘inert’ transfer. However, the impact of trace glovebox contaminants is further highlighted when storing thin (<50 nm) Li films deposited onto Li6PS5Cl and Cu foils, which are initially visible to the naked eye as reflective, silvery coatings, but within the course of a few hours disappear as they form less opaque products. On a shorter timescale (<10 min), samples transferred directly from the evaporation chamber to the lab-based XPS in a Viton® sealed vessel also failed to exhibit any metallic Li (see Fig. S3†), indicating the formation of carbonate layers that exceed 10 nm in thickness (based on an inelastic mean free path of 3.5 nm in Li2CO3).
The data presented in Fig. 1 highlight the importance of in situ deposition with UHV transfer for studying Li metal interfaces with the Li6PS5Cl surface, showing that even performing an ‘inert’ transfer can radically alter the species observed at the surface. We expect this to be equally applicable to other alkali metals, solid electrolytes and many other electrode materials. Although Li is the most reducing of the alkali metals (−3.04 V vs. the standard hydrogen electrode), it is the least chemically reactive51,52 and, thus, in situ deposition is expected to be similarly important, if not more so, for studies of electrode interfaces with metallic Na53 and K.54
In Fig. 2, we compare several methods for the in situ deposition of alkali metals on the Li6PS5Cl electrolyte: evaporation,34 Ar+ sputtering of Li metal11,34,35 and e− flood-gun irradiation.10,34 The Li6PS5Cl prior to any Li metal deposition (grey) shows the expected peaks in the Cl 2p, S 2p, P 2p, and Li 1s regions, along with common contaminants in the C 1s and O 1s regions, as discussed earlier. Comparison of the data for approximately 10 nm thick Li layers formed by e− irradiation (blue), Li sputtering (red) and evaporation (green) confirms the presence of metallic Li in all cases, as indicated by a shoulder at 52.2 eV in Fig. 2(f). We note that for the evaporated data, sample transfer involved passing through the sample introduction chamber, which is sealed by a fast access port with a polymer seal. Consequently, the Li surface is briefly exposed to higher pressures (still <5 × 10−7 mbar) which may contribute to slightly increased oxygen contamination. The Cl 2p core level spectra all show the same main features attributable to the Li6PS5Cl, but with reduced signal to noise ratio, consistent with attenuation by the Li overlayers. The S 2p core level spectra show that Li2S is the dominant S environment following Li deposition in all cases. Peak fitting finds that the attenuation of the S 2p and Cl 2p components related to Li6PS5Cl are similar for the evaporated Li surface (both with intensities of around 6% of the bare surface), indicating that the deposition method forms a continuous Li layer. In the case of the Li sputtered and e− flood-gun irradiated surfaces, following Li deposition the S 2p intensity related to Li6PS5Cl is reduced to a greater extent than the corresponding Cl 2p peak (5% vs. 14% and 8% vs. 26% of bare intensity, respectively). Although this may indicate a less continuous Li layer, the large differences more likely correspond to a loss of surface sulphur species, or an increase in surface chlorine concentration during these deposition methods.
 | ||
| Fig. 2 Photoelectron spectra collected from surfaces following the in situ preparation of Li metal surfaces on Li6PS5Cl argyrodite (grey) in a lab-based XPS through e− flood-gun irradiation (blue), Li metal sputtering (red) and thermal evaporation (green). Core level spectra are shown for (a) O 1s, (b) C 1s, (c) Cl 2p, (d) S 2p, (e) P 2p and (f) Li 1s. Data has been normalised to have equal peak areas and offset for clarity, with the binding energies calibrated such that the Cl 2p3/2 sits at 198.5 eV. | ||
Significantly, clear differences are observed in the O and C spectra (Fig. 2(a) and (b), respectively) depending on the Li deposition technique used. Whilst all spectra show some Li2O associated with the reaction between deposited Li and residual oxygen following Li metal sputtering, peaks related to Li2CO3, LiOH and adventitious C are no longer observed. For the other deposition methods, these peaks are still present, albeit with reduced signal to noise ratios following attenuation by the Li overlayer. These results therefore suggest that Li metal sputtering is causing surface adsorbed species to be removed by bombardment with the energetic Li, with similar spectral changes observed following Ar+ sputtering of both Li6PS5Cl and Li surfaces (see ESI, Fig. S2†). Differences between deposition methods are also observed in the P 2p core level, Fig. 2(e), where there appear to be different interfacial species dependent on the deposition method. The electron beam growth of Li shows the presence of Li3P at ∼126 eV, as seen in other work,12,20 whilst evaporation shows some evidence of intermediate species LixP, where the phosphorous is not fully reduced (see ESI, Fig. S1†). For Li metal sputtered surfaces, none of these interfacial species are apparent for the 10 nm thick Li film, in contrast to the findings of a previous study for 14 nm Li films,12 although we note that sample cooling to −85 °C was employed in that work to suppress the removal of volatile species, whereas deposition herein was performed at ∼20 °C. It is observed that for thinner sputtered Li films (generated by shorter sputtering times), LixP and Li3P species are initially observed to form, but then decrease in intensity relative to the Li6PS5Cl P 2p peak with increasing sputtering time. Given the sputter-induced removal of O and C containing species during Li sputtering, we attribute the loss of generated phosphorous species to a similar process. We further note that bombardment of the surface layers by energetic Li during sputtering is also likely to cause a degree of interfacial mixing, affecting the observed reaction species and surface layering.
