 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The rise of electrochemical NAPXPS operated in the soft X-ray regime exemplified by the oxygen evolution reaction on IrOx electrocatalysts
Juan Jesús
Velasco Vélez
 *ab,
Denis
Bernsmeier
c,
Travis E.
Jones
b,
Patrick
Zeller
bd,
Emilia
Carbonio
d,
Cheng-Hao
Chuang
e,
Lorenz J.
Falling
bf,
Verena
Streibel
b,
Rik V.
Mom
bg,
Adnan
Hammud
b,
Michael
Hävecker
ab,
Rosa
Arrigo
h,
Eugen
Stotz
b,
Thomas
Lunkenbein
b,
Axel
Knop-Gericke
ab,
Ralph
Krähnert
c and
Robert
Schlögl
ab
*ab,
Denis
Bernsmeier
c,
Travis E.
Jones
b,
Patrick
Zeller
bd,
Emilia
Carbonio
d,
Cheng-Hao
Chuang
e,
Lorenz J.
Falling
bf,
Verena
Streibel
b,
Rik V.
Mom
bg,
Adnan
Hammud
b,
Michael
Hävecker
ab,
Rosa
Arrigo
h,
Eugen
Stotz
b,
Thomas
Lunkenbein
b,
Axel
Knop-Gericke
ab,
Ralph
Krähnert
c and
Robert
Schlögl
ab
aDepartment of Heterogeneous Reactions, Max Planck Institute for Chemical Energy Conversion, 45470 Mülheim an der Ruhr, Germany. E-mail: velasco@fhi-berlin.mpg.de
bDepartment of Inorganic Chemistry, Fritz-Haber-Institut der Max-Planck-Gesellschaft, 14195 Berlin, Germany
cDepartment of Chemistry, Chemical Engineering Division, Technical University Berlin, 10623 Berlin, Germany
dHelmholtz-Zentrum Berlin für Materialien und Energie, BESSY II, 12489 Berlin, Germany
eDepartment of Physics, Tamkang University, New Taipei City 25137, Taiwan
fMaterials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
gLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands
hSchool of Science, Engineering and Environment, University of Salford, Manchester, M5 4WT, UK
First published on 8th February 2022
Abstract
Photoelectron spectroscopy offers detailed information about the electronic structure and chemical composition of surfaces, owing to the short distance that the photoelectrons can escape from a dense medium. Unfortunately, photoelectron based spectroscopies are not directly compatible with the liquids required to investigate electrochemical processes, especially in the soft X-ray regime. To overcome this issue, different approaches based on photoelectron spectroscopy have been developed in our group over the last few years. The performance and the degree of information provided by these approaches are compared with those of the well established bulk sensitive spectroscopic approach of total fluorescence yield detection, where the surface information gained from this approach is enhanced using samples with large surface to bulk ratios. The operation of these approaches is exemplified and compared using the oxygen evolution reaction on IrOx catalysts. We found that all the approaches, if properly applied, provide similar information about surface oxygen speciation. However, using resonant photoemission spectroscopy, we were able to prove that speciation is more involved and complex than previously thought during the oxygen evolution reaction on IrOx based electrocatalysts. We found that the electrified solid–liquid interface is composed of different oxygen species, where the terminal oxygen atoms on iridium are the active species, yielding the formation of peroxo species and, finally, dioxygen as the reaction product. Thus, the oxygen–oxygen bond formation is dominated by peroxo species formation along the reaction pathway. Furthermore, the methodologies discussed here open up opportunities to investigate electrified solid–liquid interfaces in a multitude of electrochemical processes with unprecedented speciation capabilities, which are not accessible by one-dimensional X-ray spectroscopies.
1. Introduction
In this work, different approaches are used to investigate electrochemical interfaces with photoelectron spectroscopy. These approaches will be exemplified and compared using the oxygen evolution reaction (OER) on IrOx catalysts as an experimental model. The OER is a bottleneck in electrochemical energy conversion and storage, which are crucial to the solutions to the current global energy challenges.1 Electrochemical water splitting to produce “green hydrogen” as a storable and clean fuel from renewable sources of energy plays a central role in proposed solutions to global warming, creating new opportunities to replace the use of fossil fuel technologies.2,3 The electrocatalysts used in OER conditions face two challenges: corrosion and slow kinetics,4 reducing the process efficiency.5,6 The catalysts must also remain stable under the strongly oxidizing conditions required for long term industrial applications. Iridium oxide is one of the most promising electrocatalysts for the OER in acidic solutions, because it uniquely combines both of the sought after properties in the same material: low overpotential and high corrosion resistance under harsh acidic conditions and anodic polarization.7,8 Unfortunately, iridium is among the scarcest in the Earth’s crust. This scarcity either demands strategies for its efficient usage, or alternatively electrocatalyst designs based on abundant materials that can mimic the electronic properties of the active sites of iridium. The factors in the electronic structure which make iridium oxide one of the best candidates for the OER are still under debate.9 A better understanding of the iridium chemistry of the activated catalyst is necessary in order to produce electrodes based on abundant materials that are able to match iridium’s electrocatalytic performance. To achieve this goal, it is necessary to describe the electronic properties of IrOx materials under realistic operando conditions and, in particular, with the iridium in contact with liquids.An in-depth understanding of the atomistic mechanisms underlying different electrochemical processes requires recording large sets of data collected under operando conditions to obtain key information on the electrified interface.10 Desired parameters to be known include the chemical composition at the interface, chemical states of the atoms and their variation as a result of the electrochemical processes, and the structural evolution of the material. Unfortunately, analytical techniques which are able to provide such detailed interfacial information are very limited and hardly compatible with liquids,11 usually restricting researchers to ex situ/post-mortem characterizations. This limitation leads to a loss of important information, as in many cases the intermediates and electrocatalytically active species cannot be detected in a post-process analysis, leaving us blind to key aspects ruling the reaction pathway. X-ray spectroscopic techniques are able to provide the relevant information on the electronic structure in an element specific manner but real electrochemical interfaces are buried and, most of the time, are in the presence of liquid electrolytes. Because of this fact, the electrified interfaces are inaccessible directly to common surface sensitive techniques like photoelectron spectroscopy, requiring new experimental strategies for their operation under these conditions.12 Recently, different approaches have been developed, enabling the investigation of the electronic structure variation of electrodes under potentiostatic control using near ambient pressure X-ray photoelectron spectroscopy (NAPXPS),13 from the gas phase up to bulk aqueous electrolytes.11,12,14–18 Some of the most extensively used approaches entail the use of hard X-rays to allow the photoelectrons to escape through thick electrode membranes19 (a few nanometers thick) or thin film electrolytes.20 However, the use of soft X-rays for characterizing electrified interfaces presents significant advantages with respect to hard X-ray techniques, such as higher absorption cross sections, higher surface sensitivity, higher chemical sensitivity, and fine-structure resolution.15 Unfortunately, performing in situ/operando experiments with soft X-rays is technically more challenging than in the hard X-ray regime. In this work, we will show some of the surface sensitive XPS/XAS approaches that have been established in our group in the last years, which overcome the challenge of using soft X-rays for the in situ/operando characterization of working electrocatalysts. The performance of these approaches is compared with those of well established bulk sensitive approaches in liquids based on photon-in/photo-out techniques in the fluorescence yield mode21,22 in order to validate our new approaches based on photoelectron detection. In addition, the unique capabilities of photoelectron based spectroscopy techniques will be discussed in terms of the enhanced speciation, ascribed to the capability to resolve spectra measurements with two different energies simultaneously, binding and excitation energies, using resonant spectroscopy.
In photoelectron spectroscopy, an electron is ejected almost instantaneously using a photon energy higher than the threshold of ionization. However, tuning the photon energy in the vicinity of a core level near the absorption threshold yields a selective enhancement of photoemission from the absorbing atom through the phenomenon of resonance. Thus, the excited state, with a hole in the core level, will rapidly decay by the emission of an electron, as in the Auger decay process, which is shown schematically in Fig. 1. This phenomenon is in the nature of the resonant photoemission spectroscopy (ResPES) operation. Owing to the fact that the excitation energy is close to a core level absorption threshold, the resonant process selects specific chemical elements and is sensitive to particular chemical states, meaning that the information is particularly informative about the electronic structure of the selected atoms. Note that ResPES requires a tunable source of photon energy, as is provided in a synchrotron facility, in order to be able to select the photon excitation energy close to the absorption threshold of the selected core level. This capability simultaneously yields valuable information of the occupied (PDOS) and unoccupied orbital states (XAS). Using ResPES, we find in this work that the oxygen species taking part in the electrochemical oxidation of water on iridium electrodes are more involved and complex that previously stated. It is found that the formation of single coordinated oxygen atoms to iridium (named μ1-O) yields the formation of peroxo species (named μ1-OO), which produce dioxygen (O2) as a product of the oxygen evolution reaction. This result is of prime importance and represents a significant advance in the complete understanding of this complex reaction.
