 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ surface analysis of palladium–platinum alloys in methane oxidation conditions†
Alexander I.
Large
 *a,
Roger A.
Bennett
b,
Tugce
Eralp-Erden
c and
Georg
Held
a
*a,
Roger A.
Bennett
b,
Tugce
Eralp-Erden
c and
Georg
Held
a
aDiamond Light Source, Harwell Campus, Didcot, UK. E-mail: alex.large@diamond.ac.uk
bDepartment of Chemistry, University of Reading, Reading, UK
cJohnson Matthey Technology Centre, Sonning Common, Reading, UK
First published on 29th April 2022
Abstract
Palladium and palladium–platinum foils were analysed using temperature-programmed near-ambient pressure X-ray photoelectron spectroscopy (TP-NAP-XPS) under methane oxidation conditions. Oxidation of palladium is inhibited by the presence of water, and in oxygen-poor environments. Pt addition further inhibits oxidation of palladium across all reaction conditions, preserving metallic palladium to higher temperatures. Bimetallic foils underwent significant restructuring under reaction conditions, with platinum preferentially migrating to the bulk under select conditions.
1 Introduction
Natural gas engines provide a cleaner alternative to traditional combustion engines, but emissions of unburnt potent greenhouse gas methane need to be abated with exhaust catalysts, which typically contain palladium or other precious metals on metal oxide supports. Palladium has been shown to be more active for methane oxidation than platinum, with a mixture of metallic and oxidised palladium generally required for optimal activity.1–8 Methane is the second largest contributor to radiative forcing after carbon dioxide.9 Recent international commitments made at COP26 show that reducing methane emissions is still a key component of climate change science. Countries worldwide are pledging to reduce methane emissions by 30% over the next decade, to achieve warming reduction of 0.2 °C by 2050.10Prior work in our research group focused on alumina-supported palladium and palladium–platinum nanoparticle catalysts.11,12 For complete oxidation of methane, the addition of platinum to palladium catalysts showed benefits under select reaction conditions. A key issue with palladium catalysts in methane oxidation is deactivation due to water, which is a core problem due to catalysts both operating in wet conditions, and producing water when active.13–16 Addition of platinum to palladium to form bimetallic Pd–Pt catalysts has shown activity and stability benefits across a range of oxidation reactions.17–19 Within methane oxidation, Pd–Pt alloying has often been a route to limit deactivation in wet conditions, and to improve long term catalyst stability.20–26
Near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) is a quantitative analysis tool for chemical surface states under reactive gas pressures of a few mbar. This enables operando studies of catalytic materials, to develop understanding of catalyst activity and deactivation. Over the past decade, it has become a core technique for understanding surface chemistry for heterogeneous catalysts.27–31
Analysis of “real” powder catalysts can however cause problems for NAP-XPS, such as low signal due to loadings of a few weight percent, and charging due to insulating supports. This either necessitates longer measurement times, or requires the use of smaller amounts of sample, resulting in less catalytic conversion data. Either of these is an undesirable result, given that many NAP-XPS instruments are at synchrotron facilities, where limited experimental time is available.
In this work, palladium, platinum and palladium–platinum foils were studied under methane oxidation conditions. Palladium and platinum foils have previously been studied for methane oxidation, with moderate activity observed over both.32 Due to the low metal surface area relative to nanoparticle catalysts, equivalent catalytic activity is not expected. Using metallic foils enabled us to use alloys of the same composition as the nanoparticles studied previously, but with much more intense signals in XPS, allowing for faster high quality measurements. This facilitates the use of temperature programmed NAP-XPS (TP-NAP-XPS). The palladium and platinum core levels are continually scanned in a reactive gas environment over the relevant temperature range. This ensures that the temperature ramp can still be fast enough (3.9 °C min−1) to get close to equivalent real catalyst testing ramps. It also provides sufficient temperature resolution in the data (5 °C per set of spectra) to accurately understand where changes are happening. Typically foils will show worse activity than nanoparticle samples relative to the amount of catalyst used, due to the much lower surface area. It is also important to note the structural differences between supported nanoparticles and metal foils used here. Nanoparticles for these reactions from our previous work typically had diameters of 1–10 nm, and were supported on alumina grains which are around 100 μm.12 The lack of support material could also limit the extent to the deactivation seen in wet conditions, due to prior studies showing migration of hydroxyls from the support to the metal.33
Despite the noted differences, this route still provides understanding of the behaviour of Pd and Pd–Pt systems under reaction conditions in a way that was not achievable using more realistic samples.
2 Experimental
Palladium foil (99.95%, 0.25 mm thickness) was supplied by Advent Research Materials Ltd. The foils studied here have not been characterised crystallographically, and prior experiments have shown the size of surface domains on polycrystalline Pd foils to vary significantly, over the range 10–100 μm, which provides a further variation from nanoparticle systems.34,35 Microscope images of the Pd foil are shown in Fig. S2 of the ESI.† These show a high degree of roughness and it can therefore be assumed that a variation of Pd sites is present at the surface. Palladium–platinum alloy foils (Pd65–Pt35 and Pd88–Pt12, both 0.30 mm thickness) were produced by Goodfellow Cambridge Ltd, by cold-rolling from alloy ingots. The ratios were selected to match previously tested nanoparticle catalysts of the same compositions.12 Measurements in multiple positions (with spot size of 0.1 mm × 0.1 mm) on individual samples suggest the composition was uniform across the samples both pre- and post-testing.35 Samples were cut, with 5 × 3 mm (±1 mm) pieces used for the experiments described below.All XPS data presented were collected in the Tea Pot chamber of the C branch of the B07 versatile soft X-ray beamline (VerSoX) at Diamond Light Source.35 For all TP-NAP-XPS experiments, a 400 lines per mm grating was used, with exit slit y-gap size of 25 μm. At the photon energy used throughout these experiments, 750 eV, the resolution of the beamline was 0.26 eV. Where depth profiling measurements were made, kinetic energies were kept consistent (at 415, 675 and 1500 eV) at both Pd 3d and Pt 4f. This was achieved by varying the photon energy used between 500 eV and 1835 eV. The IMFP for Pd 3d is 7.28 Å at 415 eV, 9.93 Å at 675 eV and 17.58 Å at 1500 eV. For Pt 4f the IMFP is 9.76 Å at 415 eV, 13.63 Å at 675 eV and 24.79 Å at 1500 eV.36
Prior to experiments, the foils were “cleaned” in the Tea Pot chamber in oxygen (2 mbar) to 550 °C to remove surface carbon. The chamber was then evacuated, and the sample cooled to 150 °C. This temperature is low enough to be below the relevant catalytic activity window, but high enough to ensure minimal carbon build up on the surface in vacuum (1 × 10−6 mbar). Sample temperature was measured throughout using a thermocouple mounted on the surface of the sample holder.
Core level XPS spectra (Pd 3d, Pt 4f, C 1s, O 1s, EF) were measured in vacuum at 150 °C to provide clean reference data before exposure to reaction conditions. Reaction mixtures of methane, oxygen and water were dosed at 150 °C, in the ratios and total pressures as shown in Table 1. Whilst gases were dosed, the only pumping on the chamber was via the differential pumping system of the analyser, through the analyser cone.
| Mixture | Label | CH4 | O2 | H2O | Pressure/mbar |
|---|---|---|---|---|---|
| Oxygen only | OO | — | 1 | — | 2.40 |
| Stoichiometric dry | SD | 1 | 2 | — | 0.33 |
| Stoichiometric wet low | SLW | 1 | 2 | 2 | 0.55 |
| Stoichiometric very wet | SVW | 1 | 2 | 100 | 5.20 |
| Oxygen rich dry | ORD | 1 | 120 | — | 2.90 |
| Oxygen rich wet | ORW | 1 | 120 | 100 | 5.20 |
Temperature-programmed near-ambient pressure X-ray photoelectron spectroscopy (TP-NAP-XPS) experiments were performed by repeatedly scanning Pd 3d and O 1s regions (and Pt 4f for bimetallic samples) whilst heating the foil in gas. The heating rate was constant throughout the experiments, at 3.5 °C min−1. This corresponds to one complete set of spectra every 5 °C. To compensate for the thermal expansion upon heating, the sample manipulator was moved away from the analyser in small steps at the start of each scan. Overall, this gave an approximately consistent background intensity before the Pd 3d5/2 peak throughout the temperature ramp, to allow for more reliable data analysis. The calibration for the manipulator motion was determined based on the sample motor x position during measurements at various temperatures during the initial cleaning stages for each sample (where the x direction motor moves the sample directly towards/away from the cone of the analyser). For the Pd foil, sample alignment at 150 °C and 500 °C showed a change in x position of −0.12 mm. Assuming a constant expansion rate of the sample over the time of the experiment, this would give a required movement rate of −0.000343 mm °C−1. With one set of spectra being recorded every 3.5 °C over the ramp, a step size of −0.0012 mm was used. The first two scans of a temperature-programmed experiment were collected at a fixed temperature (150 °C) to ensure everything was operating correctly before starting the temperature ramp. As a result, the starting x position was increased by double the step size, to ensure the position was correct at the start of the temperature ramp. The final temperature of the ramps varied from 420–590 °C, depending on the conditions. This variation is the result of Pd not always being fully oxidised by the initial maximum temperature of 420 °C. The spectra recorded during the TP-XPS experiments are stacked and presented in an image format, where signal intensity is plotted against both binding energy (x-axis) and temperature (y-axis), as shown in Fig. 1. This visualization method allows the temperatures at which transitions between different chemical states occur to be easily identified. Line profiles performed on this data in both axes provide further information. Line profiles collected over a range of around 10 °C provide relatively low-noise spectra from various stages during the ramp. Vertical line profiles allow changes in intensity at specific peak positions to be identified, showing clearly the temperature range over which states are stable or changing. The set of core level XPS spectra (Pd 3d, Pt 4f, C 1s, O 1s, EF) were measured again in the gas mixture to give the “post-reaction” reference, at the final temperature reached. The reaction gases were then pumped out, and the “regeneration” process was to heat in vacuum to 590 °C (where it was not already at this temperature) to reduce the Pd, before cooling back to 150 °C in vacuum for the following experiment.