Previous work comparing Li metal deposition methods on Li7La3Zr2O12 (LLZO) garnets concluded that the deposition method chosen could induce different chemical reactions at the Li metal–LLZO interface.34 In the case of the magnetron sputtering of Li, it has been argued that energetic Ar+ ions can give the Li metal atoms additional energy comparable to that of an electrochemical overpotential, thus enabling kinetic barriers for the formation of an oxygen deficient interphase (ODI) at Li–LLZO interfaces to be overcome. Li deposition by e-beam evaporation was observed to not form an ODI, until the Li layer is bombarded with Ar+ ions. “Virtual electrode” e− flood-gun deposition was also observed to form an ODI. We suggest that these differences can be described in terms of the near-surface damage induced by the different Li metal deposition methods, rather than the kinetic stability of the reaction species. The observed results in both ref. 34 and the present study are consistent with sputtering Li metal onto a surface being more energetic and, consequently, more damaging than Li deposition by evaporation. Studies have found that the energy of sputtered atoms following impact by an incident Ar+ beam can be up to 50 eV, with the most likely energy being ∼5 eV,55,56 whilst for the thermal evaporation of Li at the pressures used herein, this is <0.1 eV, according to the ideal gas law, which is consistent with the experimental observations.57,58 Electrical impedance spectroscopy studies have shown that typical activation energies for lithium plating on different electrode materials (including graphite, lithium nickel cobalt aluminate and lithium iron phosphate) can vary from ∼0.50 eV to 0.75 eV, which is an order of magnitude lower than the energy imparted by atoms during lithium sputtering.59–61 For e-beam evaporation with higher evaporation rates, the vapour becomes “less ideal”, leading to higher energy Li deposition, but this is still expected to be well below that of sputtering.
To further investigate the differences between these Li deposition techniques and the energetics involved, SLG grown by chemical vapour deposition (CVD) was transferred to SiO2(300 nm)/Si substrates and used as a model surface probe. The use of SLG allows for a sensitive and quantitative measure of the defect density generated by each of the lithium deposition methods, through the use of Raman spectroscopy, on a surface whose atomic structure is not expected to be disrupted simply by contact with Li metal,62 providing a probe of the physical damage induced by the deposition process in isolation. The Raman spectrum of the as-transferred SLG is shown in Fig. 3 (black line) with D (∼1348 cm−1), G (∼1585 cm−1) and 2D (∼2685 cm−1) peaks apparent. The measured spectrum is characteristic of SLG produced by CVD, with a 2D/G ratio of ∼6.1 ± 1.3 and a 2D peak that is well fitted by a single Lorentzian with a fwhm of ∼28 ± 4 cm−1. The D/G peak ratio provides a sensitive measure of defect density63–65 that can be used to compare the extent of physical damage induced by the different deposition processes, and it is ∼0.1 for the as-transferred SLG, consistent with a reasonably low defect density. The similarity of the green (0.6 nm Li evaporation) and black data in Fig. 3 shows that the thermal evaporation of Li does not appear to induce graphene defects, with Table 1 showing that the area ratios for D:G are very similar for both surfaces (∼0.1).
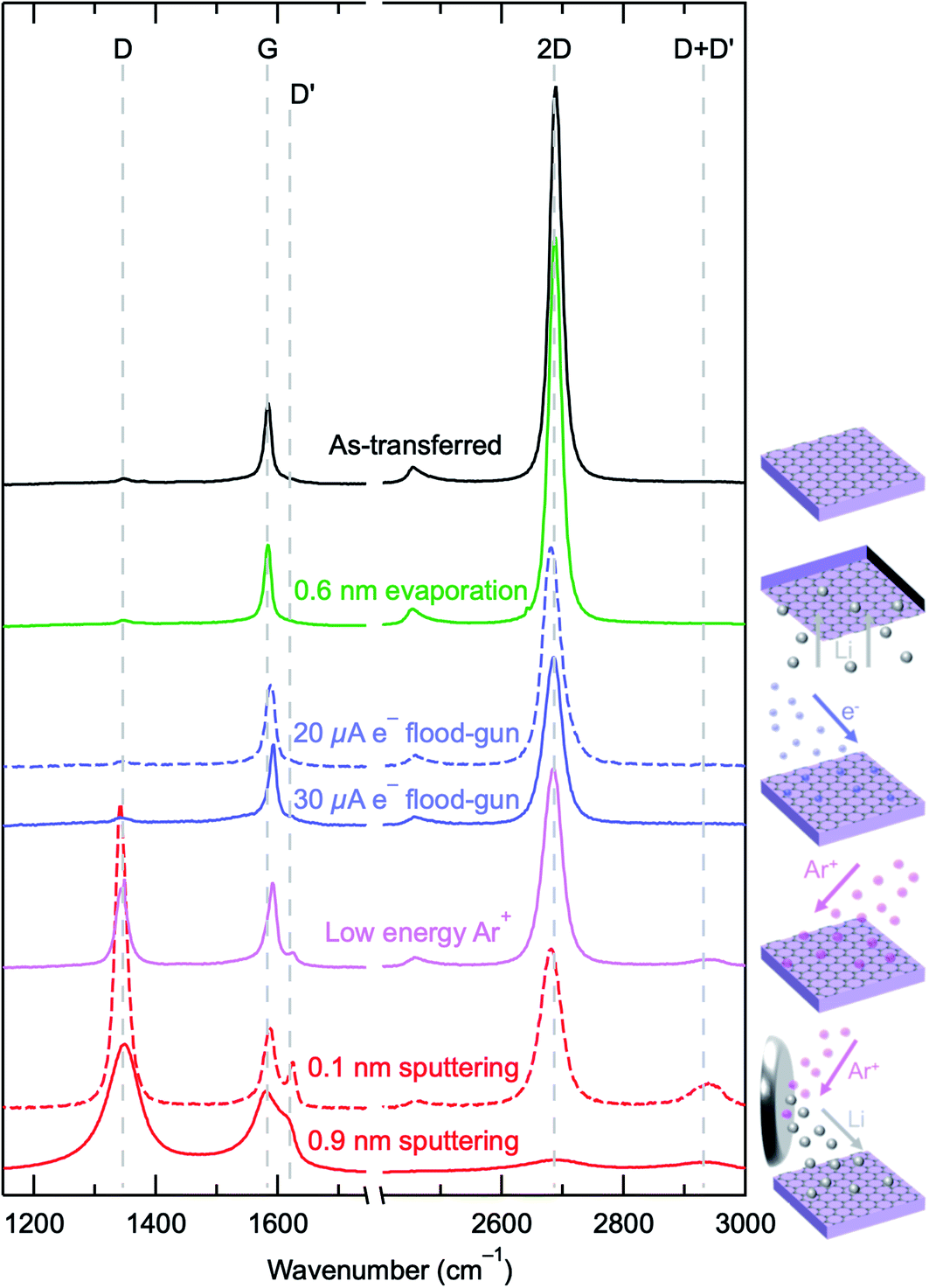 | ||
| Fig. 3 Raman spectra of SLG on SiO2(300 nm)/Si for different Li deposition methods. Data are shown for as-transferred SLG (black), 0.6 nm evaporated lithium (green), after irradiation with the e− flood-gun (blue), after irradiation with the low energy Ar+ (pink) and after sputtering with Li metal (red). All Raman spectra were collected in air, with spectra normalised to the height of the G peak. | ||