 | ||
| Fig. 1 Relevant electronic transitions for the radiative relaxation fluorescent photon emission, non-radiative Auger electron emission, photoelectron emission and resonant photoemission spectroscopy (ResPES). | ||
2. Can a bulk sensitive spectroscopy technique provide relevant information on an electrified interface?
First, we investigate the OER on an IrOx based catalyst using X-ray absorption spectroscopy (XAS), which is an element specific technique that provides information about the electronic structure of the unoccupied orbitals of a target element of interest.23 XAS is one of most widely used X-ray based spectroscopic techniques to investigate the evolution of the electronic structure of materials, such as energy materials. Note that XAS requires the use of a tunable X-ray source for scanning the energy through the absorption edge of interest. Hence, the measurements are usually performed in a synchrotron light facility. When a sample is illuminated by X-ray radiation of an appropriate energy, the interaction of this radiation with the matter is dominated by photon absorption and electron excitation from an occupied core level to unoccupied energy levels. This process yields the formation of an excited electron and a core hole where the subsequent relaxation process is through photon light emission in total fluorescence yield (TFY) or by the emission of an Auger electron and secondary electrons originating in the cascade processes. The emitted electrons can be collected as the Auger electron yield (AEY) or as the total electron yield (TEY), and also as the partial electron yield (PEY) if the electrons are selected by kinetic energy. A schematic drawing with the different relevant electronic transitions and emission processes is shown in Fig. 1. The AEY requires an electron energy analyzer for its collection, meanwhile the TEY can be collected in different ways, such as collecting the emitted electrons using a current collector electrode (Faraday cup), or grounding the sample and collecting the drain current produced as a consequence of the emission of secondary electrons (TEY-ground). The TFY signal is considered bulk sensitive, owing to the large penetration depth of the photons in the μm range in the soft X-ray regime. Meanwhile, TEY is accepted to be a surface sensitive technique due to the short total escape depth of the photoelectrons (elastic and inelastic).24,25 While useful for probing catalytic reactions, this surface sensitivity makes the collection of spectra especially challenging in the soft X-ray regime in the presence of liquids, as the photoelectrons and secondary electrons are strongly scattered in dense media.10In order to accomplish operando investigations during OER with photoelectron-based techniques, we developed a experimental setup facilitating such measurements. The setup is as follows: the electrochemical cell (EC-cell) body used for this work is made of polyether ether ketone (PEEK), which is chemically inert for most of the liquid electrolytes and electrically insulating. The cell has exchangeable working electrodes (see Fig. 2A), allowing for the use of different types of membranes (as we will discuss later). A Pt wire and a Ag/AgCl electrode (FLEXREF, sourced from WPI Florida, USA) are used as the counter and reference electrodes, respectively. The pulse-free, continuous electrolyte flow is ensured via a Perimax16 peristaltic pump from SPETEC. The total fluorescence yield (TFY) signal is acquired using a photodiode (AXUV100 Opto Diode Corp) located in the main vacuum chamber. Note that, in addition, the TEY signal can be collected using a Faraday cup or in the XPS/AEY signal using the hemispherical analyzer. More details about this setup can be found elsewhere.26 In Fig. 2, the schematic representation of the different membrane assemblies used in this work are shown. Thus, Fig. 2 shows the different approaches based on (B) a catalyst deposited on a SiNx membrane, (C) a thin film deposited on an ionomeric membrane, (D) a membrane based on free standing graphene decorated with the catalyst and (E) a catalyst on an ionomeric membrane capped with a graphene membrane.
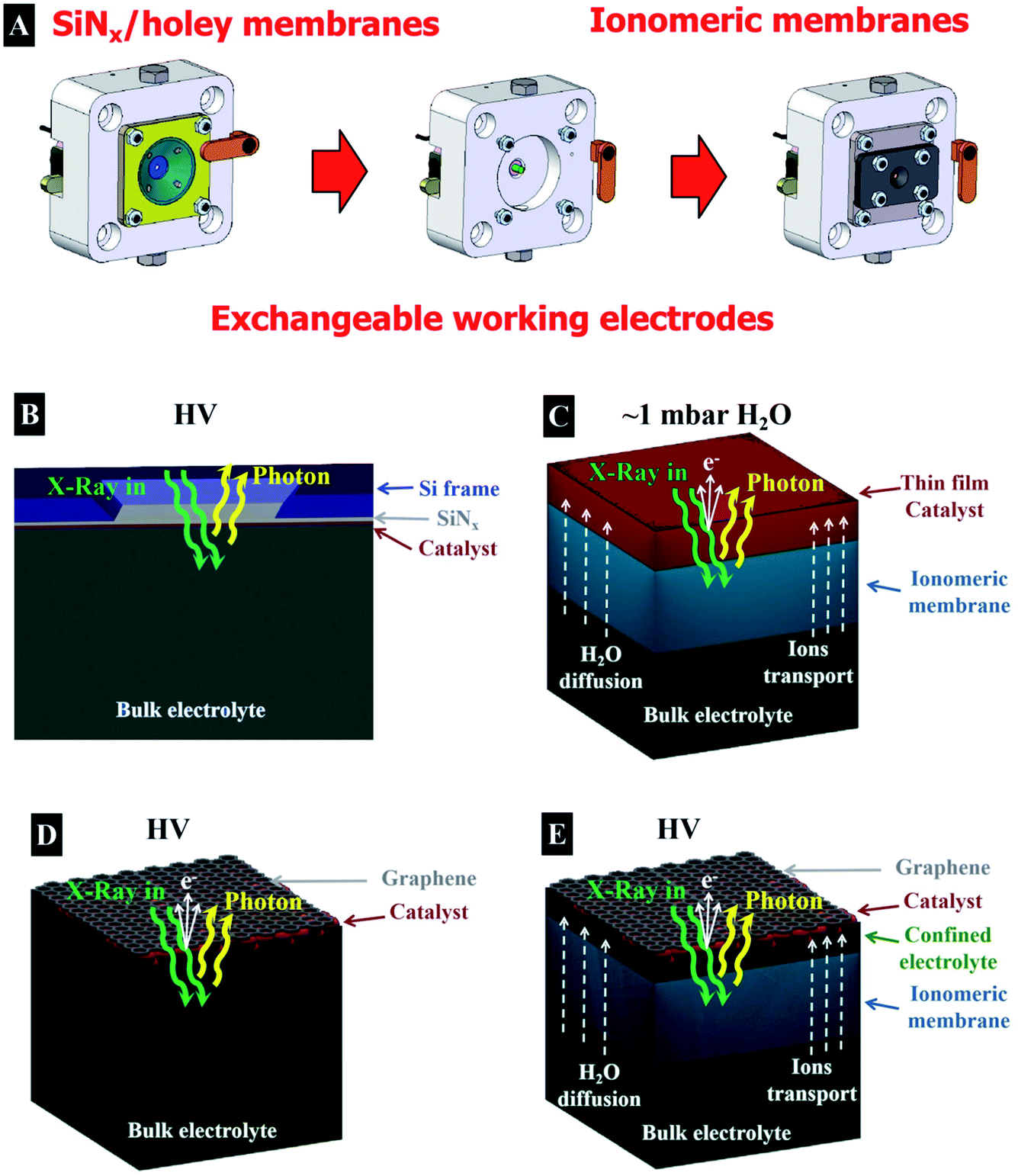 | ||
| Fig. 2 (A) Drawings of the operando EC-cell approach with exchangeable electrodes used in this work. Drawings of the different membranes used for the operando electrochemical experiments: (B) catalyst on a SiNx membrane, (C) thin film electrode on an ionomeric membrane, (D) free standing graphene decorated with the catalyst and (E) capped catalyst on an ionomeric membrane with a graphene membrane. | ||
Taking into account the information depth provided by the TEY/AEY and TFY, the simplest way to investigate electrochemical processes in the presence of a liquid is using XAS in the TFY mode.21 In order to perform measurements based on TFY, in the soft X-ray regime, usually a thin film membrane of SiNx (100 nm thick) that is quasi-transparent to the X-ray is used. The membrane separates the liquid electrolyte from the vacuum, where the X-ray comes from and where the detector is placed. A thin film is deposited onto this membrane and used as a working electrode (see Fig. 2B). The working electrode is deposited on the SiNx membrane using many different techniques, such as physical vapour deposition, sputtering, drop casting, spin coating, electrodeposition, etc. However, the collection of an O K-edge spectra of a working electrode in presence of an aqueous electrolyte is especially complicated in the TFY mode, since the aqueous electrolyte contribution is dominant due to the bulk sensitivity of this technique. Collecting an O K-edge spectrum of metal oxides in the presence of an aqueous electrolyte therefore requires extremely bulky electrodes, but this may result in a very low surface contribution from the oxide surface in the spectrum. To enhance the surface sensitivity of TFY-XAS, while still having a high material load, we make use of a mesoporous film to increase the surface to bulk ratio. For such a thick, porous electrode, the oxide contribution is dominant/comparable with respect to the aqueous electrolyte, resulting in a high surface contribution. In addition, the electrolyte can diffuse into the porous bulk, yielding higher ratios of products per gram, compared to those of compact samples. Pores of 15 nm yield a surface area of ∼140 m2 of film per m2 of substrate, compared to 20 m2 for substrates with 5 nm pores.27 Note that the surface area is a key parameter to describe the sample performance because it determines the amount of material that it is exposed to the electrolyte per mass, which has a direct influence in the overall catalytic performance.