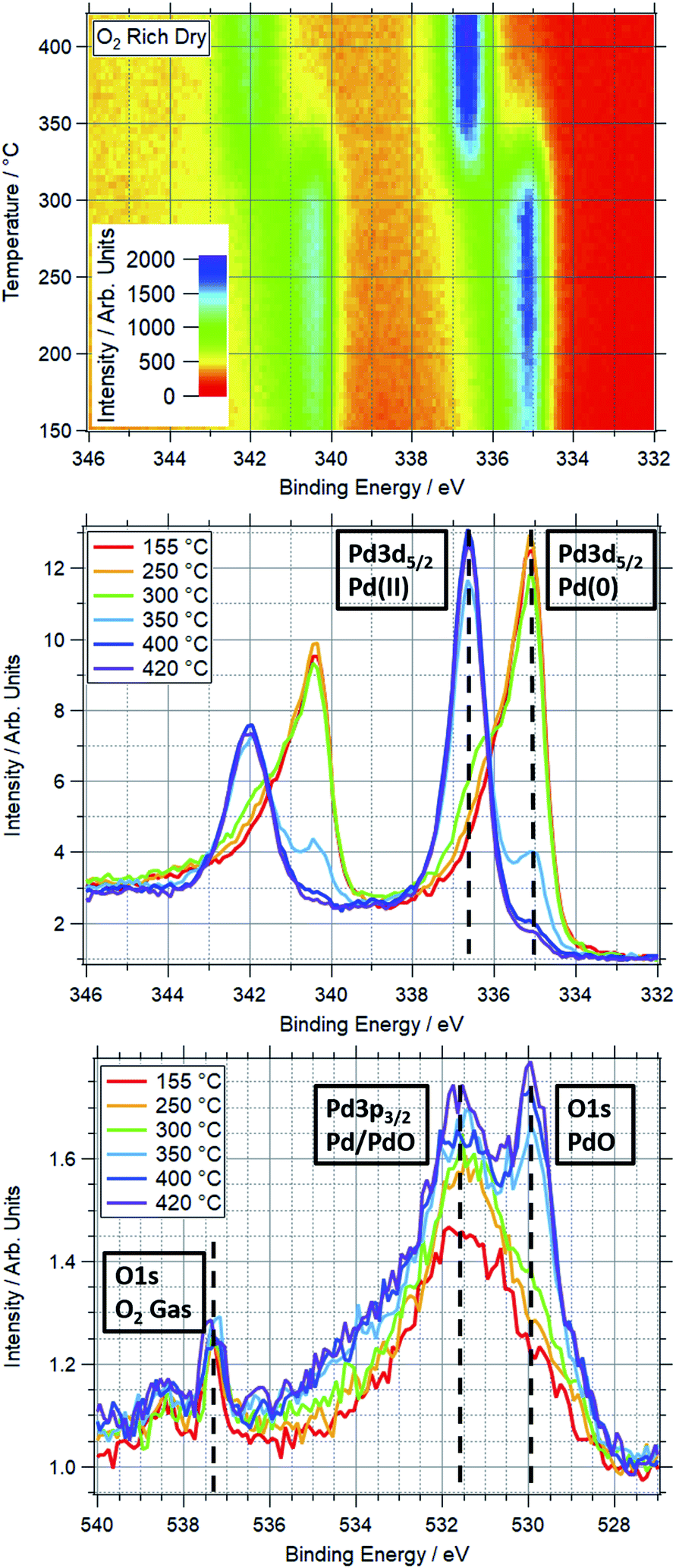 | ||
| Fig. 1 Top: TP-XPS plot covering the Pd 3d region for Pd foil in ORD conditions. Recorded with 750 eV photon energy. Energy scale corrected based on Fermi edge measured post-ramp. Middle: Pd 3d region XPS spectra taken during TP-AP-XPS ramp in ORD conditions. Each spectrum averaged over 10 °C, centred on indicated temperature. Vertical dashed lines show approximate peak positions of Pd(0) and Pd(II) components of Pd 3d5/2 peak. Unlabelled peaks on the left side of the spectra are the corresponding Pd 3d3/2 peaks. These spectra are normalised, hence scaling difference from top plot. Bottom: O 1s region XPS spectra taken during TP-AP-XPS ramp in ORD conditions. Each spectrum averaged over 10 °C at labelled temperature. Vertical dashed lines show approximate peak positions of PdO and O2 gas components of O 1s peak, and Pd/PdO component of Pd 3p3/2 peak. | ||
All Pd monometallic ramps were performed on a single Pd foil. This was due to the observed reducibility of the foil under high temperature vacuum conditions, meaning that a consistent starting point could be regenerated. All Pd–Pt bimetallic ramps were performed on one sample per Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt ratio, except for repeat experiments, where noted in the text. For each sample, all TP-XPS experiments were performed on a single sample spot in an attempt to ensure that the starting surface structure was the same for all gas conditions. Crystallographic measurements were not performed, and so the exact phase(s) of Pd and/or Pt measured in each experiment are unknown. This excludes the repeated ramps on the Pd88–Pt12 foil in stoichiometric dry conditions, which were performed on different pieces of foil.
:Pt ratio, except for repeat experiments, where noted in the text. For each sample, all TP-XPS experiments were performed on a single sample spot in an attempt to ensure that the starting surface structure was the same for all gas conditions. Crystallographic measurements were not performed, and so the exact phase(s) of Pd and/or Pt measured in each experiment are unknown. This excludes the repeated ramps on the Pd88–Pt12 foil in stoichiometric dry conditions, which were performed on different pieces of foil.
XPS cross-sections used throughout this experiment are taken or interpreted from the Elettra WebCrossSections website.37 All presented cross sections are in the units of Mbarn. Ratios are determined based on the normalised peak areas for Pd 3d and Pt 4f, along with the photoionization cross-sections for each line at the used energies. Where cross-section data were not available (i.e. for photon energies above 1500 eV) it was determined based on curve-fitting the available data and extrapolating to the relevant higher energies.