Electron irradiated SLG surfaces, with conditions that mimic the “virtual electrode” growth of Li used in Fig. 2 (4.5 hours of irradiation) for different e− flood-gun currents (20 μA, dashed blue, 30 μA solid blue), exhibit 2- to 3-fold increases in the D:G ratio compared to that of the as-transferred SLG (Table 1). High energy electrons (>100 keV) in transmission and secondary electron microscopy are known to create defects in SLG through “knock-on” processes, where an electron knocks out a carbon atom from the graphene lattice.66 However the energies used herein are well below the “knock-on” threshold.67 Other work has suggested that lower energy electrons can cause damage via radiolysis or localised heating.66 It is also observed that the low energies used in e− flood-guns (<20 eV) can cause damage on organic substrate surfaces through C–C and C–H bond scission, especially in high fluences.68,69 Additionally the low energy electron beam may induce amorphous carbon deposition through the radiolysis of residual hydrocarbons, as is routinely observed for higher energy electron beams,37,70 which has been implicated in increases in the D peak intensity for SLG surfaces.36 We therefore suggest that flood-gun irradiation may induce reactions involving adsorbed species on the SLG (e.g. polymer residues) or residual gas in the measurement chamber that leads to an increased defect concentration, either through attack of the SLG or deposition of defective carbon.
| (fwhm G)ave | (fwhm 2D)ave | (2D/G)ave | (D/G)ave | |
|---|---|---|---|---|
| As-transferred | 16.6 ± 1.4 | 27.8 ± 3.5 | 6.01 ± 1.31 | 0.096 ± 0.033 |
| 0.6 nm Li evaporation | 15.6 ± 1.4 | 28.7 ± 3.6 | 5.51 ± 1.71 | 0.103 ± 0.021 |
| 20 mA e− flood-gun | 18.5 ± 1.4 | 32.2 ± 0.5 | 4.43 ± 0.17 | 0.200 ± 0.045 |
| 30 mA e− flood-gun | 18.8 ± 0.7 | 31.7 ± 0.8 | 4.12 ± 0.67 | 0.288 ± 0.132 |
| Low energy Ar+ | 19.6 ± 0.6 | 30.3 ± 2.6 | 4.44 ± 0.25 | 1.752 ± 0.642 |
| 0.1 nm sputtering | 24.8 ± 0.5 | 37.8 ± 0.3 | 3.17 ± 0.03 | 3.431 ± 0.026 |
| 0.9 nm sputtering | 75.2 ± 11 | 186 ± 20 | 0.74 ± 0.43 | 2.644 ± 0.809 |
SLG on SiO2(300 nm)/Si was also irradiated with low energy Ar+ ions for 38 minutes, as Ar+ ion neutralisers are increasingly commonly in surface analysis instruments and often used in conjunction with e− flood-gun neutralisers for charge compensation.71 The effect of the irradiation of the graphene surface with the low energy Ar+ ions is apparent, with a clear change in the measured Raman signal (pink line, Fig. 3), now with a much more significant D peak, as well as the emergence of a D′ peak at ∼1620 cm−1 and a D + D′ peak at ∼2933 cm−1, which indicate the formation of highly defective SLG.67 These Ar+ ions have a much larger mass than the electrons (73000 times), which facilitates a more effective momentum transfer to the carbon atoms within the SLG, despite the low ion energies (∼10 eV herein), causing significant damage. Indeed SLG damage following irradiation by Ar+ ions with energies as low as 5 eV has been observed.64 It is calculated that to cause knock-on damage, an Ar+ ion needs an energy of >31 eV, however Stone–Wales defects can be generated with Ar+ ions of 5 to 10 eV.36,64 Our observations are consistent with previous reports of surface damage by the Ar+ ion and e− flood-gun71 that raises concerns around some of the observations and interpretation of data when these have been used.11,12,35
Li metal deposition resulting from Ar+ sputtering of a Li metal target, can also be observed to damage the SLG probe surface (red data, Fig. 3). In both cases, the Li deposition took place over 38 minutes (the same as the Ar+ ion irradiation), with the 0.1 nm sputtered Li data (dashed red) measured from a position on the surface that is further away from the Li metal target, whilst the 0.9 nm sputtered Li data (solid red) was closer. It is clearly seen that significant damage to the SLG has occurred, with the D peak dominating the signal and, for the 0.1 nm sputtered Li, a well-defined D′ peak is apparent. For the 0.9 nm sputtered Li surface, the extent of the damage is so great that the peaks have broadened significantly and the 2D peak has also reduced in size, indicating that the SLG has been heavily amorphized. This conclusion is supported by the observation that the D:G ratio for the 0.9 nm Li sputtered surface is smaller than that for the 0.1 nm sputtered Li case, even though a much higher defect density is expected, as established through prior observations of the amorphization trajectory of SLG using Raman spectroscopy.72 It is clear from our findings with Li evaporation that the presence of Li itself does not damage the SLG surface. However, when Li sputtering is performed, the more energetic Li atoms appear to cause significant physical damage to the SLG.