The mesoporous IrOx film was prepared on the electrolyte-exposed side of an X-ray transparent SiNx membrane by iridium acetate and micelles of amphiphilic block-copolymers using an “evaporation-induced self-assembly” (EISA) method.27Fig. 3A shows the top-down scanning electron microscopy (SEM) view of such a mesoporous IrOx-coated SiNx membrane. The image reveals an average pore size of 20 nm and distance between pores of 27 nm, with a catalyst thin film thickness of 100 nm (as shown in Fig. 3B). Note that for the experiments shown here, three consecutive mesoporous films were deposited onto the SiNx membrane, yielding a ∼300 nm thick film. The EISA method yields the formation of well-defined crystalline walls, critical for electrocatalyst stability under harsh acidic conditions in anodic faradaic processes. At the same time, the high surface to bulk area ratio is ensured by the pores, which is quite important for the electrocatalytic performance and material usage optimization. The porous film allows the diffusion of liquids (or gases) within its structure, while the surface to bulk ratio remains high compared to compact thick film electrodes (Fig. 2B shows the detection scheme). A SEM cross-section image of the SiNx membrane coated with the prepared mesoporous film is shown in Fig. 3B showing the suitability of this method to fabricate mesoporous films onto a relatively fragile thin film SiNx (100 nm thick) X-ray transparent membrane. The performance of the mesoporous membranes is compared with that of a sputtered thin film deposited onto a SiNx membrane. Fig. 3C shows the top-down SEM view of a sputtered IrOx thin film on a SiNx membrane. The cross section SEM view shows a catalyst thin film thickness of 20 nm, as seen in Fig. 3D.
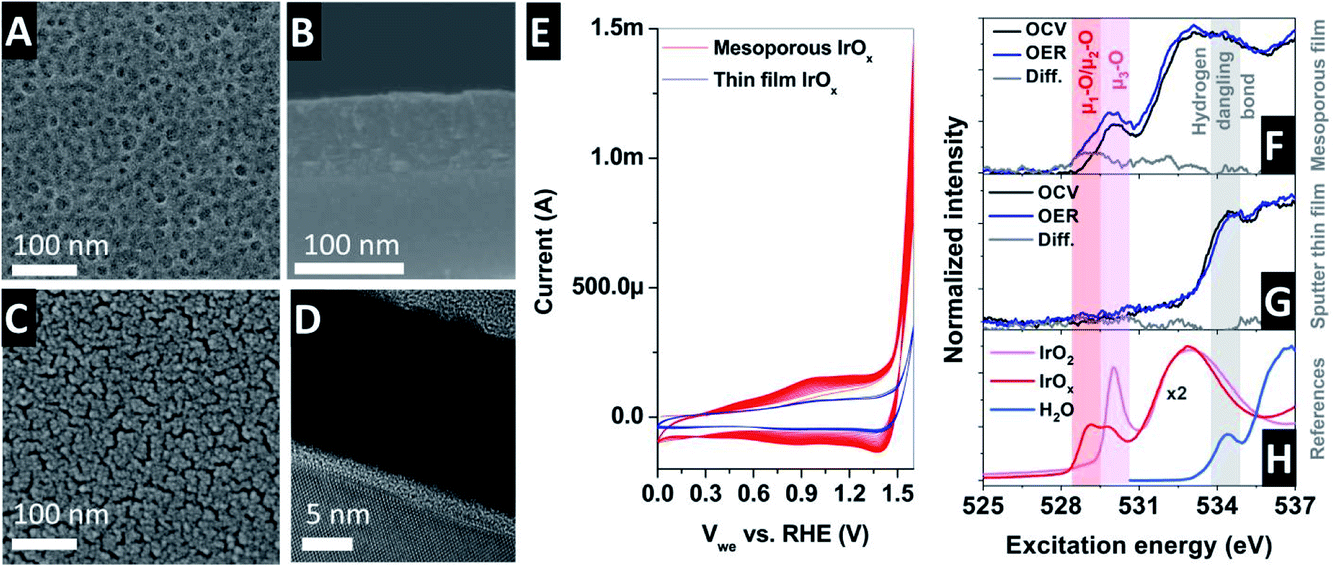 | ||
| Fig. 3 (A) Top-down SEM view of the IrOx mesoporous film on the SiNx membrane. (B) Cross-section SEM image of the IrOx mesoporous film on the SiNx membrane. (C) Top-down SEM view of the IrO2x sputtered thin film on the SiNx membrane. (D) Cross-section SEM image of the IrOx sputtered thin film on the SiNx membrane. (E) CV collected in the presence of 100 mM H2SO4 (saturated in N2) with Pt and Ag/AgCl as counter and reference electrodes, respectively, for the mesoporous (red) and sputtered (blue) thin films. Spectra collected under operando conditions with XAS-TFY under OCV and OER in 100 mM H2SO4 with Pt and Ag/AgCl as the counter and reference electrodes, respectively, for (F) mesoporous IrOx and (G) sputtered IrOx thin films. (H) Reference O K-edge spectra. | ||
The electrocatalytic performance of the thin film was investigated by means of cyclic voltammetry (CV) in 100 mM H2SO4 (saturated in N2) with Pt and Ag/AgCl as the counter and reference electrodes, respectively, with a scan rate of 20 mV s−1, as shown in Fig. 3E. The potential conversion to the reversible hydrogen electrode (RHE) was calculated using the Nernstian equation to be ERHE ≈ EAg/AgCl + 0.254 V. The cyclic voltammogram (CV) shows two oxidation and two reduction waves.28,29 In addition, the oxidation of water takes place in the region above 1.4 V vs. RHE. Continuous cycling between the open circuit voltage (OCV) and OER potentials (1.854 V vs. RHE) in 100 mM H2SO4 electrolyte at room temperature results in the development of a larger peak in the oxide region, increasing the peak height with an increasing number of cycles,30 which is ascribed to the existence of more active highly hydrated iridium (hydroxo) species.31 These trends are clearly visible in the CV in Fig. 3E, where it is generally accepted that the active sites are hydrated Ir-oxo-hydroxides formed during the OER.32–34 Note that the total current in the sputtered thin film is lower due to its lower surface to bulk ratio compared to that of the mesoporous thin film. The molecular level description of the species responsible for the high catalytic activity shown by iridium, however, is still debated9 and cannot concluded from ex situ characterizations but requires the investigation of the electrified interface under working conditions.
Thus, in order to gain deeper knowledge of the mechanism ruling the electrochemical oxidation of water on iridium oxide based catalysts, the electronic structure of the IrOx mesoporous film was investigated by O K-edge X-ray absorption spectra. The O K-edge provides information ascribed to the electronic transition from the occupied O 1s to the unoccupied O 2p hybridized with the Ir 5d orbitals. The spectrum has two features at ca. ∼530 eV and ∼533 eV ascribed to hybridization with the Ir t2g and eg orbitals, respectively35 (see the reference spectra in Fig. 3H). Different O K-edge structures were found depending on the calcination preparation temperature of the mesoporous iridium film EISA template preparation. In the present investigation, we used films prepared at 375 °C, which show higher electrocatalytic activity than those prepared at higher temperatures (500 °C and higher). More experimental details ascribed to the electrode fabrication can be found elsewhere.36 Using such electrodes, a complete set of measurements as a function of the applied potential was accomplished in the presence of 100 mM H2SO4 aqueous electrolyte de-aerated with N2 (gas). At the same time, the electrolyte was continuously refreshed by a peristaltic pump, minimizing the formation of trapped gas bubbles on the surface of the working electrode.
Fig. 3H (bottom) shows the O K-edge spectra of the reference IrO2 rutile, amorphous IrOx and water. The reference spectra are compared with the operando spectra collected at open circuit voltage (OCV) and during OER, obtained using a sputtered polycrystalline metallic iridium thin film sample of ∼20 nm (Fig. 3G) and a mesoporous film (Fig. 3F). The spectra collected with the sputtered thin film only display one dominant peak at OCV and under OER at around ∼534.5 eV, ascribed to hydrogen dangling bonds in the aqueous electrolyte. The sputtered IrOx film does not show any detectable peak at ∼530 eV. In contrast, the O K-edge spectra collected with the mesoporous electrode show a peak at ∼530 eV, ascribed to oxygen coordinated to three iridium atoms (named μ3-O).37 Upon anodic polarization at OER, a new peak appears at ∼528–529 eV, ascribed to the formation of electron deficient active oxygen species with bridging (named μ2-O) and terminal (named μ1-O) oxygen atoms.37–41 Electron-deficient oxygen species have been correlated with the formation of active centers in the electrode catalyst, however its precise atomic structure varies between different iridium (hydr)oxides.9 Thus, according to these results, a bulk sensitive technique can provide relevant atomistic information of an electrified interface if the sample has a high surface to bulk ratio.