3 Results & discussion
3.1 Surface oxidation of Pd foils in reaction conditions
An example of a two dimensional TP-XPS plot is presented in Fig. 1, showing the Pd 3d XPS spectra across a temperature ramp in ORD conditions. The figure also shows individual Pd 3d and O 1s–Pd 3p XPS spectra (averaged over 10 °C) for various key temperatures to demonstrate in more detail the changes in these regions. In Fig. 2 we present the equivalent data set for the oxygen rich wet conditions. This shows the additional presence of the water gas phase peak in the O 1s spectrum (binding energy 534 eV), whilst the Pd states remain essentially unchanged. Changes in relative peak intensity for Pd 3d and O 1s are shown in Fig. S1 in the ESI.† For all other temperature ramps involving monometallic Pd, these data are compiled into a single plot in Fig. 3. This shows the relative intensities at the Pd(0) and Pd(II) Pd 3d5/2 peak positions (solid and dashed lines at approx. 335.2 and 336.7 eV respectively) for all gas conditions. In all cases, a transition between Pd(0) and Pd(II) is observed during the temperature ramp, as indicated by the relative changes in the peak intensity of the two species. The temperatures of this change increase with the order: oxygen rich dry (ORD) < oxygen only (OO) < stoichiometric dry (SD) < oxygen rich wet (ORW) < stoichiometric wet low (SLW) < stoichiometric very wet (SVW), with cross-over temperatures of approximately 320, 332, 346, 360, 376 and 429 °C respectively.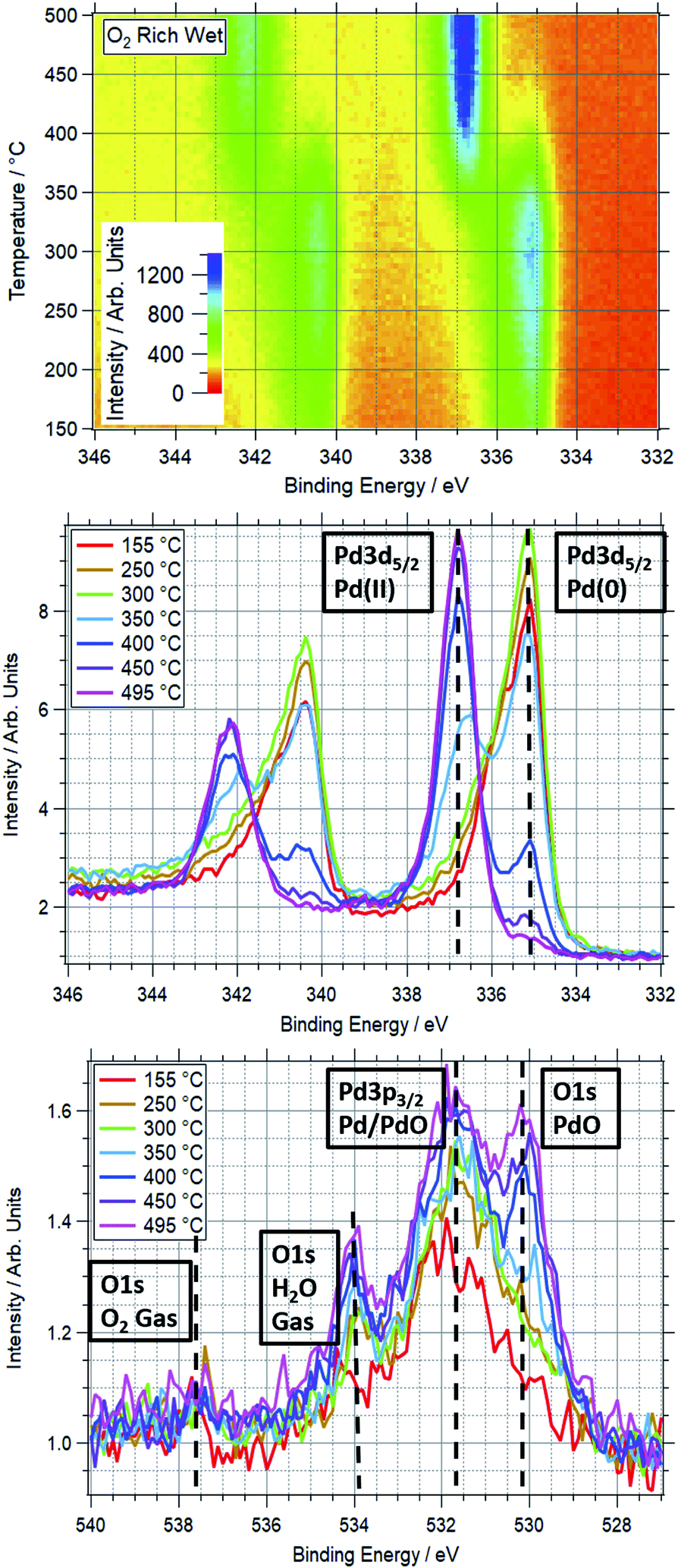 | ||
| Fig. 2 Top: TP-XPS plot covering the Pd 3d region for Pd foil in ORW conditions. Recorded with 750 eV photon energy. Energy scale corrected based on Fermi edge measured post-ramp. Middle: Pd 3d region XPS spectra taken during TP-AP-XPS ramp in ORW conditions. Each spectrum averaged over 10 °C, centred on indicated temperature. Vertical dashed lines show approximate peak positions of Pd(0) and Pd(II) components of Pd 3d5/2 peak. Unlabelled peaks on the left side of the spectra are the corresponding Pd 3d3/2 peaks. These spectra are normalised, hence scaling difference from top plot. Bottom: O 1s region XPS spectra taken during TP-AP-XPS ramp in ORW conditions. Each spectrum averaged over 10 °C at labelled temperature. Vertical dashed lines show approximate peak positions of PdO and O2/H2O gas components of O 1s peak, and Pd/PdO component of Pd 3p3/2 peak. | ||
 | ||
| Fig. 3 Intensity plots for Pd(0) and Pd(II) peak positions from Pd 3d XPS measurements in various reaction conditions. Annotated temperatures indicate the point at which the dominant Pd phase changes from reduced to oxidised. Intensity is totalled over a 0.5 eV range, with approximate peak centres of 335.4 and 336.9 eV for Pd(0) and Pd(II) respectively. All recorded with 750 eV photon energy. Within each condition, Pd(0) and Pd(II) intensities are offset by a consistent amount for clarity of presentation. | ||
In stoichiometric reaction conditions (SD, SLW, SVW, methane:oxygen 1:2, water varies), an increased water presence is shown to inhibit palladium oxidation. Without water (SD) the transition occurs around 346 °C, whilst with equal amounts of water and oxygen (SLW) this increases to around 376 °C, and further to 429 °C in excess water (SVW, methane:water 1:100).
When measured in OO, the Pd(0) to Pd(II) transition is seen to occur around 330 °C. In ORD conditions (with a similar oxygen partial pressure) this is reduced to around 315 °C. The only difference between these gas feeds is a small presence of methane (methane:oxygen 1:120). Given the ordinarily reducing nature of methane, the reduction in the oxidation temperature is not expected, as it means that Pd is more likely to oxidise in a more reducing gas mixture. When water is added to this feed (ORW), the transition temperature shifts to around 360 °C, an increase of 45 °C.
For equivalent amounts of water and methane, the inhibiting effect is much greater where there is a lower oxygen presence (+80 °C for stoichiometric, +45 °C for oxygen rich). Given that oxygen and water will be competing for surface sites, it is logical that the wet feed with less oxygen results in oxidation only occurring at higher temperatures.
Prior studies have consistently shown water presence to have a detrimental effect on methane oxidation activity over palladium catalysts.2,38,39 Here we have demonstrated that the presence of water consistently causes Pd to remain reduced at higher temperatures in reaction conditions. Similarly, our data show that higher oxygen partial pressures decrease the Pd oxidation temperature. This explains why these catalysts tend to perform better in more oxygen-rich conditions.
A platinum foil was also studied under SD conditions, matching those used for palladium. For Pt, no change was seen up to the maximum measured temperature of 550 °C. Based on peak positions, Pt remains metallic throughout the ramp, indicating Pt(0) is stable under the dry reaction conditions within the temperature window relevant to methane oxidation. An example is shown in Fig. 4, where the normalised spectra show no change in peak position or intensity between 155–530 °C. This shows the stability of the reduced Pt under a typical reaction condition, as compared to Pd, where the majority of the Pd is oxidised in these conditions below 420 °C.