We note that our results and interpretation thereof, i.e. sputter deposition of Li causes damage near surface layers of the substrate (whether it is solid electrolyte, SLG, or likely many other materials), are not consistent with the explanations provided by Connell et al.34 In their study, the differences observed between LLZO–Li interfaces produced by Li metal sputtering and e-beam evaporation are attributed to kinetic stability, requiring an activation energy to be overcome for an oxygen deficient interphase to be produced. Changes in the oxidation state of Zr were observed for surfaces where Li was sputtered on top, but not those where Li was evaporated. Furthermore, it was concluded that the same changes seen for the Li sputtered surface could be observed in the e-beam evaporated surface, if the evaporated Li film was subsequently bombarded with Ar+ ions. They argue that the Ar+ bombardment provides the Li atoms with sufficient kinetic energy to overcome the chemical activation energy barrier. We instead suggest that Li deposition by sputtering or Ar+ ion bombardment after Li evaporation transfers significant kinetic energy not just to the Li atoms, but also the other species close to the surface, hence promoting surface damage. Despite their conclusions that the lack of Zr oxidation for an Ar+ sputtered surface in the absence of Li means that changes are not solely due to sputter damage, in our view it is highly likely that sputter damage, in conjunction with the highly reducing Li environment, leads to the observed oxidation state changes in the Zr. We argue that their findings point to evidence of a physical damage process due to momentum transfer from the Li metal during sputtering, rather than the resulting sputtered Li simply having an increased kinetic energy to overcome a chemical activation energy barrier, as they propose. Consequently, the interpretation of data regarding the formation of interfacial species where either sputter deposition or activation by Ar+ bombardment has been employed must be undertaken with care to ensure that the observed effects are not significantly influenced by the energy transfer to the substrate species, potentially causing sputter damage and surface mixing.
From the above comparison of possible Li deposition methodologies, Li evaporation appears to be the most suitable for forming a well-defined interface resembling those produced by pressing Li in contact with a solid electrolyte, and subsequent interfacial studies in the absence of induced damage effects. Using SLG as a probe surface has allowed the confirmation that Li metal sputtering causes damage through a physical process, rather than just overcoming the kinetic barrier of a chemical process. In light of this, alongside the observation of the flood-gun damage of polymers observed previously,68,69 we suggest that for in situ studies of interfacial reactions at the metal–solid electrolyte interfaces, the preparation of alkali metal layers through thermal or e-beam evaporation is beneficial in avoiding physical damage to the interfacial layers, as well as avoiding the contamination associated with glovebox-based deposition.
Conclusions
Depth resolved SOXPES and HAXPES has confirmed that Li2CO3 species are present at the surface of Li6PS5Cl argyrodite, on the outermost surface of the powder grains after glovebox storage and processing, as seen for many other Li solid electrolytes. Following the deposition of metallic Li via in situ thermal evaporation, reaction of the Li metal with trace gases leads to some level of Li2O and LiOH formation. This effect is exacerbated if using a polymer-sealed ‘inert’ transfer device of the type widely used in battery-research, as residual H2O and CO2 result in significant formation of surface carbonates and bicarbonates. Surface carbonates are observed to form preferentially to lithium phosphides and lithium sulphides when Li is deposited on Li6PS5Cl, highlighting the importance of keeping battery interfaces free from CO2. These findings indicate that in situ surface preparation is key to understanding the ongoing chemistry at these reactive interfaces.The comparison of different in situ Li metal deposition methods has highlighted that this can affect the observed interfacial species. Following Li metal sputtering, the peaks due to Li2CO3, LiOH and adventitious C cease to be observed, whilst for Li evaporation and e− flood-gun growth, these environments are simply attenuated. These results are consistent with the Li metal being deposited by sputtering causing the surface adsorbed species to be removed by the energetic incident Li. Additionally differences are observed in the P 2p photoelectron peak, with the e− flood-gun growth exhibiting Li3P, whilst incomplete phosphorus reduction can be observed for thermal evaporation. Sputtering of Li initially forms reduced phosphorous species (LixP, Li3P), but increased Li sputter deposition then leads to their eventual removal. This interpretation is supported by studies using SLG as a probe surface. Thermal evaporation of Li metal does not appear to induce defects in a SLG surface, as measured by the D/G ratio compared to that of as-transferred CVD graphene. Long term irradiation (over 4 hours) with an e− flood-gun is seen to induce at least a two-fold increase in the D/G ratio, indicative of slight damage to the SLG or the deposition of defective carbon, whilst Li metal sputtering causes the carbon to amorphize almost completely after depositing as little as 1 nm of Li. Additionally, irradiation by a low energy Ar+ ion neutraliser was seen to cause significant damage to SLG, suggesting that care must be taken when choosing charge compensation methods for delicate and reactive surfaces. Caution is therefore needed when interpreting existing data that has been collected utilising low energy Ar+ charge compensation, or where in situ Li sputtering has been performed.