3. Can operando approaches based on photoelectron spectroscopy mimic the performance of bulk sensitive spectroscopies?
Bulk sensitive methods, like XAS in the TFY mode, can provide valuable information about the electronic structure of an electrified interface under electrocatalytic reaction conditions using materials with high surface to bulk ratios, like the mesoporous film used in this study. In such systems, the challenge is to compare the information acquired with the bulk sensitive XAS-TFY method to the surface sensitive spectroscopy techniques based on photoelectron spectroscopy, which can reveal information related to the electrified solid–liquid interface directly and more specifically. In this direction, different approaches have been implemented using hard/tender X-ray based methods in recent years, taking advantage of the larger inelastic mean free path (IMFP) of photoelectrons with higher kinetic energies than those generated by soft X-ray energy excitation. Thus, it is possible to investigate electrodes within a thin electrolyte meniscus using the so-called dip & pull20,42 method. Another technique entails the use of thin Si membranes for separating the liquid from the vacuum side where the analyzer is placed.19 For using this geometry in the XPS mode, the membrane must be significantly thinner and, again, hard X-rays must be used. However, in the present work, we will focus in the experimental approaches developed to be used in the soft X-ray regime, because in this energy range important elements such as carbon, nitrogen and oxygen have their absorption edge. In addition, the X-ray absorption cross-section is higher in the soft X-ray range, enhancing the signal intensity from the interface between solids and liquids with enhanced spectral resolution (i.e. enhanced Auger electron emission compared to the total fluorescence yield in the tender/hard X-ray range). Furthermore, the surface sensitivity of soft X-ray XPS is higher than in the tender/hard regime (sub-nanometer vs. several nanometers).Note that NAPXPS13 is operated in the soft X-ray regime at a partial pressure up to a few mbars. At these low partial pressures, it is not possible to allocate aqueous electrolytes in the experimental chamber. Only certain liquid electrolytes with low partial pressures, such as ionic liquids, can be placed directly in the experimental chamber and be used as electrolytes in electrochemical experiments. Taking into account these experimental limitations, the first electrochemical experiments were performed in the gas phase up to partial pressures of a few mbar. Thus, solid oxide electrolytes operating at high temperatures were first used to investigate electrochemical processes under operational conditions.43,44 Additionally, the use of ionomeric membranes allowed the investigation of electrochemical reactions, i.e. OER, at room pressure and low partial pressure of water vapor.45,46 However, this approach has significant limitations in terms of the membrane hydration (required for high current operations) and chemical potential depending on the partial pressure.
In order to improve the wetting of the ionomeric membrane, it is back-filled with an electrolyte using a peristaltic pump, while the ionomeric membrane separates the liquid electrolyte from the vacuum.47 This approach allows for an optimal membrane hydration, enabling the transport of ions but not electrons. On top of the ionomeric membrane (Nafion™ in this case), a thin film electrode is deposited. This assembly resembles industrial applications, in acidic media, in which electrochemical reactions are performed using composites of electrodes and ionomeric membranes, which separate the gaseous products from the liquid electrolyte. At the same time, the electrode faces the experimental vacuum chamber, where it is illuminated by the X-ray source and the photoelectrons are collected using the hemispherical analyzer (XPS/AEY/PEY) or with a Faraday cup (TEY). Such a membrane assembly is shown schematically in Fig. 2C. In this configuration, the working electrode is produced by sputtering a thin film directly on the ionomeric membrane. Thus, a ∼10 nm Ir thin film is sputtered onto a Nafion™ membrane, which cracks when the membrane swells or shrinks upon wetting or drying. Water can diffuse through these membranes and finally evaporates into the vacuum chamber, leaving the catalyst surface dry. However, in places in which the catalyst is in close contact with the membrane, mass transport can occur.41 A peristaltic pump refreshes the 100 mM H2SO4 electrolyte (saturated in N2) in a continuous flow behind the ionomeric membrane. The counter (Pt wire) and reference (Ag/AgCl) electrodes are immersed in the bulk liquid.
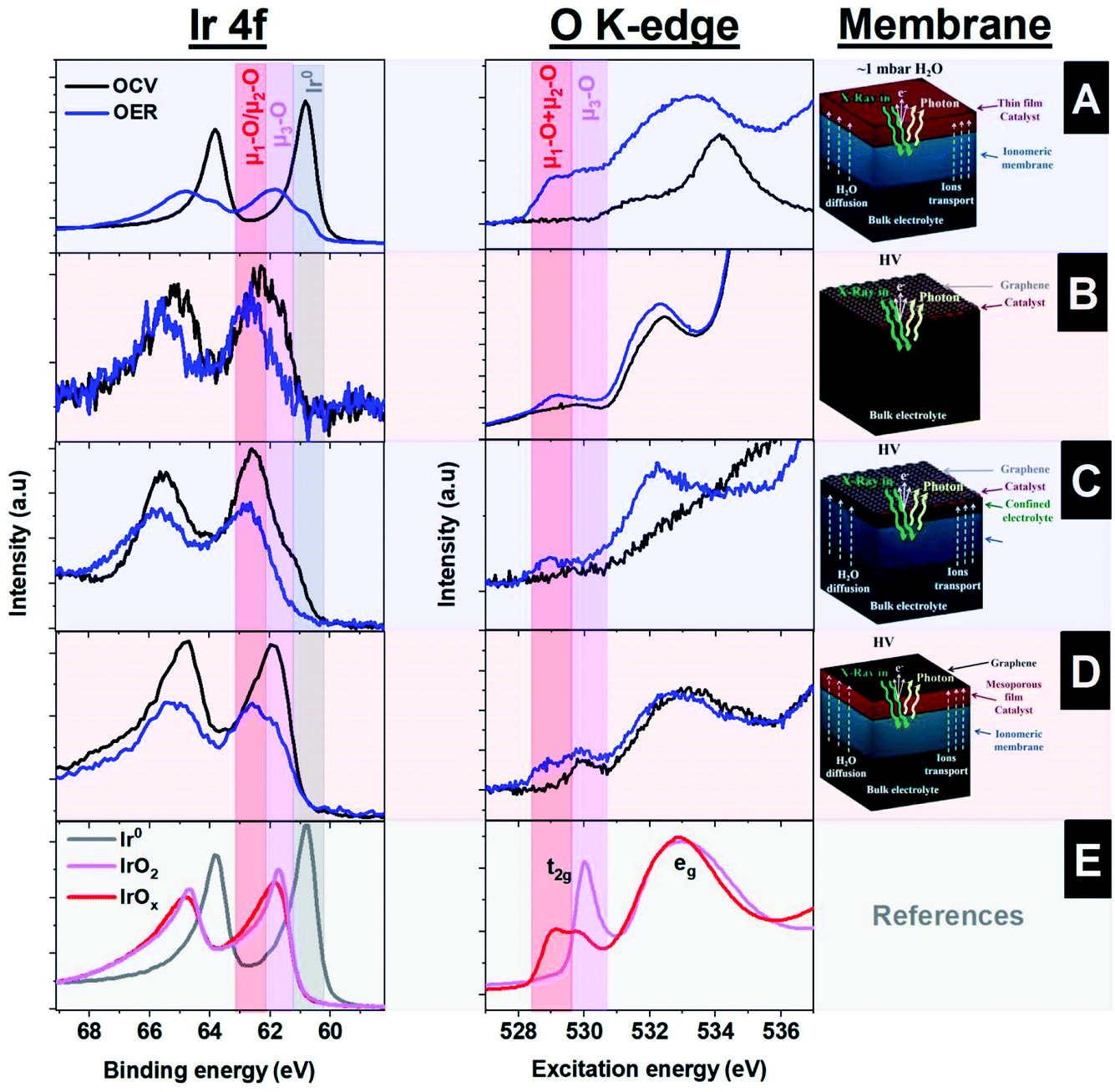
Fig. 4A shows the Ir 4f XPS and the O K-edge XAS spectra collected under OCV and OER on the surface of the sputtered thin electrode (∼10 nm). The operando collected O K-edge spectra are compared with those of the reference samples (Fig. 4E). Thus, under reaction conditions, the Ir 4f develops a shoulder at higher binding energies (62.5 eV) associated with the formation of oxidized species, including the active ones.48 At the same time, the O K-edge shows the formation of a peak at around ∼529 eV, ascribed to the formation of oxygen singly bound to iridium (μ1-O) and doubly coordinated μ2-O (bridge) species.37 Therefore, this approach, based on an ionomeric membrane with a sputtered thin film on top, provides detailed information on the active species present under the reaction conditions. However, this setup presents some limitations, since only thin films can be used as working electrodes. In principle, it is difficult to produce a reliable electrode that has good electrical conductivity between the different film patches while being thin enough to provide information of the electrified interface (iridium in close contact with Nafion™). Please note that it is important that the degree of the desiccation cracks formed in the film is a random process, which makes it quite difficult to prepare reproducible samples. Furthermore, with this approach, it is challenging to hydrate the electrode well, which adversely influences the electrochemical performance due to strong mass transport limitations. Metallic iridium present during the OER is therefore ascribed to the side of the electrode facing the vacuum chamber, i.e. no contact with either Nafion™ or the liquid electrolyte.