 | ||
| Fig. 4 Top: TP-AP-XPS plot in Pt 4f region during a ramp in stoichiometric dry conditions over platinum foil. Bottom: combined, normalised Pt 4f XPS spectra taken over 10 °C regions at various temperatures. | ||
3.2 Surface oxidation and restructuring of Pd–Pt foils in reaction conditions
Fig. 5 and 6 show the 2-dimensional TP-XPS plots for Pd 3d and Pt 4f, along with line profiles on the Pd(0)/Pd(II) and Pt(0)/Pt(II) peak positions and selected spectra taken at points throughout this ramp. This particular example shows the spectra for OO conditions for a Pd88–Pt12 foil. All other ramps are summarised in Fig. 7 (Pd) and Fig. 8 (Pt) for Pd88–Pt12 and Fig. 9 (Pd) and Fig. 10 (Pt) for Pd65–Pt35. | ||
| Fig. 5 Various Pd 3d data for Pd88–Pt12 foil in OO. Top left: TP-XPS plot. Top right: Intensity line profiles for Pd(0) and Pd(II) peak positions. Bottom: Pd 3d spectra at select temperatures throughout the ramp, combined over a 10 °C range. | ||
 | ||
| Fig. 6 Various Pt 4f data for Pd88–Pt12 foil in OO. Top left: TP-XPS plot. Top right: intensity line profiles for Pt(0) and Pt(II). Bottom: Pt 4f spectra at select temperatures, combined over a 10 °C range. | ||
 | ||
| Fig. 7 Pd 3d data for Pd88–Pt12 under various conditions. Data represent line profiles at Pd(0) and Pd(II) peak positions throughout temperature-programmed ramp. Where oxidation is seen to more than 50%, the switchover temperature is quoted. | ||
 | ||
| Fig. 8 Pt 4f data for Pd88–Pt12 under various conditions. Data represent line profiles at Pt(0) and Pt(II) peak positions throughout temperature-programmed ramp. | ||
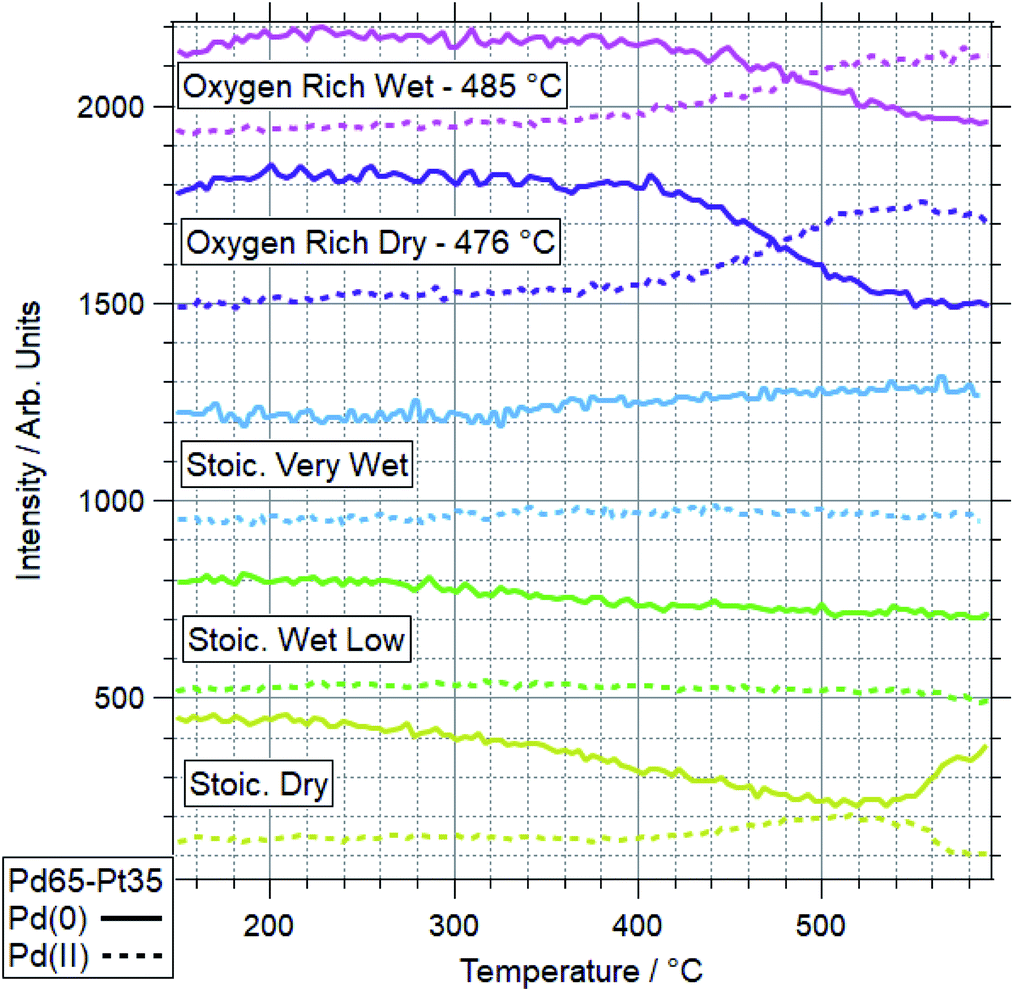 | ||
| Fig. 9 Pd 3d data for Pd65–Pt35 under various conditions. Data represent line profiles at Pd(0) and Pd(II) peak positions throughout temperature-programmed ramp. Where oxidation is seen to more than 50%, the switchover temperature is quoted. | ||
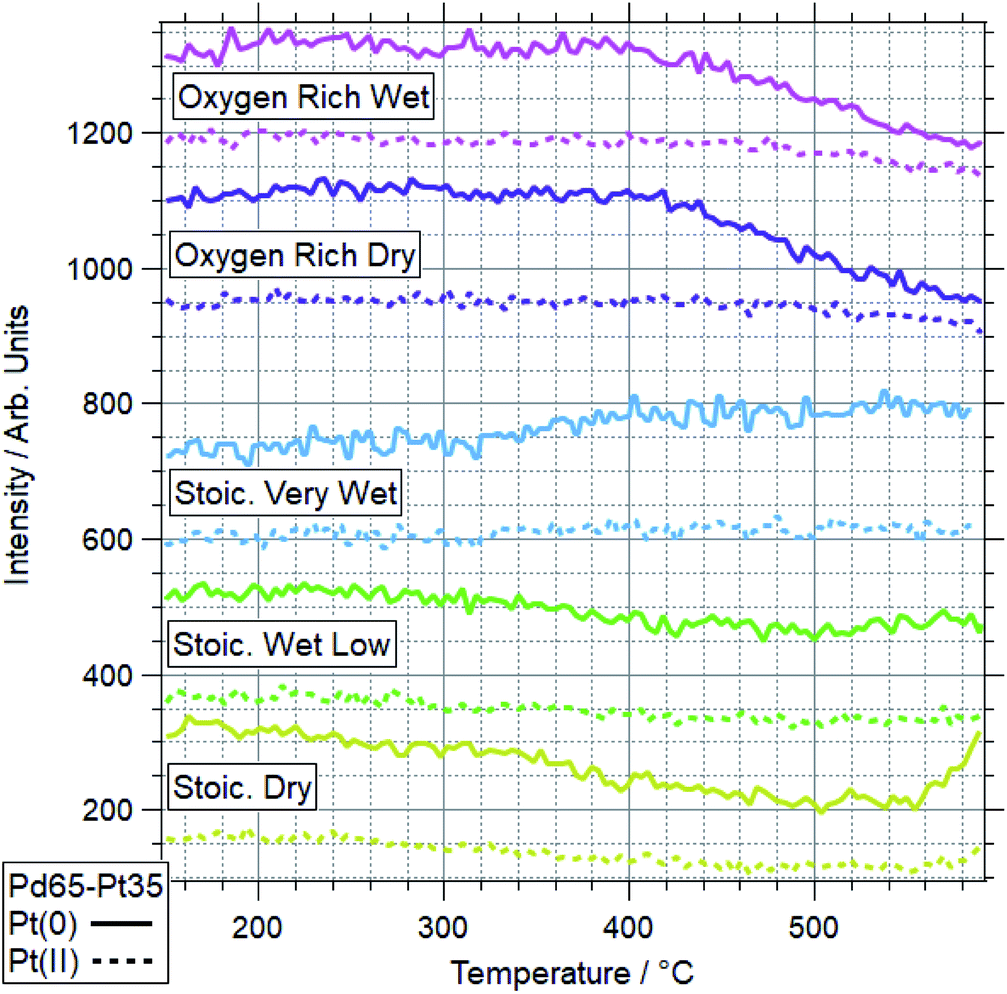 | ||
| Fig. 10 Pt 4f data for Pd65–Pt35 under various conditions. Data represent line profiles at Pt(0) and Pt(II) peak positions throughout temperature-programmed ramp. | ||
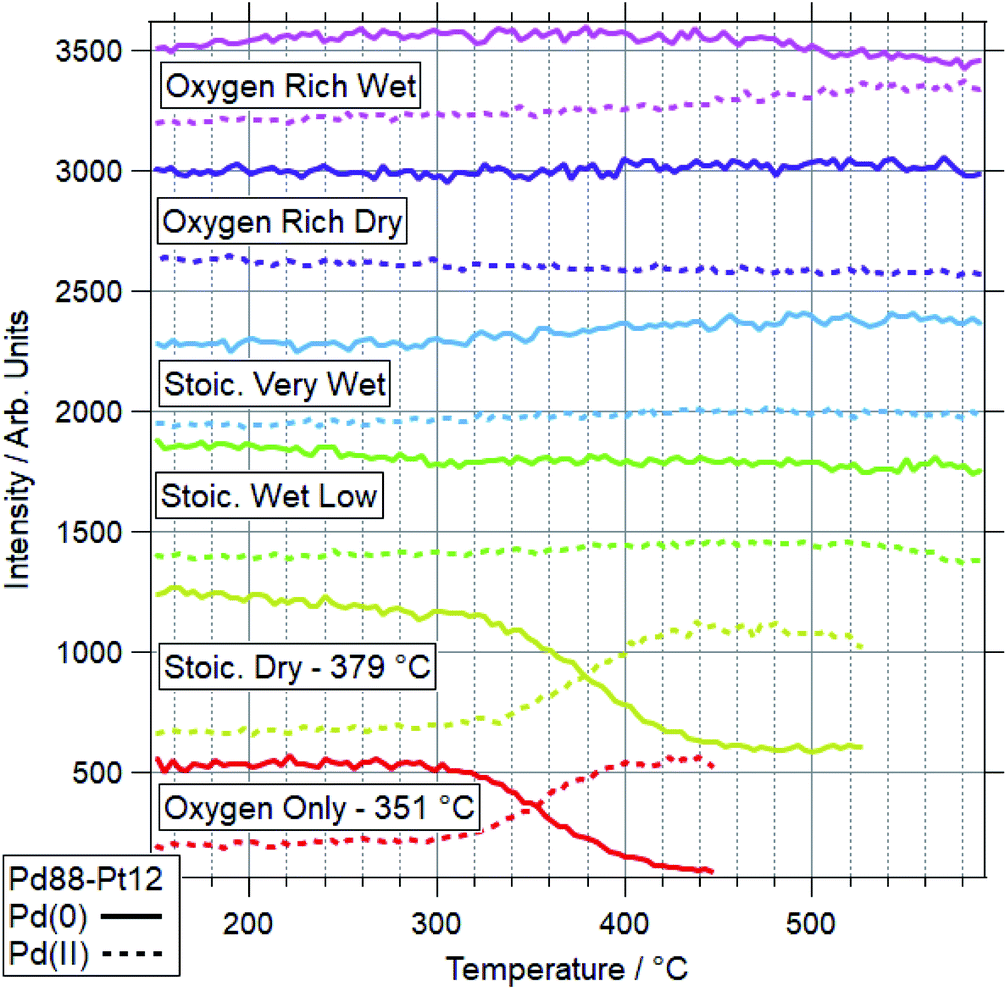
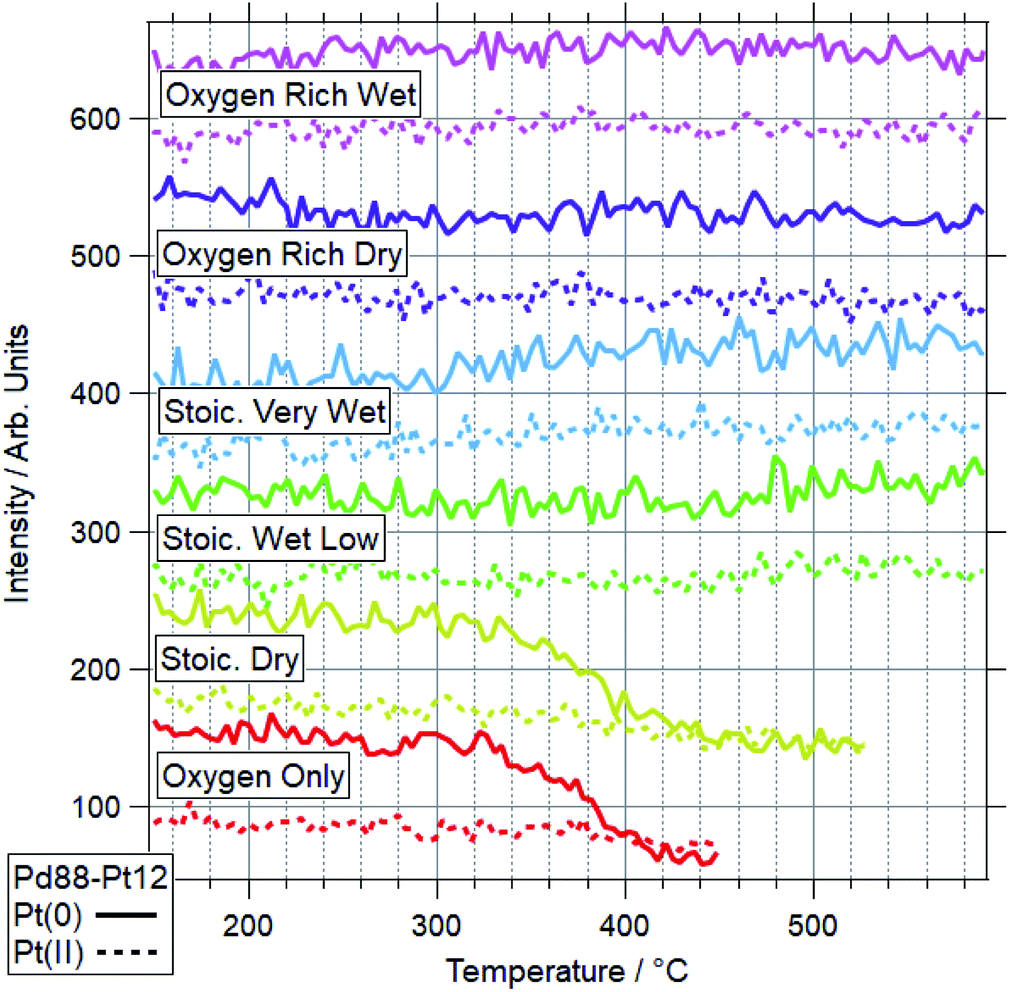
For Pd88–Pt12, only 2 conditions led to a majority of Pd(II) on the sample. These conditions were OO and SD. In both cases, the Pt(0) intensity decreases, with the Pt(II) intensity remaining the same. This shows Pt starting to migrate to the bulk, while the remaining surface Pt is partially oxidised. This platinum behaviour and the palladium oxidation begin from around the same temperature, indicating a link between the two. This suggests that the alloy is resistant to Pd oxidation whilst Pt remains well-mixed in the surface region. In both cases, the switch from Pd(0) to Pd(II) occurs at a higher temperature than for Pd foil. In all four other conditions, the dominant Pd species by 590 °C was still Pd(0). Only in the case of ORW conditions was significant oxidation seen, although the Pd remained mostly reduced. No change was seen in the Pt signal in this condition, indicating that there is likely to be a point of oxidation at which the migration of platinum occurs. With the experiments presented, the exact point of Pd oxidation at which Pt migration occurs is unclear. Significant changes in Pt(0) are generally seen at slightly higher temperatures than the start of the decrease of Pd(0), indicating the oxidation drives the migration as opposed to the inverse.
The more Pt-rich foil, Pd65–Pt35, shows some similarly varied oxidation behaviours in different conditions. There are still only two conditions which cause Pd to near-fully oxidise, ORD and ORW. These are different to the conditions which lead to oxidation for the more Pd-rich sample, Pd88–Pt12, however, for which neither oxygen rich condition caused full oxidation/restructuring. In both conditions for Pd65–Pt35, the same pattern of behaviour is seen as for the conditions in which Pd88–Pt12 changes, whereby the palladium oxidation is coupled to a reduction in intensity and partial oxidation of platinum. The temperatures