Our findings are consistent with the idea that Li metal sputtering causes physical damage, in contrast to previous work,34 which suggested that it simply overcomes kinetic barriers associated with the formation of oxygen deficient interphases at the Li metal–LLZO interface. Following our systematic study of Li deposition methods, we recommend that when studying energy materials interfaces involving alkali metals, future studies should consider deposition methods that are “in situ” and non-damaging, such as thermal evaporation.
Author contributions
RSW, MP and T-LL were responsible for funding acquisition and supervision of the work. JSG and RSW were responsible for the conceptualisation of the work. JSG, SN and JENS collected the experimental data, JSG and SN analysed the data and JSG wrote the original draft. PKT and T-LL assisted in sample preparation and data acquisition at Diamond Light Source. All authors contributed to preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge support from the Faraday Institution through Characterisation (FIRG011, FIRG020), SOLBAT (FIRG026) and LiSTAR (FIRG014) projects, as well as the Henry Royce Institute (through the UK Engineering and Physical Science Research Council grant EP/R010145/1) for capital equipment. Additionally we would like to acknowledge Diamond Light Source for synchrotron beam time (SI 25807-2, SI 25807-3).References
- M. Pasta, D. Armstrong, Z. L. Brown, J. Bu, M. R. Castell, P. Chen, A. Cocks, S. A. Corr, E. J. Cussen, E. Darnbrough, V. Deshpande, C. Doerrer, M. S. Dyer, H. El-Shinawi, N. Fleck, P. Grant, G. L. Gregory, C. Grovenor, L. J. Hardwick, J. T. S. Irvine, H. J. Lee, G. Li, E. Liberti, I. McClelland, C. Monroe, P. D. Nellist, P. R. Shearing, E. Shoko, W. Song, D. S. Jolly, C. I. Thomas, S. J. Turrell, M. Vestli, C. K. Williams, Y. Zhou and P. G. Bruce, 2020 roadmap on solid-state batteries, Journal of Physics: Energy, 2020, 2(3), 032008 CAS.
- S. Randau, D. A. Weber, O. Kötz, R. Koerver, P. Braun, A. Weber, E. Ivers-Tiffée, T. Adermann, J. Kulisch, W. G. Zeier, F. H. Richter and J. Janek, Benchmarking the performance of all-solid-state lithium batteries, Nat. Energy, 2020, 5(3), 259–270 CrossRef CAS.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Lithium metal anodes for rechargeable batteries, Energy Environ. Sci., 2014, 7(2), 513–537 RSC.
- R. H. Brugge, F. M. Pesci, A. Cavallaro, C. Sole, M. A. Isaacs, G. Kerherve, R. S. Weatherup and A. Aguadero, The origin of chemical inhomogeneity in garnet electrolytes and its impact on the electrochemical performance, J. Mater. Chem. A, 2020, 8(28), 14265–14276 RSC.
- H. Wang, D. Yu, C. Kuang, L. Cheng, W. Li, X. Feng, Z. Zhang, X. Zhang and Y. Zhang, Alkali Metal Anodes for Rechargeable Batteries, Chem, 2019, 5(2), 313–338 CAS.
- J. Liu, X. Gao, G. O. Hartley, G. J. Rees, C. Gong, F. H. Richter, J. Janek, Y. Xia, A. W. Robertson, L. R. Johnson and P. G. Bruce, The Interface between Li6.5La3Zr1.5Ta0.5O12 and Liquid Electrolyte, Joule, 2020, 4(1), 101–108 CrossRef CAS.
- M. Armand and J. M. Tarascon, Building better batteries, Nature, 2008, 451(7179), 652–657 CrossRef CAS PubMed.
- Y. Zhan, W. Zhang, B. Lei, H. Liu and W. Li, Recent Development of Mg Ion Solid Electrolyte, Front. Chem., 2020, 8(125), 1–7 Search PubMed.
- J. B. Goodenough and Y. Kim, Challenges for Rechargeable Li Batteries, Chem. Mater., 2010, 22(3), 587–603 CrossRef CAS.
- K. N. Wood, K. X. Steirer, S. E. Hafner, C. M. Ban, S. Santhanagopalan, S. H. Lee and G. Teeter, Operando X-ray photoelectron spectroscopy of solid electrolyte interphase formation and evolution in Li2S–P2S5 solid-state electrolytes, Nat. Commun., 2018, 9, 10 CrossRef PubMed.
- S. Wenzel, D. A. Weber, T. Leichtweiss, M. R. Busche, J. Sann and J. Janek, Interphase formation and degradation of charge transfer kinetics between a lithium metal anode and highly crystalline Li7P3S11 solid electrolyte, Solid State Ionics, 2016, 286, 24–33 CrossRef CAS.