 | ||
| Fig. 4 Operando spectra collected using photoelectrons from the O K-edge and Ir 4f with (A) thin film sputtered onto a Nafion™ ionomer membrane, (B) Ir NPs sputtered onto free standing graphene on a holey SiNx membrane, (C) sputtered Ir NPs “sandwiched” between Nafion™ and graphene, (D) a mesoporous IrOx thin film “sandwiched” between Nafion™ and graphene, and (E) reference spectra. | ||
Another approach for the investigation of solid–liquid interfaces with soft X-rays is based on free standing 2D (graphene) membranes.49–51 With this approach, the X-rays easily penetrate the graphene that, in turn, is also semi-transparent to photoelectrons, i.e., the photoelectrons can escape the confined environment and be detected in the vacuum side where the hemispherical analyzer is placed. To enhance the mechanical stability of larger graphene layers, the graphene is transferred onto a holey SiNx membrane with an array of micro holes.50 This approach yields a large free standing graphene area, which is larger than the X-ray beam spot size and increases the signal intensity with respect to a single small graphene covered hole. In addition, this membrane is leak tight to the electrolyte and electrically conductive, allowing for its use as a current collector. A uniform distribution of iridium nanoparticles, covering ∼20% of the surface with a size of around ∼2 nm, is deposited on top of this membrane by sputtering.29,48 A schematic representation of this approach is shown in Fig. 2D, where the electrolyte is in close contact with the iridium NPs and graphene. Using this approach, the Ir 4f and the O K-edge spectra were collected, indicating the formation of electron-deficient oxygen species present during the OER, as shown in Fig. 4B. However, the intensity of the peaks observed is lower than in the approach that uses ionomeric membranes coated with a sputtered thin film due to the lower loading of Ir NPs. In addition there is a significant part of the holey SiNx membrane that is not illuminated by the beam and is, hence, not contributing to the Ir 4f spectra. As an advantage, the graphene membrane uses a similar geometry as the bulk sensitive approaches based on TFY detection with SiNx membranes, in the sense that the electrochemical experiments are performed with bulk liquid electrolyte. However, this approach suffers from beam damage, requiring graphene of high quality to ensure stability during data acquisition. The illumination of an aqueous electrolyte with an intense X-ray beam yields the formation of radicals from water radiolysis; these radicals damage the graphene layer.15,52,53 To solve this issue, the stability of the graphene layer can be increased by stacking more layers of graphene on top of each other. However, that would require the use of higher excitation energies (in the tender X-ray regime) or the use of other stable thin membranes, i.e. coating the graphene with other materials more stable to the beam effects.54
While the free-standing graphene approach is challenged by beam damage with a risk of the electrolyte leaking into the vacuum chamber, using ionomeric membranes coated with a “thick” film is limited by wetting, mass transport, and electrical conductivity. A possible solution is the combination of both approaches based on the transparent graphene and the ionomeric membranes. This is a suitable approach to improve the stability of the free standing graphene (the ionomeric membrane holds the pressure difference) and the evaporation of water (the graphene cover is an evaporation barrier).26,37,55,56 The underlying idea is to transfer the graphene onto the ionomeric membrane and use it as a photoelectron transparent membrane, current collector and evaporation barrier in the same way as with the free standing graphene approach. There are different techniques to transfer the material of interest to the ionomeric membrane; the easiest consists of depositing NPs of Ir (of around ∼1 nm) onto the ionomer membrane by sputtering. After that, a layer or a bi-layer of graphene is transferred on top, leaving the NPs “sandwiched” between the graphene and Nafion™ (Fig. 2E shows a schematic of this approach). By doing this, an electrical contact between the different NPs with the potentiostat is ensured by graphene.
The graphene works as an evaporation barrier, yielding correct membrane wetting and the formation of a confined liquid between the Nafion™ and graphene.56 In this way, this approach allows the investigation of solid–liquid interfaces, providing similar Ir 4f and O K-edge spectra as the other approaches. However, in the graphene covered samples, the Ir 4f spectra show a much reduced contribution of metallic iridium compared to the sample without the graphene cover, indicating a better electrochemical performance because the metallic iridium is not the active phase, as shown in Fig. 4C. However, this approach has a weakness compared to the “thick” film: the ionomeric membrane itself is more sensitive to beam damage than in the case of the thicker Ir film, requiring that the spectra are collected continuously at fresh spots on the sample. In addition, the charge transport of this approach is limited by the membrane specifications (Nafion™, for example, mostly conducts protons efficiently), but this is not the case for other ions. Thus, this approach can lead to worse charge transport compared to that of free standing graphene, as well as unknown pH of the confined liquid between graphene and the ionomeric membrane, which can lead to an unknown applied potential under certain experimental conditions.
Another method employs the use of mesoporous thin films, which, unlike the sputtered films, allow electrolyte diffusion and mass transport within its volume. This assembly is shown schematically in Fig. 5A. The mesoporous Ir 100 nm film is transferred onto a Nafion™ membrane and capped with a graphene layer to improve the electrical contact and minimize water evaporation. Fig. 5B shows a SEM image of the mesoporous IrOx film on a Nafion™ membrane, meanwhile Fig. 5C shows the same type of membrane capped with graphene. The layer was prepared by EISA at 375 °C. This configuration has a clear advantage with respect to the use of NPs, as it is less sensitive to beam damage, since a large portion of the incoming radiation of the beam is blocked. Thus, the photon intensity transmitted trough a 100 nm thick iridium layer is around 10% of the original at an excitation energy of 600 eV, according to the calculation of Henke et al.57 This method therefore allows the collection of spectra over long periods of time in the same sample spot. The mesoporous films are transferred using a hot press approach between the Nafion™ membrane and the IrOx mesoporous film grown on a silicon wafer.18 It is also beneficial that this approach can be directly compared with the experiments performed in TFY with the SiNx X-ray membrane. The fact that the electrolyte diffuses through the pores of the film allows the collection of the spectra with the electrolyte in contact with the solid electrode. Thus, the Ir 4f and the O K-edge spectra resemble the spectra collected with the other approaches, with significantly higher intensities compared to those from the approach based on sputtered NPs, as shown in Fig. 4D. Therefore, these measurements confirm the presence of electron deficient oxygen species during OER. However, not every material is easily prepared as a mesoporous film using the EISA approach, which hinders the application of this approach. In addition, the mesoporous films can suffer from mass transport limitation compared to other electrode geometries.
 | ||
| Fig. 5 (A) Drawings of the capped mesoporous film on an ionomeric membrane (Nafion™) with a graphene membrane. SEM images of (B) a Nafion™ membrane coated with a mesoporous IrOx film and (C) a Nafion™ membrane coated with a mesoporous IrOx film capped with a graphene layer used in the collection of the operando X-ray spectra. | ||
Taking into account the four different approaches compatible with photoelectron spectroscopy shown in this section, it is clear that all of them provide similar information and offer different advantages and disadvantages. The spectra collected with photoelectrons are similar to those collected in the TFY mode, revealing the formation of electron deficient oxygen (μ1-O and μ2-O) during the OER on IrOx catalysts independently of the experimental approach selected. Consequently, we can conclude that similar information can be extracted using surface sensitive measurements compare to those methods based on TFY with surface enhanced sensitivity. The advantages and disadvantages of each approach discussed here are summarized in Table 1.
| Sample assembly | Preparation difficulty | Beam damage | Electrochemical performance | Spectroscopy method |
|---|---|---|---|---|
| 0.1 M H2SO4/Ir thin film/SiNx/HV | Low | Low | Excellent | Photons |
| 0.1 M H2SO4/mesoporous Ir/SiNx/HV | High | Low | Excellent | Photons |
| 0.1 M H2SO4/Nafion™/Ir thin film/HV | Low | Medium | Bad | Photons/electrons |
| 0.1 M H2SO4/Ir NPs/graphene/holey SiNx/HV | Extreme | Extreme | Excellent | Photons/electrons |
| 0.1 M H2SO4/Nafion™/Ir NPs/graphene/HV | Medium | High | Good | Photons/electrons |
| 0.1 M H2SO4/Nafion™/mesoporous Ir/graphene/HV | High | Medium | Good | Photons/electrons |