at which this change occurs are much higher however, 351 and 379 °C for Pd88–Pt12, compared to 476 and 485 °C for Pd65–Pt35. In the SD gas mixture, the Pd starts to partially oxidise, up to around 550 °C, at which point it promptly reduces back to fully Pd(0) again. This is mirrored in the Pt signal, where the reduced Pt peak decreases in intensity, before sharply increasing above 550 °C. This is another example of the strongly linked behaviours of Pd and Pt in these alloy samples. It is also in direct contrast to the behaviour of Pd88–Pt12 in ORW conditions, where partial Pd oxidation was not coupled to any changes in Pt. Both SLW and SVW conditions saw no change in Pd oxidation or relative intensity over the ramp.
When comparing the Pd65–Pt35 to Pd88–Pt12, there are a number of unusual variations. The only real similarity is the coupled behaviour of Pd (oxidation) and Pt (partial oxidation plus migration). However, the conditions in which they occur, the temperatures at which they are promoted, and the behaviour seen where only partial oxidation of palladium happens all vary with composition. Pd88–Pt12 is more likely to undergo the changes at lower temperature, and does not change significantly in either oxygen rich gas feed (ORD/ORW). Pd65–Pt35 seems to be more oxidation resistant, only oxidising in oxygen rich feeds at very high temperatures, outside of the relevant window for methane oxidation (both 475 °C and above). The greater amount of Pt can be considered as a stabilizer for metallic Pd. Given the greater presence of Pt around Pd in the Pd65–Pt35, it is understandable why this may lead to some of the relative differences seen. It is unclear, based on the information above, why the Pd88–Pt12 is resistant to oxidation in oxygen rich conditions, given that rationally these should be more likely to induce oxidation than the stoichiometric dry condition in which it was seen to oxidise.
With some unanticipated variation in Pd oxidation behaviour in the bimetallic foils, select ramps were repeated for the Pd88–Pt12 sample. First (after a cleaning cycle) was the SD experiment, in which no Pd oxidation was seen up to the maximum temperature of 590 °C – when previously this oxidation had been seen below 400 °C. After this, the sample was exposed to an OO gas feed and another ramp performed. In this case partial oxidation occurred, but again the onset of this was at a higher temperature, with the Pd 3d spectra still showing significant presence of Pd(0) at 590 °C. Following the ramp in oxygen where partial Pd oxidation was seen, and another cleaning cycle, the SD ramp was repeated again. No change was seen from the previous ramp, with palladium remaining reduced throughout. The Pd 3d spectra taken at the end of the ramps in SD conditions are shown in Fig. 11. This variation in behaviour for ramps under identical conditions pointed to a problem with relying on heating in vacuum to regenerate the bimetallic samples to a reliable starting point.
 | ||
| Fig. 11 Pd 3d spectra from 3 separate ramps performed on a Pd88–Pt12 sample in SD gas conditions, at the maximum temperatures used for each (520 °C for A, 590 °C for B1 & B2). Test A is the original, as shown previously. Tests B1 and B2 are performed on a new sample. B1 is post-cleaning only, whilst B2 followed B1, an additional ramp in OO, and additional regeneration stages. | ||
The Pd:Pt ratios were determined after each “regeneration” stage at 150 °C in vacuum and at the highest temperature of the ramp for each gas condition. The ratios were calculated from the Pd 3d and Pt 4f peak areas weighted with the respective photo-excitation cross-sections at 750 eV photon energy (1.41 and 1.49 Mbarn, respectively). All ratios are summarised in Table 2, which also includes the percentage of the theoretical value that this relates to, and the effective resultant Pd:Pt ratio. These data are presented visually in Fig. 15. Each cleaning stage involved heating to 590 °C in vacuum (where the sample was below this temperature), maintaining for 2 minutes, before cooling back to 150 °C in vacuum.