- S. Wenzel, S. J. Sedlmaier, C. Dietrich, W. G. Zeier and J. Janek, Interfacial reactivity and interphase growth of argyrodite solid electrolytes at lithium metal electrodes, Solid State Ionics, 2018, 318, 102–112 CrossRef CAS.
- A. Schwobel, R. Hausbrand and W. Jaegermann, Interface reactions between LiPON and lithium studied by in situ X-ray photoemission, Solid State Ionics, 2015, 273, 51–54 CrossRef.
- A. Sharafi, S. H. Yu, M. Naguib, M. Lee, C. Ma, H. M. Meyer, J. Nanda, M. F. Chi, D. J. Siegel and J. Sakamoto, Impact of air exposure and surface chemistry on Li–Li7La3Zr2O12 interfacial resistance, J. Mater. Chem. A, 2017, 5(26), 13475–13487 RSC.
- S. Narayanan, J. S. Gibson, J. Aspinall, R. S. Weatherup and M. Pasta, In Situ and Operando Characterisation of Li Metal–Solid Electrolyte Interfaces, Solid State Mater. Sci., 2022, 26(2), 100978 CrossRef CAS.
- A. Sharafi, H. M. Meyer, J. Nanda, J. Wolfenstine and J. Sakamoto, Characterizing the Li–Li7La3Zr2O12 interface stability and kinetics as a function of temperature and current density, J. Power Sources, 2016, 302, 135–139 CrossRef CAS.
- H. Muramatsu, A. Hayashi, T. Ohtomo, S. Hama and M. Tatsumisago, Structural change of Li2S–P2S5 sulfide solid electrolytes in the atmosphere, Solid State Ionics, 2011, 182(1), 116–119 CrossRef CAS.
- C. Dietrich, R. Koerver, M. W. Gaultois, G. Kieslich, G. Cibin, J. Janek and W. G. Zeier, Spectroscopic characterization of lithium thiophosphates by XPS and XAS – a model to help monitor interfacial reactions in all-solid-state batteries, Phys. Chem. Chem. Phys., 2018, 20(30), 20088–20095 RSC.
- Y.-C. Lu, E. J. Crumlin, G. M. Veith, J. R. Harding, E. Mutoro, L. Baggetto, N. J. Dudney, Z. Liu and Y. Shao-Horn, In Situ Ambient Pressure X-ray Photoelectron Spectroscopy Studies of Lithium–Oxygen Redox Reactions, Sci. Rep., 2012, 2(1), 715 CrossRef PubMed.
- A. Schwöbel, R. Hausbrand and W. Jaegermann, Interface reactions between LiPON and lithium studied by in situ X-ray photoemission, Solid State Ionics, 2015, 273, 51–54 CrossRef.
- H.-J. Deiseroth, S.-T. Kong, H. Eckert, J. Vannahme, C. Reiner, T. Zaiß and M. Schlosser, Li6PS5X: A Class of Crystalline Li-Rich Solids With an Unusually High Li+ Mobility, Angew. Chem., Int. Ed., 2008, 47(4), 755–758 CrossRef CAS PubMed.
- M. Chen and S. Adams, High performance all-solid-state lithium/sulfur batteries using lithium argyrodite electrolyte, J. Solid State Electrochem., 2015, 19(3), 697–702 CrossRef CAS.
- S. Yubuchi, S. Teragawa, K. Aso, K. Tadanaga, A. Hayashi and M. Tatsumisago, Preparation of high lithium-ion conducting Li6PS5Cl solid electrolyte from ethanol solution for all-solid-state lithium batteries, J. Power Sources, 2015, 293, 941–945 CrossRef CAS.
- T. Cheng, B. V. Merinov, S. Morozov and W. A. Goddard, Quantum Mechanics Reactive Dynamics Study of Solid Li-Electrode/Li6PS5Cl–Electrolyte Interface, ACS Energy Lett., 2017, 2(6), 1454–1459 CrossRef CAS.
- C. Yu, L. van Eijck, S. Ganapathy and M. Wagemaker, Synthesis, structure and electrochemical performance of the argyrodite Li6PS5Cl solid electrolyte for Li-ion solid state batteries, Electrochim. Acta, 2016, 215, 93–99 CrossRef CAS.
- M. Winter, The Solid Electrolyte Interphase – The Most Important and the Least Understood Solid Electrolyte in Rechargeable Li Batteries, Z. Phys. Chem., 2009, 223(10–11), 1395–1406 CrossRef CAS.
- R. Inada, S. Yasuda, H. Hosokawa, M. Saito, T. Tojo and Y. Sakurai, Formation and Stability of Interface between Garnet-Type Ta-doped Li7La3Zr2O12 Solid Electrolyte and Lithium Metal Electrode, Batteries, 2018, 4(2), 26 CrossRef.
- C. Wang, Y. Gong, B. Liu, K. Fu, Y. Yao, E. Hitz, Y. Li, J. Dai, S. Xu, W. Luo, E. D. Wachsman and L. Hu, Conformal, Nanoscale ZnO Surface Modification of Garnet-Based Solid-State Electrolyte for Lithium Metal Anodes, Nano Lett., 2017, 17(1), 565–571 CrossRef CAS PubMed.
- Surface Analysis by Auger and X-Ray Photoelectron Spectroscopy, D. Briggs and J. T Grant, IM Publications, Chichester, 2003 Search PubMed.