4. Can X-ray photoelectron spectroscopy provide additional speciation information not accessible by only XAS?
The well-established techniques based on bulk-sensitive methods, such as TFY, can provide similar information as X-ray photoelectron spectroscopy under certain conditions, as we have shown in this work (i.e. if a mesoporous sample is used). If that is the case, one could ask if the development of techniques compatible with photoelectron spectroscopy really deserves the time and effort needed for their implementation. To answer this question and to justify this effort, one can argue that photoelectron spectroscopy is a really surface-sensitive method, meanwhile TFY is not. Against this argument, we show in this work that, after all, in case of the IrOx system, very similar conclusions are drawn from all approaches. Another advantage of XPS is that it provides information related to other core levels. However, the TFY measurements can easily be complemented by collecting other edges of interest, for example the Ir L-edges. Hence, the additional core level information from XPS is also not a strong argument to justify the development of novel photoelectron spectroscopy approaches. Another argument in favor of photoelectron compatible techniques is that it is possible to detect changes in the potential drop induced by the ions within the electrical double layer, which is an excellent method to distinguish between the voltage and the electrochemical potential applied. XAS methods cannot provide this information. In general, though, the electrochemical performance of the approaches based on SiNx membranes are significantly better than those used for XPS and, usually, the charge transport is not an issue. Finally, if these three arguments themselves do not justify the development of photoelectron spectroscopy compatible operando strategies, what else will justify it?To answer this question, one should go one step beyond the surface sensitivity of X-ray photoelectron spectroscopy and explore other opportunities that this technique offers for the identification of the active species. ResPES is a technique which simultaneously provides information about occupied and unoccupied states of interest. Similar measurements can be done using resonant inelastic X-ray scattering (RIXS), however this technique requires substantial incident photon flux (inducing significant beam damage) to obtain the substantially scattered photons needed for collecting spectra with enough intensity and energy resolution. Therefore, RIXS cannot compete with ResPES in term of the energy resolution and signal intensity under electrochemical in situ/operando conditions. Unfortunately, ResPES is hardly compatible with liquids, requiring sophisticated technical approaches for its use under reaction conditions, whereas NAPXPS offers these capabilities. In addition, the long time necessary to collect these measurements (more than one hour) makes it necessary to use samples that are stable under harsh reaction conditions (i.e. corrosion/dissolution resistance) and X-ray beam illumination (beam damage). In order to perform ResPES measurements, the best approach is the one based on an ionomeric membrane coated with a mesoporous film capped with a graphene layer. This approach combines good electrochemical performance and long-term stability under harsh OER conditions and X-ray beam illumination. Fig. 5C shows a SEM image of the mesoporous IrOx film on Nafion™ capped with a graphene membrane. Please note that the size and density of the pores are similar to those in Fig. 3A, which correspond to the mesoporous film on the SiNx membrane prepared by EISA at 375 °C.
Using this assembly, ResPES measurements are collected for the valence band using the photon energies in the vicinity of the O 1s core level. Note that O 2p and Ir 5d orbitals are hybridized due to the covalent Ir–O bonding. Fig. 6A shows the ResPES collected through the O K-edge under OCV conditions, which is ascribed to the transition of an electron from an occupied O 1s level to the unoccupied O 2p orbitals. The ResPES of the mesoporous IrOx film36 collected in the vicinity of the O 1s shows the existence of two dominant resonant peaks, as shown in Fig. 6A. Selected valence band spectra are shown on top of the colour map and the full range AEY spectrum is shown on the right hand side. At OCV, there is a resonant peak at ∼530 eV excitation energy (EE) and a 15.6 eV biding energy (BE), highlighted in red, which is ascribed to the existence of μ3-O and μ1-OH species. Otherwise, under OER conditions, the ResPES map shows the formation of other strong resonant peaks ascribed to the formation of μ1-OO peroxo (532.5 eV EE; 14 eV BE),58 μ2-O (529.2 eV EE; 12.2 eV BE) and μ1-O (528.9 eV EE; 8.8 eV BE), as shown in Fig. 6B. The full AEY spectra collected are similar to the O K-edge absorption spectra found under OER, with a peak located at a lower excitation energy. However, in addition to the information extracted from the XAS characterization, the ResPES map shows that the absorption peak below 530 eV is a consequence of at least two different species, as indicated in Fig. 6B by the peaks at (529.2 eV EE; 12.2 eV BE) and (528.9 eV EE; 8.8 eV BE). These results indicate the existence of another important intermediate species during the reaction which is different to the singly coordinated μ1-O (terminated oxygen) and doubly coordinated μ2-O (bridge) species. Unlike previous results,37 the μ1-O active species can easily be distinguished from the μ2-O, indicating clearly that these species are key to the OER reaction. In addition, the peak appearing at (532.5 eV EE; 14 eV BE) can be ascribed for the first time to the existence of a peroxo species58 during the OER conditions. Taking into account the operando ResPES measurements, Fig. 6C shows a scheme of the reaction pathway, where under OCV conditions the dominant species are μ1-OH and μ3-O. Once the electrode is under OER conditions, the μ1-OH species undergoes deprotonation to form the active μ1-O species, which reacts to form μ1-OOH (hydroperoxo) and μ1-OO (peroxo) species. The results found in this work are of prime importance and have not been described before in the literature, highlighting the existence of different oxygen species during the OER. These results indicate that the reaction pathway yielding dioxygen is dominated by the formation of peroxo groups. The ResPES results clearly indicate that the speciation is more complex than previously thought, indicating the existence of wide ranging oxygen species under OER conditions.
 | ||
| Fig. 6 ResPES collected in the valence band using excitation energy through the O K-edge (A) at OCV and (B) under OER conditions. (C) Representation of the species found at OCV and under reaction conditions and the reaction pathway. | ||
According to these results, the ResPES characterization of solid–liquid interfaces under reaction conditions is a powerful technique to provide detailed speciation information, more than XAS alone can. The advantage of this technique, with respect to XAS, is that ResPES provides resolved spectra in binding and excitation energies simultaneously. Thus, XAS can provide 2D maps if the spectra are plotted versus another parameter, such as applied potential in this work. Otherwise, if the ResPES measurements are plotted versus the applied potential, a 3D map can be formed. Thus, ResPES map spectra allow a better speciation of the active species present during the operando reactions. Therefore, photoelectron spectroscopy based techniques have a significant advantage with respect to XAS apart from the enhanced surface sensitivity and information of the potential drop, as it is possible to simultaneously collect information on the occupied and unoccupied orbitals, which yields accurate information about the different process/species that undergo the resonance event. Information on the occupied states can be gained by collecting the ResPES through the absorption edge of interest, and information about the PDOS can be acquired by collecting the valence band spectrum while exciting at a resonant energy. This ability of the ResPES yields enhanced speciation capability, which is the cornerstone of the accurate identification of the active species present during the reactions. This is a key capability that should be systematically used in experiments that aim to understand complex electrochemical processes, and justify the last decade’s effort to get electrochemical setups running which are compatible with photoelectron spectroscopies in the soft X-ray range, as presented in this work.
5. A last reflection on the OER: how can ResPES help in interpreting the OER mechanism?
The surface oxygen species present on an IrOx/electrolyte electrified interface are directly involved in production of the dioxygen product.59,60 Thus, on an iridium (hydr)oxide catalyst, the number of singly- and doubly-coordinated oxygen atoms with iridium (μ1-O and μ2-O species, respectively) is larger for defective surfaces and more active during the OER. For structures with higher crystallinity (less defective), the dominant coordination environment is the triple iridium-coordinated oxygen atoms (μ3-O species), being less active in the OER. Thus, in the presence of an iridium active center and water molecules, the system undergoes the following reaction:| Ir* + H2O → Ir–OH + e− + H+, | (1) |
| Ir–OH → Ir–O + e− + H+, | (2) |
 | ||
| Fig. 7 Schematic representation of the simplified reaction pathway. The upper part shows the intermediates that can be considered to be known according to the operando spectroscopy and literature available (“Orbis Terrarum”). The bottom part represents part of the reaction pathway that has not been probed experimentally (“Terra incognita”) until the current ResPES experiments presented in this work. | ||
Furthermore, OER involves the adsorption of water, the transfer of four protons and electrons and the evolution of dioxygen products. This reaction is led by the formation of the O–O bond, where the exact mechanism ruling this bond formation is still under discussion in the literature. Each step of the reaction has several transition states where the oxygen–oxygen bond formation is the key factor dominating the rate step. The formation of the oxygen–oxygen bond is proposed to be produced by two different pathways: (a) through water adsorption on an active site or (b) through the oxygen–oxygen coupling of two neighbouring sites. We consider a simplified model for the absorption of water onto a deprotonated μ1-O species as:
| Ir–O + H2O → Ir–OOH + e− + H+, | (3a) |
| Ir–O + Ir–O + H2O → Ir–OOH + Ir–O–H | (3b) |
Independent of the reaction pathway (a or b), this process yields the formation of a hydroperoxo species on the surface. The hydroperoxo is transformed into a peroxo as:
| IrOOH → Ir–OO + e− + H+, | (4) |
| Ir–OO → Ir* + O2 | (5) |
Note that an alternative pathway that is still considered is the direct coupling of lattice oxygen,61 which can be written as
| Ir–O + Ir–O → 2Ir* + O2 | (3c) |
The reaction pathways involving the formation of the oxygen–oxygen bond are described schematically in Fig. 7 bottom, where the exact reaction pathway is unknown (“Terra incognita”, not mapped) in the literature and has been proposed theoretically,59,60 but not empirically. However, the operando ResPES experiments shown in this work indicate that the OER involves the formation of peroxo species, meaning that the speciation is more complex during reaction conditions involving different oxygen species. Thus, the reaction pathway yielding oxygen–oxygen bond formation is dominated by the presence of a peroxo species and does not involve the direct coupling of two oxygen neighbors in the iridium electrode lattice. Therefore, this work represents a mapping of the reaction pathway in the “Terra incognita”, going far beyond the current understanding of the OER on iridium (hydr)oxide catalysts. In short, the OER on iridium electrodes is dominated by the formation of active terminal oxygen species (μ1-O, confirmed in this work), which yield the formation of peroxo species (μ1-OO, probed for the first time in this work), controlling the reaction pathway to dioxygen (O2) evolution.