:Pt ratio based on XPS data for Pd88–Pt12 and Pd65–Pt35 foils under a range of reaction conditions, along with the % of theoretical value this equates to, and the effective alloy composition at the point of that measurement. Theoretical, based on sample composition, and average, based on the set of tested conditions, included for reference. Measurements made at end temperature of ramp in gas (varies, 450–590 °C depending on gas mixture)
| Gas mixture | Pd88–Pt12 | Pd65–Pt35 | ||||
|---|---|---|---|---|---|---|
| Ratio | % of Th. | Eff. ratio | Ratio | % of Th. | Eff. ratio | |
| Theoretical | 7.76 | 100% | 88–12 | 1.97 | 100% | 65–35 |
| Oxygen only | 18.81 | 242% | 95–5 | N/A | N/A | N/A |
| Stoichiometric dry | 29.50 | 380% | 97–3 | 3.88 | 198% | 80–20 |
| Stoichiometric wet low | 11.50 | 148% | 92–8 | 3.15 | 160% | 76–24 |
| Stoichiometric very wet | 7.47 | 96% | 88–12 | 3.00 | 153% | 75–25 |
| Oxygen rich dry | 7.99 | 103% | 89–11 | 5.29 | 269% | 84–16 |
| Oxygen rich wet | 6.83 | 88% | 87–13 | 5.13 | 261% | 84–16 |
| Average | 13.68 | 176% | 93–7 | 4.09 | 208% | 80–20 |
The analysis of the Pd–Pt foil oxidation data reveals significant alloy restructuring. For the initial experiments on the Pd88–Pt12 foil, the expected Pd:Pt ratio using 750 eV photon energy is approx. 7.75:1. Before any reaction ramps had taken place, this ratio for the clean sample was measured as 10.85:1, confirming the composition in the near-surface region was more Pd-rich than expected. High resolution spectra taken in OO conditions showed a substantial increase to 18.81:1, with this remaining broadly the same at 18.77:1 after sample regeneration. Measurements in SD methane oxidation conditions showed the highest Pd:Pt ratio, at 29.50:1, which reduced to 22.71:1 after regeneration. This represents a significant increase in surface Pd compared to the previous regeneration step. The same sample was then exposed to SLW conditions, where the ratio reduced back to 11.50:1, much closer to the clean starting point for the foil. Regeneration resulted in a minor increase in Pd:Pt – unlike both previous phases where it had resulted in a decrease – to 12.64:1. Exposure to the SVW condition further decreased the Pd:Pt ratio to 7.47:1, the lowest observed ratio for this sample in any conditions. This is within 5% of the expected value for this sample. Given the low oxygen content and high water presence, this result is not surprising. The following regeneration step resulted in an increase up to 10.75:1. The ORD experiment followed, with another reduction in Pd:Pt, to 7.99:1, which again increased after regeneration, to 10.53:1. The final ramp performed on this sample was in ORW conditions, where the Pd:Pt ratio decreased to 6.83:1.
The analysis of all data shows a general trend that wet conditions result in a higher relative Pt concentration in the near-surface region than dry conditions, with Pt concentrations increasing with the relative water content of the gas feed. With regards to oxygen levels, a greater Pd:Pt ratio was generally observed in stoichiometric conditions rather than the oxygen rich conditions. Whilst this could imply that the oxygen was effectively pulling Pt from the bulk, the Pt peaks show it to be primarily reduced in these conditions. Regenerating stages after each ramp produced different variations in relative metallic ratios. Initially there was variation, with some increases and decreases each seen. From the SLW experiment onwards, the cleaning stages tended to revert the Pd:Pt to much closer to the expected value.
In only 2 experiments did the majority of the palladium end up oxidised by the end of the ramp, with these being the oxygen only and stoichiometric dry experiments. In both cases, the total Pt signal is seen to decrease, with a significant presence of oxidised Pt by the end of the ramps. The oxygen rich wet ramp also showed partial Pd oxidation, though the majority of it remained reduced. In this experiment, no oxidation of Pt was observed. In most of the cases where no palladium oxidation occurs, no significant change in Pd:Pt is observed during the ramps. At the extreme ratios measured, the variation is effectively between Pd87–Pt13 and Pd97–Pt3, representing a 10% change in percentage points for both Pd and Pt from the original expected composition.
The same set of experiments were performed on the Pd65–Pt35 foil, with the exception that the oxygen only ramp was omitted. The expected Pd:Pt ratio for this sample, adjusted for cross sections, is 1.97:1. Post the initial cleaning, this ratio was 3.41:1, much higher than the expected value, similarly to Pd88–Pt12. The initial ramp in ORW conditions caused an increase in Pd:Pt to 5.13:1, which is the opposite to the effect caused for the more Pd-rich alloy. Regeneration increased this further to 5.83:1. The SD experiment followed, with a decrease to 3.88:1, which increased to 4.22:1 after regeneration. The ORD ramp increased this to 5.29:1, and the following regeneration cycle caused a slightly relative increase in Pt, with a ratio of 4.99:1. The SLW experiment saw a significant decrease in ratio to 3.15:1, which increased again to 3.43:1 with regeneration. The final ramp was in SVW conditions, where the Pd:Pt decreased to 3.00:1, the lowest ratio of Pd:Pt for this sample after the initial cleaning stage. This was the lowest value achieved at any stage, but still more than 50% higher than the expected ratio, confirming that after any treatments this alloy was more Pd-rich in the near-surface region. In stoichiometric conditions, the increase of water led to a relative increase in the Pt present in this near-surface area, consistent with the Pd88–Pt12 foil.
As a result of the variations in identical conditions, additional experiments were performed on various Pd–Pt foils to determine the consistency and lasting effects of exposure to reaction conditions for samples of these compositions. This included depth-profiling XPS analysis across various spots on fresh, annealed and used foils, using kinetic energies of 415, 675 and 1500 eV. This variation in energies provides more surface and bulk sensitive measurements, to provide insight into the restructuring of alloys under reaction conditions.
Fig. 12 and 13 show the Pd:Pt ratios for Pd88–Pt12 and Pd65–Pt35 foils respectively. For each, data for a “new” (as received, untreated), “cleaned” (exposed to oxygen up to 600 °C) and “tested” (exposed to multiple reaction conditions, up to 600 °C) are shown, along with the theoretical value based on the photoionization cross-sections.