- T. Waldmann, A. Iturrondobeitia, M. Kasper, N. Ghanbari, F. Aguesse, E. Bekaert, L. Daniel, S. Genies, I. J. Gordon, M. W. Löble, E. De Vito and M. Wohlfahrt-Mehrens, Review—Post-Mortem Analysis of Aged Lithium-Ion Batteries: Disassembly Methodology and Physico-Chemical Analysis Techniques, J. Electrochem. Soc., 2016, 163(10), A2149–A2164 CrossRef CAS.
- S. Wang, X. Xu, X. Zhang, C. Xin, B. Xu, L. Li, Y.-H. Lin, Y. Shen, B. Li and C.-W. Nan, High-performance Li6PS5Cl-based all-solid-state lithium-ion batteries, J. Mater. Chem. A, 2019, 7(31), 18612–18618 RSC.
- S.-K. Otto, Y. Moryson, T. Krauskopf, K. Peppler, J. Sann, J. Janek and A. Henss, In-Depth Characterization of Lithium-Metal Surfaces with XPS and ToF-SIMS: Toward Better Understanding of the Passivation Layer, Chem. Mater., 2021, 33(3), 859–867 CrossRef CAS.
- Y. Zhu, J. G. Connell, S. Tepavcevic, P. Zapol, R. Garcia-Mendez, N. J. Taylor, J. Sakamoto, B. J. Ingram, L. A. Curtiss, J. W. Freeland, D. D. Fong and N. M. Markovic, Dopant-Dependent Stability of Garnet Solid Electrolyte Interfaces with Lithium Metal, Adv. Energy Mater., 2019, 9(12), 1803440 CrossRef.
- J. G. Connell, T. Fuchs, H. Hartmann, T. Krauskopf, Y. Zhu, J. Sann, R. Garcia-Mendez, J. Sakamoto, S. Tepavcevic and J. Janek, Kinetic versus Thermodynamic Stability of LLZO in Contact with Lithium Metal, Chem. Mater., 2020, 32(23), 10207–10215 CrossRef CAS.
- S. Wenzel, T. Leichtweiss, D. Kruger, J. Sann and J. Janek, Interphase formation on lithium solid electrolytes-an in situ approach to study interfacial reactions by photoelectron spectroscopy, Solid State Ionics, 2015, 278, 98–105 CrossRef CAS.
- X. Shen, H. Wang and T. Yu, How do the electron beam writing and metal deposition affect the properties of graphene during device fabrication?, Nanoscale, 2013, 5(8), 3352–3358 RSC.
- J. C. Meyer, C. O. Girit, M. F. Crommie and A. Zettl, Hydrocarbon lithography on graphene membranes, Appl. Phys. Lett., 2008, 92(12), 123110 CrossRef.
- R. E. Honig, Vapor Pressure Data for the More Common Elements, RCA Rev., 1957, 18, 195–204 CAS.
- A. Cabrero-Vilatela, R. S. Weatherup, P. Braeuninger-Weimer, S. Caneva and S. Hofmann, Towards a general growth model for graphene CVD on transition metal catalysts, Nanoscale, 2016, 8(4), 2149–2158 RSC.
- R. S. Weatherup, 2D Material Membranes for Operando Atmospheric Pressure Photoelectron Spectroscopy, Top. Catal., 2018, 61(20), 2085–2102 CrossRef CAS.
- X. Li, Y. Zhu, W. Cai, M. Borysiak, B. Han, D. Chen, R. D. Piner, L. Colombo and R. S. Ruoff, Transfer of Large-Area Graphene Films for High-Performance Transparent Conductive Electrodes, Nano Lett., 2009, 9(12), 4359–4363 CrossRef CAS PubMed.
- A. Jablonski and C. J. Powell, NIST Standard Reference Database 71: NIST Electron Inelastic-Mean-Free-Path Database Version 1.2, 2021 Search PubMed.
- S. Tanuma, C. J. Powell and D. R. Penn, Calculations of electron inelastic mean free paths. IX. Data for 41 elemental solids over the 50 eV to 30 keV range, Surf. Interface Anal., 2011, 43(3), 689–713 CrossRef CAS.
- N. Fairley, CasaXPS, Version 2.3.17, 2021 Search PubMed.
- WaveMetrics, Igor Pro, Version 6.37, 2021.
- C. F. Mallinson, J. E. Castle and J. F. Watts, Analysis of the Li KLL Auger Transition on Freshly Exposed Lithium and Lithium Surface Oxide by AES, Surf. Sci. Spectra, 2013, 20(1), 113–127 CrossRef CAS.
- X. Wu, C. Villevieille, P. Novák and M. El Kazzi, Insights into the chemical and electronic interface evolution of Li4Ti5O12 cycled in Li2S–P2S5 enabled by operando X-ray photoelectron spectroscopy, J. Mater. Chem. A, 2020, 8(10), 5138–5146 RSC.
- S. Suga, A. Sekiyama, H. Fujiwara, Y. Nakatsu, T. Miyamachi, S. Imada, P. Baltzer, S. Niitaka, H. Takagi, K. Yoshimura, M. Yabashi, K. Tamasaku, A. Higashiya and T. Ishikawa, Do all nuclei recoil on photoemission in compounds?, New J. Phys., 2009, 11(7), 073025 CrossRef.
- S. Suga and A. Sekiyama, High energy photoelectron spectroscopy of correlated electron systems and recoil effects in photoelectron emission, Eur. Phys. J. Spec. Top., 2009, 169, 227–235 CrossRef.
- M. W. Chase Jr, NIST-JANAF Thermochemical Table, Fourth Edition, J. Phys. Chem. Ref. Data, Monogr., 1998, 9, 1–1951 Search PubMed.