6. Conclusions
Detailed insights into the electronic structure of an electrified interface requires the collection of relevant X-ray spectroscopy data (XPS, NEXAFS and ResPES) under operando conditions. Typically, the collection of this information is restricted in the soft X-ray regime to the investigation of ex situ samples, especially when using photoelectrons. In order to overcome this limitation and investigate the electronic structure of electrified solid–liquid interfaces under reaction conditions, different approaches compatible with photoelectron spectroscopy have been developed in our group in recent years. The approaches presented here, using the example of iridium oxide in a sulfuric acid aqueous electrolyte, involve a different degree of complexity and their use has different advantages and disadvantages. The operation and the atomistic information provided by each method was compared with the bulk-sensitive approach (NEXAFS photon-in/photo-out). The performance of each approach was exemplified by comparing the spectra collected during the OER on IrOx electrodes. It was found that, independent of the approach used, a peak at around ∼529 eV appears under OER conditions, which is ascribed to electron deficient oxygen species. However, by using ResPES, it was possible to prove clearly that this peak is a consequence of two different species, named μ2-O (bridge oxygen) and μ1-O (terminal oxygen). In addition, the formation of μ1-OO (peroxo) species was found under the reaction conditions, indicating that different oxygen species take part in controlling the reaction pathway. This result is of prime importance and represents a substantial advance in the understanding of the complex OER on IrOx electrodes. Even if it is accepted that the terminal oxygen atoms are the catalytically active centers, we can conclude in this work that the OER is dominated by the formation of peroxo species and not by the oxygen lattice coupling reaction. Thus, ResPES provides better speciation capabilities as far as the spectral information can be depicted simultaneously depending on the excitation and binding energies. Because of this, ResPES simultaneously provides information related to the occupied and unoccupied states. This unique capability of ResPES opens up the possibility of describing steady state intermediates more accurately. To conclude, one should remark at this point that the methodologies presented here can be implemented to investigate other relevant electrochemical processes, which shows the paramount importance of the photoelectron compatible methods discussed in this work.Author contributions
Operando mesoporous films: J. J. V. V. and R. K. conceived of the idea and designed the experiments for using mesoporous thin film electrodes on SiNx and ionomeric membranes for operando X-ray spectroscopy electrochemical characterization. D. B. fabricated the mesoporous electrodes. P. Z., E. C., C. H. C. and J. J. V. V. performed the experiments. J. J. V. V. and T. E. J. analyzed the data. Operando ionomeric membranes: R. A. conceived of and designed the idea to use ionomeric membranes coated with a thin film for operando photoelectron spectroscopy. V. P. and J. J. V. V. performed the experiments. V. P. and T. E. J. analyzed the data. Operando free standing graphene: J. J. V. V. conceived of the idea and designed the experiments for using free standing graphene on holey SiNx membranes decorated with NPs for operando photoelectron spectroscopy. J. J. V. V. and V. P. performed the operando experiments. J. J. V. V. and T. E. J. analyzed the data. Operando ionomeric membranes capped with graphene: J. J. V. V. conceived of the idea. L. J. F. and R. V. M. developed the experimental approach for transferring PMMA free graphene onto the ionomeric membranes. L. J. F. performed the operando experiments. L. J. F. and T. E. J. analyzed the data. J. J. V. V., M. H. and E. S. developed the operando X-ray spectroscopy electrochemical cell with exchangeable electrodes. A. H. and T. L. performed the electron microscopy analysis of the different samples. A. K. G. and R. S. led the project. J. J. V. V. wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank DAAD for the financial support through the framework of the Taiwanese–German collaboration (project ID 57218279 and 57392335). C.H.C. acknowledges financial support from projects 106-2112-M-032-003. We thank Helmholtz Zentrum Berlin für Materialien und Energie for allocating beamtime for our experiments within the proposal numbers 192-08521 and 191-08014.References
- R. Schlögl, ChemSusChem, 2010, 3(2), 209–222 CrossRef PubMed.
- J. A. Turner, Science, 2004, 305(5686), 972–974 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488(7411), 294–303 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110(11), 6446–6473 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323(5915), 760–764 CrossRef CAS PubMed.
- Y. Gorlin and T. F. Jaramillo, J. Appl. Chem. Sci., 2010, 132(39), 13612–13614 CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3(3), 399–404 CrossRef CAS PubMed.
- T. C. Wen and C. C. Hu, J. Electrochem. Soc., 1992, 139(8), 2158 CrossRef CAS.
- L. J. Falling, J. J. Velasco-Vélez, R. V. Mom, A. Knop-Gericke, R. Schlögl, D. Teschner and T. E. Jones, Curr. Top. Electrochem., 2021, 100842 CAS.
- J. J. Velasco-Velez, C. H. Wu, T. A. Pascal, L. F. Wan, J. Guo, D. Prendergast and M. Salmeron, Science, 2014, 346(6211), 831–834 CrossRef CAS PubMed.
- D. M. Itkis, J. J. Velasco-Velez, A. Knop-Gericke, A. Vyalikh, M. V. Avdeev and L. V. Yashina, ChemElectroChem, 2015, 2(10), 1427–1445 CrossRef CAS.
- A. Knop-Gericke, V. Pfeifer, J. J. Velasco-Velez, T. Jones, R. Arrigo, M. Hävecker and R. Schlögl, J. Electron Spectrosc. Relat. Phenom., 2017, 221, 10–17 CrossRef CAS.
- M. Salmeron and R. Schlögl, Surf. Sci. Rep., 2008, 63(4), 169–199 CrossRef CAS.
- J. J. Velasco-Velez, M. Hävecker, A. Knop-Gericke, C. Schwanke, L. Xi, K. M. Lange, J. Xiao, M. F. Tesch, R. Golnak, T. Petit, L. Puskar, U. Schade, M. Borgwardt, I. Kiyan, R. Seidel and E. F. Aziz, Synchrotron Radiat. News, 2017, 30(2), 14–19 CrossRef.
- A. Klyushin, R. Arrigo, V. Pfeifer, T. Jones, J. Velasco Velez and A. Knop-Gericke, Catalyst Electronic Surface Structure Under Gas and Liquid Environments, in Encyclopedia of Interfacial Chemistry, 2018, pp. 615–631 Search PubMed.
- V. Streibel, M. Hävecker, Y. Yi, J. J. V. Vélez, K. Skorupska, E. Stotz, A. Knop-Gericke, R. Schlögl and R. Arrigo, Top. Catal., 2018, 61(20), 2064–2084 CrossRef CAS.
- E. A. Carbonio, J. J. Velasco-Velez, R. Schlögl and A. Knop-Gericke, J. Electrochem. Soc., 2020, 167(5), 054509 CrossRef CAS.
- J. J. Velasco-Vélez, L. J. Falling, D. Bernsmeier, M. J. Sear, P. C. Clark, T. S. Chan, E. Stotz, M. Hävecker, R. Krähnert, A. Knop-Gericke, C. H. Chuang, D. E. Starr, m. Favaro and R. V. Mom, J. Phys. D: Appl. Phys., 2021, 54(12), 124003 CrossRef.
- T. Masuda, H. Yoshikawa, H. Noguchi, T. Kawasaki, M. Kobata, K. Kobayashi and K. Uosaki, Appl. Phys. Lett., 2013, 103(11), 111605 CrossRef.
- S. Axnanda, E. J. Crumlin, B. Mao, S. Rani, R. Chang, P. G. Karlsson, M. O. M. Edwards, M. Lundqvist, R. Moberg, P. Ross, Z. Hussain and Z. Liu, Sci. Rep., 2015, 5(1), 9788 CrossRef CAS PubMed.
- J. H. Guo, Y. Luo, A. Augustsson, S. Kashtanov, J. E. Rubensson, D. K. Shuh, H. Agren and J. Nordgren, Phys. Rev. Lett., 2003, 91(15), 157401 CrossRef PubMed.
- P. Jiang, J. L. Chen, F. Borondics, P. A. Glans, M. W. West, C. L. Chang and J. Guo, Electrochem. Commun., 2010, 12(6), 820–822 CrossRef CAS.
- J. Stöhr, NEXAFS spectroscopy, Springer Science & Business Media, 1992, p. 25 Search PubMed.
- S. L. Schroeder, G. D. Moggridge, E. Chabala, R. M. Ormerod, T. Rayment and R. M. Lambert, Faraday Discuss., 1996, 105, 317–336 RSC.
- B. H. Frazer, B. Gilbert, B. R. Sonderegger and G. De Stasio, Surf. Sci., 2003, 537(1–3), 161–167 CrossRef CAS.