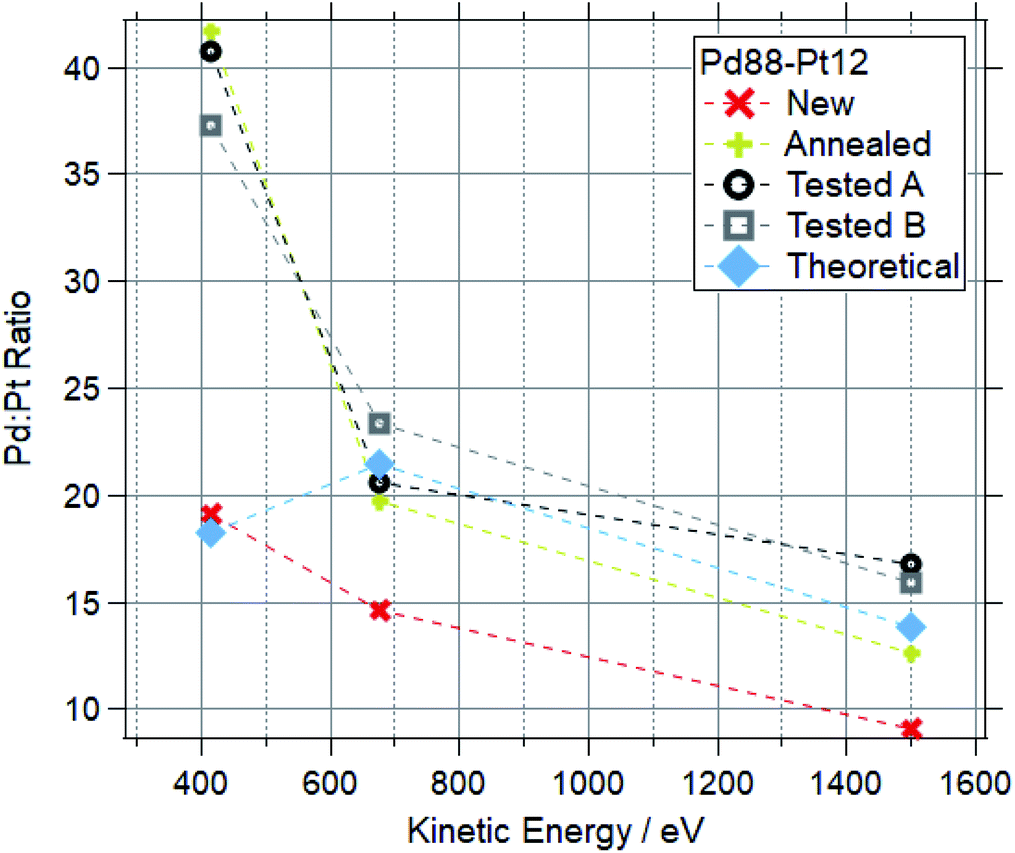 | ||
| Fig. 12 Pd:Pt ratios for Pd88–Pt12 samples at a range of kinetic energies after various treatments. | ||
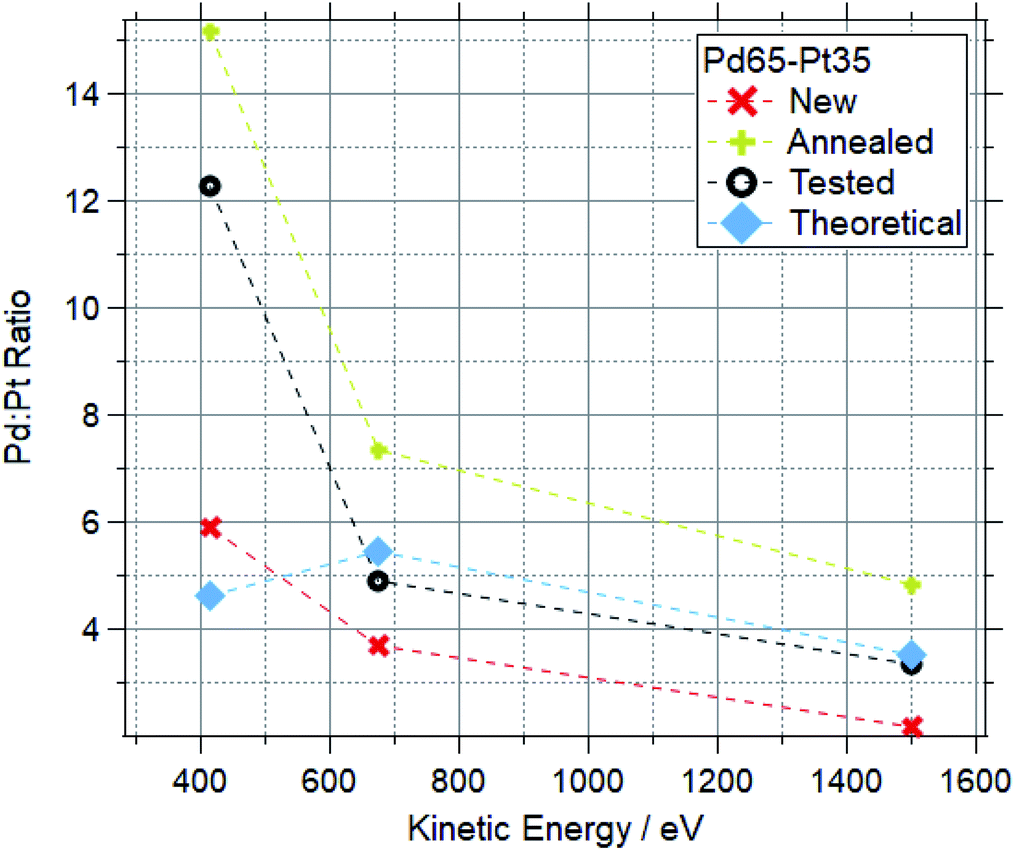 | ||
| Fig. 13 Pd:Pt ratios for Pd65–Pt35 samples at a range of kinetic energies after various treatments. | ||
For both compositions, the new foil is much more Pt rich at all measured photon energies, indicating that Pd is disproportionately buried in the bulk of the sample before treatments. For cleaned and tested samples, the more surface-sensitive measurements (415 and 675 eV kinetic energy) show a Pd-enrichment, to varying degrees. For both compositions, annealing and testing under various reaction mixtures caused an increase in Pd:Pt of around 100% in the most surface sensitive measurements (415 eV KE). The bulk changes are less extreme, with all Pd88–Pt12 samples retaining a Pd:Pt ratio between 12 and 18 when measured at 1500 eV kinetic energy. The bulk of the tested Pd65–Pt35 samples remains similar to the theoretical values, whilst annealing causes a Pd enrichment in the bulk.
To study the ratio change at different depths under heating, a Pd65–Pt35 sample was step-annealed in oxygen. Data are shown in Fig. 14. Throughout the ramp, the sample is more Pd-rich than the theoretical value at all measured depths. Between 150–450 °C the Pd:Pt ratio at 415 eV (KE) generally increases. The 675 eV scans show a smaller change, with a slight increase in relative Pt content. The most bulk sensitive spectra showed similarly small, but inconsistent changes. At 550 °C there is a clear increase in Pd:Pt at all measured depths. The low kinetic energy measurement was repeated for this temperature, approximately 1 hour after the initial measurements, and showed a significant increase in Pd:Pt ratio, from 20:1 to 71:1. The Pd 3d and Pt 4f spectra for the lowest 415 eV kinetic energy points are also shown here. This shows the gradual oxidation of palladium after 350 °C, coupled with the oxidation and decrease in intensity for Pt over the same range.
 | ||
| Fig. 14 Plots for Pd65–Pt35 foil in 2 mbar ramp in oxygen. Top left: Pd 3d region XPS data, all recorded at 750 eV photon energy, approx. 415 eV kinetic energy. Bottom left: Pt 4f region XPS data, all recorded at 490 eV photon energy, approx. 415 eV kinetic energy. Right: Pd:Pt ratios, as determined by NAP-XPS. Repeat measurement is approx. 1 hour after initial measurement. | ||
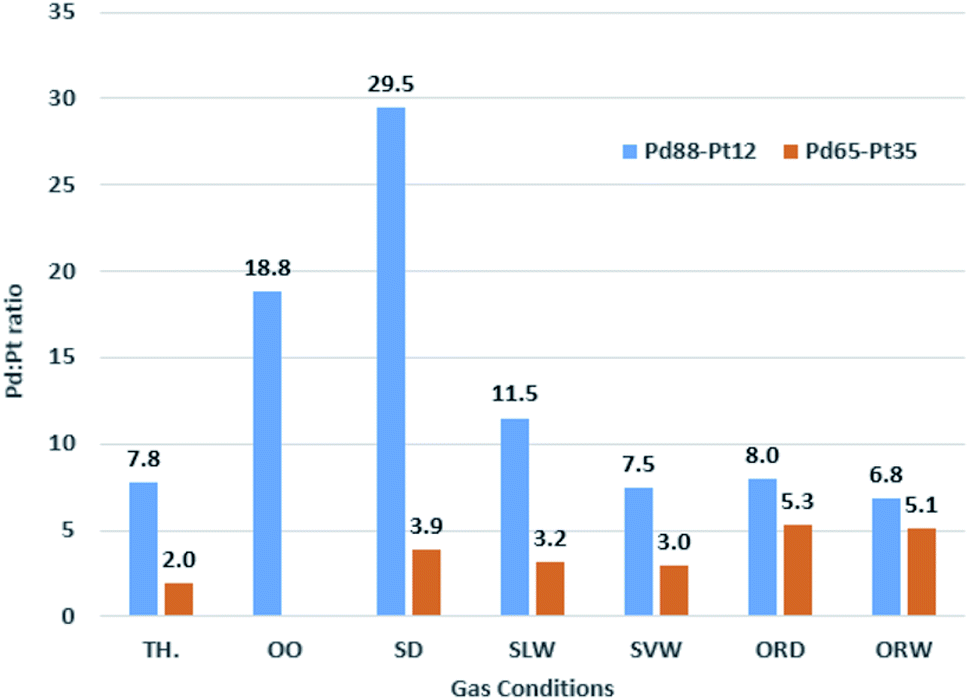 | ||
| Fig. 15 Pd:Pt ratios based on XPS data for Pd88–Pt12 and Pd65–Pt35 foils under a range of reaction conditions. Theoretical values (TH.) shown for reference. | ||
4 Conclusions
In this work we have demonstrated the effects of water and oxygen on the oxidation of palladium in Pd model catalysts under complete methane oxidation conditions. Palladium consistently underwent full oxidation from Pd(0) to Pd(II) under all tested conditions, with changes happening in the temperature range of 320–430 °C. Increased oxygen caused Pd oxidation at lower temperatures, whilst increased water inhibited the oxidation.Two bimetallic foils, Pd88–Pt12 and Pd65–Pt35, were also studied to understand previously seen benefits of bimetallic catalysts of these compositions. For these two samples, Pt inhibits Pd oxidation to varying extents. Pd88–Pt12 underwent oxidation of Pd only in pure oxygen, and dry stoichiometric reaction conditions, remaining reduced in all other tested conditions. Contrastingly Pd65–Pt35 only showed Pd oxidation in oxygen rich (dry and wet) conditions.