- M. M. Markowitz, Alkali metal–water reactions, J. Chem. Educ., 1963, 40(12), 633 CrossRef CAS.
- N. V. Sidgwick, The Chemical Elements and Their Compounds, Oxford University Press, London, 1950, vol. I Search PubMed.
- M.-C. Bay, M. Wang, R. Grissa, M. V. F. Heinz, J. Sakamoto and C. Battaglia, Sodium Plating from Na-β′′-Alumina Ceramics at Room Temperature, Paving the Way for Fast-Charging All-Solid-State Batteries, Adv. Energy Mater., 2020, 10(3), 1902899 CrossRef CAS.
- R. Rajagopalan, Y. Tang, X. Ji, C. Jia and H. Wang, Advancements and Challenges in Potassium Ion Batteries: A Comprehensive Review, Adv. Funct. Mater., 2020, 30(12), 1909486 CrossRef CAS.
- E. Franke, H. Neumann, M. Zeuner, W. Frank and F. Bigl, Particle energy and angle distributions in ion beam sputtering, Surf. Coat. Technol., 1997, 97(1–3), 90–96 CrossRef CAS.
- K. K. Kammara, R. Kumar and F. S. Donbosco, Reconsideration of metal surface sputtering due to bombardment of high-energy argon ion particles: a molecular dynamics study, Computational Particle Mechanics, 2016, 3(1), 3–13 CrossRef.
- T. Asano, N. Uetake and K. Suzuki, Mean Atomic Velocities of Uranium, Titanium and Copper during Electron Beam Evaporation, J. Nucl. Sci. Technol., 1992, 29(12), 1194–1200 CrossRef CAS.
- K. Reichelt and X. Jiang, The preparation of thin films by physical vapour deposition methods, Thin Solid Films, 1990, 191(1), 91–126 CrossRef.
- M. Koseoglou, E. Tsioumas, D. Ferentinou, N. Jabbour, D. Papagiannis and C. Mademlis, Lithium plating detection using dynamic electrochemical impedance spectroscopy in lithium-ion batteries, J. Power Sources, 2021, 512, 230508 CrossRef CAS.
- F. Katzer and M. A. Danzer, Analysis and detection of lithium deposition after fast charging of lithium-ion batteries by investigating the impedance relaxation, J. Power Sources, 2021, 503, 230009 CrossRef CAS.
- P. Shafiei Sabet, G. Stahl and D. U. Sauer, Non-invasive investigation of predominant processes in the impedance spectra of high energy lithium-ion batteries with nickel–cobalt–aluminum cathodes, J. Power Sources, 2020, 472, 228189 CrossRef CAS.
- L. G. Bulusheva, M. A. Kanygin, V. E. Arkhipov, K. M. Popov, Y. V. Fedoseeva, D. A. Smirnov and A. V. Okotrub, In Situ X-ray Photoelectron Spectroscopy Study of Lithium Interaction with Graphene and Nitrogen-Doped Graphene Films Produced by Chemical Vapor Deposition, J. Phys. Chem. C, 2017, 121(9), 5108–5114 CrossRef CAS.
- K. Murakami, T. Kadowaki and J.-i. Fujita, Damage and strain in single-layer graphene induced by very-low-energy electron-beam irradiation, Appl. Phys. Lett., 2013, 102(4), 043111 CrossRef.
- P. Ahlberg, F. O. L. Johansson, Z.-B. Zhang, U. Jansson, S.-L. Zhang, A. Lindblad and T. Nyberg, Defect formation in graphene during low-energy ion bombardment, APL Mater., 2016, 4(4), 046104 CrossRef.
- J.-H. Chen, W. G. Cullen, C. Jang, M. S. Fuhrer and E. D. Williams, Defect Scattering in Graphene, Phys. Rev. Lett., 2009, 102(23), 236805 CrossRef PubMed.
- R. F. Egerton, P. Li and M. Malac, Radiation damage in the TEM and SEM, Micron, 2004, 35(6), 399–409 CrossRef CAS PubMed.
- D. Teweldebrhan and A. A. Balandin, Modification of graphene properties due to electron-beam irradiation, Appl. Phys. Lett., 2009, 94(1), 013101 CrossRef.
- R. Havelund, M. P. Seah, A. G. Shard and I. S. Gilmore, Electron Flood Gun Damage Effects in 3D Secondary Ion Mass Spectrometry Imaging of Organics, J. Am. Soc. Mass Spectrom., 2014, 25(9), 1565–1571 CrossRef CAS PubMed.
- I. S. Gilmore and M. P. Seah, Electron flood gun damage in the analysis of polymers and organics in time-of-flight SIMS, Appl. Surf. Sci., 2002, 187(1–2), 89–100 CrossRef CAS.
- H. W. Conru and P. C. Laberge, Oil contamination with the SEM operated in the spot scanning mode, J. Phys. E: Sci. Instrum., 1975, 8(2), 136–138 CrossRef CAS.
- D. R. Baer, K. Artyushkova, H. Cohen, C. D. Easton, M. Engelhard, T. R. Gengenbach, G. Greczynski, P. Mack, D. J. Morgan and A. Roberts, XPS guide: charge neutralization and binding energy referencing for insulating samples, J. Vac. Sci. Technol., A, 2020, 38(3), 031204 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Interpretation of Raman spectra of disordered and amorphous carbon, Phys. Rev. B, 2000, 61(20), 14095–14107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1fd00118c |
| This journal is © The Royal Society of Chemistry 2022 |