- J. J. Velasco-Velez, R. V. Mom, R. V. Sandoval-Diaz, L. J. Falling, C. H. Chuang, D. Gao, T. E. Jones, Q. Zhu, R. Arrigo, B. Roldan-Cuenya, A. Knop-Gericke, T. Lunkenbein and R. Schlögl, ACS Energy Lett., 2020, 5(6), 2106–2111 CrossRef CAS PubMed.
- E. Ortel, T. Reier, P. Strasser and R. Kraehnert, Chem. Mater., 2011, 23(13), 3201–3209 CrossRef CAS.
- S. Song, H. Zhang, X. Ma, Z. Shao, R. T. Baker and B. Yi, Int. J. Hydrogen Energy, 2008, 33(19), 4955 CrossRef CAS.
- J. J. Velasco-Vélez, T. E. Jones, V. Streibel, M. Hävecker, C. H. Chuang, L. Frevel, M. Plodinec, A. Centeno, A. Zurutuza, R. Wang, R. Arrigo, R. Mom, S. Hofmann, R. Schlögl and A. Knop-Gericke, Surf. Sci., 2019, 681, 1–8 CrossRef.
- S. Gottesfeld and S. Srinivasan, J. Electroanal. Chem. Interfacial Electrochem., 1978, 86(1), 89–104 CrossRef CAS.
- L. Ouattara, S. Fierro, O. Frey, M. Koudelka and C. Comninellis, J. Appl. Electrochem., 2009, 39(8), 1361–1367 CrossRef CAS.
- E. J. Frazer and R. Woods, J. Electroanal. Chem. Interfacial Electrochem., 1979, 102(1), 127–130 CrossRef CAS.
- E. Willinger, C. Massué, R. Schlögl and M. G. Willinger, J. Appl. Chem. Sci., 2017, 139(34), 12093–12101 CAS.
- C. Massué, V. Pfeifer, X. Huang, J. Noack, A. Tarasov, S. Cap and R. Schlögl, ChemSusChem, 2017, 10(9), 1943–1957 CrossRef PubMed.
- H. M. Tsai, P.D. Babu, C. W. Pao, J. W. Chiou, J. C. Jan, K. P. Krishna Kumar, F. Z. Chien and W. F. Pong, Appl. Phys. Lett., 2007, 90(4), 042108 CrossRef.
- R. Sachse, M. Pflüger, J. J. Velasco-Vélez, M. Sahre, J. Radnik, M. Bernicke, D. Bernsmeier, V. D. Hodoroaba, M. Krumrey, P. Strasser, R. Krähnert and A. Hertwig, ACS Catal., 2020, 10(23), 14210–14223 CrossRef CAS.
- L. J. Frevel, R. Mom, J. J. Velasco-Vélez, M. Plodinec, A. Knop-Gericke, R. Schlögl and T. E. Jones, J. Phys. Chem. C, 2019, 123(14), 9146–9152 CrossRef CAS.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, C. Massué, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, M. T. Greiner, J. Allan, M. Hashagen, G. Weinwerg, S. Piccini, M. Hävecker, A. knop-Gericke and R. Schlögl, Surf. Interface Anal., 2016, 48(5), 261–273 CrossRef CAS.
- V. Pfeifer, T. E. Jones, S. Wrabetz, C. Massué, J. J. V. Vélez, R. Arrigo, M. Scherzer, S. Piccini, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2016, 7(11), 6791–6795 RSC.
- V. Pfeifer, T. E. Jones, J. J. V. Vélez, C. Massué, M. T. Greiner, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, M. T. Greiner, J. Allan, M. Hashagen, G. Weinwerg, S. Piccini, M. Hävecker, A. Knop-Gericke and R. Schlögl, Phys. Chem. Chem. Phys., 2016, 18(4), 2292–2296 RSC.
- V. Pfeifer, T. E. Jones, J. J. V. Vélez, R. Arrigo, S. Piccini, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2017, 8(3), 2143–2149 RSC.
- M. Favaro, B. Jeong, P. N. Ross, J. Yano, Z. Hussain, Z. Liu and E. J. Crumlin, Nat. Commun., 2016, 7(1), 12695 CrossRef CAS PubMed.
- C. J. Zhang, M. E. Grass, A. H. McDaniel, S. C. DeCaluwe, F. El Gabaly, Z. Liu, K. F. McCarty, R. L. Farrow, M. A. Linne, Z. Hussain, G. S. Jackson, H. Bluhm and B. W. Eichhorn, Nat. Mater., 2010, 9(11), 944–949 CrossRef CAS PubMed.
- A. Toghan, R. Arrigo, A. Knop-Gericke and R. Imbihl, J. Catal., 2012, 296, 99–109 CrossRef CAS.
- H. G. Sanchez Casalongue, S. Kaya, V. Viswanathan, D. J. Miller, D. Friebel, H. A. Hansen, J. K. Norskov, A. Nilsson and H. Ogasawara, Nat. Commun., 2013, 4(1), 1–6 Search PubMed.
- H. G. Sanchez Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, Angew. Chem., Int. Ed., 2014, 53(28), 7169–7172 CrossRef CAS PubMed.
- R. Arrigo, M. Hävecker, M. E. Schuster, C. Ranjan, E. Stotz, A. Knop-Gericke and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52(44), 11660–11664 CrossRef CAS PubMed.
- J. J. Velasco-Vélez, E. A. Carbonio, C. H. Chuang, C. J. Hsu, J. F. Lee, R. Arrigo, M. Hävecker, R. Wang, M. plodinec, F. R. Wang, A. Centeno, A. Zurutuza, L. J. Falling, R. V. Mom, S. Hofmann, R. Schlögl, A. Knop-Gericke and T. E. Jones, J. Appl. Chem. Sci., 2021, 143(32), 12524–12534 Search PubMed.
- A. Kolmakov, D. A. Dikin, L. J. Cote, J. Huang, M. K. Abyaneh, M. Amati, L. Gregoratti, S. Günther and M. Kiskinova, Nat. Nanotechnol., 2011, 6(10), 651–657 CrossRef CAS PubMed.
- J. J. Velasco-Vélez, V. Pfeifer, M. Hävecker, R. S. Weatherup, R. Arrigo, C.-H. Chuang, E. Stotz, G. Weinberg, M. Salmeron, R. Schlögl and A. Knop-Gericke, Angew. Chem., Int. Ed., 2015, 54(48), 14554–14558 CrossRef PubMed.
- S. Nemšák, E. Strelcov, H. Guo, B. D. Hoskins, T. Duchoň, D. N. Mueller, A. Yulaev, I. Vlassiouk, A. Tselev, C. M. Schneider and A. Kolmakov, Top. Catal., 2018, 61(20), 2195–2206 CrossRef.
- C. Arble, H. Guo, E. Strelcov, B. Hoskins, P. Zeller, M. Amati, L. Gregoratti and A. Kolmakov, Surf. Sci., 2020, 697, 121608 CrossRef CAS.
- T. Gupta, E. Strelcov, G. Holland, J. Schumacher, Y. Yang, M. B. Esch, V. Aksyuk, P. Zeller, M. Amati, L. Gregoratti and A. Kolmakov, ACS Nano, 2020, 14(10), 12982–12992 CrossRef CAS PubMed.
- Y. H. Lu, C. Morales, X. Zhao, M. A. Van Spronsen, A. Baskin, D. Prendergast, P. Yang, H. S. Bechtel, E. S. Barnard, D. F. Ogletree, V. Altoe, L. Soriano, A. M. Schwartzberg and M. Salmeron, Nano Lett., 2020, 20(9), 6364–6371 CrossRef CAS PubMed.
- R. Mom, L. Frevel, J. J. Velasco-Vélez, M. Plodinec, A. Knop-Gericke and R. Schlögl, J. Appl. Chem. Sci., 2019, 141(16), 6537–6544 CAS.
- L. J. Falling, R. V. Mom, L. E. Sandoval Diaz, S. Nakhaie, E. Stotz, D. Ivanov, M. Hävecker, T. Lunkenbein, A. Knop-Gericke, R. Schlögl and J. J. Velasco-Vélez, ACS Appl. Mater. Interfaces, 2020, 12(33), 37680–37692 CrossRef CAS PubMed.
- B. L. Henke, E. M. Gullikson and J. C. Davis, At. Data Nucl. Data Tables, 1993, 54(2), 181–342 CrossRef CAS.
- S. Thürmer, I. Unger, P. Slavicek and B. Winter, J. Phys. Chem. C, 2013, 117(43), 22268–22275 CrossRef.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3(7), 1159–1165 CrossRef CAS.
- J. A. Gauthier, C. F. Dickens, L. D. Chen, A. D. Doyle and J. K. Nørskov, J. Phys. Chem. C, 2017, 121(21), 11455–11463 CrossRef CAS.
- O. Kasian, S. Geiger, T. Li, J. P. Grote, K. Schweinar, S. Zhang, C. Scheu, D. Raabe, S. Cherevko, B. Gault and K. J. Mayrhofer, Energy Environ. Sci., 2019, 12(12), 3548–3555 RSC.
| This journal is © The Royal Society of Chemistry 2022 |