Under conditions where palladium does oxidise in Pd–Pt foils, platinum partially oxidises and migrates into the bulk of the foil. Depth profiling of bimetallic foils showed that the surface tends to be more Pd enriched, whilst the bulk measurements showed an increased Pt concentration. Annealing and testing in reaction conditions repeatedly caused the surface region to become much more Pd-rich than the average sample composition. Bimetallic foils which had been exposed sequentially to multiple reaction conditions showed relatively small differences in Pd:Pt ratio to those which had only been annealed, as compared to the fresh samples. Combining these results with prior studies which showed Pd–Pt catalysts were more stable long term than Pd equivalents, we suggest that the key to long term bimetallic catalyst stability is the retention of reduced Pd at the catalyst surface. Based on the simultaneous Pd oxidation and Pt migration observed throughout, we suggest that keeping Pd reduced is a result of Pd and Pt being well mixed in the surface region of the catalyst.
Author contributions
A. I. L was responsible for investigation, data analysis and writing. R. A. B. was responsible for investigation. T. E.-E was responsible for conceptualization. G. H. was responsible for conceptualization, investigation and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. I. L. acknowledges support from a studentship from Johnson Matthey and University of Reading. This work was carried out with the support of Diamond Light Source, instrument B07-C (proposals SI27121 and SI24584).Notes and references
- K. I. Muto, N. Katada and M. Niwa, Appl. Catal., A, 1996, 134, 203–215 CrossRef.
- R. Burch, F. J. Urbano and P. K. Loader, Appl. Catal., A, 1995, 123, 173–184 CrossRef CAS.
- P. Gélin and M. Primet, Appl. Catal., B, 2002, 39, 1–37 CrossRef.
- M. Monai, T. Montini, R. J. Gorte and P. Fornasiero, Eur. J. Inorg. Chem., 2018, 2018, 2884–2893 CrossRef CAS.
- J. G. Firth and H. B. Holland, Trans. Faraday Soc., 1969, 65, 1121–1127 RSC.
- J. G. McCarty, Catal. Today, 1995, 26, 283–293 CrossRef CAS.
- Y. Pan, C. Liu and P. Shi, Appl. Surf. Sci., 2008, 254, 5587–5593 CrossRef CAS.
- Y. H. C. Chin, M. García-Diéguez and E. Iglesia, J. Phys. Chem. C, 2016, 120, 1446–1460 CrossRef CAS.
- T. F. Stocker, D. Qin, G.-K. Plattner, M. M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, IPCC, 2013: Climate Change 2013: the Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013, pp. 1–1535 Search PubMed.
- U. D. of State, United States, European Union, and Partners Formally Launch Global Methane Pledge to Keep 1.5C Within Reach, 2021, https://www.state.gov/united-states-european-union-and-partners-formally-launch-global-methane-pledge-to-keep-1-5c-within-reach/ Search PubMed.
- R. Price, T. Eralp-Erden, E. Crumlin, S. Rani, S. Garcia, R. Smith, L. Deacon, C. Euaruksakul and G. Held, Top. Catal., 2016, 59, 516–525 CrossRef CAS.
- A. Large, J. Seymour, W. Quevedo Garzon, K. Roy, F. Venturini, D. C. Grinter, L. Artiglia, E. Brooke, M. B. De Gutierrez, A. Raj, K. R. Lovelock, R. A. Bennett, T. Eralp-Erden and G. Held, J. Phys. D: Appl. Phys., 2021, 54, 174006 CrossRef CAS.
- C. F. Cullis, T. G. Nevell and D. L. Trimm, J. Chem. Soc., Faraday Trans. 1, 1972, 68, 1406–1412 RSC.
- R. Burch and F. J. Urbano, Appl. Catal., A, 1995, 124, 121–138 CrossRef CAS.
- J. C. Van Giezen, F. R. Van Den Berg, J. L. Kleinen, A. J. Van Dillen and J. W. Geus, Catal. Today, 1999, 47, 287–293 CrossRef CAS.
- F. Zhang, C. Hakanoglu, J. A. Hinojosa and J. F. Weaver, Surf. Sci., 2013, 617, 249–255 CrossRef CAS.
- H. Zhang, M. Jin, H. Liu, J. Wang, M. J. Kim, D. Yang, Z. Xie, J. Liu and Y. Xia, ACS Nano, 2011, 5, 8212–8222 CrossRef CAS PubMed.
- Y. Han, J. Kim, S. U. Lee, S. I. Choi and J. W. Hong, Chem. – Asian J., 2020, 15, 1324–1329 CrossRef CAS PubMed.
- A. P. Wong, E. A. Kyriakidou, T. J. Toops and J. R. Regalbuto, Catal. Today, 2016, 267, 145–156 CrossRef CAS.
- K. Narui, H. Yata, K. Furuta, A. Nishida, Y. Kohtoku and T. Matsuzaki, Appl. Catal., A, 1999, 179, 165–173 CrossRef CAS.
- K. Persson, A. Ersson, S. Colussi, A. Trovarelli and S. G. Järås, Appl. Catal., B, 2006, 66, 175–185 CrossRef CAS.
- A. I. Osman, J. K. Abu-Dahrieh, F. Laffir, T. Curtin, J. M. Thompson and D. W. Rooney, Appl. Catal., B, 2016, 187, 408–418 CrossRef CAS.
- G. Lapisardi, L. Urfels, P. Gélin, M. Primet, A. Kaddouri, E. Garbowski, S. Toppi and E. Tena, Catal. Today, 2006, 117, 564–568 CrossRef CAS.
- S. A. Yashnik, Y. A. Chesalov, A. V. Ishchenko, V. V. Kaichev and Z. R. Ismagilov, Appl. Catal., B, 2017, 204, 89–106 CrossRef CAS.
- R. Abbasi, G. Huang, G. M. Istratescu, L. Wu and R. E. Hayes, Can. J. Chem. Eng., 2015, 93, 1474–1482 CrossRef CAS.
- P. Xu, Z. Wu, J. Deng, Y. Liu, S. Xie, G. Guo and H. Dai, Chin. J. Catal., 2017, 38, 92–105 CrossRef CAS.
- H. Ali-Löytty, M. W. Louie, M. R. Singh, L. Li, H. G. Sanchez Casalongue, H. Ogasawara, E. J. Crumlin, Z. Liu, A. T. Bell, A. Nilsson and D. Friebel, J. Phys. Chem. C, 2016, 120, 2247–2253 CrossRef.
- W. C. Chueh, A. H. McDaniel, M. E. Grass, Y. Hao, N. Jabeen, Z. Liu, S. M. Haile, K. F. McCarty, H. Bluhm and F. El Gabaly, Chem. Mater., 2012, 24, 1876–1882 CrossRef CAS.
- D. A. Svintsitskiy, T. Y. Kardash, O. A. Stonkus, E. M. Slavinskaya, A. I. Stadnichenko, S. V. Koscheev, A. P. Chupakhin and A. I. Boronin, J. Phys. Chem. C, 2013, 117, 14588–14599 CrossRef CAS.
- M. F. Lichterman, S. Hu, M. H. Richter, E. J. Crumlin, S. Axnanda, M. Favaro, W. Drisdell, Z. Hussain, T. Mayer, B. S. Brunschwig, N. S. Lewis, Z. Liu and H. J. Lewerenz, Energy Environ. Sci., 2015, 8, 2409–2416 RSC.
- L. Lukashuk, N. Yigit, R. Rameshan, E. Kolar, D. Teschner, M. Hävecker, A. Knop-Gericke, R. Schlögl, K. Föttinger and G. Rupprechter, ACS Catal., 2018, 8, 8630–8641 CrossRef CAS PubMed.
- M. Aryafar and F. Zaera, Catal. Lett., 1997, 48, 173–183 CrossRef CAS.
- P. Velin, M. Ek, M. Skoglundh, A. Schaefer, A. Raj, D. Thompsett, G. Smedler and P. A. Carlsson, J. Phys. Chem. C, 2019, 123, 25724–25737 CrossRef CAS.
- D. Vogel, C. Spiel, M. Schmid, M. Stöger-Pollach, R. Schlögl, Y. Suchorski and G. Rupprechter, J. Phys. Chem. C, 2013, 117, 12054–12060 CrossRef CAS PubMed.
- G. Held, F. Venturini, D. C. Grinter, P. Ferrer, R. Arrigo, L. Deacon, W. Q. Garzon, K. Roy, A. Large, C. Stephens, A. Watts, P. Larkin, M. Hand, H. Wang, L. Pratt, J. J. Mudd, T. Richardson, S. Patel, M. Hillman and S. Scott, J. Synchrotron Radiat., 2020, 27, 1153–1166 CrossRef CAS PubMed.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1991, 17, 911–926 CrossRef CAS.
- Atomic Calculation of Photoionization Cross-Sections and Asymmetry Parameters, https://vuo.elettra.eu/services/elements/WebElements.html Search PubMed.
- A. Raj, Johnson Matthey Technol. Rev., 2016, 60, 228–235 CrossRef CAS.
- N. Sadokhina, G. Smedler, U. Nylén, M. Olofsson and L. Olsson, Appl. Catal., B, 2017, 200, 351–360 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1fd00113b |
| This journal is © The Royal Society of Chemistry 2022 |